microbial genomics, pan-genomics, and metagenomics · pdf filemicrobial genomics,...
TRANSCRIPT

Microbial Genomics, Pan-genomics, and Metagenomics in disease and in
health
Talk dedicated to Francis Ouellette and all the VanBUG organizers/volunteers current and past!
William Hsiao BCCDC Public Health Labs [email protected]

Microbes Germs

Microbes – learn to love them
§ Microbes harbour much higher genetic diversity than eukaryotic organisms
§ Less than 0.5% of the microbial species have been identified – huge potential for discovery of new genes and new functions
§ Most Microbes (>99.9%) do not cause diseases in human
§ Microbes can be engineered to act as little factories for energy production, drug production, immune system booster (probiotics), pollution clean up, and environmental sensors
The Bacterial/Archaeal Genome
§ Typically contained within a single large, circular chromosome (some are linear)
§ Haploid genomes
§ May contain plasmids (extrachromosomal DNA)
§ No introns in the genes
§ Genome size range from 0.5Mb to ~10Mb (average is about 3 - 5Mb and contain about 3000-5000 genes)
§ Much easier than eukaryotic genomes to assemble and to annotate
§ First free-living organism sequenced is a bacterium – Haemophilus influenzae in 1995

Shotgun Sequencing – 90s style
Fraser et al 2000, Nature
contigs
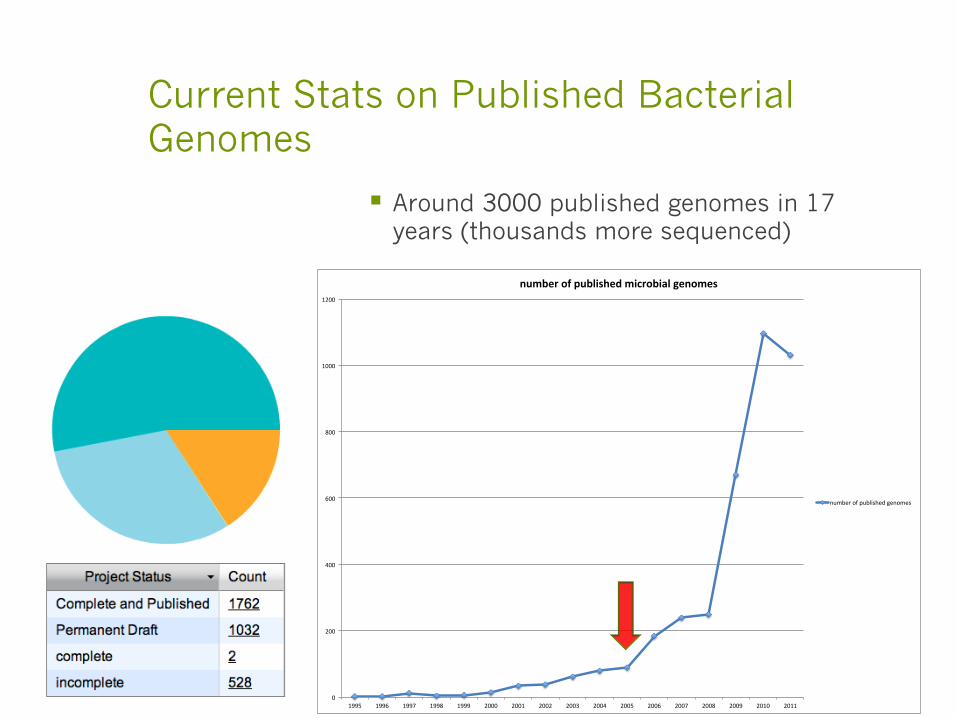
Current Stats on Published Bacterial Genomes
§ Around 3000 published genomes in 17 years (thousands more sequenced)
0"
200"
400"
600"
800"
1000"
1200"
1995" 1996" 1997" 1998" 1999" 2000" 2001" 2002" 2003" 2004" 2005" 2006" 2007" 2008" 2009" 2010" 2011"
number'of'published'microbial'genomes'
number"of"published"genomes"
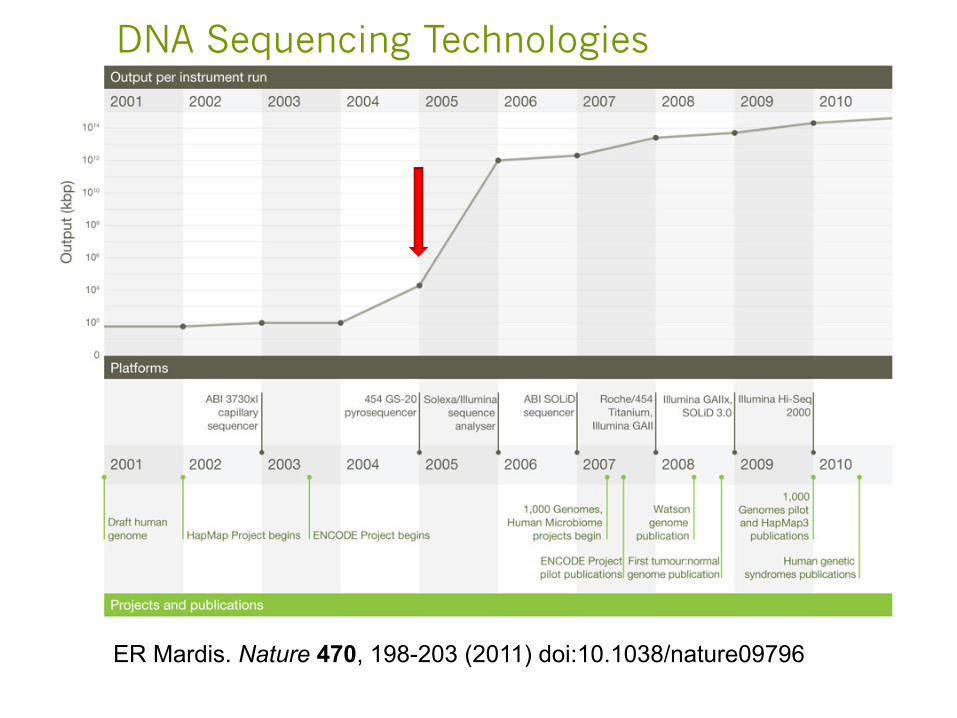
ER Mardis. Nature 470, 198-203 (2011) doi:10.1038/nature09796
DNA Sequencing Technologies
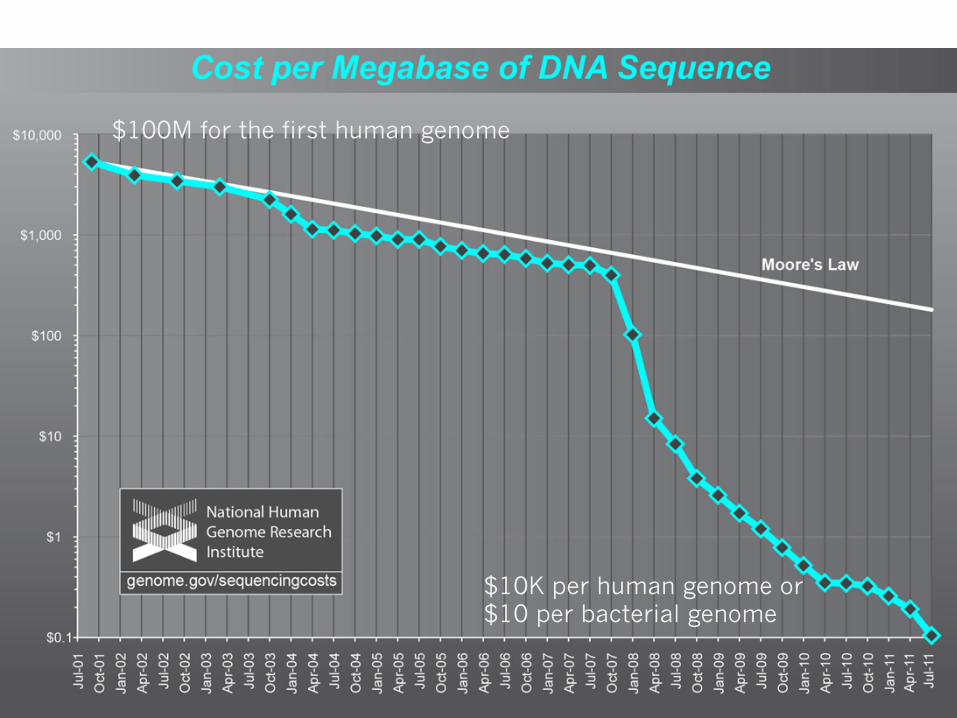
$10K per human genome or $10 per bacterial genome
$100M for the first human genome
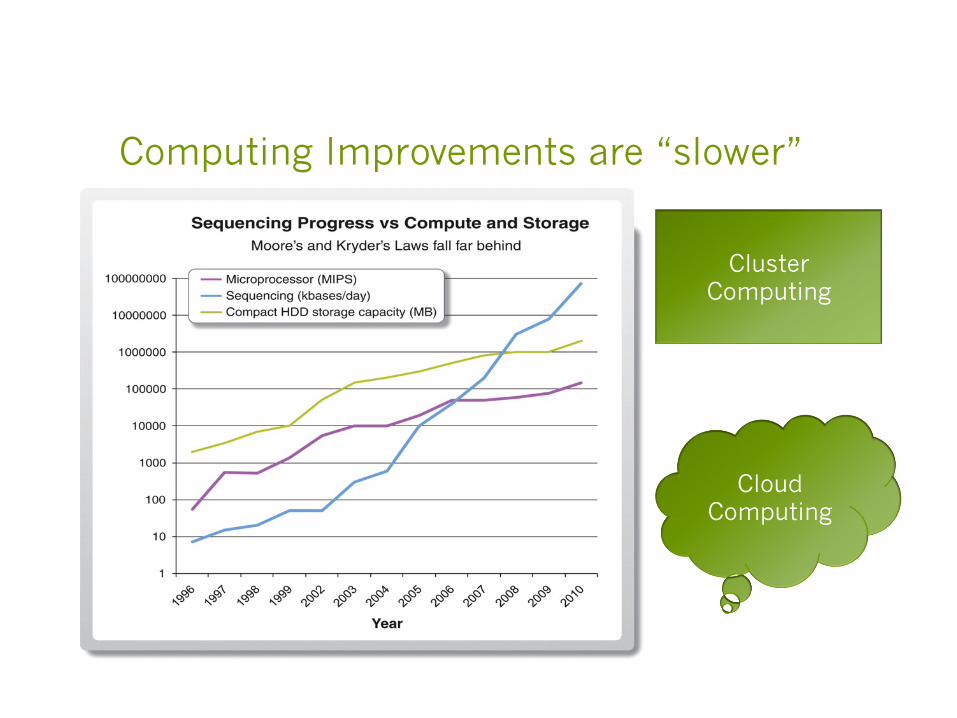
Computing Improvements are “slower”
Cloud Computing
Cluster Computing
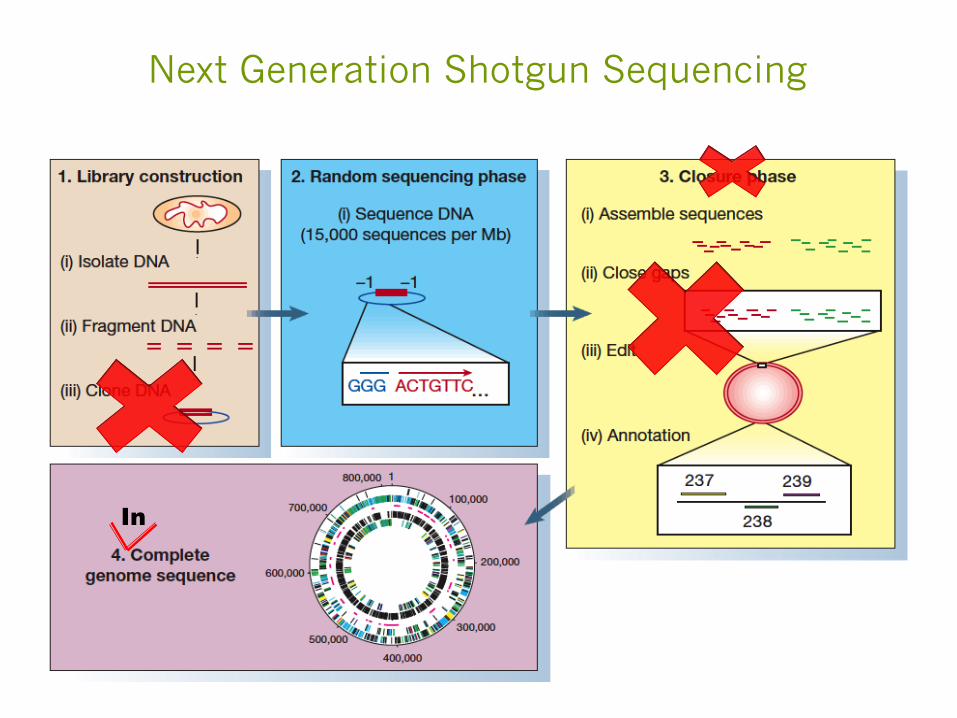
Next Generation Shotgun Sequencing
In
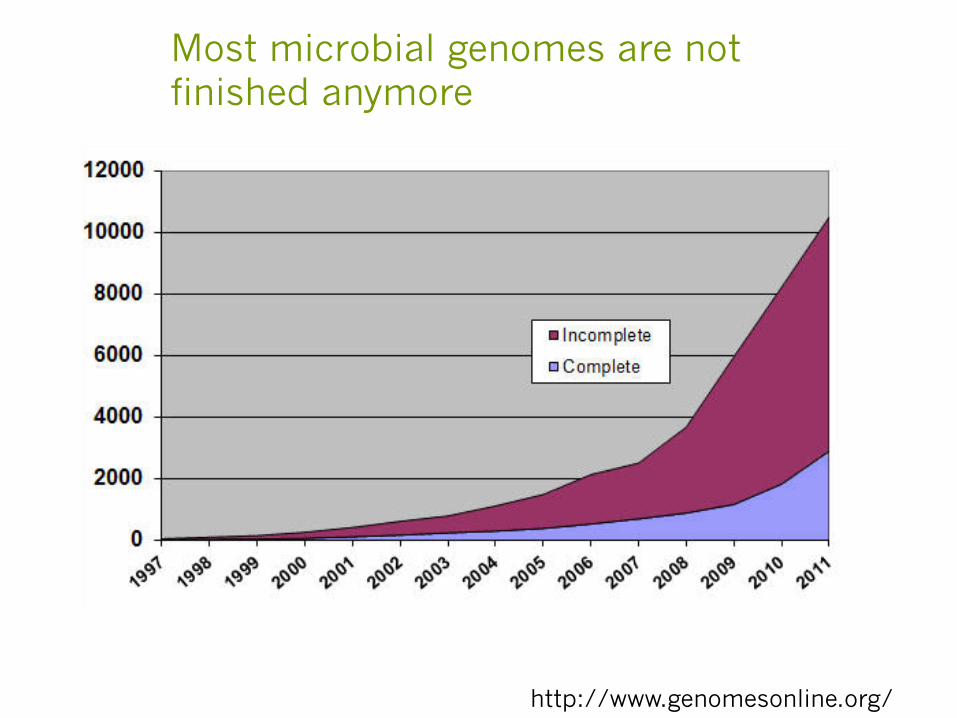
Most microbial genomes are not finished anymore
http://www.genomesonline.org/

Improving Assembly –paired-end and optical map
§ With high depth coverage from next generation sequencers, the gaps in unfinished genomes are usually due to unresolved repeats. So by incorporating long range information, we can order the contigs better and close the gaps
§ One of my post-doc projects involves in sequencing several bacterial genomes and since we got back incomplete genomes with a few hundred contigs, we explored other way to improve the genome assembly
§ We decided to use optical maps (high density, whole genome restriction maps aka fingerprints) to help us assemble the genome

Theodore Assembler
Hsiao et al, in preparation
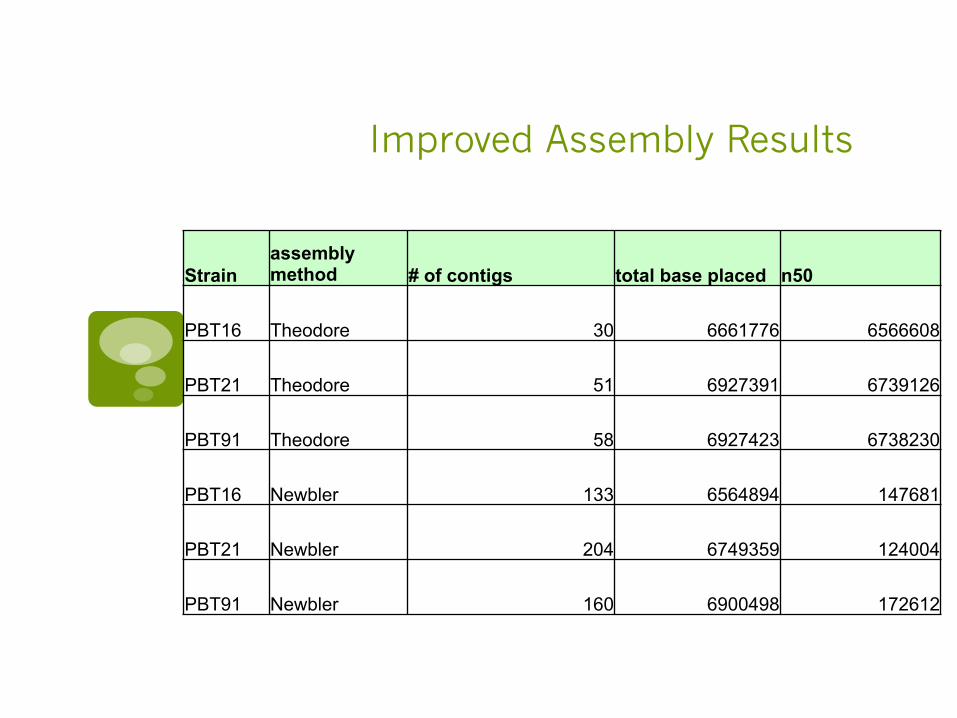
Improved Assembly Results
Strain assembly method # of contigs total base placed n50
PBT16 Theodore 30 6661776 6566608
PBT21 Theodore 51 6927391 6739126
PBT91 Theodore 58 6927423 6738230
PBT16 Newbler 133 6564894 147681
PBT21 Newbler 204 6749359 124004
PBT91 Newbler 160 6900498 172612

Automated Genome Annotation
§ Several systems available to public, each with sophisticated approaches to assign functions to predicted genes / proteins
§ BASys (http://basys.ca)
§ Prok-annotation pipeline (http://ae.igs.umaryland.edu/cgi/intro_info.cgi)
§ IMG-ER (https://img.jgi.doe.gov/cgi-bin/er/main.cgi)
§ RAST (http://rast.nmpdr.org/rast.cgi)
§ Most of the systems run on large clusters of computers and take less than a day to annotated a genome
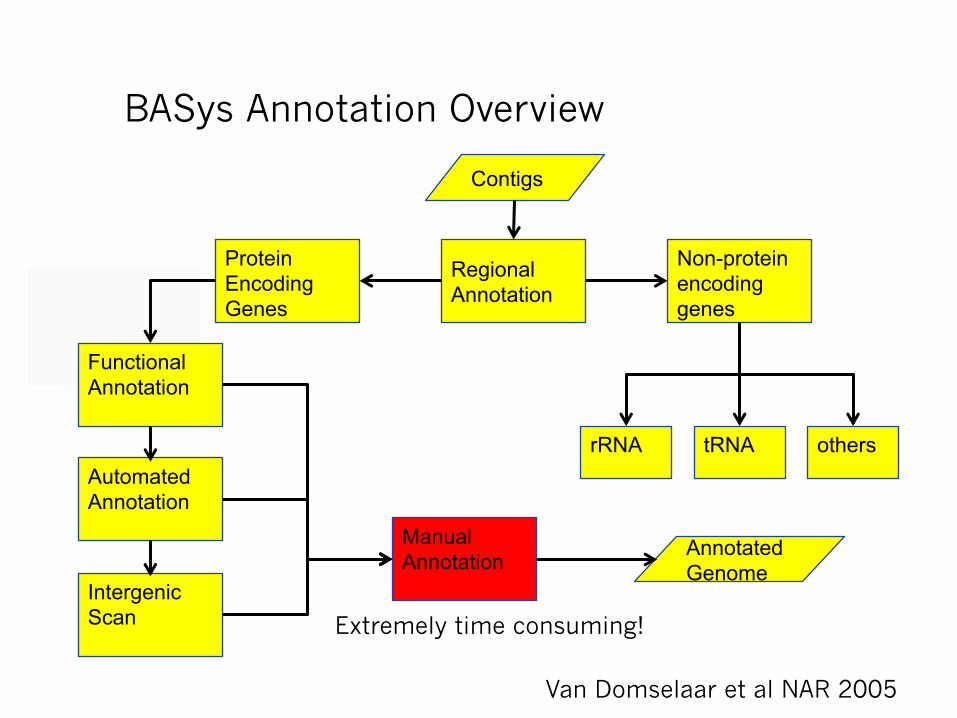
BASys Annotation Overview
Contigs
Regional Annotation
Non-protein encoding genes
Protein Encoding Genes
rRNA tRNA others
Manual Annotation
Functional Annotation
Automated Annotation
Intergenic Scan
Annotated Genome
Van Domselaar et al NAR 2005
Extremely time consuming!
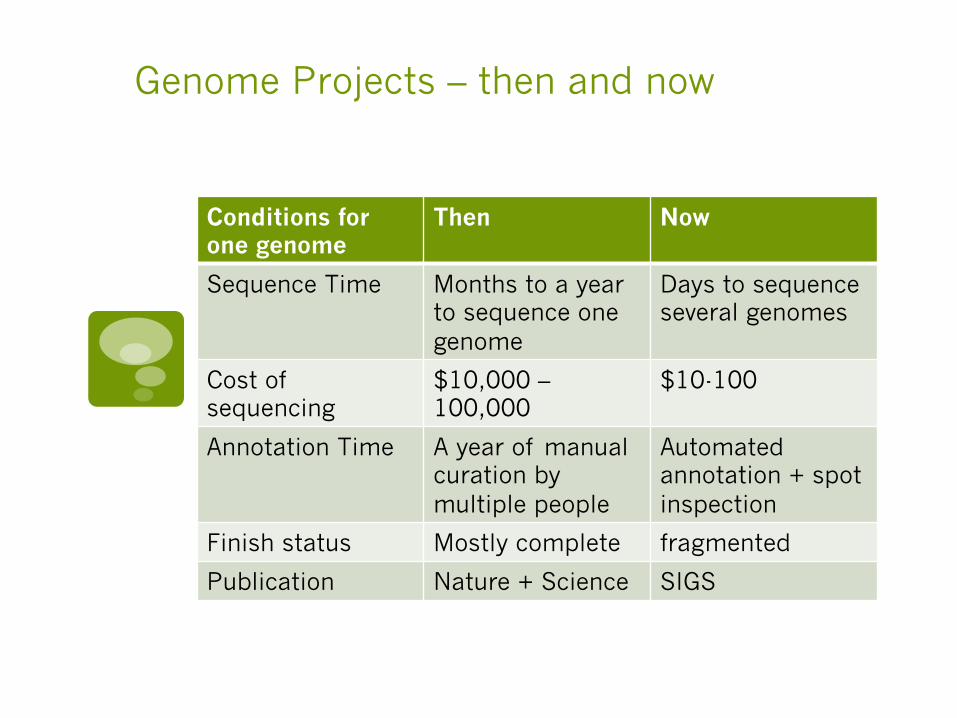
Genome Projects – then and now
Conditions for one genome
Then Now
Sequence Time Months to a year to sequence one genome
Days to sequence several genomes
Cost of sequencing
$10,000 – 100,000
$10-100
Annotation Time A year of manual curation by multiple people
Automated annotation + spot inspection
Finish status Mostly complete fragmented
Publication Nature + Science SIGS

Comparative Genomics
§ First Comparative Genomic paper published in 1999
§ 2 Helicobacter pylori genomes isolated 7 years apart were compared
Found more than half of the strain specific genes are clustered in hyper variable regions This observation soon was consistently observed in many other species
Alm et al, Nature 1999

Tools to detect Genomic Islands
§ In Fiona’s Lab, we developed several tools to aid the identification of genomic islands (genomic regions that are likely to be horizontally acquired from another species) § IslandPath – based on DNA signatures
of the genomes and other features associated with islands (Hsiao et al Bioinformatics, 2003)
§ IslandPick – based on comparative genomics (Langille et al BMC Bioinformatics, 2008)
§ IslandViewer – integrated approach to identify and view genomic islands (Langille et al Bioinformatics, 2009)
20
Ba
cill
us s
ub
tilis
16
8
Bo
rre
lia b
urg
do
rfe
ri B
31
Bu
ch
ne
ra s
p. A
PS
Ch
lam
yd
ia t
rach
om
atis D
Clo
str
idiu
m a
ce
tob
uty
licu
mA
TC
C8
24
Esch
erich
ia c
oli
K1
2
Esch
erich
ia c
oli
O1
57
Ha
em
op
hilu
s in
flu
en
za
e R
d-K
W2
0
He
lico
ba
cte
r p
ylo
ri 2
66
95
Lis
teria
in
no
cu
a C
lip11
26
2
Myco
ba
cte
riu
m le
pra
e
Myco
ba
cte
riu
m t
ub
erc
ulo
sis
CD
C1
55
1
Myco
pla
sm
a p
ne
um
on
iae
M1
29
Ne
isse
ria
me
nin
gitid
is M
C5
8
Pse
ud
om
on
as a
eru
gin
osa
PA
O1
Sa
lmo
ne
lla t
yp
him
uriu
m L
T2
Sta
ph
ylo
co
ccu
s a
ure
us N
31
5
Str
ep
toco
ccu
s p
ne
um
on
iae
TIG
R4
Su
lfo
lob
us s
olfa
taricu
s
Vib
rio
ch
ole
rae
ch
rom
oso
me
I
Vib
rio
ch
ole
rae
ch
rom
oso
me
II
Ye
rsin
ia p
estis C
O9
2
0.00%
10.00%
20.00%
30.00%
40.00%
50.00%
60.00%
70.00%
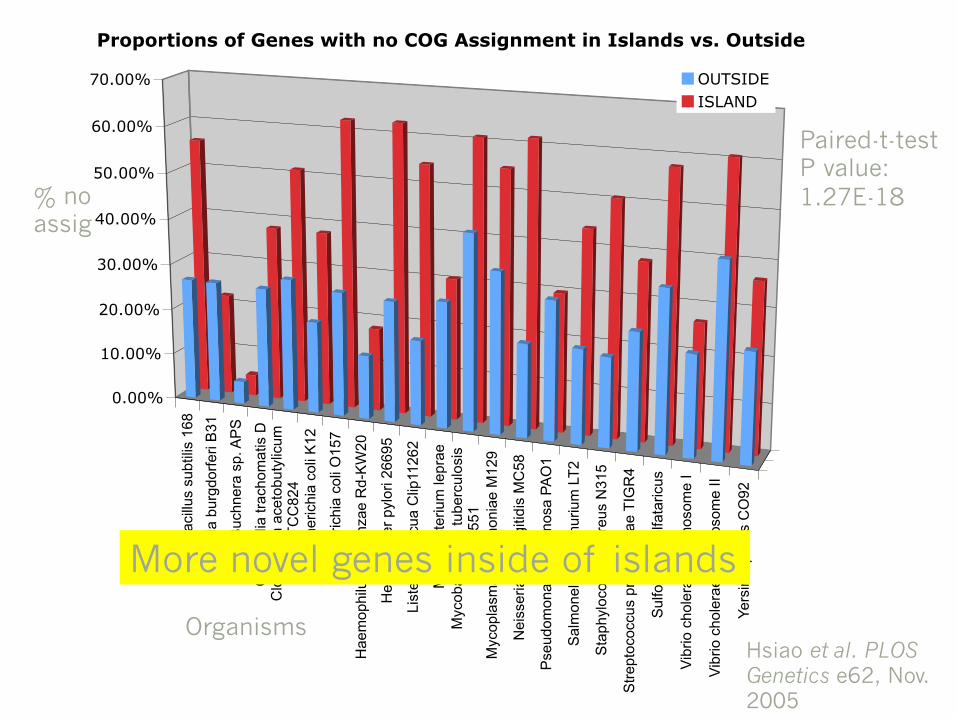
Proportions of Genes with no COG Assignment in Islands vs. Outside
OUTSIDE
ISLAND
Paired-t-test P value: 1.27E-18
More novel genes inside of islands
Hsiao et al. PLOS Genetics e62, Nov. 2005
% no assig
Organisms

Pan-genomes
§ Comparative Genomics and gene-gain and gene-loss in microbes lead to the idea of pan-genomes
§ The term first coined in 2005 in a paper by Tettelin et al., in which they compared sequenced genomes from six S. agalactiae.
§ Pan-genome consists of the core (shared) genes of a species + its strain-specific (dispensable) genes
§ Pan-genome calculation extrapolates observations based on a limited number of strains to come up with the theoretical number of genomes required to fully capture the pan-genome of a species
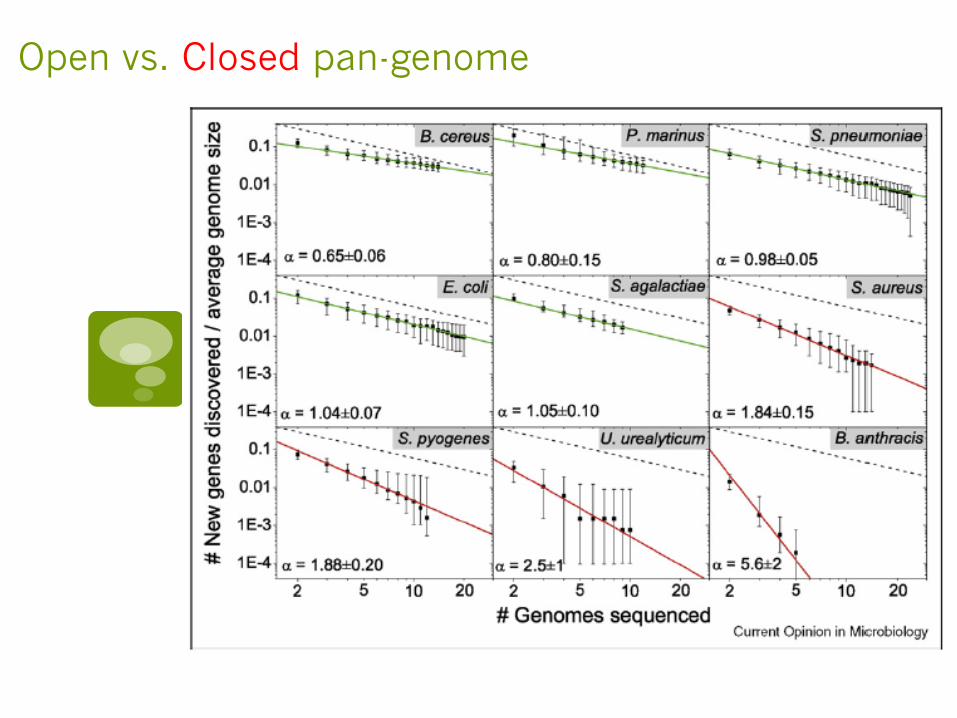
Open vs. Closed pan-genome

SNP-phylogeny for very closely related genomes
§ For very closely related isolates or very slowly evolving species, sometimes there is very little gene-gain and gene-loss.
§ In these cases, SNPs detected by aligning these genomes can be used as basis for comparison and phylogenetic tree reconstruction of the evolutionary history of the species
§ Whole Genome SNPs and Social Network Questionnaire used to reconstruct a TB outbreak in BC

Pangenome + Metadata!
§ A TB outbreak occurred in a BC community over a 3 year period
§ Molecular marker suggests that the outbreak is clonal but traditional contact tracing couldn’t identify a source
§ Whole genome sequencing and social network questionnaires (include location information) provide higher resolution data to allow a reconstruction of a likely scenario for the outbreak events.
§ Further epidemiological investigation point to increased crack cocaine usage (common locations) in the community
Gardy, Johnston, Ho Sui et al NEJM 2011
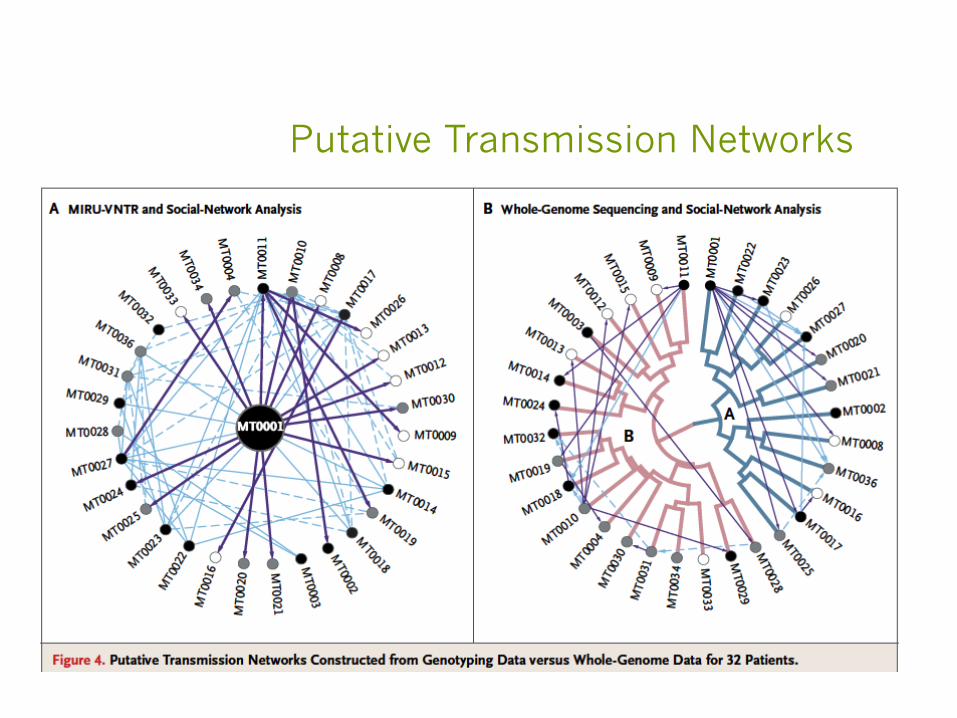
Putative Transmission Networks

Pangenome + Metadata!
§ This paper really demonstrated the power of whole genome sequencing
§ But, the availability of the metadata (disease conditions, locations, contacts, dates, etc) that facilitated the interpretation of the whole genome data

Biodiversity § In a recent global ocean survey study, ~4000 novel
protein families were detected, a significant addition to ~13,000 known protein families (Yooseph et al, PLoS Biology, 03/2007)
§ Sampling human gut, >3 million non-redundant bacterial genes and >1000 prevalent species identified (Qin et al, Nature, 03/2010)
§ In environmental surveys to date, 30% - 70% of the genes identified in the samples are novel
§ >90% of all genetic diversity comes from non-eukaryotic organisms
§ How can we begin to study this diversity and identify important microorganisms?

What is Metagenomics?
§ Meta = beyond
§ Coined by Jo Handelsman (environmental microbiologist) in 1998
§ Has taken a more precise definition as studies to analyze genetic material from a mixed population living in the same environment
§ Who’s there? What do they do?
§ How do they interact with each other and with the environment?

Typical Experimental Protocols
Samples from Environment or hosts Enriched for
microbes
Extract DNA or RNA from mixed population (no culturing & cloning!)
Targeted Sequencing • Use PCR primers to target specific
regions of genome • E.g. 16S rRNA, capsid,18S • Able to sequence deeper and broader • No metabolic functional information • Good for finding out “Who’s there”
Shotgun Sequencing • Sequence randomly all the DNA that are
in the sample (RNA is reverse transcribed first)
• Obtain functional information • Don’t know the exact host of each gene • Good for finding out “What is the
community doing”

Taxonomic Binning
§ After obtaining the 16S or other amplicon sequences, taxonomic binning based on sequence similarity or based on k-mer frequency similarity is carried out to assign a read to a taxon
§ Alternatively, reads are clusters to form OTUs (operational taxonomic unit) since many reads can not be assigned to a taxon
§ In the end, we obtain a matrix of count data associated with each taxa/OTU
Taxon E. coli OTU 1 B. theta P. aeruginosa
Sample 1 5 8 77 23
Sample 2 11 34 3 12

International Human Microbiome Consortium
§ International efforts to characterize the bacteria associated with human body sites
§ Systemic survey of the bacteria found in each site in healthy individuals – metagenomics
§ Sequencing of reference genomes of bacteria isolated from human – genomics and pangenome
§ Targeted study of microbiomes associated with various diseases
§ More information: http://www.hmpdacc.org and http://commonfund.nih.gov/hmp/

Endodontics 16S Microbiome
§ Root canal infections are a leading cause of oro-facial pain and tooth loss in western countries
§ No clear etiology; polymicrobial factors
§ Patients with root canal infections and periapical abscess were studied for the transition of microbiota from healthy oral sites to root canal and abscess
§ 3 samples (normal oral, infected root canal, and abscess) were obtained from each of 8 individuals undergone treatment
§ First study we know to sample healthy and diseased oral microbiota from the same individuals
Hsiao et al, submitted
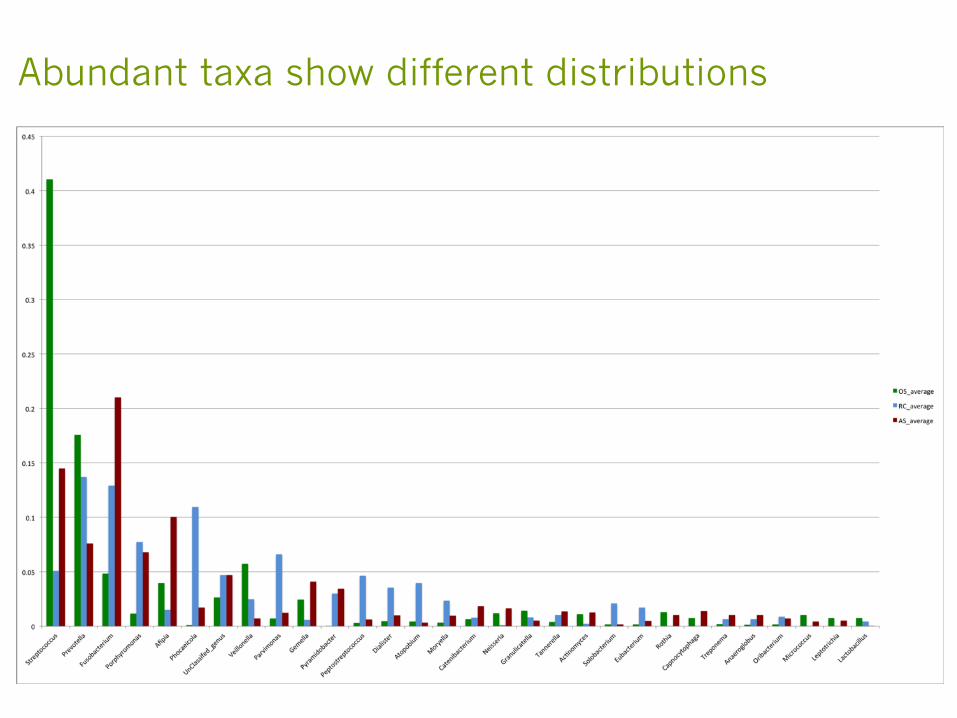
Abundant taxa show different distributions

Diseased sites have lower diversity
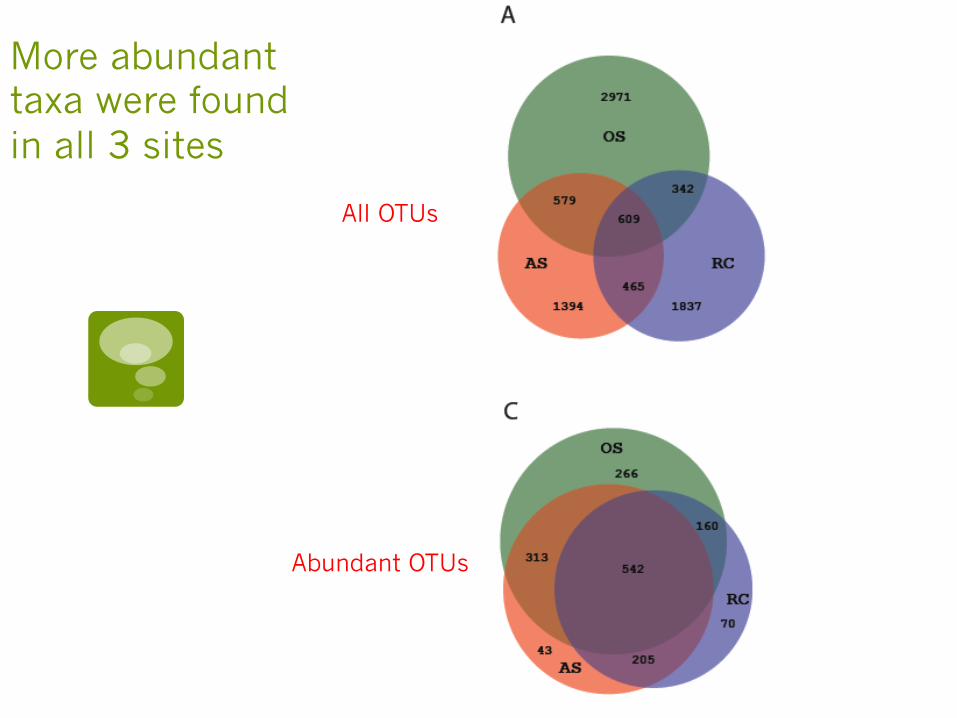
More abundant taxa were found in all 3 sites
All OTUs
Abundant OTUs

Differentially Distributed Bacteria may be associated with disease
§ We were interested to know which organisms are found differentially distributed in healthy vs. diseased sites
§ So after adjusting the count data for variance and sampling depth, we used paired-t tests and ANOVA tests to identify OTUs that are differentially distributed
§ We are especially interested in organisms that are found more abundant in diseased samples
§ In short, we were able to identify specific bacteria (some known opportunistic pathogens) to have higher relative abundance in diseased samples
§ These include: Granulicatella adiacens, Eubacterium yurii, Prevotella melaninogenica, Prevotella salivae, Streptococcus mitis, and Atopobium rimae

Watershed Microbiome
§ Genome BC project
§ Project leaders Dr. Patrick Tang and Dr. Judith Isaac-Renton
§ Two major goals are
§ 1) To use metagenomics to identify novel microbial biomarkers of watershed health
§ 2) Develop tools to match the microbial fingerprint of a contaminated watershed to the specific source of pollution

Current Water Quality Monitoring Problems
“The most significant problems associated with pathogen
measurement are the lag time involved in testing and… the large
number of false results… The absence of E. coli does not assure
the absence of more resistant fecal pathogens… source protection
planning must be carried out on an ecologically meaningful scale – that
is, at the watershed level.” The Honourable Dennis R. O’Connor
Walkerton Inquiry Commissioner 38

1. We need better tests § Water quality test: Is fecal pollution present? § Pollution attribution test: Which species is the cause?
2. The tests need better indicators § New bacterial, viral, and potentially protozoan markers
3. An environmental survey is needed to find these novel indicators § Metagenomics is the only tool that can do this survey
Metagenomics will Provide the Solutions “DNA analysis offers promise for the future” Walkerton Inquiry Report
39

Pilot study looking at 16S microbiome at different sites under wet and dry conditions
§ Two Watershed sites
§ Two different conditions (wet day vs. dry day)
§ Multiple different time points throughout a day
§ Two replicated samples per sampling event
§ 16S sequences from the samples amplified and sequenced
§ Microbiome profile generated based on 16S sequences
§ Clustering of the samples based on relative abundance of the species (OTUs)
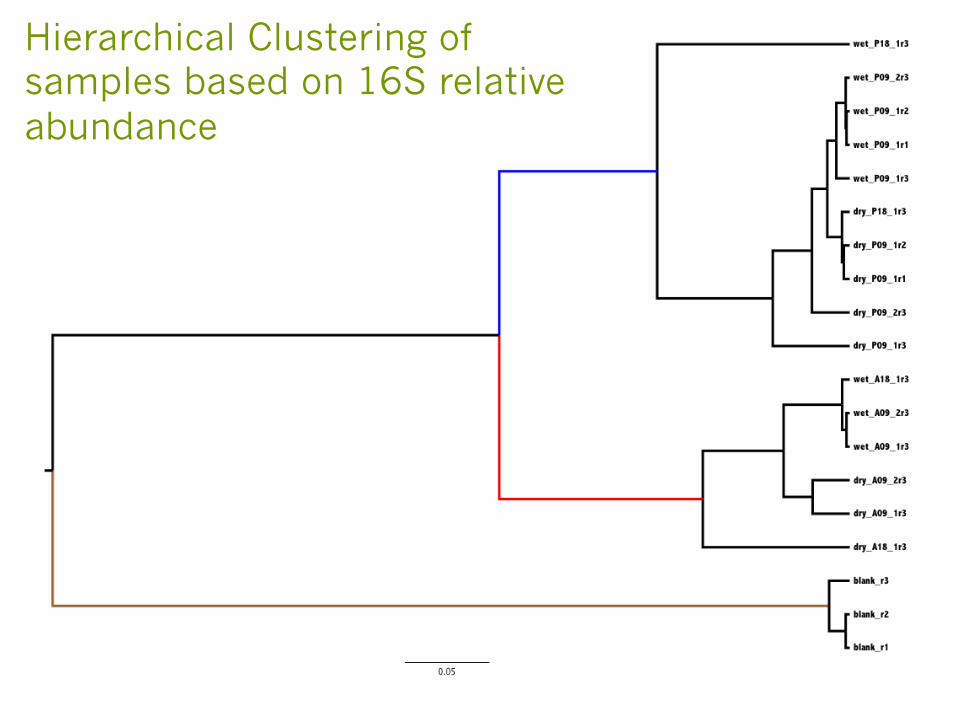
Hierarchical Clustering of samples based on 16S relative abundance

Systems Approach – Mouse Gut Model
§ Host is a dynamic system just like the microbiota and it’s the interaction between host and microbes that really produce the observed outcome.
§ So, we want to be able to study the host gene expression changes and the microbiota changes simultaneously.
§ Immunity vs. metabolism in the gut: a trialogue between B lymphocytes, microbiota and the intestinal epithelium
Shulzenko, Morgun, Hsiao et al, Nat. Med, 2011
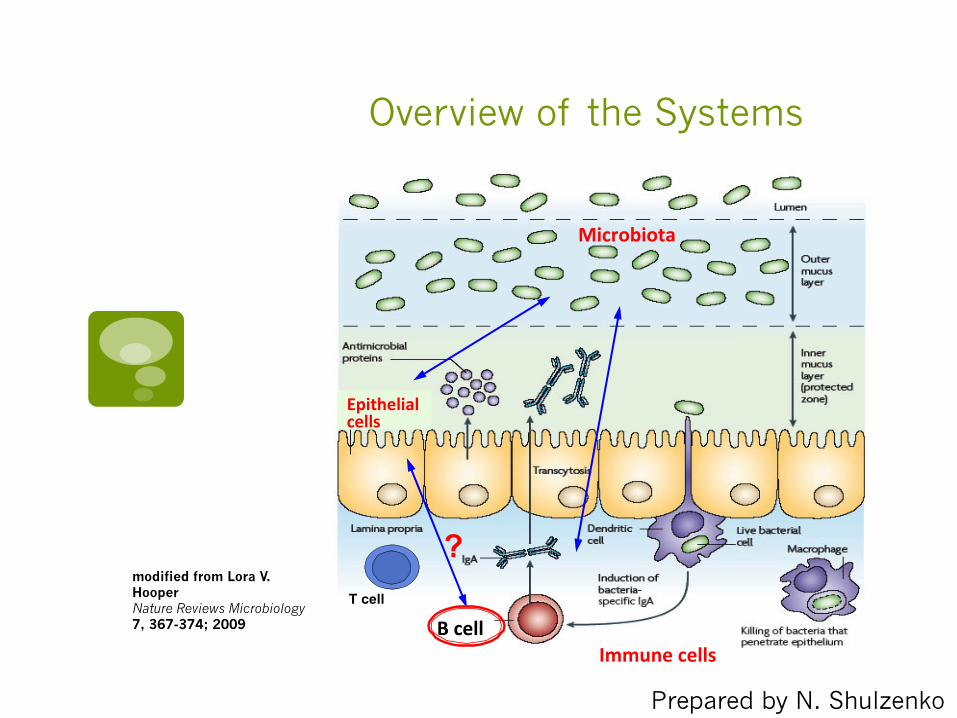
Overview of the Systems
T cell
B cell
Epithelial cells
Microbiota
? modified from Lora V. Hooper Nature Reviews Microbiology 7, 367-374; 2009
Immune cells
Prepared by N. Shulzenko
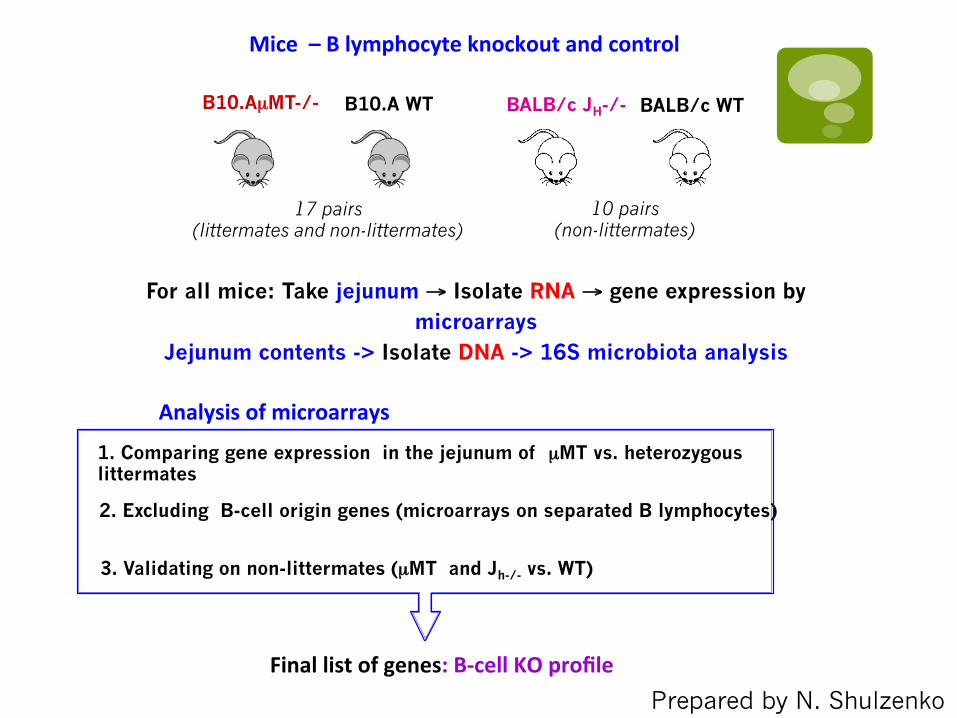
BALB/c JH-/-
10 pairs (non-littermates)
Mice – B lymphocyte knockout and control
For all mice: Take jejunum → Isolate RNA → gene expression by microarrays
Jejunum contents -> Isolate DNA -> 16S microbiota analysis
BALB/c WT B10.A WT
17 pairs (littermates and non-littermates)
B10.AµMT-/-
1. Comparing gene expression in the jejunum of µMT vs. heterozygous littermates
3. Validating on non-littermates (µMT and Jh-/- vs. WT)
2. Excluding B-cell origin genes (microarrays on separated B lymphocytes)
Analysis of microarrays
Final list of genes: B-‐cell KO profile Prepared by N. Shulzenko
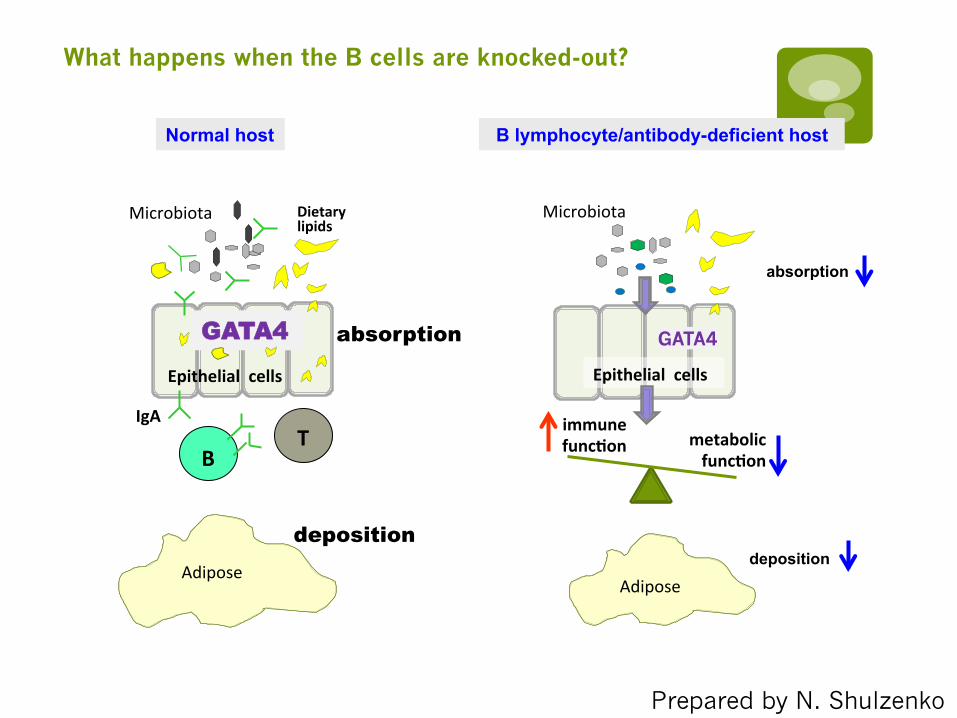
B
GATA4
Dietary lipids
Microbiota
IgA
Epithelial cells
Normal host B lymphocyte/antibody-deficient host
GATA4
Microbiota
Epithelial cells
T metabolic funcDon
immune funcDon
absorption
deposition
absorption
deposition Adipose
Adipose
Prepared by N. Shulzenko
What happens when the B cells are knocked-out?
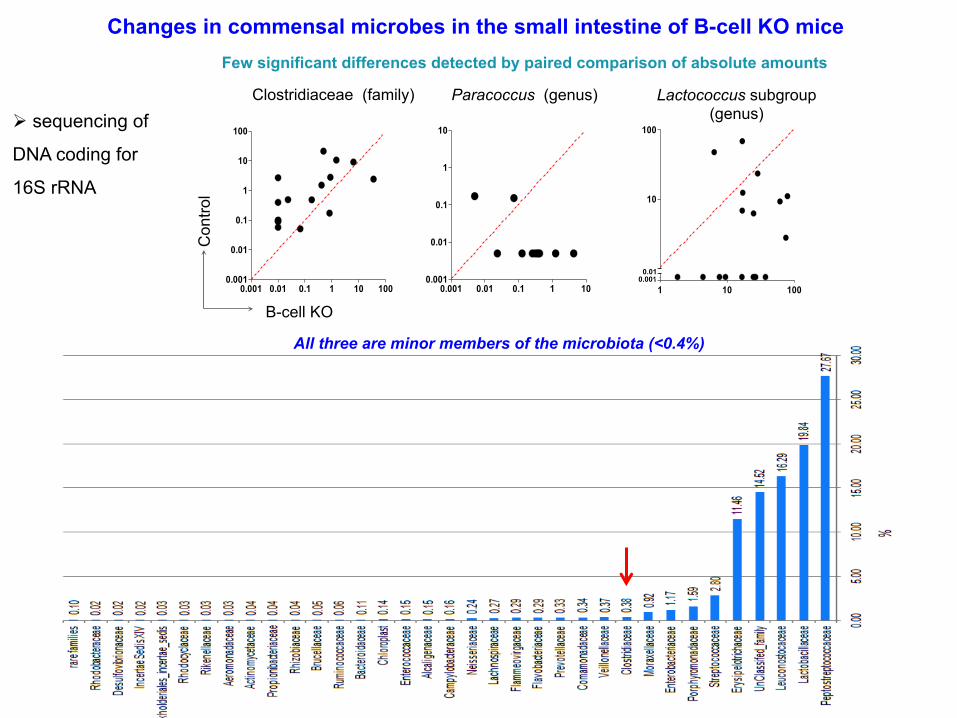
Clostridiaceae (family) Paracoccus (genus) Lactococcus subgroup (genus)
0.001 0.01 0.1 1 10 1000.001
0.01
0.1
1
10
100
0.001 0.01 0.1 1 100.001
0.01
0.1
1
10
1 10 1000.00010.0010.011
10
100
1
0.001 0.01
Changes in commensal microbes in the small intestine of B-cell KO mice Few significant differences detected by paired comparison of absolute amounts
All three are minor members of the microbiota (<0.4%)
B-cell KO
Con
trol
Ø sequencing of
DNA coding for
16S rRNA

Do microbiota really play a role in the changes?
Germ-free vs. conventional B-cell KO
Microbiota has a major role in “B-‐cell KO” intesDnal profile No difference in gene expression between BcKO and control mice under germ-‐free condiDons
Prepared by N. Shulzenko
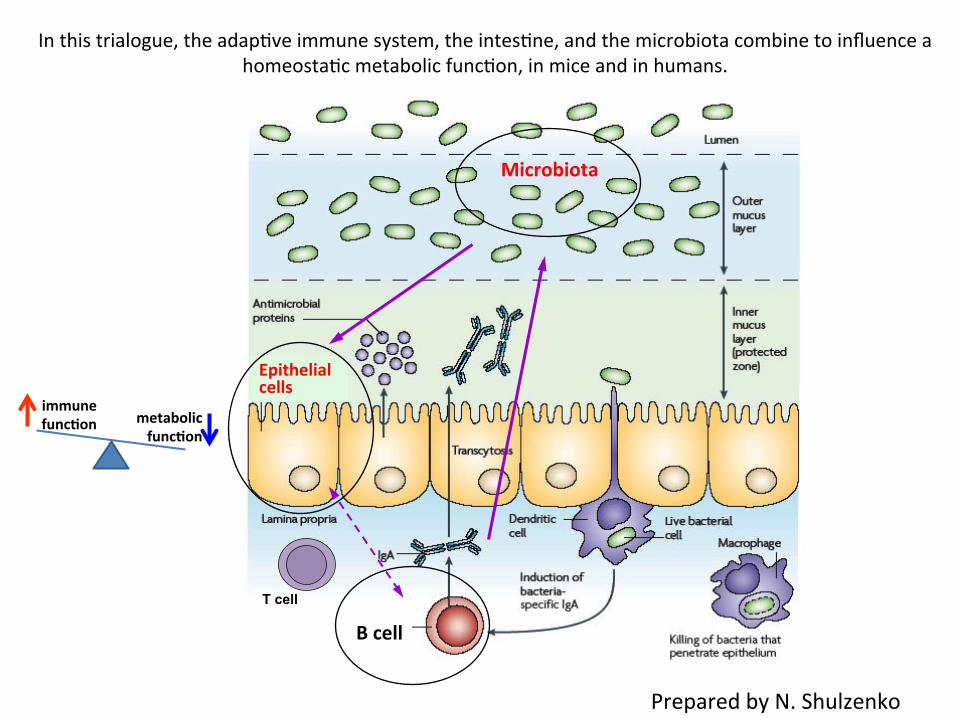
T cell
B cell
Epithelial cells
Microbiota
In this trialogue, the adap=ve immune system, the intes=ne, and the microbiota combine to influence a homeosta=c metabolic func=on, in mice and in humans.
metabolic funcDon
immune funcDon
Prepared by N. Shulzenko
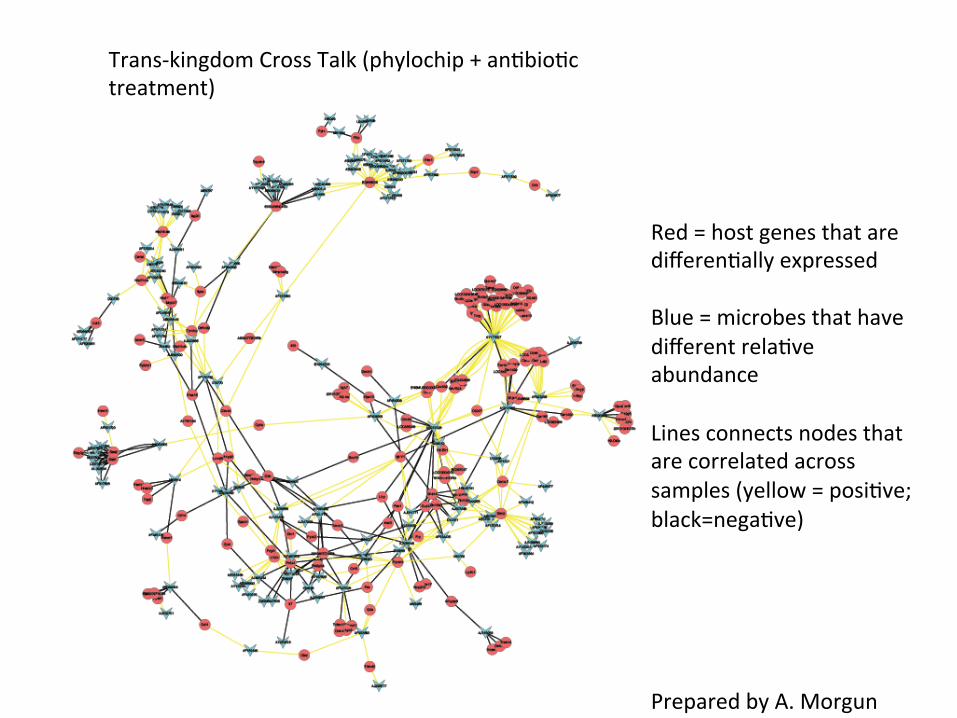
Trans-‐kingdom Cross Talk (phylochip + an=bio=c treatment)
Red = host genes that are differen=ally expressed Blue = microbes that have different rela=ve abundance Lines connects nodes that are correlated across samples (yellow = posi=ve; black=nega=ve)
Prepared by A. Morgun

Future and Wishes
§ Microbial genomics with its rapid advances in the past two decades has a bright future in helping us to understand the world’s most dominating life forms better!
§ Many diseases and health issues have polymicrobial origins and pan-genome and metagenomics can help us solve these mysteries
§ Combination of different data types is key to interpret genomic data
§ “World Peace” – combating microbes with broad-spectrum antibiotics = last resort and is often counter-productive (we need our microbiota for health)
§ With increasing number of genomes available, tools for comparative microbial genomics and good comparative genome browser capable of handling hundreds of incomplete genomes will be very useful
§ Better statistical tools to integrate the data and to help interpret the results are also needed

Acknowledgements
§ Claire Fraser-Liggett
§ Art Delcher
§ Elliott Drábek
§ Zhenqiu Liu
§ Cheron Jones
§ Brandi Cantarel
§ Institute for Genome Sciences (sequencing, annotation)
§ Ashraf Fouad
§ Andrey Morgun
§ Natalia Shulzhenko
§ Jeffrey Gordon (and his lab)
§ Patrick Tang
§ Judy Isaac-Renton
§ Fiona Brinkman
§ Natalie Prystajecky
§ Miguel Uyaguari
§ Jennifer Gardy
§ Michael Chan
§ Stephen Pleasance

Outline
§ Progression from Microbial Genomics, Pangenomics, and Metagenomics
§ Bioinformatics tools used for these analyses
§ My own projects and HMPs as examples § Health § Diseases
§ Tool developments § Database management § Assemblers
§ Classifier
§ Future of the field and Wish list for tools