meldingen 30 november 2011 anja van haren eudravigilance coördinator informatiebijeenkomst nieuwe...
Post on 19-Dec-2015
223 views
TRANSCRIPT

Meldingen
30 november 2011
Anja van HarenEudraVigilance Coördinator
Informatiebijeenkomst nieuwe wetgeving farmacovigilantie

2
The information on these slides is for general information purposes only and presents the state of knowledge at November 30, 2011.
No rights can be derived from this information. The Medicines Evaluation Board accepts no liability for direct or consequential damage resulting from the use of, reliance on or action taken on the basis of information provided at this session.
Disclaimer

Content
1. Key changes expedited reporting- some changes highlighted
2. Artikel 57(2)
3. Future of expedited reporting in the Netherlands
4. Closure

4
- New broad definition of Adverse Reaction(Adverse Drug Reaction; ADR)
- Better data collection:– Expedited requirements for non-serious reports– Patient reporting– Literature monitoring by EMA– Medication errors that result in ADRs are collected
Key changes (1/2)

5
- Simplified logistics for reporting(after transitional period)- All ADRs from MAHs and MSs sent to EV only– MSs are ‘auto-forwarded’ their national data– MAHs access reports in EudraVigilance (EV)
- Use of internationally agreed standards (proposed to use ISO ICSR and IDMP standards from Jan 2015)
Key changes (2/2)

6
New definition of Adverse Reaction:
Reporting of ADRs is not limited to adverse effects in normalconditions of use, but also from :
- uses outside terms of Marketing Authorisation (including misuse and abuse)- medication error- overdose- occupational exposure
New ADR definition
‘a response to a medicinal product which is noxious and unintended’

Misuse, abuse, medication error, etc
More guidance will be provided in GVP Module VI:
“Reports with no associated adverse reaction should not be reported on expedited basis” - these should be considered in PSUR and RMP
Proposals for definitions:• misuse• abuse• medication error• overdose • occupational exposure
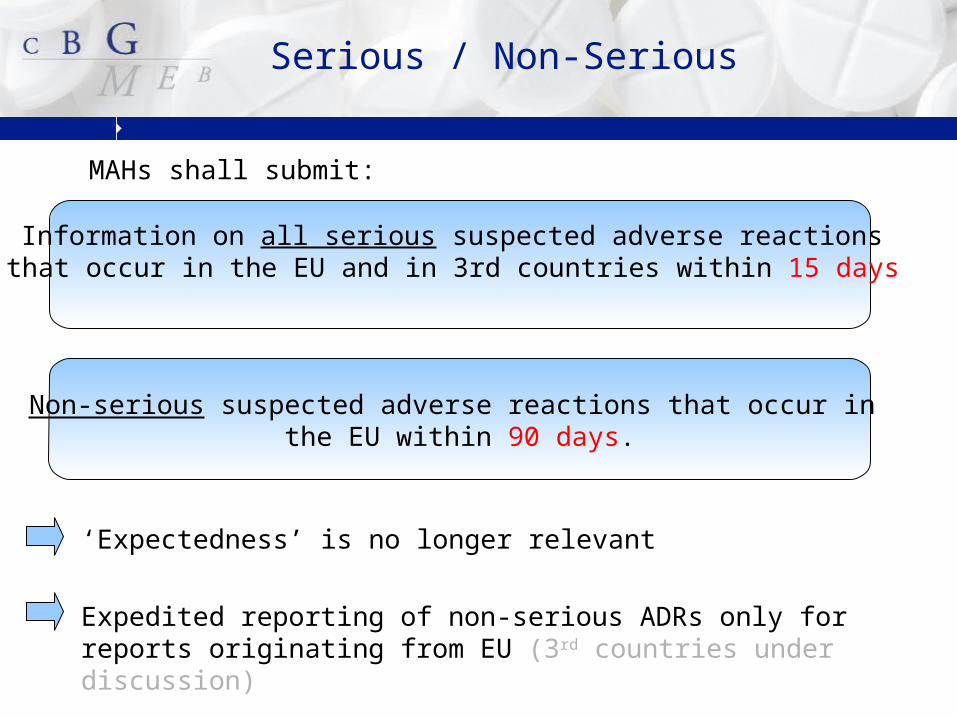
MAHs shall submit:
Serious / Non-Serious
Non-serious suspected adverse reactions that occur in the EU within 90 days.
Information on all serious suspected adverse reactions that occur in the EU and in 3rd countries within 15 days
‘Expectedness’ is no longer relevant
Expedited reporting of non-serious ADRs only for reports originating from EU (3rd countries under discussion)

More guidance provided in GVP Module VI
Patient reports (1/3)
The MAH shall not refuse to consider reports of suspected adverse reactions received electronically or by
any other appropriate means from patients and HCPs
MAH shall record all suspected adverse reactions in the EU or in 3rd countries, whether reported spontaneously by patients or HCPs, or occurring in the context of a post-authorisation study
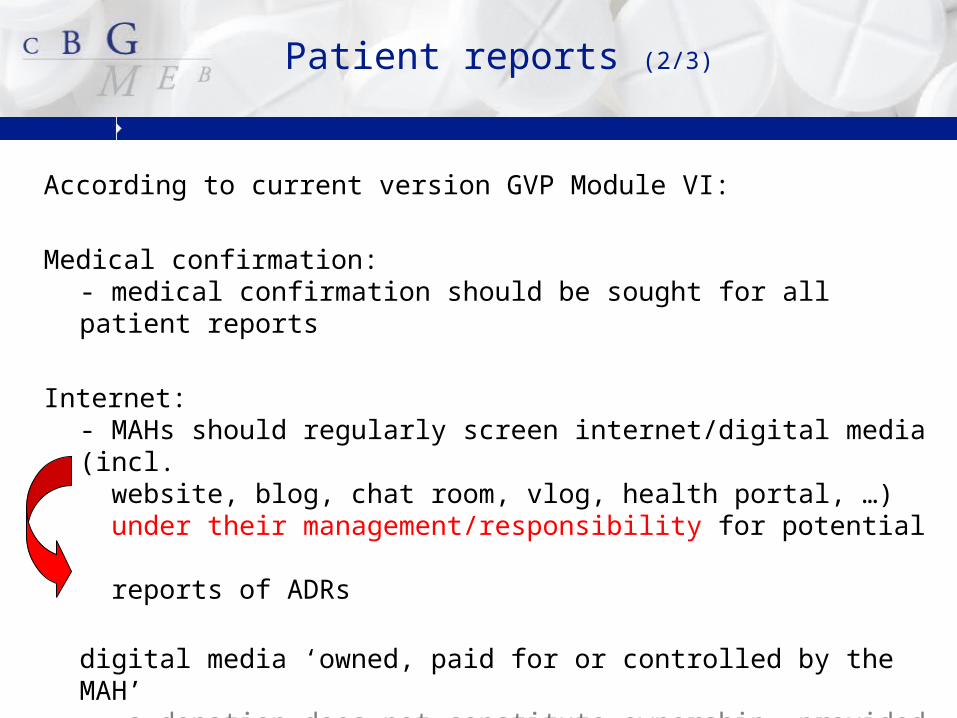
According to current version GVP Module VI:
Medical confirmation:- medical confirmation should be sought for all patient reports
Internet: - MAHs should regularly screen internet/digital media (incl. website, blog, chat room, vlog, health portal, …) under their management/responsibility for potential reports of ADRs
digital media ‘owned, paid for or controlled by the MAH’ a donation does not constitute ownership, provided that the MAH does not control the content of the site
Patient reports (2/3)

According to current version GVP Module VI:for reports from Patient Support Programmes
Patient reports (3/3)
Solicited reports
e.g. MAH asks if adverse eventswere noted with use of product
=> Submit to authorities only when there is possible causal relationship as per reporter or MAH
Not-solicited reports
e.g. MAH contacts patient for a refill and is informed of a suspected adverse reaction
=> Submit to authorities as this should be considered as a spontaneous report of suspected adverse reaction
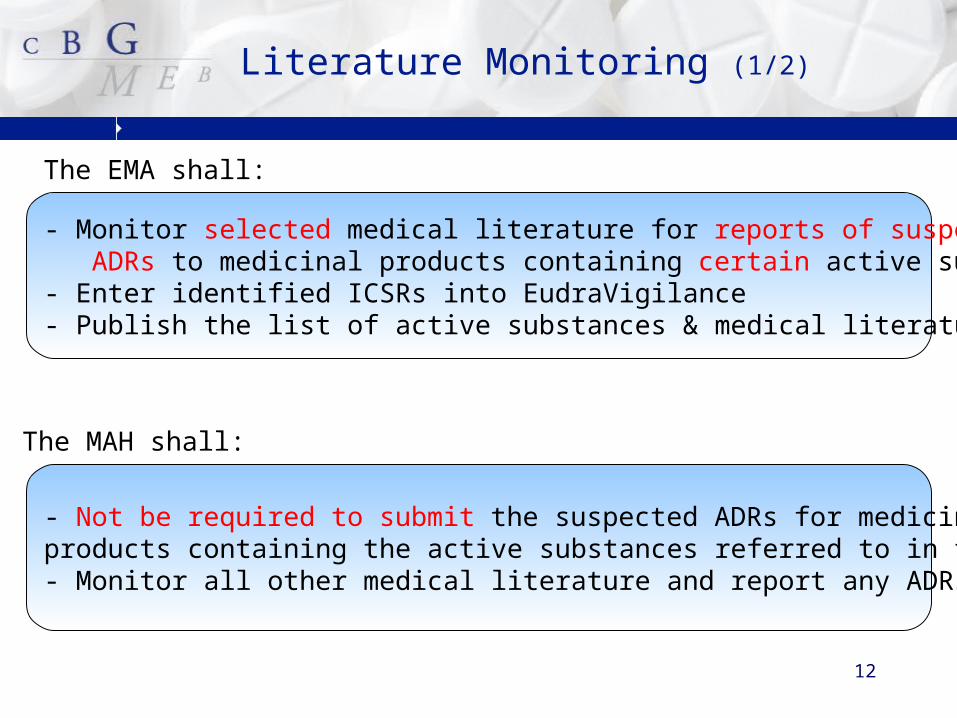
12
Literature Monitoring (1/2)
The MAH shall:
- Monitor selected medical literature for reports of suspected ADRs to medicinal products containing certain active substances- Enter identified ICSRs into EudraVigilance - Publish the list of active substances & medical literature
- Not be required to submit the suspected ADRs for medicinal products containing the active substances referred to in the list - Monitor all other medical literature and report any ADRs
The EMA shall:

13
Literature Monitoring (2/2)
Purpose of literature monitoring by EMA is to avoid duplicate reports in EudraVigilance
Timeline publication of list selected literature & substances: ??(possibly reference will be made to databases such as Medline, Embase, etc rather than listing medical journals)
MAH access to EudraVigilance planned for 2014/2015MAH access to literature articles: ??
MAHs will still need to conduct literature screening:- for the purpose of ongoing safety review - to identify ICSRs
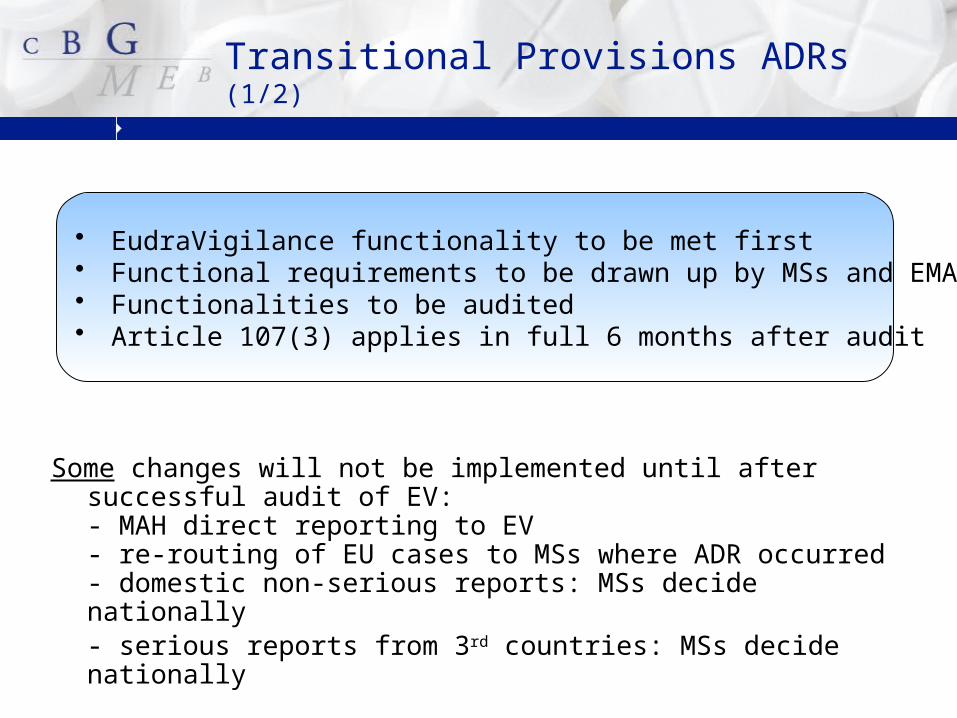
Transitional Provisions ADRs (1/2)
Some changes will not be implemented until after successful audit of EV: - MAH direct reporting to EV- re-routing of EU cases to MSs where ADR occurred- domestic non-serious reports: MSs decide nationally- serious reports from 3rd countries: MSs decide nationally
• EudraVigilance functionality to be met first• Functional requirements to be drawn up by MSs and EMA• Functionalities to be audited• Article 107(3) applies in full 6 months after audit

Latest position: EV ready for audit late 2014The changes to ADR reporting will be implemented in full
6 months after the audit (meaning mid 2015)
However, as from July 2012: - requirements related to patient reports apply- expectedness no longer relevant: submit ALL serious cases (serious cases from 3rd countries depend on MS)- submission of domestic non-serious cases depend on MS
Transitional Provisions ADRs (2/2)

Content
1. Key changes expedited reporting
2. Artikel 57(2)
3. Future of expedited reporting in the Netherlands
4. Closure
Aim of new legislation

Identification of medicinal products
Identification of medicinal products in ADR reports is key to pharmacovigilance activities:Need reliable information for signal detection/analysis and regulatory actions
EV access policy:MAH access to ICSRs related to their medicinal products
art 57(2) amended

“EudraPharm has been established to fulfil Articles 57(1)(l) and 57(2) of Regulation (EC) No 726/2004.”
Article 57(2) implementation (1/4)

The new Article 57(2), 2nd subparagraph of Regulation (EU) 726/2004 requires:
• The EMA to make public a format for the electronic submission of information on medicinal products for human use by 2 July 2011
• MAHs to electronically submit information to the EMA electronicallyon all medicinal products for human use authorised or registered in the EU, by 2 July 2012 at the latest, using this format
• MAHs to inform the EMA of any new or varied marketingauthorisations granted in the EU using this format
Article 57(2) implementation (2/4)

21
Stepwise ‘upgrade’ of the EudraVigilance Medicinal Product Dictionary (EVMPD):
- Initial format: EudraVigilance Medicinal Product Report Message - updated format by end of 2014 to be fully aligned with the 5 ISO Identification of Medicinal Products (IDMP) standards
MAHs are populating EVMPD, not EudraPharm
Article 57(2) implementation (3/4)

Information to be provided to EudraVigilance Medicinal Product Dictionary initially will include:
• Description of the (invented) name• Description of the therapeutic area(s) e.g. ATC Code• The designation of additional monitoring where applicable• Description of the clinical particulars i.e. therapeutic indication(s)• Details of the MAH • Details of the marketing authorisation, including authorisation date, marketing
status, Marketing authorisation procedure• Country of marketing authorisation + Marketing authorisation number• Mutual-recognition procedure number/decentralised- procedure number• Orphan drug designation• Detailed description of the active substance(s), excipient(s), adjuvant(s) and
their specific characteristics • Strength of the active substance(s)• Description of the medical device(s) if applicable• Pharmaceutical dose forms• Route(s) of administration • Description of the packaging information• Electronic copy of the SPC, Package Leaflet (and annexes for CAPs)
MAHs submit more information to EVMPD than currently available in EudraPharm
SPC/PIL is used to validate information submitted
Article 57(2) implementation (4/4)
Content
1. Key changes expedited reporting
2. Artikel 57(2)
3. Future of expedited reporting in the Netherlands
4. Closure

In anticipation of the new legislation…
MAHs no longer need to send suspected ADRs requiring expedited reporting according to Reg (EC) No 726/2004 and Directive 2001/83/EC to the MEB, but should report instead directly to EV
Timeline for direct reporting to EV: - may start on 1 December 2011 - Should be done at the latest by 7 February 2012
Until the new EU pharmacovigilance legislation enters into force, these expedited reporting requirements only apply to serious cases, regardless of authorisation procedure

By 7 Feb 2012 at the latest:
For domestic cases requiring expedited reports according to
Reg (EC) 726/2004 & Directive 2001/83/EC:
- Report directly to EV
- Lareb and RIVM reports should not be transmitted to EV, as this will create duplicates
- ‘non-serious’ Lareb/RIVM cases which have been upgraded to ‘serious’ by the MAH should not be submitted to EV
- For literature cases a copy of the relevant published article should be sent to EMA in line with instructions Vol9A

For EEA cases requiring expedited reports according to Reg (EC) 726/2004 & Directive 2001/83/EC:
EEA cases will be made available to the MEB through EudraVigilance, according to current procedures in occurrence countries.
To avoid duplicate reports MAHs should NOT submit these EEA cases to EV directly
By 7 Feb 2012 at the latest:

For non-EEA cases requiring expedited reports according to Reg (EC) 726/2004 & Directive 2001/83/EC:
Non-EEA cases should already be submitted to EV according to current rules
There is no longer a need to apply for the non-EEA waiver
By 7 Feb 2012 at the latest:

The procedures for MAHs for receiving Lareb cases are not affected
Until the new legislation enters into force, there is no requirement to submit non-serious reports from the Netherlands- harmonised approach under discussion in the PhVWP
In case of technical problems with ICH E2B(R2) reporting to EV, reports should be sent to the EMA in an alternative way in line with the instructions given by the EMA
MAHs who do not yet have ICH E2B(R2) reporting in place can send CIOMS forms until further notice;- expected deadline July 2012
No changes to reporting of SUSARs from Clinical Trials (according to Directive 2001/20/EC)
Further details

Health Care Professional
MAH
Patient
Future flow of NL reports
Lareb report
NL Industry report
NL Industry report
Lareb report
for expedited reports according to Reg (EC) 726/2004 & Directive 2001/83/EC
Content
1. Key changes expedited reporting
2. Artikel 57(2)
3. Future of expedited reporting in the Netherlands
4. Closure

Closing Remarks
Many details in Concept Paper on Implementing Measures (!)
GVP Module VI will provide further guidance - 2 months consultation expected Jan 2012
Benefits for MAHs to have a single reporting point will depend on agreement of ‘fully functional EV system’
New reporting procedures for direct reporting to EV will be published on MEB website(s)
Thank you for your attention

33
• ADR = Adverse Drug Reaction• EMA = European Medicines Agency• EV = EudraVigilance• GVP = Good Vigilance Practice• ICSR = Individual Case Safety Report• IDMP = Identification of Medicinal Products• MA = Marketing Authorisation• MAH = Marketing Authorisation Holder• MS = Member State• PhVWP = Pharmacovigilance Working Party• PSUR = Periodic Safety Update Report• RMP = Risk Management Plan
Abbreviations