laboratory diagnosis of thalassemia screening for...
TRANSCRIPT

1
Laboratory Diagnosis of Laboratory Diagnosis of ThalassaemiaThalassaemiaand Abnormal and Abnormal HaemoglobinsHaemoglobins
Pranee Pranee WinichagoonWinichagoon FucharoenFucharoenThalassemia Research CenterThalassemia Research Center
Institute of Institute of Molecular BiosciencesMolecular BiosciencesMahidol UniversityMahidol University
Disorders of Hemoglobin Synthesis
Hereditary Persistence of Fetal
Hemoglobin (HPFH)
Persistence in the synthesis of γ-globin chain
4.
Thalassemia-hemoglobinopathy
Synthesis of an abnormal globinchain with reduced synthesis of the Hb variant: quantitative & qualitative disorder
3.
Abnormal Hb(Hb Variant)
Synthesis of an abnormal globinchain (amino acid change): qualitative disorder
2.
ThalassemiaDecreased or absence of globinchain synthesis of hemoglobin: quantitative disorder
1.
Laboratory Diagnosis of ThalassemiaRBC IndicesRBC Osmotic FragilityRBC MorphologyIntraerythrocytic Inclusion BodiesHb TypingHb A2 QuantitationHb F QuantitationAcid Elution StainingSerum FerritinGlobin Chain SynthesisDNA AnalysisFamily Study
@ One tube osmotic fragility (OF) testor MCV/MCH
@ Dichlorophenolindophenol (DCIP) precipitation test
@ Detection of Hb Bart’s
SCREENING FOR THALASSEMIA
0.36% buffered saline 0.36% buffered saline
Normal β-thalassemia/HbE disease

2
Red Blood Cell IndicesRed Blood Cell Indices
• Mean Corpuscular Volume (MCV) 87+5 fL
• Mean Corpuscular Hemoglobin (MCH) 29+2 pg
• Mean Corpuscular Hemoglobin Concentration (MCHC) 34+2 %
Mean Corpuscular Volume (MCV)Mean Corpuscular Volume (MCV)
65+366+1081+564+574+368+583+788+6
MCV (fL)
23+9Hb E with α-Thal 1 trait
23+5α-Thal 1 trait22+1Homozygous α-Thal 2 20+3β-Thal trait
27+3α-Thal 2 trait
20+1Homozygous HbE
26+2Hb E trait
29+2NormalMCH (pg)
DichlorophenolindophenolDichlorophenolindophenol (DCIP) (DCIP) precipitation testprecipitation test
Hb E (α2β2Glu->Lys) เปนฮีโมโกลบินผิดปกติ เกิดจาก
กรดอะมิโนตําแหนง 26 ในสาย β-globin เปล่ียนจาก glutamic เปน lysine เปนผลใหการรวมกันของสาย α-globin และ β-globin ไมเสถียรและเผยหมู sulfhydryl(-SH) อิสระออกมา จึงถูก oxidized ดวยสาร DCIP ไดงายและเร็วกวาฮีโมโกลบินชนิดอ่ืนและตกตะกอนลงมา
α β
δγ
NormalHb A=α2β2HbA2=α2δ2Hb F=α2γ2
α β
δγ
α-Thalassemiaβ4=HbH
γ4=HbBart’s
α β
δγ
β-ThalassemiaIncreased HbA2
(4-6%)
ImmunodetectionImmunodetection of of HbHb BartBart’’ssImmunodetection of Hb Bart’s
1 2 3
4 5 6
Screening for α-thalassemia
Positive for: α-thalassemia 1 heterozygote, α-thalassemia 2 homozygote, Hb H disease, EA Bart’s disease and Hb Bart’s hydrops fetalis

3
iron supplementation x 3mo.Repeated study after
Dx : Hb E trait
Normal
Dx : Normalα-Thal 2 traitHb CS trait
Negative Positive
DCIP
Hb Bart’s
Dx : α-Thal 1 traitβ-Thal trait
Fe deficiency
Decrease
PositiveNegative
DCIP
- Clinical- Rbc morphology- Inclusion body
(Hb H disease)- F cell stain
(β-thal/Hb E)
Dx : Hb E traitHb E Homozygoteβ-Thal/Hb EHb H disease
Negative PositiveDx : β-Thal trait
Fe deficiencyDx : α-Thal 1
trait
MCV or One tube osmotic fragility
Routine Laboratory Diagnosis of Thalassemia
@ Hb Concentration@ RBC Indices and Morphology@ Hb Typing@ Hb F and A2 Quantitation@ RBC Inclusion Bodies
Hb Bart’s in Bart’s hydrops fetalis
Orig
in
A2(
E)
F A Bar
t’sH
+-
B/E diseaseHomo EE trait
B traitE traitB/E disease
Homo EHomo E
Cellulose acetate electrophoresis
- Cellulose acetate electrophoresis and elution
- Microcolumnchromatography
- Automated HPLC, LPLC
- Capillary electrophoresis
Hb analysis and quantification Cellulose Acetate Electrophoresis

4
Hemoglobin Separation by Ion Exchange Chromatography Variant Variant HbHb testing systemtesting system
• Automated high performance liquid chromatography system (HPLC) improves resolution quality (sharp peaks)
• Cation exchange analytical cartridge (30 mm, ID 4.6mm) / particle size ~ 6-9 microns
• 5 μL of blood only. Blood stable for testing ~30 days
• Fast analysis intervals of 6.5 minutes
Principle: HPLC as a Hb testing system
2. Phosphate buffer at different concentration (mobile phase) pass under pressure (either high or low) through the ionic exchange column.
3. Due to the buffer gradient of increasing ionic strength and pH, Hbs are separated according to their ionic interaction with the stationary phase.
1
2
3 4 1. Hemoglobins in the hemolysate are adsorbed on a resin of anionic or cationic particles column (stationary phase).
Principle: HPLC as a Hb testing system
4. Separated Hbs pass through the flow cell of photometer where changes in the absorbance at 415 nm are measured.
5. The assays provide quantitative results for percent HbA2 and HbF, while detecting the most commonly occurring abnormal hemoglobins.
Windows are the time ranges in which Hbs are eluted.
- Common Hbs: F, A, A2
- Abnormal Hbs: S, C, D
Retention time is measured from the time of sample injection to maximum point of each elution peak.
RT (retention time) values Interpretation of results : RT
RT Window
• F 1.15 (1.00-1.30) mins• P2 1.45 (1.30-1.60) mins• P3 1.75 (1.60-1.90) mins• A0 2.60 (2.20-3.30) mins• A2 (E) 3.83 (3.68-3.98) mins• D-window 4.05 (3.98-4.12) mins• S-window 4.27 (4.12-4.42) mins• C-window 5.03 (4.88-5.18) mins
Hb variants have specific RT values

5
Capillary Electrophoresis Principle
The Capillarys® system uses the principle of capillary electrophoresis in free solution, charged molecules are separated by their electrophoreticmobility in an alkaline buffer with a specific pH.
- Separation occurs according to the electrolyte pH and electro-osmotic flow, from cathode to anode.- Hemoglobins were separated within 4 minutes, according to isoelectricpoint, by migration in an electric field in the liquid phase. Direct detection of the hemoglobin is made at an absorbance wavelength of 415 nm at their cathodic end
Positives charges from buffer solution
Negatives charges from capillary wall
Electro-osmotic flow carries allproteins towards the cathodic side
This interaction leads to electro-osmotic flow (EOF) from theanode to the cathode
Control AFSC
Capillarys Electrophoresis PrincipleThermic bridgeTemperatureControlled byPeltier device
Anode +Cathode -
Detector Deuterium lamp
MigrationMigration
High High VoltageVoltage
A
FS
A2C
Normal Hb Type
α β
δγ
NormalHb A=α2β2HbA2=α2δ2Hb F=α2γ2
α β
δγ
α-Thalassemiaβ4=HbH
γ4=HbBart’s
α β
δγ
β-ThalassemiaIncreased HbA2
(4-6%)
α-Thalassemia
α-globin Gene Deletions
ζ2 α1α2 θ1
0kb 10kb 20kb 30kb
ψζ1ψα2 ψα1inter- ζ HVR 3'HVR
--MED
--20.5
--SEA
-α3.7
-α4.2
α-Globin Genotype
Hb Bart’s hydropsfetalis
Hb H diesease
Normal α-Thalassemia 2 α-Thalassemia 1
Hb ConstantSpring
Hb H-CS disease HomozygousHb CS
−CS −CSCS− CS−
6
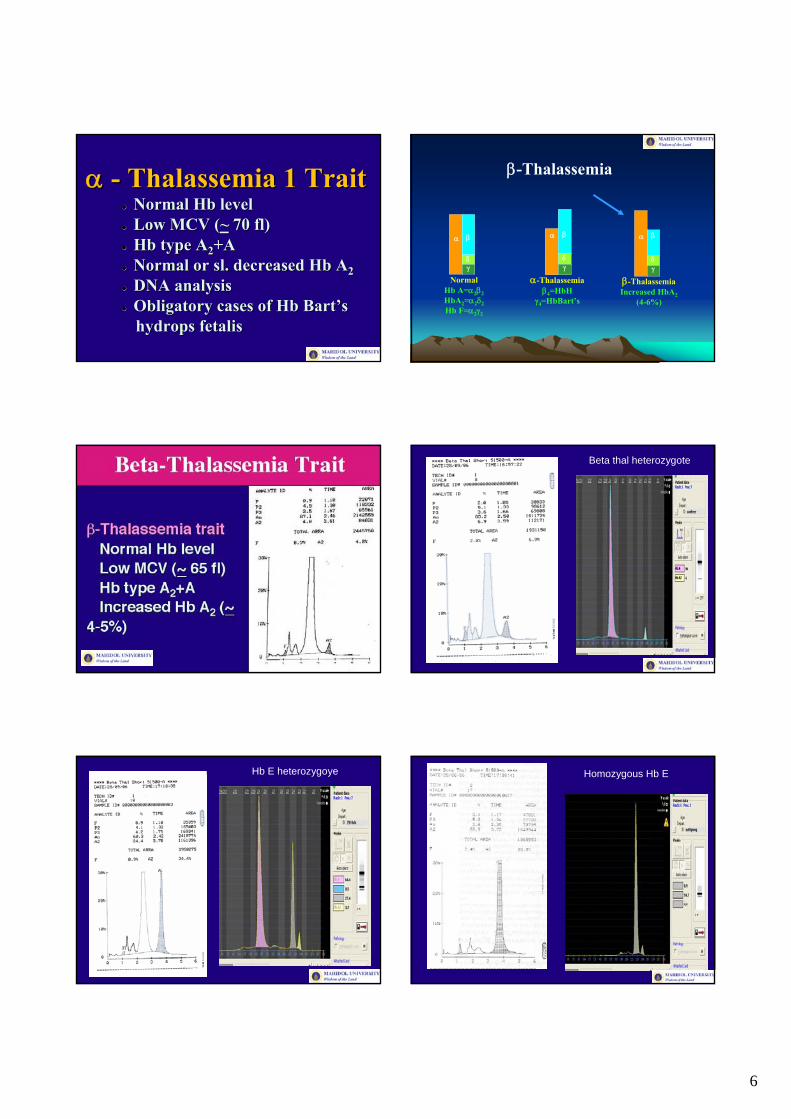
αα -- Thalassemia 1 TraitThalassemia 1 TraitNormal Normal HbHb levellevelLow MCV (Low MCV (~~ 70 fl)70 fl)HbHb type Atype A22+A+ANormal or Normal or slsl. decreased . decreased HbHb AA22DNA analysisDNA analysisObligatory cases of Obligatory cases of HbHb BartBart’’s s hydropshydrops fetalisfetalis
α β
δγ
NormalHb A=α2β2HbA2=α2δ2Hb F=α2γ2
α β
δγ
α-Thalassemiaβ4=HbH
γ4=HbBart’s
α β
δγ
β-ThalassemiaIncreased HbA2
(4-6%)
β-Thalassemia
Beta thal heterozygote
Hb E heterozygoye Homozygous Hb E
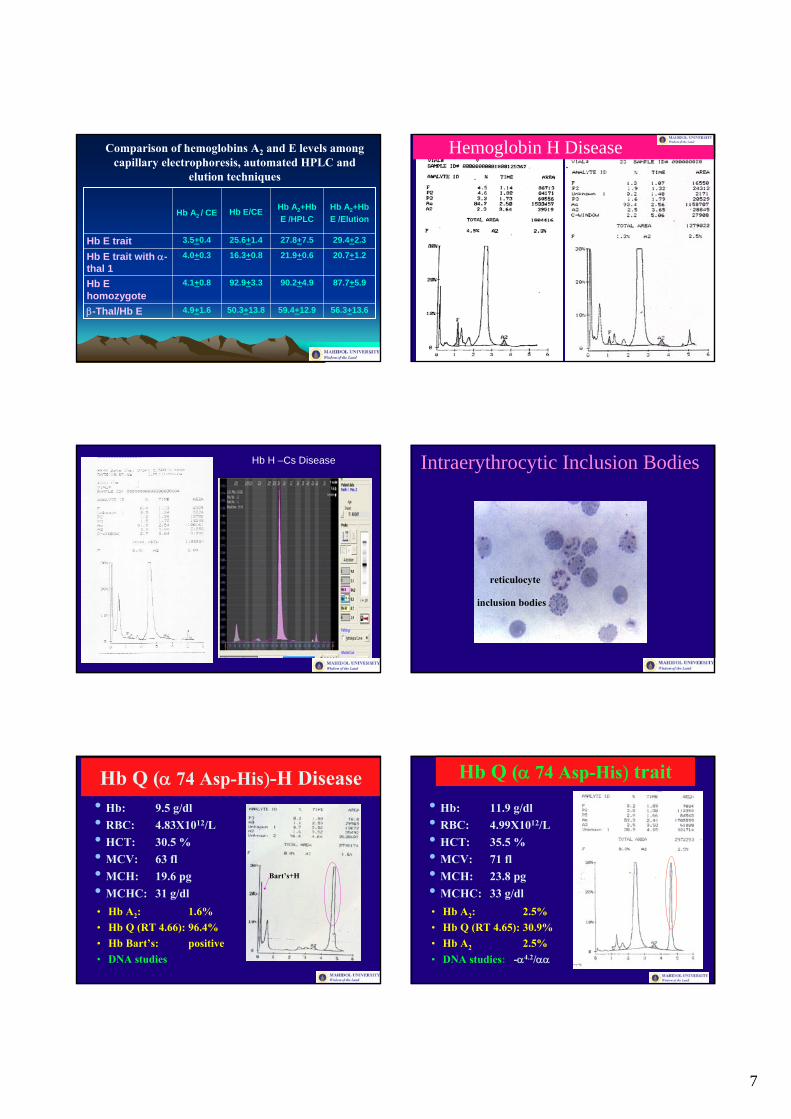
7
56.3+13.659.4+12.950.3+13.84.9+1.6β-Thal/Hb E
87.7+5.990.2+4.992.9+3.34.1+0.8Hb E homozygote
20.7+1.221.9+0.616.3+0.84.0+0.3Hb E trait with α-thal 1
29.4+2.327.8+7.525.6+1.43.5+0.4Hb E trait
Hb A2+Hb E /Elution
Hb A2+Hb E /HPLC
Hb E/CEHb A2 / CE
Comparison of hemoglobins A2 and E levels among capillary electrophoresis, automated HPLC and
elution techniques
Hemoglobin H Disease
Hb H –Cs Disease Intraerythrocytic Inclusion Bodies
reticulocyte
inclusion bodies
Hb Q (α 74 Asp-His)-H Disease• Hb: 9.5 g/dl• RBC: 4.83X1012/L• HCT: 30.5 %• MCV: 63 fl• MCH: 19.6 pg• MCHC: 31 g/dl• Hb A2: 1.6%• Hb Q (RT 4.66): 96.4%• Hb Bart’s: positive• DNA studies
Bart’s+H
Hb Q (α 74 Asp-His) trait
• Hb: 11.9 g/dl• RBC: 4.99X1012/L• HCT: 35.5 %• MCV: 71 fl• MCH: 23.8 pg• MCHC: 33 g/dl• Hb A2: 2.5%• Hb Q (RT 4.65): 30.9%• Hb A2 2.5%• DNA studies: -α4.2/αα
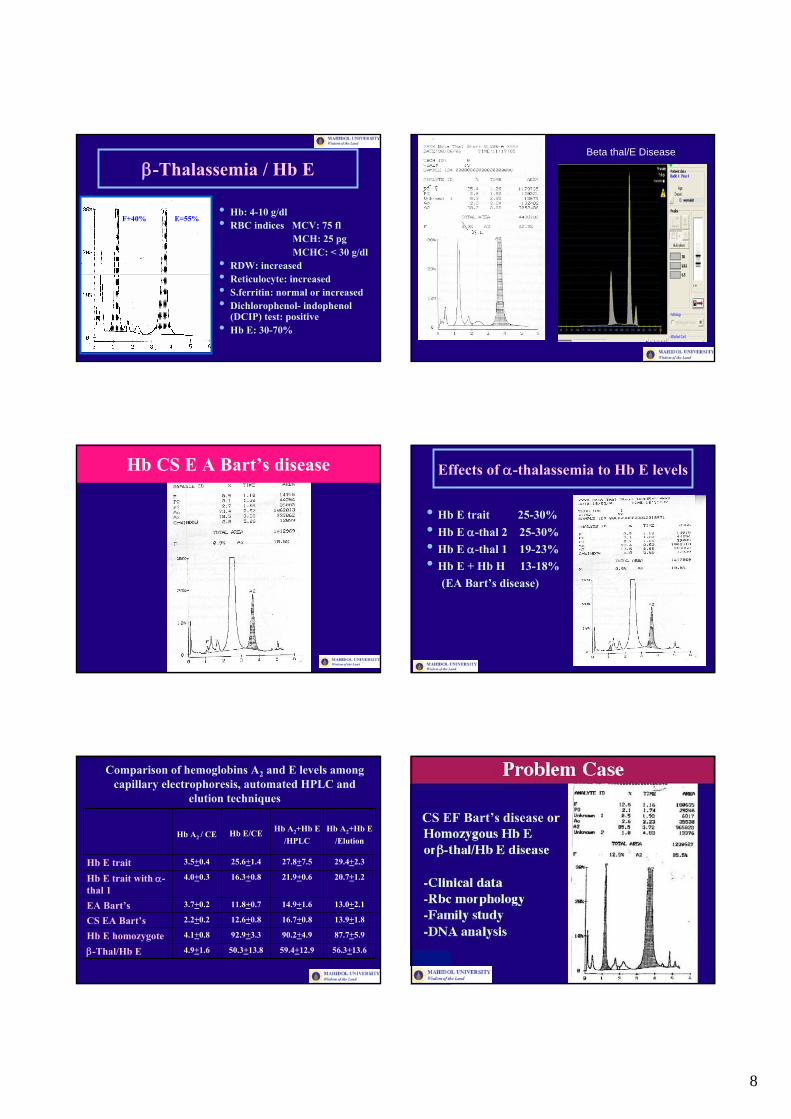
8
β-Thalassemia / Hb E
• Hb: 4-10 g/dl• RBC indices MCV: 75 fl
MCH: 25 pgMCHC: < 30 g/dl
• RDW: increased• Reticulocyte: increased• S.ferritin: normal or increased• Dichlorophenol- indophenol
(DCIP) test: positive• Hb E: 30-70%
E=55%F+40%
Beta thal/E Disease
Hb CS E A Bart’s disease Effects of α-thalassemia to Hb E levels
• Hb E trait 25-30%• Hb E α-thal 2 25-30%• Hb E α-thal 1 19-23%• Hb E + Hb H 13-18%
(EA Bart’s disease)
56.3+13.659.4+12.950.3+13.84.9+1.6β-Thal/Hb E
87.7+5.990.2+4.992.9+3.34.1+0.8Hb E homozygote
13.9+1.816.7+0.812.6+0.82.2+0.2CS EA Bart’s
13.0+2.114.9+1.611.8+0.73.7+0.2EA Bart’s
20.7+1.221.9+0.616.3+0.84.0+0.3Hb E trait with α-thal 1
29.4+2.327.8+7.525.6+1.43.5+0.4Hb E trait
Hb A2+Hb E /Elution
Hb A2+Hb E /HPLC
Hb E/CEHb A2 / CE
Comparison of hemoglobins A2 and E levels among capillary electrophoresis, automated HPLC and
elution techniques
CS EF Bart’s disease or

9
Hb E/β+-thalassemia vsHb E/Hb Malay
• Hb: 10 g/dl• RBC: 5.3X1012/L• Hct: 31 %• MCV: 62 fl• MCH: 19 pg• MCHC: 32 g/dl• Hb E+ Hb A2: 58.9 %• Hb F: 5.4 %• Hb A (Hb Malay): 26.5 %• Parents: Hb Malay and Hb E
trait• DNA studies
Hb E/Hb Dhonburi (β126 Val-Gly)• Hb: 12.1 g/dl• RBC: 5.8X1012/L• Hct: 44 %• MCV: 77 fl• MCH: 21 pg• MCHC: 27 g/dl
Triton urea gel: the abnormal fraction migrates more rapidly than βA-chain
Hb type E +“A”Hb E+ Hb A2: 38.3 %Hb F: 0.8 %Hb Dhonburi (“A”) 42.6 %DNA studies
α2γ2(HbF)α2γ2 (HbF)
α2δ2 (HbA2)α2β2 (HbA)
Hb Gower II α2 ε2Hb Portland ζ2γ2Hb Gower I ζ2ε2
Embryo Fetus Adult
δ
εε GGγ AAγ δ βψβψβ
ζ2
α-like globin gene (Ch16)
β-like globin gene(Ch11)
α2 α1
γ γ δ β
Postnatal Age (Weeks )Postconceptual Age (Weeks )
Site oferythropoisis
Cell
6 12 18 24 30 36 0 6 12 18 24 30 36 42 48
Postnatal Age (Weeks )Postconceptual Age (Weeks )
0
10
20
30
40
50
60
% o
f tot
al g
lobi
n syn
thes
is
Bone marrowspleenliver
Yol
k sa
c
Site oferythropoisis
type megaloblast macrocyte normocyte
αα
γγ
ββεε
ζζ
αα
γγ
ββ
δ
* Acquired :- Physiology : pregnancy (HbF ~ 3-7%)- Recovery from marrow hypoplasia :
after BM transplant,chemotherapy
- Fetal erythropoiesis : juvenile chronic myeloid leukemia
- Miscellaneous : aplastic anemia, PNH, leukemia, hepatoma
Increased hemoglobin FIncreased hemoglobin F
Normal adult: Hb F ≤ 1%Increased Hemoglobin F
* Genetic : Thalassemia: Hereditary persistence of
fetal hemoglobin (HPFH)- pancellular- heterocellular
Hemoglobin FHemoglobin F
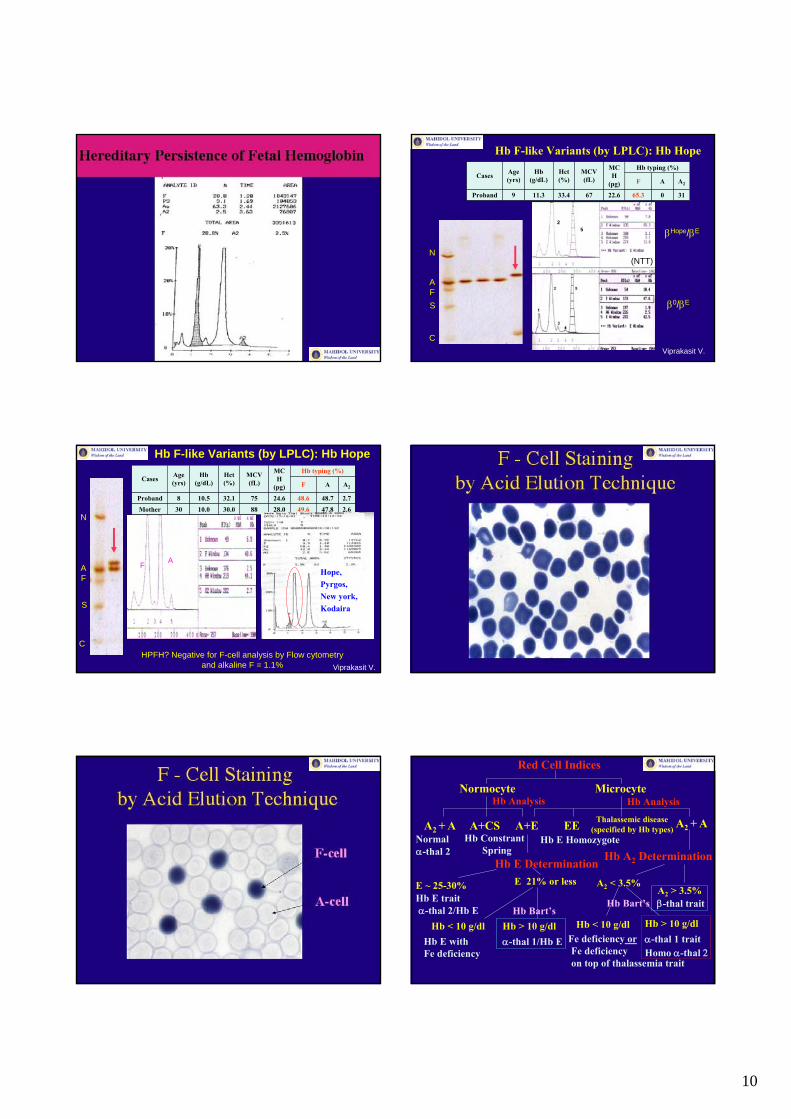
10
Hb F-like Variants (by LPLC): Hb Hope
AFS
C
N(NTT)
βHope/βE
β0/βE
31
A2
0
A
65.322.66733.411.39Proband
F
Hb typing (%)MCH
(pg)
MCV (fL)
Hct(%)
Hb(g/dL)
Age (yrs)Cases
25
Viprakasit V.
Hb F-like Variants (by LPLC): Hb Hope
2.647.849.628.08830.010.030Mother
2.7
A2
48.7
A
48.624.67532.110.58Proband
F
Hb typing (%)MCH
(pg)
MCV (fL)
Hct(%)
Hb(g/dL)
Age (yrs)Cases
HPFH? Negative for F-cell analysis by Flow cytometryand alkaline F = 1.1%
(SPW)
N
AF
S
C
Hope,Pyrgos,New york, Kodaira
F A
Viprakasit V.
Red Cell Indices
Normocyte MicrocyteHb Analysis Hb Analysis
A2 + A A+CS A+E EE A2 + ANormalα-thal 2
Thalassemic disease(specified by Hb types)
Hb ConstrantSpring
Hb E Determination Hb A2 DeterminationHb E Homozygote
E ~ 25-30% E 21% or less A2 < 3.5%A2 > 3.5%
Hb E traitα-thal 2/Hb E
β-thal trait
Hb < 10 g/dl Hb > 10 g/dl Hb < 10 g/dl Hb > 10 g/dl
Hb E withFe deficiency
α-thal 1/Hb E Fe deficiency orFe deficiencyon top of thalassemia trait
α-thal 1 traitHomo α-thal 2
Hb Bart’sHb Bart’s
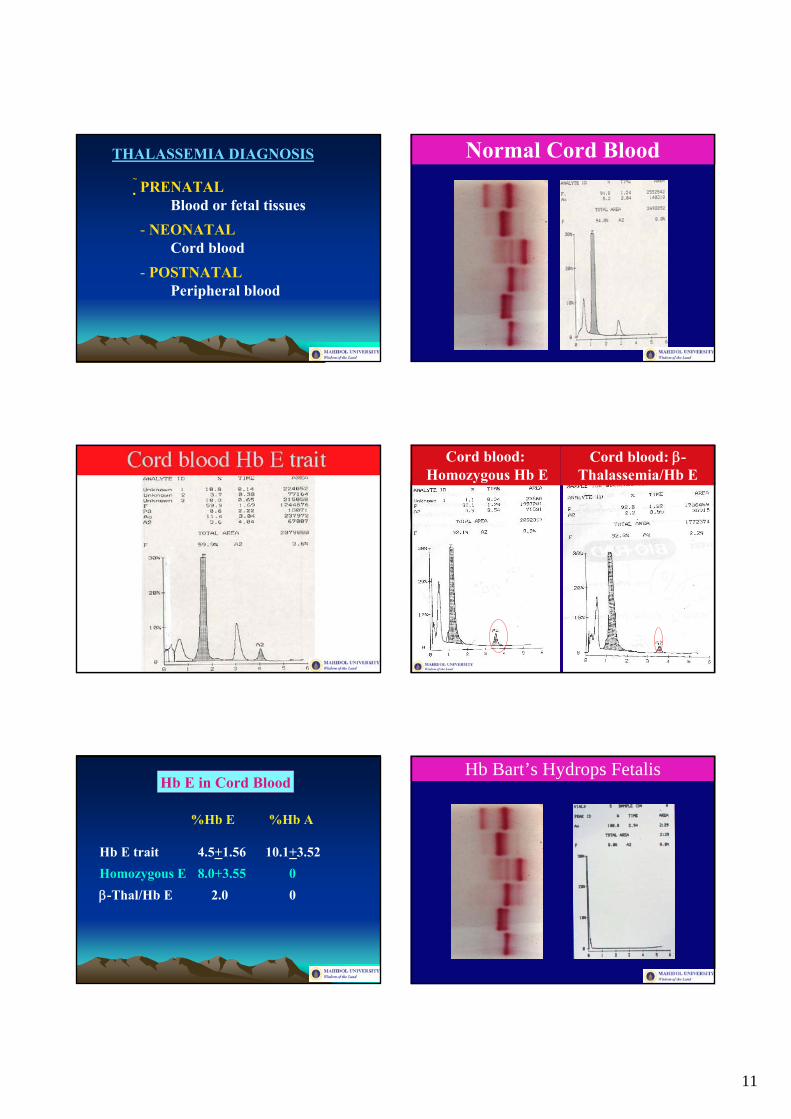
11
THALASSEMIA DIAGNOSIS
PRENATALBlood or fetal tissues
- NEONATALCord blood
- POSTNATALPeripheral blood
Normal Cord Blood
Cord blood:Homozygous Hb E
Cord blood: β-Thalassemia/Hb E
Hb E in Cord Blood
%Hb E %Hb A
Hb E trait 4.5+1.56 10.1+3.52Homozygous E 8.0+3.55 0β-Thal/Hb E 2.0 0
Hb Bart’s Hydrops Fetalis

12
α-Thalassemia%Hb Bart’s
α-Thalassemia 2 trait 1.9+0.71Homozygous α-thalassemia 2 8.0+2.01 α-Thalassemia 1 trait 9.2+1.09Hb CS trait 2.4 + 0.31Homozygous Hb CS 15Hb H disease (include EABart’s
and EFBart’s disease) 20-30
Laboratory Investigation*Subjects:1.Adults: Child
AdolescentReproductivePregnancy
2. Neonatal (Cord blood)3. Prenatal (Fetal blood)4. Family members
INTERPRETATIONINTERPRETATION
Clinical dataLaboratory dataFamily studyHistory : Previous blood transfusion
Type of specimens (cord/fetal blood, child/adult blood)
HEMOGLOBINOPATHIES IN THAILAND (1)
1. HEMOGLOBIN VARIANTS
1.1 COMMON- Hb E: β 26 Glu-Lys- Hb Constant Spring: elongated α-chain1.2 RARE ALPHA MUTANTS- Anantharaj: α 11 Lys-Gln - Siam: α 15 Gly-Arg- Queens: α 34 Leu-Arg - Thailand: α 56 Lys-Thr- J-Buda: α 61 Lys-Asn - Mahidol: α 74 Asp-His- Suan Dok: α 109 Leu-Arg
1.3 RARE BETA MUTANTS
- Hb C: β 6 Glu-Lys - Siriraj: α 7 Glu-Lys- Malay: β 19 Asn-Ser - G-Hsin Chu: β 22 Glu-Ala- J-Bangkok: β 56 Gly-Asp - Pyrgos: β 83 Gly-Asp
- New York: β 113 Val-Glu - D-Punjab: β 121 Glu-Gln- Dhonburi: β 126 Val-Gly - Tak: elongated β-chain- Lepore (Boston)
HEMOGLOBINOPATHIES IN THAILAND (2)

13
RT Values for Hb VariantspH 8.5
+_
A2 (3.6)
Tak (4.3)D Punjab (4.2)
S (4.4)Montgomery (4.6)Q-Mahidol (4.6)
C (5.0)O-Arab (4.8)
E (3.6)Lepore (3.6)D-Iran (3.6)
Hb Constant Spring (5.1)
A (2.4)
Hb Malay (2.4)Hb Dhonburi
H (0.5)
J Bangkok (1.9)
New York (1.4)Hope (1.38)
Pyrgos (1.38)Kodaira (1.4)
F (1.2)
Bart’s (0.2)
Abnormal Hbs
Hb G-Coushattaα 22 Glu-Ala
Hb Siamα 15 Gly-Arg
Hb J-Budaα 61 Lys-Asn
Hb Hope(β136 Gly->Asp)
Hope,Pyrgos,New york, Kodaira
Abnormal Hb at P2 Hb D/Hb E
HbTak
Hb Tak / Hb E
Hb Tak (β147 +AC) trait• Hb: 16.6 g/dl• RBC: 5.6X1012/L• Hct: 48 %• MCV: 87 fl• MCH: 30 pg• MCHC: 34 g/dl• RDW: 12.3• Hb F 0.4 % RT 1.11• Hb A 55.7% 2.46• Hb A2 4.0% 3.6• Hb Tak 34.4% 4.23
Hb E/Hb Tak• Hb: 13.7 g/dl• RBC: 5.7X1012/L• Hct: 40 %• MCV: 69 fl• MCH: 24 pg• MCHC: 33 g/dl• RDW: 15.8 %• Hb E+ Hb A2: 47.3%• Hb F: 1.9 %• Hb Tak: 46.4%• DNA studies
HbTak
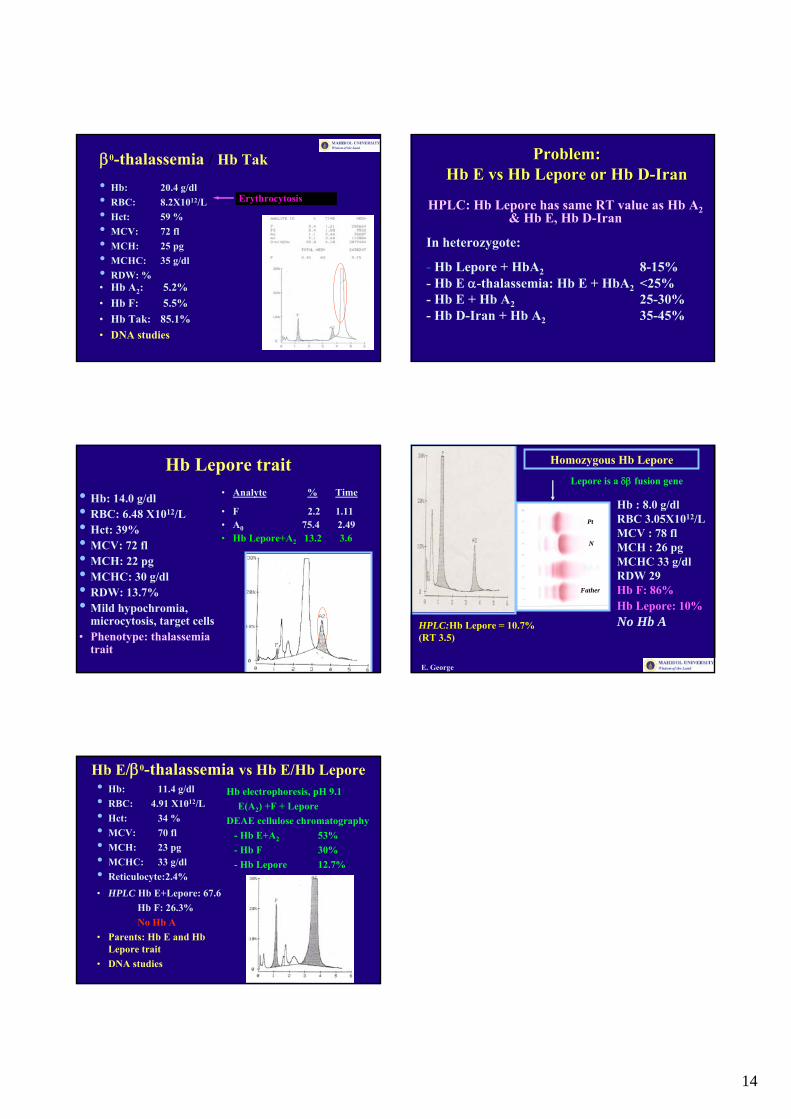
14
β0-thalassemia / Hb Tak
• Hb: 20.4 g/dl• RBC: 8.2X1012/L• Hct: 59 %• MCV: 72 fl• MCH: 25 pg• MCHC: 35 g/dl• RDW: %• Hb A2: 5.2%• Hb F: 5.5%• Hb Tak: 85.1%• DNA studies
Erythrocytosis
Problem: Problem: HbHb EE vsvs HbHb LeporeLepore or or HbHb DD--IranIran
HPLC: Hb Lepore has same RT value as Hb A2& Hb E, Hb D-Iran
In heterozygote:
- Hb Lepore + HbA2 8-15%- Hb E α-thalassemia: Hb E + HbA2 <25% - Hb E + Hb A2 25-30%- Hb D-Iran + Hb A2 35-45%
• Hb: 14.0 g/dl• RBC: 6.48 X1012/L• Hct: 39%• MCV: 72 fl• MCH: 22 pg• MCHC: 30 g/dl• RDW: 13.7%• Mild hypochromia,
microcytosis, target cells• Phenotype: thalassemia
trait
Hb Lepore trait• Analyte % Time
• F 2.2 1.11• A0 75.4 2.49• Hb Lepore+A2 13.2 3.6
S,F,A
Pt
Father
N
E. George
Homozygous Hb Lepore
Hb : 8.0 g/dlRBC 3.05X1012/LMCV : 78 flMCH : 26 pgMCHC 33 g/dlRDW 29Hb F: 86%Hb Lepore: 10%No Hb AHPLC:Hb Lepore = 10.7%
(RT 3.5)
S,F,A
Pt
Father
N
Lepore is a δβ fusion gene
Hb E/β0-thalassemia vs Hb E/Hb Lepore• Hb: 11.4 g/dl• RBC: 4.91 X1012/L• Hct: 34 %• MCV: 70 fl• MCH: 23 pg• MCHC: 33 g/dl• Reticulocyte:2.4%• HPLC Hb E+Lepore: 67.6
Hb F: 26.3% No Hb A
• Parents: Hb E and HbLepore trait
• DNA studies
Hb electrophoresis, pH 9.1E(A2) +F + Lepore
DEAE cellulose chromatography- Hb E+A2 53%- Hb F 30%- Hb Lepore 12.7%