investigator-held ind studies jeffrey w. clark, md df/hcc medical director for clinical trials...
TRANSCRIPT

Investigator-held IND StudiesJeffrey W. Clark, MD
DF/HCC Medical Director for Clinical Trials Operations
April 25, 2008

2
• Regulations allow an individual to be both study sponsor (IND holder) and clinical investigator

3
Objectives
• Describe factors that influence whether an IND is required
• Explain the regulatory requirements for an IND
• Identify strategies for fulfilling IND holder (sponsor-investigator) obligations

4
Role of the FDA
• Federal agency responsible for ensuring safe and accurately represented products• Includes drugs, biologics, medical devices, and
radiation-emitting products
• Oversees clinical investigations of FDA-regulated products• Any funding source
• Academic center or other U.S. location
• Purpose of marketing or scientific knowledge

5
FDA Centers
• Center for Drug Evaluation and Research (CDER)
• Center for Biologics Evaluation and Research (CBER)
• Center for Devices and Radiologic Health (CDRH)
Divided into three centers relevant to clinical research

6
Center for Drug Evaluation and Research (CDER)
• Responsible for regulating drugs, chemical entities and proteins
• Allows shipment of investigational products for research upon submission of an Investigational New Drug application (IND)

7
Center for Biologics Evaluation and Research (CBER)
• Responsible for regulating biological and related products
• Blood, vaccines, tissues, cellular and gene therapies
• Allows the study of biological products upon submission of an Investigational New Drug application (IND)

8
Center for Devices and Radiologic Health (CDRH)
• Responsible for regulating medical devices and radiation-emitting products
• Allows the clinical study of medical devices upon submission of an Investigational Device Exemption (IDE)

When is an IND required?
Unapproved Product(s)
• Required for new drug or biologic use in a clinical trial
Approved Product(s)
• Usually required to study a new aspect of an approved product
• Different indication
• Different administration or
dosage level
• New drug combination
• Different population
9
Source: 21 CFR 312.2

10
Off-label Use in the Practice of Medicine
• Approved products may be used by physicians outside of labeled indications for the practice of medicine
• No IND is needed
Source: 21 CFR 312.2

11
When is an IND not required?
• Generally not required when all criteria met:• No intent to support new use or labeling change
• No intent to support change in advertising
• No factor such as route of administration, dosage, or study population significantly increases risk
• Compliance with FDA informed consent and IRB review requirements
• No promotion or representation of product as safe or effective treatment for condition under study
Source: 21 CFR 312.2

12
Best Practice
• It’s a much safer path to file an IND application and have it deemed exempt than not to file and later be subject to a determination that an IND should have been requested

13
Never Forget the IRB
• Whether an IND is or is not required, all clinical research must have IRB review and approval

14
Study Start-up Procedures
• Discuss the study concept with the Disease Program
• Draft the protocol
• Use the biomedical protocol template and guidance
documents created by DF/HCC

15
IND Submission Process
• FDA Form-1571
• FDA Form-1572
• FDA Form-3674
Sponsor-Investigator prepares and submits in triplicate to appropriate FDA Center:
Helpful hint: A single IND may be utilized for one or more phases (I, II, or III) of an investigation, or for multiple protocols related to the IND purpose and indication.

16
Form FDA-1571
• Wait 30 days post IND submission before beginning research
• Unless earlier notification indicates research can begin
• Not begin or continue the research if placed on clinical hold
• Use an IRB for initial and ongoing review and approval of the research
• Conduct research in accordance with all applicable regulatory requirements
• Including oversight of all work performed under the IND
Contractual agreement between Sponsor-investigator and FDA to:

The Importance of Form FDA-1572
• Statement of Investigator
• Legally binding contract with FDA
• Personally conduct study in
accordance with protocol
• Don’t make changes to the
research without IRB approval
• Promptly report any changes
and unanticipated risks to IRB

18
Form FDA-3674
• Certifies enrollment of referenced studies in clinicaltrials.gov
• Refers to “applicable” clinical trials upon which the submission directly relies or which may not yet be published, not to literature references
• Example: application uses data from a previous
clinical trial to support going from Phase I to Phase II
Important: Make sure you have the appropriate information to complete this form. Willful and knowing false statements may be viewed as a criminal offense.

19
IND Application Approval Process
• FDA Acknowledgment Letter
• Arrives 1-2 weeks after FDA receipt of IND submission
• Assigns IND number, gives date of receipt, reminds
sponsor-investigator of obligations under the IND
• NOT an approval to begin• May not start until 30 days after IND receipt date
• Unless earlier notification indicates otherwise

20
Possible FDA Actions
• Request additional information or place on clinical hold• Research cannot begin until all concerns are
addressed in ways acceptable to FDA
• Conclude project is exempt • Research may be conducted without an IND
• Passive Activation• Allowing 30 days from filing to pass without comment
Helpful hint: Confirm FDA’s non-objection prior to starting any clinical trials.

21
Clinical Hold
• May apply to more than one study under an IND
• May occur at the time of IND proposal or during clinical investigation
• No new accrual
• Existing participants may not receive study product
Source: 21 CFR 312.42
Legal order to delay or suspend research

22
IND Post Approval Obligations
• Selecting qualified investigators and monitors
• Providing necessary information to investigators
• Monitoring the research
• Controlling the investigational product
• Reporting significant adverse events to FDA, IRB, and investigators
• Maintaining the IND
• Maintaining and Retaining Accurate Records

23
Initiate National Protocol Registration• Register all research under the IND with
clinicaltrials.gov
• Contact the Clinical Trials Education Office (CTEO)
for guidance• [email protected] or 617-582-8480
More information: Topic covered in more detail in the National Protocol Registration module.

24
Manufacture and/or Label Investigational Product
• Keep work areas clean and free of:
• Dirt, dust, and vermin
• Objects not required for manufacturing
• Label drug(s) as follows:
Caution: New Drug—Limited by Federal [or United States] law to investigational use.
Source: 21 CFR 210, 312.6, 610

25
Select Qualified Investigators and Study Monitors
• Base selection on training and experience
• Obtain the following from participating investigators
• Form FDA-1572
• CV or other evidence indicating expertise in the
clinical area of study
Source: 21 CFR 312.53

26
Provide Necessary Information to Investigators
• Disseminate information with respect to adverse events and safe use of the investigational product
• Protocol
• Investigator’s brochure
• Safety reports (SAEs, IND safety reports)
• Progress reports
• Publications
Source: 21 CFR 312.55

27
Train Participating Investigators
• Train at the beginning and at intervals during the trial
• Study protocol and study-specific procedures
• Adverse event reporting
• Protocol deviation and violation reporting
• Establish procedures for training new investigators
• Document protocol and study-specific training
Source: 21 CFR 312.55

28
Establish Regular Communications with Participating Investigators
• Schedule progress reports with participating investigators
• Suggested timelines• Weekly (phase I)
• Monthly (phase II)
• At least every 3-6 months (phase III)
• Maintain documentation• Minutes from face-to-face meetings and teleconferences,
or email updates

29
Monitor the Research
• Adhere to the data and safety monitoring plan
• Review the site monitoring reports for protocol and regulatory compliance
• End participation of non-complying investigators
• Discontinue shipments of study drug
• Report serious non-compliance to FDA (and IRBs)
Source: 21 CFR 312.56

30
Control Distribution and Return of Investigational Product
• Permit shipment of study product only to investigators participating in the study
• Remind participating sites to return or properly destroy any unused investigational products
• Maintain written records of the disposition
Source: 21 CFR 312.59

31
Evaluate Adverse Events (AEs)
• Review safety and efficacy data as it is obtained from participating investigators
• Identify all safety reports previously filed concerning a similar event
• Analyze the significance of the event in light of previous similar reports
• Determine if any corrective actions should be taken as a result of the event
Source: 21 CFR 312.32

32
Report Adverse Events to DFCI IRB
• Report AEs and SAEs from any location• Use the appropriate internal or external event report
form
• Amend the protocol and/or revise the consent form as necessary
More information: General adverse event reporting is covered in more detail in the Monitoring Data and Participant Safety module.
Source: DF/HCC SOPs PM-402, PM-407, PM-408, AE-601

33
Report Adverse Events to FDA
• Report via an IND Safety Report any AE that meets all three criteria:• Serious
• Unexpected • Not in the drug brochure, protocol or consent form
• Study treatment related
• Report via an IND Safety Report all SAEs that are fatal or life-threatening• Associated with the use of the drug/biologic
Source: 21 CFR 312.32; DF/HCC SOPs PM-408, AE-601

34
Report Events to Participating Investigators
• Notify participating investigators of any event submitted to FDA
• Provide same information submitted to FDA
• Forward any DFCI IRB-approved corrective actions that must be taken as a result of the event
• Amended protocol and/or revised consent form
Source: 21 CFR 312.32, 312.55; DF/HCC SOPs PM-408, AE-601
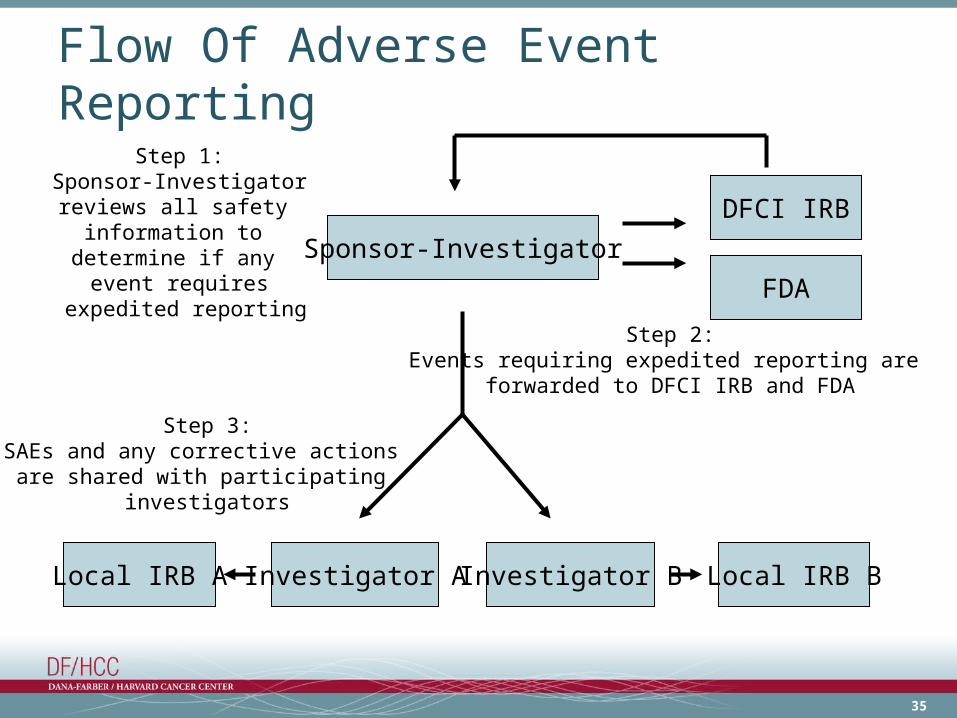
35
Flow Of Adverse Event Reporting
Sponsor-Investigator
Investigator ALocal IRB A Local IRB BInvestigator B
DFCI IRB
Step 1:Sponsor-Investigator
reviews all safety information to
determine if any event requires
expedited reporting
Step 3:SAEs and any corrective actions
are shared with participating investigators
FDA
Step 2:Events requiring expedited reporting are
forwarded to DFCI IRB and FDA

36
Summary of Adverse Event Notification
Who Circumstance Timeline
DFCI IRB Reportable event from any study location
Within 10 days of notification
FDA SAEs that are unexpected and related (or possibly related) to study
7-15 days from notification, depending on nature of event
Participating Investigators
SAEs that are unexpected and related (or possibly related) to study
After DFCI IRB review and response
Others As appropriate According to guidelines
Important: Reporting requirements for FDA or other oversight bodies may differ from the DFCI IRB. As Sponsor-Investigator, you must comply with all reporting requirements.

37
Maintain Accurate Study Records
• Describe in writing all tasks delegated to others
• Document the receipt, shipment, and disposition of the study product
• Retain all study records in a secure facility
• At least 2 years after study completion/discontinuation
• For 6 years following study completion for HIPAA
compliance
Helpful hint: Use the DF/HCC Delegation of Responsibility and Training Log available on the DF/HCC website under Clinical Research Unit.
Source: 21 CFR 312.59

38
Submit Records, Reports and Final Results
• Allow authorized FDA representatives to have access to records/reports relating to the research
• Submit safety and progress reports to FDA as required
Important information: Sponsor-investigators are obligated to allow FDA to inspect study conduct and documentation at any time. Contact QACT if your IND research is chosen for an FDA inspection.
Source: 21 CFR 312.30-312.33, 312.38, 312.58; DF/HCC SOP OV-101

39
FDA Reporting Requirements under an IND• An IND is a living
document
• Protocol Amendments
• Information Amendments
• IND Safety Reports
• Annual Reports
• Final Study Report
Important information: Submit to FDA a new version of form 1571 with each amendment or report. Submit a new version of form 1572 with the appropriate protocol amendments.

40
Protocol Amendments
• Notify FDA of any protocol changes under the IND as they are proposed and IRB approved
• New protocol
• Change to an existing protocol that affects:• Rights, safety or welfare of participants
• Scope or scientific quality of the study
• New investigator
• Identify amendment contents
• Example: “Protocol Amendment: New Protocol”
Source: 21 CFR 312.30

41
Changes to Existing Protocols that Require FDA Protocol Amendments
• Increase in dose, duration of exposure, sample size
• Addition/deletion of study group
• New test or procedure to improve monitoring, reduce side effects or adverse events
• Elimination of immediate hazard to study participants or tests that monitor safety

42
Information Amendments
• Notify FDA of any other essential protocol information on the IND• Response to FDA comments
• New toxicology, chemistry or technical information
• Discontinuance of a study
• Limit frequency to once every 30 days • Submit significant manufacturing changes in real time
• Identify amendment contents• Example: “Information Amendment: Response ”
Source: 21 CFR 312.31

43
IND Safety Reports
• Notify FDA of any AE associated with the study product that is both serious and unexpected• Include findings from tests in laboratory animals that
suggest a significant risk to humans
• Submit FDA Form 3500A or in narrative form• As soon as possible but no later than 15 calendar
days from initial knowledge of the event
• Notify all participating investigators
Reporting serious and unexpected study-related AEs
Source: 21 CFR 312.32

44
IND Safety Reports
• Notify FDA of any unexpected fatal or life-threatening events related to the study product
• Submit telephone and facsimile transmissions
• As soon as possible but no later than 7 calendar days from initial knowledge of the event
• Provide follow up information as soon as available
• Notify all participating investigators
Reporting unexpected fatal/life-threatening study-related AEs
Source: 21 CFR 312.32

45
Annual Reports
• Submit to FDA within 60 days of IND anniversary
• Due each successive year
• Report on status of each clinical study
• Summarize data obtained in last year
• Include IRB documentation
Helpful hint: Submit the IND Annual Report to FDA when filing the Continuing Review with DFCI IRB.
Source: 21 CFR 312.33
Brief report of the progress of the clinical investigation

46
Final Study Report
• Submit to FDA as soon as each clinical study concludes
• No later than six months from study completion
• Summarize results of each study as completely as possible

47
Discontinuation of an Investigation
• When?
• IND presents unreasonable or significant risk to
participants
• Submit an information amendment to FDA within 5 working days after determining the IND should be discontinued
• Notify all participating investigators and IRBs
Source: 21 CFR 312.56

48
Withdrawal of an IND
• May withdraw an effective IND at any time without prejudice
• Submit an information amendment to FDA
• Notify all participating investigators and IRBs
Source: 21 CFR 312.38

49
Summary of Periodic Submissions to FDA
Submission Type Timeframe
Protocol Amendment: New Protocol After IRB approval but before implementation
Protocol Amendment: Change in Protocol After IRB approval but before implementation
Protocol Amendment: New Investigator Within 30 days of being added
Information Amendments At time of occurrence
IND Safety Report (serious & unexpected) Within 15 calendar days of notice
IND Safety Report (fatal or life threatening) Within 7 calendar days of notice
Annual Report Within 60 days of anniversary
Discontinuation of investigation Within 5 working days of decision
Withdrawal of IND At time of occurrence

50
The Challenge
• A sponsor-investigator must be aware of his or her responsibilities as the sponsor and attentive to the reporting standards of IND study conduct

51
Caveats about Undertaking Sponsor Investigator Trials
• “Where problems have come in recent years, the majority have come in studies where the investigator was also the sponsor.”
David Lepay
FDA’s senior advisor on clinical science
Guide to Good Clinical Practice January 2005

52
Tales from FDA Warning Letters
• You failed to submit an IND but followed subjects through August 2004 when you informed the IRB that the study
was closed. 21 CFR 312.20(a)
• As a sponsor, you failed to submit protocol amendments to the FDA before study revisions were implemented. 21 CFR 312.30 (a) and (b)
• No records available for any study monitoring prior to the monitoring report of xx. 21 CFR 312.50
Mistakes made by investigators acting as IND holders

53
How DF/HCC Can Help
• Supply templates for investigator-initiated IND research• Initial IND submission template
• IND annual report template
• Various transmittal letter templates
• Provide institutional contacts for IND-related questions
• Assist with FDA audit preparation

54
For More Information
• Templates and Institutional Contacts
• Contact the Clinical Trials Education Office (CTEO)• [email protected] or 617-582-8480
• FDA Audit Preparations
• Contact the Quality Assurance Office for Clinical Trials (QACT)• [email protected] or 617-632-3761
• FDA Documents, Forms and Warning Letters
• Refer to www.fda.gov

55
Summary
• There is only one code of federal regulations regarding IND research
• FDA applies the same rules to every IND holder
• Be familiar with your obligations under the IND
• Seek guidance when necessary
• Use DF/HCC and external resources to navigate the regulatory process and maintain an effective IND

Conclusion
• Be in charge of your project, give it your unswerving attention, and see it through to completion
• Remember that absolute responsibility and accountability are required
• Communication with all investigators and study teams is imperative
• Monitoring and quality assurance are essential
56
Do I really want to hold an IND? If yes, then: