ivd inspections technical update 2018 · • iso 14971:2007 (2012) medical devices-application of...
TRANSCRIPT

IVD Inspections
Technical Update 2018
Dr Dragana Milic
Medical devices inspections
1 Copenhagen, Denmark 24-28 September 2018

2 Copenhagen, Denmark 24-28 September 2018
• Harmonisation - internal and external
(PQ Medicines/Vaccines/Vector Control and MDSAP)
• Transparency (clarify rules and requirements)
• Are we hitting the mark, what can we do better together?
Objectives

3 Copenhagen, Denmark 24-28 September 2018
• Requirements of mature national regulatory authorities
including: USA FDA, Australian TGA, Health Canada,
Japanese PMDA, Brazilian ANVISA
– These are MDSAP Authorities
– (Medical Devices Single Audit Program)
Observers
– WHO - Diagnostic Prequalification Program
– European Union (a decision about full participation?)
MDSAP

4 Copenhagen, Denmark 24-28 September 2018
Recognized Auditing Organisations conduct audits of a
medical device manufacturers that satisfy the QMS
requirements of 5 Regulatory Authorities. WHO requirements
about to be added.
– ‘Single Audit Program’
Initiated in 2012 by the International Medical Device
Regulators Forum - IMDRF - ex GHTF
Pilot ended 2016, transition until end 2018
MDSAP

Assess and recognize
Make regulatory decisions – Market Authorisation
Audit and certify
Share audit report and certificate
Auditing
Organisations
Medical
Device
Manufacturers
Regulatory
Authorities CONCEPT
5

6 Copenhagen, Denmark 24-28 September 2018
• PQDx_014 Information for Manufacturers on the Inspection of
Manufacturing Site(s)
• ISO 13485:2003 and 2016 Medical Devices-Quality
management systems – Requirements for regulatory purposes
• ISO 14971:2007 (2012) Medical devices-Application of risk
management to medical devices
• IMDRF and GHTF Documents
Reference and Guidance documents

7 Copenhagen, Denmark 24-28 September 2018
Assessment Types
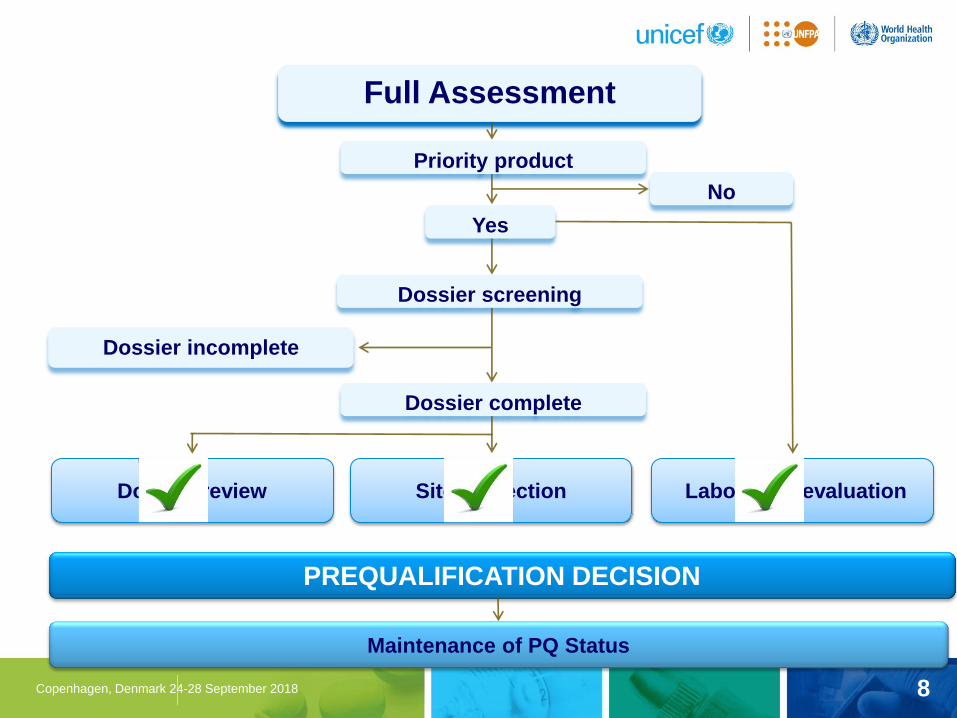
8 Copenhagen, Denmark 24-28 September 2018
Full Assessment
Dossier review Site inspection Laboratory evaluation
Dossier incomplete
PREQUALIFICATION DECISION
Dossier complete
Dossier screening
Priority product
Yes
No
Maintenance of PQ Status
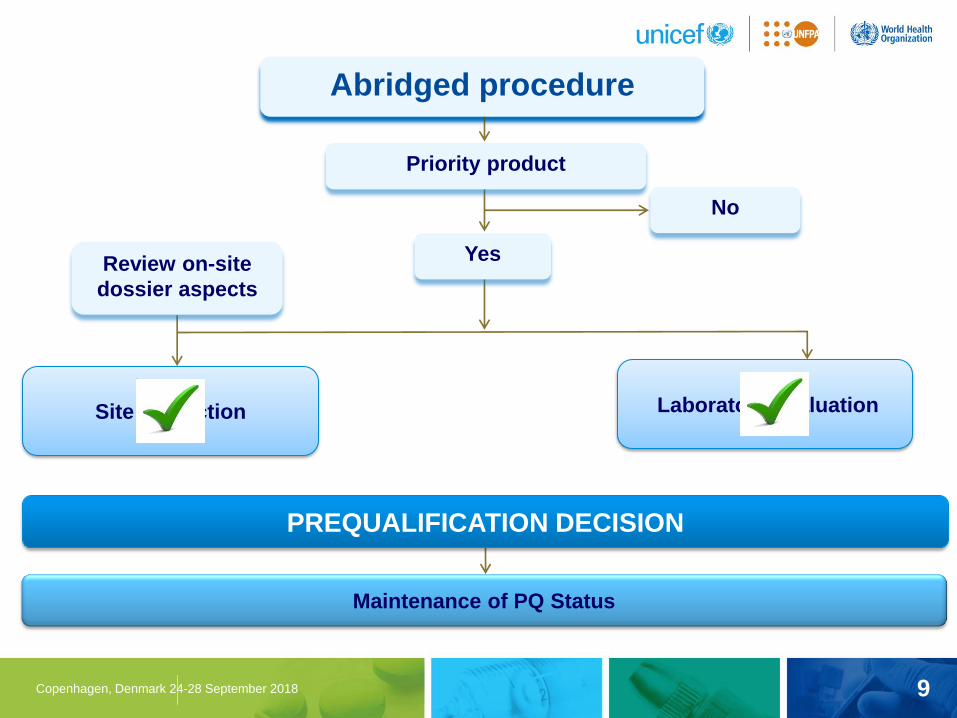
9 Copenhagen, Denmark 24-28 September 2018
Abridged procedure
Site inspection Laboratory evaluation
Priority product
Yes
No
Maintenance of PQ Status
PREQUALIFICATION DECISION
Review on-site
dossier aspects
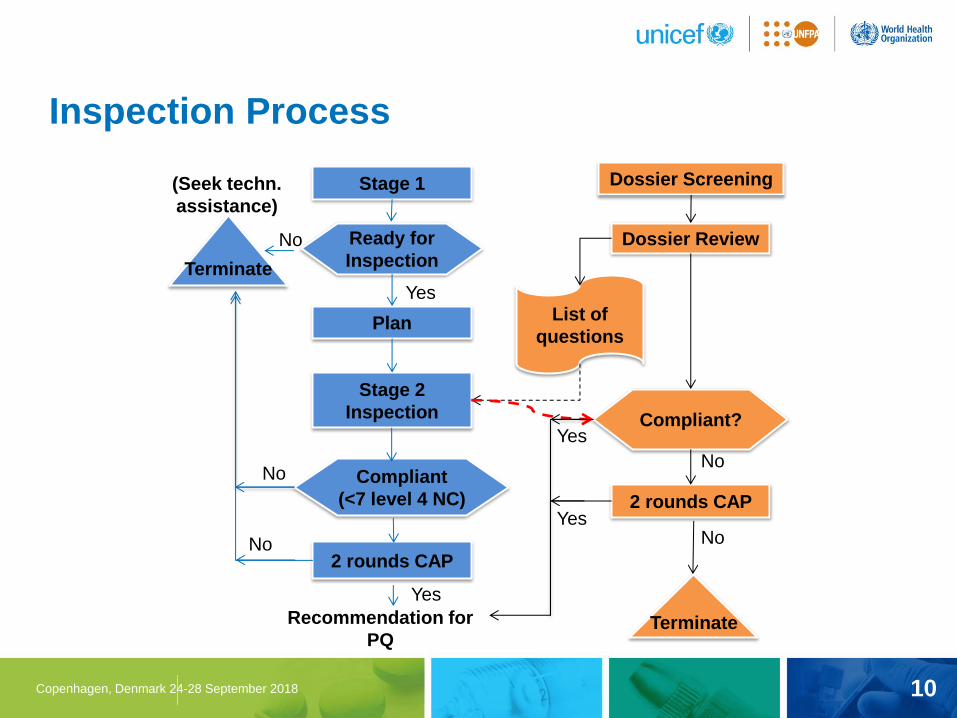
10 Copenhagen, Denmark 24-28 September 2018
Inspection Process
Stage 1
Plan
Stage 2
Inspection
2 rounds CAP
Ready for
Inspection
Compliant
(<7 level 4 NC)
(Seek techn.
assistance)
Recommendation for
PQ
No
Yes
Yes
No
Dossier Screening
Dossier Review
List of
questions
Compliant?
2 rounds CAP
Yes
No
No
No
Terminate
Yes
Terminate

11 Copenhagen, Denmark 24-28 September 2018
Element Required documents
Quality
Management
System
Quality manual including staff organogram
List of current quality management procedures (SOP)
Standard operating procedures for:
Complaint handling and vigilance
Control of nonconforming goods/processes
Change control/change notifications (product and
processes)
Risk management
Supplier evaluation and control, verification of purchased
product
Design and development
Audit report of the most recent full regulatory inspection/audit
and all subsequent surveillance inspections/audits
Any valid quality management system certificate(s) (e.g. ISO
13485)
Name and contact details of the responsible person at the site
of manufacture regarding the inspection
Required information – Full and Abridged
Inspection

12 Copenhagen, Denmark 24-28 September 2018
Element Required documents
Product Labelling (instructions for use (IFU),
component and box labels
Photographs of kit, box including contents, kit components
Accessories (including photographs)
Copy of current product regulatory approval certificate(s)
Summary of changes initiated or applied to the product
subsequent to the above regulatory approvals
Required information – Full and Abridged
Inspection

13 Copenhagen, Denmark 24-28 September 2018
Element Required documents
Manufacturing
Full address, including latitude and longitude of the
manufacturing facility(s)
Site floor plan
Manufacturing flowchart including in-process control points
List of critical raw materials ( including details of the
supplier of each material)
List of outsourced processes with direct product impact
(e.g. outsourced manufacturing of components (conjugated
antibodies, strips, reagents…), outsourced laboratory testing,
packaging, printing, etc.) including details of the supplier for
each process
Required information – Full and Abridged
Inspection

– WHO is taking into account the outcomes of MDSAP
inspections.
– WHO is reviewing MDSAP reports and where they include
the evidence of manufacturer‘s compliance with relevant
requirements a WHO inspection may be:
• Waved
• Shortened
• Undertaken as planned
– Exceptions - for couse inspection; or requested during
screening or dossier assessment process.
25/09/2018 14
On site inspections and MDSAP

WHO is worknig with MDSAP, AOs and manufacturers
– Content of the MDSAP report to enable utilisation in lieu of
WHO inspection:
• Includes relevant requirements of ISO 13485
• Includes eidence of compliance with WHO specific
requirements
• Evidence of implemenetation of TSS, TGS, standrads
• Product specification requirements
25/09/2018 15
International Alignment – WHO‘s activities

– Content of the report continued:
• Risk management (appropriate for intended use)
• Design inputs/outputs including TSS
• Performance requirements,
• Design validation: Usability in resource limited settings
Clinical data, performance data
Stability
Labeling, IFU
QC requirements
• Feedback provisions
• Other
25/09/2018 16
International alignment

Prerequisite for an onsite inspection
17 Copenhagen, Denmark 18-21 September 2017

18 Copenhagen, Denmark 24-28 September 2018
• Fully implemented quality management system (design &
development, manufacturing including quality control, storage,
distribution)
• Meets ISO 13485:2003 requirements with the transition to 2016
version still often delayed ( external reasons seem preveiling)
• Product design,
• Validation of processes
• Products undergoing prequalification must be in routine
manufacturing
• Sufficient capacity to ensure reliable delivery
• Quality control processes follow risk principles, performance tested
according to claims in instruction for use
• Storage conditions, temperature and humidity, validated for
intermediates, components and kit, real time data required
Quality Management System
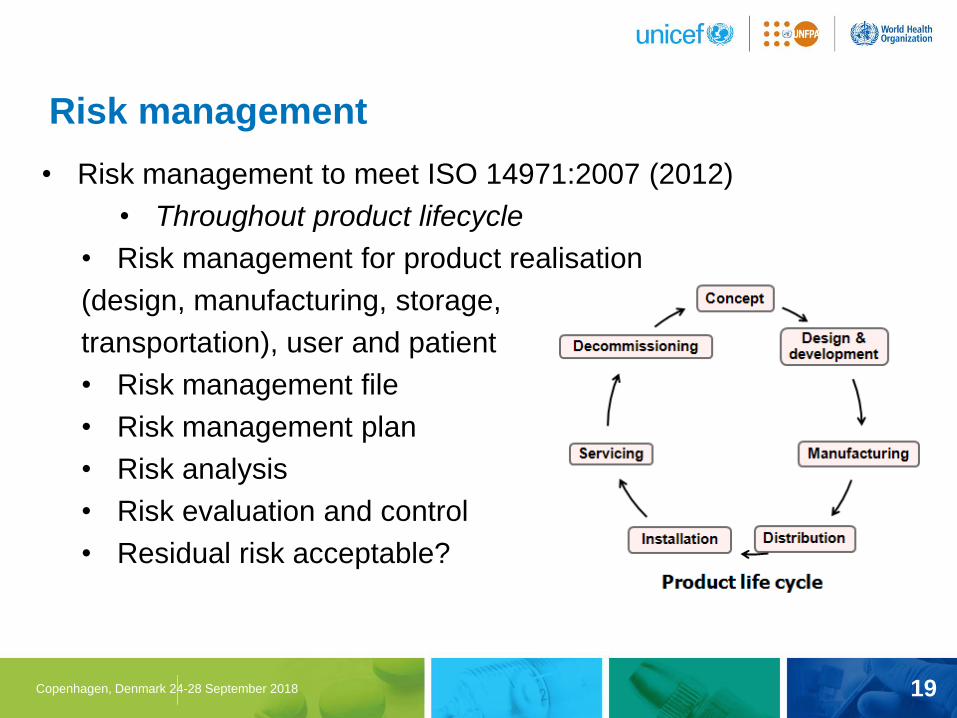
19 Copenhagen, Denmark 24-28 September 2018
• Risk management to meet ISO 14971:2007 (2012)
• Throughout product lifecycle
• Risk management for product realisation
(design, manufacturing, storage,
transportation), user and patient
• Risk management file
• Risk management plan
• Risk analysis
• Risk evaluation and control
• Residual risk acceptable?
Risk management

20 Copenhagen, Denmark 24-28 September 2018
• Product stable to meet challenging conditions (hot, wet,
dry, dusty)
• Stability ensured by design
• Stabilty ensured through teh control of critical
processes
• Transportation studies (simulate "worst" conditions)
• Long term stability at limiting conditions
• In-use stability studies (open kit, strip, vial)
• Data to support all claims available on-site
• Consistency of product labelling (component, kit and
shipping box)
Product Stability

21 Copenhagen, Denmark 24-28 September 2018
• Transfer from R&D to production completed
• Evaluated and approved suppliers (think of services!)
• Validated processes (acceptance ranges determined, in-process controls established)
• Standardized batch sizes
• Established non-conformance management and "out-of-specification" processes
• Batch manufacturing records established (include all manufacturing information, full traceability of material and equipment)
• Trained personnel (requirements determined, training plan, records)
Routine Manufacturing

22 Copenhagen, Denmark 18-21 September 2017
Abridged Inspection

23 Copenhagen, Denmark 24-28 September 2018
Abridged Inspection
• Aim:
• To avoid duplication
• Reduce time
• Reduce cost
• When:
• If a stringently assessed regulatory version is submitted for
prequalification
• If a non-stringently assessed (rest of the world) regulatory
version of the product is submitted for prequalification
assessment but a stringently assessed regulatory version
also exists (no difference between the two versions)

24 Copenhagen, Denmark 24-28 September 2018
Stringently Assessed
• Approved by a GHTF/IMDRF founding member regulatory authority and assessed at a level comparable to that required by WHO
• Competent Authorities from the Member States of the European Union
• Directive 98/79/EC on in vitro diagnostic medical devices
• Associated Notified Bodies
MDSAP authorities
• US FDA
• Health Canada
• Japanese Ministry of Health, Labour and Welfare
• Australian TGA
• Brazilian ANVISA

25 Copenhagen, Denmark 24-28 September 2018
PQ Stage
Full assessment
Abridged assessment
Dossier review Yes No
Site inspection Full Inspection Abridged inspection (information
package requested)
Laboratory evaluation Yes Yes
Full and Abridged Inspections

26 Copenhagen, Denmark 24-28 September 2018
• Less time spent on site
• Fewer inspectors (usually one inspector and one technical
expert)
Objectives of Abridged Inspections

27 Copenhagen, Denmark 24-28 September 2018
• Inspection team determined
• Communication with manufacturer (suitable inspection dates,
required documentation)
• Manufacturer to agree on the inspection team (Brief CVs
provided)
• Inspection plan provided one to two week prior to the
inspection
Inspection planning

28 Copenhagen, Denmark 24-28 September 2018
• Opening meeting, debriefings, full transparency
• Open exchange of information between WHO and
Manufacturer throughout the entire process
• Inspection of the full site to review the manufacturing process
• Manufacturing of the WHO/or a similar product is
required
• Access to all requested documents and records
• Sampling process with risk based emphasis
• Review and confirmation of raw data related to the dossier
submission including all validations, stability studies and
performance data
The onsite Inspection

29 Copenhagen, Denmark 24-28 September 2018
• Follows MDSAP model
• Utilises MDSAP report format and Ninconformities Grading and
Exchange (NGE) form.
The onsite Inspection

Ris
k M
anagem
ent
Purc
hasin
g
30
Management
Device Marketing
Authorisation and
Facility
Registration
MD Adverse
Events and
Advisory Notice
Reporting
Device Marketing
Authorisation and
Facility
Registration
Measurement,
Analysis and
Improvement
Design and
Development
Production and
Service
Controls
MDSAP
Audit
Sequence
of ISO
13485
and
Regulatory
Processes

• Design and Development
• Priority criteria for selection of design files for review:
• Complaints of known problem with a particular device,
• Product risk
• Recent design changes particularly design changes
made to correct quality/design problems
• Reviewing complaints and NCs enables this selection
31
MDSAP Model, its application by WHO and
how/why it makes sense?

• Production and Service Controls
• Priority criteria for selection of production processes for
review:
• Corrective and preventive action indicators of process
problems…
• Production processes for higher risk products…
• Processes with direct impact on devices quality….
32
MDSAP Model, its application by WHO and
how/why it makes sense?

• Production and Service Controls continued:
• Purchasing: criteria for selection of suppliers:
• Indications of problems with supplied products or
processes from audit of the Measurement, analysis and
Improvement process……
33
MDSAP Model, its application by WHO and
how/why it makes sense?

34 Copenhagen, Denmark 24-28 September 2018
• Closing meeting to discuss any findings
• A list of findings is left onsite for the manufacturer to begin CAPA preparation whenever possible.
• A report is issued within 30 days of the site inspection unless otherwise informed. (Of note MDSAP reports are required to be issued by AOs within 90 days).
• WHO challenge - aligning MDSAP reporting format with WHO’s reporting timelines.
• Grading of nonconformities is automated and independently reviewed before the final report is released.
End of Inspection

35 Copenhagen, Denmark 24-28 September 2018
• Based on the GHTF Model
GHTF/SG3/N19:2012
‘Quality management system – Medical devices – Nonconformity
Grading System for Regulatory Purposes and Information
Exchange’.
Grading nonconformities - Levels 1 to 5.
Grading of nonconformities

Nonconformity grading
•GHTF N19 - Nonconformity Grading System for Regulatory Purposes
•NCs are assigned a grade, 1 – 5
•Calculated in two steps
– Grading Matrix
– Escalation Rules
No Documented Process, +1
Release of nonconforming medical device, +1
3 4
1 2
Occurrence
Imp
act
First Repeat
Indirect
Cl 4
.1 –
6.3
Direct
Cl 6
.4 –
8.5
.3
36

37 Copenhagen, Denmark 24-28 September 2018
• Required to be submitted within 30 days of receipt of report
• Root cause analysis
• Correction
• Corrective action
• Timeline and responsible person/department
• Evaluation of the effective implementation of the
corrective action
• Two rounds of CAPA are allowed
Corrective action plan

38 Copenhagen, Denmark 24-28 September 2018
14
33
11
25
16
59
16
50
0
10
20
30
40
50
60
70
Number of Inspection Number of products reviewed
2015
2016
2017
2018
Inspection statistics

39 Copenhagen, Denmark 24-28 September 2018
Next presentation:
Inspection findings
What worries us (and you)
Expectations

Where to find information
40 Copenhagen, Denmark 24-28 September 2018
• Contact us by email
• Sign up for our mailing list
By emailing [email protected]
• Check our website
http://www.who.int/diagnostics_lab
oratory/evaluations/en/