introduction 1.1: structure of hemoglobin
TRANSCRIPT

1
00
1. INTRODUCTION 1.1: Structure of Hemoglobin
Hemoglobin contains two pairs of unlike polypeptide chains, one chain of each pair is α
or α - like chain while the other is non α (β, γ or δ ) chain. α chains of all human
hemoglobins are alike. Non - α chains include β chains of normal adult hemoglobin
(α2 β2), γ chains of fetal hemoglobin (HbF) α2 γ2 and δ chains of hemoglobin A2 (Helen
and Sharma 2001).
1.2: Globin gene clusters:
Genes that regulate the synthesis and structure of different globins are organized in two
separate clusters, the α or α - like globin genes and β and β allied globin genes.
1.3: α globin genes cluster:
α - like globin genes are encoded on chromosome 16 and are found in the order
5’ - ζ - ψζ - ψα2 - ψα1 - α2 - α1-θ - 3’. The α Globin gene cluster occupies a region of
70 kilobases close to the short arm of chromosome 16 band p13.3. The CAP site of ζ
gene is designated 0. α2 gene lies 20 kb away from the ζ - gene on the centromeric
side, a further 3.7 kb away. 40kb upstream of the ζ globin gene lies HS (Hypersensitive
sites) which is the α globin gene regulatory elements. In addition to the three functional
α - like genes, the cluster also (Bunn 1986) contains three pseudogenes (ψζ,ψα2 and
ψα1) and gene θ (Clegg 1987). α globin gene cluster lies between 170 and 430 kb from
the telomere (Flint et al 1997)(Fig.1.1).

2
Fig: 1.1. Organization of human α and β- globin gene clusters on chromosome 16 and 11.The genetic control of various embryonic, fetal and adult haemoglobins is also shown. Hatched boxes in the individual genes represent 3’ and 5’ untrasnlated regions, black boxes represent exons and the white boxes represent intervening sequences (IVS) (Weatherall 1987).
1.4: Normal Structural Variation
From the 16p telomeric repeats there are four polymorphic, subtelomeric alleles in which
α - globin genes lie 170kb (A), 245kb (D), 350 kb (B) and 430kb (C) (Wilkie et al 1991a
and Higgs et al 1993).
As a result of unequal genetic exchange, normal persons may have 4,5 or 6 α - globin
genes (Goossens et al. 1980; Higgs et al, 1980: Galanello et al . 1983; Gu et al 1987) and
2,4,5 or 6 ζ like genes (Winichagoon et al 1982; Felice et al . 1986; Trent et al 1986 and
Titus et al 1988).
Around the α - globin locus 10 variable number of tandom repeats (VNTRs) have been
identified (Flint et al 1997). They produce highly polymorphic segments of cluster and
can be used as genetic markers throughout the genome and have been used to produce
individual – specific finger prints (Jeffreys 1987 and Fowler et al 1988). In α - globin

3
gene, cluster restriction fragment length polymorphisim is produced by a large number of
single base – polymorphic sites (Higgs et al 1986)
1.5: β – globin gene cluster
β – globin gene cluster is on the short arm of chromosome 11 (Fritsch et al 1980; Spritz
et al 1980, Baralle et al 1980, Slightom et al 1980). The β - like globin gene cluster
consist of the embryonic ε gene (Baralle et al 1980), duplicated γ – globin gene
(Slightom et al 1980) and δ (Spritz et al 1980) and β – globin genes (Lawn et al: 1980).
The two γ – globin genes are identical except at codon 136, where the Gγ gene contains a
glycine and the Aγ gene an alanine residue unequally expressed during fetal development
(Alter 1979). γ genes are duplicated one codes for glycine Gγ and the other for alanine
Aγ (Fig. 1.1).
The two fetal γ genes lie 15 and 20 kb downstream from the embryonic ε gene, while the
δ and β genes are 35 and 43 kb further downstream (Weatherall and Clegg 2001b) Locus
control region (LCR) is the regulatory region that is essential for the expression of all the
genes in the complex. It is present upstream of the ε gene and spans ∼ 15kb. (Weatherall
and Clegg 2001a) it contains four elements, HS1 to HS 4.
β, γ, δ or ε chains have 146 amino acid; valine and histidine are at the beginning of β
genes while Tyr β 145 and His β 146 at the c- terminal residues. δ chain differs from β
chain only 10 residues γ chains differ by 39 residues. β gene cluster contains a series of
single – point RFLP (Jaffrey 1987 and Antonarakis et al 1982).

4
Globin gene structure, function and regulation
Globin gene contains three trans regions called the exons which are separated by two
introns or intervening sequences (IVS) of variable length. Transcription begins at Cap
site, immediately after this is the promoter region that consists of 100 base– pairs. Three
short sequences within this region bind RNA polymerase that catalyzes messenger RNA
synthesis (Dierks 1983).
Two sequences are important for the initiation of gene transcription, these are called
TATA box and CAT box. Mutations involving these sequences reduce enzyme binding
and thereby limit mRNA transcription. AATAAA is the sequence present downstream
from the third exon, it tiggers the enzyme process that cuts mRNA at an appropriate point
and terminates gene transcription (Richard, L.et al 1993).
Two other promoter elements, CCAT box and CACC homology α box on the upstream
from CAP site are also required for optimal transcription (Weatheral and Clegg 2001b).
From the Cap site the first exon encompasses ∼ 50 bp of 5’ untranslated sequences (UTR)
and codons for amino acids 1- 31 in α and 1- 29 together with two bases of codon 30 in
the β- globin gene. Exons 2 encode amino acids 32 – 99, the portions of the globin
polypeptide that is involved in haem binding and amino acids 31 – 104 that is α1 β2
( α2β1) contacts.
Remaining amino acids 100 – 141 for α, 105 – 146 for β and 3’ untranslated region of
100 bp are encoded on exon 3. The IVSI intron varies in length from one allele to
another. Intervening sequences are removed from the initial transcript and the exon
sequences are joined with mRNA. This process is dependent on sequences at the border
between the exons and introns which are

5
( ) AG/ GT ( ) AGT at the 5’ end and ( ) N ( ) AG /G at the 3’ end of intron. GT and
AG dinucleotides are maintained in all cases, mutation in these sequences frequently
leads to thalassemia as reported by (Weatherall 2001b).
1.6: Globin gene expression
Hemoglobin synthesis is heterogeneous at all stages of development. It is confined to
yolk sac in the embryo, where hemoglobin Gower 1 (ζ2ε2) Gower 2 (α2ε2) and Portland
(ζ2γ2) are produced at 5 weeks gestation the ζ/ζ + α chains synthesis ratio is 0.82 ± 0.04.
By the 6th week of gestation it declines to 0.03 during development there is transition to
fetal hemoglobin (HbF, α2γ2) to adult hemoglobin HbA1 (α2β2) and HbA2 (α2δ2)
(Peschle et al 1985). α - globin gene expression remains constant throughout life.
Whereas the protein products of α1 and α2 genes are identical, however the steady state
level of α2 – mRNA predominates over the α1 – mRNA by approximately 3:1 (Liebhaber
et al 1986). β- gene is expressed in yolk sac cells and stays at a steady level throughout
development. Presence of a β gene downstream may be important in suppressing γ gene
expression (Behringer et al 1990 and Enver et al 1990). γ gene expression is
incompletely switched off in adults (Lloyd et al . 1992; Roberets et al 1997a and
Stamatoyanopoulos et al 1997). A sindicated in a study CH haplotypes are useful genetic
determinants for beta-thalassemia major and intermedia patients, while the 3'HS1 (+179
C →T) mutation may have functional consequences in gamma-globin genes expression
(Papachatzopoulou et al 2007). It has been reported recently that cAMP-dependent
pathway, the activity of which is augmented by multiple cytokines, plays a role in
regulating HBG expression in beta-thalassaemia (Bailey et al 2007).
C T
AG
CT
CT

6
1.7: Temporal Control:
Expression of globin genes is sequential during fetal development. In the early embryo
erythropoiesis mainly takes place in the yolk sac. It shifts to the liver in fetal life and then
to the bone marrow in the late prenatal and postnatal life (Weatherall and Clegg 1981).
More than 20kb 5’ to the ε globin gene, cis – activating erythroid specific DNAase – I
Hypersensitive sites are present (Tuan et al 1985). This region confers a high level,
position independent expression of like globin genes (Grosveld et al 1987). This
cis – acting sequences responsible for this effect are called Locus control region (LCR).
Orkin (1982) and Behringer et al (1990) postulated that temporal regulation of β –like
globin genes results from competition between embryonic fetal and adult globin genes for
interaction with a common LCR.
It was described that trans- acting factors like GATA binding protein, synthesized in the
yolk sac, fetal liver and bone marrow may bind to a DNA sequence motif (T/A) GATA
(A/G) present in ε, γ and β-globin promoters for the order by expression of the respective
genes (Orkin 1982) expression. GATA – 1 and GATA -2 have been shown to be essential
(Shivdasani and Orkin 1996) for the transcriptional control of erythroid specific gene.
LCR for α - globin cluster has been suggested in the sequences upstream from the ζ-
globin gene (Higgs et al, 1990). Addition of a poly (A) tract at the 3’ end of the mRNA is
involved in the processing and stability of mRNA. A poly (A) additional signal,
AAUAAA is conserved in the 3’untranslated region of the RNA approximately10 – 30
nucleotides upstream of where the initial transcript is cut and the poly (A) additional tract
is added. In only θ genes this signal sequence is AGUAA.

7
1.8: Regulation of globin – gene function
Regulatory sequence for the globin genes include the promoters series of enhancer
elements on a master regulatory region
1.8.1: Promotors:
DNA sequences present upstream of transcriptional start sites where the transcription
complex including RNA polymerase binds are called promotors. The first 5’ untranslated
sequences is the TATA and CCAAT homology boxes found 30 and 70 bp upstream of
mRNA CAP site (Anagnou et a. 1985; Myers et al 1986; de Boer et al, 1988. and
Antoniou and Grosveld 1990) and are critical for correct siting of initiation and high level
of transcription CCAAT site is duplicated in two γ – globin genes, both are necessary for
maximum rates of initiation. 90pb upstream from the initiation site the GGGGYG (Y: a
pirimidin nucleotide) or the invertal type “CRCCC” (R: a purine nucleotide) (Collins and
Weisman1984). Low levels of transcription of the δ – globin is partially due to the
modification of CCAAT sequence to CCAAC. Many erythroid specific genes have in
addition CACCC box in the promoter upstream of the CCAAT box. CACCC box
homologies are found in most of the β – gene promoters but are not found in the
promoters of δ and α - genes (Donz et al 1996).
More distal regions of the promoters of these genes includes GATA – 1 & NF – E2 for
erythroid transcription factors and site for the ubiquitous factors YY1, Sp1 and Oct – 1.
Though not fully understood, these factors are necessary for maximum rate of
transcription (Fig: 1.2).

8
1.8.2: Enhancers
In addition to the promoter sequences more distal sequences are also present. These
sequences increase the level of gene transcription and are called “enhancers”. These
enhancers may lie 5’ or 3’ to the gene or within the gene itself. These include the
regulatory region of α - cluster HS – 40 and elements β LCR HS2. A small region of 800
bp lying 3’ of the Aγ gene (Bodine & Ley 1987; Purucker et al .1990; Balta et al 1994)
and two segments of β – globin gene, one in the large intervening sequences and one 3’ to
the gene (Behringer et al 1987; Kollias et al 1987) have enhancing properties. No effect
of Aγ enhancers loss is observed (Liu et al 1998) while deletion of enhancer to 3’ to gene
significantly reduces the expression of the β gene in this system (Liu et al 1997). A 30 to
100 kb 3’ to the β cluster, additional enhancer sequence are identified 3’ deletion of 3’
breakpoint causes Hereditary Persistence of Fetal Hemoglobin (Feing E.A & Forget
1989, Anagnou et al 1995)
1.8.3: β - globin locus control region
Control of β - globin gene resides in the locus control region (LCR) which consists of
five DNase – hypersensitive sites that lie upstream of β - globin genes (Talbot et al 1989;
Collins et al 1990).
1.8.4: Transcription
The multi protein complex required for transcription of globin genes includes an enzyme
RNA polymerase II. Which transcribes DNA into a mRNA copy. Some of the
transcription factors involved in the regulation of erythropoiesis and globin synthesis are

9
GATA – 1, FOG (Friend of GATA – 1), NF – E 2, EKLF SSP (Weatherall 2001b)
(Fig: 1.2).
1.8.5: RNA processing
Primary RNA transcription of globin gene has a half life of about 5 minutes and requires
further processing including capping and sequencing (Fig: 1.2).
1.8.6: Capping
Addition of 7 – methylguanosine residue at 5’ end of mRNA is known as capping. It
prevents the exonneucleulytic degradation of nascent transcript and in ribosomal binding
to mRNA during translation (Weatherall 2001b).
1.8.7: Poly(A) addition
3’ end of the primary transcript is cleaved at a specific point that is usually 10 – 25
nucleotides downstream of a highly conserved AAVAA motif. This is followed by the
addition of 200 – 300bp long tract of poly (A) residue. Addition of Poly (A) ensures the
stability of mRNA (Weatherall 2001b).
1.8.8: Translation
Transcription of the globin gene is initiated at the “Cap Site” which is located 50bp
upstream of the initiation codon (AUG). As transcription proceeds, exons and introns are
included and extends well beyond the highly conserved 3’ AATAA polyadenylation site
(Collins and Weisman 1984) (Fig: 1.2).

10
Fig 1.2: Typical mammalian gene and steps entailed in its transcription and translation. Exons are shown in black and introns (intervening sequence, IVS) unshaded. Regions of gene which code for untranslated portions of messenger RNA are indicated as NC (non-coding regions). Position of 5' regulatory boxes are indicated (Weatherall 1987).
1.8.9: Splicing
Removal of intervening sequences is carried out initially by cleaving of 5’ splice site after
a nucleophilic attack by 2’OH group on an A residue 10 – 60 nucleotides upstream of 3’
acceptor site. This forms a 2’ – 5’ phosphodiester bond to produce a ‘lariant’ structure
containing the intron and the 3’ exon. 3’ OH group now attacks 3’ splice site and joins
two exons and releases free lariant intron (Weatherall 2001b). The intron sequences are
thus excised and the donor and the acceptor sites of exons are sealed. Donor sites are

11
identified by the nucleotides GT at the 5’ end of intron and acceptor sites by AG at the 3’
end. In addition to these dinucleotides, nucleotide sequences adjacent to them called the
consensus sites, are required for accurate and efficient splicing (Mount 1982) (Fig: 1.2).
After the processing of primary transcript to mRNA it is exported from nucleus to the
cytoplasm. Amino acids are transported to mRNA template on carriers called transfer
RNAs. The order of amino acids in a globin chain is determined by a triplet code. tRNA
carries amino acid to the template and finds the position. mRNA is translated from 5’ to
the 3’ end. There are specific initiation (AUG) and termination (UAA, UAG, UGA)
Codons (Hoffbrand A.2005).
1.8.10: Switching over of globin gene
Developmental hemoglobin switching involves sequential globin gene activations and
repressions that are incompletely understood. As an adaptation of changing oxygen
requirements, different hemoglobins, all composed of two different pairs of globin chains
each attached to a heme moiety, are synthesized in embryo, fetus and adult (Wood and
Weatherall 1983). Molecular investigations of the last 20 years have delineated the two
basic mechanisms that control globin gene activity during development – autonomous
silencing and gene competition. Studies of hemoglobin switching have provided major
insights on the control of gene loci by remote regulatory elements (Stamatoyannopoulos.
2005). The β - like genes undergo two switches (embryonic → fetal → adult). At 6
months after birth, HbF comprises less than 5% of the total hemoglobin and continues to
fall until reaching the adult level of < 1% at 2 years of age. It is at this stage that
mutations affecting the β gene become clinically apparent. The switch from fetal (γ) to

12
adult (β) hemoglobin production is not complete since small amounts of β expression
persist in adult life. The residual amount of fetal hemoglobin (α2γ2) is presenting a sub-
set of erythrocytes called F cells which also contain adult (α2β2) hemoglobin. The tissue
and developmental – specific expression of the individual globin genes is governed by
the direct physical interactions between the globin promoters and the β - LCR (Carter et
al .2002 and Tolhuis et al. 2002), the interaction is mediated through binding of
tissue-restricted and ubiquitous transcription factors. Sever β - thalassemia usually
manifest as a result of the decline in the synthesis of fetal hemoglobin (α2γ2) during the
first year of life (Olivieri,1999).As the developmental expression is thought to rely on
mechanisms of gene slicing and gene silencing and gene competition, mediated by the
different transcription factors in embryonic, fetal and adult cells, the ξ- and γ- globin
genes are autonomously silenced at the appropriate developmental stage, expression of
the adult β globin gene depends on lack of competition from the γ gene for the LCR
sequences (Wood, 2001). Previous studies have shown that developmental regulation of
globin genes is complex, involving chromatin remodeling as well as interactions among
multiple trans-acting factors. These include GATA-1, NF-E2, SSP/NF-E4, erythroid
Krüppel-like factor (EKLF), and other proteins. For example, EKLF, a positive regulator
specific for the adult β-globin promoter, requires posttranslational modification and/or
interaction with other factors to mediate a hemoglobin switch; in K562 cells, transected
EKLF activates a contransfected β-globin promoter but is unable to activate the
endogenous, chromosomally located β-globin gene (Robert et al 2001).

13
1.9: Historic Background
Cooley and Lee in 1925 described a severe form of anemia with spleenomegaly and bone
changes (Cooley and Lee 1925). In 1932 George H published a comprehensive account
of the pathologic changes in this disease in Whipple and William L.Bradford (Whipple &
Bradford 1932). Originally called thalassemic anemia, it was abbreviated to thalassemia
from θαλασσα, “the sea” by Whipple. Fernando Rietti of Ferrara described a mild form
of hemolytic jaundice in which the red cells showed increased osmotic resistance (Rietti
1925). Similar descriptions were published subsequently by other Italian workers (Greppi
1928 and Micheli et al 1935). Thus the condition was known as La Malattia di Rietti –
greppi – Micheli, or haemolytic jaundice with decreased red-cell fragility. By 1949 it
became apparent that thalassemia was not a single disease but a complex syndrome
characterized by wide phenotypic diversity. First genetic evidence of Cooley’s anemia
was determined by Caminopetros (1938), Neel (1950) and Bianco et al (1952) alluded
was that this was a homozygous state of a recessive trait resulting in decreased
intracellular hemoglobin content (Hypochromia) and small sized red cells (Microcytosis).
Inherited disorders of haemoglobin are the commonest monogenic diseases. It is
estimated that about 7 % of world's population are the carriers of thalassemic gene. These
disorders fall into two groups: the structural variants of haemoglobin and the
thalassaemias (Weatherall 2000a).
1.10: Thalassemias:
Thalassemia are defined as a group of inherited hematological disorders characterized by
early onset of anemia resulting from reduced rate of synthesis of one or more globin
chains caused by globin chain mutations (Low 2005). Thalassemias are the commonest

14
monogenic syndromes (Thein 1992), characterized by decreased synthesis of one or the
other polypeptide chains resulting by decreased intracellular hemoglobin content
(hypochromia) and small size of red cells (microcytosis). Because of continued normal
production of unaffected globin chain the imbalanced globin – chain synthesis leads to
the accumulation of unstable aggregates of these unpaired globin chains leading to
oxidative membrane damage and premature destruction of erythrcytes in the peripheral
circulation and also at earlier stages of maturation in the bone marrow (Forget and Olvieri
2003). This decreases the hemoglobin level in the blood and oxygen carrying capacity of
the red blood cells.
1.11: Genetic classification
Depending on the type of the effected globin chain, thalassemias can be classified into
α-,β-, γδβ thalssemia. (Table -1).
1.11.1: β - Thalassemia
Two main types are described; βo Thalssemia in which no β globin chain is produced and
β+ thalassemia in which some β- globin chains are produced. Less sever forms of β
thalassemia are sometimes designated β++ to indicate that the defect in β- chain
production is particularly mild (Weatherall 2001b).
1.11.1.1: Molecular Pathology of β- Thalassemia
Majority of β – thalassemias are caused by point mutations. Almost 200 β – thalassemia
alleles have now been characterized (Weatheral 2001b). Studies on the molecular
genetics of thalassemia in various ethnic groups have shown that each group tends to

15
have its own set of common mutations (Kazazian et al, 1990). These mutations affect the
gene expression by a variety of mechanisms (Table1.1).
Table -1.1: Thalassemias and related disorders (Weatherall 2001b). α- thalassemia
αo
α+
Deletion (-α ) Non deletion (-αT)
β thalassemia βo β+ Normal Hb A2 Type 1 (Silent )
Type 2 δβ thalassemia (δβ)+ (δβ)o
(Aγδβ)o γ Thalassemia δ thalassemia δo δ+ εγδβ Thalassemia HPFH Deletion (δβ)o, (A γδβ)o Non deletion Linked to β – globin genes Gγβ+, A γ β+ Unlinked to β – globin genes
1.11.1.2: Deletion restricted to the β globin genes
Fourteen deletions affecting only the β – gene have been described. Of these the 619 – bp
deletion is common, and is restricted to Sindhi and Punjabi population of India and

16
Pakistan accounting for about 20% of β – thalassemia alleles (Thein et al 1984 Varawalla
et al 1991). These deletions vary widely in size, but remove always a region from
position – 125 to +78 relative to the mRNA CAP site in the β promoter, which includes
the CACCC, CCAAT and TATA elements (Weatherall 2001b). Dominantly inherited
beta thalassaemia intermedia is caused by a new single nucleotide deletion in exon 2 of
the beta globin gene: Hb Morgantown (beta91 CTG→CG) (Luo et al 2005).
1.11.1.3: Mutations affecting β – globin gene transcription
Mutations affecting beta globin transcription are Promoter Mutations. A group of 19 such
mutations have been described which are single base substitutions in the conserved DNA
sequences that form the β – globin promoter. These mutations reduce the binding of RNA
polymerase and lower the level of mRNA to 10 to 25 % of normal; this is compatible
with relatively mild phenotype of β+ thalassemia (Treisman et al 1983)
1.11.1.4: Transcriptional mutations:
Several different base substitutions have been found that involve the conserved
sequences upstream from β – globin gene (Weatherall 2000 and Huisman et al 1997).
Despite of considerable variability in the clinical severity associated with mutations of
this type in, the phenotype is β+ thalassemia.
Several of these mutations are close to CCAT box as exemplified by C-T substitution at
position -88 and – 87 (Orkin et al 1984, Orkin et al 1982).While the others lie within the
ATA box homology (Ponez 1983). C→ T substitution at position – 101 which involves
one of the promoter elements is characterized by “Silent” β thalassemia (Gonzalez –
Redando et al 1989). A→ C substitution at the CAP site (+1) even in homozygous state
may show the clinical features of β – thalassemia trait (Wong et al 1987).

17
1.11.1.5: RNA – Processing mutations
Ends of exons and introns are marked by the presence of dinucleotide, GT at the 5’
(donor site) and AG at the 3’ (receptor site). Single – base changes that involve either of
these splice junctions abolish normal RNA splicing and cause βo – thalassemia (Huisman
et al 1997).
Single – base substitution with in the consensus sequence of the IVS – 1 donor site
surrounding the invariant dinucleotide at the splice junctions show remarkable variability
in their associated phenotypes (Orkin et al 1982). C → T substitution at position 5 of
IVS – I produces abnormal spliced RNA and causes sever β+ - thalassemia phenotype
(Orkin et al 1982) T→ C substitution at position 6, commonly found in
Mediterranean region produces a very mild form of β+ thalassemia (Tamgagnini
et al 1983) G- C substitution at position 5 has been found in Melanesia and causes
β thalassemia in New Guinea (Hill et al 1988). Mutations creating new splice sites
within either introns or exons affect RNA processing and cause variable
phenotype effects. G →A substitution at position 110 of IVS – I is the most
common form of β – thalassemia in Mediterranean region. It causes 10 percent
slicing and results in sever β+ thalassemia (Spiritz et al 1980, Busslinger et al
1981). Mutation at 116 in IVS -I produces a new acceptor site and causes βo
thalassemia phenotype (Weatherall et.al. 1985). Activation of donor sites within
exons may result in abnormal splicing. Within exon 1 there is a cryptic donor site in the
region of codons 24 through 27. G T dinucleotide sites are present at this site. Several

18
mutations can activate this site so that it is utilized during RNA processing within
production of abnormal mRNA (Weatherall et al 1985).
Amino acid substitution like A→ G in codon 19, G → A in codon 26, and G→T in codon
27 mutations produces hemoglobins Malay, Hemoglobin E and Knossos. At least 38
mutations result in abnormal RNA splicing and cause thalassemia ranging in severity
from β+ to βo – thalassemia (Baysal and Carver 1995). Some mutations involve poly
adenylation signal site AAUAAAA in the 3’ untranslated region of β – globin mRNA
(Orkins et al 1985) like T→ C substitution in this sequence leads to reduction in the
transcription length of normal β -globin mRNA and result in sever β+ thalassemia
phenotype (Orkin et al 1985).
1.11.1.6: Mutations causing Abnormal Translation of Messenger
RNA
Chain termination mutation
Some substitutions of the base change an amino acid codon into nonsense codon and
result in termination of chain and prevent translation of mRNA resulting in βo
thalassemia. Many such mutations have been described (Huisman 1997). These include
codon 36 mutation which is common in the Mediterranean region (Treeartin et al 1981,
Rosatelli et al 1987) and codon 17 mutation that is common in southeast Asia (Chang and
Kan. 1979).
Insertion or deletion of one two or four nucleotides in the coding region of β – globin
gene disrupts the normal reading frame. Translation of mRNA takes place in the addition

19
of anomalous amino acids until a termination codon is reached in the new reading frame.
Several frameshift mutations of this type have been described (Huisman et al 1997).
Insertion of one nucleotide between codons 8 and 9, and deletion of four nucleotides in
codons 41 and 42 are common Asian Indian mutations (Kazazian et al 1984).
Combination of frameshift 41/42 and βo-thalassemia or Hb E produced mild to moderate
symptoms with thalassemia intermedia phenotype and severe symptoms with thalassemia
major phenotype (Laosombat et al 2001).
Unstable β - globin variants
In spite of being highly unstable, some β – globin chain variants are capable of forming
tetramers that precipitate in the red cells precursors or in the mature erythrocyte and give
rise to a blood dyscrasia ranging from dominantly inherited β – thalassemia to a
hemolytic anemia. Unstable hemoglobin may produce thalassaemia intermedia phenotype
(Dash et al 2006).
Hemoglobinopathies caused by unstable beta-chain variants have a dominant
thalassemia-like phenotype in which carriers have the clinical expression of thalassemia
Intermedia. Highly unstable alpha-globin variants, on the other hand become
phenotypically apparent only when they interact with other alpha-thalassemia mutations
(Traeger-Synodinos et al 2000).
Thalassemia syndromes and unstable hemoglobins traditionally represent two
phenotypically separate disorders of hemoglobin molecule. Highly unstable hemoglobin
variants, often have phenotypic characteristics associated with ineffective erythropoiesis
(thalassemias) as well as peripheral hemolysis (unstable hemoglobins). Many highly
unstable beta chain variants cause a dominant thalassemia-like phenotype in which

20
heterozygotes for such mutations have a clinical expression similar to thalassemia
intermedia. Phenotypic expression of highly unstable alpha-globin variants is usually less
severe, due mainly to a gene dosage effect. They are often characterized only on
interaction with other alpha-thalassemia mutations. It is for this reason that they are
classified as nondeletional alpha-thalassemia determinants (Traeger-Synodinos et al
1999). Heterozygous genotype for a novel 6 bp (TGGTCT) deletion of beta-globin gene
involves codons 33-35. This deletion results in the removal of two valine residues from
beta-globin chain at position 33/34.
(B15/B16) and the substitution of the tyrosine residue at position 35 (C1) by an aspartic
acid (beta 33-35 [B15-C1] Val-Val-Tyr→0-0-Asp). This abnormal haemoglobin is
called Hb Dresden and is found to be exquisitely unstable.
Mediterranean beta-thalassemia (thal) mutation, IVS-I-110 (G→A), in trans position to a
beta-globin gene mutation at codon 107 (GGC→>GAC), gives rise to a rare unstable
beta chain variant Hb Lulu Island or beta107 (G9) Gly→Asp (Papassotiriou I et al
2006).
Silent β thalassemia
A number of extremely mild β – thalassemia alleles are either silent or almost
unidentifiable in heterozygotes. Some involve the region of the promoter boxes of
β – globin genes, others involve the CAP sites or the 5’ or 3’ untranslated regions
(Weatherall 2000b, Huisman 1997).
β-thalassaemia heterozygotes with C → T substitution at nucleotide position -101 from
the Cap site, in the distal CACCC box of the beta-globin gene promoter is the most

21
common silent beta-thalassaemia mutation in the Mediterranean population
(Maragoudaki et al 1999). These alleles usually cause thalassemia Intermedia.
Dominantly inherited β thalassemia
Mutations inherited in the dominant fashion causing symptomatic β – thalassemia have
been identified (Thein 1992). Many of them involve exon III of β globin gene and include
frame shift premature termination mutation, and complex rearrangements leading to
elongation of β globin gene and highly unstable β – globin gene products (Higgs et al
1986).27 such mutations have been identified (Baysal and Carver 1995). The most
common of this type is a GAA → TAA change at codon 121 leading to truncated
β – globin chain (Kazazian et al 1986). In the heterozygous state dominant thalassemia
mutations form hyper – unstable haemoglobin variants that precipitate in the erythroid
cells and cause thalassemia Intermedia (Thein 1992).
β – Thalassemia due to unknown mutation
Some typical β – thalassemias are observed with out any detectable mutation in the β –
globin gene or its immediate flanking regions (Semenza et al 1984, Kazazian et al 1990)
Compound heterozygosity for two new mutations in the beta-globin gene [codon 9 (+TA)
and polyadenylation site (AATAAA→AAAAAA)] cause thalassemia intermedia in a
Tunisian patient (Jacquette A et al 2004).
Variant forms of β thalassemia
Several forms of β – thalassemia in heterozygous state with normal Hemoglobin A2 are
identified (Weatherall et al 2000b). Some of them are due to “silent” β – thalassemia
alleles others reflect the coinheritance of β and δ Thalassemia gene.

22
δβ thalassemia
The δβ–thalassemia may be divided into δβ+ and δβº based on the residual output of the
δ- and β-chains from the affected chromosome. δβ+ thalassemia is due to the presence of
two different mutations within the same β-like gene cluster. δβº Thalassemia on the other
hand are due to large deletions involving the εγδβ-gene cluster. Nondeletion δβº
thalassemia is a rare form of δβ thalassemia in the Sardinian population. Homozygous
state for nondeletion δβº thalassemia, which produced a symptomless clinical phenotype
with a peculiar Hb pattern has been reported (Galanello et al 2002).
DUCTION Inherited disorders of δ and β chain synthesis are of two main types, the δβ thalassemia
and the Hb Lepore syndromes. A classification and description of the main forms of δβ
thalassemia is shown in the table 1.2.
Many of the δβ thalassemia are produced by the deletions of δ and β globin genes. These
are associated with persistent synthesis of γ chain at a much higher level than is observed
in β thalassemia. Production of high levels of γ chain leads to relatively a mild degree of
globin chain imbalance and hence these conditions are much milder than the β
thalassemia. Gγδβ thalassemia is characterized by the deletion of Aγ genes, and synthesis
of Gγ chains only. If the production of Aγ is preserved in the mutation than both type of
γ chains are produced and the condition is called Gγ Aγδβ thalassemia. At the molecular
level these disorders are very heterogeneous and require the determination of underlying
defects.

23
Table 1.2: The main groups of disorder of δ and β chain production
Condition Homozygote Heterozygote
Gγ Aγδβ Thalassemia Thal Intermedia 100% HbF
Thal minor Hb F 5 – 20%; HbA2 1.5 -2 % α /non- α1.3 – 1.8 /1
Gγδβ Thalassemia As above As above
δβ Lpore Thalassemia Thal major or intermedia 80% HbF ; 20% HbLepore
Thal minor Hb Lepore8 – 20%
Aγ Gγ HPFH Thal minor 100% Hb F
Normal blood picture, 15 – 30 % HbF; 1.5 – 2 % HbA2
Other forms of δβ thalassemia are Hb Lepore disorders. These disorders are
characterized by δβ fusion genes directing the synthesis of δβ chain fusion. These are
produced by the unequal crossing over of δ and β globin genes. Depending on the exact
position of crossing over resulting in the δβ fusion several different forms of hemoglobin
Lepore have been found. Most common being Hb Lepore Boston found commonly in
parts of Italy and Yugoslavia. Hemoglobin Lepore produce much more sever clinical
phenotype than δβ thalassemia because of less output of γ chains to compensate for the
deficiency of δ and β chains.

24
Gγ Aγδβ Thalassemia
Homozygous forms of this conditions have a mild anemia with hemoglobin values in the
8 – 11 g/dl range. Red cells have typical thalassemic changes with low MCV and MCH
values. Hb A and A2 are absent and Hemoglobin F is 100%. Marked imbalance with α/ γ
synthesis ratios of approximately 3 are observed.
Hetrozygous forms are similar to β thalassemia heterozygotes although red cell changes
are less marked. It contains both Gγ and Aγ chains in heterozygous and homozygous state.
Hb F levels are 5 – 20% range and A2 is normal or slightly reduces.
Gγ δβ Thalassemia Clinical and hematological finding are same as Gγ AγHPFH, although homozygotes are
slightly more severely affected. These conditions can be differentiated by chemical
analysis of HbF which shows only the presence of Gγchains.
Sardinian δβ Thalassemia Clinical and hematological findings are similar to other forms of δβ thalassemia.
However they have HbF that is mainly nearly all Aγ type.
INTERACTIONS OF δβ THALASSEMIA AND LEPORE HEMOGLOBIN Compound heterozygous state of δβ thalassemias and Lepore hemoglobin with βO or β+
thalassemia results in the phenotype of thalassemia Intermedia.

25
γ δβ THALASSEMIA This is a rare form of thalassemia, not found in homozygous state. Adults heterozygotes
have hematologic changes similar to those of β thalassemia heterozygotes. The condition
is associated with a hemolytic anemia, jaundice in newborn period. α and non α chain
production imbalances are seen at birth and adult life. Diagnosis require globin gene
analysis (Weatherall .1983)
1.11.2: Incidence and population genetic:
Initially thalassemia was thought to be confined to Mediterranean, African and Asian
ancestry but sporadic cases have been reported in many ethnic groups. It is now thought
that malaria has played a role in the propagation of thalassemia genes.
The eight most frequent mutations encountered in Iraqi population are IVS-II-1 (G→A),
codon 44 (-C), codon 5 (-CT), IVS-I-1 (G→A), codon 39 (C→T), IVS-I-6 (T→C),
codons 8/9 (+G) and IVS-I-5 (G→C). These mutations accounted for 81.7% of the
thalassemic defects in this population. The less frequent mutations are codon 8 (-AA),
IVS-I-110 (G →A), codon 30 (G→C) and codon 22 (-7 bp).Genetic abnormality in 11.5
of β - Thalassemia remain un characterized. This is the first study of beta-thal mutations
from Iraq (Al-Allawi et al 2006).
1.11.3: Thalassemia Intermedia
1.11.3.1: Definition
Thalassemia Intermedia is the term used to describe the clinical and hematological
findings in patients with β - Thalassemia. Although not transfusion dependent, these

26
patients manifest a more sever degree of anemia than that found in heterozygous carriers
for α - or δ thalassemia (Weatherall 1996). Beta(0)-thalassaemia intermedia (beta(0)-TI)
describes patients who lack beta-globin synthesis yet manifest a non-transfusion-
dependent form of beta-thalassaemia (Chang et al 2001).
The proportion of patients with homozygous thalassemia intermedia is strikingly different
among different ethnic groups. About 10% of patients of Mediterranean ethnicity who are
homozygous can be classified as intermediates. In contrast, more than 70% of African-
American patients who are homozygous may be classified as such. This difference
reflects the kinds of thalassemia mutations, especially so-called “mild mutations,” that
are prevalent in these ethnic groups (Pearson et al 1996)
1.11.3.2: History
La – Malattia di Rietti – greppi – Micheli (Hemolytic jaundice with reduced red cell
fragility). This condition was characterized by much more sever anemia, jaundice and
splenomegaly.
In 1940 Wintrobe (1940) described a milder form of thalassemia. In Italy many patients
with Mediterranean anemia of intermediate severity have been reported (Marmont and
Bianchi 1948) Chini and Valeri (1949) called this condition ‘Mediterranean haemopathic
syndromes’.
In 1940 , Silvestroni and Bianco describeded anemia microcitica costituzionale
(Silvestroni & Bianco 1944 – 45). A mild form of haemolytic jaundice in which red cells
showed increased osmotic resistance was reported in 1925 by Fernando Rietti of Ferrara
(Rietti 1925). Many description were published shortly afterwards by other Italian

27
workers including Greppi (1928) and Micheli et al (1935). This form of anemia was
described by Rietti, Greppi and Michli and was reviewed by Chini and Valeri (1949).
The earliest reports of milder forms of thalassemia called it thalassemia Intermedia. The
term thalassemia intermedia appeared in the literature in the 1950s (Sturgeon et al 1995).
The term thalassemia intermedia is a useful descriptive title for clinical phenotype that
can result from the interaction of many different thalassemia alleles, either among
themselves or with those for structural hemoglobin variants.
Thalassemia Intermedia entails considerable clinical and genetic heterogeneity. Anemia
is variable and may patients have splenomegaly.They are clinically as well as genetically
variable. Parents may have mild thalassemia or a single gene may be inherited in the
families (Weatherall 2001b).
1.11.3.3: Clinical Presentation
Thalassemia Intermedia present with extraordinary diverse clinical spectrum from almost
complete health to a condition characterized by severe growth retardation and skeletal
deformities requiring transfusion therapy. Typical features of patients with thalassemia
are skeletal changes, particularly in the skull and in the malar bones. Patients develop
oro-facial deformities and subsequently demonstrated clinical and laboratory
abnormalities consistent with thalassemia intermedia (Ficarra et al 1987). Skeletal
abnormalities in beta-thalassemia are widening of medullary spaces, rarefaction of bone
trabeculae, thinning of cortical bone, and perpendicular periosteal spiculation. Premature
epiphyseal fusion (PEF) is found though more rarely (Colavita et al 1987).
Thalassemia intermedia have a later clinical onset and a milder anemia than thalassemia
major, characterized by high output state, left ventricle remodeling, and age-related

28
pulmonary hypertension. Bone deformities, extramedullary hematopoiesis (EMH), and
spleen and liver enlargement are the consequences of hypoxia and enhanced
erythropoiesis. Afebrile, EMH-related pleuritis represents a potentially life-threatening
complication in thalassemia (Aessopos 2006). Two main factors determine cardiac
disease in this form. One is the high output state that results from chronic tissue hypoxia
and from hypoxia-induced compensatory reactions. The other is the vascular involvement
that leads to an increased pulmonary vascular resistance and an increased systemic
vascular stiffness (Aessopos et al 2007a).
The clinical picture of TI patients who have not received transfusions or have
occasionally received transfusions is dominated by the consequences of chronic
hemolytic anemia, tissue hypoxia, and their compensatory reactions, such as bone
deformities and fractures, extramedullary hemopoiesis, spleen and liver enlargement,
hypercoagulability, and pulmonary hypertension. These complications, especially the
latter two, are getting more frequent and severe over the years. Nowadays, although TI
patients have almost no changes in the course of the disease, well-treated TM patients
with regular transfusion-chelation therapy showed suppression of the anemia-related
disorders in parallel to prolongation of life. The new oral iron chelators and the magnetic
resonance imaging application for early detection of heart iron load are promising for
further improvement on Survival (Aessopos 2007b).
Mild anemia is one of the hematological findings in heterozygous β thalassemia. Patients
with β– thalassemia Intermedia have hemoglobin level usually below 9 – 10 g/dl
particularly if there is associated splenomegaly. At the other end of the spectrum, there
are patients with miserable childhood gross skeletal deformities. They are periodically

29
transfused to avoid these distressing complications. Osteoporosis and osteopenia are
frequent complications of thalassemia major (TM) and thalassemia intermedia (TI)
(Origa et al 2005). Fractures are frequent among the aging patients with β - TM (Vogiatzi
et al 2006) and TI. Spinal cord compression due to extramedullary hematopoiesis is a
rare complication of thalassemia (Saghafi et al 2005).
Another group has hemoglobin values between 6 and 9 g/dl. They grow reasonably well
and reach adult life. They may become transfusion dependent if they develop
complications like hypersplenism or folic acid deficiency, nutritional deficiency or inter
current infection which may exacerbate anemia.
Thalassemia intermedia patients usually do not require blood transfusion however all
patients show variable degrees of erythropoietic marrow expansion to compensate for
anemia. This is the major cause of complications in untransfused individuals
(Camaschella et.al.1996). Kinetic studies clearly separate cases that, or will be, clinically
intermediate, showing a higher medullary uptake of radioactive iron, a less ineffective
erythropoiesis than that seen in Cooley's disease and a greater peripheral haemolysis. In
our study, no overlap was seen between the two groups. Iron kinetic studies are then of
prognostic interest and may help in therapeutic decisions, transfusion regimen, iron
chelation and splenectomy (Najean Y et al 1985).
One case in English literature of crystal proven gout in thalassemia intermedia was
reported that indicates the relative rarity of gout in this clinical setting despite evidence of
urate over production in one report. Long survival and renal insufficiency may have
contributed to the patients' tophaceous gout (Kumar and Gruber 2003).

30
Hypocholesterolemia accompanies anemias with high-erythropoietic activity. We suggest
that the high-erythropoitic activity-associated hypocholesterolemia is due to increased
cholesterol requirements by the proliferating erythoid cells (Shalev et al 2006). Patients
with thalassemia may suffer from a sensory polyneuropathy especially as they grow
older, this is particularly so and if they are not optimally treated (Sawaya et al 2006).
Pulmonary hypertension (PHT) is part of the cardiopulmonary complications of
thalassemia lack of systematic treatment in TI leads to a cascade of reactions that
compensates for chronic anemia but at the same time allow the development of PHT
(Aessopos and Farmakis 2005).
Low transfusion regimen may cause a decrease in serum concentration of EPO, which is
independent of the level of hemoglobin (Dore et al 1993).
Venous thromboembolic events such as pulmonary embolism, deep venous thrombosis
patients with and portal vein thrombosis have been observed in adult thalassemia
patients, mainly in beta-thalassemia intermedia. Clinical findings are consistent with
alterations that indicate a state of activation of the haemostatic mechanisms in
thalassemias. These alterations are related to high platelet counts due to splenectomy
and/or liver dysfunction (Cappellini et al 2005, Ciceri et al 2000). Low plasma heparin
cofactor II levels are related to increased red cell turnover and can be normalized once
the increased turnover has been suppressed by hypertransfusion. Thrombotic tendencies
in patients with low Heparin co factor II levels in the presence of haemolysis might in
principle decreased by blood transfusion (O'Driscoll et al 1995).
Some occular abnormalities like degeneration of retinal pigment epithelium, lenticular
opacities, vascular abnormalities, and angioid streaks have been reported in TI

31
(Gartaganis et al 1989, Aessopos et al 1989). An acquired diffuse elastic tissue defect
that resembles inherited pseudoxanthoma elasticum (PXE) has been noticed with a
significant age-related frequency in hemoglobin disorders, especially beta-thalassemia
and has been held responsible for a number of complications observed in these cases,
some of which are quite severe. Patients with beta-thalassemia intermedia, who presented
with severe visual acuity impairment associated with angioid streaks, the typical ocular
manifestation of PXE has been reported (Aessopos et al 2008).
1.11.3.4: Clinical Grading
Clinical phenotype of homozygous beta thalassemia varies in severity from mild
thalassemia intermedia to the severe thalassemia major (Galanello et al 2002). Three
grades of the disease are described by Ho et al (1998) these are mild, moderate and
severe.
Severe:
If the transfusion started before the age of 4 years, it is usually required every 3 and 4
months. The patients with the transfusion requirement between these extremes were
classified as “severe”.
Moderate:
Severe if transfusion was started at the age of 4 years or above and frequency between 6
– 16 weeks.

32
Mild
Patients in the “mild” group maintain their hemoglobin at 7.5 g/dl or higher with out
transfusion. They are transfused less than once every 2 years if transfusion is started
before the age of 10 years transfusion interval is less than 6 months if transfusion is
started after 10 years.
1.11.3. 5: genotypes
Based on the molecular genetics five different genotypes are identified in
thalassaemia intermedia patients:
Group I: homozygosity for mild mutations Group II: combinations of mild/severe mutations Group III: homozygosity or double heterozygosity for severe mutations Group IV: heterozygosity -87/IVS1-6 Group V: IVS1-6/CD 6-A This is of particular importance for genotype-phenotype correlation, carrier
detection, genetic counseling and prenatal diagnosis (Rigoli et al 2003).
1.11.3.6: Age at presentation
One of the most useful indicators of thalassemia Intermedia is the age at presentation.
Mean age of presentation is 13.1 months with a range of 2 to 36 months (Kattimis et al
1975). In a study 11% of patients presented in the first year while 30% presented in the
second year and 59% presented after the age of 2 years. However, the socioeconomic
environment and other factors influence the age at presentation (Modell and Berdaukas

33
1984). Thalassemia Intermedia with βo thalassemia homozygousity may also present late
(Cao 1988). Those who were transfusion dependent presented at mean age of 8.5 ± 9.1
months, while transfusion independent patients presented at mean age of 17.4 ± 11.8
months. All these patients maintained their hemoglobin and were homozygous for the
CD39 C → T nonsense mutation.
It was suggested that bone marrow erythroblast may mature further and generate more
hemoglobin. These patients may have hyperbilirubinemia due to increased catabolism of
hemoglobin (Berdoukas 1993).
Thalassemia Intermedia shows diverse spectrum of clinical heterogeneity where patient
may be anemic in early child hood while others may not be diagnosed until adolescence
or later age (Ahern et al 1975). Spleen may be enlarged, occasionally massively
(Farhangi Sass and Bank 1970).
Regular transfusions are usually not required. Growth and development are usually
normal and skeletal changes of β- thalassemia major types are rarely seen (Erlandson et
al 1964). Chronic hypersplenism is common (Rapaport et al 1957) and gallstones
frequently form (Erlandson et al 1964). Unconjugated hyperbilirubunema resulting from
ineffective erythropoiesis is common (Weatherall and Clegg 1981). Bone
demineralization in adult thalassaemic patients due to the contribution of growth
hormone and insulin-like growth factor I at different skeletal sites has been reported by
Scacchi et al (2008).
Other complications include diabetes mellitus secondary to iron overload (Erland et al
1964) and leg ulcers (Weatherall and Clegg 1981), Hyperuricaemia (March et al 1952)
gout (Weatherall and Clegg 1981) . Pancytopenia (Schiliro et al 1983) has been reported

34
but uncommon. Anemia may worsen in pregnancy (Walker et al 1969). Iron overload can
develop even in patients who have not been transfused, this may cause heart failure
hepatomegaly (Celada 1982) hypopituitarism and prophyria (Bannarman et al 1967).
Tumor – like masses of bone marrow are common (Knoblich 1960). Folate deficiency
may worsen anemia and megaloblastic erythropoiesis (Erlandson et al 1964).
1.11.3.7: Molecular basis of β- Thalassemia Intermedia phenotype
Thalassemia intermedia is a clinical entity characterized by moderate, non-transfusional
anemia and hepatosplenomegaly. This phenotype can result from different genetic
combinations and is sometimes present in patients with only one parent showing the
thalasemia minor phenotype (Martinez-Lopez J et al 1998). Many Interactions and
mutations can give rise to this phenotype (table 1.3).
1.11.3.8: Interactions of silent β thalassemias
β - 10 C → T
This mutation which occurs in distal CACC box, to down regulate globin gene
transcription very slightly and causes extremely mild form of thalassemia intermedia
(Gonzalez – Redondo et al 1988).
β 5’ untranslated region (UTR ) + 10 (- T)
This mutation with compound heterozygous state with β CD 39 C → T is characterized
by a mild form of thalassemia intermedia.
β CAP + 33 C → G
This is almost silent in the heterozygous state although the HbA2 level may be slightly
elevated and the α/β - globin synthesis ratio varies from 1.5 to 1.8. in the heterozygous

35
state with several beta thalassemia alleles including IVSI – I G → A and IVSI – 110 G →
A (Ho et al 1996) mutations TI is usually very mild.
IVS 2 844 C → G
In homozygous form, this mutation (Murru et al; 1991) causes extremely mild form of
β - thalassemia Intermedia. Interaction with splice mutation β IVS2 – 745 C → G also
causes mild form of thalassemia Intermedia (Rosatelli et al 1994). Splicing mutations are
common causes of beta-thalassemia. Some mutations permit normal splicing as well as
aberrant splicing. Reduced level of normal beta-globin synthesis produces a mild disease
(thalassemia intermedia) (Vadolas et al 2006).
β CAP + 1 A → C
Observed in Asian Indians, this mutation can cause thalassemia Intermedia of varying
severity (Wong et al 1987).
β Termination codon + 6 C → G
With IVSI – I (G → A) causes moderately sever thalassemia Intermedia (Maragaudaki et
al 1998).
Codon 104(-G)
Codon 104(-G), a heterozygous frameshift mutation in exon 2 of HBB, resulted in a
dominantly inherited beta0-phenotype with mild anemia in a German kindred, and
thalassemia intermedia in the index patient. A co-inherited a gene triplication, long-term
transfusion therapy, and ineffective erythropoiesis were confounding factors (Lahr et al
2007) (table 1.4).

36
β - 88 C → T
This mutation decreases the rate of transcription. It is common among Africans and Afro
– Americans and may cause mild form of Thalassemia Intermedia (Weatherall 2001b).
Compound heterozygote for two beta-globin gene promoter mutations, like nucleotide
(nt)-88 C→T mutation from the cap site, and two-nucleotide (AA) deletion between nt -
29 and -26 within the TATA box of the beta-globin gene causes TI phenotype (Basran et
al 2006).
β - 87 C → G
This mutation reduces the binding of transcription factors to β- globin gene (Treisman et
al 1983) and produces thalassemia intermedia (weatherall 2001b). -87 were found in
association with haplotype VIII (beta-87/VIII) or V (beta-87/V). β beta-87/VIII showed a
configuration of rare polymorphisms in the 5' sub-haplotype. This has been reported to
exert an increasing effect on Hb F synthesis (De Angioletti et al 2004, Rosatelli et al
1989).
β - 30 T → A
This is common in Turkish, Macedonian and Tunisian population and causes mild form
of thalassemia intermedia (Fei et al 1988)
β - 29 A→ G
This is common in Africans and Afro – Americans and produces fairly mild thalassemia
Intermedia (Weatherall 2001b).

37
Table 1.3: Thalassemia intermedia (Weatherall D.J.2001)
1. Mild defects in β – globin production Homozygous mild β+ thalassemia Compound heterozygous for severe βo or β+ and mild β+ thalassemia Interactions of βo with ‘silent’ or ‘mild’ β thalassemia Homozygous for ‘silent’ β thalassemia
2. Reduced globin imbalance due to co-inheritance of α and β thalassemia Homozygous or compound heterozygous βo or β+ thalassemia with two or three α gene Homozygous or compound heterozygous severe βo or β+ thalassemia with non – deletion α2 – gene mutation Homozygous or compound heterozygous severe β+ thalassemia with one or two α – gene deletions
3. Sever β thalassemia with increased capacity for γ – chain synthesis homozygous or compound heterozygous βo or β+ thalassemia with heterocellular HPFH
Homozygous or compound heterozygous βo or β+ Thalassemia with a particular β – globin RFLP haplotype Mechanism unknown 4. Deletion forms of δβ thalassemia and HPFH
Homozygous (δβ)o or (A γ δβ) thalassemia Compound heterozygous for βo or β+ and (δβ)o or (A γ δβ) thalassemia Homozygosity for Hb Lepore (some cases) Compound heterozygosity for (δβ)o, G γ β+ or A γ β+ HPFH and βo or
β+thalassemia Compound heterozygosity for (δβ)o thalassemia and (δβ)o HPFH
5. Compound heterozygousity for β or δβ thalassemia and β- chain structural variants Hbs S/, C/ E/β or δβ thalassemia Many other rare interactions
6. Other β thalassemia alleles or interactions Dominant β thalassemia β thalassemia Trait associated with αααα – gene arrangements Highly unstable β – globin chain variants

38
Table-1.4: Some interactions of silent, mild and severe β Thalassemia alleles as the basis of thalassemia intermedia (TI). Where data is available the severity is classified as mild (MTI) or severe (STI). TI indicates a variable phenotype, or insufficient information.Data from Huisman et al. (1997). Ho et al (1998) and references cited in text.
Mut
atio
ns
-101
(C→
T)
-92
(C→
T)
-88(
C→
T)
-87(
G→
C)
-87(
C→
T)
-86(
C→
A)
-30
(T→
C)
-29
(A→
G)
5’U
T R
+10
- T
5’U
T R
+22
(G-A
)
5’ U
TR +
33 (C
-G)
CA
P+1
(A→
C)
CD
19 (A
→G
)
CD
26(G
→A
)
CD
27 (
G→
T)
IVSI
-6 (
T→C
)
IVS
2-84
4 (→
G)
β te
rm +
6 (C
-G)
AA
TAA
A→
A
ATA
AG
AA
TAA
A→
A
AC
AA
A
AA
TAA
A→
A-A
AA
CD8 - AA STI TI TI
CD8/9+G STI
CD15 G-A TI
IVSI-I G-A TI TI TI
IVSI-5 G-C TI TI/STI
IVSI-5 G-T MTI
IVSI-110 G-A MTI TI STI TI MTI MTI TI STI
CD36/37-T MTI

39
Mut
atio
ns
-101
(C
→T)
-92
(C→
T)
-88
(C→
T)
-87
(G→
G)
-87
(C→
T)
-86
(C→
A)
-30
(T→
C)
-29
(A
→G
)
5’U
T R
+10
-T
5’U
T R
+22
(G-
A)
5’ U
TR +
33 (C
-G
)
CA
P+1
(A→
C)
CD
19 (A
→G
)
CD
26 (
G→
A)
CD
27 (
G→
T)
IVSI
-6 (
T→C
)
IVS
2-84
4 (C
→G
)
β Te
rm +
6 (C
-G)
AA
TAA
A→
A
ATA
AG
AA
TAA
A→
A
AC
AA
A
AA
TAA
A→
A- A
AA
CD39C-T TI STI TI TI MTI STI
CD41-C MTI
CD41/42 -TTCT MTI /STI
CD44-C MTI STI
IVS2-1 G-A MTI TI MTI STI
IVS2-654 C-T TI
IVS2-745 C -G MTI TI MTI
IVS2-745 C-G
IVS2-748 C-A
IVS2-849 A-G

40
β Codon 19AAC → AGC ; β 19 Asn → Ser, Hemoglobin Malay
This mutation is found amongst Southeast Asians of Malaysian origin. In homozygous
state, it causes mild form of thalassemia Intermedia (Yang et al 1989).
β codon 27 (GCC → TCC ; β 27 Ala →Ser; Hb Knossos )
A codon 27 change activates an alternative splice site, resulting in a slight reduction in
the quantity of normal β – globin messenger RNA. More over δ – globin in cis position
has a deletion of a single A in codon 59 leading to premature termination at codon 60. It
is found in Mediterranean and adjacent regions (Weatherall 2001b). Compound
heterozygous form with IVSI – 6 T → C, Codon 8 – AA, IVSI →110 G→A, IVSI – I G
→ A and IVS2 – I G → A results in mild to moderate thalassemia intermedia.
An A→G transition at the usual intervening sequence 2 (IVS2) acceptor splice site has
been described. Functional analysis of transcripts produced by this mutant gene in a
transient expression vector indicates that the mutation inactivates the normal acceptor
splice site and results in some utilization of a cryptic splice site near position 580 of
IVS2. This mutation would be expected to produce a beta-globin gene which results in no
normal beta-globin mRNA (Atweh GF et al 1985).
β IVS I → 6 T → C
Common in Mediterranean populations, a splice mutation results in mild to moderate
form of thalassemia Intermedia (Weatherall 2001b).
β 87 C → T
In compound heterozygous form with allele IVSI – 110 G → A demonstrate moderately
severe form of thalassemia Intermedia (Weatherall 2001b). PolyA; AATAA →

41
AATAAG, found in Kurdish Jewish families, poly A; AATAAA→ AATGAA
demonstrates mild form of Thalassemia Intermedia, where as poly (A): AATAAA →
AACAA in combination with IVS2-1 G → A causes moderately severe form of
thalassemia Intermedia. Poly A: AATAAA → AATAGA observed in Malaysia is
associated with an extremely mild interaction with HbE and there fore is probably a mild
β – thalassemia allele (Weatherall 2001b).
G →T CHANGE IN CODON 121
A child with severe beta – thalassemia intermedia has been described, born to a Greek-
Cypriot with hematological findings of beta-thalassemia trait, and a Polish father who is
hematologically normal. G→T change in codon 121 of the beta-globin gene in the child
was the result of a spontaneous mutation that occurred during spermatogenesis in a
paternal germ cell (Kazazian Jr. et al 1986).
β -101 C → T SUBSTITUTION This mutation was observed in patients with mild thalassaemia intermedia, `her
haemoglobin levels were around 9.5 g/dl and haemoglobin F levels < 25% (Maragoudaki
et al 1999).
HB KNOSSOS (BETA 27 (B9) ALA→SER)
Hb Knossos (beta 27 (B9) Ala→Ser) is a hemoglobin variant that can cause beta (+)
thalassemia intermedia syndrome (Baklouti et al 1986). Hb Knossos is characterized by
reduced synthesis and by interaction with beta-thalassemia, in which the double
heterozygotes display typical features of thalassemia intermedia. Hb Lepore and Knossos
(beta 27 Ala→Ser) have been found to be associated with beta-thalassemia intermedia

42
picture with total absence of Hb A2. This indicates that beta Knossos gene is most
probably flanked by a delta (0)-thalassemia gene. Hb Knossos, representing 70% of total
hemoglobin in this study, displayed decreased affinity for oxygen (P50 = 35 mm Hg), a
fact presumably accounting for the relatively good tolerance of the condition (Morle et al
1984).
BETA + THALASSAEMIA--PORTUGUESE TYPE Clinically, the homozygotes range from asymptomatic to thalassaemia intermedia and
they are characterized by low levels of HbF (less than 20%) indicating only a mild deficit
in beta globin production. Heterozygotes are indistinguishable from those with the more
common types of beta thalassaemia as regards red cell morphology, haemoglobin
analysis and globin chain synthesis. Globin gene mapping excluded the presence of alpha
thalassaemia in these patients and demonstrated no abnormalities in the beta-like globin
gene cluster. Restriction enzyme site polymorphisms around the beta gene cluster are
identical on both chromosomes in all of the homozygotes, confirming their homogeneity
(Tamagnini et al 1983).
HEMOGLOBIN MISSISSIPPI (HBMS: BETA 44SER→CYS)
Hemoglobin Mississippi has anomalous properties that include disulfide linkages with
normal beta-, delta-, gamma-, and alpha-chains and formation of high molecular weight
multimers. HbMS-beta +-thalassemia may result from proteolytic digestion of HbMS, as
well as excessive alpha-chains characteristic of beta +-thalassemia. This causes
excessive of cellular damage that results in the phenotype of thalassemia intermedia
(Steinberg et al 1987).

43
Hb Dhofar
Hb Dhofar is a variant haemoglobin (beta(29 (GGC-GGT) gly-gly), beta(58 (CCT-CGT)
pro-arg)) associated with a thalassaemic phenotype and unique to the Sultanate of Oman.
Clinical and haematological data suggest that this mutation behaves like a moderately
severe beta(+) thalassaemia allele resulting in a thalassaemia intermedia phenotype (Daar
et al 2008)
1.11.3.9: INSERTION/FRAMESHIFT MUTATION This mutation is characteraized by an insertion of eight nucleotides into exon 2 of beta-
globin gene. As a result of the shift in the protein reading frame, this gene codes for an
elongated beta-globin chain (159 amino acids) with an abnormal amino acid sequence
beyond residue beta 99. Patients with this mutation present with a mild form of beta-
thalassemia intermedia with moderate anemia, evidence of iron overload, severe red cell
morphological changes, significant reticulocytosis, and marked increase in the proportion
of fetal hemoglobin (Williamson et al 1997).
1.11.3.10: MICROSATELLITES AND THEIR FUNCTIONAL PRELEVANCE. Short tandem repeats are abundantly present within the genome. They are commonly
used as polymorphic markers but their potential functional role is poorly understood.
Several of these microsatellites have been described within the beta-globin locus, some
could be involved in controlling gene expression. (TG)n (CG)m dinucleotide repeat
polymorphisms in the two gamma-globin gene IVS2s has been observed and in vivo and
in vitro data demonstrates a possible contribution of the gamma-gene IVS2s polymorphic

44
microsatellites to the variable Hb F synthesis in major haemoglobinopathies
(Lapoumeroulie et al 1999).
1.12: α -Thalassemia
There are two α-globin genes on each chromosome 16 (αα/αα) and, in α+-thalassemia,
one gene of the pair is deleted (−α). Clinical effects are modest as reduced α-globin
chain synthesis in homozygotes (−α/−α) causes only mild anemia (average hemoglobin
level 1–2 g/dl lower than normal), hemoglobin level and red cell indices in heterozygotes
(−α/αα) are often indistinguishable from normal (Weatherall 1981).
Mutations affecting almost every stage of globin gene expression has been described. The
only lesion not yet characterized is the one affecting enhancing sequences, although such
sequences have not yet been identified in the globin gene system. Clinically important
alpha-thalassemias are the deletion types that occur at a much higher frequency than the
nondeletion lesions. In contrast, apart from one deletion beta-thalassemia lesion found in
Pakistan, clinically significant beta-thalassemia lesions are not caused by gene deletion.
The common beta-thalassemia lesions in the Mediterranean region and Asia are caused
by defective mRNA synthesis processing or translation (Kan 1985).
There are less common, “nondeletional” forms of α+-thalassemia caused by mutations
that reduce the output of one or the other α-globin genes. Both variants are associated
with increased levels of the γ4 tetramer (Hb Bart’s) in the neonatal period as a reflection
of excess production of γ chains of fetal hemoglobin (α2γ2) (Allen et al 1997).
In the South West Pacific region, the striking geographical correlation between the
frequency of α+-thalassemia and the endemicity of Plasmodium falciparum suggests that
this hemoglobinopathy provides a selective advantage against malaria in the South West

45
Pacific region, the striking geographical correlation between the frequency of α+-
thalassemia and the endemicity of Plasmodium falciparum suggests that this
hemoglobinopathy provides a selective advantage against malaria. Paradoxically, α+-
thalassemia increases the incidence of contracting mild malaria in the first 2 years of life
(Sammy Wambua et al 2006). The mechanism whereby α+-thalassemia protects against
malaria may have an immunological basis. Parasitised α+-thalassaemic erythrocytes
bound greater levels of antibody from malaria endemic sera and were more readily
phagocytosed by blood monocytes compared with control cells. A greater frequency of
malaria has been found in young children with thalassemia than normals children both in
Papua New Guinea and Vanuatu. In the latter study infestation with, Plasmodium vivax
was increased particularly in children aged <30 months. It was proposed that this increase
may act as a natural vaccine against Plasmodium falciparum. These findings were
confirmed by S. J. Allen and his colleagues in their study Plasmodium vivax infection in
the community control children was more common in homozygous α+-thalassemia.
Increased reticulocyte count in α+-thalassemia may underlie the increased susceptibility
to Plasmodium vivax infection because this parasite only infects reticulocytes (Allen et
al 1997). Although it has been recognized for many years that symptomless carriers are
more resistant to malaria. Retrospective serological analysis has shown a relatively high
frequency of exposure to both Plasmodium falciparum and Plasmodium vivax (Anuja
Premawardhena et al 2004).
1.12.1: Molecular Pathology of Alpha thalassemia
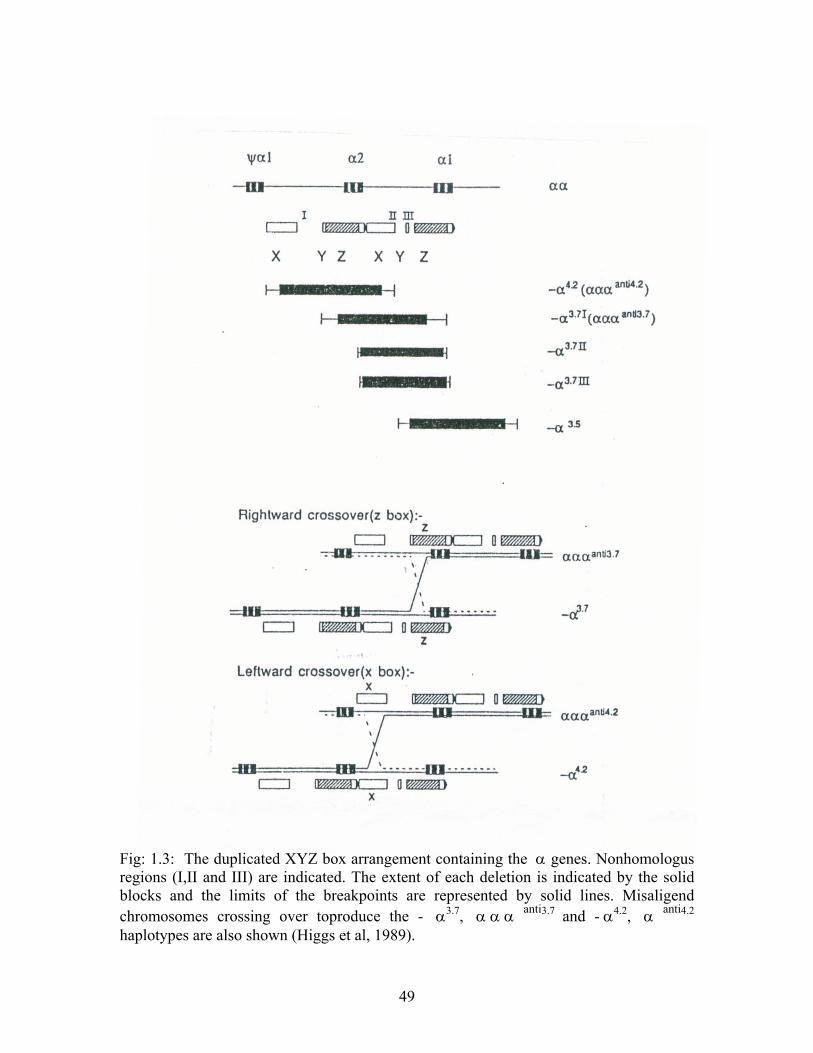
α-2 and α-1 genes are embedded within two highly homologous 4 – kb duplicated
segments. The sequence homology of these regions has been conserved throughout

46
evolution by gene conversion and unequal crossover events (Fig1.3). They are further
subdivided into smaller homologous segments X,Y and Z separated by the non
homologous regions I, II and III (Higgs et al 1989, Higgs et al 1984).
The chromosome with a hybrid α2/ α1 gene (-α 3.7 deletion) results from reciprocal
recombination between Z segments, which are 3.7 kb apart, whereas recombination
between homologous X segments, which are 4.2 kb apart , results in a single α 1 gene ,
thus deleting the entire α 2 gene ( - α 4.2 deletions).
Seven types of α - thal- 2 determinants have been reported to date. The - α3.7 deletion is
the most common and is divided into three type I, II and III, depending on the precise
location of the homologous recombination between the Z boxed (Higgs et al 1984). The
second most common α - thal – 2 is the - α 4.2 determinant which is found at high
frequencies in South china, mainly in the Guangxi (58%) and Jiangxi (29%) (Baysal &
Huisman 1994).
Other less common α - thal – 2 determinants are the - α 2.7 and -α 3..5 deletions. The
former deletes only the α - 1 – globin gene, leaving the α 2 locus intact, while in the
- α 3.5 type, the deletion extends from the 5’ end of the α1 gene to the 5’ end of the θ 1
gene (Baysal and Huisman 1994).
Deletion of proline at alpha37(C2) is predicted to result in severe instability of the variant
hemoglobin, which on interaction with a synthesis-deficient alpha-thalassemia mutation
causes a relatively severe dyserythropoietic anemia, representing an alternative
phenotype associated with highly unstable alpha-chain variants (Traeger-Synodinos et al
2000).

47
According to the output of α - chain, α - thalassemias are classified into αo –
thalassemia with no output of α- globin chains, α- thalassemia – 1, and α + - thalassemia
with a reduced α- chain out put α-thalassemia – 2 (Weatherall 2001b). α - thalassemias
are most frequently due to deletion of the genes and less frequently it results from
mutations involving one or some nucleotides within the structural gene, so called
nondeletional α-thalassemia (αT α or ααT) (Reviewed in Higgs et al 1989).
α - thalassemia – 2 occurs more frequently than any other type of thalassemia and has a
frequency of upto 30% in certain parts of Africa (While in other parts of the world such
as Polynesia mainly Melanesia and remote parts of India α - thalassemia - 2 is inherited
by more than 50 % of all individuals (Baysal and Huisman 1994).
Individuals who inherit two or three functional α-genes (-α/αα, -α/-α or - - /α α) have α
thalassemia trait with a mild hypochromic microcytic anemia (Higgs et al 1989). Those
who inherit one α gene (- - / - α) have HbH disease, a moderately sever hemolytic
anemia with a variable clinical course (Wasi et al , 1974). Those who inherit no α genes
(--/--) develop sever intra – utrine anemia which in the absence of intensive neonatal care
and life – long transfusion (Bianchi et al 1986) results in death at or around the time of
birth, a condition known as the Hb Barts (γ4) hydrops fetalis syndrome (Lie – Injo & Hie,
1960: Higgs et al 1989). Sever determinants (- -) only occur at high frequency in
Southeast Asia, 3.45% to 5.3% of the population are carriers (Hundrieser et al 1988). In
Mediterranean basin less than 1% of the population are carriers (velati et al 1986). Hb
Barts or hydrops fetalis syndrome is observed in 1:300 hospital births (Thumasathit et al ,
1968) and may account for up to 26% of prenatal deaths (Cong & Shong 1982).

48
Alpha-Thalassemia mutations are one of the most common mutations in Man, and they
cause Hb H disease and Hb Barts hydrops fetalis. Hb H disease is not necessarily a
benign disorder as has been generally thought (Chui 2005).
1.12.2: Heterozygous β thalassemia with ααα/αα
This interaction result in a variable phenotype displaying a spectrum from β thalassemia
trait alone, mild type of TI to sever TI. Additional alpha-genes may increase the severity
of heterozygous beta-thalassemia. Co-existence of -alpha3.7 mutations with homozygous
beta-thalassemia may convert a transfusion-dependent thalassemia major to a non
transfusion-dependent thalassemia intermedia (Al Qaddoumi et al 2006). Alpha globin
gene triplication is an important genetic determinant underlying thalassemia intermedia in
North Indians (Panigrahi et al 2006a). However presence of extra copies of alpha-globin
gene has been shown to worsen the degree of anemia in beta-thalassemia heterozygotes.
Therefore presence of triplicated alpha-globin genes should always be considered in
apparent beta-thalassemia carriers who were more symptomatic than expected (Ma et al
2001, Oggiano et al 1992 and Beris et al 1991).
49
Fig: 1.3: The duplicated XYZ box arrangement containing the α genes. Nonhomologus regions (I,II and III) are indicated. The extent of each deletion is indicated by the solid blocks and the limits of the breakpoints are represented by solid lines. Misaligend chromosomes crossing over toproduce the - α3.7, α α α anti3.7 and - α4.2, α anti4.2 haplotypes are also shown (Higgs et al, 1989).

50
1.12.3: Homozygous β thalassemia with ααα/ααα
A triplicated α- gene arrangement behaving as an α - thalassemia alleles was reported by
Higgs and Pressley (1980) from Saudi Arabia.
1.12.4: Heterzygous β thalassemia with ααα/ααα
Homozygous state for the triplicated α -globin arrangement in association with
heterozygous β thalassemia has been observed in the Mediterranean population only.
Patients heterozygous for βo – or sever β+ - thalassemia mutations had clinical picture of
moderate to sever form of thalassemia intermedia (Galanello et al 1983).
1.12.5: Heterozygous β – thalassemia associated with αααα/αα
Heterozygous β thalassemia with inheritance of six α - globin genes in the homozygous
inheritance with the triplicated α globin gene or heterozygous inheritance of the
quadruplicated and normal α - globin – gene complement has been reported (Thompson
et al 1989). This genotype is associated with mild to moderate form of thalassemia
intermedia. Individuals heterozygous for codon 39 CAG-to-TAG mutation are also
heterozygous for a triplicate alpha-globin gene locus (alpha alpha alpha anti 3.7). Thus
compound heterozygous condition of a beta39 C-to-T mutation and triplicate alpha-
globin gene increases alpha:beta-globin chain imbalance and accounts for the presence of
beta-thalassemia intermedia (Rhodes et al 1999, De Angioletti M et al 1992, Advani et al
1992). The greater degree of globin chain imbalance resulting from two additional alpha
chain genes is the likely mechanism for clinically severe phenotype of thalassaemia
intermedia (Thein et al 1984).

51
1.12.6: Dominant β thalassemia associated with ααα/αα
Severe effect of this particular β- thalassemia allele together with the usual degree of
imbalanced globin synthesis results in severe form of TI. In dominant β thalassemia
associated with ααα/αα interaction (Thein et al 1990). Beta-thalassemia heterozygotes
conjuncted with alpha-globin gene triplication was the major cause of beta-thalassemia
intermedia in a Korean family (Chen et al 2000). However combination of a triplicated
alpha globin locus with heterozygous beta-thalassaemia produces a clinical phenotype of
thalassaemia intermedia (Camaschella et al 1987).
1.12.7: Phenoptype effect of Interactions of α and β thalassemia
Relative excess of alpha- over beta-globin chains in the erythroid precursors is the major
pathophysiological feature of homozygous beta-thalassemia. Its clinical picture is usually
characterized by a transfusion-dependent dyserythropoietic anemia (thalassemia major).
However, some patients present with moderate anemia that does not require regular blood
transfusions (thalassemia intermedia). Molecular heterogeneity of beta-thalassemia
mutations and alpha- and gamma-globin gene expression play an important role in
modifying the clinical phenotype. Thus genetic factors do not significantly alter the
clinical phenotype when present alone but ameliorate the course of homozygous beta-
thalassemia when inherited in combination (Kulozik et al 1993).
Excess alpha-globin chains play a major role in the pathophysiology of homozygous
beta-thalassaemia. In beta-thalassaemia carriers a minor effect of alpha-globin chain
excess is reflected in a minimal or mild anaemia without clinical symptoms. Factors that
increase alpha-chain excess in heterozygotes are expected to accentuate the severity of
the clinical and haematological phenotype. Homozygous alpha-gene triplication interacts

52
with a severe beta-thalassaemia mutation to cause an alpha-chain excess equivalent to
that observed in homozygous beta-thalassaemia intermedia. In heterozygotes for severe
beta-thalassaemia mutations with one additional alpha-globin gene, the alpha-chain
excess causes a more pronounced degree of anaemia than is seen in beta-thalassaemia
heterozygotes (Traeger-Synodinos et al 1996).
Major determinants of disease severity in beta thalassaemia, the beta thalassaemia
mutations, with co-inheritance of alpha thalassaemia trait and high HbF determinants act
as ameliorating factors. Presence of an alpha thalassaemia deletion significantly reduces
initial disease severity, although the effect on pubertal development is less clear
(Gringras et al. 1994) Co inheritance of different α - thalassemia alleles modifies clinical
picture of sever β – thalassemia. Significant amelioration in homozygous or compound
heterozygous state (Weatherall et al 1981) is observed when there is coinheritance of one
or more α - thalassemia determinants. How ever co – inheritance of one or more α -
thalassemia alleles less effectively ameliorate thalassemia (Weatherall 2001).
1.12.8: β – thalassemia trait modified by the co – existence of α
thalassemia
Co existence of α thalassemia is important in the context of thalassemia Intermedia.
Significant effect on red cell indices are noted by the co – inheritance of β – thalassemia
with α+ thalassemia (-α / - α ). β – thalassemia with homozygosity for α+ thalassemia
and heterozygosity for αo Thalassemia can result in normal or almost normal
hematological picture with an elevated hemoglobin A2 level. It is assumed that in
heterozygotes the compensatory increase in β - chain production from the β locus in

53
transposition to β chain - thalassemia gene locus is mediated at the transcriptional or
translational levels. These changes are observed in hemoglobinization of the red cells in
βo heterozygous for the deletional forms of α+ thalassemia. It appears that there is a
major increase in β and α globin production from the normal loci. It may however be
appreciated that α and β globin gene loci do not achieve their full capacity for
compensating the defective output of their partner (Weatherall 2001b).
Frameshift mutation in exon 3 of the beta-globin gene, a single nucleotide deletion (-C)
in between codons 140/141 (GCC/CTG→GCC/TG), was found in an 8-year-old
Argentinean girl with clinical picture of thalassemia intermedia. It leads to a beta-chain
that is elongated to 156 amino acids [(141)Trp-Pro-Thr-Ser-Ile-Thr-Lys-Leu-Ala-Phe-
Leu-Leu-Ser-Asn-Phe-(156)Tyr-COOH]. The resulting hemoglobin was named
Hemoglobin Florida (Weinstein et al 2006).
1.12.9: Co-inheritance of homozygous or compound heterozygous β – thalassemia gene with different varieties of α - thalassemia Inheritance of one or more α- thalassemia alleles might reduce the severity of
homozygous or compound heterozygous β - thalassemia. In a study on moderately severe
β- thalassemia intermedia with homozygous or compound heterozygous inheritance of
β- thalassemia 3 were heterozygous for α+ thalassemia deletion (-α/αα) while two were
heterozygous for non- deletion α+ thalassemia alleles (Weatherall et al 1981). A genetic
combination of silent beta-thalassaemia, high Hb A2 beta-thalassaemia and single alpha
globin gene deletion may cause mild thalassaemia intermedia. Reduced alpha globin
chain output results in a more balanced globin chains synthesis which in turn accounts for

54
the mild clinical phenotype (Galanello et al 1984). simple heterozygosity for the common
beta degrees -thalassemia mutation beta39 (C→T), both presenting with a thalassemia
intermedia phenotype has also been reported by Harteveld et al (2008)
βo – Thalassemia
In Cypriot, Sardinian and Asian patients that are homozygotes for βo thalassemia with co
inheritance of two α - globin genes loss, (-α / - α) or non deletional form of α+
thalassemia involving α2 – globin genes more likely to have clinical phenotype of
thalassemia Intermedia than homozygous βo – thalassemia patients with co inheritance of
single α- globin gene are deletion usually have thalassemia major with slightly late onset
clinical presentation.
β+ - thalassemia
Homozygous or compound heterozygous β+ thalassemia with co inheritance of two
α - globin genes and some times with single α globin gene deletion is associated with
milder clinical phenotype. Inheritance of single α-globin gene deletion and non deletional
α thalassemia allele produce contrasting effects. This is because non – deletional
α thalassemia alleles causes greater reduction in α - chain production (Thein et al 1988,
Galaenello et al 1989). Co – inheritance of α thalassemia varies in different population.
In Italian patients co inheritance of deletional forms of α+ thalassemia causes thalassemia
Intermedia (Camaschella and Cappelini 1995) whereas mild form of β - thalassemia
rarely occurs in Israel. This reflects a low frequency α - thalassemia in this population.
Co inheritance of heterozygous state for α+ thalassemia with compound heterozygous
state for IVSI- 6 T→ C / IVSI-110 G → A respectively produces mild and moderately

55
severe phenotypes respectively. Co inheritance of heterozygous state for α+ thalassemia
with homozygousity for IVSI – 110 G→A mutation causes sever form of thalassemia
intermedia.
Co inheritance of single missing α gene with heterozygosity for IVSI – 5G →C / Fr 8 – 9
+ G are associated with extremely mild phenotype (Ho et al 1998). Mutation of the beta-
globin gene initiation codon (ATG→AAG) which should give rise to beta (0)-thalassemia
trait (Waye et al 1996) Point mutation in the polyadenylation signal (AATAAA-
AATAAG) is known to cause a moderately severe beta-thalassaemia phenotype (Rund et
al 1993).
Reasonable correlation exists between the mildness of phenotype and homozygous βo,
severe β+ thalassemia and homozygous α+ thalassemia. However single α - globin gene
has minimal phenotype effect on homozygosity of βo thalassemia.Whereas effect on
compound heterozygous β+ or βo or homozygous β+ has unpredictable effect due to loss
of single α- globin gene. Role of α thalassemia in the generation of thalassemia
Intermedia is dependent upon its relative frequency in the population (Kuloziki et al
1993).
1.12.10: Homozygous β+ or βo thalassemia associated with compound heterozygosity for α+ and αo thalassemia Loukopoulos et al (1978) described the occurrence of moderately severe form of β
thalassemia intermedia by the combination of β thalassemia homozygous with HbH
disease. Furbetta et al (1979) described the association of βo thalassemia homozygous
with HbH disease. Extremely high hemoglobin A2 in these patients as reported may be

56
due to the high levels of δ - chain production, which normally do not reach the peripheral
blood in homozygous β thalassemia.
1.12.11: Pathophysiology
Globin imbalance is central to importance in the severity of HbH disease and
heterozygous αo, α+ and β+ thalassemia show similar degree of anemia and
morphological changes of their red cells and almost the same abnormalities of membrane
function showing the same rate of potassium leak but they show different red cell
survival time. Normal red cell survival is demonstrated in individuals with balanced
globin synthesis but those having unbalanced globin synthesis and HbH disease had
shortened red cell survival (Knox – Macauly et al 1972). This is because excess α -
chains are more injurious to erythroid precursors and their progeny than excess β chains.
Homozygous β thalassemia together with HbH genotype may show grossly hypochromic
red cells with severe degree of potassium leak, almost normal red cell survival, mild
degree of ineffective eryhtropoisis and myeloid / erythroid ratio of 0.55 (Loukopoulos et
al 1978). Membrane association of abnormal or partially oxidized alpha-globin chains has
a more deleterious effect on the membrane skeleton than do beta-globin chains.
Accumulation either of unmatched alpha or beta globin chains in turn causes the
intramedullary and peripheral hemolysis that leads to varying degree of anemia (Yuan et
al. 1995). Hyperactivity of macrophages to chronic hemolysis causes an increase in
cytokines, such as IL-8, found in thalassemia syndromes (Dore et al 1995). Severity of
the clinical course in βO-thalassemia does not correlate with an imbalance between alpha
and gamma chain synthesis in the peripheral blood but is determined by the synthetic

57
ratio in the bone marrow cells where the bulk of hemoglobin synthesis takes place
(Cividalli et al 1978).
Iron overload is an important factor in cellular damage. Studies have shown an enhanced
lysosomal fragility due to increased iron storage (Frigerio et al 1984). Iron-dependent
oxidative reactions in beta-thalassaemic erythrocyte membranes are involved in
premature cell removal and anaemia. Membrane-bound free iron significantly correlates
with bound haemichromes suggesting a causal relation.It is how ever poorly related to
serum non-transferrin iron which seems to contribute little to the damage from outside
the cells. Spleen plays an important role in the removal of cells with more membrane iron
(Tavazzi D et al 2001).
Since the only other route of glucose metabolism in erythrocytes is the pentose phosphate
pathway (PPP), these results indicate that PPP is more active in beta-thalassaemia
intermedia erythrocytes, perhaps as a consequence of their elevated intracellular
oxidative state (Ting et al 1994). Red blood cell membranes from patients with beta-
thalassemia major and intermedia had an average of 25% less sialic acid and a 50%
decrease in titratable SH groups indicating a change in lipid-to-protein ratio. The
difference being due, in part, to increased oxidative stress (Kahane and Rachmilewitz
1976)
Oxidative stress causes the attachment of haemichrome to membrane, thus aggregation of
band 3 and erythrophagocytosis removes RBC (Cappellini et al 1999). Membrane
association of abnormal or partially oxidized alpha-globin chains has a more deleterious
effect on the membrane skeleton than do beta-globin chains (Yuan et al. 1995). Loss of
phospholipid asymmetry, and consequently exposure of phosphatidylserine (PS), is

58
thought to play an important role in the cell pathology. Thus hemichrome binding to band
3, hemichrome-mediated oxidation of band-3 cytoplasmic domains, generation of high-
molecular-weight band-3 aggregates, and enhanced opsonization by anti-band-3
antibodies is a possible sequence of events leading to phagocytic removal of erythrocytes
in thalassemia (Mannu et al 1995, Scott et al 1992,Schrier et al 1989).
Anemia in thalassemias is caused by a combination of ineffective erythropoiesis
(intramedullary hemolysis) and a decreased survival of adult RBCs in the peripheral
blood. This premature destruction of the thalassemic RBC could in part be due to a loss
of phospholipid asymmetry, because cells that expose PS are recognized and removed by
macrophages. Presence of PS-exposing subpopulations of thalassemic RBC are most
likely important, because they could provide a surface for enhancing hemostasis reported,
and mediate the rapid removal of these RBCs from the circulation (Kuypers et al 1998).
Many factors such as iron overload, liver injury, hormonal disturbances and aging affects
lipids and LP pattern in patients with major and intermedia form of beta-thalassaemia
(Papanastasiou et al 1996). It was suggested in a study carried out in France that Anemia
in beta-thalassemia patients targets hepatic hepcidin transcript levels independently of
iron metabolism genes controlling hepcidin expression (Camberlein et al 2008).
Since beta-TI RBCs show essentially normal levels of [Ca2+]i and normal Ca influx,
their high total Ca content should not be associated with any of the deleterious effects
observed in vitro with increased levels of [Ca2+]i (Bookchin et al 1988). The short
platelet life span in addition to reported increased circulating platelet aggregates and
decreased response of TM platelets to aggregating agents suggests in vivo platelet
activation in thalassemic patients (Eldor et al 1989).

59
In transfusion-dependent patients, increased values of serum transferrin correspond to
lesion worsening (Sergiacomi et al 1993). Abnormal metabolism of iron with its
deposition in the tissues and impairment of the vessels of the microcirculatory bed results
in osteoarthropathy in beta-thalassemia (Musaev et al 1991) Extramedullary hemopoiesis
in the lower spinal column may cause low back pain (Gouliamos et al 1991, Papavasiliou
et al 1990, Sakai et al 1990).
Pseudo-Gaucher cells were reported in a bone marrow biopsy of hemoglobin E disease
and a spleen from thalassemia intermedia patients (Sharma et al 2007).
Evidence for the existence of a chronic hypercoagulable state observed in patients with
beta thalassemia intermedia, suggests the expression of a procoagulant surface by
thalassemia intermedia red blood cells may be the major underlying factor giving rise to
platelet and coagulation inhibitor abnormalities in these patients. These alterations were
not related to iron overload or hepatic dysfunction (Bhattacharyya et al 2007). Protein Z,
thrombin-antithrombin complexes, Protein C concentration (PC:Ag) and activity
(PC:Act) were measured. PZ, PC:Ag and PC:Act were significantly lower in thalassemia
major and thalassemia intermedia subjects than in 30 healthy controls (p < 0.001), while
thrombin-antithrombin complex levels were significantly increased and related to PC
levels but not to PZ levels. PZ and PC levels are reduced in thalassemia but only PC has
an effect on the thalassemia hypercoagulable state (Del Vecchio et al 2007). Lipid profile
in TI patients is not influenced by age, sex, liver injury, hemoglobin or ferritin levels; the
higher erythroid bone marrow activity with the enhanced cholesterol consumption could
be the dominant mechanism implicated in the lipid abnormalities of TI patients
(Amendola 2007).

60
1.12.12: Role of Hb F in generating thalassemia Intermedia
Co-inheritance of alpha-thalassaemia, certain variants of the beta-like globin gene cluster
and elevated fetal haemoglobin (HbF) production are all associated with beta(0)-TI
(Chang 2001). Variation in the ability to produce HbF in the postnatal period might be a
major factor in the generation of the phenotype of β - thalassemia Intermedia. Parents of
Thalassemia Intermedia patients had elevated HbA2 levels, but co – existence of an
α - thalassemia gene seemed unlikely (Weatherall et al 1981).
Clinical manifestations of beta-thalassemia (beta-thal) intermedia phenotypes are
influenced by the persistence of fetal hemoglobin (HbF) and by several polymorphisms
located in the promoters of A- and beta-globin genes. The -158GgammaT and the (AT)9
(T)5 alleles were found to be associated with increased levels of HbF in beta-thal carriers,
but not in wild-type subjects (Guida et al 2006).
Mild form of β+ thalassemia Hereditary persistent of fetal hemoglobin appeared to be
segregating (Knox – Macaulay et al 1973). Detailed studies of parents or relatives did not
always evidence for a second determinant of this type (Weatherall 1981).
1.12.13: DETERMINANTS WITH IN β - GLOBIN GENE
CLUSTER
Production of Hb F is related to a particular RFLP haplotype (Labie et al 1985a, Thein et
al 1987) 5’ haplotypes Hind, IIE , Hind III Gγ, Hind III Aγ, Hind II ψβ, and Hind II 3’
ψβ related HbF production in thalassemia Intermedia was determined (table 13. 6 p 566).
In Asian Indians strong association of - + - ++ with β thalassemia Intermedia was found .

61
In Asians it was found in 14 out of 28 β - globin chromosomes from thalssemia
intermedia patients and mainly 10 out of 84 chromosomes from thalassemia major.
Relatively mild clinical course was shown with homozygosity for this haplotype.Where
as in thalassemia major patient’s homozygosity of this haplotype was associated with late
presentation. Absolute level of HbF and heterozygousity and homozygocity for this
haplotype has a likely relationship.
1.12.14: Xmn- I polymorphism
This haplotype is in linkage desequilibrium with Xmn –I polymorphism ( C →T - 158 )
and has been associated with increased expression of Gγ gene in Sickle cell anemia or
β thalassemia of diverse origin (Gilman & Huisman 1985; Harano et al 1985; Labie et al
1985a). Thus homozygosity and heterozygosity for Gγ Xmn I (+) polymorphism might
play an important role in underlying the mild form of homozygous βo thalassemia in
Afro – Asian population and less in Italians. In Sardinia majority of the patients showed
thalassemia intermedia who were also homozygous or heterozygous for the
Mediterranean haplotype IX which contains - + - + + 5’ subhaplotype, Frameshift (- 1bp)
at codon 6 has been associated with this haplotype in this population (Galanello et al
1989). Homozygous state for Gγ Xmn – I (+) site ameliorates the clinical picture of
homozygous β – thalassemia mutation whereas heterozygousity for this state has more
limited effect (Galanello et al 1989).
In Asian Indians a strong association of Gγ Xmn-I (+) polymorphism with the βo-
thalassemia IVSI – I G → T mutation is observed (Ho et al 1998a). Homozygoutes for
this mutation have a slightly milder disease than other forms of βo thalassemia. Many of

62
them become transfusion dependent only in later life. It is believed that the XmnI (+)
polymorphism in the promoter region of the Gγ gene is associated with an increased
capacity for fetal hemoglobin production. In conditions of erythroid “stress”
homozygocity for this polymorphism may sometimes be associated with a milder clinical
course in homozygous or compound heterozygous β thalassemia. However there may be
another region in the β –globin gene cluster that is related to an increased propensity for
fetal hemoglobin production. Phenotypic heterogeneity among patients who carry this
polymorphism is not clear. In sickle cell anemia, different chromosomes carrying Xmn I
site change are associated with widely different levels of fetal hemoglobin production
(Weatherall 2001b). Apart from the deletional and nondeletional forms of HPFH and
occasional cases of γ – globin gene triplication or quadruplication (Yang et al. 1986)
there is no definite evidence that regions other than Gγ – 158 C → T XmnI
polymorphism play a major role in elevating γ – chain synthesis in β thalassemia
intermedia (Weatherall 2001b). Xmn I polymorphism was found in association with this
prevalent mutation and was detected in the homozygous state in majority of the of the
patients homozygous for the IVS-II-1 (G → A) mutation in a study carried out in Iran
(Karimi et al 2002).
C→ T change at position -158 in the promoter of the Gγ gene creates an XmnI cleavage
site (Labie et al 1985b). This substitution causes a high proportion of Gγ chains in HbF.
HbF levels were found to be highest with Xmn – I +/+ individuals. Xmn I +/+ or I
genotype have been associated with increased HbF levels in Yugoslav patients also
(Efremov et al 1987).

63
β –thalassemia Mutations and hemoglobin F production
In rare forms partial or complete deletion of β globin gene involving β – globin gene
promoters may cause elevated HbF level to a degree to produce a mild phenotype.
Severity of beta-thal intermedia and the increased Hb F level are strictly dependent upon
the type of beta-globin gene mutation. No relation is found between Hb F synthesis and
Epo secretion. Mutation Gγ -158 C →T, which is common among patients with beta-thal
intermedia and very rare in thal major patients does not seem, to influence the Hb F
content in beta thal intermedia patients (Mastropietro et al 2002).
1.13: CORFU δβO THALASSEMIA
The Corfu delta betao thalassemia clinically resembles thalassemia intermedia. In the
homozygous state there is a complete absence of hemoglobin (Hb) A and Hb A2 and a
high level of Hb F.β globin gene contains G→A mutation at IVS 1 position 5. Mutation
in the beta globin gene is not the sole cause of the absence of Hb A in Corfu delta beta
zero thalassemia it is more likely that deletion of a 7.2 kilobase (kb) segment containing
part of the delta globin gene and sequences upstream which contains sequences necessary
for the normal activation of the beta globin gene are at fault (Kulozik et al 1988).
1.14: Deletion form of β thalassemia
Usually high levels of HbA2 and significantly increased levels of HbF in heterozygotes
have been associated to at least 17 different β thalassemia mutations particularly if they
involve the β – globin – gene promoter region. It is thought that the absence of β –globin
gene promoters causes upregulation of δ and γ gene by freeing certain particular DNA

64
binding proteins which may be rate limiting (Huisman et al 1997). Del 619 pb at 3’ end
encountered in Northern India gives rise to a mosaic of cells with either one or no
functional beta-globin gene. It extends to a region of frequent loss of heterozygosity
called LOH11A, which is located close to the beta-globin locus. Thus, Loss of
heterozygosity can be a cause of non-malignant genetic disease (Badens et al 2002).
1.15: Point Mutation of the β- Globin gene
γ chain production may be increased in the presence of even very mild β- thalassemia
allele. Fetal hemoglobin production is also increased with mutation on chromosome with
Gγ XmnI (+) polymorphism (Gonzalez- Redando et al 1988). Heterozygotes for the
promoter gene mutations tend to have a slightly higher level of fetal hemoglobin than
those for other forms of β thalassemia.
1.16: HPFH
The most common forms of hereditary persistence of fetal hemoglobin synthesis (HPFH)
and delta betao -thalassemia result from simple deletions of the beta-globin gene cluster
or point mutations in the gamma-globin gene promoters or deletions of 11.5 kb and 1.6
kb or an inversion of 7.6 kb. Larger deletions remove both the delta-and the beta-globin
genes with 3' flanking sequences. While the smaller deletions affect DNA of unknown
function. The Hematologic phenotype and molecular structure of the rearranged
beta-globin gene cluster are consistent with a competitive relationship between fetal and
adult globin genes and/or with translocation of enhancer sequences into gamma-globin
gene region (Kulozik et al 1992). Interaction of hereditary persistence of fetal
hemoglobin HPFH-6 with beta-thalassemia causes thalassemia Intermedia. Much milder

65
phenotype with with Hb E IVS1→5 G-C mutation and G insertion between codons 8/9
and the beta (E)-gene have also been illustrated (Fucharoen S et al 2002).
1.17: Homozygous or compound heterozygous β thalassemia with
heterocellular HPFH
Hereditary persistence of fetal hemoglobin (HPFH) is a condition whereby a
continuously active gamma-globin gene expression leads to elevated fetal hemoglobin
(Hb F) levels in adult (Stamatoyannopoulos and Grosveld. 2001). HbF can be raised in
certain forms of heterocellular HPFH with β thalassemia or Sickle cell gene. In
heterozygous state either alone or together with β thalassemia the augmentation of fetal
haemoglobin production is extremely small. Yet when it interacts in severely affected
homozygous or compound heterozygous state determinants the fetal hemoglobin by
several g/dl (Weatherall 2001b). Genetic determinant(s) of high HbF in the absence of
HPFH is linked to intergenic haplotype T and does not disrupt intergenic transcription
(Papachatzopoulou et al 2006).
1.18: Heterozygous β thalassemia with thalassemia intermedia
phenotype
There are three main mechanisms whereby β – thalassemia heterozygous can be
associated to thalassemia intermedia.
1) β-thalassemia complicated by the inheritance of more than the usual number of
α - globin genes.

66
2) β-globin gene mutations producing products of unusual properties and giving rise
to heterozygous phenotypes now called dominant β thalassemia.
3) Both duplicated and quadruplicated α - globin gene arrangements have little
effect on normal individuals but when inherited with a single β – thalassemia
allele sufficient globin imbalance produces β- thalassemia which is more severe
than heterozygous β – thalassemia.
1.19: Dominant forms of β thalassemia
This group of β thalassemia is transmitted in an autosomal dominant fashion. It is
associated with clinical features and inclusions in the normoblasts and peripheral red cells
after splenectomy (Weatherall et al 1973).
1.19.1: Mechanism of dominant β thalassemia phenotype.
Heterozygous forms of β thalassemia are mild while these are more severe. Nonsense or
frameshift mutations that produce truncated β chains upto 72 residues in length are
usually associated with a mild phenotype which produces long unstable products
reflecting their heme biding properties and molecular instability. In heterozygotes,
mRNAs associated with these mutation are not transported to the cytoplasm and hence no
gene product is produced. mRNA with mutations in exon 3 are transported and translated
normally. Lack of helix H expose one of the batches of helix G and also of helix E and
F.This leads to aggregate and hence precipitates of β chain products in the form of
inclusion are present in the progenitors of these patients (Thein et al 1990). Therefore
these conditions are characterized by ineffective erythropoiesis.

67
1.19.2: Thalassaemia-like carriers not linked to beta-globin
gene cluster
This genetic determinant appears haematologically heterogeneous, displaying either a
silent beta-thalassaemia-like phenotype or a typical beta-thalassaemia carrier-like
phenotype in different families. Compound heterozygosity for both beta-thalassaemia-
like determinant and typical beta-thalassaemia allele result either in thalassaemia
intermedia or thalassaemia major. By linkage analysis both the silent and the typical beta-
like determinants are not linked to the beta-globin cluster. Sequence analysis of the
hypersensitive site cores of locus control region and of the genes coding for the
transcription factors erythroid, Kruppel-like factor and nuclear factor (erythroid-derived
2) were normal. Beta-globin mRNA levels determined by real-time polymerase chain
reaction were reduced in both types of beta-like carriers. These results indicate the
existence of causative genetic determinants that are not yet molecularly defined. They
most likely, result from either a reduction or loss of function of a gene that codes for
unknown transcriptional regulator(s) of the beta-globin gene. The knowledge of these
rare beta-thalassaemia-like determinants have implications for clinical as well as prenatal
diagnosis of beta-thalassaemia (Faa et al 2006).
1.20: Haemoglobin Lepore
Homozygous Haemoglobin Lepore (Hb Lepore) can also cause thalassemia intermedia
(Pasangna et al 2005).

68
1.21: Hemoglobinopathies
Although over 400 variants of structural haemoglobin have been identified, only,
haemoglobins S, C, and E, reach high frequencies in certain populations (Weatherall
2000b). Structural hemoglobin variants typically are caused by a point mutation in a
globin gene that produces a single amino acid substitution in a globin chain.Although
most of them are of limited clinical significance, a few subtypes have been identified
with frequency. Homozygous Hb C and Hb S produce significant clinical manifestations,
whereas Hb E and Hb D homozygotes may be mildly symptomatic. Although
heterozygotes for these variants are typically asymptomatic, their diagnosis may be
important for genetic counseling.Thalassemia, in contrast, results from quantitative
reduction in globin chain synthesis. Those with diminished beta-globin chains are termed
beta-thalassemias while those with decreased alpha-chain production are called alpha-
thalassemias. Severity of clinical manifestations in these disorders relates to the amount
of globin chain produced and the stability of residual chains present in excess. The
thalassemia minor syndromes are characterized clinically by mild anemia with persistent
microcytosis. Thalassemia intermedia (i.e., Hb H disease) is typified by a moderate
variably compensated hemolytic anemia that may present with clinical symptoms during
a period of physiologic stress such as infection, pregnancy, or surgery (Clarke and
Higgins 2000).
Hemoglobin E (beta26Glu → Lys) is the most common hemoglobin (Hb) variant in
Southeast Asia and the second most prevalent worldwide. However in India, it is
prevalent in Bengal and the north-eastern region, but relatively rare in the rest of the
country (Kishore et al 2007).

69
Milder phenotype of HbE/beta-thalassaemia cannot be attributed to co-precipitation of
HbE and excess free alpha-globin chains (Wickramasinghe, Lee et al 1997). Hemoglobin
E beta-thalassemia is an important cause of childhood chronic disease in Southeast Asia.
It is characterized by the presence of hemoglobin E and F and amount of hemoglobin E
ranges from 35% to 75%. These patients are generally classified as having thalassemia
intermedia because they have inherited a beta-thalassemia allele and hemoglobin E which
acts as a mild beta+thalassemia. However, a remarkable variability in the clinical
expression ranging from a mild form of thalassemia intermedia to transfusion-dependent
conditions is observed. Severe hemoglobin E beta-thalassemia may have clinical features
of thalassemia major. Phenotypes of thalassemia major can be predicted from the early
onset of clinical symptoms and the requirement of regular blood transfusion from infancy
(Fucharoen and Winichagoon 2000).
Hemoglobin E is a beta chain variant that has its most significant interaction with
thalassaemia. The compound heterozygous state, thus produced, can result in a
thalassaemia intermedia/major phenotype with affected individuals being transfusion
dependent (Kakkar 2005). Haemoglobin E beta thalassemia (HbE beta thalassemia) has a
remarkable variability in clinical expression ranging from a mild form of thalassemia
intermedia to a transfusion dependent condition (Tyagi et al 2004). Patients with Hb beta
+ [IVS 1-5 (G→C)] clinically present as beta-thalassaemia intermedia and remain
asymptomatic in the absence of blood transfusions. Children who receive blood
transfusion develop progressive iron loading with age. Serum ferritin and serum alanine
transaminase levels are significantly raised in patients who receive blood transfusions. In
the presence of blood transfusion but with out adequate iron chelation therapy,

70
splenectomy becomes an inevitable procedure at some stage of the disease (George and
Wong 1993)
Since Hb Malay migrates with HbA on electrophoresis and chromatography, this variant
ought to be included in the differential diagnosis of beta-thalassemia major or intermedia
in subjects of Southeast Asian descent who have HbA on hemoglobin electrophoresis.
The possible presence of this mutation should also be considered for genetic counseling
in couples at risk (Ma 2000). Hb O-Arab (beta 121 Glu→Lys) and a beta
zero-thalassaemia trait has also been found to be associated with thalassemia Intermedia
(Morle et al 1984). Retrospective serological analysis has shown a relatively high
frequency of exposure to both Plasmodium falciparum and P. vivax (Anuja
Premawardhena1 et al 2004).
1.22:EF Bart's disease EF Bart's disease is an uncommon form of thalassaemia intermedia resulting from the
co-inheritance of alpha-thalassaemia and haemoglobin E in the same subject. DNA
mapping and haemoglobin electrophoresis indicate that there are four genotypes
involving 5 abnormal globin genes, responsible for this thalassaemia syndrome
(Fucharoen et al 1988)
1.23: Hb Vicksburg
A new hemoglobin variant, Hb Vicksburg, produces the phenotype of thalassemia
intermedia in patients who are doubly heterozygous for this variant and
betao-thalassemia. Structural analysis of Hb Vicksburg demonstrate deletion of leucine at
beta 75 (E19). Hb Vicksburg is expected to comprise the major portion of the hemolysate

71
in the patients because of the presence of beta 0-thalassemia on the trans chromosome,
but it comprises only 7.6%. Thus, Hb Vicksburg is synthesized at a rate lower than that
expected on the basis of gene dosage. Deletion of beta 75, for a number of reasons, is not
expected to lead to diminished synthesis of the variant. The most plausible explanation
for the low output of Hb Vicksburg is that a mutation for beta +thalassemia is present in
cis to the structural mutation (Adams and Steinberg 1981).
1.24: Pregnancy
Intrauterine growth restriction (IUGR) complicates more than half of the pregnancies
with TI. Red cell transfusion is needed in most cases even in non-transfusion-dependent
patients. Postpartum splenectomy might be necessary in some patients (Nassar et al
2006). How ever in one case pregnancy could not follow its normal course and was
interrupted during the 24th week because of intrauterine death of the fetus due to
haemolytic anaemia following transfusional, alloimmunisation and portal vein
thrombosis. Therefore all thalassemic patients should be tested for various blood antigen
systems so that transfusion reactions can be avoided (Bianconcini et al 1993).
1.25: Genotype phenotype relationship
Prediction of beta-thalassaemia major phenotype from beta-genotype is generally
relatively straightforward. However, despite the ability to accurately define the beta-
thalassaemia mutations, prediction of a beta-thalassaemia intermedia phenotype from the
genotype sometimes remains problematic this has important implications in genetic
counseling and prenatal diagnosis. The clinical expression of beta-thalassemia
intermedia is variable and complications are more frequent than in the minor form.
Thromboembolism risk increase after splenectomy (Pierre 2006).

72
Classical phenotype of heterozygous beta-thalassemia may be modified by a number of
environmental and genetic interacting factors. Some of these are (1) coinheritance of
alpha-thalassemia which may normalize the red blood cell indices; (2) presence of a mild
beta-thalassemia mutation; (3) cotransmission of delta-thalassemia which may normalize
the raised HbA2 typical of heterozygous beta-thalassemia and (4) presence of a silent
mutation which can be defined only by imbalanced beta-globin chain synthesis. A
number of molecular mechanisms can produce non transfusion dependent thalassemia
syndromes referred to as thalassemia intermedia. The most common are homozygosity
for mild beta-thalassemia mutations, coinheritance of alpha thalassemia with
homozygous beta-thalassemia or genetic determinants that ensure continuous production
of HbF in adult life or the presence of heterozygosity for hyperunstable globin variants
(Cao A et al 1994). Molecular basis of thalassemia intermedia, are very heterogeneous,
but in general any factor that is capable of reducing the globin-chain imbalance results in
a milder form of thalassemia. These factors include the presence of a silent or mild beta-
thalassemia allele associated with a high residual beta-globin chain production.
Coinheritance of alpha-thalassemia or genetic determinants that increase the gamma-
chain production may cause mild thalassemia. Less frequent mechanisms are double
heterozygosity for beta-thalassemia and triplicated alpha genes, and the presence of a
hyperunstable hemoglobin variant. For significant number of βo thalassemia
homozygotes with a thalassemia intermedia phenotype the modifying factors have not
been defined. In contrast, there are simple beta-thalassemia carriers who, for unknown
reasons, have an unusually severe clinical phenotype (Galanello and Cao 1998) In a
patients with nondeletion genotype the analysis of hematological values revealed lower

73
levels of RBC as well as HbA2 with significantly higher levels of Hb H. Clinical
variability was remarkable ranging from totally asymptomatic conditions to severe
thalassemia Intermedia characterized by marked hemolytic crises, liver and spleen
enlargement and the necessity for frequent transfusions. The genotype do not justify the
gravity of the phenotype in every case, differences in clinical manifestations are notable
and are not easily explainable in subjects apparently having the same genotype (Mirabile
et al 2000). Individuals heterozygous for beta +33 C-G mutation alone are clinically and
hematologically silent with normal red blood cell indices and normal levels of
hemoglobin (Hb) A2. A direct relationship between genotypic and phenotypic severity is
clearly demonstrated in these cases with obvious implications for prenatal diagnosis (Ho
et al 1996).
Alpha-thalassemia is phenotypically and genotypically heterogeneous. DNA analysis is
invaluable as it provides a specific diagnosis and enables reliable genetic counseling.
Clinical presentation of individuals carrying two or more alpha-globin lesions was highly
variable. In general, the severity correlates inversely with the number of functional alpha-
globin genes. In some cases, impairment of two alpha-globin genes by point mutations
may lead to a thalassemia-intermedia-like picture which could be misdiagnosed as beta-
thalassemia (Oron-Karni et al 2000). The severity correlated inversely with the number
of functional alpha-globin genes.
1.26: Geographical distribution
Thalassemia Intermedia is observed in Indians and Pakistanis (Weatherall et al 1981b),
Arabs (Cividalli et al 1978; Weatherall et al 1981a), Britons (Knox – Macaulay et al
1973), Algerians (Godet et al 1977) and Italians (Bianco et al 1977).

74
1.27: Thalassemia Intermedia In India
Pattern of mutations in Uttar Pradesh differed from those in other Indian states and in
families who migrated from Pakistan. Frequency of IVS-I-5 (G→C) and 619 bp deletion
mutations was 64.3 and 2.5% respectively in families from Uttar Pradesh compared to a
prevalence of 37.5 and 27.5% respectively in Pakistani immigrants. Of the 10 common
Asian Indian mutations, eight were observed in subjects studied from different parts of
India (Agarwal et al 2000). In another study carried out by Verma et al 91.8% of the
subjects had one of the five commonest mutations [IVS-I-5 (G→C), 34.1%; 619-bp
deletion, 21.0%; IVS-I-1 (G→T) 15.8%; codons 8/9 (+G), 12.1%, and codons
41/42 (-CTTT), 8.7%. 5.9% of the subjects had a less common mutation(Verma et al
1997, Agarwa et al 1994) In a study carried out by Panigrahi et al, the possible molecular
basis was (i) co-existent α-deletions (n=16/50), (ii) homozygous XmnI polymorphism
(n=17/50), (iii) both factors (n=3/50), and (iv) milder beta-alleles (n=9/50) in
homozygous beta-thalassemia (total 50 cases). In heterozygous beta-thalassemia, alpha
alpha alpha anti-3.7 triplication was the predominant factor (Panigrahi I et al 2006b).
Unstable hemoglobin should be suspected in a patient with thalassaemia Intermedia
phenotype even if both parents are haematologically normal (Dash et al 2006).
Hemoglobin E is very common in north-east India with relatively fewer reports from rest
of the country. Reports of hemoglobin E in the Punjabi population are even rarer.
Hemoglobin E is a beta chain variant that has its most clinically significant interaction
with thalassaemia. The compound heterozygous state thus produced can result in a
thalassaemia intermedia/major phenotype with affected individuals being transfusion
dependent (Kakkar 2005). Alpha globin gene triplication is an important genetic

75
determinant underlying thalassemia intermedia in North Indians. Patients with alpha-
triplication may develop jaundice with marked increase in serum bilirubin following
antecedent aggravating factors (Panigrahi et al 2006a). Most common
hemoglobinopathies observed in 1015 cases were sickle cell trait (29.8%), sickle cell
disease (7.5%), sickle cell-beta-thalassemia (1.7%), beta-thalassemia trait (18.2%),
thalassemia major (5.3%), thalassemia intermedia (0.9%), Hb E trait (0.9%), Hb E
disease (0.3%), E-beta-thalassemia (0.7%), Hb D trait (0.2%) and SD disease (0.2%).
Sickle cell disorders with high level of fetal hemoglobin were common in general castes
(0.3-20.7%), scheduled castes (0-8.9%) and scheduled tribals (0-5.5%).Transfusion
dependent beta-thalassemia syndrome was prevalent in Brahmin, Karan, Khandyat, Teli,
etc. Most of the cases belonged to Anugul district, followed by Khurda, Nayagarh,
Phulbani, Cuttack, Jajpur, Dhenkanal, Ganjam, Keonjhar, Mayurbhanj, etc. The
heterogeneous population harbors almost all major hemoglobinopathies in general castes,
scheduled castes and tribes that belong to Coastal and South-Western regions of Orissa
(Balgir 2005).
Nucleotide -88 (C→T) promoter mutation is a common beta-thalassemia mutation in the
Jat Sikhs of Punjab, India (Garewal et al 2005). Hb Hofu [beta 126(H4)Val→Glu]- βo-
thalassemia [codons 8/9 (+G)] combination were identified in a Thalassemia Intermedia
patient (Pande et al 1995). The six severe and common Indian mutations seen in
Thalassemia Intermedia patients are IVS 1-5 (G→C), 619 bp deletion, IVS 1-1 (G→T),
codons 8/9 (+G), codon 15 (G→A), codons 41/42 (-CTTT). Majority of the severe and
mild TI group, IVS 1-1 (G→T), codon 30 (G→C), capsite +1 (A→C), poly A (T→C),
-28(A→G), and -88 (C→T). Four mild mutations in combination with other severe β+ or

76
βo mutations resulted in a very variable clinical presentation. This indicates that, in
majority of Indian patients beta genotype cannot predict the phenotype (Colah et al
2004). Other mutations found in India are frameshift codon 55 (+A) in Maharashtrans
and a frameshift codons 47- 48 (+ATCT) in Punjabis (Garewal et al 1994).
It was observed that the nature of beta-thalassemia mutations was not very different
between the beta-thalassemia major and beta-thalassemia intermedia groups. How ever
co-inheritance of one or more alpha-globin gene deletions (-alpha 3.7) and the presence of
the XmnI polymorphism were associated with less severe disease in Indians (Nadkarni et
al 2001). Nine different types were found, of which six were associated with betao, one
with severe beta+ and two with mild beta+ thalassaemia. Comparison of the beta-globin
gene cluster haplotypes, alpha globin genotypes and beta gene mutations of the
thalassaemia major group with the thalassaemia intermedia group suggests that the
co-inheritance of a high Hb F determinant associated with the - + - + + 5' beta haplotype
and the inheritance of a mild beta-thalassaemia mutation are the major ameliorating
factors of disease severity in Asian Indians (Thein et al 1988). Coexistence of a novel
beta-globin gene deletion (codons 81-87) with codon 30 (G→C) mutation was also
identified in an Indian patient with βo-thalassemia (Shaji 2002). A 10329 base pair
deletion which results in the loss of 5' beta promoter region and the entire beta-globin
gene is associated with unusually high levels of Hb A2 in the heterozygous state (Craig et
al 1992). Hb Lepore has also been reported in the Indian population (Shaji et al 2003).
Alpha Thalassemia alpha globin gene triplication is an important genetic determinant underlying thalassemia
intermedia in North Indians (Panigrahi et al 2006a) In heterozygous beta-thalassemia,

77
alpha alpha alpha anti-3.7 triplication was the predominant factor (Panigrahi I et al
2006b). One of the possible molecular mechanism of the effects caused by Yisui
Shengxue Granules is that it can up-regulate the expression levels of alpha-hemoglobin
stabilizing protein (AHSP) and erythroid transcription factor GATA-1 mRNAs, enhance
the protein synthesis of AHSP which can bind the relative excess free alpha-globin,
prevent the formation of alpha -globin-cytotoxic precipitates in red blood cells and
decrease the hemolysis (Liu 2006).
Unstable Hemoglobin Hemolytic anaemia due to unstable hemoglobins arising from spontaneous mutation has
been reported (Dash et al 2006)
Hemoglobin E Hemoglobin E is very common in north-east India (Kakkar 2005). In one study most
common hemoglobinopathies observed amongst 1015 cases were: sickle cell trait
(29.8%), sickle cell disease (7.5%), sickle cell-beta-thalassemia (1.7%), beta-thalassemia
trait (18.2%), thalassemia major (5.3%), thalassemia intermedia (0.9%), Hb E trait
(0.9%), Hb E disease (0.3%), E-beta-thalassemia (0.7%), Hb D trait (0.2%) and SD
disease (0.2%). Sickle cell disorders with high level of fetal hemoglobin were common in
general castes (0.3-20.7%), scheduled castes (0-8.9%) and scheduled tribals (0-5.5%).
Heterogeneous population harbors almost all major hemoglobinopathies in general castes,
scheduled castes and tribes that belonging to Coastal and South-Western regions of
Orissa (Balgir 2005).

78
δβ thalassemia
Deletion within the beta globin gene complex starts 3 kilobases from the 3' end of the
Aγ gene. The deletion removes the delta and the beta globin genes and continues to a
variable extent in the 3' direction. Heterozygotes for this deletion have about 25% Hb F
with a G gamma:A gamma ratio of 70:30. Iinteraction with beta+ thalassaemia results in
the clinical picture of thalassaemia Intermedia (Wainscoat et al 1984).
1.28:Thalassemia Intermedia In China
Seven different mutations have been identified and the A to G substitutions in the TATA
box of beta-globin gene account for 42% of these mutant beta-globin genes. Most
patients have a beta(+) thalassaemia and one copy of TATA box mutation.
β0 thalassaemia intermedia the mild phenotype may be explained either by the presence
of the - + - + + 5' beta-globin gene cluster haplotype which contains the XmnI site -158 nt
to the Gγ-globin gene or by the number of alpha-globin genes present (Antonarakis et al
1988). Codons 41/42 (-CTTT) beta0-thalassemia and nt - 28 (A → G)beta(+)-thalassemia
mutations together with concurrent (SEA) alpha-thalassemia (SEA) deletion has been
identified in China. A novel mutation of -73(A→T) in the CCAAT box of the beta-
globin gene has been identified in a patient with the mild beta-thalassemia intermedia
(Chen et al 2007).
Hb Westmead is a common alpha-globin gene mutation in this population apart from Hb
Constant Spring and Hb Quong Sze. (Wong et al 2004).
(G)gamma((A)gammadeltabeta)(o)-deletion and beta-thalassemia (cd 41-42), Hb H

79
disease (genotype -alpha(3.7)/-(SEA) have also been identified. Patients
possessing(G)gamma((A)gamma delta beta)(o)/beta-thalassemia were not transfusion
dependent. This could be due to co-inheritance of alpha-thalassemia-2 (genotype-alpha
(3.7)/alphaalpha) in children together with their compound heterozygous condition (Tan
Jin Ai et al 2003). In a study carried out in Hong Kong 5.0 percent of the subjects were
carriers of alpha-thalassemia of whom 4.5 percent were carriers of the Southeast Asian
type of deletion in which both alpha-globin genes on the same chromosome 16 are
deleted. 3.4 percent were carriers of either beta-thalassemia or a mutation that codes for
hemoglobin E. 0.3 percent were carriers of both alpha- and beta-thalassemias (Lau et al.
1997). Another mutation (GAA-TAA) in codon 121 that produces a thalassemia
intermedia phenotype with inclusions in erythrocytes has also been identified in Hong
Kong (Wakamatsu et al 1994). The molecular defects of beta- thalassemia intermedia in
Guangdong Province were highly heterogeneous and its spectrum was different from the
reports from other provinces of China (Zhang L et al 2007).
Alpha Thalassemia
Marked genetic and clinical variability of Hb H syndrome is due to the molecular
heterogeneity of alpha-thalassemia (thal). The hallmark is the presence of excess beta
chains forming Hb H (β tetramer). Classical Hb H disease in Chinese presents as "alpha-
thalassemia intermedia" and is due to a double heterozygosity for two deletional forms of
alpha-thal, alpha-thal-1 and alpha-thal-2. Majority of cases with alpha-thal-1 defect have
a deletion of at least 18.1 kb starting 3' to the zeta 1 gene which includes the psi alpha
and the two alpha genes; it is similar to that described in Thais. However deletion of the
entire zeta-alpha gene cluster (zeta-alpha-thal-1) has also been described. In alpha-thal-2

80
defects, the rightward deletion (alpha -3.7 kb, all type I defects) is more common than the
leftward deletion (alpha - 4.2 kb); one of the latter is associated with Hb Q. A few alpha-
thal defects belong to the nondeletion type, the most common being Hb Constant Spring
(CS). This anomaly, when coinherited with alpha-thal-1, produces Hb H-CS disease
which is associated with marked anemia and splenomegaly due to the instability of alpha-
CS chain. Hb Quong Sze produces alpha-thal-2 phenotype because of the unstable alpha-
Quong Sze chain. Classical Hb H disease and Hb New York (NY) [alpha
113(G15)Val→Glu] with severe anemia, requiring frequent blood transfusions due to the
deleterious effect of increased alpha-NY chain turnover has also been described (Chan et
al 1988).
A rare thalassemia intermedia case caused by co-existence of Hb H disease (--(SEA)/-
alpha(4.2)) and beta-thalassemia major (beta (CD17A)>T/beta (IVS2-654C)>T) has also
been indentified. The practice of prenatal diagnosis in this case may also provide
reference for diagnosis of similar cases (Li et al 2008).
Hemoglobin Constant Spring (Hb CS) and Hb Paksé, two abnormal Hbs characterized
by elongated alpha-globin chains resulting from mutations of the termination codon in the
alpha2-globin gene, are the most prevalent nondeletion alalpha-thalassemias in Southeast
Asia. Hematological findings confirm the mild thalassemia intermedia phenotypes for
pure homozygous Hb CS and homozygous Hb CS with Hb E heterozygote and Hb E
homozygote (Singsanan et al 2007).

81
1.29: Thalassemia Intermedia In Japan
Thalassemia is relatively rare in northeast Asia including Japan where malaria is
uncommon. However, thalassemia in Japan has peculiar mutation spectrum and
characteristics. Most β-thal patients in Japan are heteorozygotes and have thal minor
phenotype. They are many a times misdiagnosed as iron deficiency anemia. Thirty-four
mutations of β-thal have thus far identified, ten of which accounts for 80% of beta-thal
carriers. Among them 60% are native to Japanese while 40% are probably from abroad.
One exception is homozygosity for -31G-A which leads to thal Intermedia. More than
half of the patients with alpha-thal are of Southeast Asian type, but mutations in the
remaining patients seem to be unique to the patients of Japanese descend. Thus Japanese
thalls have dual origin. Frequency of beta-thal is 1 in 600 to approximately 1 in 1,000 and
that of alpha (+)-thal (- alpha/) is 1 in 400, alpha-thal trait (- alpha/- alpha) is therefore
extremely rare. Another alpha-thal trait (- -/alpha alpha) is one fifth of beta-thal.
Seventeen families of HbH disease (- -/- alpha) were found. Many of them are related to
Southeast Asia (Hattori 2002).
In one study that was carried out in 100,000 samples 29 contained electrophoretically
detectable abnormalities in the heterozygous state condition. 12 had beta-thalassemia, 3
alpha-thalassemia, 1 delta-thalassemia. Among 45 carriers of beta-thalassemia from 12
families, 5 were noted to have thalassemia intermedia (Imamura et al 1980).
1.30: Thalassemia Intermedia In Lebanon
Three factors: mild beta-globin gene mutations, deletions in the alpha-globin gene and
the presence of a polymorphism for the enzyme Xmn I in the Ggamma-promoter region

82
have been observed.: 68% of patients have a mild beta+ mutation (IVSI-6, cd29, -88 or
-87), while 26% of patients are positive for the Xmn I polymorphism associated with
increased production of HbF, which shows strong linkage to particular mutations
(IVSII-1, cd8 and cd30).
1.31: Thalassemia Intermedia In Thailand
Haemoglobin (Hb) Hope [beta136(H14)Gly→Asp(GGT→ GAT)] is one of the unstable
haemoglobin variants of the beta-globin chain, which is demonstrated in people of
various ethnic backgrounds. This hemoglobin was reported in a Thai female patient with
clinical thalassaemia intermedia since childhood. Homozygous Hb Hope without
abnormal alpha globin chain involvement, heterozygous Hb Hope in association with
-alpha(3.7) mutation, in clinically silent individuals were identified (Sura et al 2007).
Phenotypes with globin genotypes in patients with HbH disease in northeast Thailand
was studied. It was found that most prevalent molecular defect was interaction of
alpha-thalassemia 1 (SEA type) with the Hb Constant Spring followed by deletion of
three alpha-globin genes with the SEA type alpha-thalassemia 1 and the 3.7- or 4.2-kb
deletion of alpha-thalassemia 2 (14 of 52 patients) and the interaction of the SEA alpha-
thalassemia, Hb Pakse, Hb CS. (Boonsa et al 2004).
An unusual form of thalassemia intermedia caused by interaction of the hemoglobin
Constant Spring (Hb CS), homozygous Hb E and alpha degrees -thalassemia found in
two unrelated pregnant Thai women has been reported (Fucharoen et al 2007).
1.32: Thalassemia Intermedia In Brazil
Main hereditary hemoglobin disorders of clinical significance in Brazil are sickle cell
disease and beta-thalassemia. Sickle gene was introduced by the slave trade whereas

83
beta-thal was introduced later due to a massive migration (mostly by the Italians)
between 1870 and 1953 to the southeast Brazil. Molecular studies performed in the
southeast of the country showed a marked prevalence (47 – 54%) of the nonsense
mutation at codon 39 (C → T), leading to severe forms of betao-thal. However, the
northeast region of the country has a different demographic history characterized by the
insignificant Italian migration. Owing to this and since the majority of cases of beta-thal
in Pernambuco (a state located in the northeast of the country) have mild or intermediate
clinical and laboratory features a different spectrum of beta-thal mutations in this region
is likely as reported by the fact. 33.3% had beta-thal intermedia, 33.3% had HbS/beta-thal
and 23% were beta-thal trait individuals. IVS-I-6 (T →C) 62.8%, IVS-I-1 (G →A)
15.1%, IVS-I-5 (G → C) 9.3%, IVS-I-110 (G → A) 8.2%, codon 39 (C → T) 3.5%, and
codon 30 (AGG → AGC) 1.1% are amongst the common mutation (Araujo et al 2003).
1.33: Thalassemia Intermedia in Greece
A major factor modifying the clinical expression of homozygous high-HbA2
beta-thalassemia in Greece is the inheritance of mild beta-thalassemia mutations.
Although there is not always a complete correlation of genotype with clinical phenotype,
the inheritance of two mild beta-thalassemia alleles results in almost all cases (11 of 12
cases in one study) in thalassemia intermedia phenotype (Kanavakis et al 1995). In
another study carried out in Greek patient two mutations, IVS-I-1 (G→A) and a C→G
mutation at position 6 3' to the terminating codon (term + 6) were identified suggesting
that C→G mutation in this untranslated region of the beta-globin gene causes a slight
decrease in the stability of the mRNA which becomes clinically important only in
situations where beta chain synthesis in trans is eliminated (Jankovic et al 1991).

84
Homozygous beta ++ has also been found to be associated with thalassemia Intermedia
(Kattamis et al 1982).
1.34: Thalassemia In Pakistan Pakistan has a population of more than 160 million people with an overall carrier
frequency of approximately 5.6% for beta-thalassemia. Punjab is the largest province of
the country with more than 50% of the population. The state of beta-thalassemia is
alarming as consanguinity rate is very high (>81%) and the literacy rate is low in South
Punjab. A thalassemia prevention program is the need of the hour in this part of Pakistan
(Baig et al 2006). It is observed that 58% of the siblings of beta-thalassaemia major
children in Hazara division of Pakistan had beta-thalassaemia trait (Anjum et al 2001).
Serum ferritin estimation in carriers of beta- thalassaemia trait is important (Qureshi et al
1995). Two forms of hypochromic microcytic anaemia i.e. iron deficiency and
beta-thalassaemia trait are common in Pakistan. Iron deficiency was found in 9% while
beta-thalassaemia was seen in 3% in the study carried out by Afroz et al (1998). Ratio
between the percentages of microcytic hypochromic cells as a screening test is an easy,
reliable and sensitive index which can be used for mass screening of beta thalassaemia
trait particularly in a population where iron deficiency is also prevalent (Saleem et al
1995). Thalassemia trait is found in 11% of the Pakistani population (Molla et al 1992).
Khattak MF and Saleem M. reported the incidence to be 5.4%. In Pathans the incidence
was 7.96% while in Punjabis it was 3.26% prevalence rate (Khattak and Saleem 1992)
most frequent mutations were IVS-1 position 5 (G-C), codons 8/9 (+G), IVS-1 position 1
(G-T), codons 41/42 (-CTTT) and the 619 bp deletion at the 3' end of the gene. Mutations
at IVS-2 position 1 (G-A) and codon 30 (G-C), previously were described in Asian

85
Indians in 1991 (Varawalla et al 1991). Two other mutations IVS-1 nt.5 (G-C) and codon
8-9 (+G) mutations were identified in 1995 (Khan et al 1995).
Five most common mutations identified in Pakistan are IVS1-5 (G-C), IVS1-1 (G-T), Fr
41-42 (-TTCT) Fr 8-9 (+G) and deletion 619 bp (Khateeb et al 2000). Molecular basis of
beta-thalassemia in Thailand, Pakistan, Sri Lanka, Mauritius, Syria, and India, has been
studied. The results confirm and extend earlier findings for Thailand, Pakistan, India,
Mauritius and Syria. Two novel mutations were identified, codon 55 (-A) and IVS-I-129
(A→C), both found in Sri Lankan patients. Two beta-thalassemia mutations were found
to coexist in one beta-globin gene: Sri Lankan patients homozygous for the betao codon
16 (-C) frameshift were also homozygous for the beta+ codon 10 (C →A) mutation.
Studies of Sri Lankan, Pakistani, and Indian carriers suggest that codon 10 (C →A)
mutation is a rare polymorphism on an ancestral allele in which betao codon 16 (-C)
mutation has arisen. Each country was found to have only a few common mutations
accounting for 70% or more of the beta-thalassemia alleles (Old et al 2001). For
β - thalassemia a gene over 4000 homozygotes are born each year in Pakistan.
β-thalassaemia alleles from five major ethnic groups of Pakistan have been
characterized. The complete spectrum comprises of 19 mutations. There are important
ethnic and regional differences in the prevalence of mutations. The five most common
mutations, IVSI-5 (G-C) (37.3%), Fr 8-9 (+G) (25.9%), del 619 (7.0%), Fr 41-42 (-
TTCT) (6.7%) and IVSI-1 (G-T) (5.4%), constitute 82.3% of the total thalassemia
population in Pakistan. Fr 8-9 (+G) is the most common mutation in Northern Pakistan
(41.3%), whereas IVSI-5 (G-C) is the most frequent mutation in Southern Pakistan
(52.2%). A novel 17 bp deletion involving Cd126-13 was also identified (Ahmed et al

86
1997). The IVS-I-5 (G→C) Asian Indian mutation was the most frequent mutation
reported from United Arab Emirates (el-Kalla and Mathews 1993) Determination of
beta-globin gene haplotypes in North-west Pakistan, Gujarat, Punjab and Sindh suggest
that high frequency alleles i.e. intervening sequence 1 (IVS-1) nucleotide 5 (G-C) and
codons 41/42 (-CTTT) are older mutations as determined by multiple haplotype
associations and widespread geographical distribution. Microepidemiology of
beta-thalassaemia in this region reflects considerable ethnic diversity, gene flow from
population migration and natural selection by malaria infection (Varawalla et al 1992).
Spectrum of mutations is heterogeneous in this population and 19 different mutations in
all ethnic groups are identified. The four most common mutations, IVS-I-5 (G→C)
(37.7%), codons 8/9 (+G) (21.1%), 619 bp deletion (12.4%), and IVS-I-1 (G→T) (9.5%),
account for 80.7% of the alleles. There are differences between ethnic groups and also
between provinces. Amongst the four provinces of Pakistan, IVS-I-5 (G→C) mutation is
more prevalent in Sindh and Balochistan, bordering India in the south and Iran in the
southwest, while codons 8/9 (+G) mutation is more common in Punjab and the North
West Frontier Province, bordering India in the northeast and Afghanistan, respectively.
619 bp deletion is (46%) in Gujratis and Memons residing in the Province of Sindh,
neighboring the Indian Gujrat (Khan and Riazuddin. 1998). β-thalassemia mutation
codon 45(-T) has also been identified in Pakistan (El-Kalla and Mathews 1997).
Population and genetic studies suggest the origin for the Indian deletion beta thalassaemia
(600 bp deletion involving the 3' end) in people from Sindh and the adjacent area of
Gujarat (Thein et al 1984).

87
Alpha-Thalassaemia
Frequency of –alpha (3.7) allele was found to be 8.3%. Ethnic differences were
statistically significant for Pashtoons vs. Balochis and Pashtoons vs. Sindhis. In this
group, 24.6% of the patients had one or two alpha genes deleted. Prevalence –alpha (4.2)
was found to be 0.2% while that of alpha alpha alpha (anti3.7) allele was 0.9%.
The –alpha (4.2) allele was found only in Sindhis, while alpha alpha alpha (anti3.7) was
present in Punjabis, Sindhis and Balochis. 22.4% of the patients were with triplicated
alpha genes (Khan et al 2003).
Level of Hb Bart's is directly related to the inheritance of alpha-thalassaemia gene. Hb
electrophoresis for Hb Bart's in cord blood is a very simple method of determining out
the prevalence of alpha-thalassaemia gene in a given population. 2.4% of the general
population was found carrier of alpha-thalassaemia gene of these. 75% had alpha-
thalassaemia-2 gene and 25% were carriers of alpha-thalassaemia-1 gene (Rehman et al
1991). Prevalence and molecular basis of alpha thalassaemia in the British South Asian
population was determined. Of the 266 subjects in whom gene mapping was performed,
28 had a single alpha+ thalassaemia deletion and one was homozygous for this deletion
(gene frequency 0.056). Half of the heterozygotes had normal mean cell haemoglobin
(MCH) values. 16 subjects had probable non-deletional alpha+ thalassaemia none had
alphao thalassaemia (Hassall et al 1998). The common beta-thalassemia lesions in the
Mediterranean region and Asia are caused by defective mRNA synthesis, processing, or
translation (Kan 1985).

88
A rare alpha2-globin chain variant, Hb Sallanches [alpha104(G11) Cys→Tyr], in a
Pakistani family having three homozygous patients with transfusion-dependent Hb H
disease were identified. This variant, previously reported in a French patient and a West
Indian homozygous patient with Hb H disease, is due to a mutation at codon 104
(TGC→TAC) (Khan 2000).
δβ-Thalassaemia
Delta beta-thalassaemia is a rare disorder in Pakistan. Although the clinical picture is
very mild its combination with beta-thalassaemia trait can produce a sever transfusion
dependent thalassaemia. DNA based diagnosis is possible in the prenatal as well as the
postnatal period. Haematological and genetic features of δβ-thalassaemia in Pakistani
patients were characterized. In the study all heterozygotes and 4/6 homozygotes were
asymptomatic. One homozygote had thalassaemia intermedia while another had
transfusion dependent anaemia. Mean Hb, TRBC, MCV, MCH, Hb-F and Hb-A2 in delta
beta-thalassaemia heterozygotes were 11.6 g/dl, 5.37 x 1012/L, 70.9 fl, and 21.7 pg, 14%
and 2.6% respectively. Similar values in the four untransfused homozygotes were 10.6
g/dl, 5.34 x 1012/L, 69.2 fl, and 20.8 pg, 100% and 0% respectively. Mutation analysis
revealed that all 13 individuals had the same Inv/Del (G)gamma(Agammadelta beta)
(Ahmed and Anwar 2006b).
XmnI Ggamma-polymorphism The XmnI Ggamma-polymorphism (C-T polymorphism at position -158 to the Ggamma-
globin gene) was studied in 13 individuals from six unrelated Pakistani families with
delta beta-thalassemia. All subjects had the Asian-Indian Inv/Del (GγAγδβ)o that

89
included six heterozygotes, six homozygotes, and one compound heterozygote of delta
beta- and beta-thalassemia. All seven delta beta-thalassemia heterozygotes (including one
compound heterozygote) had -/+ genotype, all six homozygotes had +/+ genotype. The
results strongly suggest a tight linkage between the XmnI Ggamma-polymorphism and
the Asian-Indian Inv/Del (GγAγδβ)o degrees. This finding could explain the unusually
well-preserved capacity to produce fetal hemoglobin in delta beta-thalassemia (Ahmed
and Anwar 2005b).
Hemoglobinopathies Hemoglobin D-Iran (Hb D-Iran, beta 22 Glu→Gln) is a beta-chain variant that was first
described in 1973. Hb D-Iran in combination with normal Hb A (Hb D-Iran trait) is a
benign condition. Hb D-Iran has also been described in combination with sickle
hemoglobin and beta thalassemia, but never as a homozygous mutation. One case of
homozygous Hb D-Iran in an infant of Pakistani descent has been described, hematologic
values, hemoglobin electrophoresis, peripheral blood smear, and clinical course suggest
that homozygous Hb D-Iran is a relatively benign condition with mild microcytic anemia,
poikilocytosis, and minimal hemolysis (Thornburg CD et al 2002). D-Los Angeles (alpha
2 beta 3 121 (glutamine→glycine) and thalassemia trait has also been detected in a
Pakistani family (Dawod et al 1988).
1.35: Treatment
Treatment of β -thalassemia depends on the clinical severity and ranges from no
treatment, in cases of β-thalassemia traits or some mild thalassemia Intermedia , to
frequent transfusions with chelation therapy and augmentation of fetal-hemoglobin
synthesis, in cases of thalassemia major or intermedia (Robin et al 2006). Pleurodesis

90
seems to be an effective and safe therapeutic option for exudative effusions, while
transfusion-chelation therapy combined with hydroxyurea may be helpful in suppressing
increased erythropoiesis (Aessopos 2006a). HU appears suitable for the treatment of leg
ulcers unresponsive to conventional treatment in patients with thalassaemia intermedia
(Gamberini 2004). Due to genetic heterogeneity of beta-thalassemia (beta-thal) patients,
several efforts have been undertaken to determine the efficacy of hydroxyurea treatment.
The results indicated that erythroid progenitor cells treated with 30 mumol/l hydroxyurea
for 96 h preferentially enhanced (G) gamma-and (A)gamma-globin mRNA (Hydroxyurea
2006). In addition to acting in synergy with the XmnI polymorphism, alpha deletions
may be an independent factor predicting good response to HU in thalassemia intermedia
(Panigrahi 2005). Hydroxyurea may be an alternative to transfusions for TI patients as
well as for TM patients in countries that have limited blood supplies (Bradai M et al
2007)
The treatment of thalassemia mainly includes blood transfusion which has its own
consequences. Growth retardation in regularly transfused thalassemic children has been
observed. Hypothyroidism is unlikely to be the sole cause of growth retardation; it may
however have a potentiating or permissive role. Strong association of transfused iron load
and decreased thyroid function stresses the need for intensive chelation therapy (Jain et al
1995). It was observed that Pakistani thalassaemic patients are significantly iron
overloaded due to improper iron chelation because of socioeconomic reasons. To
overcome this problem iron chelaters were used. In a study carried out at Armed forces
institute of Pathology, Rawalpindi the efficacy and adverse effects of Deferiprone (DFP,
L1)FP in Pakistani thalassaemic patients were studied. Deferiprone (l1, 1, 2- dimethyl-3-

91
hydroxypyrid-4-one) is a bidentate oral iron chelater that binds iron in a 3:1 ratio. It also
was concluded that DFP was well tolerated and caused fewer side effects. It had much
better patient compliance and was effective in lowering serum ferritin level in previously
most poorly chelated patients. It also has the potential advantage of economy and
increased compliance (Ayyub et al 2005). Studies of the standardized, 3D, 16-segments
map of the circumferential distribution of T2* values, of cardiovascular magnetic
resonance (CMR) in thalassemia major (TM) and thalassemia intermedia (TI) patients
and of electrocardiogram (ECG) changes associated with TM, have been carried out.
Similarly, the segment-dependent correction map of the T2* values and the artifactual
variations in normal subjects and the T2* correction map to correct segmental
measurements in patients with different levels of myocardial iron burden have been
evaluated. Cardiovascular magnetic resonance can be a suitable guide to cardiac
management in TI, as well as in TM (Ramazzotti et al 2008)
Exposure to hepatitis C virus (HCV) and its effect on ALT levels was studied in
transfusion dependent cases of thalassaemia major. 60% cases were anti HCV positive
and also showed raised Alanine Transaminase (ALT) levels. Of 14 anti HCV negative,
Hepatitis B Surface Antigen (HBs Ag) negative patients, seven showed raised ALT,
indicating the possible chances of acute viraemia (Bhatti et al 1995). Another hazard of
transfusion is the Red cell immunization. Red cell alloantibodies were detected in 4.97%
patients, and belonged mainly to Rh system, with one example each of anti-K, anti-Jsb
and anti-Jka. Rate of red cell alloimmunization in beta-thalassaemia major is relatively
low in our setup and may be related to red cell homogeneity between the donor and
recipient population. Routine pre-transfusion matching of blood, other than ABO and Rh

92
"D" antigens is not recommended because of low rate of red cell alloimmunization, and
high costs associated with such testing. Hyperhaemolysis, due to acquired red cell
antibodies was found to be an important complication. Patients who develop this
complication should be tested for presence of underlying antibodies and considered for
immunosuppressive treatment (Bhatti et al 2004).
Transmission rate of hepatitis C virus (HCV) infection through household contacts is to
be very low but may play a role in HCV spread by infected persons (Akhtar et al 2004).
In HCV seropositive thalassaemic children 20.5%were positive for anti-HCV antibodies.
HCV genotype 3a and 3b were found in 89% and 11% respectively (Akhtar et al 2002).
Transfusion transmitted hepatitis G virus in polytransfused β- thalassemia major children
was found in 21%, however no causal relationship between HGV and hepatitis was
observed (Moatter et al 1999).
Stem cell transplantation facility became available in Pakistan in 1999. Since then both
allogeneic and autologous procedures have been carried out for severe aplastic anaemia,
β-thalassaemia major and certain haematological malignancies. Allogeneic peripheral
blood stem cell transplantation is also feasible and life saving in otherwise fatal disorders.
This could be carried out effectively in Pakistan (Farzana et al 2003). Allogeneic BMT is
the only curative therapy for beta-Thalassaemia patients. Success rate can be increased if
patients are selected carefully and transplanted at an early age (Hashmi et al 2004). GHD
is present in up to a quarter of adult beta-thalassemia Patients (Vidergor et al 2007).
Acute GvHD developed in 68% of the patients with in beta-Thalassaemia who underwent
allogeneic stem cell transplantation. Morbidity and mortality due to severe acute and
chronic GvHD remains high despite standard prophylaxis against GvHD. New strategies

93
are needed to prevent and treat GvHD (Hashmi et al 2005). In developing countries
where consanguinous marriages are common gene variants are trapped within extended
families so that an affected child is a marker of a high risk group.
In one of the studies carried out in such families, 31 percent of the persons with an index
case were carriers; carriers had a 25 percent risk of producing an affected child. 8 percent
of the married couples consisted of two carriers. Carriers married to carriers with two or
more healthy children avoided further pregnancy and most such couples with one or no
healthy children used prenatal diagnosis. Testing of extended families is therefore a
feasible way of deploying DNA-based genetic screening in communities in which
consanguinous marriage is common (Ahmed et al 2002). Frequency of consanguineous
marriages among British Pakistanis was found to be 55 percent (Darr and Modell 1988).
Therapy of thalassemia has in the past been confined to transfusion and chelation. In
order to develop an objective test for discriminating between patients with thalassaemia
intermedia requiring blood transfusion and those not likely to require transfusion, the
medullary width (MW) in the midpoint of the second left metacarpal and the bone mass
were measured. Changes were visible radiologically and was concluded, therefore, the
measurement of MW seems to be an objective, simple test for discriminating between
patients requiring or not requiring blood transfusions. It was also observed that bone
deformities were reversible if transfusions were instituted using MW(greater than 0.5 cm)
regardless of the age or the haemoglobin concentration. This test may help clinicians to
decide about the optimal time for institution of regular transfusion in patients with
thalassaemia intermedia (Sbyrakis et al 1987). Evaluate quality of life (QOL) in
transfusion-independent patients with thalassemia (non-Tx) compared with that in

94
transfused patients (Tx) and to identify the factors that affect QOL in thalassemia. The
most commonly reported affected domains were feelings such as anxiety, depression, and
concern of overall health status or indications of recent deterioration in health. In contrast
with previous beliefs, transfusion-independent thalassemia patients also suffer serious
impairment in QOL (Pakbaz 2 005).
Recently, novel modes of therapy have been developed for thalassemia based on the
pathophysiology and molecular pathology of the disease, both of which have been
extensively studied. This includes transfusion, chelation (intravenous and oral),
antioxidants and various inducers of fetal hemoglobin (hydroxyurea, erythropoietin,
butyrates, hemin). Most of the newer therapies are suitable primarily for thalassemia
intermedia patients (Rund and Rachmilewitz 2000, Hoppe et al 1999, Bachir and
Galacteros 1994 and Karimi et al 2006). Therapeutic approaches for homozygous beta-
thalassemia entail blood transfusions and iron chelation therapy with deferoxamine or
deferiprone for preventing tissue hemosiderosis. Multiple transfusions may modulate the
response of serum EPO to the degree of anemia, resulting in increased EPO levels and
independent anemia in the TM patients (Chaisiripoomkere et al 1999 and de
Montalembert et al 1989). Continuous Increase in Erythropoietic Activity despite the
Improvement in Bone Mineral Density by Zoledronic Acid in Patients with Thalassemia
Intermedia-Induced Osteoporosis(Voskaridou et al 2008). Rgular chelation therapy with
deferoxamine is effective in patients with secondary hemochromatosis. Self-administered
deferoxamine subcutaneously once or twice a day seems to be the most practical method
to protect against the progression of hemochromatosis (Kobayashi et al 1996 and de
Montalembert et al 1989). Recently, much effort has been focused on various inducers of

95
fetal hemoglobin (HbF) such as recombinant human erythropoietin (rHuEPO), especially
in beta-thalassemia intermedia. HbF values registered a slight, non-significant increase.
rHuEPO treatment has a beneficial effect in transfusion-dependent beta-thalassemia
patients. Although a slight increase in HbF levels was observed, other possible
mechanisms are probably involved (Wong et al 2004). Viral-mediated globin gene
transfer in hematopoietic stem cells effectively treats a severe hemoglobin disorder (May
et al 2002). Administration of recombinant human erythropoietin (rHuEpo) in
combination with iron and folic acid may ameliorate blood indices as an alternative
choice to blood transfusion (Lialios et al 2000, Bourantas et al 1997, Leroy-Viard et al
1991 and Chaidos et al 2004). Thus recombinant erythropoietin seems to be an effective
treatment for anaemia in beta-thalassemia intermedia but long term randomized trials are
needed especially for patients with beta thalassemia major (Nisli et al 1996). Increasing
the level of haemoglobin F (HbF) by pharmacological agents has been proposed to
ameliorate the severity of the disease by improving the balance in globin chain synthesis.
Hydroxyurea, as an effective agent with low toxicity for activating gamma-globin gene,
has been shown to enhance HbF synthesis (Zeng et al 1995).
Hydroxyurea stimulating fetal hemoglobin (Hb) production. can eliminate transfusion
needs in children with beta-thalassemia major. This could be particularly useful in
countries where supply of blood and chelating agents are limited (Bradai et al 2003, de
Paula et al 2003, Cario et al 2002 and Cao et al 2005). Hydroxyurea may be an
alternative to transfusions for TI patients as well as for TM patients in countries that have
limited blood supplies (Bradai 2007).

96
Butyrate derivatives can stimulate fetal hemoglobin in patients with intermediate
thalassemia Intermedia (Domenica Cappellini et al 2000) Reduced serum or erythrocyte
Mg levels have been reported in human beta thalassemia. These deficiencies may play a
role in the cellular abnormalities characteristic of this disorder. Dietary Mg
supplementation improves some of the cellular function abnormalities of β thalassemia
intermedia (De Franceschi et al 1998).
Fetal hemoglobin switching agents have been proposed to treat genetic blood disorders,
such as sickle cell anemia and beta-thalassemia, in an effort to compensate for the
dysfunctional form of the beta-globin chain in adult hemoglobin. The rationale behind
this approach is to pair the excess normal alpha-globin chain with the alternative fetal
gamma-chain to promote red blood cell survival and ameliorate the anemia.
Reprogramming of differentiation in intact, mature, adult white blood cells in response to
inclusion of monoclonal antibody CR3/43 has been described. This form of retrograde
development has been termed "retrodifferentiation", with the ability to re-express a
variety of stem cell markers in a heterogeneous population of white blood cells. This
form of reprogramming, or reontogeny, to a more pluripotent stem cell state ought to
recapitulate early hematopoiesis and facilitate expression of a fetal and/or adult program
of hemoglobin synthesis or regeneration on infusion and subsequent redifferentiation.
This novel clinical procedure may profoundly modify the devastating course of many
genetic disorders in an autologous setting, thus paving the way to harnessing pluripotency
from differentiated cells to regenerate transiently an otherwise genetically degenerate
tissue such as thalassemic blood (Abuljadayel et al 2006).

97
1.36: Gene therapy in Thalassemia Intermedia
Gene therapy approaches to these disorders envision stem cell targeted gene transfer,
autologous transplantation of gene-corrected stem cells, and functional, phenotypically
corrective globin gene expression in developing erythroid cells has been adopted.
Lentiviral vector systems potentially appear to afford adequately efficient gene transfer
into stem cells and are capable, with appropriate genetic engineering, of transferring a
globin gene with the regulatory elements required to achieve high-level, erythroid-
specific expression. Results obtained in use of lentiviral vectors to insert a γ-globin gene
into murine stem cells with phenotypic correction of the thalassemia phenotype are
uncertain and suggest evaluation of the risk of gene therapy strategies for the treatment of
the hemoglobin disorders (Arthur et al 2003).
Gene transfer for beta-thalassemia requires gene transfer into hematopoietic stem cells
using integrating vectors that direct regulated expression of beta globin at therapeutic
levels. Among integrating vectors, oncoretroviral vectors carrying the human beta-globin
gene and portions of the locus control region (LCR) have suffered from problems of
vector instability, low titers and variable expression. In recent studies, human
immunodeficiency virus-based lentiviral (LV) vectors were shown to transmit human
beta-globin gene and a large LCR element, resulting in correction of beta-thalassemia
intermedia in mice (Malik & Arumugam 2005). Thus lentiviral vectors are very
promising drugs for the treatment of β-thalassemia intermedia and β-thalassemia major
(Stefano et al 2003).

98
1.37: Prenatal diagnosis Religion is believed to have a significant impact on individuals from minority ethnic
groups when making decisions about prenatal genetic screening, prenatal diagnosis and
termination of pregnancy. They generally considered religion and faith as an important
factor in the decision-making process, but the perceived severity of the condition would
play a more important role (Ahmed et al 2006c) Two renowned Islamic scholars ruled
that a pregnancy could be terminated if the fetus was affected by a serious genetic
disorder, and if termination was done before 120 days (17 weeks) of gestation. Prenatal
diagnosis of beta-thalassaemia was introduced in Pakistan in May 1994. During the first 3
½ years of the service 300 couples availed the test. This study demonstrates that prenatal
diagnosis is feasible and acceptable in a Muslim country such as Pakistan (Ahmed et al
2000) Pakistani women's attitudes towards prenatal diagnosis and termination of
pregnancy are influenced by various factors, and therefore their religion should not be
taken as a proxy for their attitudes either for or against termination of pregnancy (Ahmed
et al 2006a)
1.38: Diagnosis Thalassemia is usually diagnosed in infancy and is characterized by ineffective
erythropoeisis, bone marrow expansion and rapid destruction of erythrocytes. The
curative treatment available for this disease is either blood transfusion and chelation or
bone-marrow transplantation which is too expensive. Therefore, the only option is to
prevent the birth of an affected child by Carrier testing and prenatal diagnosis. Carrier
screening refers to identification of heterozygotes (carriers). In obstetrics, screening

99
provides the prospective parents with information about whether their children could
inherit a genetic disorder so that they could consider reproductive alternatives. It can be
made retrospectively, following the birth of an affected child or prospectively. Carrier
detection is based on the full blood counts and Hb electrophoresis and estimation of Hb
A2 and F levels followed by definitive diagnosis (mutation detection) by DNA analysis
which is guided by the results of hematological parameters. Prenatal diagnosis is one of
the most effective and direct approaches for prevention of thalassemia. The object of
prenatal diagnosis is to provide an accurate and rapid result as early in the pregnancy as
possible. It is offered in the I, II and III trimester of pregnancy accomplished by mutation
analysis on PCR-amplified DNA from chorionic villi sampling, amniocentesis and fetal
cordocentesis.
Sample selection in prenatal diagnosis: First trimester: Chorionic Villi Sampling Second trimester (15-19 weeks): Amniocentesis Third trimester (>22 weeks): Fetal Cordocentesis
α+-thalassemias are characterized by red cells which are often not microcytic, HbA2 and HbF levels are always normal therefore reliable diagnosis can only be achieved by DNA analysis. Silent β-thalassemias are characterized by normal MCV, MCH, HbA2, HbF,and total haemoglobin, and is solely defined by the slight imbalance in the α/β-globin synthesis ratios.(Table 1.5 and 1.6 and 1.7)

100
Table 1.5: Laboratory parameters of alpha , Beta and delta beta thalassemia
Parameters α–thalassemia (αº-thal)
β–thalassemia (Heterozygous)
δβ–thalassemia
Red Blood Cell Count
Milder hematological changes
High
Milder hematological Changes
Mean Corpuscular Volume (MCV)
Reduced
Reduced (60-70fl)
70fl
Mean Corpuscular Hemoglobin (MCH)
Reduced
Reduced (19-23pg)
24pg
Red Blood Cell Morphology
Microcytosis, Hypochromia
Microcytosis, Hypochromia
Microcytosis, Hypochromia
Hb concentration
Slight Reduction
Less than Normal (2g/dl)
Normal or slightly reduced
HbA2
Low to Normal (1.5-2.5%)
Elevated (4-6%)
Normal or slightly reduced
HbF
---
Elevated (2 - 1-3%)
Increased (5-20%)
Osmotic fragility
Decrease
Decrease
---
α/β-globin biosynthetic ratios
0.7
1.5 – 2.5
1.5

101
Table 1.6: Clinical and Hematological features of the β- Thalassemia Intermedia Syndromes (Richard et al 1993) Features Major Intermedia Minor Minima
Severity of manifestations ++++ ++ +, ± ± , 0
Genetics Homozygotes, Double Heterozygotes
Homozygotes, double heterozygotes, rarely heterozygotes
Heterozygotes Heterozygotes
Splenomegaly ++++ ++ , +++ +. 0 0
Jaundice +++ ++.+ 0 0
Skeletal changes ++++, ++ +,0 +,0 0
Hb,g/dl <7 7 – 10 >10 Normal
Hypochromia ++++ +++ ++ +
Microcytosis +++ ++ + 0
Target cells 10 – 35% ++ + ±
Basophilic stippling ++ + + 0, +
Reticulocyte (%) 5 – 15 3 – 10 2 – 5 1 – 2
Nucleated red cells +++ +, 0 0 0
±: little or no abnormality, +: mild abnormality, ++++: prominent abnormality
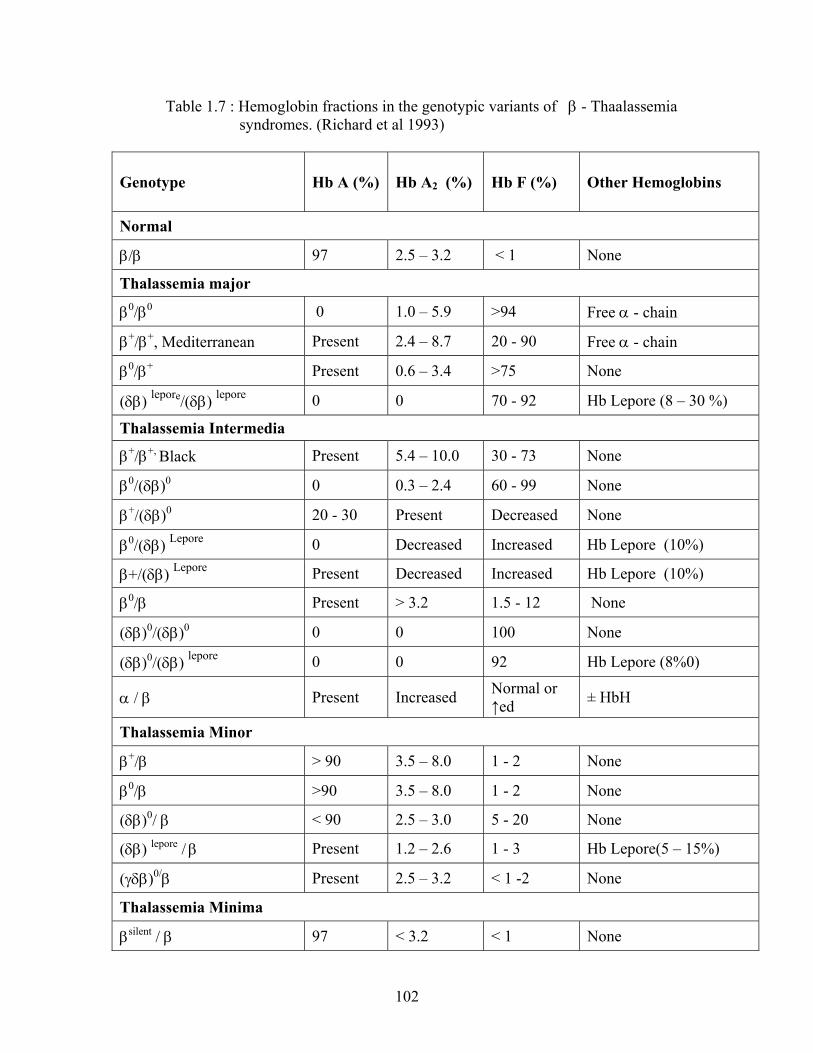
102
Table 1.7 : Hemoglobin fractions in the genotypic variants of β - Thaalassemia syndromes. (Richard et al 1993)
Genotype Hb A (%) Hb A2 (%) Hb F (%)
Other Hemoglobins
Normal
β/β 97 2.5 – 3.2 < 1 None
Thalassemia major
β0/β0 0 1.0 – 5.9 >94 Free α - chain
β+/β+, Mediterranean Present 2.4 – 8.7 20 - 90 Free α - chain
β0/β+ Present 0.6 – 3.4 >75 None
(δβ) lepore/(δβ) lepore 0 0 70 - 92 Hb Lepore (8 – 30 %)
Thalassemia Intermedia
β+/β+, Black Present 5.4 – 10.0 30 - 73 None
β0/(δβ)0 0 0.3 – 2.4 60 - 99 None
β+/(δβ)0 20 - 30 Present Decreased None
β0/(δβ) Lepore 0 Decreased Increased Hb Lepore (10%)
β+/(δβ) Lepore Present Decreased Increased Hb Lepore (10%)
β0/β Present > 3.2 1.5 - 12 None
(δβ)0/(δβ)0 0 0 100 None
(δβ)0/(δβ) lepore 0 0 92 Hb Lepore (8%0)
α / β Present Increased Normal or ↑ed ± HbH
Thalassemia Minor
β+/β > 90 3.5 – 8.0 1 - 2 None
β0/β >90 3.5 – 8.0 1 - 2 None
(δβ)0/ β < 90 2.5 – 3.0 5 - 20 None
(δβ) lepore / β Present 1.2 – 2.6 1 - 3 Hb Lepore(5 – 15%)
(γδβ)0/β Present 2.5 – 3.2 < 1 -2 None
Thalassemia Minima
βsilent / β 97 < 3.2 < 1 None