lysosomal acid lipase deficiency: biology, disclosure · lysosomal acid lipase deficiency: biology,...
TRANSCRIPT

9/26/2014
1
Lysosomal Acid Lipase Deficiency: Biology, Clinical Manifestations, Diagnosis, and Novel
Approach to Treatment
Mark A. Goldberg, M.D.Medical and Regulatory Strategy
Synageva Biopharma Corp.
Associate Clinical Professor of MedicineHarvard Medical School
September 26, 2014
DisclosureMark Goldberg, M.D. discloses the following relationships with commercial companies:
• Employee and shareholder of Synageva Biopharma
Learning ObjectivesAt the end of this presentation the
participant will be able to:
1. Understand the biology and the clinical manifestations of lysosomal acid lipase deficiency
2. Know how to test for lysosomal acid lipase deficiency
3. Understand treatment options for patients with lysosomal acid lipase deficiency
Lysosomal Acid Lipase (LAL) Deficiency: A single disease, with marked clinical heterogeneity
Lysosomal Acid Lipase (LAL) Deficiency: A single disease, with marked clinical heterogeneity
Historical terms to describe the disease
– “Wolman disease” - 1956 by Dr. Wolman
• Infant who died at the age of ~two months: GI symptoms, hepatosplenomegaly, poor weight gain, and bilateral adrenal calcifications
– “Cholesteryl Ester Storage Disease (CESD)” - 1963 by Dr. Fredrickson
• 12y/o with hypercholesterolemia + hepatomegaly (300-500x increase in CE on biopsy)
Underlying cause is the same1,2
– Autosomal recessive disease affecting lipid metabolism due to mutations in the LIPA gene encoding lysosomal acid lipase
– Results in lysosomal accumulation of lipids (cholesteryl esters and triglycerides)
1. Patrick and Lake : “Deficiency of an acid lipase in Wolman’s disease”, Nature, 19692. Burke and Schubert : “Deficient activity of hepatic acid lipase in cholesterol ester storage disease”, Science, 1972Cortner et al: “Genetic variation of lysosomal acid lipase”, Pediatric Research, 1976Goldstein, et al: “Role of lysosomal acid lipase in the metabolism of plasma Low density lipoprotein,” JBC, 1975
Biology of Lysosomal Acid Lipase (LAL)Biology of Lysosomal Acid Lipase (LAL)
5
Healthy Individuals LAL Deficient Patients
Nucleus
LDL particle
Hepatocyte
• Normal lipid homeostasis
• Hydrolysis yielding free cholesterol and free fatty acids
LAL
Normal Lysosome
Nucleus
LDL particle
Hepatocyte
• Disruption of lipid homeostasis
• Accumulation of lipid in lysosome
Enlarged Lysosome
LAL
LAL Deficiency GeneticsLAL Deficiency Genetics
Mutations have variable expression of protein– Distinction on clinical progression is not based on enzyme activity –
variable assay methods/substrate.
Common mutation (splice mutation) E8SJM-1
• Autosomal recessive
• LIPA gene maps to chromosome 10q23.2iq23.3

9/26/2014
2
Lysosomal Acid Lipase Deficiency (LAL D) presents across a clinical continuum
Lysosomal Acid Lipase Deficiency (LAL D) presents across a clinical continuum
Affected liver removed during transplant surgery age 9
Often fatal within first 6 months of life
Marked growth failure in first few months of life AND
Rapidly progressive liver disease
Atherosclerosis
Infants Children & Adults
Complications of chronic liver disease (bleeding varices and ascites)
Dyslipidemia: Increased LDL, and decreased HDL
Common Aspects
Autosomal recessive disorder of lipid metabolism
Increased morbidity and early mortality
Fatty liver, elevated transaminases fibrosis/cirrhosis
7
Disease Spectrum
Rapid Disease Progression in LAL D Infants
Rapid Disease Progression in LAL D Infants
Rapidly progressive and fatal
Prominent hepatic and GI manifestations– Persistent vomiting, diarrhea
– Abdominal distension
– Profound growth failure
– Hepatomegaly and liver failure
– Splenomegaly
Adrenal calcification frequently present
Incidence: ~1/500,000
Treatment options:
– No safe and effective therapies
– HSCT (and liver transplant) have limited success and high mortality
1Data on File, Synageva BioPharma Corp.
Kaplan-Meier Estimate: Survival in LAL D Infants with Growth Failure* Kaplan-Meier Estimate: Survival in LAL D Infants with Growth Failure*
*Population shown are subjects who did not undergo HSCT or liver transplantSource: Jones et al, WORLD 2014 poster #113
Upper 95% Confidence Limit
Survival Function Estimate
Lower 95% Confidence Limit
n is number of patients at risk
No untreated subject with growth failure (presenting
before 6 months) survived to 12 months of age
9
Time Point1 Median Age
Age at Symptom Onset 1.0 Month
Age at Diagnosis 2.6 Months
Age at Death 3.7 Months
LALD Presenting in InfantsTypical Sign/Symptoms & Differential LALD Presenting in InfantsTypical Sign/Symptoms & Differential
Clinical Age <1 year Vomiting and/or Diarrhea Abdominal distention Hepatomegaly and/or splenomegaly Growth failure
Laboratory Elevated transaminases Elevated ferritin Elevated triglycerides Coagulopathy Cytopenias
Imaging Hepatomegaly and/or splenomegaly Adrenal calcifications (may not be present)
10
Prolonged "gastroenteritis" with growth
failure
Hemophagocytic lymphohistiocytosis
(HLH)
Glycogen storage disease
Cryptogenic liver cirrhosis
Niemann-Pick disease Type C
Chanarin Dorfman syndrome
Galactosemia
Fructose intolerance and other amino acid
metabolism disorders
Typical Findings Differential Diagnosis
Shortened lifespan and morbidity
Prominent hepatic manifestations
– Fatty liver (microvesicular steatosis)
– Elevated transaminases
– Fibrosis and cirrhosis
– Liver failure (often early in life)
Cardiovascular involvement– Elevated LDL cholesterol– Low HDL frequently observed– Variably elevated triglycerides– Accelerated atherosclerosis
Other manifestations:– splenomegaly– GI manifestations– Lymphadenopathy
Prevalence: 1:40,000 – 1/300,000
Affected liver removed during transplant surgery age 9
LAL Deficiency presenting in childhood or adulthood: A rare disease with a common phenotypeLAL Deficiency presenting in childhood or adulthood: A rare disease with a common phenotype
Presentation in Children & AdultsPresentation in Children & Adults
More frequent presentation of LAL Deficiency
Historically known as Cholesteryl Ester Storage Disease
2013 literature review1
– >80% of patients presented before 12 years of age – Death due to progressive liver disease occurred as early as 7 years of age
– 64% had fibrosis and/or cirrhosis
Disease presentation is variable– Hepatic manifestations and dyslipidemia dominate the clinical picture.
– Some are diagnosed in childhood, while others remain undiagnosed until adulthood
High potential for delayed or misdiagnosis– Metabolic syndrome: combination of fatty liver, elevated serum
transaminases, and dyslipidemia
– Focusing on non-obesity may increase the suspicion for LAL D
9/26/2014
3
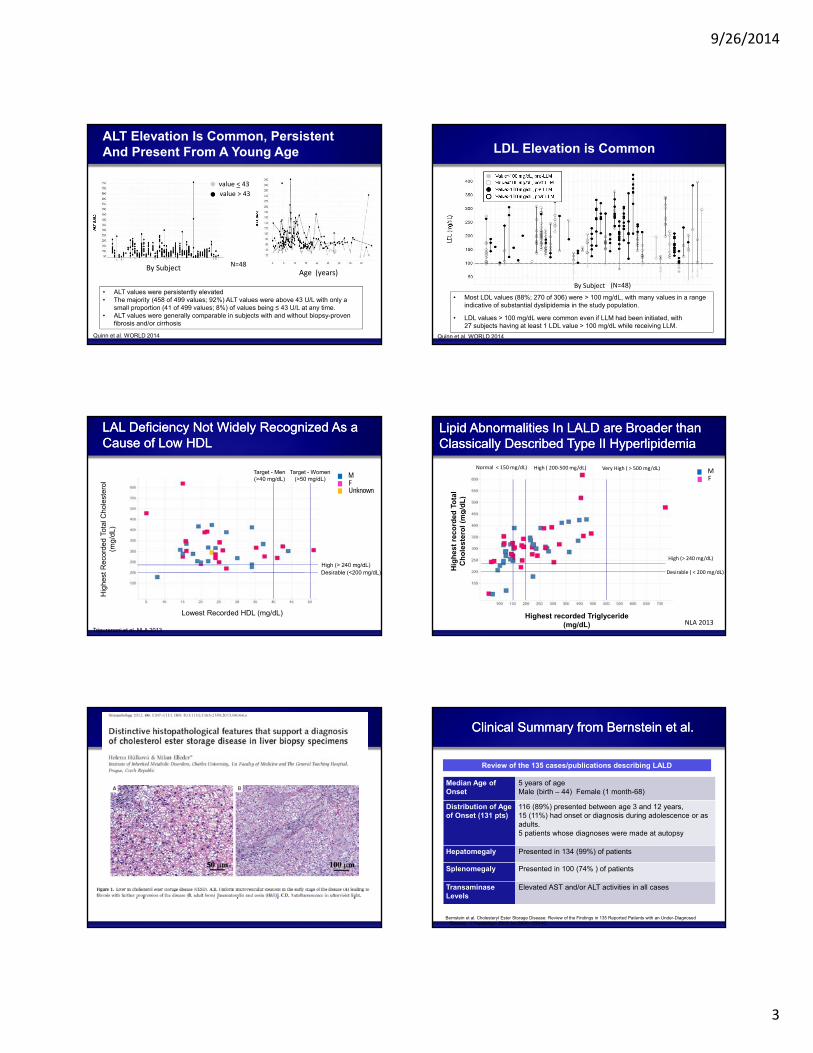
ALT Elevation Is Common, Persistent And Present From A Young Age
• ALT values were persistently elevated• The majority (458 of 499 values; 92%) ALT values were above 43 U/L with only a
small proportion (41 of 499 values; 8%) of values being ≤ 43 U/L at any time.• ALT values were generally comparable in subjects with and without biopsy-proven
fibrosis and/or cirrhosis
Age (years)By Subject
value < 43
value > 43
Quinn et al. WORLD 2014
N=48
LDL Elevation is Common
• Most LDL values (88%; 270 of 306) were > 100 mg/dL, with many values in a range indicative of substantial dyslipidemia in the study population.
• LDL values > 100 mg/dL were common even if LLM had been initiated, with 27 subjects having at least 1 LDL value > 100 mg/dL while receiving LLM.
By Subject
Quinn et al. WORLD 2014
(N=48)
Hig
hest
Rec
orde
d To
tal C
hole
ster
ol
(mg
/dL)
LAL Deficiency Not Widely Recognized As a Cause of Low HDLLAL Deficiency Not Widely Recognized As a Cause of Low HDL
Lowest Recorded HDL (mg/dL)
High (> 240 mg/dL)Desirable (<200 mg/dL)
Target - Men(>40 mg/dL)
FM
Unknown
Target - Women(>50 mg/dL)
Tripuraneni et al. NLA 2013
Hig
he
st r
eco
rded
To
tal
Ch
ole
ster
ol
(mg
/dL
)
Lipid Abnormalities In LALD are Broader than Classically Described Type II HyperlipidemiaLipid Abnormalities In LALD are Broader than Classically Described Type II Hyperlipidemia
Highest recorded Triglyceride (mg/dL)
High (> 240 mg/dL)
Desirable ( < 200 mg/dL)
High ( 200‐500 mg/dL)Normal < 150 mg/dL) Very High ( > 500 mg/dL)
FM
NLA 2013
Clinical Summary from Bernstein et al. Clinical Summary from Bernstein et al.
Median Age of Onset
5 years of ageMale (birth – 44) Female (1 month-68)
Distribution of Age of Onset (131 pts)
116 (89%) presented between age 3 and 12 years, 15 (11%) had onset or diagnosis during adolescence or as adults. 5 patients whose diagnoses were made at autopsy
Hepatomegaly Presented in 134 (99%) of patients
Splenomegaly Presented in 100 (74% ) of patients
TransaminaseLevels
Elevated AST and/or ALT activities in all cases
Review of the 135 cases/publications describing LALD
Bernstein et al. Cholesteryl Ester Storage Disease: Review of the Findings in 135 Reported Patients with an Under-Diagnosed Disease. J Hepatology. 2013 Jun;58(6):1230-43

9/26/2014
4
Clinical Summary from Bernstein et al. (cont)Clinical Summary from Bernstein et al. (cont)
Liver Injury and/or Liver Failure (135 pts)
• Occurred in all patients• Death due to liver disease progression: 7- 56 years old• 50% of deaths were in patients under 21 years of age.
Liver Biopsy(112 (83%) pts)
• A striking orange-yellow in color • Diffuse, uniform microvesicular steatosis • 72 (64%) had fibrosis and/or cirrhosis
• Findings were consistent among patients, and appeared independent of age, genotype, or other factors
Bernstein et al. Cholesteryl Ester Storage Disease: Review of the Findings in 135 Reported Patients with an Under-Diagnosed Disease. J Hepatology. 2013 Jun;58(6):1230-43
Management Options & Clinical TrialsManagement Options & Clinical Trials
Infants– Electrolyte replacement, parenteral nutrition, formula modifications, etc.
Children & Adults– Statins/lipid lowering agents – but liver disease progression can still occur1
Hematopoietic stem cell or liver transplantation– Has been associated with serious complications (e.g., death, graft-versus-host disease)
Enzyme replacement therapy (ERT) – Sebelipase alfa is an investigational ERT
– Reported encouraging phase ½ results in LAL deficient adults2
– Ongoing trials for LAL deficient infants (LAL-CL03) and children/adults (ARISE)
20
1. Bernstein et al. Cholesteryl Ester Storage Disease: Review of the Findings in 135 Reported Patients with an Under-Diagnosed Disease. J Hepatology. 2013 Jun;58(6):1230-432. Balwani, et al. Clinical effect and safety profile of recombinant human lysosomal acid lipase in patients with cholesteryl ester storage disease. Hepatology. 2013 Sept: 58 (3) : 950–57
Sebelipase Alfa: Pre-Clinical and Clinical Development
Sebelipase Alfa: Pre-Clinical and Clinical Development
21
Sebelipase alfa
Targeting and ActivitySebelipase alfa
Targeting and Activity
Terminal mannose/GlcNac and mannose-6-phosphate (M6P) for targeted delivery
Uptake into key cells
Lysosomal localization demonstrated
Corrects enzyme deficiency
Lysotracker red Overlap imagesOregon green-labeled sebelipase alfa
0
1
2
3
4
5
6
0 0.16 0.5 1.6 5
Cel
lula
r L
AL
Act
ivit
y (n
Un
its/
cell)
sebelipase alfa (ug/ml)
Normal Human Fibroblasts
LD Fibroblasts
0 0.16 0.5 1.6 5.0
LAL Deficiency Rat ModelReproduces Key Aspects of Human DiseaseLAL Deficiency Rat ModelReproduces Key Aspects of Human Disease
Accumulation Lipid Substrate in Liver, Spleen and Gut
Growth failure Increased Mortality
Maximal life span in LAL-/- rats is approximately 14 weeks
Rat model 1st described Japan 1990 (Yoshida H, Kuriyama M. Lab Animal Sci. 1990;40(5):486-9.
Sebelipase alfaPreclinical Targeting and ActivitySebelipase alfaPreclinical Targeting and Activity
In Vivo Activity
* sebelipase alfa 5mg*kg-1 once weekly for 4 weeks

9/26/2014
5
Sebelipase alfaPreclinical Targeting and ActivitySebelipase alfaPreclinical Targeting and Activity
In Vivo Activity
* sebelipase alfa 5mg*kg-1 once weekly for 4 weeks
sebelipase alfa (SBC-102) Restores Normal Growth And Increases Survival in Preclinical Disease Modelsebelipase alfa (SBC-102) Restores Normal Growth And Increases Survival in Preclinical Disease Model
26
Improvements In Growth And Organ Size Is Associated With Correction Of Underlying PathologyImprovements In Growth And Organ Size Is Associated With Correction Of Underlying Pathology
SBC-102
Placebo
Liver Sebelipase alfa
Sebelipase alfa
LAL-CL03 (Phase 2/3 Trial)LAL-CL03 (Phase 2/3 Trial)
LAL D infants with growth failure first 6 months of life Open label Intra-patient dose
escalation of sebelipase alfa – 0.35 mg/kg to 3 mg/kg
Once weekly dosing Multicenter N= 8 - 10
Primary Endpoint: Survival at 12 months of age
Key Secondary Endpoints: Safety and tolerability Survival beyond 12 months of
age Growth Liver parameters (AST, ALT,
GGT, Alk Phos, Bilirubin) Lipids Development (Denver II) Pharmacokinetics
EndpointsTrial Design
28
Valayannopoulos et al. WORLD 2014
Key Inclusion & Exclusion CriteriaKey Inclusion & Exclusion Criteria
Inclusion:– Confirmed diagnosis of LAL D
– Growth failure with onset before 6 months of age
• Weight decreasing across at least 2 of the 11 centile lines on WHO weight for age chart OR
• Body weight in kg below the 10th centile AND no weight gain for the 2 weeks prior to screening OR
• Loss of ≥ 5% of birth weight in a child older than 2 weeks
– Infants with rapidly progressive course if no growth failure*
Exclusion:– Myeloablative preparation, or other systemic pre-transplant
conditioning
– Previous hematopoietic stem cell or liver transplant*Exceptional circumstance and requires review with the safety committee prior to enrollment
29Valayannopoulos et al. WORLD 2014
Patient Disposition & Baseline CharacteristicsPatient Disposition & Baseline Characteristics
Data as of June 2014
9 patients enrolled
Median age at time of first infusion: 3.0 months – Range 1.1 – 5.8 months
Range of time in the trial: 0.6 to 31.8 months
Laboratory Values:Parameter Median Range
ALT 96 U/L 16-297 U/L
AST 125 U/L 71-716 U/L
Bilirubin 7 umol/L 2.4-464 umol/L
Hemoglobin 9.3 g/dL 7.2-10.2 g/dL
Platelets 173 x109/L 39-563 x109/L
Signs and Symptoms:• Diarrhea or Vomiting (6 of 9)
• Hepatomegaly (7 of 9)
• Splenomegaly (6 of 9)
• Adrenal calcification (4 of 9)
30Valayannopoulos et al. WORLD 2014

9/26/2014
6
Kaplan-Meier Estimate of Survival in LAL D Infants with Growth Failure*: Results of Natural History StudyKaplan-Meier Estimate of Survival in LAL D Infants with Growth Failure*: Results of Natural History Study
*Population shown are subjects who did not undergo HSCT or liver transplantSource: Jones et al, LDN 2014 poster #113
Upper 95% Confidence Limit
Survival Function Estimate
Lower 95% Confidence Limit
n is number of patients at risk
No untreated subject with growth failure (presenting
before 6 months) survived to 12 months of age
31
0 6 12 18 24 30
Patient Age (Months)
Alive and continuing on sebelipase alfa
Deceased (not related to sebelipase alfa)
Survival (12 months of age)
Initial Survival Data with Sebelipase Alfa
Sebelipase Alfa Phase 2/3 Data in Infants: EfficacySebelipase Alfa Phase 2/3 Data in Infants: Efficacy
Patient 1
Patient 2
Patient 6
Patient 7
Patient 8
Patient 9
Patient 3
Patient 4
Patient 5
36 40
Source: Synageva as of June 2014
32
Infusion HistoryAge at Death
Relationship to sebelipase alfa
Cause of Death
1 infusion (0.35 mg/kg/wk)3
monthsNot related
Complications of disease
1 infusion (0.35 mg/kg/wk)2
monthsNot related
Complications of non-protocol specified
abdominal paracentesis
4 infusions • 2 infusions (0.35 mg/kg/wk)• 2 infusions (1.0 mg/kg/wk)
4months
Not relatedComplications of
disease
33
6 Subjects continue on sebelipase alfa3 Deceased Subjects
• Not related to sebelipase alfa
Dosing StatusDosing Status
Valayannopoulos et al. WORLD 2014
Growth Curve of Subject 1Growth Curve of Subject 1
Subject 1 (M)
Age (months)
We
igh
t (k
g)
34
Survival at 12 months of age
Growth Curves of the Other Surviving Subjects
Growth Curves of the Other Surviving Subjects
Subject 9 not shown (received only a single infusion at time of data cut)Valayannopoulos et al. WORLD 2014
35
SafetySafety
Majority of SAEs:– Were not related to sebelipase alfa– Related to documented central line infections or hospitalizations for
empirical treatment with antibiotics
3 related SAEs – Occurred in a single subject in association with the same infusion
• Fever (malaise)• Malaise with tachycardia • Tachycardia
– Resulted in overnight hospitalization• Treated with IV antibiotics empirically for possible line bacteremia (all cultures
negative)
• Majority of IRRs have been mild • Fever• Vomiting
36
Valayannopoulos et al. WORLD 2014

9/26/2014
7
Anti-Drug AntibodyAnti-Drug Antibody
4 subjects developed anti-drug antibody (ADA)– Subject 3 - ADA positive after 7 weeks on treatment
– Subject 5 - ADA positive after 8 weeks on treatment
– Subject 6 - ADA positive after 16 weeks on treatment
– Subject 1 - ADA positive after 58 weeks on treatment
No apparent change in clinical response after ADA development
37Valayannopoulos et al. WORLD 2014
Sebelipase Alfa: Experience in Adults
Sebelipase Alfa: Experience in Adults
102 wk data
R. Tripuraneni et al. Effect of Sebelipase in Adults with Lysosomal Acid Lipase Deficiency Oral presentation EAS 2014
38
Phase 1/2 Trial Design with Sebelipase alfa in Adults with LAL DeficiencyPhase 1/2 Trial Design with Sebelipase alfa in Adults with LAL Deficiency
40
Characteristics and Baseline Test ResultsCharacteristics and Baseline Test Results
Parameter Population
Age (median, yrs) 29
Male:Female (n) 6:3
White (n) 9
Weight (median, kg) 72
BMI (median, kg/m2)BMI ≥ 30 kg/m2 (n)
25.21
Hepatomegaly (n) 8
Lipid lowering meds (n)Statins (n)
76
Elevated* ALT or AST (n) 2
*Elevated: ≥1.5xULN and <3xULN.
RED = abnormal value
Lab Analyte Reference Range
Median (Range)
ALT (U/L) ≤67 76 (22 to 119)
AST (U/L) ≤50 56 (37 to 69)
Total Chol (mg/dL) 69-232 182 (116 to 391)
LDL (mg/dL) ≤162 135 (70 to 300)
HDL (mg/dL) ≥35 39 (22 to 49)
Triglycerides (mg/dL)
≤199 108 (80 to 277)
Improvement in TransaminasesImprovement in TransaminasesMean percent decrease: 58% ALT and 28% AST (wk 104)
n=8n=9
41
Improvement in Dyslipidemia ProfileImprovement in Dyslipidemia Profile
n=8n=9
Mean percent decrease: 54% LDL, 31% TG (wk 104)Mean percent increase: 18.4% HDL (wk 104)
42

9/26/2014
8
Rapid, Sustained Reduction inLiver Volume and Fat Fraction (MRI)Rapid, Sustained Reduction inLiver Volume and Fat Fraction (MRI)
Per
cen
t C
han
ge
Fro
m C
L04
Bas
elin
e
-9% -14%
-35%
-55%
Liver Volume Liver Fat Fraction
-42%
‐60
‐50
‐40
‐30
‐20
‐10
0
10
Week 10Week 24Week 52
‐10%‐12%
‐9%
‐42%
‐35%
‐55%
n=8n=8 n=7 n=7n=7 n=6
Safety ProfileSafety Profile
No drug-related serious adverse events (SAE) in this trial
One unrelated SAE: cholecystitis and cholelithiasis– The subject underwent elective cholecystectomy.
– The subject has continued in the study.
No evidence of anti-drug antibodies in subjects tested to date in this study
Most infusion-related reactions (IRR) were mild, mainly GI related (diarrhea, abdominal cramping).
44
ARISE Trial ARISE Trial
* Age- and gender-specific ULN provided by the central laboratory performing the assayhttp://www.clinicaltrials.gov/ct2/show/NCT01757184
Study Design• Randomized, double-blind, placebo
controlled• Multi-center global trial• N = 50
Endpoints• Normalization in ALT*• Change in LDL, non-HDL, TG, HDL • Normalization in AST• Change in liver volume and liver fat
content• Improvement in liver histopathology
June 30, 2014
Specialty Patient Populations
Hepatologists • All non-obese* patients with
• Persistent hepatomegaly OR
• Unexplained elevation in transaminase
• Cryptogenic cirrhosis
• Microvesicular steatosis or macro/microvesicular steatosis • can also do additional IHC staining
Lipidologists • Non–obese: LDL >=160mg/dL & HDL <40mg/dL (males) or <50mg/dL(females)
• Presumed familial hypercholesterolemia (FH) patients with unclear family history
• Presumed FH patients who have negative genetic testing for the genes encoding LDLR, APO B, and PCSK9 genes
Which Patient to Test for LAL D?
*Non obese definition: adults <30 kg/m2 or <95th percentiles in those less than 18 years of age
Blood Test for LAL DBlood Test for LAL D
LAL activity is determined by via a LAL specific inhibitor
Allows for the possibility of testing high risk populations
47
Hamilton J, et al. Clin Chim Acta. 2012;413:1207-10.
Recent Development of Dry Blood Spot Assay Allows for Easy Testing
LAL Activity
Labs That Perform LAL D Testing (US)Labs That Perform LAL D Testing (US)
48
Laboratory Contact InformationDBS
MethodGenetic
SequencingLeukocyte
AssayMassachusetts General Hospital (MGH)*Boston, MA
Phone: 1-617-967-2045E-mail: [email protected] site: www.massgeneral.org/research/resourcelab.aspx?id=43
x x –
Seattle Children’s Hospital and University of Washington*Seattle, WA
Rhona Jack, PhD Phone: 1-206-987-2216Web site: www.seattlechildrens.org/labman x x –
Baylor College of MedicineHouston, TX
Phone: 1-800-411-GENE (4363)Website: www.bcm.edu/research/medical-genetics-labs/ – x x
GeneDxGaithersburg, MD
Phone: 1-301-519-2100E-mail: [email protected] site: www.genedx.com/
– x x
PreventionGenetics, Marshfield, WI
Phone: 1-715-387-0484E-mail: [email protected]: http://preventiongenetics.com/
– x x
Thomas Jefferson University,Philadelphia, PA
David A. Wenger, PhD Phone: 1-215-955-4923E-mail: [email protected] site: www.jefferson.edu/jmc/departments/neurology/programs/neurogenetics/lysosomal_diseases.html
– – x
Laboratory Corporation of AmericaResearch Triangle Park, NC
Phone: 1-800-345-4363Web site: www.labcorp.com/wps/portal/provider/testmenuLAL Assay test code: 402300
x – –
HIBM*Encino, CA
Phone: 1-818-789-1033Web site: http://hibm.org/hrg/www/pages/ x x –
48*accepts samples from around the world

9/26/2014
9
SummarySummary
LAL deficiency is an under-recognized cause of cirrhosis, accelerated atherosclerosis, and early death
It usually presents in childhood
Key signs in children and adults include: (not all are required)
– Elevated transaminases, hepatomegaly and/or microvesicular steatosis
– Dyslipidemia (elevated LDL and low HDL)
Diagnosis can be made via a simple blood test
Analysis of sebelipase alfa in the ongoing clinical trials
– Improved growth & survival in infants
– Produces sustained improvements in the biochemical markers of liver damage, and the dyslipidemia in the adults
– Safety and tolerability profile is encouraging after administration of more than >300 infusions.
4950
US and European Guidelines: Endorse Testing to Rule Out LAL D
ESPGHAN = European Society for Pediatric Gastroentereology, Hepatology and NutritionChalasani et al. Gastroenterol. 2012;142:1592-1609Vajro P, et al. JPGN. 2012;54:700-13
• Symptoms overlap with hereditary disorders• Rule out LAL D in microvesicular steatosis cases
• Nonspecific symptoms of NAFLD overlap with genetic-metabolic causes
• Rule out LAL D before diagnosing NAFLD
Liver Disease: Two Rare DiseasesLiver Disease: Two Rare Diseases
Wilson disease
Symptoms >3y of age
Acute presentation at any age >3y
Chronic progressive disease
Autosomal recesive
LAL Deficiency
Symptoms from infancy
Acute presentation in infancy
Chronic progressive disease >2y of age
Autosomal recesive
51
Is tested in each child with liver disease >3y
Should we routinely test for LAL D in children with liver disease?
Thank you!