mesenchymal stem cells from human fat engineered to...
TRANSCRIPT

Cancer Therapy: Preclinical
Mesenchymal Stem Cells from Human Fat Engineered toSecrete BMP4 Are Nononcogenic, Suppress Brain Cancer,and Prolong Survival
Qian Li1, Olindi Wijesekera1, Sussan J. Salas7, Joanna Y. Wang1, Mingxin Zhu1,8, Colette Aprhys1,Kaisorn L. Chaichana1, David A. Chesler1,2,5, Hao Zhang3, Christopher L. Smith4, Hugo Guerrero-Cazares1,Andre Levchenko6, and Alfredo Quinones-Hinojosa1
AbstractPurpose:Glioblastoma is themost common adult primarymalignant intracranial cancer. It is associated
with poor outcomes because of its invasiveness and resistance to multimodal therapies. Human adipose-
derived mesenchymal stem cells (hAMSC) are a potential treatment because of their tumor tropism, ease of
isolation, and ability to be engineered. In addition, bone morphogenetic protein 4 (BMP4) has tumor-
suppressive effects on glioblastoma and glioblastoma brain tumor–initiating cells (BTIC), but is difficult to
deliver to brain tumors.We sought to engineer BMP4-secretinghAMSCs (hAMSCs-BMP4) and evaluate their
therapeutic potential on glioblastoma.
Experimental Design: The reciprocal effects of hAMSCs on primary human BTIC proliferation,
differentiation, and migration were evaluated in vitro. The safety of hAMSC use was evaluated in vivo by
intracranial coinjections of hAMSCs and BTICs in nude mice. The therapeutic effects of hAMSCs and
hAMSCs-BMP4 on the proliferation and migration of glioblastoma cells as well as the differentiation of
BTICs, and survival of glioblastoma-bearingmice were evaluated by intracardiac injection of these cells into
an in vivo intracranial glioblastoma murine model.
Results: hAMSCs-BMP4 targeted both the glioblastoma tumor bulk andmigratory glioblastoma cells, as
well as induced differentiation of BTICs, decreased proliferation, and reduced the migratory capacity of
glioblastomas in vitro and in vivo. In addition, hAMSCs-BMP4 significantly prolonged survival in a murine
model of glioblastoma. We also demonstrate that the use of hAMSCs in vivo is safe.
Conclusions: Both unmodified and engineered hAMSCs are nononcogenic and effective against
glioblastoma, and hAMSCs-BMP4 are a promising cell-based treatment option for glioblastoma. Clin
Cancer Res; 20(9); 2375–87. �2014 AACR.
IntroductionGlioblastoma is the most common and aggressive malig-
nant primary intracranial neoplasm in adults (1). Themedian survival for patients with glioblastoma is approx-
imately 14.6 months, despite aggressive combinatorial tre-atment (1). The malignant nature of glioblastoma and itsability to resist multimodal treatments have been attributedto its highly proliferative and migratory ability as well as itsheterogeneous cell composition (2–6). This heterogeneityis theorized to be because of a small population of stem-likeprogenitor cells called brain tumor initiating cells (BTIC;refs. 2–6). BTICs are highly resistant to chemotherapy andradiation therapy, andmayunderlie thehigh recurrence rateand treatment failure observed in patients with glioblasto-ma (2–6). Therefore, therapies directly targeting BTICsmight be more effective than current therapies.
Mesenchymal stem cells (MSC) are clonally expansivewith the capacity todifferentiate intoosteocytes, adipocytes,and chondrocytes under specific in vitro stimuli (7, 8).Commonly used types of MSCs are bone marrow–derivedMSCs (BM-MSC) and human adipose–derived MSCs(hAMSC; refs. 7 and 9). MSC’s intrinsic ability to home totumors, ease of isolation from various tissues, and abilityto readily expand in vitromake them attractive candidates todeliver specific, targeted cancer therapeutics (9–15). The
Authors' Affiliations: 1Department of Neurosurgery and Oncology; 2Divi-sion of Pediatric Neurosurgery; 3Department of Molecular Microbiologyand Immunology, Bloomberg School of Public Health; 4Department ofBiomedical Engineering, Johns Hopkins University School of Medicine;5Department of Neurosurgery, University of Maryland, Baltimore, Mary-land; 6Department of Biomedical Engineering, Yale University, NewHaven,Connecticut; 7Department of Neurosurgery, Jefferson Medical College,Philadelphia, Pennsylvania; and 8Department of Neurosurgery, TongjiHospital, Tongji Medical College, Huazhong University of Science andTechnology, Wuhan, Hubei, People's Republic of China
Note: Supplementary data for this article are available at Clinical CancerResearch Online (http://clincancerres.aacrjournals.org/).
CorrespondingAuthors:AlfredoQuinones-Hinojosa, Department of Neu-rosurgery, Johns Hopkins University, 1550 Orleans St, CRB-II, Room 247,Baltimore, MD 21201. Phone: 410-502-2869; Fax: 410-502-7559; E-mail:[email protected]; and Andre Levchenko, [email protected]
doi: 10.1158/1078-0432.CCR-13-1415
�2014 American Association for Cancer Research.
ClinicalCancer
Research
www.aacrjournals.org 2375
on March 22, 2019. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from

effects of MSCs on tumor cells in vivo, however, remainincompletely characterized, and seem to depend heavily oncancer type and source of MSCs (14, 15). Unlike BM-MSCs,hAMSCs are easier to obtain, more genetically and mor-phologically stable in long-term culture, have a lowersenescence ratio, and have a greater proliferative capacity(7, 9). Of the limited number of studies that have evaluatedthe effect of hAMSCs on commercial glioblastoma cell lines,some found these cells reduced tumor recurrence and hadan overall tumor-suppressive effect (11, 16, 17). However,there have also been reports of MSCs transforming intotumor-associated fibroblasts (TAF), which can potentiallysupport tumor growth and promote amalignant phenotype(18–20). Yet, no studies have evaluated the effects ofhAMSCs on human BTICs in vivo with a primary cell line.Furthermore, no studies have reported the changes thatmayoccur in hAMSCs after they interact with human BTICs.
Because of their capability to target glioblastoma cells,hAMSCs can be used to deliver therapeutic agents to glio-blastoma (9, 21–23). Bone morphogenetic protein 4(BMP4) is a potential therapeutic agent that has been shownto have an antiproliferative effect on neural progenitor cells(24–28), and, more recently, has been shown to signifi-cantly decrease the proliferation of stem-like, tumor-initi-ation precursors of glioblastomas as well as drive the dif-ferentiation of these cells toward a predominantly glial fate(29). These findings make BMP4 a promising treatment forglioblastoma, but no studies thus far have investigated itstherapeutic potential or its ability to be delivered via stemcells (29). The goals of this study were to investigate the
interaction between BTICs and hAMSCs-BMP4 and thereciprocal effects of each cell type on the other’s prolifera-tion, differentiation, andmigration. Furthermore, we inves-tigated the effect of hAMSCs-BMP4 on survival in a mousemodel of glioblastoma. These interactions are paramount tounderstanding the utility of hAMSCs and BMP4 to treatglioblastoma in human clinical trials.
Materials and MethodsCell lines
Early passage hAMSCs and BTIC cultures were used andauthenticated by Johns Hopkins Genetic Resources CoreFacility. hAMSCs (Invitrogen, R7788-115) were cultured inMesenPRO complete media (1% antibiotic/antimycotic(Invitrogen, 15240-062), 1% Glutamax (GIBCO, 35050-061), 1 vial of MesenPRO RS growth supplement (GIBCO,12748-018), and MesenPRO RS basal media (GIBCO,12747-010)). Human BTIC cultures (276 and 612) wereobtained from intraoperative tissue (as approved by JohnsHopkins Institutional Review Board) and cultured in lam-inin-coated flasks (Sigma, L2020, 1 mg/cm2) with stem cellmedia (30). As previously validated and shown by ourgroup, the human BTIC cultures are able to form onco-spheres, are multipotential, and form tumors when imp-lanted into animal models (30). To evaluate the tumori-genic capacity of BTICs in vivo, BTIC line 276 was injectedintracranially in mice by our group resulting in the forma-tion of solid tumors, whereas 612 formed diffuse tumors(30, 31). The molecular subtype of BTIC culture 276 ismesenchymal and 612 is proneural, which was determinedusing a metagene score based approach for subtype desig-nation, assessing 4 mesenchymal and 2 proneural genesusing a microfluidics based quantitative PCR assay (32).Commercial U87 cells (ATCC, HTB-14) were cultured inDulbecco’s modified Eagle medium media with 10% FBS.
Retroviral production, lentiviral production, andinfection
To induce the expression of BMP4 in hAMSCs, an MFG-based retroviral vector systemwas combinedwith a BMP2/4hybrid (33). To identify hAMSCs and BTICs in our in vitrococulture and in vivo mouse experiments, we transducedthese cells with lentiviral vectors coding for GFP, td-tomato,or GFP/bioluminescent proteins. Viral vectors were pack-aged fromHEK293 cells. After collection and concentration,hAMSCs (hAMSCs-Vector, hAMSCs-BMP4, GFP/biolumi-nescent-hAMSCs, and td-tomato-hAMSCs) and BTICs(GFP-276 and GFP-612) were infected and sorted by aMoFlo cytometer (Beckman Coulter).
Coinjection in vivo studiesTo investigate the effect and the safety of coinjected
hAMSCs on glioblastoma cell proliferation in vivo, 6- to8-week NOD/SCID mice were stereotactically injectedwith 0.5 � 106 GFP-276 (n ¼ 5), 0.5 � 106 td-tomato-hAMSCs (n ¼ 5), 0.5 � 106 GFP-276 mixed with 0.5 � 106
td-tomato-hAMSCs (coinjection; n ¼ 5), and 1.0 � 106
Translational RelevanceGlioblastoma is the most common primary intracra-
nial malignant cancer in adults, and is associated withpoor outcomes despite multimodality therapy. This lackof effectiveness of current therapies is presumablybecause of a small subset of tumor cells, so-called braintumor initiating cells (BTIC), with self-renewal capabil-ities and stronger tumor-initiating capacities. Mesenchy-mal stem cells (MSC) have an endogenous tropismtoward certain cancers, and adipose tissue provides afeasible and less invasive source of MSCs. Moreover,bone morphogenetic protein (BMP4) has been shownto decrease proliferation by inducing BTIC differentia-tion. Therefore, adipose-derived MSCs engineered tosecrete BMP4 (hAMSCs-BMP4)may be a potential effec-tive treatment option for glioblastoma. In this study,we show that engineered hAMSC-BMP4 cells reduce theproliferation and migration of glioblastoma and inducedifferentiation of BTICs in vitro and in vivo. Furthermore,a single, cardiac injection of these cells into a mousemodel of glioblastoma significantly prolongs survival.These findings suggest that hAMSCs-BMP4 are a prom-ising novel cell-based therapy for patients with glioblas-toma and potentially other metastatic cancers.
Li et al.
Clin Cancer Res; 20(9) May 1, 2014 Clinical Cancer Research2376
on March 22, 2019. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from

GFP/bioluminescent-hAMSCs into the right striatum(L: 1.34 mm, A: 1.5 mm, D: 3.5 mm; n ¼ 5). Followinginjection, mice in the GFP/bioluminescent-hAMSC groupwere imaged using an IVIS small animal imaging system(Perkin Elmer) at different time periods (7, 14, and 28 dayspostinjection). After 4 weeks, animals were euthanized andperfused with 4% PFA. Brains were extracted, cryo-sec-tioned, and immunostained for human nuclei (Millipore,MAB4383). To quantify tumor area, tumor mass was out-lined based on 40,6-diamidino-2-phenylindole (DAPI)staining and calculated using Image J. To quantify glioblas-toma cell migration, the distance from the tumor margin,determined by DAPI staining, to each human nucleiþ
/DAPIþ/td-tomato� cell was quantified. A blinded observerperformed all counting.
hAMSCs-BMP4 in vivo studiesTo determine the effect of hAMSCs-BMP4 on the malig-
nancy of orthotopic glioblastoma tumors in vivo, 1 � 106
BTIC 276 were suspended in 2 mL PBS and stereotacticallyinjected into the right striatum (L: 1.34 mm, A: 1.5 mm, D:3.5 mm) of immunosuppressed nude mice. Two weekspostinjection, 0.5 � 106 GFP-hAMSCs-Vector (n ¼ 7),GFP-hAMSCs-BMP4 (n ¼ 5), or equal volume of PBS(n ¼ 5, 100 mL) were systemically injected into the leftcardiac ventricle (34). After 2 weeks, the mice brains wereperfused, fully cryo-sectioned at a 10 mm thickness, andimmunostained for GFP, BMP4 (Abcam, ab93939), Ki67(Thermo, RM-9106-s1), Nestin (Abcam, ab5922), Tuj1,GFAP, TNF-a (Abcam, ab6671), VEGF (Abcam, ab46154),and human nuclei. Tumor mass was outlined and cellnumber of positive staining inside the tumor mass wascounted and normalized relative to DAPI inside the tumorbulk. The ratio of Ki67þ/DAPI was used to measure prolif-eration; Nestinþ/DAPI, Tuj1þ/DAPI, and GFAPþ/DAPI tomeasure differentiation; and TNF-aþ/DAPI and VEGFþ
/DAPI tomeasure tumor necrosis and angiogenesis. Immu-nostaining for Nestin was used as a BTICmarker, as this hasbeen validated in several studies (35, 36). To quantifyglioblastoma cell migration, human nuclei antibodies wereused. Tumor mass was outlined by DAPI and the center oftumormass was calculated using Image J. The distance fromthe center of tumor mass to each human nucleiþ/DAPIþ/GFP� cell was quantified based on 110 to 250 cells pergroup. A blinded observer performed all counting. 41, 38,and 41 slides have been analyzed in PBS, hAMSCs-Vector,and hAMSCs-BMP4 groups, respectively. All in vivo proce-dures were approved by the Johns Hopkins UniversityAnimal Care and Use Committee.
Survival studyTo determine the effect of hAMSCs-BMP4 on the survival
of orthotopic glioblastoma tumors-bearingmice in vivo, 0.5� 106 U87 were suspended in 2 mL PBS and stereotacticallyinjected into the right striatum (L: 1.34 mm, A: 1.5 mm, D:3.5 mm) of immunosuppressed nude mice. Ten days post-injection, 0.5� 106 hAMSCs-Vector (n¼ 7), GFP-hAMSCs-BMP4 (n¼5), or equal volumeof PBS (n¼10, 100mL)were
systemically injected into the left cardiac ventricle. Micewere followed for 125 days to monitor survival. A Kaplan–Meier survival analysis was performed with results reportedas the median and mean survival times with a 95% confi-dence interval. The statistical difference between the 3conditions was determined by log-rank analysis.
Statistical analysisResults are reported as mean � SEM. Comparisons were
done using two-way ANOVA for MTS assays, one-wayANOVA (Kruskal–Wallis test follow up an Dunns posttest)for transwell and nanopattern assays, A Kaplan–Meier sur-vival analysis was performed with results reported as themedian survival times with a 95% confidence interval. Thestatistical difference between the 3 conditions was deter-mined by log-rank analysis, and t tests (Mann–Whitney) forother experiments using GraphPad Prism 5 software. Sta-tistical significance was defined as P < 0.05.
ResultshAMSCs-BMP4 decrease migration of BTICs byreducing both migration and migration speed in vitro
Prolific migration of glioblastoma cells and BTICs greatlycontributes to the mortality of the disease. Human MSCshave been shown to have intrinsic tumor suppressive affectsin somemodels (37), and the effects of hAMSCs and BMP4on glioblastoma and BTIC migratory capacity has not yetbeen evaluated. Thus, we investigated whether unmodifiedhAMSCs and BMP4 secreting-hAMSCs (hAMSCs-BMP4)could affect the migratory capabilities of BTICs in vitro.
hAMSCs-BMP4 cells were able to synthesize and releaseBMP4as shownbyWesternblots performedwith cell lysatesand conditionedmedia from hAMSCs-BMP4 and hAMSCs-Vector. The majority of mature BMP4 is extracellular, andthere is no visible mature BMP4 in the hAMSCs-Vector cells(Fig. 1A). As shown in Supplementary Fig. S1A, 1 � 106
hAMSCs-BMP4 cells secrete approximately 74 ng after 3days of culturing.
Using in vitro Boyden chamber transwell assays, theeffect of hAMSCs-Vector, hAMSCs-BMP4, and an exoge-nous 50 ng/mL BMP4 dose on BTIC migration wasassessed (Fig. 1C). Conditioned media from empty vec-tor–infected hAMSCs [hAMSC-Vector-conditioned media(CM)], hAMSCs-BMP4 (hAMSC-BMP4-CM), and BMP4-supplemented media resulted in a 2-fold decrease in thenumber of migrating BTICs (Fig. 1C, P < 0.001). However,there were no significant differences between these 3treatments (P > 0.05). Similar findings were seen whenusing a different BTIC line (BTIC 612; Supplementary Fig.S1B and S1D).
To assess the effects of hAMSCs and BMP4 on BTICmigration speed, a nanopattern chamber was used (Fig.1D). BTIC migration speed decreased when cultured inhAMSC-Vector-CM and hAMSC-BMP4-CM, as well as in theBMP4 treatment group (50ng/mL)byalmost 2-fold for both612 (Fig. 1E,P < 0.01) and276 (Supplementary Fig. S1E andS1F, P < 0.05) BTICs. There was no significant difference inthe ability of hAMSC-Vector-CM, hAMSC-BMP4-CM, and
BMP4-Secreted hAMSCs Repress Glioblastoma In Vivo
www.aacrjournals.org Clin Cancer Res; 20(9) May 1, 2014 2377
on March 22, 2019. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from

BMP4 to decrease the migration speed of BTICs (Fig. 1E, P >0.05).
hAMSCs-BMP4 decrease proliferation of BTICs in vitroAnother key feature underlying the malignant nature of
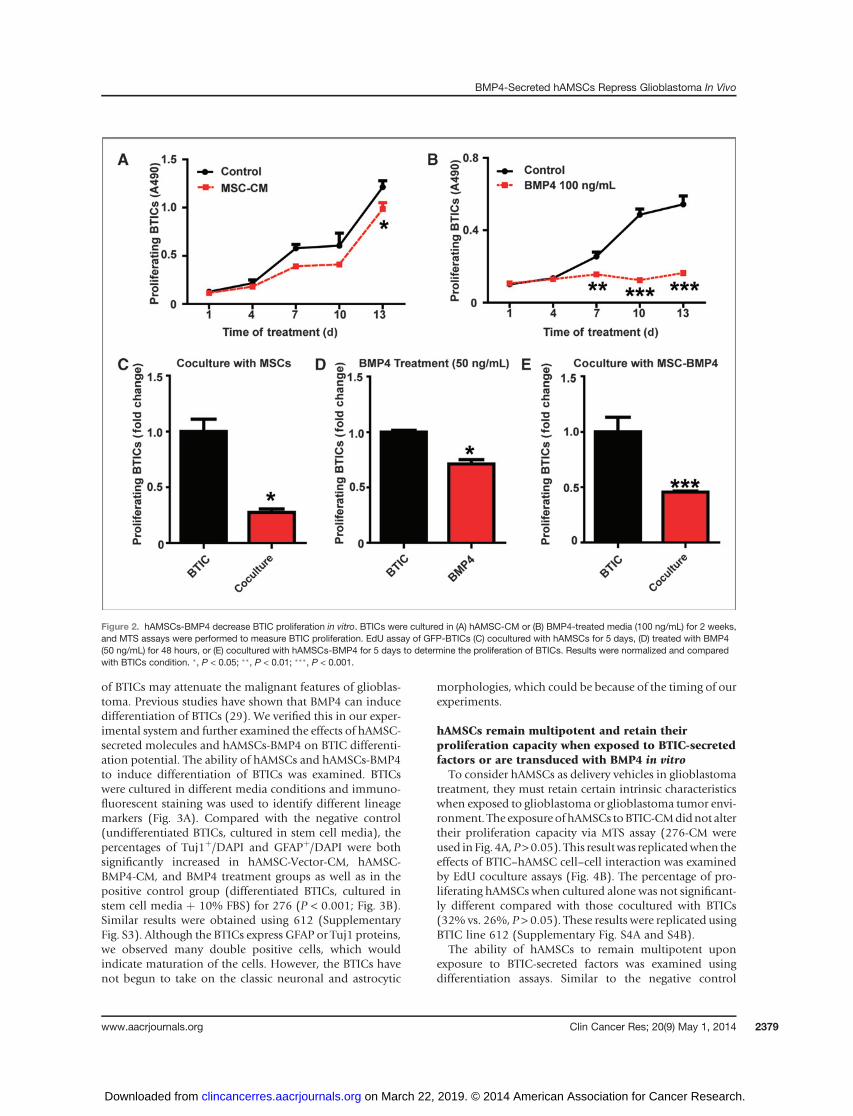
glioblastoma is its capacity for unlimited and rapid prolif-eration. Previous studies have demonstrated the effects ofBMP4 in reducing the proliferative capabilities of BTICs(29), but there are none that report the effects of hAMSCsonBTIC proliferation. Therefore, MTS (to test cell viability andproliferation as a function of time) and EdU assays (toexamine cell proliferation at specific time points) were usedto evaluate the effects of BMP4, hAMSC conditionedmedia,and hAMSC-BMP4 conditioned media on BTICs. Usingthe MTS assay, hAMSC-CM treatment resulted in no statis-tically significant difference in proliferation between thehAMSC-CM and the control groups during the first 10 days(P > 0.05; Fig. 2A). However, hAMSC-CM decreased prolif-erationofboth 276(Fig. 2A) and612BTICs (SupplementaryFig. S2A) significantly at day 13 (P < 0.05). In comparison,exogenous BMP4 (100 ng/mL) demonstrated a significantdecrease in BTIC proliferation after day 7 for 276 (Fig. 2B)
and at day 13 for 612 (Supplementary Fig. S2B, P < 0.01). Toverify these results and further investigate whether cell–cellinteractions between hAMSCs and BTICs (coculture EdUassay) could affect the proliferation of BTICs, we coculturedhAMSCs with BTICs and found the proliferation of276 cells decreased significantly (Fig. 2C andSupplementaryFig. S2C, 3-fold decrease at both day 5 and day 13, P < 0.05).Furthermore, when treated with exogenous BMP4 (50 ng/mL) for 48 hours, proliferation of 276 cells decreasedsignificantly (Fig. 2D, 30% decreased, P < 0.05). BTICproliferation also decreased significantly when coculturedwith hAMSCs-BMP4 (Fig. 2E; for 276, 2-fold decrease at5 days, P < 0.001). Similar effects were seen using the 612BTIC line (Supplementary Fig. S2D and S2E). These experi-ments demonstrate that BMP4 and hAMSCs-BMP4 decreasethe proliferation of BTICs effectively in vitro, and that unal-tered hAMSCs can also decrease the proliferation of BTICsvia secreted proteins and cell–cell contact.
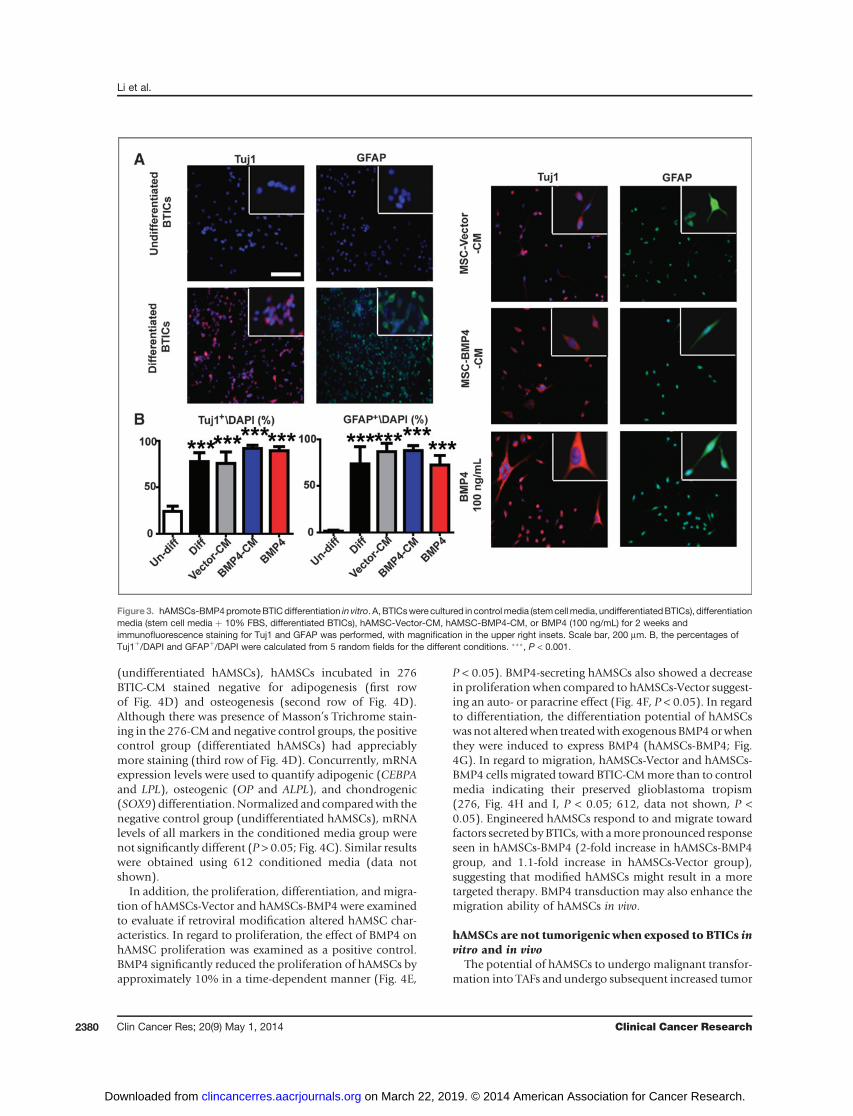
hAMSCs-BMP4 induce differentiation of BTICs in vitroBTICs seem to underlie the ability for glioblastoma
migration and proliferation; inducing the differentiation
Figure 1. hAMSCs-BMP4 decrease migration of BTICs in vitro. A, hAMSCs were infected with BMP4 or vector retroviruses. Western blots were performedusing cell lysates and concentrated media. b-Actin served as a loading control. B, schematic of conditioned media (CM) collection and coculturing methods.C, BTICs were treated with 50 ng/mLBMP4, or were cultured in hAMSC-Vector-CM, hAMSC-BMP4-CM, or control media for 24 hours and a Boydentranswell assay was performed. Results were normalized and compared with the control media condition. D, schematic of cells migrating on a nanopattern.E, nanopattern assay of BTICs cultured in hAMSC-Vector-CM, hAMSC-BMP4-CM, control media, or treated with 50 ng/mL BMP4. ��, P < 0.01;���, P < 0.001.
Li et al.
Clin Cancer Res; 20(9) May 1, 2014 Clinical Cancer Research2378
on March 22, 2019. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
of BTICs may attenuate the malignant features of glioblas-toma. Previous studies have shown that BMP4 can inducedifferentiation of BTICs (29). We verified this in our exper-imental system and further examined the effects of hAMSC-secreted molecules and hAMSCs-BMP4 on BTIC differenti-ation potential. The ability of hAMSCs and hAMSCs-BMP4to induce differentiation of BTICs was examined. BTICswere cultured in different media conditions and immuno-fluorescent staining was used to identify different lineagemarkers (Fig. 3A). Compared with the negative control(undifferentiated BTICs, cultured in stem cell media), thepercentages of Tuj1þ/DAPI and GFAPþ/DAPI were bothsignificantly increased in hAMSC-Vector-CM, hAMSC-BMP4-CM, and BMP4 treatment groups as well as in thepositive control group (differentiated BTICs, cultured instem cell media þ 10% FBS) for 276 (P < 0.001; Fig. 3B).Similar results were obtained using 612 (SupplementaryFig. S3). Although the BTICs express GFAP or Tuj1 proteins,we observed many double positive cells, which wouldindicate maturation of the cells. However, the BTICs havenot begun to take on the classic neuronal and astrocytic
morphologies, which could be because of the timing of ourexperiments.
hAMSCs remain multipotent and retain theirproliferation capacity when exposed to BTIC-secretedfactors or are transduced with BMP4 in vitro
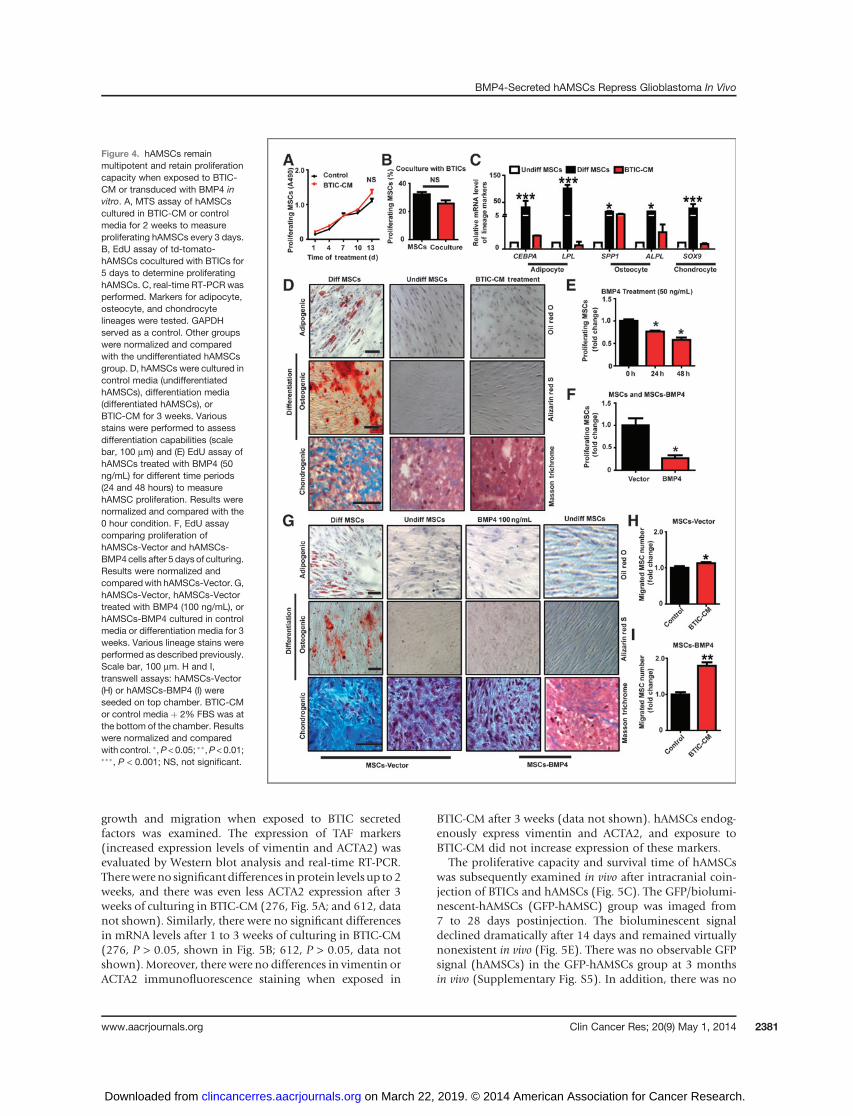
To consider hAMSCs as delivery vehicles in glioblastomatreatment, they must retain certain intrinsic characteristicswhen exposed to glioblastoma or glioblastoma tumor envi-ronment. The exposure of hAMSCs toBTIC-CMdidnot altertheir proliferation capacity via MTS assay (276-CM wereused in Fig. 4A,P>0.05). This resultwas replicatedwhen theeffects of BTIC–hAMSC cell–cell interaction was examinedby EdU coculture assays (Fig. 4B). The percentage of pro-liferating hAMSCs when cultured alone was not significant-ly different compared with those cocultured with BTICs(32% vs. 26%, P > 0.05). These results were replicated usingBTIC line 612 (Supplementary Fig. S4A and S4B).
The ability of hAMSCs to remain multipotent uponexposure to BTIC-secreted factors was examined usingdifferentiation assays. Similar to the negative control
Figure 2. hAMSCs-BMP4 decrease BTIC proliferation in vitro. BTICs were cultured in (A) hAMSC-CM or (B) BMP4-treated media (100 ng/mL) for 2 weeks,and MTS assays were performed to measure BTIC proliferation. EdU assay of GFP-BTICs (C) cocultured with hAMSCs for 5 days, (D) treated with BMP4(50 ng/mL) for 48 hours, or (E) cocultured with hAMSCs-BMP4 for 5 days to determine the proliferation of BTICs. Results were normalized and comparedwith BTICs condition. �, P < 0.05; ��, P < 0.01; ���, P < 0.001.
BMP4-Secreted hAMSCs Repress Glioblastoma In Vivo
www.aacrjournals.org Clin Cancer Res; 20(9) May 1, 2014 2379
on March 22, 2019. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
(undifferentiated hAMSCs), hAMSCs incubated in 276BTIC-CM stained negative for adipogenesis (first rowof Fig. 4D) and osteogenesis (second row of Fig. 4D).Although there was presence of Masson’s Trichrome stain-ing in the 276-CM and negative control groups, the positivecontrol group (differentiated hAMSCs) had appreciablymore staining (third row of Fig. 4D). Concurrently, mRNAexpression levels were used to quantify adipogenic (CEBPAand LPL), osteogenic (OP and ALPL), and chondrogenic(SOX9) differentiation.Normalized and comparedwith thenegative control group (undifferentiated hAMSCs), mRNAlevels of all markers in the conditioned media group werenot significantly different (P > 0.05; Fig. 4C). Similar resultswere obtained using 612 conditioned media (data notshown).
In addition, the proliferation, differentiation, and migra-tion of hAMSCs-Vector and hAMSCs-BMP4 were examinedto evaluate if retroviral modification altered hAMSC char-acteristics. In regard to proliferation, the effect of BMP4 onhAMSC proliferation was examined as a positive control.BMP4 significantly reduced the proliferation of hAMSCs byapproximately 10% in a time-dependent manner (Fig. 4E,
P < 0.05). BMP4-secreting hAMSCs also showed a decreasein proliferationwhen compared to hAMSCs-Vector suggest-ing an auto- or paracrine effect (Fig. 4F, P < 0.05). In regardto differentiation, the differentiation potential of hAMSCswasnot alteredwhen treatedwith exogenousBMP4orwhenthey were induced to express BMP4 (hAMSCs-BMP4; Fig.4G). In regard to migration, hAMSCs-Vector and hAMSCs-BMP4 cells migrated toward BTIC-CMmore than to controlmedia indicating their preserved glioblastoma tropism(276, Fig. 4H and I, P < 0.05; 612, data not shown, P <0.05). Engineered hAMSCs respond to and migrate towardfactors secretedbyBTICs,with amore pronounced responseseen in hAMSCs-BMP4 (2-fold increase in hAMSCs-BMP4group, and 1.1-fold increase in hAMSCs-Vector group),suggesting that modified hAMSCs might result in a moretargeted therapy. BMP4 transduction may also enhance themigration ability of hAMSCs in vivo.
hAMSCs are not tumorigenic when exposed to BTICs invitro and in vivo
The potential of hAMSCs to undergo malignant transfor-mation into TAFs and undergo subsequent increased tumor
Figure3. hAMSCs-BMP4promoteBTICdifferentiation in vitro. A, BTICswere cultured in controlmedia (stemcellmedia, undifferentiatedBTICs), differentiationmedia (stem cell media þ 10% FBS, differentiated BTICs), hAMSC-Vector-CM, hAMSC-BMP4-CM, or BMP4 (100 ng/mL) for 2 weeks andimmunofluorescence staining for Tuj1 and GFAP was performed, with magnification in the upper right insets. Scale bar, 200 mm. B, the percentages ofTuj1þ/DAPI and GFAPþ/DAPI were calculated from 5 random fields for the different conditions. ���, P < 0.001.
Li et al.
Clin Cancer Res; 20(9) May 1, 2014 Clinical Cancer Research2380
on March 22, 2019. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
growth and migration when exposed to BTIC secretedfactors was examined. The expression of TAF markers(increased expression levels of vimentin and ACTA2) wasevaluated by Western blot analysis and real-time RT-PCR.Therewereno significant differences inprotein levels up to2weeks, and there was even less ACTA2 expression after 3weeks of culturing in BTIC-CM (276, Fig. 5A; and 612, datanot shown). Similarly, there were no significant differencesin mRNA levels after 1 to 3 weeks of culturing in BTIC-CM(276, P > 0.05, shown in Fig. 5B; 612, P > 0.05, data notshown).Moreover, there were no differences in vimentin orACTA2 immunofluorescence staining when exposed in
BTIC-CM after 3 weeks (data not shown). hAMSCs endog-enously express vimentin and ACTA2, and exposure toBTIC-CM did not increase expression of these markers.
The proliferative capacity and survival time of hAMSCswas subsequently examined in vivo after intracranial coin-jection of BTICs and hAMSCs (Fig. 5C). The GFP/biolumi-nescent-hAMSCs (GFP-hAMSC) group was imaged from7 to 28 days postinjection. The bioluminescent signaldeclined dramatically after 14 days and remained virtuallynonexistent in vivo (Fig. 5E). There was no observable GFPsignal (hAMSCs) in the GFP-hAMSCs group at 3 monthsin vivo (Supplementary Fig. S5). In addition, there was no
Figure 4. hAMSCs remainmultipotent and retain proliferationcapacity when exposed to BTIC-CM or transduced with BMP4 invitro. A, MTS assay of hAMSCscultured in BTIC-CM or controlmedia for 2 weeks to measureproliferating hAMSCs every 3 days.B, EdU assay of td-tomato-hAMSCs cocultured with BTICs for5 days to determine proliferatinghAMSCs. C, real-time RT-PCR wasperformed. Markers for adipocyte,osteocyte, and chondrocytelineages were tested. GAPDHserved as a control. Other groupswere normalized and comparedwith the undifferentiated hAMSCsgroup. D, hAMSCs were cultured incontrol media (undifferentiatedhAMSCs), differentiation media(differentiated hAMSCs), orBTIC-CM for 3 weeks. Variousstains were performed to assessdifferentiation capabilities (scalebar, 100 mm) and (E) EdU assay ofhAMSCs treated with BMP4 (50ng/mL) for different time periods(24 and 48 hours) to measurehAMSC proliferation. Results werenormalized and compared with the0 hour condition. F, EdU assaycomparing proliferation ofhAMSCs-Vector and hAMSCs-BMP4cells after 5 days of culturing.Results were normalized andcomparedwith hAMSCs-Vector. G,hAMSCs-Vector, hAMSCs-Vectortreated with BMP4 (100 ng/mL), orhAMSCs-BMP4 cultured in controlmedia or differentiation media for 3weeks. Various lineage stains wereperformed as described previously.Scale bar, 100 mm. H and I,transwell assays: hAMSCs-Vector(H) or hAMSCs-BMP4 (I) wereseeded on top chamber. BTIC-CMor control media þ 2% FBS was atthe bottom of the chamber. Resultswere normalized and comparedwith control. �,P < 0.05; ��,P < 0.01;���, P < 0.001; NS, not significant.
BMP4-Secreted hAMSCs Repress Glioblastoma In Vivo
www.aacrjournals.org Clin Cancer Res; 20(9) May 1, 2014 2381
on March 22, 2019. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from

observable td-tomato (hAMSCs) in the coinjection group(data not shown). Even after staining for GFP (data notshown) and human nuclei, colocalization of human nucleiand GFP was only found in the GFP-276 and coinjectiongroups and no signals in GFP-hAMSCs or PBS groups (datanot shown). After 4 weeks, the coinjection group had asmallermean tumor area of 135,700 mm2 as compared withGFP-BTIC group, with a mean tumor area of 209,800 mm2
(P ¼ 0.0189; Fig. 5D). When measuring individual celldistance from the tumor margin, there was no significantdifference between the BTICs-only and coinjection groups(P ¼ 0.3442; Supplementary Fig. S5C). In addition, asshown in Fig. 5F, tumors in the BTIC group seem largerthan the coinjection group, suggesting that the presence of
additional hAMSCs does not contribute to rampant tumorprogression in vivo.
hAMSCs-BMP4 increase the median survival time ofglioblastoma bearing mice, drive differentiation, anddecrease proliferation and migration of glioblastomacells in vivo
To examine the effects of hAMSCs-BMP4onglioblastomacell-proliferative capacity and migratory ability, and onstem-ness of BTICs in vivo, a mouse model of glioblastomawas created as described previously by our group (38). Inthis model, hAMSCs-Vector (n ¼ 7), hAMSCs-BMP4 (n ¼5), or PBS (n ¼ 5) were administered via cardiac injectionafter glioblastoma tumor formation (Fig. 6F). As shown
Figure 5. hAMSCs are not tumorigenic and do not transform into TAFs in vitro or in vivo. A and B, hAMSCs were cultured in BTIC-CM or control mediafor 1 to 3 weeks and (A) Western blots (b-actin served as a control) and (B) real-time RT-PCR (GAPDH served as a control) were performed to quantifyTAF markers (vimentin and ACTA2). C, schematic of the coinjection experiment where PBS, GFP-BTICs, GFP/bioluminescent-hAMSCs (GFP-hAMSCs), orGFP-BTICs mixed with td-tomato-hAMSCs (td-hAMSCs) were injected into mice and sacrificed 4 weeks later. D, quantification of mean tumor area ofthe GFP-BTIC and coinjection groups using DAPI staining. The coinjection group had a smaller mean tumor area of 135,700 mm2 as compared with theGFP-BTIC group, with a mean tumor area of 209,800 mm2 (P ¼ 0.0189). �, P < 0.05. E, live animal imaging of the GFP-hAMSCs condition. At 14 daysafter GFP-hAMSCs injection, the hAMSC signal drastically decreases. Each mouse brain represents the counts of bioluminescent signal at each time point.F, DAPI and human nuclei stain for GFP-BTICs and coinjection groups (n ¼ 5). Larger tumors were only seen in the GFP-BTICs condition comparedwith the coinjection condition. Brain sections and tumor mass are outlined. Scale bar, 200 mm. NS, not significant.
Li et al.
Clin Cancer Res; 20(9) May 1, 2014 Clinical Cancer Research2382
on March 22, 2019. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from

Figure 6. hAMSCs-BMP4 increase the median survival time of glioblastoma-bearing mice, drive differentiation, and decrease proliferation andmigration of glioblastoma cells in vivo. A, immunoreactivity for GFP and BMP4 to test the expression of BMP4. Scale bars, 200 mm. B, GFP-hAMSCs-BMP4cells were seen near satellite Nestinþ cells away from the main tumor bulk. Scale bars, 200 mm. C, representative pictures and quantification of GFPand Ki67 staining to test the proliferation of glioblastoma cells. Scale bars, 200 mm. D, representative pictures and quantification of GFP, nestin, GFAP,and Tuj1 staining to test the differentiation of BTICs. Arrowheads in the GFP-hAMSC-BMP4 GFAP staining correspond to magnified insets of GFP-hAMSC-BMP4 and GFAPþ cells at the tumor center, and a GFAPþ cell with mature astrocytic morphology at the tumor periphery. Magnified pictures areshownon the left. Scale bars, 200 mm.E, representative pictures (right hemisphere) and quantification ofmigratory glioblastomacells. The average distance ofmigrated glioblastoma cells, identified as human nucleiþ/DAPIþ/GFP� cells outside tumor bulk, from the center of tumor mass (outlined) was measured.Scale bars, 200 mm. �, P < 0.05; ��, P < 0.01; ���, P < 0.001. F, schematic of the in vivo experiment for which immunofluorescence staining wasperformed in A to E: BTIC culture 276 was intracranially injected into 6- to 8-week-old nude mice. At 4 weeks after injection, GFP-hAMSCs-Vector (n ¼ 7),GFP-hAMSCs-BMP4 (n¼ 5), or equal volumes of PBS (n¼ 5) were injected intracardially. Mice were sacrificed 2 weeks later. G, U87 cells were intracraniallyinjected into 6- to 8-week-old nude mice. Ten days after injection, GFP-hAMSCs-Vector (n ¼ 7), GFP-hAMSCs-BMP4 (n ¼ 5), or equal volumes ofPBS (n¼ 10) were injected intracardially. Mice were followed for 125 days to monitor survival. Kaplan–Meier survival analysis resulted in the median survivalof mice treated with hAMSCs-BMP4 (undefined) being significantly greater than that of mice treated with hAMSCs-Vector (P ¼ 0.01; 76 days) andcontrol mice (P ¼ 0.002; 52 days), with no significant difference between the PBS and hAMSCs-Vector groups (P ¼ 0.09).
BMP4-Secreted hAMSCs Repress Glioblastoma In Vivo
www.aacrjournals.org Clin Cancer Res; 20(9) May 1, 2014 2383
on March 22, 2019. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from

in Fig. 6A, therewas onlyBMP4(human specific antibodies)seen in mice injected with hAMSCs-BMP4. Subsequently,GFP staining confirmed the homing of the hAMSCs-Vectorand hAMSCs-BMP4 groups to the tumor bulk. As shownin Fig. 6A, C, and D, hAMSCs-Vector and hAMSCs-BMP4migrated to the tumor bulk (defined by DAPI density).Interestingly, in only the hAMSCs-BMP4 group, GFP signalswere found around migratory BTICs (defined by Nestinþ
cells not part of the tumor bulk; Fig. 6B). Human-specificKi67 stainingwas used to assess proliferation, and no Ki67þ
cells were observed colocalizing with GFPþ hAMSCs (Fig.6C). To quantify the effect of hAMSCs-Vector and hAMSCs-BMP4 on glioblastoma cell proliferation, Ki67þ cells werenormalized with corresponding DAPIþ cells in the tumormass. Therewas a 2-fold decrease in the ratio of Ki67þ/DAPIin the hAMSCs-BMP4 group compared with both the PBSgroup and hAMSCs-Vector group (Fig. 6C, P < 0.05). Inaddition, immunofluorescence staining for TNF-a andVEGFwere performed to investigate characteristics ofmalig-nant tumors (TNF-a is a marker for necrosis, and VEGF is aproangiogenicmolecule; TNF-a andVEGF secretion are alsoknownassociatedwith TAFs). As seen in Supplementary Fig.S6, there were decreased TNF-a and VEGF staining in thehAMSCs-Vector and hAMSCs-BMP4 groups (2-fold ofdecrease, P < 0.05)
The ability of hAMSCs-Vector and hAMSCs-BMP4 toinduce differentiation of BTICs in vivo were evaluated bystaining cells for Nestin, GFAP, and Tuj1. There was anincreased number of Nestinþ cells in the PBS group, and anincreased number ofGFAPþ and Tuj1þ cells in the hAMSCs-Vector and hAMSCs-BMP4 groups (P < 0.05; Fig. 6D). Wealso observed that hAMSCs-BMP4 can decrease the numberof Nestinþ cells compared with the hAMSCs-Vector group.
In addition, to determine if hAMSCs-Vector andhAMSCs-BMP4 can affect the migration of glioblastomacells in vivo, the tumor bulk was outlined utilizing DAPIstaining. The average distance of glioblastoma cells(human nucleiþ/DAPIþ/GFP� cells) that migrated fromthe center of tumor bulk was calculated based on humannuclei staining (Fig. 6E). As shown in Fig. 6E, hAMSCs-Vector and hAMSCs-BMP4 both inhibited the migratoryability of glioblastoma cells significantly (P < 0.001). Inaddition, as compared with the hAMSCs-Vector group,hAMSCs-BMP4 significantly decreased the migration ofglioblastomas (Fig. 6E, P < 0.001).
To investigate if hAMSCs-Vector and hAMSCs-BMP4 canaffect the survival of glioblastoma bearing mice, 0.5 � 106
U87 cells were stereotactically injected into immunosup-pressed nude mice. Ten days postinjection, 0.5 � 106
hAMSCs-Vector (n ¼ 7), GFP-hAMSCs-BMP4 (n ¼ 5), orequal volume of PBS (100 mL, n ¼ 10) were systemicallyinjected into the left cardiac ventricle. Mice were followedfor 125 days to monitor survival. As shown in Fig. 6G, themedian survival ofmice treatedwith hAMSCs-BMP4 (unde-fined) was significantly greater than that of mice treatedwith hAMSCs-Vector (P ¼ 0.01; 76 days) and control mice(P ¼ 0.002; 52 days). There was no significant differencebetween the PBS and hAMSCs-Vector groups (P ¼ 0.09).
DiscussionGlioblastoma is the most common and aggressive malig-
nant primary intracranial neoplasm in adults, with a medi-an survival of approximately 14.6 months despite combi-natorial treatments of surgical resection, chemotherapy,and radiotherapy (39). Glioblastoma has heterogeneousgenetic alterations in pathways associated with prolifera-tion, survival, invasion, and angiogenesis. Glioblastomacells are known to use white matter tracts and microvascu-lature basement membranes to migrate long distances,making complete surgical resection of the tumor difficult,almost inevitably leading to recurrence (40). The well-known glioblastoma molecular subtype classifications areproneural, neural, classical, and mesenchymal (41). The 2primary glioblastoma cell lines used in this study, 276 and612, belong to mesenchymal and proneural subtypes,respectively. hAMSCs-BMP4 treatment was able to attenu-ate malignant tumor characteristics of 2 different subtypesof glioblastoma in this study, reinforcing the therapeuticeffect of hAMSCs-BMP4 in potential future clinical trials.
Although human MSCs have been manipulated toexpress a wide variety of anticancer therapeutic factorsbecause of their tropism toward inflammation and tumorcells (17, 42–44), the effects of hAMSCs on glioblastomaand BTICs have not been fully described. This study was thefirst to find that hAMSCs inhibit proliferation and inducedifferentiation of BTICs, as well as confirm that hAMSCsdecrease the migration of BTICs in vitro. Furthermore,hAMSCs can induce differentiation, reduce proliferationand migration, and may even diminish angiogenesis ofglioblastoma in vivowhen injected intracardially. However,when intracranially coinjected with BTICs, we did notobserve a difference in the extent of cell migration in vivo,but we did find a reduction in tumor size. These resultsindicate that hAMSCs can have intrinsic antitumor effectsand are promising for glioblastoma treatment.However, wefound that unmodified hAMSCs have a limited effect oninhibiting glioblastoma cell proliferation and survival, andthey can only track glioblastoma tumor bulk but lack theability to home to migratory glioblastoma cells in vivo.Enhancement of the hAMSCs by engineering them to deliv-er specific agents may augment their anticancer effects (37).
BMPs are known to play a role in the differentiation ofadult neural stem cells into different mature cell types (45,46). Recently, BMP4 has been shown to reduce glioblasto-ma tumor burden in vivo and improve survival in a mousemodel of glioblastoma by potentially reducing the frequen-cy of symmetric cell divisions or by blocking proliferationand inducing differentiation of BTICs (29). One of thechallenges to effective treatment of glioblastoma is thetargeting of BTICs, which seem to underlie the ability ofthe tumor to recur (2). BMP4 is an ideal therapeutic can-didate because of its affect on BTICs; however, optimizingits delivery is critical (47). Aside from local delivery withpolyacrylic beads (29), there are no reports describing stemcell-based vehicles for BMP4delivery. The goal of the in vitroexperiments was to demonstrate the potential therapeuticeffects hAMSCs-BMP4have on glioblastoma.We found that
Li et al.
Clin Cancer Res; 20(9) May 1, 2014 Clinical Cancer Research2384
on March 22, 2019. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from

BMP4 treatment, whether exogenously administered orreleased by genetically modified hAMSCs, can decrease theproliferation of BTICs and make BTICs commit to maturelineages in vitro and in vivo. Most of these effects are alsoobserved with unmodified hAMSCs, but to a lesser extent invivo. Although there were no significant differences betweenthehAMSCs-Vector andhAMSCs-BMP4 treatment groups inregard to migration, proliferation, and differentiations invitro, differences were noted in vivo with regard to glioblas-toma proliferation, nestin expression, and glioblastomamigration (Table 1). Interestingly, hAMSCs-BMP4andexog-enous BMP4 can reduce migration and migration speed ofBTICs, unlike the effect of BMP4 on other types of cancers(24–28). In addition, hAMSCs-BMP4 not only display tro-pism toward glioblastoma tumorbulk, but can alsohome tomigratory glioblastoma cells in vivo. Most importantly, asingle, cardiac injection of 1 million hAMSCs-BMP4 signif-icantly increases survival of glioblastoma-bearingmice com-pared with the hAMSCs-Vector and PBS treatments. Com-mercial glioblastoma cell line U87 was used for the survivalstudy because it is well-established and commonly used inglioblastoma survival studies and has been shown to beextremely aggressive with a low survival rate,making it idealto study survival in a murine glioblastoma model (48–50).Future studies would be interested to use different subtypesof patient-derived BTICs, including proneural, mesenchy-mal, classical, and neural, to perform survival studies and toinvestigate themolecularmechanisms behind the therapeu-tic effect of hAMSCs-BMP4. These studies will be a prom-ising step toward personalized glioblastoma therapy.The present literature raises several additional concerns.
The effects of cancer cells on the proliferative capacity andmalignant potential of human MSCs is a critical consider-ation for its potential utility in clinical trials (9–11, 14). It hasalso been proposed that cancer cells may be able to induceMSCs to form TAFs, which can then support and stimulatetumor growth andmigration aswell as promote amalignantphenotype (18–20). We found that hAMSC proliferationdoes not increase in response to BTIC-secreted factors orcoculturing with BTICs in vitro. When cultured in condi-tioned media from BTICs in vitro, hAMSCs did not upregu-
late their expressionoffibroblastmarkers, suggesting that theBTIC–hAMSC interaction does not foster the adoption of aTAF phenotype. In vivo, hAMSCswere not detectable 14 daysafter intracranial injection, suggesting that they are function-al for only a short window of time. This has both advantagesand disadvantages. The advantage of having this window oftime is minimization of potential deleterious effects of celltherapy, including oncogenesis, tumor induction, and neo-vascularization. The disadvantages includeneed for repeatedcell injections, similar to chemotherapy. Despite the shortwindow of time, however, a single cardiac injection of 1million hAMSCs-BMP4 was able to significantly prolongsurvival of glioblastoma-bearing mice. When injected aloneand with BTICs, hAMSCs also did not demonstrate anincreased proliferative capacity and no hAMSCs remainedin the brain 3 months after injection. Furthermore, in ourmodel of glioblastoma, hAMSCs delivered to establishedtumors did not demonstrate colocalization with Ki67 andmarkers of tumor growth (TNF-a and VEGF; refs. 51 and52)were attenuated in the presence of hAMSCs-BMP4. Theseresults suggest that hAMSCs are neither tumor-supportivenor tumorigenic. In addition, we demonstrated that BTICsand BMP4 do not alter the stem cell properties, tumortropism, or induce differentiation of hAMSCs in our experi-ments. Moreover, these cells retained their stem cell–likecharacteristics and tumor tropism in vivo, which is funda-mental to their utility as a vehicle for antitumor agents. Tworecent studies found that hAMSCs promoted growth andangiogenesis of glioblastoma cells in vivo (53). However, wediscovered that hAMSCs-BMP4 decrease the proliferation ofglioblastoma cells, and hAMSCs-Vector and hAMSCs-BMP4decrease the expression of the angiogenesis markers, VEGFand CD31 (data not shown) in vivo, although there was notsufficient quality and number of cells immunoreactive toCD31 to allow for quantification and statistical analyses.hAMSCs may therefore contribute to better outcomes in amultifactorial fashion, of which, angiogenesis is one com-ponent. Other components include tumor proliferation,migration, and differentiation, among others. Differencesbetween our tumor models and those of Akimoto andcolleagues in cell type,MSC source, injection site, and timing
Table 1. Summary of hAMSCs and hAMSCs-BMP4 effects on BTICs
Control condition vs.
BTIC behaviorhAMSCtreatment
hAMSC-BMP4treatment
hAMSC treatment vs.hAMSC-BMP4 treatment
In vitro Proliferation a a N.S.Differentiation a a N.S.Migration/migration speed a a N.S.
In vivo Proliferation a a a
Differentiation a a a
Migration a a a
Survival N.S. a a
aRepresents statistical significance, P < 0.05; N.S. represents no significance, P > 0.05.
BMP4-Secreted hAMSCs Repress Glioblastoma In Vivo
www.aacrjournals.org Clin Cancer Res; 20(9) May 1, 2014 2385
on March 22, 2019. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from

of MSC injections may account for these contrasting results.Notably, this prior study cotransplantedMSCs and glioblas-toma cells subcutaneously. Our experiments with intracra-nial injections were meant to be a proof the principle thatensured delivery of hAMSCs to the tumormass and evaluatethe safety of intracranial injection. We also delivered MSCssystemically through a single, cardiac injection to an estab-lished intracranial glioblastoma tumor mass, which isthought to be a more accurate model for glioblastoma inhuman patients.
In conclusion, our results demonstrate the extraordinaryability of hAMSCs-BMP4 to decrease the proliferative andmigratory capacity of glioblastoma cells, induce differenti-ation of BTICs in vitro and in vivo, and ultimately prolongsurvival glioblastoma-bearing mice with a single, cardiacinjection of one million cells. In addition, our findingsdemonstrate the safety and efficacy of engineered hAMSCsin delivering targeted therapy in a mouse model of glio-blastoma. Both unmodified hAMSCs and hAMSCs-BMP4do not undergomalignant transformationwhen exposed toglioblastoma cells, and do not support tumor growth.Further advances with hAMSCs-BMP4 to create a moresophisticated delivery system may include engineeringthese cells to control the secretion of BMP4. TGF-b andothermarkers specific to glioblastoma cells within the brain(54, 55) may serve as molecular switches to induce thecontextually specific release of BMP4. Basedonourfindings,we are optimistic that engineered hAMSC-based anticancertherapies will continue to demonstrate their promise inclinical trials for glioblastoma. In the future, we predict thisstem cell–based approach will have wide-reaching poten-tial, including autologous hAMSCs from adipose tissue andthe treatment of other primary and secondary brain cancers.
Disclosure of Potential Conflicts of InterestNo potential conflicts of interest were disclosed.
Authors' ContributionsConception and design: Q. Li, S.J. Salas, D.A. Chesler, C.L. Smith,H. Guerrero-Cazares, A. Quinones-HinojosaDevelopment of methodology: S.J. Salas, M. Zhu, C. Aprhys, K.L. Chai-chana, D.A. Chesler, C.L. Smith, A. Levchenko, A. Quinones-HinojosaAcquisitionofdata (provided animals, acquired andmanagedpatients,provided facilities, etc.):Q. Li, O.Wijesekera, S.J. Salas, J.Y. Wang, M. Zhu,K.L. Chaichana, H. Zhang, C.L. Smith, A. Levchenko, A. Quinones-HinojosaAnalysis and interpretation of data (e.g., statistical analysis, biosta-tistics, computational analysis):Q. Li, O. Wijesekera, S.J. Salas, J.Y. Wang,M. Zhu, K.L. Chaichana, D.A. Chesler, C.L. Smith, H. Guerrero-Cazares,A. Levchenko, A. Quinones-HinojosaWriting, review, and/or revision of themanuscript:Q. Li, O.Wijesekera,J.Y. Wang, K.L. Chaichana, D.A. Chesler, C.L. Smith, H. Guerrero-Cazares,A. Levchenko, A. Quinones-HinojosaAdministrative, technical, or material support (i.e., reporting or orga-nizingdata, constructingdatabases):Q.Li, S.J. Salas,M.Zhu, A. Levchenko,A. Quinones-HinojosaStudy supervision: S.J. Salas, H. Guerrero-Cazares, A. Levchenko, A. Qui-nones-Hinojosa
AcknowledgmentsWe thankDr. K.H.William Lau from Loma LindaUniversity for the gift of
the BMP2/4 DNA plasmid, Bryant Beltran for designing the MATLABprogram to track cells using the nanopattern assay, and Dr. K. Aldape andDr. K.Wani from theUniversity of TexasMDAndersonCancer Center for theglioblastoma cell classification.
Grant SupportThis research was supported by the National Institutes of Health (RO1,
NS070024) and the Maryland Stem Cell Research Fund (A. Quinones-Hinojosa).
The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely to indicatethis fact.
Received May 22, 2013; revised February 7, 2014; accepted February 12,2014; published online May 1, 2014.
References1. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn
MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomidefor glioblastoma. N Engl J Med 2005;352:987–96.
2. Quinones-Hinojosa A, ChaichanaK. The human subventricular zone: asource of new cells and a potential source of brain tumors. Exp Neurol2007;205:313–24.
3. Bleau AM, Hambardzumyan D, Ozawa T, Fomchenko EI, Huse JT,Brennan CW, et al. PTEN/PI3K/Akt pathway regulates the side pop-ulation phenotype and ABCG2 activity in glioma tumor stem-like cells.Cell Stem Cell 2009;4:226–35.
4. Eramo A, Ricci-Vitiani L, Zeuner A, Pallini R, Lotti F, Sette G, et al.Chemotherapy resistance of glioblastoma stem cells. Cell Death Differ2006;13:1238–41.
5. Murat A, Migliavacca E, Gorlia T, LambivWL, Shay T, HamouMF, et al.Stem cell-related "self-renewal" signature and high epidermal growthfactor receptor expression associated with resistance to concomitantchemoradiotherapy in glioblastoma. J Clin Oncol 2008;26:3015–24.
6. Sakariassen PO, Immervoll H, Chekenya M. Cancer stem cells asmediators of treatment resistance in brain tumors: status and contro-versies. Neoplasia 2007;9:882–92.
7. Strioga M, Viswanathan S, Darinskas A, Slaby O, Michalek J. Same ornot the same? Comparison of adipose tissue-derived versus bonemarrow-derivedmesenchymal stem and stromal cells. StemCells Dev2012;21:2724–52.
8. Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F,Krause D, et al. Minimal criteria for defining multipotent mesenchymal
stromal cells. The International Society for Cellular Therapy positionstatement. Cytotherapy 2006;8:315–7.
9. Lamfers M, Idema S, van Milligen F, Schouten T, van der Valk P,Vandertop P, et al. Homing properties of adipose-derived stem cells tointracerebral glioma and the effects of adenovirus infection. CancerLett 2009;274:78–87.
10. Kosztowski T, Zaidi HA, Quinones-Hinojosa A. Applications of neuraland mesenchymal stem cells in the treatment of gliomas. Expert RevAnticancer Ther 2009;9:597–612.
11. Lee DH, Ahn Y, Kim SU, Wang KC, Cho BK, Phi JH, et al. Targeting ratbrainstem glioma using human neural stem cells and human mesen-chymal stem cells. Clin Cancer Res 2009;15:4925–34.
12. Xu F, Zhu JH. Stem cells tropism for malignant gliomas. Neurosci Bull2007;23:363–9.
13. Augello A, Kurth TB, DeBari C.Mesenchymal stemcells: a perspectivefrom in vitro cultures to in vivo migration and niches. Eur Cells Mater2010;20:121–33.
14. Momin EN, Mohyeldin A, Zaidi HA, Vela G, Quinones-Hinojosa A.Mesenchymal stem cells: new approaches for the treatment of neu-rological diseases. Curr Stem Cell Res Ther 2010;5:326–44.
15. Momin EN, Vela G, Zaidi HA, Quinones-Hinojosa A. The oncogenicpotential of mesenchymal stem cells in the treatment of cancer:directions for future research. Curr Immunol Rev 2010;6:137–48.
16. Kucerova L, Matuskova M, Hlubinova K, Altanerova V, Altaner C.Tumor cell behaviour modulation by mesenchymal stromal cells. MolCancer 2010;9:129.
Clin Cancer Res; 20(9) May 1, 2014 Clinical Cancer Research2386
Li et al.
on March 22, 2019. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from

17. Nakamura K, Ito Y, Kawano Y, Kurozumi K, Kobune M, Tsuda H, et al.Antitumor effect of genetically engineeredmesenchymal stemcells in arat glioma model. Gene Ther 2004;11:1155–64.
18. Spaeth EL, Dembinski JL, Sasser AK, Watson K, Klopp A, Hall B, et al.Mesenchymal stem cell transition to tumor-associated fibroblastscontributes to fibrovascular network expansion and tumor progres-sion. PLoS ONE 2009;4:e4992.
19. Mishra PJ, Humeniuk R, Medina DJ, Alexe G, Mesirov JP, Ganesan S,et al. Carcinoma-associated fibroblast-like differentiation of humanmesenchymal stem cells. Cancer Res 2008;68:4331–9.
20. Hall B, Dembinski J, Sasser AK, Studeny M, Andreeff M, Marini F.Mesenchymal stem cells in cancer: tumor-associated fibroblasts andcell-based delivery vehicles. Int J Hematol 2007;86:8–16.
21. Pendleton C, Li Q, Chesler DA, Yuan K, Guerrero-Cazares H, Qui-nones-Hinojosa A. Mesenchymal stem cells derived from adiposetissue vs bone marrow: in vitro comparison of their tropism towardsgliomas. PLoS ONE 2013;8:e58198.
22. Lee HK, Finniss S, Cazacu S, Bucris E, Ziv-Av A, Xiang C, et al.Mesenchymal stem cells deliver synthetic microRNAmimics to gliomacells and glioma stem cells and inhibit their cell migration and self-renewal. Oncotarget 2013;4:346–61.
23. Altanerova V, Cihova M, Babic M, Rychly B, Ondicova K, Mravec B,et al. Human adipose tissue-derived mesenchymal stem cellsexpressing yeast cytosinedeaminase::uracil phosphoribosyltrans-ferase inhibit intracerebral rat glioblastoma. Int J Cancer 2012;130:2455–63.
24. Chiu CY, Kuo KK, Kuo TL, Lee KT, Cheng KH. The activation of MEK/ERK signaling pathway by bone morphogenetic protein 4 to increasehepatocellular carcinoma cell proliferation and migration. Mol CancerRes 2012;10:415–27.
25. Guo D, Huang J, Gong J. Bone morphogenetic protein 4 (BMP4) isrequired for migration and invasion of breast cancer. Mol Cell Biochem2012;363:179–90.
26. Deng H, Makizumi R, Ravikumar TS, Dong H, YangW, YangWL. Bonemorphogenetic protein-4 is overexpressed in colonic adenocarcino-mas and promotes migration and invasion of HCT116 cells. Exp CellRes 2007;313:1033–44.
27. Virtanen S, Alarmo EL, Sandstrom S, Ampuja M, Kallioniemi A. Bonemorphogenetic protein-4 and -5 in pancreatic cancer–novel bidirec-tional players. Exp Cell Res 2011;317:2136–46.
28. Braig S, Mueller DW, Rothhammer T, Bosserhoff AK. MicroRNA miR-196a is a central regulator of HOX-B7 and BMP4 expression inmalignant melanoma. Cell Mol Life Sci 2010;67:3535–48.
29. Piccirillo SG, Reynolds BA, Zanetti N, Lamorte G, Binda E, Broggi G,et al. Bone morphogenetic proteins inhibit the tumorigenic potential ofhuman brain tumour–initiating cells. Nature 2006;444:761–5.
30. Chaichana KL, Guerrero-Cazares H, Capilla-Gonzalez V, Zamora-Berridi G, Achanta P, Gonzalez-Perez O, et al. Intra-operativelyobtained human tissue: protocols and techniques for the study ofneural stem cells. J Neurosci Methods 2009;180:116–25.
31. Guerrero-Cazares H, Chaichana KL, Quinones-Hinojosa A. Neuro-sphere culture and human organotypic model to evaluate brain tumorstem cells. Methods Mol Biol 2009;568:73–83.
32. Colman H, Zhang L, Sulman EP, McDonald JM, Shooshtari NL, RiveraA, et al. Amultigene predictor of outcome inglioblastoma.NeuroOncol2010;12:49–57.
33. Peng H, Chen ST, Wergedal JE, Polo JM, Yee JK, Lau KH, et al.Development of an MFG-based retroviral vector system for secretionof high levels of functionally active human BMP4. Mol Ther 2001;4:95–104.
34. Kang Y. Analysis of cancer stem cell metastasis in xenograft animalmodels. Methods Mol Biol 2009;568:7–19.
35. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al.Identification of human brain tumour initiating cells. Nature 2004;432:396–401.
36. Tomita T, Akimoto J, Haraoka J, Kudo M. Clinicopathological signif-icance of expression of nestin, a neural stem/progenitor cell marker, inhuman glioma tissue. Brain Tumor Pathol 2013 Nov 8. [Epub ahead ofprint]
37. Shah K. Mesenchymal stem cells engineered for cancer therapy. AdvDrug Deliv Rev 2012;64:739–48.
38. Garzon-Muvdi T, Schiapparelli P, ap Rhys C, Guerrero-Cazares H,Smith C, Kim DH, et al. Regulation of brain tumor dispersal by NKCC1through a novel role in focal adhesion regulation. PLoS Biol 2012;10:e1001320.
39. Fuller GN. The WHO classification of tumours of the central nervoussystem, 4th edition. Arch Pathol Lab Med 2008;132:906.
40. Erpolat OP, Akmansu M, Goksel F, Bora H, Yaman E, Buyukberber S.Outcome of newly diagnosed glioblastoma patients treated by radio-therapy plus concomitant and adjuvant temozolomide: a long-termanalysis. Tumori 2009;95:191–7.
41. VerhaakRG,HoadleyKA,PurdomE,WangV,Qi Y,WilkersonMD, et al.Integrated genomic analysis identifies clinically relevant subtypes ofglioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR,and NF1. Cancer Cell 2010;17:98–110.
42. Nakamizo A, Marini F, Amano T, Khan A, Studeny M, Gumin J, et al.Human bone marrow-derived mesenchymal stem cells in the treat-ment of gliomas. Cancer Res 2005;65:3307–18.
43. Sasportas LS, Kasmieh R, Wakimoto H, Hingtgen S, van de Water JA,Mohapatra G, et al. Assessment of therapeutic efficacy and fate ofengineered human mesenchymal stem cells for cancer therapy. ProcNatl Acad Sci U S A 2009;106:4822–7.
44. Alieva M, Bago JR, Aguilar E, Soler-Botija C, Vila OF, Molet J, et al.Glioblastoma therapy with cytotoxic mesenchymal stromal cells opti-mized by bioluminescence imaging of tumor and therapeutic cellresponse. PLoS ONE 2012;7:e35148.
45. Lim DA, Tramontin AD, Trevejo JM, Herrera DG, Garcia-Verdugo JM,Alvarez-Buylla A. Noggin antagonizes BMP signaling to create a nichefor adult neurogenesis. Neuron 2000;28:713–26.
46. Panchision DM, Pickel JM, Studer L, Lee SH, Turner PA, Hazel TG,et al. Sequential actions of BMP receptors control neural precursor cellproduction and fate. Genes Dev 2001;15:2094–110.
47. Westphal M, Lamszus K. The neurobiology of gliomas: from cellbiology to the development of therapeutic approaches. Nat RevNeurosci 2011;12:495–508.
48. Martuza RL,Malick A,Markert JM,Ruffner KL, CoenDM. Experimentaltherapy of human glioma by means of a genetically engineered virusmutant. Science 1991;252:854–6.
49. Menon LG, Kelly K, Yang HW, Kim SK, Black PM, Carroll RS. Humanbonemarrow-derivedmesenchymal stromal cells expressing S-TRAILas a cellular delivery vehicle for human glioma therapy. Stem Cells2009;27:2320–30.
50. Santra M, Zhang X, Santra S, Jiang F, Chopp M. Ectopic double cortingene expression suppresses themalignant phenotype in glioblastomacells. Cancer Res 2006;66:11726–35.
51. van Horssen R, Ten Hagen TL, Eggermont AM. TNF-a in cancertreatment: molecular insights, antitumor effects, and clinical utility.Oncologist 2006;11:397–408.
52. Ellis LM, Hicklin DJ. VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat Rev Cancer 2008;8:579–91.
53. Akimoto K, Kimura K, NaganoM, TakanoS, To'a Salazar G, YamashitaT, et al. Umbilical cord blood-derived mesenchymal stem cells inhibit,but adipose tissue-derived mesenchymal stem cells promote, glio-blastoma multiforme proliferation. Stem Cells Dev 2013;22:1370–86.
54. Penuelas S, Anido J, Prieto-Sanchez RM, Folch G, Barba I, Cuartas I,et al. TGF-b increases glioma-initiating cell self-renewal through theinduction of LIF in human glioblastoma. Cancer Cell 2009;15:315–27.
55. WuA,Wei J, Kong LY,Wang Y, PriebeW, QiaoW, et al. Glioma cancerstem cells induce immunosuppressivemacrophages/microglia. NeuroOncol 2010;12:1113–25.
www.aacrjournals.org Clin Cancer Res; 20(9) May 1, 2014 2387
BMP4-Secreted hAMSCs Repress Glioblastoma In Vivo
on March 22, 2019. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from

2014;20:2375-2387. Clin Cancer Res Qian Li, Olindi Wijesekera, Sussan J. Salas, et al. SurvivalBMP4 Are Nononcogenic, Suppress Brain Cancer, and Prolong Mesenchymal Stem Cells from Human Fat Engineered to Secrete
Updated version
http://clincancerres.aacrjournals.org/content/20/9/2375
Access the most recent version of this article at:
Material
Supplementary
http://clincancerres.aacrjournals.org/content/suppl/2015/07/17/20.9.2375.DC1
Access the most recent supplemental material at:
Cited articles
http://clincancerres.aacrjournals.org/content/20/9/2375.full#ref-list-1
This article cites 54 articles, 10 of which you can access for free at:
Citing articles
http://clincancerres.aacrjournals.org/content/20/9/2375.full#related-urls
This article has been cited by 2 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://clincancerres.aacrjournals.org/content/20/9/2375To request permission to re-use all or part of this article, use this link
on March 22, 2019. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from