jun liu - harvard universityjunliu/sequence_analysis.pdf · biological sequence analysis 17 the...
TRANSCRIPT

Biological Sequence Analysis 1
Biological Sequence Analysis and Motif Discovery
Introductory Overview LectureJoint Statistical Meetings 2001, Atlanta
Jun LiuDepartment of Statistics
Harvard Universityhttp://www.fas.harvard.edu/~junliu

Biological Sequence Analysis 2
Topics to be covered• Basic Biology: DNA, RNA, Protein; genetic code.• Biological Sequence Analysis
– Pairwise alignment --- dynamic programming• Needleman-Wunsch• Smith-Waterman• Blast …
– Multiple sequence alignment• Heuristic approaches • The hidden Markov model
– Motif finding in DNA and protein sequence

Biological Sequence Analysis 3
Central Paradigm
Courtesy of Doug Brutlag

Biological Sequence Analysis 4
Buliding Blocks of Biological Systems: nucleotides and amino acids
• DNA (nucleotides, 4 types): information carrier/encoder.• RNA: bridge from DNA to protein.
• Protein (amino acids, 20 types): action molecules.
• Genetic code: deciphering genetic information.atgaatcgta ggggtttgaa cgctggcaat acgatgactt ctcaagcgaa
cattgacgac ggcagctgga aggcggtctc cgagggcgga ……
MNRRGLNAGNTMTSQANIDDGSWKAVSEGG ……

Biological Sequence Analysis 5
genome
gene
intron exon
splicing
protein
DNA sequences
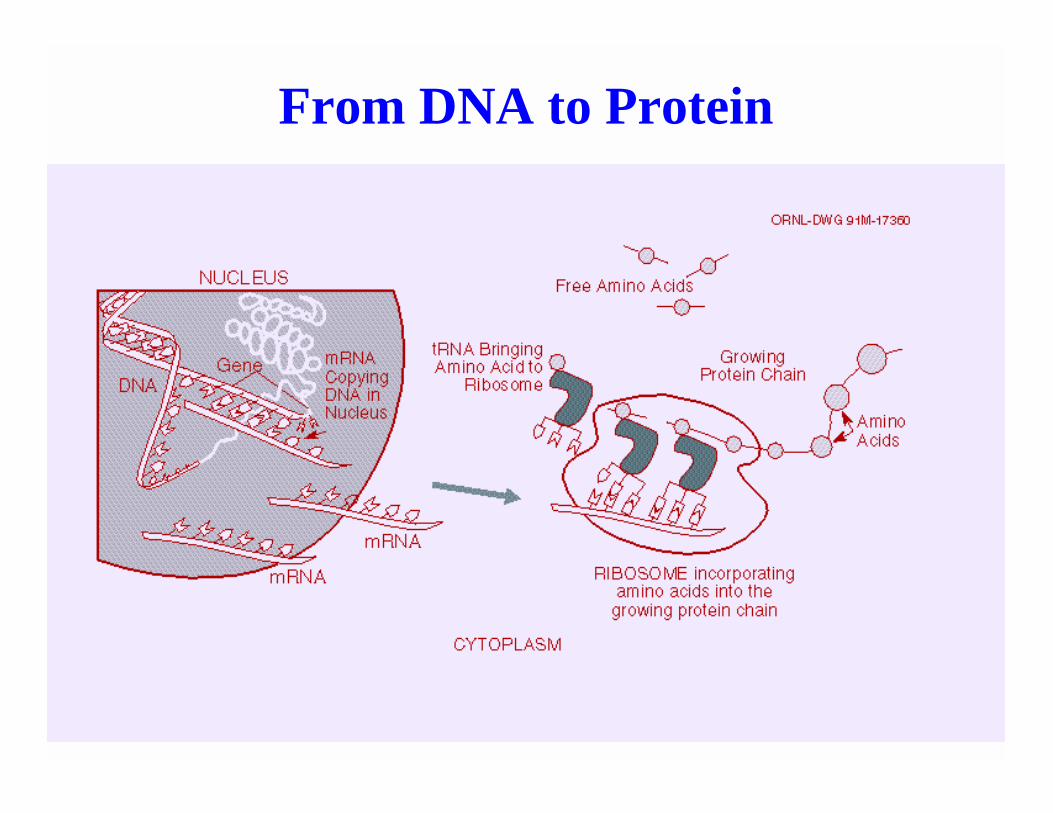
Biological Sequence Analysis 6
From DNA to Protein

Biological Sequence Analysis 7
Genetic Code

Biological Sequence Analysis 8
Main Resources for Data• NCBI --- national center for biotechnology
information.– GenBank maintainance– BLAST searching and servers– Entrez database– Taxonomy database– Structure database– Bankit and SEQUIN submission software– Web access http://www.ncbi.nlm.nih.gov
• EMBL --- European equivalent of NCBI.– Web access http://www.embl-heidelberg.de– EBI --- outstation of EMBL. http://www.ebi.ac.uk/

Biological Sequence Analysis 9
Finding “Patterns” inBiological Sequences
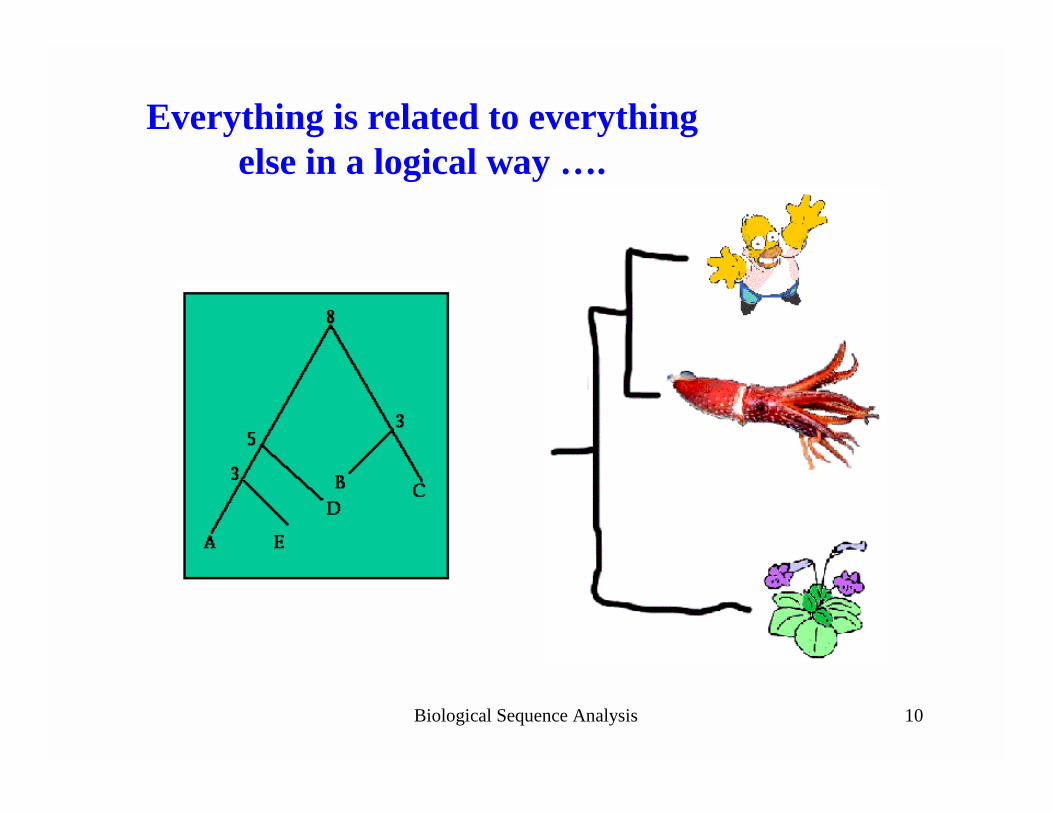
Biological Sequence Analysis 10
Everything is related to everything else in a logical way ….
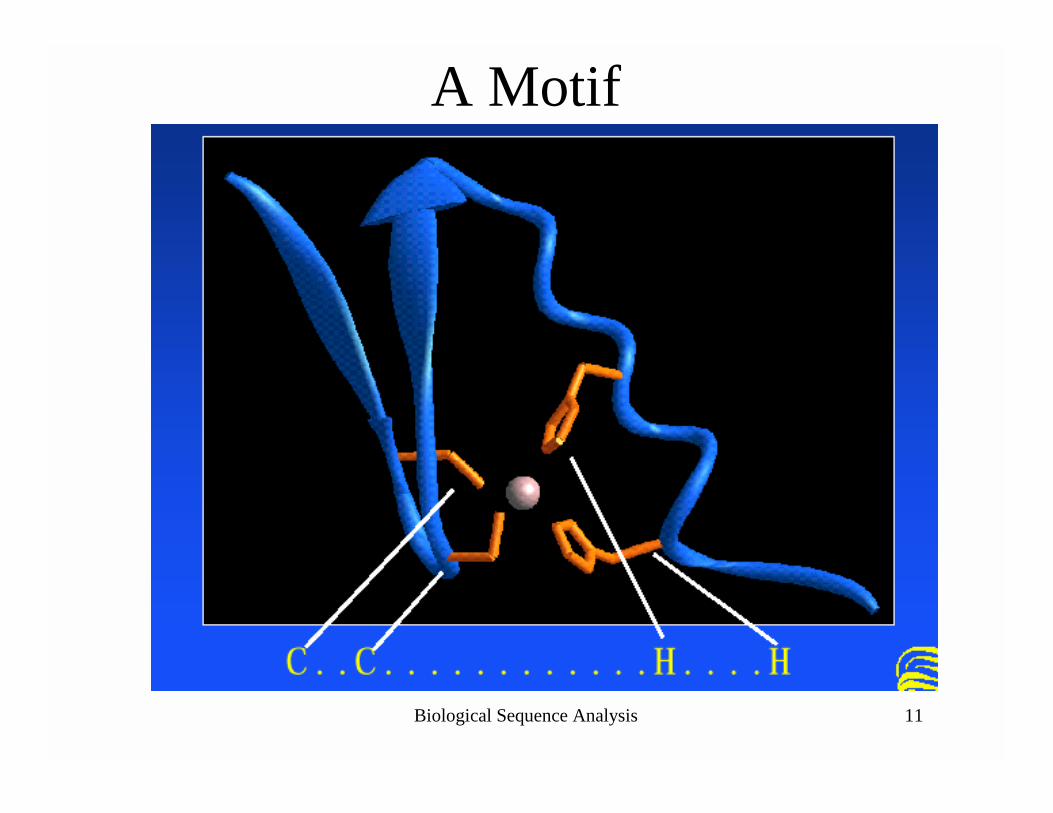
Biological Sequence Analysis 11
A Motif

Biological Sequence Analysis 12
Brute-forceStringSearch

Biological Sequence Analysis 13
Boyer-Moore String Search
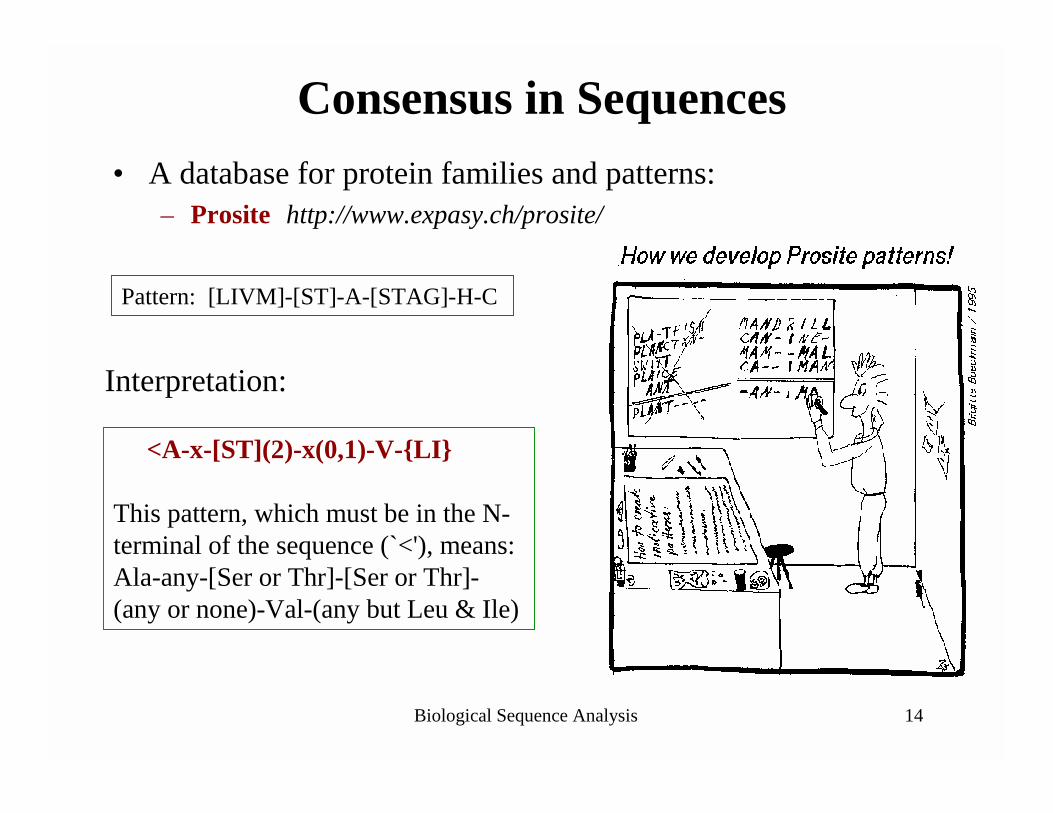
Biological Sequence Analysis 14
Consensus in Sequences• A database for protein families and patterns:
– Prosite http://www.expasy.ch/prosite/
<A-x-[ST](2)-x(0,1)-V-{LI}
This pattern, which must be in the N-terminal of the sequence (`<'), means: Ala-any-[Ser or Thr]-[Ser or Thr]-(any or none)-Val-(any but Leu & Ile)
Pattern: [LIVM]-[ST]-A-[STAG]-H-C
Interpretation:

Biological Sequence Analysis 15
Pattern Finding Programs• Prosite web program (http://www.expasy.ch/prosite/)
– ScanProsite - Scan a sequence against PROSITE or a pattern against SWISS-PROT
– ProfileScan - Scan a sequence against the profile entries in PROSITE
• Motif web programs ( http://motif.stanford.edu/identify/)– IDENTIFY– SCAN
• GCG SEQWEB programs (commercial)– Stringsearch– Findpatterns– Motifs

Biological Sequence Analysis 16
Pairwise Sequence Alignment Methods
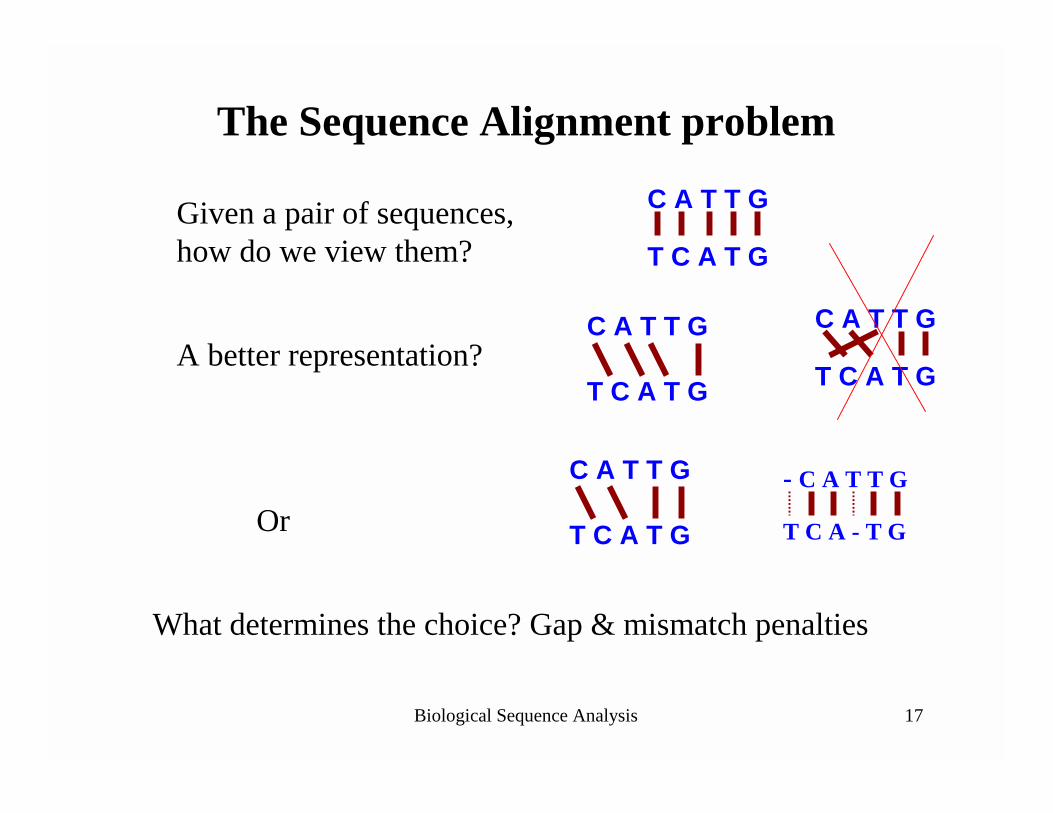
Biological Sequence Analysis 17
The Sequence Alignment problem
C A T T G
T C A T GGiven a pair of sequences, how do we view them?
C A T T G
T C A T GA better representation?
C A T T G
T C A T GOr
What determines the choice? Gap & mismatch penalties
- C A T T G
T C A - T G
C A T T G
T C A T G

Biological Sequence Analysis 18

Biological Sequence Analysis 19
Global: Needleman-Wunsch Algorithm• Finding the optimal alignment via dynamic programming• Example alignment
C A T T G …TCATG
o
o
C A T T - - G
T C - - A T G

Biological Sequence Analysis 20
Alignment Recursion
F(i,j)F(i-1,j)
F(i,j-1)F(i-1,j-1)
−−−−
+−−=
)1,( ),1(
),()1,1(max),(
γγ
jiFjiF
yxsjiFjiF
ji C A T T G
TCATG
0 -1 -2 -3 -4 -5-1
1-2-3-4-5
E.g., 1 ,2),( }{ == = γyxIyxs 0-1-2
0 -10332
554
0-12 1
46
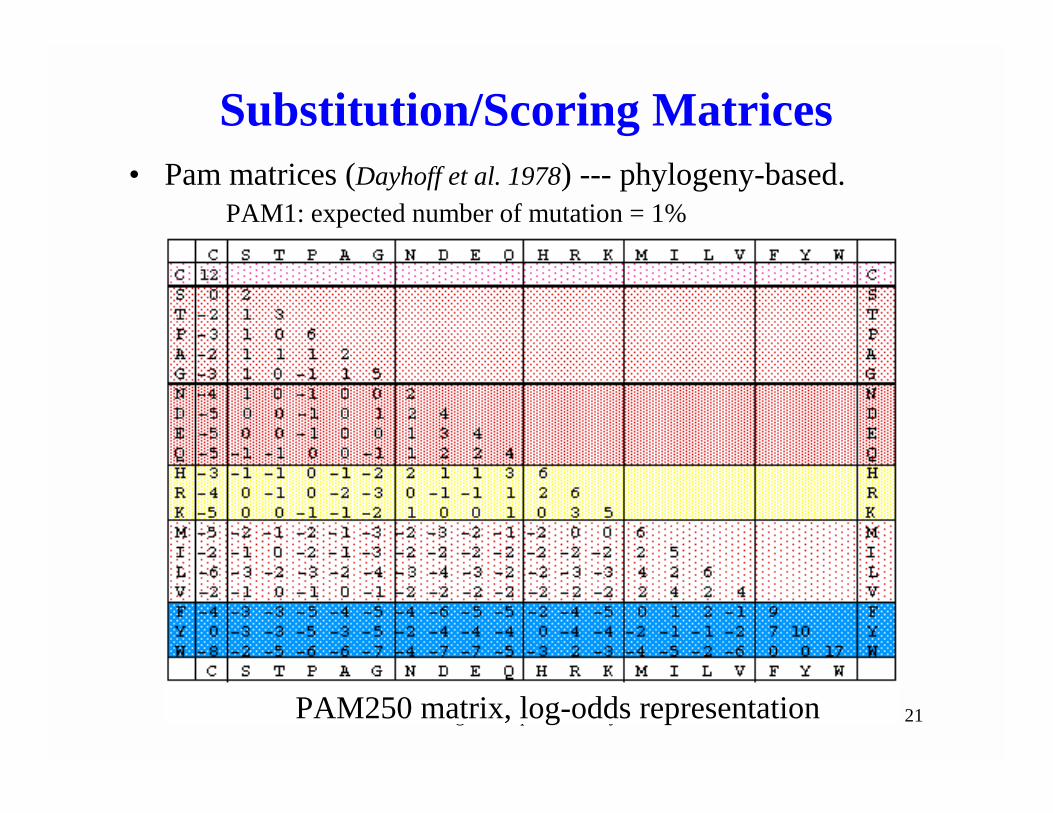
Biological Sequence Analysis 21
Substitution/Scoring Matrices• Pam matrices (Dayhoff et al. 1978) --- phylogeny-based.
PAM250 matrix, log-odds representation
PAM1: expected number of mutation = 1%

Biological Sequence Analysis 22
BLO(ck)SU(bstitution)M(atrix) (Henikoff & Henikoff 1992)
• Derived from a set (2000) of aligned and ungapped regions from protein families; emphasizing more on chemical similarities (versus how easy it is to mutate from one residue to another). BLOSUMx is derived from the set of segments of x% identity.
BLOSUM62 Matrix, log-odds representation

Biological Sequence Analysis 23
Gap Penalties• Linear score: �(g)=-gd
– Typically: d=8, in unit of half-bits (=4log2)
• Affine score: �(g)=-d-(g-1)e– d: gap opening penalty; e: gap extension penalty– Typical d=12, e=2 (in unit of half-bits)
• Gap penalty corresponds to log-probability of opening a gaps. For example, under the standard linear score, P(g=k)=exp(-dk)=2-4k

Biological Sequence Analysis 24
Local Alignment: Smith-Waterman Algorithm
−−−−
+−−=
)1,( ),1(
),()1,1( 0
max),(
djiFdjiF
yxsjiFjiF ji

Biological Sequence Analysis 25
Sequence comparison and data base search

Biological Sequence Analysis 26
BLAST(Altschul et al. 1990)
• Create a word list from the querry; – word length =3 for protein and 12 for DNA.
• For each listed word, find “neighboring words” (~ 50),• For each sequence in the database, search exact matches to each
word in the set. • Extend the hits in both directions until score drops below X• No gap allowed; use Karlin-Altschul statistics for significance• New versions (>1.4) of BLAST gives gapped alignments.• Compute Smith-Waterman for “significant” alignments• BLASTP (protein), BLASTN (DNA), BLASTX (pr �DNA).
TWWS >)',(
QTVGLMIVYDDA

Biological Sequence Analysis 27
BLAST 2.0• Two word hits must be found within a window of A
residues in order to trigger extension• Gapped extension from the middle of ungapped HSPs
• Position-specific iterative (PSI-) BLAST.– Profile constructed on the fly and iteratively refined.– Begin with a single query, profile constructed from those
significant hits; use the profile to do another search, and iterate the procedure till “convergence”

Biological Sequence Analysis 28
A Bayesian Model for Pairwise Alignment
Missing data --- Alignment matrix
A i,j = 1 if residue i of sequence 1 aligns with residue j of sequence 2, 0 otherwise.
Observed data pair of sequence R(1), R(2)
ΨR R1 2, PAM or Blosum A Ai j
ii j
j, ,≤ ≤∑ ∑1 1
)2()1()2()1( ,,)2()1( ),|,(
jiji RRjiRRji AARRP Ψ+Θ+Θ=Θ

Biological Sequence Analysis 29
Multiple Alignment

Biological Sequence Analysis 30
ClustalW Step 2: bBuild the Tree

Biological Sequence Analysis 31

Biological Sequence Analysis 32
However ….
• No explicit model to guide for the alignment --- heuristic driven.
• Tree construction has problems.• Overall, not sensitive enough for remotely
related sequences.

Biological Sequence Analysis 33
The Hidden Markov Model
• For given zs, ys ~ f(ys| zs, θ), and the zs follow a Markov process with transition ps(zs | zs-1, φ).
z1 z2 z3 ztzs…... …...
y1 y2 y3 ys yt
π φ θt t t tz p z z y y( ) ( , , | , , ; , )==== 1 1l l
“The State Space Model”
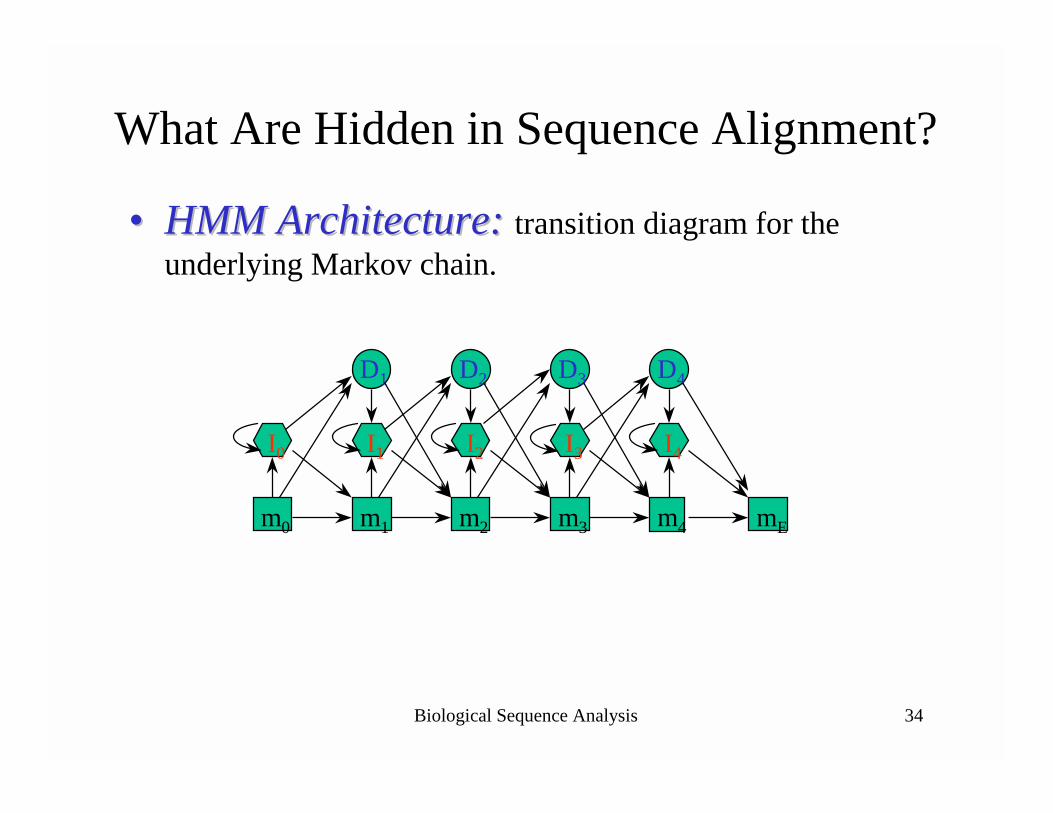
Biological Sequence Analysis 34
What Are Hidden in Sequence Alignment?
•• HMMHMM Architecture: Architecture: transition diagram for the underlying Markov chain.
m1 mEm0 m2 m3 m4
I0 I1 I2 I3 I4
D1 D2 D4D3

Biological Sequence Analysis 35
Gi ====
01
if generated by insertion if generated by a match
δ i = o f d e le t io n s#
Dashed path:
(δ1,G1)
r1 r6r2
(δ2,G2)
r3
(δ3,G3) (δ6,G6)
(0,1) (2,0) (2,0) (2,0)
r4
(δ4,G4)
r5
(δ5,G5)
(2,1) (2,0)Solid path:(0,1) (0,1) (0,1) (0,1) (0,0) (0,0)
The PathPath
m0
m1
m2
m3
m4
r0 r1 r2 r3 r4 r5 r6

Biological Sequence Analysis 36
Pfam alignment view (http://pfam.wustl.edu/browse.shtml)

Biological Sequence Analysis 37
More Restricted ModelsMore Restricted Models(Liu et al. 99, (Liu et al. 99, Neuwald Neuwald et al. 97)et al. 97)
•• Block Motif Propagation Model:Block Motif Propagation Model: limit the total number of gaps but no deletions allowed.
Motif 1 Motif 2 Motif 3 Motif 4
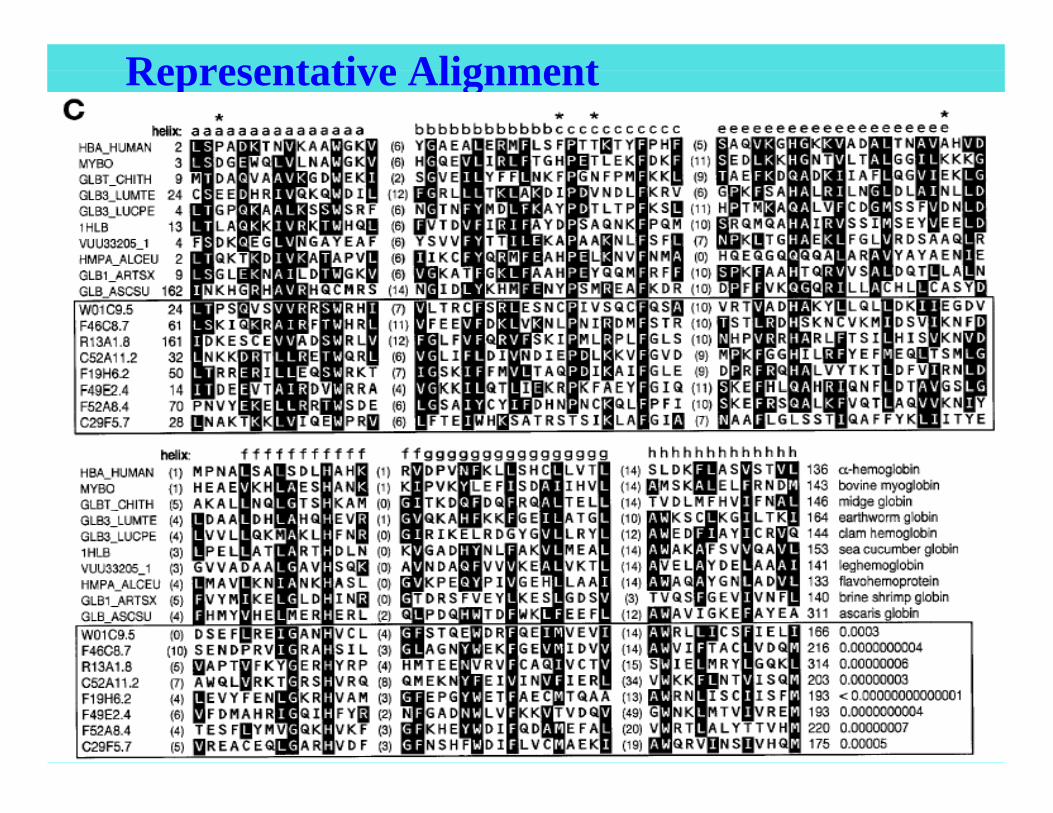
Biological Sequence Analysis 38
Representative Alignment

Biological Sequence Analysis 39
Finding Repetitive Patterns

Biological Sequence Analysis 40
Motif Alignment ModelMotif
width = w
length nk
a1
a2
ak
The missing data: Alignment variable: A={a1, a2, …, ak}
• Every non-site positions follows a common multinomialwith p0=(p0,1 ,…, p0,20)
• Every position i in the motif element follows probabilitydistribution pi=(pi,1 ,…, pi,20)

Biological Sequence Analysis 41
The Algorithm• Initialized by choosing random starting positions
• Iterate the following steps many times:– Randomly or systematically choose a sequence, say,
sequence k, to exclude.
– Carry out the predictive-updating step to update ak
• Stop when no more observable changes in likelihood• Available Programs:
– Gibbs motif sampler (http://www.wadsworth.org/res&res/bioinfo)
– Bioprospector (http://bioprospector.stanford.edu)
– MACAW (http://www.ncbi.nlm.nih.gov)
)0()0(2
)0(1 ,......,, Kaaa

Biological Sequence Analysis 42
a2
a1The PU-Step
ak ?
a3
1. Compute predictive frequencies of each position i in motifcij= count of amino acid type j at position i.c0j = count of amino acid type j in all non-site positions.qij= (cij+bj)/(K-1+B), B=b1+ • • •+ bK “pseudo-counts”
2. Sample from the predictive distriubtion of ak .
P a lqqk
i R l i
R l ii
wk
k
( ) , ( )
, ( )==== ++++ ∝∝∝∝ ++++
++++====∏∏∏∏1
01
Biological Sequence Analysis 43
Using MACAW

Biological Sequence Analysis 44
Idea 2: Mixture modeling
• View the dataset as a long sequence with k motif types:
• Idea: partition the input sequence into segments that
correspond to different (unknown) motif models.• It is a mixture model (unsupervised learning).
• Implement a predictive updating scheme.

Biological Sequence Analysis 45
Special Case: Bernoulli Sampler
• Sequence data: R = r1 r2 r3…… rN
• Indicator variable: ∆∆∆∆
= δ1δ2δ3 .. .. .. δΝ
• Likelihood: π(R, ∆ | Θ, ε), ε is the prior prob for δi=1• Predictive Update:
δ i ====
10
,,
if it is the start of an elementif not.
π δπ δ
εε
( | , )( | , )
>
>
>
>
[ ]
[ ]
,
,
k k
k k
i r
ri
wRR
pp
k i
k i
========
====−−−−
−−−−
−−−− ====
++++ −−−−
++++ −−−−
∏∏∏∏10 1
1
101
∆∆
parameter for the motif model

Biological Sequence Analysis 46
References (self)• Durbin R. et al. (1997). Biological Sequence Analysis, Cambridge
University Press, London.• Liu, J.S. (2001). Monte Carlo Strategies in Scientific Computing, Springer-
Verlag, New York.• Liu, J.S. and Lawrence, C.E. (1999). Bayesian analysis on biopolymer
sequences. Bioinformatics, 138-143.• Liu, J.S., Neuwald, A.F., and Lawrence, C.E. (1999) . Markovian
structures in biological sequence alignments. J. Amer. Statist. Assoc., 1-15.• Neuwald, A.F., Liu, J.S., Lipman, D.J., and Lawrence, C.E. (1997) .
Extracting protein alignment models from the sequence database. Nucleic Acids. Res. 25, 1665-1677.
• Liu, J.S., Neuwald, A.F., and Lawrence, C.E. (1995) . Bayesian models for multiple local sequence alignment and Gibbs sampling strategies. J. Amer. Statist. Assoc. 90, 1156-1170.
• Lawrence, et al. (1993). Science 262, 208-214.