killing of kras-mutant colon cancer cells via rac-...
TRANSCRIPT

Therapeutic Discovery
Killing of Kras-Mutant Colon Cancer Cells via Rac-Independent Actin Remodeling by the bGBP Cytokine, aPhysiological PI3K Inhibitor Therapeutically Effective In Vivo
Livio Mallucci1, Dong-yun Shi1, Derek Davies2, Peter Jordan2, Alastair Nicol2, Lavinia Lotti4,Renato Mariani-Costantini5, Fabio Verginelli5, Valerie Wells3, and Daniel Zicha2
AbstractActivating mutations in Kras are the most frequent mutations in human cancer. They define a subset of
patients who do not respond to current therapies and for whom prognosis is poor. Oncogenic Kras has been
shown to deregulate numerous signaling pathways of which the most intensively studied are the Ras/
extracellular signal–regulated kinase cascade and the phosphoinositide 3-kinase (PI3K)/Akt cascade. How-
ever, to date, there are no effective targeted therapies in the clinic against Kras-mutant cancers. Here, we report
that the b-galactoside–binding protein (bGBP) cytokine, a physiologic inhibitor of class I PI3Ks, is a potent
activator of apoptosis in Kras-mutant colorectal cancer cells, even when coharboring mutant-activated
PIK3CA. Our study unveils an elective route to intrinsic and extrinsic apoptosis, which involves the
cytoskeleton. Early events are inhibition of PI3K activity and Rac-independent actin rearrangement assignable
to phosphoinositide changes at the plasma membrane. Cyclin E deregulation, arrest of DNA synthesis, and
checkpoint kinase 2 activation underscore events critical to the activation of an intrinsic apoptotic program.
Clustering of CD95/Fas death receptors underscore events critical to the activation of extrinsic apoptosis. In
nudemice,wepresent thefirst evidence that xenograft tumordevelopment is strongly inhibitedbyHu-r-bGBP.
Taken together, our results open a new therapeutic opportunity to a subset of patients refractive to current
treatments. This first demonstration of therapeutic efficacy against Kras-mutant colon cancer suggests thatHu-
r-bGBPmay also be therapeutically effective against other cancers harboring activating Ras mutations as well
as PIK3CA mutations. Mol Cancer Ther; 11(9); 1884–93. �2012 AACR.
IntroductionActivating mutations in the Ras genes (H,K,N; ref. 1),
themost commononcogenicmutations in human cancers,are dominant determinants of drug resistance. Most fre-quent in Kras, Kras mutations define tumors refractory toconventional chemotherapy and radiation therapy (2, 3)and to more recently introduced therapies based onEGF receptor tyrosine kinase inhibitors (4–7). Locked inthe GTP-bound mode, oncogenic Ras constitutively acti-vates the Raf/MEK/extracellular signal–regulated kinase(ERK) cascade and an integrated signaling network,
which controls cell proliferation and cell survival (8, 9)to be a major mediator of tumorigenesis.
The importance of activating mutations in the Rasoncogenes in human cancers has made Ras and down-stream effectors elective targets for therapeutic inter-vention, but the value of targeting inhibitors in the clinicremains doubtful. For example, prevention of Ras anchor-age at the cell membrane by farnesyltransferase inhibi-tors has been shown to be effective in mice expressingoncogenic Ras but not in patients with solid tumors(10). Similarly, in more recent studies, CI-1040, a MEKinhibitor, was found to induce significant shrinkage ofKras-driven lung carcinomas in mice (11), but had nosignificant effect in patients with advanced lung, breast,colon, and pancreatic cancers (12). Using a differentapproach, a number of compounds with efficacy againstcells with mutant-activated Kras have been identified byhigh-throughput screening (13–16) and shown to inhibitxenograft growth (13, 15, 16); however, the mechanismsby which these compounds operate are not fully under-stood and their anticancer efficacy in the clinic remainsuncertain.
Recently, the necessary role played by the phosphoi-nositide 3-kinase (PI3K)/Akt pathway in maintainingtumor growth has made this pathway a target for small
Authors' Affiliations: 1School of Biomedical and Health Sciences, King'sCollege London; 2CancerResearchUKLondonResearch Institute; 3NYU inLondon, London, United Kingdom; 4Department of ExperimentalMedicine,La Sapienza University, Rome; and 5Unit of General Pathology, AgingResearch Center, G. D'Annunzio University Foundation, Chieti, Italy
Note: Current address for D.-y. Shi: Department of Biochemistry andMolecular Biology, Shanghai Medical College of Fudan University,200032 Shanghai, PR China.
Corresponding Author: Livio Mallucci, School of Biomedical and HealthSciences, King's College London, Franklin Wilkins Building, 150 StamfordStreet, SE1 9NH London, United Kingdom. Phone: 44-207-848-4257; Fax:44-207-848-4500; E-mail: [email protected]
doi: 10.1158/1535-7163.MCT-11-1041-T
�2012 American Association for Cancer Research.
MolecularCancer
Therapeutics
Mol Cancer Ther; 11(9) September 20121884
on May 28, 2018. © 2012 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst July 2, 2012; DOI: 10.1158/1535-7163.MCT-11-1041-T

molecule inhibitors of the PI3K cascade (17–21).However,the efficacy of these inhibitors as single agents in Kras-driven oncogenesis is disputable. For example, in micethat had developed lung tumors in response to Krasactivation, no significant tumor regression was foundupon treatment with a dual pan-PI3K/mTOR inhibitor,which had instead shown efficacy in lung tumors drivenby the expression of activated PI3K (22). In other experi-ments, the efficacy of the PI3K inhibitor PX-866 wassignificantly less in xenografts bearing Kras and PIK3CAmutations than in those having PIK3CA mutations alone(23). On the other hand, Kras-mutant lung tumors didregress when the pan-PI3K/mTOR inhibitor was used incombinationwith anMEK inhibitor (22). Similarly, in cellsexpressing Kras or Hras mutants, resistance to PI3Kinhibitors could be reversed by inhibitors of the Ras/ERKpathway (24). Despite these positive results, it remains tobe established whether in the human system Kras onco-genicity could be overcome by the combined targeting ofthe PI3K and the Ras pathways and be beneficial as, inaddition to toxicity, a disadvantage of this approach is thatthese pathways are also required for the proliferation ofnormal cells and for the maintenance of their homeostaticbalance.In previous studies, we have shown that monomeric
b-galactoside–binding protein (bGBP), an antiprolifera-tive cytokine (25) that in normal cells participates inthe negative regulation of the cell cycle (26), operatesthrough mechanisms that involve high-affinity receptor
binding (Kd � 1.5 � 10�10 mol/L; ref. 26) and molecularinteractions leading to downregulation of class IA andclass IB PI3Ks (27). Inhibition of PI3K activity by Hu-r-bGBP was found to have two major outcomes: sup-pression of Ras-GTP loading, leading to a block of ERKactivation (27) and negation of Akt gene expressionleading to loss of Akt (21), conditions that either byblocking the ability of cancer cells to proliferate or byimpairing their ability to survive can block oncogenic-ity, thus highlighting a selective anticancer effect as innormal cells, bGBP-enforced cell cycle restriction isreversible (26).
Wenowreport that in colorectal carcinomacells bearingoncogenic Kras downregulation of PI3K activity by Hu-r-bGBP at doses that did not inhibit canonical signalingdownstreamofRas or PI3Kpromoted events critical to theactivation of an intrinsic and an extrinsic cell death pro-gram initiating with Rac-independent actin reorganiza-tion assignable to phosphoinositide changes at the plasmamembrane.We also report that Hu-r-bGBP had therapeu-tic efficacy in vivo and that the added presence of activat-ing mutations in PIK3CA did not confer resistance totreatment with Hu-r-bGBP.
Materials and MethodsCells
SW480, SW620, and LoVo cells were acquired fromthe American Type Culture Collection and cultured in
Figure 1. Treatment withHu-r-bGBP inhibits cell proliferationand induces apoptosis. A, rate of cellproliferation as related to doseresponse. Values are means oftriplicate cultures � SEM. Insetsshow cell-cycle phase distribution atday 3 of treatment with 4 nmol/L Hu-r-bGBP. Hu-r-bGBP was added 6hours after seeding. B, apoptoticevents at day 3 of treatment. Top tobottom, mitochondrial membranepotential assessed bytetramethylrhodamine ester (TMRE)staining; phosphatidylserineorientation at the plasma membraneassessed by Annexin V staining;caspase-3 activity assessed usingPhiPhiLux. Inset values arepercentages of cells committed toapoptosis (encircled area). Hu-r-bGBP 4 nmol/L added 6 hours afterseeding. Data are from parallelrepresentative experiments.
1 110 00
2020
2010
4040
4030 60
60
6050
80
80100120
2 22 3 33 4 44 5 55DayDay Day
4
2
0
1
42
TMRETMRE TMRE
Annexin Annexin
Caspase-3 Caspase-3Caspase-3
DA
PI
DA
PI
DA
PI
DA
PI
DA
PI
DA
PI
DA
PI
DA
PI
DA
PI
0
105103101
105
103
101
Annexin
0
1
42
A
B
SW480 SW620 LoVo
1.9 9.2
1.7 4.6
1.0 4.5
2.2 38.8
2.2 23.6
0.9 23.9
4.7 25.0
1.3 13.6
0.4 8.3
105103101
105
103
101
105103101
105
103
101
105103101
105
103
101
105103101
105
103
101
105103101
105
103
101
105103101
105
103
101
105103101
105
103
101
105103101
105
103
101
105103101
105
103
101
105103101
105
103
101
105103101
105
103
101
105103101
105
103
101
105103101
105
103
101
105103101
105
103
101
105103101
105
103
101
105103101
105
103
101
105103101
105
103
101
0 0 0
Cel
l num
ber
× 1
0-5
βGBPnmol/L
βGBPnmol/L
βGBPnmol/L
βGBP βGBP βGBPControl ControlControl
βGBP βGBP βGBPControl Control Control
Day 3 Day 3 Day 3
Killing Kras-Mutant Cancer Cells
www.aacrjournals.org Mol Cancer Ther; 11(9) September 2012 1885
on May 28, 2018. © 2012 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst July 2, 2012; DOI: 10.1158/1535-7163.MCT-11-1041-T
Leibovitz’s L-15 medium with 10% fetal bovine serum.HCT116wt, HCT116mut, DLD1wt, and DLD1mut cells, agift from Bert Vogelstein (John Hopkins University, Bal-timore,MD),were cultured as originally reported (28). Allcell lines were authenticated. Cell population distributionwas assessed by propidium iodide staining and fluores-cence-activated cell-sorting analysis as reportedprevious-ly (26). Epithelial cells of the normal colon mucosa fromsurgical interventions were obtained with informed con-sent and the study was approved by the Medical School’sEthics Committee.
Recombinant bGBPHu-r-bGBP was expressed in Escherischia coli BL21
(DE3) using hGal-1 cDNA in PET21a, purified by lac-tose–agarose (Sigma) affinity chromatography and purityassessed by matrix-assisted laser desorption/ionization–time-of-flight (MALDI-TOF).
Apoptosis assaysTetramethylrhodamine ethyl ester (Molecular Probes/
Invitrogen) staining, Annexin V (Pharmingen) staining,and caspase-3 activity (OncoImmunin) were assessed
_ _+ +βGBP
βGBP
GAPDH
ERK2
SW480 SW620 LoVo_ _
+ +_ _
+ +
_ _+ +
_ _+ +
_ _+ +
Akt1/2
Day
pAkt
GAPDH
1 2 1 2 1 2
Day 1 2 1 2 1 2
0 6 12 18 0 12 24 360
0 6 12 18
10
20
30
40
50
60
70
80
90
100
PIP
3 %
of
con
tro
l
SW480 SW620 LoVo
Hours HoursHours
A B
SW480 SW620 LoVoControl βBGP Control βBGP Control βBGP
C
* * ** * *
*
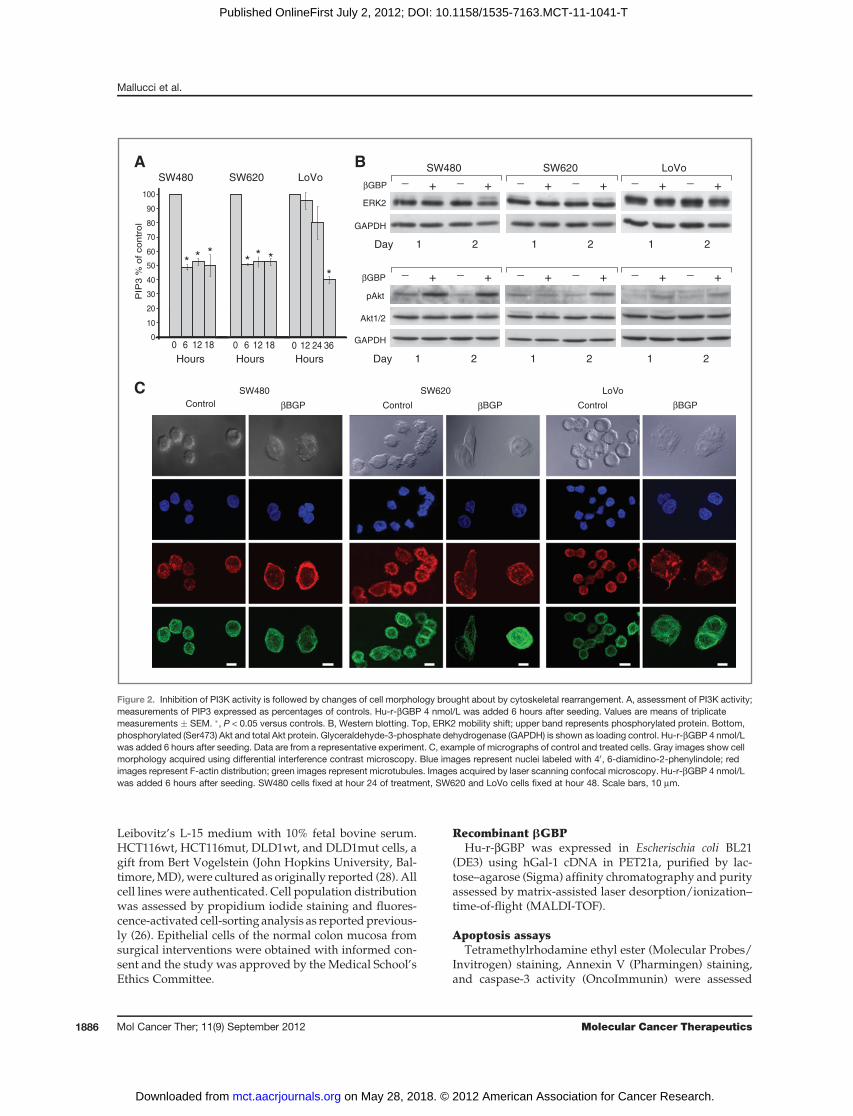
Figure 2. Inhibition of PI3K activity is followed by changes of cell morphology brought about by cytoskeletal rearrangement. A, assessment of PI3K activity;measurements of PIP3 expressed as percentages of controls. Hu-r-bGBP 4 nmol/L was added 6 hours after seeding. Values are means of triplicatemeasurements � SEM. �, P < 0.05 versus controls. B, Western blotting. Top, ERK2 mobility shift; upper band represents phosphorylated protein. Bottom,phosphorylated (Ser473) Akt and total Akt protein. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is shown as loading control. Hu-r-bGBP 4 nmol/Lwas added 6 hours after seeding. Data are from a representative experiment. C, example of micrographs of control and treated cells. Gray images show cellmorphology acquired using differential interference contrast microscopy. Blue images represent nuclei labeled with 40, 6-diamidino-2-phenylindole; redimages represent F-actin distribution; green images represent microtubules. Images acquired by laser scanning confocal microscopy. Hu-r-bGBP 4 nmol/Lwas added 6 hours after seeding. SW480 cells fixed at hour 24 of treatment, SW620 and LoVo cells fixed at hour 48. Scale bars, 10 mm.
Mallucci et al.
Mol Cancer Ther; 11(9) September 2012 Molecular Cancer Therapeutics1886
on May 28, 2018. © 2012 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst July 2, 2012; DOI: 10.1158/1535-7163.MCT-11-1041-T

according to manufacturers’ instructions and analyzedusing an LSRII (Becton Dickinson).
PI3K assayStep-by-step description of the method for assessment
of PI3Kactivity has been reportedpreviously (21). In brief,the immmunoprecipitated class I enzyme complex (27)was incubated in a kinase reaction for 3 hours with40 pmol phosphatidylinositol (4,5)-biphosphate (PIP2)substrate and the phosphatidylinositol (3,4,5)–triphos-phate (PIP3) generated assayed in a competitive ELISA(Echelon Biosciences). Differences were tested using Stu-dent t test and P < 0.05 was considered statisticallysignificant.
Western blottingERK2 phosphorylationwas visualized bymobility shift
usinganti-p42polyclonal anitibodies (SantaCruzBiotech-nology). Phosphorylated Akt was detected by antiphos-pho-Akt (Ser473) antibody (Cell Signaling Technology)and total Akt1/2 protein probed with anti-Akt1/2 anti-bodies (Santa Cruz Biotechnology). E2F1 was detectedusing anti-E2F1 polyclonal antibodies (Santa Cruz Bio-technology) and phosphorylated checkpoint kinase (Chk)2 detected with antiphospho-Chk2 (Thr 68) polyclonalantibodies (Cell Signaling Technology). Secondaryantibodies conjugated to horseradish peroxidase (GEHealthcare) were used for visualization by enhancedchemiluminescence (GEHealthcare). Blotswere reprobedwith anti-glyceraldehyde-3-phosphate dehydrogenaseantibodies (Santa Cruz Biotechnology) or with monoclo-nal anti-b-actin (Sigma). Active Rac1 levels were assessedby affinity precipitation using PAK1 p21 binding domainagarose (Millipore) according to the manufacturer’sinstructions, followed by immunoblot analysis usinganti-Rac1 monoclonal antibody (Cell Signaling) andOdyssey fluorescent secondary antibodies. Total Rac wasassessed using the same antibodies.
Fluorimetric quantitation of cyclin ECells fixed in cold 70% ethanol were treated with
fluorescein isothiocyanate (FITC)-labeled monoclonalanti-cyclin E antibody (Becton Dickinson), washed,stainedwith propidium iodide containingRNase (Sigma),
and analyzed using an LSRII (Becton Dickinson). FITCfluorescence was collected using a 530/30 filter and pro-pidium iodide fluorescence using a 610/20 filter. At least20,000 events were recorded.
DNA synthesisCells incubated with bromodeoxyuridine (BrdU; Sig-
ma) for 30 minutes were fixed in cold 70% ethanol andstained for BrdUrd uptake using a monoclonal anti-BrdUantibody (BectonDickinson), followedbyanFITC-labeledmonoclonal goat anti-mouse antibody (Dako). DNA wascounterstained with propidium iodide (Sigma) contain-ingRNase (Sigma). Sampleswere analyzedusinganLSRII(Becton Dickinson) and FITC fluorescence collected usinga 530/30 filter and propidium iodide fluorescence using a610/20 filter. At least 20,000 events were recorded.
MicroscopyCells on coverslips fixed in 3% paraformaldehyde/0.2%
gluteraldehyde solution were stained with Texas Redphalloidin (Molecular Probes) for F-actin detection. Fortubulin detection, cells were treated with a monoclonalanti-a-tubulin antibody (Sigma) followed by anti—mousesecondary antibody labeled with Alexa Fluor 488 (Molec-ular Probes). DNA was stained with 40, 6-diamidino-2-phenylindole (DAPI, Sigma). Imaging was carried outusing a Zeiss LSM 510 confocal system. Analysis of theF-actin–labeled regions was conducted in an automatedmanner using journals in MetaMorph software (UniversalImaging) and statistical significance tested by ANOVA(29). For CD95/Fas and FasL imaging with a Zeiss Axio-phot microcope, cells were fixed in 4% paraformaldehyde,incubated with a monoclonal antibody to CD95/Fas(Abcam) followed by FITC-conjugated goat anti-mouseimmunoglobulin G (IgG; Cappel) and rabbit polyclonalFasL antibody (Santa Cruz Biotechnology), followed byTexas red–conjugated goat anti-rabbit IgG (Jackson Immu-noresearch Laboratories). Time-lapse imaging by differen-tial interference contrast microscopy was used to monitorperipheral dynamics at the plasma membrane edge.Recordings on sets of 150 cells from 3 randomly selectedfields fromdouble experimentswere analyzed and gradedon a 0 to 3 arbitrary scale by 2 independent observers andstatistical significance determined by c2 test.
Table 1. Quantitation of cell spread area as defined by F-actin staining
F-Actin spread area at 24 h, mm2 F-Actin spread area at 48 h, mm2
Control bBGP P Control bBGP P
SW480 187.2 � 4.0 (278) 216.7 � 4.9 (229) <0.01 192.3 � 3.6 (414) 236.6 � 9.0 (117) <0.001SW620 190.7 � 3.4 (330) 219.3 � 4.9 (208) <0.01 212.8 � 4.3 (289) 245.1 � 5.8 (219) <0.01LoVo 183.4 � 4.0 (221) 224.8 � 4.0 (342) <0.001 193.1 � 2.7 (707) 250.4 � 6.7 (179) <0.001
NOTE: Values aremeans�SEM.Data acquired usingMetaMorph software and analyzed by ANOVA (29) inMathematica 4.2. Values inbrackets represent the number of cells measured.
Killing Kras-Mutant Cancer Cells
www.aacrjournals.org Mol Cancer Ther; 11(9) September 2012 1887
on May 28, 2018. © 2012 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst July 2, 2012; DOI: 10.1158/1535-7163.MCT-11-1041-T
Tumor growth in miceThymectomized female nude CD-1mice (Charles River
Laboratory) were implanted subcutaneously in the rightflankwith 5� 106 SW620 cells in 150 mL PBS. Controlmice
received an equal volume of PBS. Mice were ear markedand randomized in groups of 7 and treatment with Hu-r-bGBP dissolved in PBS started at a tumor volume ofapproximately 40 mm3. Treatment was carried out by
0
25
50
75
100
125
_ _ _ _ _ _+ + + + + +βGBP
1 12 2GAPDH
Day
E
E2F1
SW480 SW620 LoVo
1 2
E2F1CM 12 h 24 h 48 h
SW480
Actin
D
SW480 SW620 Lo Vo
0 50 150 2500
50
150
250
Propidium iodide
Day
11
94
750 27
38
8 67
84
Propidium iodide
Propidium iodide
Propidium iodide
Propidium iodide
Propidium iodide
Brd
Urd
FIT
CB
rdU
rd F
ITC
60.1 41.6
0 50 150 250
48.5 0.6
0 50 150 250 0 50 150 2500
50
150
250
60.5 42.2
49.6 1.1
050
150
250
050
150
250
0 50 150 250 0 50 150 250
0 50 150 250 0 50 150 250
55.0 35.9
47.4 2.2
0 50 150 250 0 50 150 250
0 50 150 250 0 50 150 250
A
B
_ _ _ _ _+ + + + +βGBP HU
LoVo
1 2 3 4 24 h
Thr68P-Chk2
GAPDH
Day
_ _ _ _ _+ + + + +βGBP HU
SW620
1 2 3 4 24 h
Thr68P-Chk2
GAPDH
Day
_ _ _ _ _+ + + + +βGBP HU
SW480
1 2 3 4 24 h
Thr68P-Chk2
GAPDH
Day
C
1 2 3 Day1 2 3 Day1 2 30
25
50
75
100
125
0
25
50
75
100
125
Day
1D
ay 2
050
150
250
050
150
250
Flu
ores
cenc
e in
tens
ity
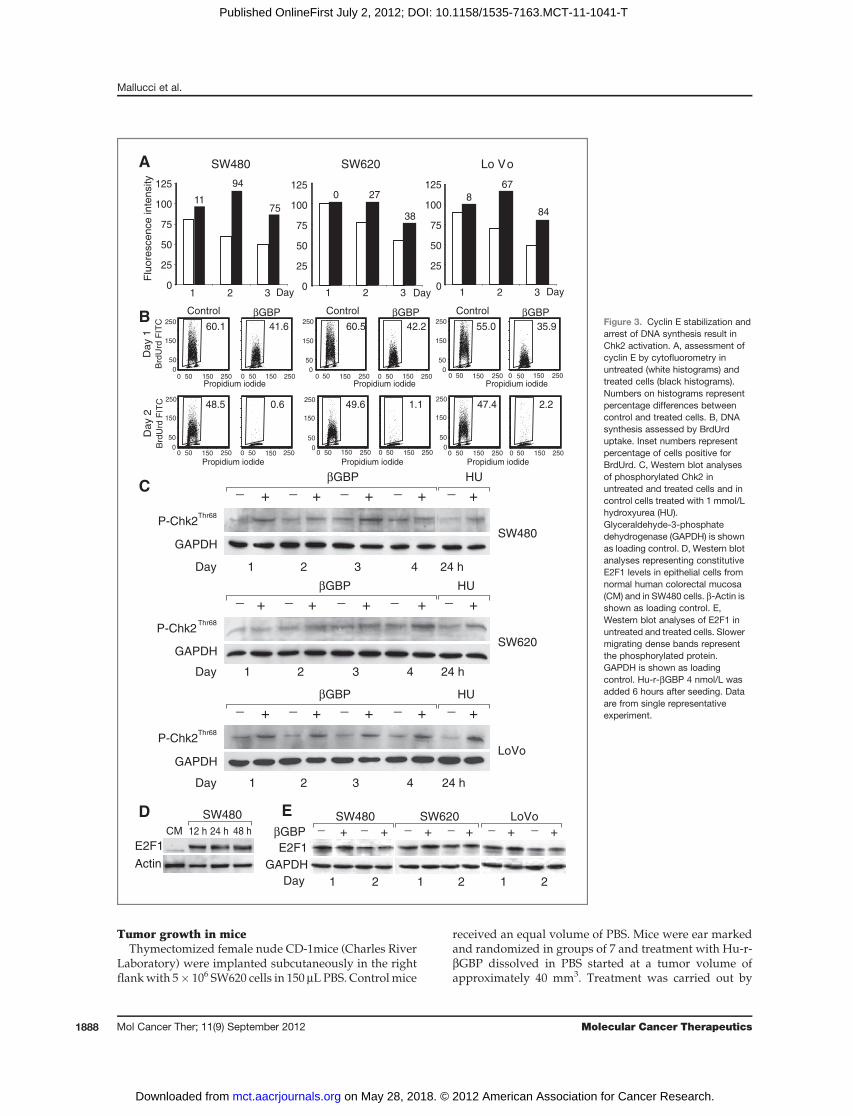
βGBPControl βGBPControl βGBPControlFigure 3. Cyclin E stabilization andarrest of DNA synthesis result inChk2 activation. A, assessment ofcyclin E by cytofluorometry inuntreated (white histograms) andtreated cells (black histograms).Numbers on histograms representpercentage differences betweencontrol and treated cells. B, DNAsynthesis assessed by BrdUrduptake. Inset numbers representpercentage of cells positive forBrdUrd. C, Western blot analysesof phosphorylated Chk2 inuntreated and treated cells and incontrol cells treated with 1 mmol/Lhydroxyurea (HU).Glyceraldehyde-3-phosphatedehydrogenase (GAPDH) is shownas loading control. D, Western blotanalyses representing constitutiveE2F1 levels in epithelial cells fromnormal human colorectal mucosa(CM) and in SW480 cells. b-Actin isshown as loading control. E,Western blot analyses of E2F1 inuntreated and treated cells. Slowermigrating dense bands representthe phosphorylated protein.GAPDH is shown as loadingcontrol. Hu-r-bGBP 4 nmol/L wasadded 6 hours after seeding. Dataare from single representativeexperiment.
Mallucci et al.
Mol Cancer Ther; 11(9) September 2012 Molecular Cancer Therapeutics1888
on May 28, 2018. © 2012 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst July 2, 2012; DOI: 10.1158/1535-7163.MCT-11-1041-T

s.c. injection of 150 mL in the tumor area for 6 consecutivedays with 1 day interval, up to 35 days. Control micereceived an equal volume of PBS. Experiments werecarried out in accordance with the U.K. CoordinatingCommittee on Cancer Research guidelines and approvedby the local responsible authorities.
ResultsArrest of cell replication and apoptosis is preceded bycytoskeletal rearrangementWe tested Hu-r-bGBP against SW480 and SW620 colo-
rectal cancer cells, of primary and metastatic derivation,both harboring KrasG12V, and against LoVo cells, also ofmetastatic origin, which harbor KrasG13D (COSMIC data-base). We found that treatment with Hu-r-bGBP at con-centrations of 2–4 nmol/L, some 10-fold lower than thoserequired for other cancer cells (21, 30), resulted in growtharrest and accumulation of cells in S-phase, a state revers-ible in normal cells (26) but followed in the cells of thisstudy by total cell loss attributable to the activation of anapoptotic program documented by changes in mitochon-drial membrane potential, functional alteration of theplasma membrane, and caspase-3 activation (Fig. 1Aand B).We next investigated whether as in other cell contexts
(21, 27) PI3K was a responder to the action of Hu-r-bGBP.As cell phosphoinositide levels do not directly representthe functional state of the PI3K enzymes, but are the resultof PI3K and PTEN activity, to estimate PI3K activity weisolated the PI3K enzyme complex (27) by immunopre-cipitation and assessed its ability to convert PIP2 into PIP3in a kinase reaction by measuring the generated PIP3 in acompetitive ELISA (21). PIP3 quantitation (Fig. 2A) showsthat downregulation of PI3K activity was a prime event,although delayed in the LoVo cells. However, contrary to
previous evidence from other cell types (21, 27), we foundno indication, after PI3K inhibition, of loss of ERK or Aktfunction in which phosphorylation state in the cells pro-grammed to death was instead increased (Fig. 2B), aresponse conceivably similar to that seen in human cancercells in which inhibition of mTORC1 by a rapamycinderivative resulted in increase ERK and Akt activity viaa feedback loop (31). We discovered instead that thetreated cells had undergone a profound change of shapecharacterized by F-actin rearrangement, spreading of themicrotubular network, and an enlargement of the cellspread area in which changes in time were quantitatedin terms of F-actin spatial distribution (Fig. 2C and Table1), changes that can block cancer cellmotility and invasion(32) but in our case followed by apoptosis (Fig. 1B).
To investigate whether actin remodeling related tochanges in Rac activity, a key regulator of actin dynamics,we assessed active Rac1 levels by immunoblot analysisand examinedprotrusive activity at themembrane edge, aRac-mediated process (33), by time-lapse imaging withinthe 24 hours time span, leading to cytoskeletal changes.We found that in the cells in which PI3K activity had beeninhibited, Rac1 levels remained unchanged and edgemembrane activity was not affected by treatment withHu-r-bGBP or by treatment with wortmannin (10 mM), apharmacologic p110 inhibitor. These results, consistentwith the finding that edge ruffling induced by constitu-tively active Rac is not blocked upon PI3K inhibition (34),suggest that in cells harboring oncogenic Ras, Rac activa-tion is maintained through Ras-mediated signaling in aPI3K independent manner (35).
Cyclin E stabilization and arrest of DNA synthesisresult in Chk2 activation
To determine which biochemical events consequent toPI3K inhibition and cytoskeletal changes may play a part
Figure 4. Localization of CD95/Fasand FasL in controls and in treatedcells. Control cells show punctatestaining for both CD95/Fas (green)and FasL (red) with low levels ofcolocalization (bottom, merge).Treated cells show clustering ofCD95/Fas (green) and clustering ofFasL (red), which can be frequentlyfound at the same localization on cellsurfaces (bottom, merge). Examplesof this clustering are indicated byarrowheads. Hu-r-bGBP 4 nmol/Lwas added 6 hours after seeding.SW480 cells fixed at hour 24 oftreatment, SW620 and LoVo cellsfixed at hour 48. Scale bars, 10 mm.
SW480 SW620 LoVo
Control βBGP Control βBGP Control βBGP
Fas
Fas
LM
erge
Killing Kras-Mutant Cancer Cells
www.aacrjournals.org Mol Cancer Ther; 11(9) September 2012 1889
on May 28, 2018. © 2012 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst July 2, 2012; DOI: 10.1158/1535-7163.MCT-11-1041-T
in cell-cycle arrest and in the activation of an apoptoticprogram, we examined parameters involved in cellgrowth restriction.
Following immunoblot analysis, which in thearrested cells revealed no changes in cyclin D1 levelsbut raised and persistent levels of cyclin E and agradual, and expected, decline of cyclin A, we quan-titated cyclin E by cytofluorometry and found thatwhile decreasing in the replicating cells, cyclin E levelsin the arrested cells remained ectopically high, withresulting relative values greater than those of controlsby about 60% to 90% (Fig. 3A). As ectopic expressionof cyclin E can cause impairment of DNA replicationdue to defects in replication initiation (36), we inves-tigated whether in the arrested cells DNA synthesishad been inhibited. We found that by day 2 of treat-ment, DNA synthesis had come to a halt (Fig. 3B).Because processes that interfere with DNA synthesismay result in the activation of DNA damage check-points and death by apoptosis (37), we turned ourattention to the downstream kinase effectors of theDNA damage response pathways (37–39). Chk1 wasnot detectable but we found that Chk2, which isrequired for responses to DNA damage and replicationblock (37, 39), had been activated (Fig. 3C). This is ofrelevance as Chk2 can phosphorylate E2F-1, regulatingits stability and transcriptional activity (38, 40), and bea cause of apoptotic induction in cells in which, incontrast to normal cells, E2F-1 is overexpressed (41, 42),as can be seen when comparing epithelial cells from thenormal colon mucosa and the colon cancer cells of thisinvestigation (Fig. 3D and E).
Involvement of death receptorsFurther to the rearrangement of cytoskeletal architec-
ture (Fig. 2B), it is conceivable that rearrangement in theorganization of subcortical actin may affect macromo-lecular mobility within the plane of the plasma mem-brane. We therefore investigated whether treatmentwith Hu-r-bGBP would affect the distribution patternof the CD95/Fas death receptor which, as well as otherdeath receptors (28), is a mediator of apoptosis, andexamined whether the distribution pattern of Fas Lwould change accordingly. The evidence collected (Fig.4) shows that as suggested by their colocation, CD95/Fas-Fas L clustering, a prime condition for the activationof an extrinsic apoptotic program, was detectable ineach cell line.
Hu-r-bGBP has efficacy in vivoNext, we tested whether the therapeutic efficacy
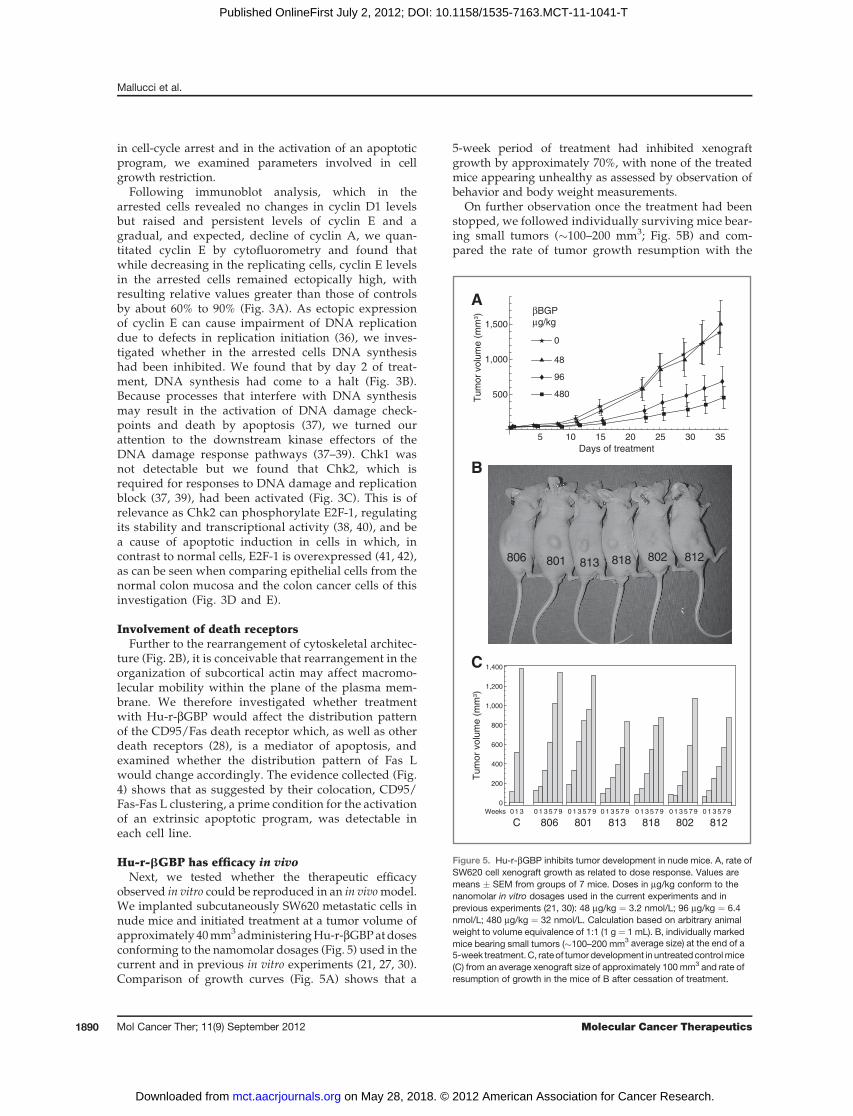
observed in vitro could be reproduced in an in vivomodel.We implanted subcutaneously SW620 metastatic cells innude mice and initiated treatment at a tumor volume ofapproximately 40mm3administeringHu-r-bGBPatdosesconforming to the namomolar dosages (Fig. 5) used in thecurrent and in previous in vitro experiments (21, 27, 30).Comparison of growth curves (Fig. 5A) shows that a
5-week period of treatment had inhibited xenograftgrowth by approximately 70%, with none of the treatedmice appearing unhealthy as assessed by observation ofbehavior and body weight measurements.
On further observation once the treatment had beenstopped, we followed individually surviving mice bear-ing small tumors (�100–200 mm3; Fig. 5B) and com-pared the rate of tumor growth resumption with the
βBGPμg/kg
Days of treatment
Tum
or v
olum
e (m
m3 )
Tum
or v
olum
e (m
m3 )
806 801 813 818 802 812
0
200
400
600
800
1,000
1,200
1,400
01 3 0 1 3 5 7 9 0 1 3 5 7 9 0 1 3 5 7 9 0 1 3 5 7 9 0 1 3 5 7 9 0 1 3 5 7 9Weeks
806 801 813 818 802 812C
5 10 15 20 25 30 35
500
1,000
1,500
480
96
48
0
A
B
C
Figure 5. Hu-r-bGBP inhibits tumor development in nude mice. A, rate ofSW620 cell xenograft growth as related to dose response. Values aremeans � SEM from groups of 7 mice. Doses in mg/kg conform to thenanomolar in vitro dosages used in the current experiments and inprevious experiments (21, 30): 48 mg/kg ¼ 3.2 nmol/L; 96 mg/kg ¼ 6.4nmol/L; 480 mg/kg ¼ 32 nmol/L. Calculation based on arbitrary animalweight to volume equivalence of 1:1 (1 g ¼ 1 mL). B, individually markedmice bearing small tumors (�100–200 mm3 average size) at the end of a5-week treatment.C, rateof tumor development in untreatedcontrolmice(C) from an average xenograft size of approximately 100 mm3 and rate ofresumption of growth in the mice of B after cessation of treatment.
Mallucci et al.
Mol Cancer Ther; 11(9) September 2012 Molecular Cancer Therapeutics1890
on May 28, 2018. © 2012 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst July 2, 2012; DOI: 10.1158/1535-7163.MCT-11-1041-T

growth rate of control xenografts from an average vol-ume of approximately 100 mm3 hence. Histogramsshow that resumption of tumor development proceededat a slower rate and that in only 2 instances within a 9-week period xenograft size had approached the sizeattained in 3 weeks by the xenografts of the control mice(Fig. 5C).
Mutant PIK3CA does not confer resistance toHu-r-bGBPIn addition to oncogenic Kras, in which frequency in
human colorectal cancer is about 40%, activating muta-tions in the PIK3CA gene, also frequent in colorectalcancer (43), can induce oncogenicity (44) and tumorinvasion (45). To investigate whether Hu-r-bGBP wouldbe therapeutically effective in colorectal cancer cellsbearing both mutations, we examined HCT116 andDLD1, both mutated at Kras-G13D (13) in which eitherthe mutant or the wild-type PIK3CA allele had beendeleted by homologous recombination to express 1 of 2major hot spot mutations (44). In a tightly controlledexperiment, we examined HCT116G13D cells harboringan H1047R alteration in exon 20 (kinase domain) andDLD1G13D cells, which have an E545K alteration in exon9 (helical domain) paired with their isogenic wild-typecounterparts. Our results show that compared with thewild type, the growth rate of cells harboring mutantPIK3CA was increased and apoptosis was delayed by 1day (DLD1mut; Fig. 6A and B), but the response to Hu-r-bGBP was fundamentally similar in all cells whetherPIK3CAwild type or mutant and similar to the responseof the colorectal carcinoma cells of Figure 1, which areexempt of PIK3CA mutations. Inhibition of PI3K activ-ity was followed by inhibition of cell proliferation and
cell death assignable to the activation of an apoptoticprogram.
DiscussionMutant-activated Kras defines a subset of patients with
poor prognosis. We show that the PI3K inhibitor used inthis study was remarkably efficacious. Hu-r-bGBP as asingle agent induced apoptotic death in cancer cells har-boringmutant-activatedKras, evenwhencoexpressing anactivating mutation in PIK3CA, and was therapeuticallyeffective in vivo.
Unlikemolecules designed to target critical hubswithinsignaling pathways, the bGBPmolecule operates throughmechanisms, which involve high-affinity receptor bind-ing (26) and molecular interactions leading to functionalinhibition of the p110 catalytic subunit of class IA andclass IB PI3Ks with consequent loss of ERK (27) and Aktfunction (21). Surprisingly, in the cancer cells of this study,inhibition of PI3K activitywas not characterized by loss ofactive ERK or active Akt, key prognostic therapeuticmarkers to bGBP response (46), whose phosphorylationstate was instead increased but characterized by actinreorganization in a Rac-independent manner.
Although our data provide no indication that changesin actin organization were related to loss of Rac activity,the fact that phosphoinositides and PIP2 in particularhave an important role in the binding and control of actinregulatoryproteins (47) lendsground to infer that changesin PIP2 levels, turnover, or spatial distribution broughtabout by molecular interactions involved in bGBP recep-tor–PI3K communication (27) may affect plasma mem-brane–cytoskeletal linkages and have a causative role inthe activation of 2 distinct sets of events critical to the
1 2 3 4 5Day
00
6
24
81012
0
42
8
HCT116wt
βGBPnmol/L
βGBPnmol/L βGBP
nmol/L
βGBPnmol/L
Cel
l num
ber
×10
-5
DA
PI
105
103
101
105
103
101
105103101 105103101
TMRE
11.2 90.2
100
50
0 24 48 72 96Hour0
PIP
3 %
A
B βGBPControl
0
4
2
8
HCT116mut
1 2 3 4 5Day
00
6
24
81012
105103101
105
103
101
105
103
101
105103101
DA
PI
TMRE
13.9 79.5
100
50
0 24 48 72 96Hour0
PIP
3 %
βGBPControl
DLD1wt
0
48
1 2 3 4 5Day
0 60
6
24
81012
Cel
l num
ber
×10
-5
105
103
101
105
103
101
105103101 105103101
DA
PI
TMRE
16.2 78.9
100
50
0 24 48 72 96Hour0
PIP
3 %
βGBPControl
DLD1mut
0
48
121 2 3 4 5
Day0 6
0
6
24
81012
105
103
101
105
103
101
105103101 105103101
DA
PI
TMRE
13.4 47.5
100
50
0 24 48 72 96Hour0
PIP
3 %
βGBPControl
****
****
* * * ***
Figure 6 Hu-r-bGBPovercomesKras andPIK3CAmutations. A, growth curves show rate of cell proliferationas related todose response inPIK3CAwild-type (wt)and in PIK3CAmutant (mut) colorectal cancer cells. Values aremeans of triplicate cultures�SEM. Inset histograms represent PI3K activity asmeasurements ofPIP3 expressed as percentages of controls. Values aremeans of triplicatemeasurements� SEM. �,P < 0.05 versus controls. Hu-r-bGBP8 nmol/L or 12 nmol/L(DLD1mut) was added 6 hours after seeding. B, loss of mitochondrial membrane potential assessed by tetramethylrhodamine ester (TMRE) staining at day4 or 5 (DLD1mut). Inset values are percentages of cells committed to apoptosis (encircled area). Hu-r-bGBP was added 6 hours after seeding.
Killing Kras-Mutant Cancer Cells
www.aacrjournals.org Mol Cancer Ther; 11(9) September 2012 1891
on May 28, 2018. © 2012 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst July 2, 2012; DOI: 10.1158/1535-7163.MCT-11-1041-T

initiation of an apoptotic program. One relating to ageneral rearrangement of the cytoskeleton, consequentcell-cycle checkpoint restrictions, and the ectopic accu-mulation of cyclin E whose bearing on the impairment ofDNA replication provides a potential for errors in DNArepair and for the activation of an intrinsic apoptoticprocess. The other set of events relates, conceivably, tochanges in cortical actin organization, increased macro-molecular mobility within the plane of the plasma mem-brane, and clustering of death receptors, a critical condi-tion for the activation of an extrinsic apoptotic process.
Two other aspects of our study deserve mention. Onepertains to the importance that activatingmutations in thePIK3CAgene have in conferring advantages that facilitatecell growth and invasion (44). Our data (Fig. 6) providestrong evidence that in colorectal carcinoma cells harbor-ing PIK3CA further to oncogenic Ras, inhibition of PI3Kactivity was followed by the activation of a cell deathprogram, underscoring that as single agent, Hu-r-bGBPhad full therapeutic efficacy where combination targetingof PI3K and of Ras downstream effectors may insteadbe required (22, 24). The other key aspect relates to theability of Hu-r-bGBP to strongly reduce tumor growthand to convert tumors that had survived a 5-week periodof treatment from a faster to a slow developing (Fig. 5BandC) and, conceivably, amore benign tumor phenotype.On the basis of amodel discussed in aprevious report (21),this result may be interpreted as proof-of-principle evi-dence that cancer vulnerability in response to Hu-r-bGBPchallenge is greater in the aggressive cancer phenotype, asa contained tumor growth after the end of treatment issuggestion of clonal selection through elimination of themore aggressive cells.
Recently, a number of compounds identified by high-throughput screening have been reported to have thera-peutic efficacy against Kras-mutant cancer cells (13–16);however, among all candidate agents for the therapy ofcancers harboring mutant-activated Ras, the bGBP mol-ecule, a cytokine, by virtue of its physiologic nature is theonly one, to our knowledge, which in the clinic would be
implicitly exempt from drug toxicity and drug resistance.As, further to colorectal carcinoma, mutations in the Rasoncogenes and in genes involved in PI3K regulationextend to other cancers, our results suggest that Hu-r-bGBP is, potentially, a therapeutic agent that could pro-vide benefit to a large number of patients.
Disclosure of Potential Conflicts of InterestNo potential conflicts of interest were disclosed.
Authors' ContributionsConception and design: L. Mallucci, D.-y. Shi,Development of methodology: D.-y. Shi, P. Jordan, L. Lotti, R. Mariani-CostantiniAcquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): D.-y. Shi, P. Jordan, L. Lotti, R. Mariani-Costan-tini, F. Verginelli, V. WellsAnalysis and interpretation of data (e.g., statistical analysis, biostatis-tics, computational analysis): L. Mallucci, D.-y. Shi, A. Nicol, L. Lotti, R.Mariani-Costantini, V. Wells, D. ZichaWriting, review, and/or revision of the manuscript: L. Mallucci,Administrative, technical, or material support (i.e., reporting or orga-nizing data, constructing databases): R. Mariani-Costantini, D. ZichaStudy supervision: L. Mallucci, V. Wells, D. Zicha
AcknowledgmentsPI3-Kinase ELISA Kits were generously provided by Echelon Bios-
ciences. The authors are indebted to Bert Vogelstein (John HopkinsUniversity) for generously providing cell lines HCT116 wt/null, HCT116mut/null, DLD1 wt/null, and DLD1 mut/null; Julian Downward andEsther Castellano for advice and expertise about Rac assessment; CosmoRossi for management of xenograft study; Simone Martino for technicalassistance; Roger Morris for his support; Paul Nielsen for his interest; andGiampietro Schiavo for critical reading of the manuscript. They alsoacknowledge with gratitude the artwork and kindness of the late KateKirwan.
Grant SupportThis work was supported by a KCL/PR China KC Wong fellowship
award (D.-y. Shi), by 2MIURgrants (L. Lotti andR.Mariani-Costantini), byAIRC grant # IG 9168 (R.Mariani-Costantini), and by funding fromCancerResearch UK (D. Davies, P. Jordan, A. Nicol, and D. Zicha).
The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely to indicatethis fact.
Received December 21, 2011; revised June 11, 2012; accepted June 12,2012; published OnlineFirst July 2, 2012.
References1. Barbacid M. Ras genes. Annu Rev Biochem 1987;56:779–827.2. Cengel KA, Voong KR, Chandrasekaran S, Maggiorella L, Bruner S,
Stanbridge S, et al. Oncogenic K-Ras signals through epidermalgrowth factor receptor and wild-type H-Ras to promote radiationsurvival in pancreatic and colorectal carcinoma cells. Neoplasia2007;9:341–8.
3. Mascaux C, Iannino N, Martin B, Paesmans M, Berghmans T, DusartM, et al. The role of RAS oncogene in survival of patients with lungcancer. A systematic review of the literature with meta-analysis. BrJ Cancer 2005;92:131–9.
4. Pao W, Wang T, Riely GJ, Muiller VA, Pau Q, Ladanyi M, et al. KRASmutations andprimary resistance of lung adenocarcinomas to gefitinibor erlotinib. PLoS Med 2005;2:e17.
5. Massarelli E, Varella-Garcia M, Tang X, Xavier AC, Ozburn NC, LiuDD, et al. KRAS mutation is an important predictor of resistance totherapy with epidermal growth factor receptor tyrosine kinase
inhibitors in non–small-cell lung cancer. Clin Cancer Res 2007;13:2890–6.
6. Jhawer M, Goel S, Wilson AJ, Montagna C, Ling Y-H, Byun D-S, et al.PIK3CA mutation/PTEN expression status predicts response of coloncancer cells to the epidermal growth factor receptor inhibitor cetux-imab. Cancer Res 2008;68:1953–61.
7. VanCutsemE, KohneC-H, Hitre E, Zaluski J, ChineC-RC,MakhsonA,et al. Cetuximab and chemotherapy as initial treatment for metastaticcolorectal cancer. N Engl J Med 2009;360:1408–17.
8. Downward J. Targeting Ras signaling pathways in cancer therapy. NatRev Cancer 2003;3:11–22.
9. Gupta S, Ramjaun AR, Haiko P, Wang Y, Warne PH, Nicke B, et al.BindingofRas to phosphoinositide 3-kinase p110a is required for Ras-driven tumorigenesis in mice. Cell 2007;129:957–68.
10. Kohl NE, Omer CA, Conner MW, Anthony NJ, Davide JP, Desolms SJ,et al. Inhibition of farnesyltransferase induces regression of mammary
Mallucci et al.
Mol Cancer Ther; 11(9) September 2012 Molecular Cancer Therapeutics1892
on May 28, 2018. © 2012 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst July 2, 2012; DOI: 10.1158/1535-7163.MCT-11-1041-T

and salivary carcinomas in ras transgenic mice. Nat Med 1995;1:792–7.
11. Ji H, Wang Z, Pereira S, Li D, Liang M-C, Zaghlul S, et al. Mutations inBRAF and KRAS converge on activation of the mitogen-activatedprotein kinase pathway in lung cancer mouse models. Cancer Res2007;67:4933–9.
12. Rinehart J, Adjei AA, LoRusso PM, Waterhouse D, Hecht JR, NataleRB, et al.Multicellular phase II study of theoralMEK inhibitor, C1040, inpatients with advanced non-small-cell lung, breast, colon, and pan-creatic cancer. J Clin Oncol 2004;22:4456–62.
13. Torrance CJ, Agrawal V, Vogelstein B, Kinzler KW. Use of isogenichuman cancer cells for high-throughput screening and drug discovery.Nat Biotech 2001;19:940–5.
14. Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification ofgenotype-selective antitumor agents using synthetic lethal chemicalscreening in engineered human tumor cells. Cancer Cell 2003;3:285–96.
15. Guo W, Wu S, Liu J, Fang B. Identification of a small molecule withsynthetic lethality for K-Ras and protein kinase C iota. Cancer Res2008;68:7403–8.
16. Shaw AT, Winslow MM, Magendantz M, Ouyang C, Dowdle J, Sub-ramanian A, et al. Selective killing of K-rasmutant cancer cells by smallmolecule inducers of oxidative stress. Proc Natl Acad Sci U S A2011;108:8773–8.
17. Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/Akt pathway for drug discovery. Nat Rev Drug Discov 2000;4:988–1004.
18. Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase-Akt path-way in human cancer. Nat Rev Cancer 2002;2:489–501.
19. Lim KH, Counter CM. Reduction in the requirement of oncogenic Rassignaling to activation of PI3K/AKT pathway during tumor mainte-nance. Cancer Cell 2005;8:381–92.
20. Shaw R, Cantley LC. Ras, PI(3)K and mTOR signaling controls tumourcell growth. Nature 2006;441:424–30.
21. Wells V, Mallucci L. Phosphoinositide 3-kinase targeting by theb-galactoside binding protein cytokine negates akt gene expressionand leads aggressive breast cancer cells to apoptotic death. BreastCancer Res 2009;11:R2.
22. Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R,et al. Effective use of PI3K and MEK inhibitors to treat mutant KrasG12D and PIK3CA H1047R murine lung cancers. Nat Med 2008;14:1351–6.
23. Ihle NT, Lemos R Jr, Wipf P, Yacoub A, Mitchell C, Siwak D, et al.Mutations in the phosphatidylinositol-3-kinase pathway predict forantitumor activity of the inhibitor PX-866 whereas oncogenic Ras is adominant predictor for resistance. Cancer Res 2009;69:143–50.
24. Torbett NE, Luna-Moran A, Knight ZA, Houk A, Moasser M, Weiss W,et al. A chemical screen in diverse breast cancer cell lines revealsgenetic enhancers and suppressors of sensitivity to PI3K isoform-selective inhibition. Biochem J 2008;415:97–110.
25. Blaser C, Kaufman M, Muller C, Zimmerman C, Wells V, Mallucci L,et al. b-galactoside binding protein secreted by activated T cellsinhibits antigen-induced proliferation of T cells. Eur J Immunol1998;28:2311–9.
26. Wells V, Mallucci L. Identification of an autocrine negative growthfactor: mouse-b-galactoside binding protein is a cytostatic factor andcell growth regulator. Cell 1991;64:91–7.
27. Wells V, Downward J, Mallucci L. Functional inhibition of PI3K by thebGBP molecule suppresses Ras-MAPK signaling to block cell prolif-eration. Oncogene 2007;26:7709–14.
28. Cummins JM, Kohli M, Rago C, Kinsler KW, Vogelstein B, Bunz F. X-linked inhibitor of apoptosis protein (XIAP) is a nonredundant mod-ulator of tumor necrosis factor-related apoptosis-inducing ligand(TRAIL)-mediated apoptosis in human cancer cells. Cancer Res 2004;64:3006–8.
29. Zicha D, Genot E, Dunn GA, Kramer IM. TGFb1 induces a cell cycle-dependent increase in motility of epithelial cells. J Cell Sci 1999;112:447–54.
30. Ravatn R, Wells V, Nelson L, Vettori D, Mallucci L, Chin K-V.Circumventing multidrug resistance in cancer by b-galactosidebinding protein, an antiproliferative cytokine. Cancer Res 2005;65:1631–4.
31. Carracedo A, Ma L, Teruya-Felstein J, Roojo F, Salmena L, Alimonti A,et al. Inhibition of mTORC1 leads toMAPK pathway activation througha PI3K-dependent feedback loop in human cancer. J Clin Invest2008;118:3065–74.
32. Rosenthal DT, Iyer H, Escudero S, Bao L,Wu Z, Ventura AC, et al. p38gpromotes breast cancer cellmotility andmetastasis through regulationof RhoC GTPase, cytoskeletal architecture, and a novel leading edgebehaviour. Cancer Res 2011;71:6338–49.
33. Burridge K, Wennerberg K. Rho and Rac take center stage. Cell2004;116:167–79.
34. Nobes D, Hawkins P, Stephens L, Hall A. Activation of the small GTP-binding proteins rho and rac by growth factor receptors. J Cell Sci1995;108:225–33.
35. LambertM, Lambert QT, ReutherGW,Malliri A, Siderovski DP, SondekJ, et al. Tiam1mediates Ras activation of Rac by a PI(3)K-independentmechanism. Nat Cell Biol 2002;4:621–5.
36. Ekholm-Reed S, Mendez J, Tedeco D, Zetterberg A, Stillman B, ReedSI. Deregulation of cyclin E in human cells interfereswith prereplicationcomplex assembly. J Cell Biol 2004;165:789–800.
37. ZhouBB, Elledge SJ. TheDNAdamage response: putting checkpointsin perspective. Nature 2000;408:433–9.
38. Lin WC, Lin FT, Nevins JR. Selective induction of E2F1 in response toDNA damage mediated by ATM-dependent phosphorylation. GenesDev 2001;15:1833–44.
39. Kastan MB, Bartek J. Cell cycle checkpoints and cancer. Nature2004;432:316–23.
40. StevensC, Smith L, La ThangueNB.Chk2 activates E2F-1 in responseto DNA damage. Nat Cell Biol 2003;5:401–9.
41. Krek W, Xu G, Livingston D. Cyclin A-kinase regulation of E2F-1 DNAbinding function underlies suppression of an S phase checkpoint. Cell1995;83:1149–58.
42. Fueyo J, Gomez-Manzano C, Jung WK, Liu TJ, Alemany R, McDon-nell TJ, et al. Overexpression of E2F-1 in glioma triggers apoptosisand suppresses tumor growth in vitro and in vivo. Nat Med1998;4:685–90.
43. Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, et al. Highfrequency ofmutations of thePIK3CAgene in humancancers. Science2004;304:554.
44. Bader AG, Kang S, Vogt P. Cancer specific mutations in PIK3CA areoncogenic in vivo. Proc Natl Acad Sci U S A 2006;103:1475–9.
45. Samuels Y, Diaz L Jr, Schmidt-Kittler O, Cummins JM, Delong L,Cheong I, et al. Mutant PIK3CA promotes cell growth and invasion ofhuman cancer cells. Cancer Cell 2005;7:561–73
46. Mallucci L, Wells V. bGBP, compositions comprising bGBP, andrelated methods and uses thereof. United States patent US7994113. 2011 Aug 9.
47. Di Paolo G, De Camilli P. Phosphoinositides in cell regulation andmembrane dynamics. Nature 2006;443:651–7.
Killing Kras-Mutant Cancer Cells
www.aacrjournals.org Mol Cancer Ther; 11(9) September 2012 1893
on May 28, 2018. © 2012 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst July 2, 2012; DOI: 10.1158/1535-7163.MCT-11-1041-T

2012;11:1884-1893. Published OnlineFirst July 2, 2012.Mol Cancer Ther Livio Mallucci, Dong-yun Shi, Derek Davies, et al.
In VivoInhibitor Therapeutically Effective GBP Cytokine, a Physiological PI3KβActin Remodeling by the
Killing of Kras-Mutant Colon Cancer Cells via Rac-Independent
Updated version
10.1158/1535-7163.MCT-11-1041-Tdoi:
Access the most recent version of this article at:
Cited articles
http://mct.aacrjournals.org/content/11/9/1884.full#ref-list-1
This article cites 46 articles, 17 of which you can access for free at:
Citing articles
http://mct.aacrjournals.org/content/11/9/1884.full#related-urls
This article has been cited by 1 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://mct.aacrjournals.org/content/11/9/1884To request permission to re-use all or part of this article, use this link
on May 28, 2018. © 2012 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst July 2, 2012; DOI: 10.1158/1535-7163.MCT-11-1041-T