metabolic networks john pinney theoretical systems biology group [email protected] 341...
TRANSCRIPT

Metabolic networks
John Pinney
Theoretical Systems Biology group
341 Introduction to Bioinformatics: Biological Networks
25th February 2010

Part 1: Constructing metabolic networks

What is metabolism?
“Metabolism is the set of chemical reactions that occur in living organisms in order to maintain life.”
Image: section through an Escherichia coli cell
by David Goodsell

What is metabolism?
Key classes of biochemicals:
amino acids• proteins
carbohydrates• bacterial envelope
nucleotides• genetic material
lipids• membranes
coenzymes • transfer chemical groups
minerals• assist in biochemical transformations

glucose
glucose 6-phosphate
Enzymes
Metabolic reactions are catalysed by proteins called enzymes.

Metabolic pathways
Traditionally, biochemists consider a series of consecutive metabolic reactions to form a pathway.
Image: CK12.org

Metabolic networks
However, pathways often overlap so much that it is more accurate to consider the set of all metabolic reactions as forming a network.
Image: Wikipedia

How should we represent metabolic networks?
Traditional textbook representation:
Compounds are shown as boxes.
Arrows connect compounds to show interconversions.
Arrows are labelled with the name of the associated enzyme.
Cofactors (commonly-used compounds) included with curved arrows.
Image: Michal, G. (1993). Biochemical Pathways Poster. Boehringer Mannheim GmbH

Why should we study metabolic networks?
Fundamental to lifeSince enzymes are encoded in the genome, metabolism is one mechanism by which an organism’s genotype (specific set of genes) is connected to its phenotype (how it behaves). Many metabolic processes are common to all forms of life.
BiotechnologyDeep understanding of the metabolic networks of bacteria is needed if they are to be genetically modified to produce a desired product with maximum yields.
MedicineAberrations in human metabolism are fundamental to diseases such as diabetes and some types of cancer.Knowledge of the metabolic networks of pathogens and parasites can help to select drug targets (or target combinations) that will be most effective.

How should we represent metabolic networks?
Traditional textbook representation:
Compounds are shown as boxes.
Arrows connect compounds to show interconversions.
Arrows are labelled with the name of the associated enzyme.
Cofactors (commonly-used compounds) included with curved arrows.
Image: Michal, G. (1993). Biochemical Pathways Poster. Boehringer Mannheim GmbH

Representing metabolic networks for systems biology
simple graph
metabolite
digraph bipartite digraph
reaction
or morecomplex still..?
enzyme

Metabolic reconstruction
Task:
Given the genome sequence for an organism, find its metabolic network.
Resources:Sequence databasesGenome annotationsDatabases of metabolic reactions
Tools:Sequence similarity searchesText extractionMachine learningExperimental data (high- and low-throughput)
Francke C et al. (2005)
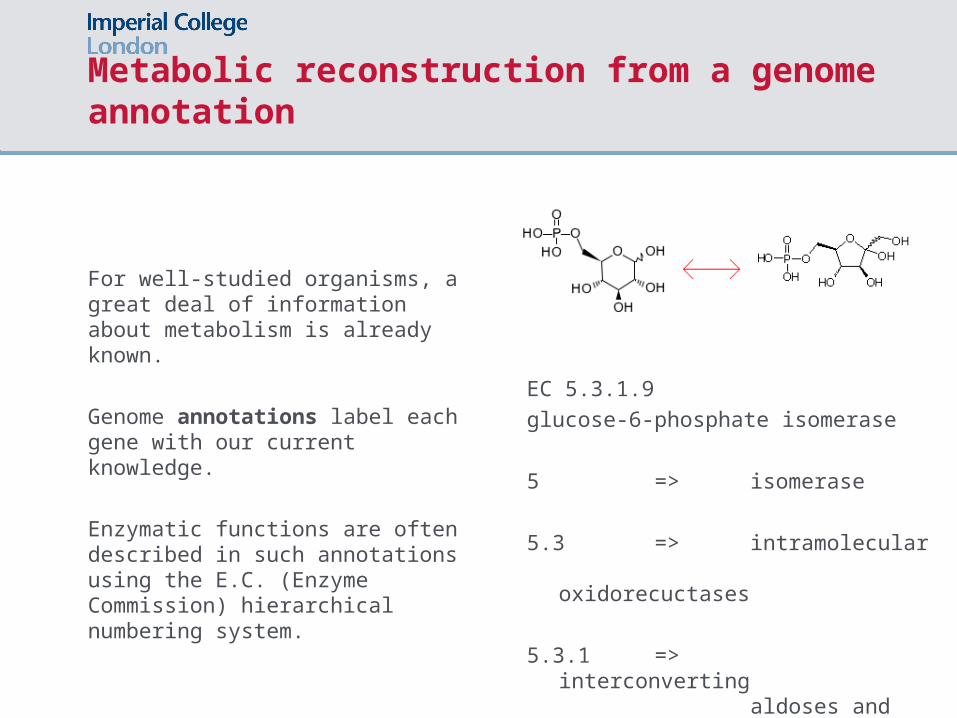
Metabolic reconstruction from a genome annotation
For well-studied organisms, a great deal of information about metabolism is already known.
Genome annotations label each gene with our current knowledge.
Enzymatic functions are often described in such annotations using the E.C. (Enzyme Commission) hierarchical numbering system.
EC 5.3.1.9
glucose-6-phosphate isomerase
5 => isomerase
5.3 => intramolecular
oxidorecuctases
5.3.1 => interconverting aldoses
and ketoses
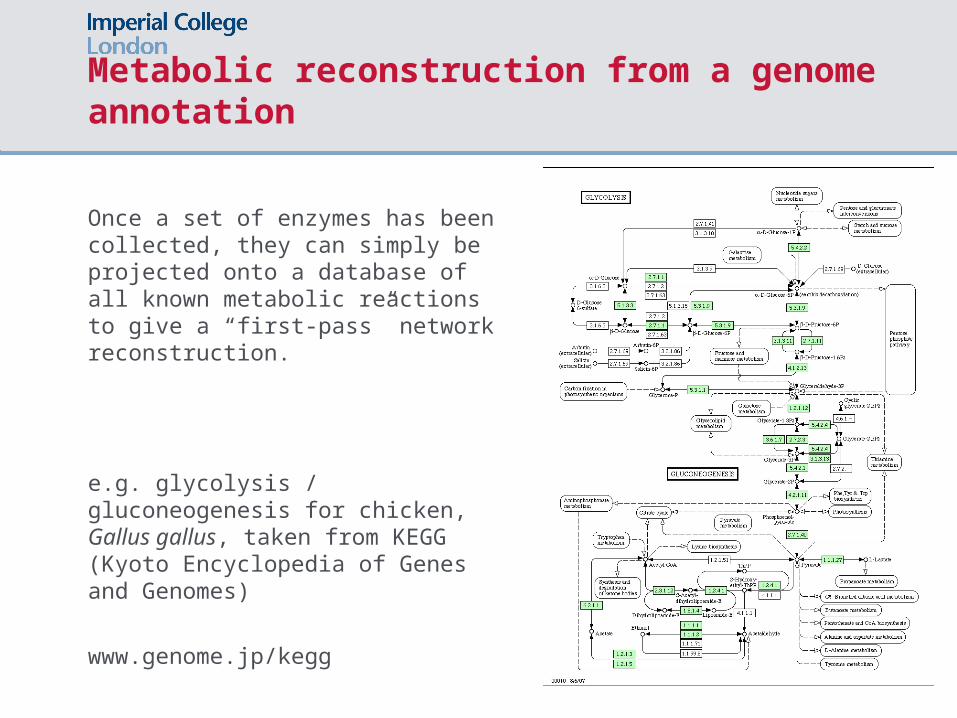
Metabolic reconstruction from a genome annotation
Once a set of enzymes has been collected, they can simply be projected onto a database of all known metabolic reactions to give a “first-pass” network reconstruction.
e.g. glycolysis / gluconeogenesis for chicken, Gallus gallus, taken from KEGG (Kyoto Encyclopedia of Genes and Genomes)
www.genome.jp/kegg

Metabolic reconstruction from a proteome
Often a well-curated genome annotation is unavailable, but we have a good idea of where the protein-coding genes are on the genome so can extract a predicted proteome (set of all protein sequences encoded by the genome).
The task is now to assign enzymatic functions to these protein sequences.
genome sequence with known protein-coding regions.
predicted proteins

Metabolic reconstruction from a proteome
If a closely-related organism has a good annotation, it may be possible to identify orthologous (i.e. functionally equivalent) proteins using basic sequence alignment methods such as BLAST.
More sophisticated methods for orthology assignment are also available.
annotated proteome
Functional assignment by sequence similarity (e.g. BLAST)
new proteome

Metabolic reconstruction from a proteome
However, using profile models for enzyme domains is a more sensitive way to detect sequence similarities, especially across large evolutionary distances.
multiple alignment of enzyme domains from many species
Highly-conserved amino acids
profile model (position-specific scoring matrix / profile HMM)
library of models for all enzyme functions with known sequences

Metabolic reconstruction from a proteome
Known ligand-binding residues from bacterial structure
EPSP synthase
shikimate kinase
McConkey GA et al. (2004)
ATP/GTP binding motif

Limitations of sequence-based methods
Large evolutionary distancesTransfer of function from a distant sequence may not be reliable.Enzyme may be too divergent to be recognised from sequence.
Multiple functionsSome enzymes have multiple protein domains that have different functions.An enzyme may “moonlight” - i.e. catalyse several different reactions using the
same active site.
Reactions with unknown sequencesThere are several known metabolic reactions for which no example enzyme
sequences are known.
Unknown reactionsAcross all kingdoms of life, there are many hundreds of metabolic reactions that
are as yet completely uncharacterised!
Manual curation
Computational assignment of gene function is not 100% accurate!
It will always be important to examine and refine initial automated metabolic reconstructions carefully before attempting to analyse the resulting network.
Comparative genomics can be a powerful tool in network curation.
By comparing genomes between different species, we attempt to use their shared evolutionary histories to help us identify gene functions more accurately.
What genes are close to this gene?
Has this gene ever fused with another one?
Which genes tend to be present in the same organisms as this one?
Which genes control whether this one is switched on?
What experimental evidence is there?

Gaps in a reconstructed network
Even after curation, a network may still contain obvious gaps, also known as pathway holes.
source
sink
consumed but not produced
produced but not consumed
intermediate reaction missing

Phylogenetic profiling (evidence for functionally associated genes)
Anticorrelation analysis (evidence for functionally analogous genes)
Methods for gap-filling
g1 g2 g3 g4 g5 g6 g7 g8 g9 g10
s1 + + + + + +
s2 + + + + + +
s3 + + +
s4 + + + + + +
s5 + + + + +
s6 + + + +
s7 + + + +
s8 + + + +
shared pattern anticorrelated pattern
species
gene
Osterman A and Overbeek R (2003); Pellegrini M et al. (1999)
?
?

Methods for gap-filling
Evidence from various sources can be integrated using machine learning to give an overall likelihood that a particular gene might fill a particular pathway hole.
For parasitic or symbiotic organisms, we also need to consider the possibility of metabolite exchange with the host or subversion of host enzymes.
Green ML et al. (2004)

Part 2: Metabolic network analysis

Analysis of metabolic networks
Metabolic networks can be analysed on several different levels.
Topologically
Basic network structure
Stoichiometrically
Considering the numbers of molecules of each type consumed and produced by each reaction.
Dynamically
Considering the rates of each reaction and variations in metabolite concentrations over time.

Topological analysis
Metabolic networks can be studied purely from the point of view of their graph properties.
Degree distributionClustering coefficientShortest path lengthModularityetc.
These types of investigations may (or may not!) provide useful insights into how metabolic networks have evolved.
Wagner A and Fell DA (2001)

Topological analysis
Chokepoint analysis can help to reveal potential drug targets
highlighted squares are all chokepoint reactions, as they have unique substrates and/or products
Yeh I et al. (2004)

Petri net representations
metabolite
bipartite digraph
reaction
The bipartite digraph representation of a metabolic network is very close to a modelling paradigm from computer science called a Petri net.
Various forms of Petri net representation have been successfully used in the analysis of many biological networks, especially for gene regulation, signal transduction and metabolic systems.
Petri net

Petri nets for metabolic systems
Image: I. Barjis and V. Gehlot, SCSC 2007

Petri Nets
A tool for modelling a system:
• simple.• easy to represent graphically.• represents concurrent processes.• mathematically rigorous.• large theoretical framework has been developed.
Peterson JL (1981) Petri Net theory and the modeling of systems Prentice-Hall, NJ

Introduction to Petri Nets
Generic features of a system
Composite:• A system is considered to be made up of separate, interacting
components.
State:• Each component has its own state of being, which determines its future
actions.
Concurrency:• Components in two or more parts of the system may be simultaneously
active.

Petri nets are usually described mathematically using matrix notation.
However, they can also be represented as directed graphs with two types of node: places and transitions.
Introduction to Petri Nets
place
transition
arc

Introduction to Petri Nets
Transitions
Each transition has a set of input places and a set of output places.
input place
output place

Introduction to Petri Nets
Places
Places may be marked by tokens. Each place may hold an integer number of tokens.
A particular distribution of tokens over a net is called a marking. This represents the state of the system.
marked places

Introduction to Petri Nets
enabled transition
sFiring transitions
Transitions whose input places are all marked by at least one token are said to be enabled.
A transition fires by removing one token from each of its input places and creating new tokens at its output places.

Introduction to Petri Nets
Firing transitions
Transitions whose input places are all marked by at least one token are said to be enabled.
A transition fires by removing one token from each of its input places and creating new tokens at its output places.

Introduction to Petri Nets
Firing transitions
Transitions whose input places are all marked by at least one token are said to be enabled.
A transition fires by removing one token from each of its input places and creating new tokens at its output places.

Introduction to Petri Nets
Firing transitions
Firing may continue until no transition is enabled, at which point execution halts.
Although the initial marking determines the possible future behaviour of the net, the order in which transitions are fired is not fixed: the same initial marking may lead to different final states.

Introduction to Petri Nets
Firing transitions
Firing may continue until no transition is enabled, at which point execution halts.
Although the initial marking determines the possible future behaviour of the net, the order in which transitions are fired is not fixed: the same initial marking may lead to different final states.

Introduction to Petri Nets
Firing transitions
Firing may continue until no transition is enabled, at which point execution halts.
Although the initial marking determines the possible future behaviour of the net, the order in which transitions are fired is not fixed: the same initial marking may lead to different final states.
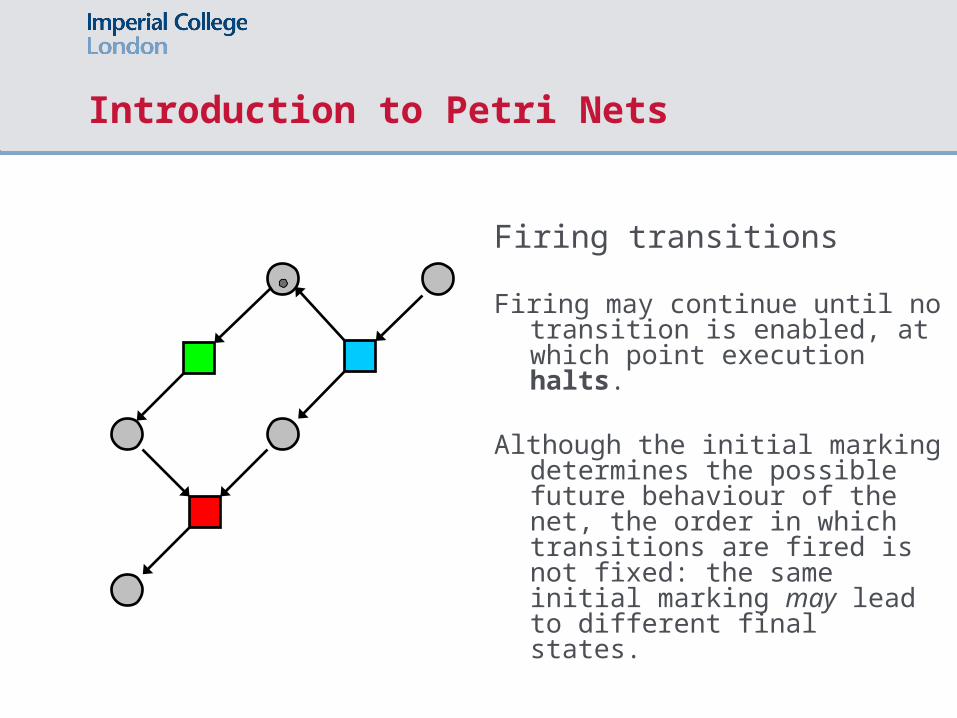
Introduction to Petri Nets
Firing transitions
Firing may continue until no transition is enabled, at which point execution halts.
Although the initial marking determines the possible future behaviour of the net, the order in which transitions are fired is not fixed: the same initial marking may lead to different final states.

Introduction to Petri Nets
Firing transitions
Firing may continue until no transition is enabled, at which point execution halts.
Although the initial marking determines the possible future behaviour of the net, the order in which transitions are fired is not fixed: the same initial marking may lead to different final states.
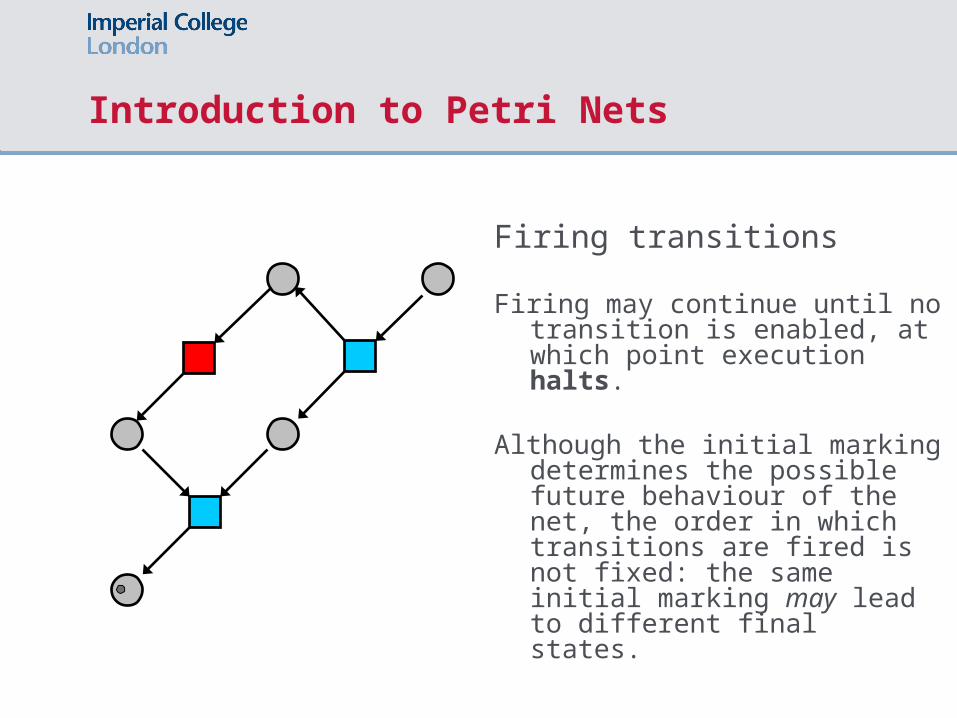
Introduction to Petri Nets
Firing transitions
Firing may continue until no transition is enabled, at which point execution halts.
Although the initial marking determines the possible future behaviour of the net, the order in which transitions are fired is not fixed: the same initial marking may lead to different final states.

Introduction to Petri Nets
Firing transitions
Firing may continue until no transition is enabled, at which point execution halts.
Although the initial marking determines the possible future behaviour of the net, the order in which transitions are fired is not fixed: the same initial marking may lead to different final states.

Matrix notation for Petri nets

Stoichiometric analysis
Part of E. coli metabolism
Elementary Flux Modes are formal definitions of minimal pathways that can operate independently at steady state.
They are equivalent to the set of minimal T-invariants of the Petri net incidence matrix describing the system.
Schuster S et al. (1999)

Stoichiometric analysis
Schuster S et al. (1999)

Stoichiometric analysis
Flux balance analysis (FBA) is a widely used stoichiometric analysis technique.
For a given growth condition (e.g. known input nutrients):
Assume that metabolic system operates in a steady state.
Assume certain constraints on system (mass-balance, flux limitations).
Assume an “objective” that is expected to be maximised by evolution (e.g. biomass production).
FBA can be used to predict reaction fluxes and essential enzymes under a given growth condition.

FBA example
anoxic (no oxygen)
hypoxic (limited oxygen)
aerobic (unlimited oxygen)
Grafahrend-Belau E et al. (2008)
Pathways of starch storage at different phases of development in barley seeds

Metabolic control analysis
Given kinetic parameters, we can calculate sensitivity of the flux through a given pathway to the inhibition of any enzyme involved.
This replaces the concept of a “rate-limiting step” in a pathway with the idea of control being shared to some degree between all enzymes, represented by each enzyme’s flux control coefficient, C.
Requires detailed kinetic model: currently limited to a few very well characterised pathways in specific organisms.
Bakker BM et al. (2000)
C=1
C=0
0<C<1

Metabolic control analysis
Bakker BM et al. (2000)
The human trypanosome parasite Trypanosoma brucei has a unique organelle called the glycosome, which carries out the glycoloysis that is essential for its survival.
MCA has been applied to the glycolytic pathway in T. brucei to determine which of these enzymes would be the best drug targets.
MCA is potentially very helpful in drug target investigations because it allows us to consider the likely effects of incomplete inhibition of enzyme function.

Dynamic modelling approaches
There are many general software packages available for systems biology that can be used to model and simulate the dynamic behaviour of metabolic networks and to integrate them with processes such as gene regulation and protein interactions.
Metabolic models can often be shared between different software using Systems Biology Markup Language (SBML).
(see sbml.org for examples)
Modelling could be
Deterministic e.g. ordinary differential equations (ODEs)
or
Stochastic e.g. Gillespie algorithm, Petri net simulation

Systems Biology Markup Language

Summary
Metabolic networks are central to much of systems biology and have important applications in biotechnology and medicine.
They can be reconstructed to some extent from genome sequences, but a complete and accurate metabolic model is difficult to achieve and requires a great deal of manual curation.
Metabolic networks may be analysed at various degrees of detail, using topological, stoichiometric and/or dynamic approaches.

References
•Oberhardt MA et al. Applications of genome-scale metabolic reconstructions. Mol Syst Biol (2009) 5:320
•Francke C et al. Reconstructing the metabolic network of a bacterium from its genome. Trends Microbiol (2005) 13:550-8
•Bakker BM et al. Metabolic control analysis of glycolysis in trypanosomes as an approach to improve selectivity and effectiveness of drugs. Molecular and Biochemical Parasitology (2000) 106:1-10
•Grafahrend-Belau E et al. Flux balance analysis of barley seeds: a computational approach to study systemic properties of central metabolism. Plant Physiol (2008)
•Green ML et al. A Bayesian method for identifying missing enzymes in predicted metabolic pathway databases. BMC Bioinformatics (2004) 5:76
•McConkey GA et al. Annotating the Plasmodium genome and the enigma of the shikimate pathway. Trends Parasitol (2004) 20:60-5
•Osterman A and Overbeek R. Missing genes in metabolic pathways: a comparative genomics approach. Current Opinion in Chemical Biology (2003) 7:238-51
•Pellegrini M et al. Assigning protein functions by comparative genome analysis: protein phylogenetic profiles. Proc Natl Acad Sci USA (1999) 96:4285-8
•Schuster S et al. Detection of elementary flux modes in biochemical networks: a promising tool for pathway analysis and metabolic engineering. Trends Biotechnol (1999) 17:53-60
•Wagner A and Fell DA. The small world inside large metabolic networks. Proc Biol Sci (2001) 268:1803-10
•Yeh I et al. Computational analysis of Plasmodium falciparum metabolism: organizing genomic information to facilitate drug discovery. Genome Res (2004) 14:917-24