molecular characterisation of c11orf67 in luminal breast ...bacterial empyema pathogens, induces...
TRANSCRIPT

Molecular Characterisation of C11orf67 in Luminal Breast Cancer
Rabab Rashwan
MBBS, MSc
School of Anatomy, Physiology and Human Biology
2016
This thesis is presented for the degree of Doctor of Philosophy
University of Western Australia


i
Abstract
The recent integration of both genomic and transcriptomic datasets have added a further
dimension to the landscape of breast cancer subtyping, defining novel functional
subgroups with distinctive oncogenic drivers that carry important implications for
therapy. This integrative clustering has unveiled a novel subtype of hormone receptor
positive (HR+) breast cancer associated with high proliferation and very poor survival
characterised by copy number amplification and overexpression of a cluster of candidate
oncogenic drivers at 11q13.5-q14 amplicon.
At the heart of this amplicon we have demonstrated the selective overexpression of
C11orf67/AAMDC (Adipogenesis associated Mth938 domain containing) which encodes
a hypothetical protein of 122 amino acids with unknown function. In a pilot tissue
microarray of 75 breast cancer cases C11orf67 amplification and expression were
significantly correlated with hormone receptor positivity, these positive cases also
demonstrated high risk features with, in particular high grade of the tumour.
In functional elucidation studies, knockdown of C11orf67 in the highly expressing T47D
cell line resulted in decreased cell proliferation, cell migration, anchorage independent
cell growth and induction of cellular senescence. T47D xenografts with stable shRNA-
induced C11orf67 knockdown injected into BALB/c mice showed significantly lower
tumour volumes relative to T47D with empty vector. A genome wide analysis of these
T47D C11orf67 shRNA cells compared to T47D empty vector cells using the Illumina
HumanHT-12 platform demonstrated 40 differentially expressed genes. Network analysis
revealed a proliferation node, enriched in cell cycle proteins, and a metabolic node
comprising several biosynthetic enzymes such as MTHFD1L involved in one-carbon
folate metabolism. Supporting this link and pointing to potential utility in chemotherapy
selection, induction of ectopic C11orf67 expression in MCF7 cells increased sensitivity
to 5-fluorouracil and methotrexate but not to taxol.

ii
Investigating potential novel binding partners and effectors, in yeast two hybrid screening
C11orf67 was found to associate strongly with RABGAP1L, a protein involved in
controlling GTPase signalling, protein trafficking, and autophagy.
Exploring the molecular cues that control C11orf67 expression, our data suggest the locus
is regulated by transcription factors associated with high proliferation and metabolic
control, notably Myc and NF-κB, as well as HRs. Estrogen leads to a significant
downregulation of C11orf67 in T47D cells, which was reversed by the antiestrogen drug
tamoxifen, whereas Progesterone significantly increased C11orf67 levels. In keeping
with this, MCF7 cells ectopically expressing C11orf67 were resistant to the anti-
proliferative effects of tamoxifen compared to the parent cell line.
These observations endorse C11orf67 as a novel oncogenic driver with exciting
therapeutic potential, which could serve to distinguish the HR+ tumours at high risk of
relapse and guide both the selection of current chemotherapeutical and endocrine
treatments as well as the design of future precision therapeutics, notably anti-folate one-
carbon drugs and novel endocrine agents.

iii
Acknowledgements
All praises to Allah for giving me the strength to completing this thesis.
Special appreciation goes to my supervisor, Associate Professor Pilar Blancafort.
Conducting this PhD project and writing up this thesis would be totally impossible if it is
not for her. I would like to express my heartfelt thanks and appreciation to her for her
invaluable guidance, inspiration, help, support and advice provided throughout the entire
years.
Big thanks to Dr Jeremy Parry and Mr Nathan Acott for the great help with the TMA
staining. Thanks to Dr Iwona Kardas and Dr Magda Ratajska for setting up the FISH
protocol, Dr Piotr Kozlowski for the cBioPortal analyses, and Mr Peter Fleming for
helping me with the immunohistochemistry staining. I also wish to thank Dr Anabel
Sorolla, for helping me with the western blot analyses and for her friendship, enduring
positivity and encouragement especially at times when I have needed it most. Sabine,
Ben, Colette, Mahira, Edi, and Agustin, thank you all for all the love, laughs, support, and
friendship through my PhD years.
I would like to extend my sincerest thanks to my siblings and my parents, Aida and
Kamal, thank you for your emotional support, and prayers. Thanks for believing in me
and being proud of me no matter what path I choose.
Words cannot express my appreciation and love for my lovely kids, Sama and Mostafa,
who have given me much happiness and keep me hopping. I hope I have been a good
mother and that I have not lost too much during the time of my study.
Saving the most important for last, I wish to give my heartfelt thanks to my husband,
Ebrahim, you have continued to love and support me when I have been at my worst during
this process. Thank you for being my rock throughout my PhD, for taking care of me, for
your encouragement and motivation.

iv
Publications
Journal Articles
Stolzenburg S, Beltran AS, Swift-Scanlan T, Rivenbark AG, Rashwan R, Blancafort P.
Stable oncogenic silencing in vivo by programmable and targeted de novo DNA
methylation in breast cancer. Oncogene 2015, 34:5427-35.
Sorolla A, Ho D, Wang E, Evans CW, Ormonde CF, Rashwan R, Singh R, Iyer KS,
Blancafort P. Sensitizing basal-like breast cancer to chemotherapy using nanoparticles
conjugated with interference peptide. Nanoscale 2016, 8:9343-53.
Rashwan R, Sorolla A, Parry J, Redfern A, Blancafort P. Characterization of the novel
oncogenic role of C11orf67 in luminal breast cancer. In Preparation (2016).
Rashwan R, Varano J, Lansley S, Lee G. Streptococcus pneumoniae, but not other
bacterial empyema pathogens, induces mesothelial cell death. In Preparation (2016).
Conferences
Rashwan R, Sorolla A, and Blancafort P (2014) “Characterization of the novel oncogenic
role of C11orf67 in luminal breast cancer”. Poster Presentation, 7th Cairo International
Biomedical Engineering Conference CIBEC, Cairo, Egypt. December 2014.
Rashwan R, Sorolla A, and Blancafort P (2014) “Characterization of the novel oncogenic
role of C11orf67 in luminal breast cancer”. Oral Presentation, Cancer Council 5th
Biennial Research Symposium, Perth, Australia. October 2014.
Rashwan R, Sorolla A, and Blancafort P (2014) “Characterization of the novel oncogenic
role of C11orf67 in luminal breast cancer”. Poster Presentation, 26th Lorne Cancer
Conference, Melbourne, Australia. February 2014.

v
Rashwan R, and Blancafort P (2013) “Characterization of the novel oncogenic role of
C11orf67 in luminal breast cancer”. Oral Presentation, 8th State Cancer Conference,
Perth, Australia. October 2013.
Rashwan R, and Blancafort P (2013) “Characterization of the novel oncogenic role of
C11orf67 in luminal breast cancer”. Oral Presentation, COMBIO, Perth, Australia.
October 2013.
Rashwan R, Varano J, Lansley S, and Lee G (2013) “Streptococcus pneumoniae, but not
other bacterial empyema pathogens, induces mesothelial cell death”. Poster Presentation,
Thoracic Society of Australia and New Zealand TSANZ, Darwin, Australia. March 2013.

vi
Awards
2015
UWA Completion scholarship, University of Western Australia
To support my thesis completion and submission
2014
UWA Graduate Research School Travel Award, University of Western Australia
To support my travel to the 7th CIBEC meeting, Egypt 2014
2012
LIWA PhD Top-Up scholarship award (2012-2013), Lung Institute of Western
Australia
To support my postgraduate studies at the University of Western Australia
2011
International Egyptian scholarship award (2011-2015), Mission department and
scholarship office, Egypt
To support my postgraduate studies at the University of Western Australia

vii
Table of Contents
Chapter 1: General Introduction .......................................................... 1
1.1 Breast cancer ................................................................................................... 2
1.1.1 Incidence and mortality ................................................................................. 2
1.1.2 Normal mammary gland development .......................................................... 2
1.1.3 Breast cancer heterogeneity .......................................................................... 4
1.1.4 Prognosis of breast cancer ............................................................................. 6
1.1.4.1 Histological classification ..................................................................... 6
1.1.4.2 The Nottingham histological grade ....................................................... 6
1.1.4.3 Tumour Node Metastasis Staging ......................................................... 6
1.1.4.4 Immunohistochemical markers ............................................................. 7
1.1.5 Management of breast cancer........................................................................ 7
1.1.5.1 Surgery .................................................................................................. 7
1.1.5.2 Radiotherapy ......................................................................................... 8
1.1.5.3 Chemotherapy ....................................................................................... 8
1.1.5.4 Targeted therapies ................................................................................ 9
1.2 Breast cancer subtypes ................................................................................. 10
1.2.1 Clinical and molecular classification of breast cancer ................................ 10
1.2.2 Intrinsic breast cancer subtypes .................................................................. 11
1.2.2.1 Luminal A breast cancer ..................................................................... 12
1.2.2.2 Luminal B breast cancer ..................................................................... 12
1.2.2.3 Basal-like breast cancer ...................................................................... 13
1.2.2.4 HER2-enriched breast tumours........................................................... 14
1.2.2.5 Claudin-low subtype ........................................................................... 14
1.2.3 Inter-cluster breast cancer subtypes ............................................................ 15
1.3 The ER pathway and tamoxifen resistance ................................................ 17
1.3.1 Estrogen Receptor (ER) .............................................................................. 17
1.3.2 ER domain structure .................................................................................... 18
1.3.3 ER signalling pathway ................................................................................ 18
1.3.4 Transcriptional output of ER signalling ...................................................... 21
1.3.5 Endocrine therapy and ER+ breast cancer ................................................... 21

viii
1.3.6 Mechanisms of tamoxifen resistance .......................................................... 21
1.3.6.1 Coregulators of the ER ....................................................................... 22
1.3.6.2 Loss of ER and activation of growth factor receptor pathways .......... 23
1.3.6.3 Cell cycle signalling regulators .......................................................... 23
1.4 Molecular pathogenesis of breast cancer .................................................... 25
1.4.1 Oncogenes and tumour suppressor genes ................................................... 25
1.4.2 The “case” of the 11q13-q14 amplicon in breast Cancer ............................ 26
1.4.2.1 CCND1 ................................................................................................ 29
1.4.2.2 EMSY ................................................................................................... 29
1.4.2.3 PAK1 ................................................................................................... 29
1.4.2.4 AQP11 ................................................................................................. 30
1.4.2.5 RSF1 .................................................................................................... 30
1.4.2.6 GAB2 ................................................................................................... 30
1.4.2.7 C11orf67 ............................................................................................. 31
1.5 Statement of aims .......................................................................................... 32
Chapter 2: C11orf67 as a Novel Biomarker in Breast Cancer ......... 34
2.1 Introduction ................................................................................................... 35
2.2 Materials and Methods ................................................................................. 38
2.2.1 Cell lines and cell culture ............................................................................ 38
2.2.2 RNA extraction ........................................................................................... 38
2.2.3 Reverse transcription and cDNA synthesis ................................................. 39
2.2.4 Real-time Polymerase Chain Reaction ....................................................... 39
2.2.5 Total protein extraction and western blotting ............................................. 39
2.2.6 Tissue Microarrays ...................................................................................... 40
2.2.7 Immunocytochemistry ................................................................................ 41
2.2.8 Fluorescence microscopy ............................................................................ 41
2.2.9 Fluorescence In Situ Hybridization ............................................................. 42
2.2.10 Statistical analysis ................................................................................... 43
2.3 Results ............................................................................................................ 44
2.3.1 Bioinformatic and structural analyses of C11orf67 .................................... 44
2.3.2 Identification of the full-length cDNA sequence and spliced transcripts of
C11orf67. ................................................................................................................ 44
2.3.3 Subcellular localization of Flag-tagged C11orf67 Isoform_2 .................... 49

ix
2.3.4 The pattern of C11orf67 gene alteration in cancer...................................... 50
2.3.5 C11orf67 expression profile in breast cell lines .......................................... 52
2.3.6 C11orf67 is overexpressed in HR+ and high-grade breast cancer ............... 56
2.3.7 Copy number amplification of C11orf67 in HR+ breast cancer .................. 60
2.1 Discussion ............................................................................................................. 63
Chapter 3: Functional Consequences of C11orf67 Gene Alteration 67
3.1 Introduction ................................................................................................... 68
3.2 Materials and Methods ................................................................................. 71
3.2.1 Reagents and Antibiotics............................................................................. 71
3.2.2 Lentiviral C11orf67 shRNA infection ........................................................ 71
3.2.3 Lentiviral C11orf67 cDNA infection .......................................................... 72
3.2.4 MTT cell viability assays ............................................................................ 73
3.2.5 Immunofluorescence ................................................................................... 73
3.2.6 Anchorage-independent colony formation assays ...................................... 74
3.2.7 In Vitro cell migration assay ....................................................................... 74
3.2.8 TUNEL assay .............................................................................................. 75
3.2.9 In vivo tumourigenicity assays.................................................................... 76
3.2.10 Immunohistochemistry of tumour sections ............................................. 76
3.2.11 Senescence-associated β-galactosidase staining ..................................... 77
3.2.12 Illumina microarray analysis ................................................................... 77
3.2.12.1 RNA preparation ............................................................................. 77
3.2.12.2 Labelling and Purification .............................................................. 78
3.2.12.3 Hybridization and data export ........................................................ 78
3.2.12.4 Raw data preparation and statistical analyses ............................... 78
3.2.13 Statistical analysis ................................................................................... 79
3.3 Results ............................................................................................................ 80
3.3.1 Functional consequences of C11orf67 ablation in T47D cells ................... 80
3.3.1.1 Knockdown of C11orf67 by shRNAs ................................................... 80
3.3.1.2 C11orf67 ablation reduces the proliferative potential of T47D ......... 83
3.3.1.3 C11orf67 knockdown decreases tumourigenic phenotype of T47D cells
in vitro ............................................................................................................. 85
3.3.1.4 C11orf67 promotes migratory behaviour in ER+ breast cancer cells 87

x
3.3.1.5 Downregulation of C11orf67 expression inhibits tumour growth in a
xenograft model................................................................................................... 88
3.3.1.6 C11orf67 knockdown is not involved in cellular apoptosis ................ 90
3.3.1.7 C11orf67 knockdown induces cellular senescence ............................. 92
3.3.1.8 C11orf67 knockdown deactivates AKT/mTOR and MAPK pathways 92
3.3.1.9 Identification of genes and biological pathways downstream of
C11orf67 ............................................................................................................. 95
3.3.2 Functional analyses of C11orf67 overexpression in breast cell lines ......... 97
3.3.2.1 C11orf67 activates genes involved in metabolism .............................. 99
3.3.2.2 C11orf67 overexpression increases sensitivity to anti-metabolites .. 101
3.4 Discussion ..................................................................................................... 103
Chapter 4: Regulation and Binding Partners of C11orf67 ............. 107
4.1 Introduction ................................................................................................. 108
4.2 Materials and Methods ............................................................................... 111
4.2.1 Reagents and Antibodies ........................................................................... 111
4.2.2 Cell Culture ............................................................................................... 111
4.2.3 siRNA Transfection .................................................................................. 112
4.2.4 Rat AAMDC qRT-PCR ............................................................................ 112
4.2.5 Chromatin immunoprecipitation assay ..................................................... 113
4.2.6 Luciferase assay ........................................................................................ 113
4.2.7 Yeast Two-Hybrid Analysis...................................................................... 114
4.2.8 Statistical analysis ..................................................................................... 115
4.3 Results .......................................................................................................... 116
4.3.1 11q13.5-q14 locus is enriched in transcription factor binding sites ......... 116
4.3.2 Estrogen modulates C11orf67 gene expression. ....................................... 117
4.3.3 Estrogen downregulates C11orf67 expression in an ER-dependent manner ..
................................................................................................................... 121
4.3.4 Tamoxifen upregulates expression of multiple oncogenes in the 11q13.5-
q14 cluster ............................................................................................................. 121
4.3.5 Aberrant expression of C11orf67 alters the sensitivity to tamoxifen ....... 124
4.3.6 C11orf67 is upregulated in pregnancy ...................................................... 126
4.3.7 NF-κB regulates the expression of C11orf67 ........................................... 128
4.3.8 C11orf67 acts as a regulator of NF-κB transcriptional activity ................ 130

xi
4.3.9 RABGAP1L as a binding partner of C11orf67 ......................................... 132
4.3.10 RABGAP1L expression co-localises with C11orf67 in ER+ breast
cancers ............................................................................................................... 133
4.4 Discussion ..................................................................................................... 136
Chapter 5: General Discussion .......................................................... 141
Chapter 6: References ........................................................................ 153

xii
List of Figures
Figure 1-1 Schematic representation of the mammary gland ........................................... 3
Figure 1-2 Hormonal control of female mammary gland development ........................... 4
Figure 1-3 Possible models of origin of breast cancer subtypes ....................................... 5
Figure 1-4 The clinical outcomes of the inter-cluster subgroups .................................... 16
Figure 1-5 Schematic representation of ER protein and ER signalling pathways .......... 20
Figure 1-6 Detailed schematic diagram of the 11q13-q14 amplicon .............................. 27
Figure 2-1 Selective overexpression of C11orf67 in breast cancer subtypes ................. 36
Figure 2-2 Phylogenetic analysis and structural similarity of C11orf67 ........................ 46
Figure 2-3 Alternative splicing of the C11orf67 gene .................................................... 47
Figure 2-4 Cellular localization of C11orf67 Isoform_2 ................................................ 49
Figure 2-5 Alteration frequency and survival probability of C11orf67 in different
cancers ............................................................................................................................. 51
Figure 2-6 Expression pattern of C11orf67 in breast cell lines ...................................... 55
Figure 2-7 IHC staining of C11orf67 in 75 cases of breast cancer TMA ....................... 58
Figure 2-8 C11orf67 amplification detected by FISH in ER+ breast cancer .................. 62
Figure 3-1 Illustration of the PI3K/Akt/mTOR pathway ................................................ 69
Figure 3-2 Knockdown of C11orf67 by shRNAs in T47D cells .................................... 82

xiii
Figure 3-3 C11orf67 knockdown decreases cellular proliferation .................................. 84
Figure 3-4 C11orf67 knockdown decreases cellular colony formation .......................... 86
Figure 3-5 C11orf67 knockdown decreases cellular migration ...................................... 87
Figure 3-6 C11orf67 knockdown inhibits in vivo tumour growth .................................. 89
Figure 3-7 C11orf67 knockdown is not associated with cellular apoptosis.................... 91
Figure 3-8 C11orf67 knockdown induces cellular senescence ....................................... 93
Figure 3-9 C11orf67 knockdown deactivates Akt/mTOR and MAPK pathways .......... 94
Figure 3-10 Expression profiling of C11orf67 knockdown cells ................................... 96
Figure 3-11 C11orf67 overexpression does not increase cellular proliferation .............. 98
Figure 3-12 Changes in gene/protein expression in response to C11orf67 overexpression
....................................................................................................................................... 100
Figure 3-13 C11orf67 overexpression increases the sensitivity to one- carbon folate
antagonists ..................................................................................................................... 102
Figure 4-1 Canonical and alternative pathways of NF-κB activation ........................... 109
Figure 4-2 Transcription factor binding sites of the 11q13.5-q14 amplicon ................ 118
Figure 4-3 C11orf67 is regulated by Estrogen .............................................................. 120
Figure 4-4 Estrogen regulates C11orf67 expression in an ER dependent manner ....... 122
Figure 4-5 Effect of estradiol and tamoxifen on the expression of multiple oncogenes at
11q13.5-q14 .................................................................................................................. 123

xiv
Figure 4-6 Changes in C11orf67 mRNA levels alter the sensitivity of T47D cells to
tamoxifen ...................................................................................................................... 125
Figure 4-7 Effect of pregnancy hormones on C11orf67 expression ............................. 127
Figure 4-8 C11orf67 is regulated by NF-κB ................................................................. 129
Figure 4-9 Effect of C11orf67 knockdown on NF-κB activity ..................................... 131
Figure 4-10 Y2H screen reveals RABGAP1L and SF3B1 as binding partners of
C11orf67 ....................................................................................................................... 134
Figure 4-11 The intracellular pattern of RABGAP1L expression corresponds to that of
C11orf67 expression in breast cancer sections and cell lines ....................................... 135
Figure 5-1 Proposed mechanisms underlying C11orf67-induce tamoxifen resistance in
breast cancer .................................................................................................................. 148

xv
List of Tables
Table 1-1 Potential oncogenes residing in the 11q13.5-q14 cis-acting amplicon........... 28
Table 2-1 Characteristics of the C11orf67 isoforms ....................................................... 48
Table 2-2 Source, clinical and pathological features of breast cancer cell lines used in
this study ......................................................................................................................... 53
Table 2-3 C11orf67 expression in 75 cases of breast cancer patients............................. 59
Table 3-1 Nucleotide sequences and exon number of C11orf67-specific shRNAs ........ 72

xvi
List of abbreviations
4EBP1 Eukaryotic initiation factor 4E binding protein
5-FU 5-Fluorouracil
aa Amino acid
AAMDC Adipogenesis associated Mth938 domain containing
AD Activating domain
AF-1 Activation function-1
AF-2 Activation function-2
AIHW Australian Institute of Health and Welfare
AIs Aromatase inhibitors
APQ 6Amino-4(4-phenoxyphenylethylamino) quinazoline
AR Androgen receptor
bp Base pair
BSA Bovine Serum Albumin
CCND1 Cyclin D1
CCNE1 Cyclin E1
CDK Cyclin-dependent kinase
ChIP Chromatin immunoprecipitation
CMF Cyclophosphamide, Methotrexate and 5-Fluorouracil
CNA Copy number amplification
CSS Charcol-stripped seum
DBD DNA binding domain
DCIS Ductal Carcinoma In Situ
DHFR Dihydrofolate reductase
DMEM Dulbecco’s Modified Eagle Medium
DMSO Dimethylsulphoxide
dUTP Deoxyuridine triphosphate
E2 17β-Estradiol
EDTA Ethylenediaminetetraacetic acid
EGF Epidermal growth factor
EGFR1 Epidermal growth factor receptor 1
ER Estrogen receptor

xvii
ER+ Estrogen receptor positive
ERBB2 Erythroblasts leukemia viral oncogene homolog 2
ERE Estrogen Response Elements
FBS Fetal Bovine Serum
FFPE Formalin-Fixed, Paraffin-Embedded
FISH Fluorescence In Situ Hybridization
GHR Growth hormone receptor
HER2 Human epidermal growth factor receptor-2
HR Hormone receptor
HR+ Hormone receptor positive
HuMEC Human mammary epithelial cells
ICC Immunocytochemistry
IDC Invasive ductal carcinoma
IHC Immunohistochemistry
IKKα/β Inhibitor of nuclear factor kappa-B kinase subunit alpha and
beta
ILC Invasive lobular carcinoma
IntClust 2 Inter-cluster 2 subtype
IκBα Inhibitor kappa B alpha
LBD Ligand-binding domain
LN Lymph node
MAPK Mitogen-activated kinases
Mif Mifepristone
Mth938 Methanobacterium thermoautotrophicum
MTHFD2 Methylenetetrahydrofolate dehydrogenase
mTOR Mammalian target of rapamycin
MTX Methotrexate
MYC v-myc avian myelocytomatosis viral oncogene homolog
NAT Normal adjacent breast tissue
NF-κB Nuclear factor Kappa B
NHG Nottingham Histological Grade
NLS Nuclear localisation signal
NSCLC Non-small cell lung cancer
ORF Open reading frame
P13K Phosphoinositide 3-kinase

xviii
PAK1 p21-activated kinase 1
PBS Phosphate Buffered Saline
PCR Polymerase chain reaction
Pen/Strep Penicillin-Streptomycin
PFA Paraformaldehyde
PG Progesterone
PIP2 Phosphatidylinositol 4,5 bisphosphates
PIP3 Phosphatidylinositol 3,4,4-triphosphate
PPARGC1 Peroxisome proliferator-activated receptor gamma,
coactivator 1
Ppp4 Phosphoprotein phosphatase 4
PR Progesterone receptor
PR+ Progesterone receptor positive
qRT quantitative Real-time
RABGAP1L Ras-related in brain GTPase-activating protein 1-like
RB Retinoblastoma
RSF1 Remodeling and spacing Factor 1
S6K1 40S ribosomal protein S6 kinase 1
SC Subcutanous
SD Standard deviation
SDS Sodium dodecyl sulfate
SE Standard error
SERDs Selective estrogen receptor downregulators
SERMs Selective estrogen receptor modulators
SFM Serum-free media
sh1 C11orf67 shRNA 1
sh2 C11orf67 shRNA 2
sh5 C11orf67 shRNA 5
SHMT Serine hydroxymethyl transferase
shRNA Short hairpin RNA
SM Second messengers
SOP Standard operating procedures
Tam Tamoxifen
TBS-T Tris-buffered saline/Tween
TCGA The Cancer Genome Atlas

xix
TDLU Terminal ductal lobular units
TdT Terminal deoxy-transferase
TF Transcription factor
TFBS Transcription factor binding site
TMA Tissue microarray
TNBC Triple-negative breast cancer
TNFα Tumour necrosis factor alpha
TNM Tumour Node Metastasis
TS Thymidylate synthase
TSS Transcription start site
TUNEL Terminal deoxynucleotidyl transferase-dUTP nick end
labeling
UAS Upstream activation sequences
VEGFA Vascular endothelial growth factor A
Y2H Yeast two-hybrid
β-gal β-galactosidase


Chapter 1:
General Introduction

General Introduction
2
1.1 Breast cancer
1.1.1 Incidence and mortality
Breast cancer is the most common invasive tumour in women and one of the leading
causes of cancer-related deaths worldwide (Jemal et al., 2011, Torre et al., 2015).
According to the Australian Institute of Health and Welfare (AIHW) Report 2014, breast
cancer is the third most commonly diagnosed cancer in Australia. It is expected that
15,740 new cases of breast cancer will be detected in 2015, with an age-standardised
incidence rate of 59 cases per 100,000 of the population (AIHW, 2014).
In Australia, 2015 statistical records show that breast cancer remains the fourth most
common cause of death from cancer (AIHW, 2014). The mortality rate from breast cancer
in Australia is predicted to increase from 2,819 deaths in 2012 to 3,065 deaths in 2015
(25 males and 3,040 females). Although the number of breast cancer cases are increasing
each year, the five-year relative survival from breast cancer improved from 72% between
1982-1986 to 90% between 2007-2011 (AIHW, 2014).
1.1.2 Normal mammary gland development
It is important first to introduce the structure of the normal mammary gland and its
hierarchical tissue organization to understand the pathogenesis of breast cancer. The
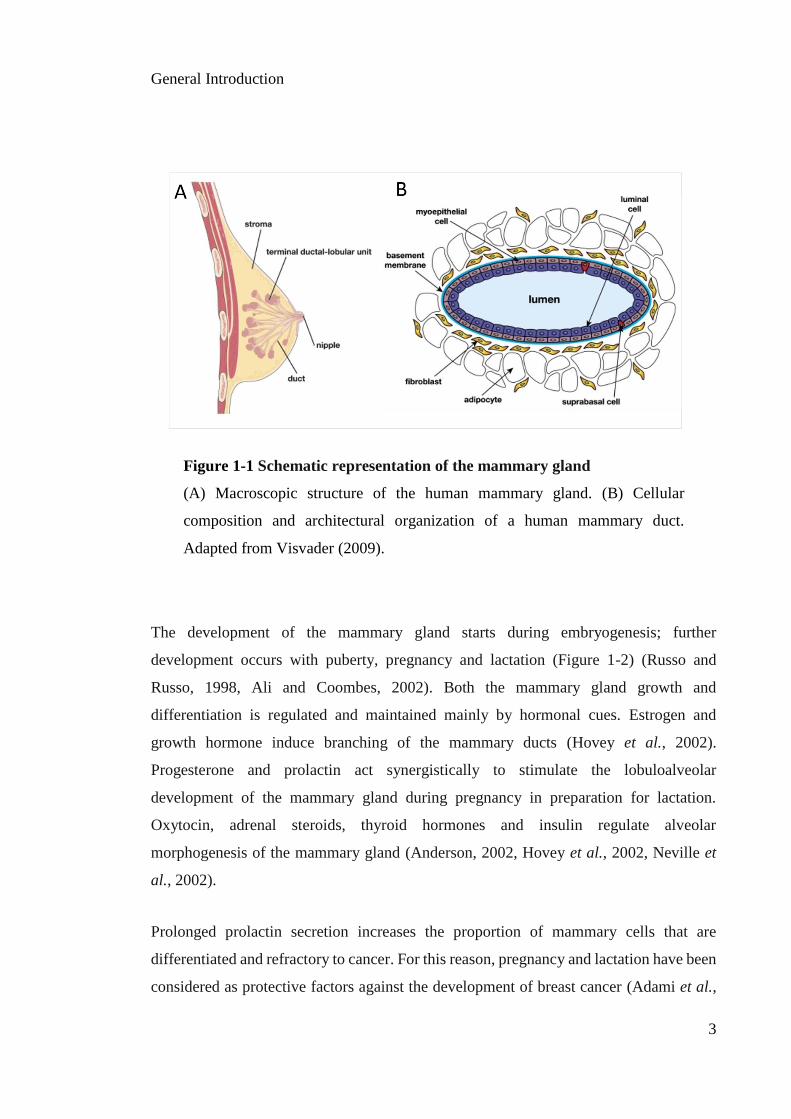
human breast is a tubuloalveolar gland characterized by a branching network of ducts.
Clusters of small ducts constitute the terminal ductal lobular units (TDLUs) (Visvader,
2009) (Figure 1-1A). The TDLUs eventually mature into a more complex structure
containing several lobules per lobe, with each lobe functioning as a separate gland (Mills
et al., 2011, Lanfranchi, 2014). The cellular epithelial architecture is composed of a
bilayer of luminal cells surrounding an inner lumen and an external layer of myoepithelial
cells that contact the basement membrane (Figure 1-1B). These epithelial cells are
surrounded by fibroblasts and adipocytes, which compose the stroma of the mammary
gland (Anderson, 2002, Visvader, 2009).
General Introduction
3
The development of the mammary gland starts during embryogenesis; further
development occurs with puberty, pregnancy and lactation (Figure 1-2) (Russo and
Russo, 1998, Ali and Coombes, 2002). Both the mammary gland growth and
differentiation is regulated and maintained mainly by hormonal cues. Estrogen and
growth hormone induce branching of the mammary ducts (Hovey et al., 2002).
Progesterone and prolactin act synergistically to stimulate the lobuloalveolar
development of the mammary gland during pregnancy in preparation for lactation.
Oxytocin, adrenal steroids, thyroid hormones and insulin regulate alveolar
morphogenesis of the mammary gland (Anderson, 2002, Hovey et al., 2002, Neville et
al., 2002).
Prolonged prolactin secretion increases the proportion of mammary cells that are
differentiated and refractory to cancer. For this reason, pregnancy and lactation have been
considered as protective factors against the development of breast cancer (Adami et al.,
Figure 1-1 Schematic representation of the mammary gland
(A) Macroscopic structure of the human mammary gland. (B) Cellular
composition and architectural organization of a human mammary duct.
Adapted from Visvader (2009).

General Introduction
4
1998). However, elevated circulating prolactin is associated with higher risk of breast
cancer development with poorer patient outcomes (Swaminathan et al., 2008).
1.1.3 Breast cancer heterogeneity
Heterogeneity is one of the hallmarks of breast cancer (Kim et al., 2012). Intratumoural
heterogeneity accounts for the intrinsic differences within individual tumours, leading to
extensive variation in phenotypic properties and a different pattern of expression of
molecular markers (Visvader, 2011). Intertumoural heterogeneity leads to the
classification of different tumour subtypes with different morphology, molecular profile
and expression of specific biomarkers (Skibinski and Kuperwasser, 2015).
Two hypothetical mechanisms have been proposed to explain the intertumoural
heterogeneity of breast cancer. Different genetic events may be concurrent within the
Figure 1-2 Hormonal control of female mammary gland development
All hormone receptors (pink boxes) are required in the mammary epithelium.
The growth hormone receptor (GHR) is required in the stroma. Adapted from
Brisken and O'Malley (2010).
General Introduction
5
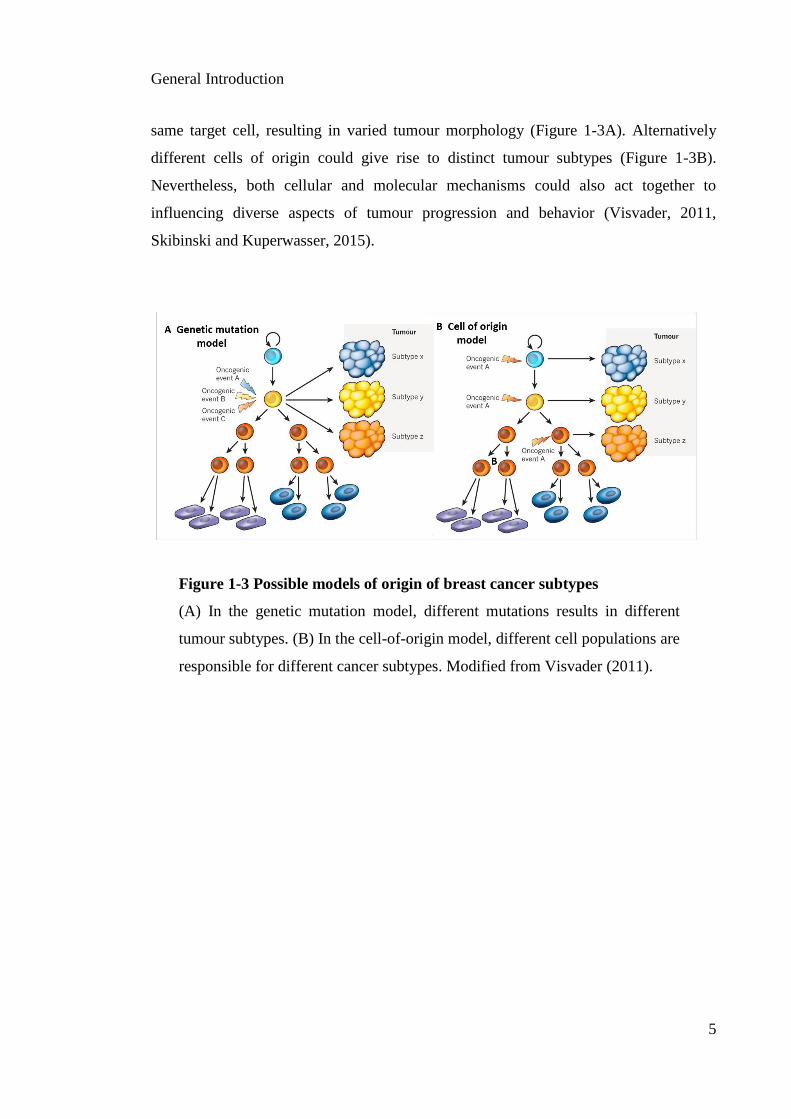
same target cell, resulting in varied tumour morphology (Figure 1-3A). Alternatively
different cells of origin could give rise to distinct tumour subtypes (Figure 1-3B).
Nevertheless, both cellular and molecular mechanisms could also act together to
influencing diverse aspects of tumour progression and behavior (Visvader, 2011,
Skibinski and Kuperwasser, 2015).
Figure 1-3 Possible models of origin of breast cancer subtypes
(A) In the genetic mutation model, different mutations results in different
tumour subtypes. (B) In the cell-of-origin model, different cell populations are
responsible for different cancer subtypes. Modified from Visvader (2011).

General Introduction
6
1.1.4 Prognosis of breast cancer
1.1.4.1 Histological classification
Breast cancer can be classified histologically as non-invasive (referred to in situ) and
invasive. Ductal Carcinoma In Situ (DCIS) is the most common type of non-invasive
cancer where cancer cells are restricted to the basement membrane (Malhotra et al., 2010,
Weigelt et al., 2010). Invasive breast cancer occurs when cancer cells spread beyond the
basement membrane, and is associated with a high risk of distant metastasis involving
the spread of cancer from the breast to secondary sites).
The most common histological type is invasive ductal carcinoma (IDC), which accounts
for about 50-80% of all breast cancers (Zheng et al., 2013). Invasive lobular carcinoma
(ILC) comes next and is found in 5-15% of breast malignancies (Rakha et al., 2008b).
Histological classifications, however, are purely descriptive and in most cases do not
confer prognostic information.
1.1.4.2 The Nottingham histological grade
Grading tumours according to the Nottingham histological grade (NHG) has proven very
useful for predicting disease aggressiveness. In this type of classification tumours are
assigned a grade from I (well differentiated) to III (poorly differentiated), by evaluating
morphological features of the tumour such as tubule formation, nuclear polymorphism,
and mitotic index (Rakha et al., 2008a). The prognostic implication of NHG has been
validated in several independent patient cohorts (Fong et al., 2015, Santos et al., 2015b).
1.1.4.3 Tumour Node Metastasis Staging
The Tumour Node Metastasis (TNM) staging system is a standard method used to classify
breast cancer patients in the routine clinic (Sobin, 2003). In this system, tumour size (T),
lymph node involvement (N) and distant metastasis (M) are taken into account. By

General Introduction
7
combining three factors which each contributes to prognostic information, an estimate of
the clinical stage of the disease is obtained (Kwan et al., 2012).
1.1.4.4 Immunohistochemical markers
Several immunohistochemical markers are assessed clinically to provide prognostic
information, and most importantly, predictive treatment information. Molecular
biomarkers, such as the estrogen receptor (ER), progesterone receptor (PR), and human
epidermal growth factor receptor-2 (HER2), will be discussed in detail in the following
sections.
The proliferative marker Ki-67 is evaluated immunohistochemically to determine the
proliferative activity of a tumour. However, the clinical use of Ki67 is limited, partly due
to the lack of a clearly defined cut-off value, and difficulties in standard operating
procedures (SOP) of the immunohistochemical method (Yerushalmi et al., 2010, Luporsi
et al., 2012).
1.1.5 Management of breast cancer
1.1.5.1 Surgery
Surgery is the principal treatment modality used for breast cancer treatment. It is
conducted either as modified radical mastectomy, partial mastectomy or breast-
conserving surgery. Breast-conserving surgery is the primary choice for patients with
early stage breast cancer (Fisher et al., 2002, Veronesi et al., 2002). During the surgical
procedure, a sentinel lymph node (LN) biopsy is performed to determine the potential
presence of malignant cells disseminating from the primary site of the breast lesion. In
the case of a negative biopsy, further resection of the LNs is not necessary, sparing the
patient from side effects associated with LN removal (Senkus et al., 2013, Gherghe et al.,
2015).

General Introduction
8
1.1.5.2 Radiotherapy
Postoperative radiotherapy is administered to eradicate possible residual microscopic
disease at and around the localization of the tumour (Veronesi et al., 1993). Postoperative
radiotherapy of the breast and thoracic wall is administered when the risk of local
recurrence within the next ten years is greater than 20% (Darby et al., 2011).
Radiotherapy is recommended for breast cancer patients who undergo breast-conserving
therapy, mastectomy, or if the tumour is larger than 20 mm (Del Barco et al., 2013,
Senkus et al., 2013).
Loco-regional radiotherapy is recommended for all patients with metastases in four or
more LNs (Del Barco et al., 2013, Senkus et al., 2013). Radiotherapy induces acute side
effects, such as erythema of the skin and pneumonitis, and late side effects, including
neuropathy of the affected brachial plexus, lymphedema of the upper extremities and
increased mortality from cardiac disease. However, it seems that the improved precision
of modern techniques has reduced the risk of developing these side effects (Clarke et al.,
2005, Darby et al., 2011).
1.1.5.3 Chemotherapy
Chemotherapy is unselective and targets all dividing or proliferating cells. The synergic
effect of several cytotoxic agents is achieved by targeting multiple pathways (Cazzaniga
et al., 2006, Sukel et al., 2008, Greenberg et al., 2011, Del Barco et al., 2013). A
combination of cyclophosphamide, methotrexate and fluorouracil (CMF) was the
standard regimen during the late 1970s and the first half of the 1980s. Further
improvement of breast cancer treatment outcomes was observed after the addition of the
taxanes (Gines et al., 2011). Both methotrexate (MTX) and 5-fluorouracil (5-FU) target
the one-carbon metabolic pathway and consequently inhibit the cells from synthesizing
nucleic acids inducing cell cycle arrest and apoptosis (Xu and Chen, 2009). MTX and 5-
FU interfere with the biosynthesis of thymidylate and purine by inhibiting the
mitochondrial enzyme dihydrofolate reductase (DHFR) and the thymidylate synthase

General Introduction
9
(TS) respectively. Therefore, these two agents show selectivity to rapidly proliferating
cancer cells overexpressing genes coding for folate metabolism enzymes (Vazquez et al.,
2013).
The standard chemotherapy regimens used today include different combinations of
anthracyclines, taxanes, cyclophosphamide, MTX and 5-FU (Gianni et al., 2011).
However, because of the lack of cellular selectivity, chemotherapy regimens are
associated with significant toxicities. For some patients, chemotherapy is effective at the
beginning of the treatment, but later resistance is observed, particularly in the metastatic
setting (when the tumour has already spread from the site of origin). This is a particular
problem for aggressive breast cancers that present with recurrence. For these patients, the
development of targeted therapeutic approaches would represent a very significant
advance over non-specific approaches.
1.1.5.4 Targeted therapies
Targeted therapies offer the possibility of efficient and tailored treatment based on the
molecular profile of the tumour (Saji and Kimura-Tsuchiya, 2015). The combination of
targeted therapy with either chemotherapy or endocrine therapy based on patient
predictive biomarkers has been shown to significantly enhance the overall survival in
patients with breast cancer (Kawalec et al., 2015).
An example of a novel targeted agent is the mammalian target of rapamycin (mTOR)
inhibitor everolimus (Fedele et al., 2012). Adding everolimus to endocrine therapy has
improved the survival of hormone receptor (HR) positive patients compared to endocrine
therapy alone (Bachelot et al., 2014, Lin et al., 2015, Xie et al., 2015).
However, targeted therapies are not yet standard strategies for the management of breast
cancer patients. Better trial designs, and tumour and patient selection criteria will be
critical to understanding the complexity of the targeted therapy (Fedele et al., 2012). In
any event, stratification of breast cancers in molecular subtypes represents the first critical
step in the development of novel tailored treatments.

General Introduction
10
1.2 Breast cancer subtypes
1.2.1 Clinical and molecular classification of breast cancer
Three well-understood molecular markers with established prognostic and predictive
outcomes are routinely used in the clinic: ER, PR, and HER2 (Davies et al., 2011). Based
on the expression levels of these receptors in breast tumour biopsies breast cancers are
clinically classified as hormone receptor-positive (HR+), HER2+, or triple-negative if they
lack the expression of all three receptors (ER-, PR-, and HER2-) (Cianfrocca and
Goldstein, 2004). Receptor levels are scored by a pathologist using standardized
immunohistochemical procedures.
The nuclear receptors ER and PR facilitate cellular growth and proliferation by binding
with their cognate ligands estrogen and progesterone. Ligand-bound receptors
consequently act as transcriptional regulators that stimulate the expression of pro-
proliferative genes in mammary epithelial cells. Thus, disruption of the molecular
mechanisms of regulation of these receptors can contribute to carcinogenesis. In general,
HR expression is associated with a good prognosis and predicts a positive response to
hormonal treatment (Utsumi et al., 2007). In contrast, HER2 overexpression is associated
with copy number amplification and, to a lesser extent, chromosomal polysomy, is a
marker for a poor prognosis and indicates treatment with HER2 inhibitors such as
trastuzumab (Hudis, 2007).
Triple-negative breast cancer (TNBC) which lacks expression of ER, PR and HER2,
represents 30% of primary breast cancers and is usually associated with larger tumour
size, lymph nodal positivity, and poor outcome (Bae et al., 2015). Due to its molecular
nature and lack of well-studied targets, neither antibody nor hormone therapies benefit
patients with TNBC (Han et al., 2015). Because of its poor prognosis, this type of breast
cancer is in great need of novel therapeutic strategies.

General Introduction
11
1.2.2 Intrinsic breast cancer subtypes
The development of DNA Microarray technology has greatly expanded understanding of
cancer biology at both the molecular and transcriptional level through the interrogation
of tens of thousands of expressed genes simultaneously (Creighton, 2012).
Hierarchical clustering of breast cancer patients based on differentially expressed intrinsic
genes has led to the identification of novel molecular subclasses of breast cancer. Intrinsic
molecular classification of human breast cancer includes luminal A, luminal B, basal-
like, normal-type and HER2+ subtypes (Perou et al., 2000). Results from these seminal
studies have shown strong reproducibility across many different datasets. Therefore, these
five defined subtypes are usually referred to as the “intrinsic subtypes of breast cancer”
(Santos et al., 2015a).
Importantly, the molecular subtyping of breast tumours mostly reflects the established
clinical and histopathological-based classifications, with the basal-like subtype
representing ER-/HER2- diseases, HER2-enriched representing ER-/HER2+, and the
normal-like and luminal A/B subtypes representing ER+ (Creighton, 2012). More
recently, larger dataset studies have identified additional molecular subtypes including
the interferon-rich, claudin-low and molecular apocrine subtypes (Hu et al., 2006,
Herschkowitz et al., 2007, Reis-Filho and Pusztai, 2011). Larger and independent cohorts
of patients have been used to validate the clinical value of this classification (Sorlie et al.,
2003).
Although gene expression analyses do reflect outcomes, patterns of clinical behavior and
response to therapy, genome-wide gene expression analyses are not sufficiently cost
effective to be implemented in a routine test for breast cancer patients. Instead, a much
smaller gene set predictor comprising 50 genes (PAM50) has been used to stratify breast
cancer samples into the intrinsic subtypes using a quick and cost-effective quantitative
real-time Polymerase Chain Reaction (qRT-PCR) assay (Parker et al., 2009). The PAM50
assay also provides a risk of relapse score for each patient and is highly predictive of
treatment response (Nielsen et al., 2010, Dowsett et al., 2013).

General Introduction
12
1.2.2.1 Luminal A breast cancer
Luminal-like breast cancer derives its name from the finding that these tumours show
similar expression profiles of keratins 8/18 typically associated with normal luminal
epithelial cells (Perou et al., 2000).
Luminal A breast cancers are ER+, HER2- and represent 40% of all breast cancers (Guiu
et al., 2012). The molecular profiling of this subtype has shown that these tumours have
relatively lower rates of proliferation and high expression of ER-regulated genes (Cancer
Genome Atlas, 2012). Luminal A tumours are generally sensitive to anti-hormonal
therapy and patients tend to have better clinical outcomes than patients with other intrinsic
subtypes (Parker et al., 2009, Prat and Perou, 2011).
1.2.2.2 Luminal B breast cancer
Luminal B breast cancers represent 20% of all breast malignancies and 30% of ER+ breast
cancers. These luminal tumours are highly proliferative (mostly Ki-67 positive) and
generally do not benefit from anti-hormonal therapy or chemotherapy and consequently
have a high rate of relapse after treatment (Allred et al., 2004, Cui et al., 2005).
Luminal B breast tumours are generally more heterogeneous than the luminal A subtype.
They characteristically express lower levels of ER-related genes than luminal A, show
variable HER2 expression and in many cases these tumours lose PR expression, and
become dependent on other signalling pathways, such as high epidermal growth factor
receptor 1 (EGFR1) pathway (Prat and Perou, 2011, Guiu et al., 2012, 2012).
Luminal B tumours have also been shown to have frequent focal regions of chromosomal
amplification, p53 mutations, and aneuploidy (presence of an abnormal number of
chromosomes in a cell) (Cancer Genome Atlas, 2012, Habashy et al., 2012).
Consequently, patients with luminal B malignancies have significantly worse outcomes
and a relatively higher risk of relapse than luminal A patients, and treatment protocols
vary significantly on a tumour to tumour basis (Prat and Perou, 2011).

General Introduction
13
The Cancer Genome Atlas (2012) (TCGA) consortium has yielded a catalogue of genes
that are amplified or deleted in luminal B cancers and many of these aberrations may have
significant roles in breast cancer development and progression. Having mapped potential
genomic aberrations in these tumours, the real challenge is to distinguish the genomic
alterations that drive the development and progression (Creighton, 2012). As drivers can
evolve during disease progression; another challenge is to distinguish mutations that are
important during tumourigenesis but are dispensable during the clonal evolution of the
tumour (Koch, 2014).
1.2.2.3 Basal-like breast cancer
Basal-like tumours include an extremely heterogeneous group representing
approximately 15% of all breast cancer cases. Approximately 80% of basal tumours are
TNBCs. These tumours have high proliferative rates and show gene expression patterns
more similar to that of basal mammary epithelial cell populations, as opposed to the
luminal epithelium (Cancer Genome Atlas, 2012). Basal-like tumours display high rates
of p53 mutations (84%), and have high expression of DNA repair proteins.
Unsurprisingly, they have very high genomic instability, and most samples show
aneuploidy (Jiao et al., 2014).
Patients with basal-like tumours have significantly poorer outcomes than patients with
other intrinsic types (Prat and Perou, 2011). Many of the established molecular therapies
that work in other intrinsic subtypes are minimally effective in basal-like tumours due to
the high percentage that are TNBCs. However, novel treatments for basal-like and TNBC
tumours are continually being tested both at the bench and clinically, employing both
traditional DNA-damaging chemotherapeutic agents as well as targeted molecular
therapies for other pathways associated with TNBC (Griffiths and Olin, 2012, Shastry
and Yardley, 2013).

General Introduction
14
1.2.2.4 HER2-enriched breast tumours
HER2 is a member of the EGFR family, consisting of four different receptor tyrosine
kinases: HER1 (EGFR), HER2, HER3, and HER4. Upon ligand binding, EGFRs dimerise
and cross-phosphorylate each monomer to initiate downstream signalling pathways,
facilitating cellular proliferation, differentiation, adhesion, and migration. Unlike other
EGFR family members, HER2 does not require ligand binding to dimerise and initiate
downstream signalling (Ciardiello and Tortora, 2008). Instead, it forms heterodimers with
the other HER receptors, thereby extending ligand interaction and prolonging pathway
activation (Harari and Yarden, 2000, Schmitt, 2009, Barros et al., 2010).
Overexpression of HER2 occurs in 15% of breast cancers and is associated with poor
clinical outcomes (Slamon et al., 1987, Borg et al., 1990, Tolaney et al., 2015).
Trastuzumab selectively antagonizes HER2 proteins and improves overall survival
(Barros et al., 2010, Giampaglia et al., 2010). The level of HER2 expression is determined
by immunohistochemistry (IHC), and scored either 0, 1+, 2+ or 3. Gene amplification is
evaluated using in situ hybridization techniques in the cases with high IHC scores
(Piccart-Gebhart et al., 2005, Romond et al., 2005, Gianni et al., 2011).
1.2.2.5 Claudin-low subtype
The claudin-low group was identified by Herschkowitz et al. (2007). This subtype has
low expression of claudin genes which are associated with tight junctions and cell-cell
adhesion, and therfore referred to as mesenchymal cancers. Other hallmarks of these
tumours include the enrichment of stem cell-like markers (high CD44/CD24 ratios) and
high lymphocyte infiltration (Prat and Perou, 2011). Patients with, these tumours show
poor prognosis, reduced survival curves, and variable response to therapy that is
intermediate between basal and luminal subtypes (Prat et al., 2010).

General Introduction
15
1.2.3 Inter-cluster breast cancer subtypes
Integrated genomic and transcriptomic dataset analyses of breast tumour samples has
helped define novel functional intrinsic subtypes of breast cancer and determine possible
somatic drivers in the process of breast cancer development. Curtis et al. (2012) defined
levels of gene expression and both copy number variants and somatic variants in a group
of primary breast cancer patients.
The clustering analyses suggested ten integrative clusters (labeled IntClust 1-10). Some
of these IntClust groups correspond to distinct intrinsic subtypes, but many split the
sample into unique groups. For example, IntClust 3 is composed predominantly of
luminal A tumours that have a good prognosis. IntClust 4 includes both ER+ and ER-
tumours, a variety of intrinsic subtypes, and is characterized by a favorable outcome.
IntClust 5 contains HER2 enriched and luminal tumours (ER+) that may benefit from
targeted therapy. This subtype exhibits the worst disease-specific survival at both five and
15 years, possibly because trastuzumab was not availabile at the time patients enrolled in
the study (Curtis et al., 2012) (Figure 1-4). IntClust 10 includes the majority of all basal-
like tumours and patients with these tumours have relatively good long-term outcomes
after five years. IntClust 1, 6, and 9 are several intermediate prognosis groups composed
mainly of ER+ cancers. IntClust 7 and 8 include luminal A patients with similar profiles
and good outcomes.
Of central interest for this thesis is the IntClust 2 subtype that represents a new ER+
subgroup that is characterized by poor prognosis, a steep mortality curve and elevated
hazard ratios. This represents an especially high-risk subgroup (Figure 1-4). One of the
hallmarks of this subgroup is the copy number amplification (CNA) and overexpression
of a cluster of candidate oncogenic drivers at the 11q13-q14 amplicon. Several driver
genes located in this region have been linked to endocrine resistance and poor breast
cancer prognosis (Hughes-Davies et al., 2003, Santarius et al., 2010) and ovarian cancer
(Brown et al., 2008).

General Introduction
16
The following section describes the ER pathway and its role in the development of
endocrine resistance as well as the mechanisms of resistance to endocrine therapy. The
significance of 11q13-q14 amplification in IntClust 2 subtype of breast cancer is then
presented, followed by the clinical importance of ER+ IntClust 2 subtype.
Figure 1-4 The clinical outcomes of the inter-cluster subgroups
Kaplan-Meier plot of disease-specific survival for the ten inter-cluster
subtypes. The IntClust 2 subtype is represented by a green line (marked red),
showing steep mortality curve and poor specific survival probability. For each
cluster, the number of samples at risk is indicated as well as the total number
of deaths (in parentheses). Adapted from Curtis et al. (2012).

General Introduction
17
1.3 The ER pathway and tamoxifen resistance
Estrogen is essential in women for a diversity of physiological processes. It influences
the growth, differentiation, and the function of tissues in the reproductive system,
including the breast, uterus, vagina, and ovaries (Schwabe et al., 1993). It also induces
expression of immediate and delayed hormone-responsive genes in normal and
transformed mammary epithelial cells (Altucci et al., 1996). However, prolonged
exposure to high levels of estrogen increases the risk of breast cancer by constitutively
activating the transcription of genes mainly involved in metabolism and cell cycle
regulation (Hervouet et al., 2013).
The ER was first described in the 1960s by Jensen and Jacobson when tissue uptake and
retention of radiolabeled estradiol was detected in the uterus of rats (Jensen, 1962). By
the end of the 1970s it was established that patients with ER+ tumours were more likely
to respond to endocrine treatment compared to patients with ER- tumours (Jensen et al.,
1968, McGuire, 1975).
While ER is still considered a critical predictive marker for the response to endocrine
treatment, some patients with ER+ cancer will eventually present with recurrent disease.
For this reason, many researchers are focusing on finding additional markers for
predicting endocrine response. Below is an introduction to the structure and function of
ER followed by a brief overview of what is known thus far of the mechanisms of action
of the anti-hormonal therapy, and why certain tumours develop resistance.
1.3.1 Estrogen Receptor (ER)
The ER is a nuclear hormone receptor belonging to the steroid nuclear receptor
superfamily of transcription factors (Parker, 1993). ER exists in two different isoforms,
ERα, and ERβ, transcribed from two distinct genes located on separate chromosomes
(Menasce et al., 1993, Enmark et al., 1997).

General Introduction
18
ERα is the predominant isoform expressed in the uterus, mammary gland, testis, pituitary,
liver, kidney, heart and skeletal muscle. In contrast, the expression of the ERβ transcript
is restricted to the ovary and prostate (Kuiper et al., 1997, Couse and Korach, 1999).
These two transcripts are rarely expressed within the same cell type, indicating distinct
functions of the two isoforms (Kuiper et al., 1996, Couse and Korach, 1999).
Studies have shown that ERα knockout mice display impaired mammary gland
development with a morphology a lifelong resembling that of newborn mice.
(Bocchinfuso and Korach, 1997). In contrast, ERβ knockout mice show normal ductal
structure of the mammary glands, which appear to undergo normal differentiation during
pregnancy and lactation. These studies suggest that ERα is the predominant receptor
during normal mammary gland development and regulation (Couse and Korach, 1999,
Gustafsson and Warner, 2000). In this thesis, ER will refer to ERα if not otherwise
specified.
1.3.2 ER domain structure
The ER protein consists of six functional domains (Figure 1-5A) The activation function-
1 (AF-1) is located within domains A and B, which in conjunction with the activation
function-2 (AF-2) of domain E is involved in mediating transcription. The ligand-binding
domain (LBD) is located in the same region as AF-2. The DNA binding domain (DBD)
of region C is required for the activated receptor to bind to specific DNA elements for
transcription initiation. Domain D functions as a flexible hinge between regions C and E
and contains several nuclear localisation signals (NLS) (Kumar et al., 1987, MacGregor
and Jordan, 1998).
1.3.3 ER signalling pathway
Estrogen and the ER are crucial regulators of complex biological networks controlling
cellular proliferation, apoptosis, invasion and angiogenesis (Rochefort et al., 1998, Ali et
al., 2000). The ER is activated by three major forms of estrogen in the human body,
namely estrone, estradiol, and estriol. In premenopausal women, ovaries produce 17β-

General Introduction
19
estradiol (E2), which is the more potent activating ligand of ER (Kuiper et al., 1997,
Cheskis et al., 2007). Upon binding of E2 to the LBD, the ER undergoes an allosteric
change into a dimerised active receptor complex that translocates into the nucleus. Several
co-factors are also recruited to the complex (McKenna et al., 1999). The various pathways
through which ER may activate transcription are outlined below and illustrated in Figure
1-5B.
In the classical ligand-dependent pathway, the activated ER complex binds directly to
DNA motifs known as estrogen response elements (ERE) in the proximity of target gene
promoters. However, in the non-classical ligand dependent pathway, the activated ER
complex tethers to already bound transcription factors (TFs) acting as a co-regulator
(Klein-Hitpass et al., 1988, Kushner et al., 2000). The ER has also been suggested to be
activated near the plasma membrane where it may modulate and interact with several
different pathways in a non-genomic mode. This is followed by initiating signalling
cascades via second messengers (SM), eventually leading to a rapid physiological
response that does not involve gene regulation (Levin, 1999). This interaction is most
likely a factor contributing to the resistance to tamoxifen frequently observed in ER+ and
HER2-overexpressing tumours (Shou et al., 2004, Li et al., 2012). Additionally, the ER
might be activated in a ligand-independent manner. In the absence of the ligand, growth
factor signalling leads to activation of kinases that may phosphorylate and activate the
ER. This pathway is thought to explain the hormone-independent growth observed in
some tumour subtypes (Lee et al., 2000, Campbell et al., 2001, Razandi et al., 2003,
Arpino et al., 2009).

General Introduction
20
Figure 1-5 Schematic representation of ER protein and ER signalling
pathways
(A) Modular organisation of the ER protein consisting of six functional
domains termed A, B, C, D, E and F. Modified from Hervouet et al. (2013). (B)
Distinct molecular pathways involved in regulatory actions of ER. Adapted
from Heldring et al. (2007).
AF-1: activation function-1, DBD: DNA binding domain, LBD: ligand-binding
domain, AF-2: activation function-2, ER: estrogen receptor, TF: transcription
factor, SM: second messengers, GF: growth factor.

General Introduction
21
1.3.4 Transcriptional output of ER signalling
More than 1000 genes are thought to be regulated by the ER (Kok and Linn, 2010). Gene
expression profiling of E2-stimulated breast cancer cells has shown downregulation of
the majority of the transcripts. Nevertheless, the net result is an increase in proliferation-
associated processes and suppression of apoptosis (Frasor et al., 2003). Well-known
upregulated transcripts upon E2 stimulation include Myc, CCND1 and PR (Dubik et al.,
1987, Altucci et al., 1996, Flototto et al., 2004).
1.3.5 Endocrine therapy and ER+ breast cancer
Endocrine therapy is the first line of medical treatment for ER+ breast cancer. This therapy
involves the manipulation of the endocrine system through the administration of agents
that inhibit the downstream activity or production of estrogen. Three categories of anti-
estrogen drugs are used in the clinic: selective estrogen receptor modulators (SERMs),
selective estrogen receptor downregulators (SERDs) and aromatase inhibitors (AIs)
(Bean et al., 2014).
SERMs affect ER directly by direct competition with the ligand for binding to ER.
Binding of a SERM to ER leads to insufficient conformation changes of the receptor and
inhibition of transcription. This group includes tamoxifen, raloxifen, and toremifine
(Jordan, 2004). SERDs (ICI182780, and CI164384) act by inducing a conformational
change of ER that promotes ER for degradation by the proteasome (Dauvois et al., 1992).
AIs (Letrozole) inhibit the enzyme aromatase, which prevents the conversion of
androgens into estrogens leading to inhibition of estrogen synthesis (Goss et al., 2003,
Kalidas and Brown, 2005).
1.3.6 Mechanisms of tamoxifen resistance
Tamoxifen is widely used in both the treatment of breast cancer and in the preventive
setting for patients with a high risk of developing breast cancer. The use of tamoxifen has

General Introduction
22
reduced breast cancer recurrence and thus, significantly increased survival rates (Brown
and Lippman, 2000, Early Breast Cancer Trialists' Collaborative, 2005). However, one-
third of women treated with the recommended five-year course of tamoxifen will relapse
within 15 years. Identifying novel biomarkers that can predict response to tamoxifen is
challenging (Early Breast Cancer Trialists' Collaborative, 2005).
Resistance to tamoxifen can be described as either intrinsic (de novo) or acquired. De
novo resistance exists before any treatment is given, while the acquired resistance
develops during tamoxifen administration after an initial period of response (Osborne and
Schiff, 2011). Some of the mechanisms that have been suggested to contribute to
endocrine resistance will be addressed in the following sections.
1.3.6.1 Coregulators of the ER
Coactivators and corepressors of the ER constitute a group of proteins that have been
repeatedly associated with tamoxifen resistance. The ER coactivator protein SRC-3 is
frequently amplified and overexpressed in breast cancer (Anzick et al., 1997, Bautista et
al., 1998). Studies of this co-activator both in vitro and in xenograft models have linked
its overexpression to tamoxifen resistance. High SRC-3 levels have been associated with
an impaired tamoxifen response in patients (Osborne et al., 2003). Another ER
coactivator is SRC-1 which has also been clinically associated with mediating tamoxifen
resistance and with reduced disease-free survival (Fleming et al., 2004, Redmond et al.,
2009).
Corepressors recruited to the tamoxifen-bound receptor, such as N-CoR, are thought to
have the opposite effect and play an important role in mediating the inhibitory effect of
tamoxifen. Low N-CoR mRNA expression has been significantly associated with
decreased relapse-free survival in a patient cohort and a xenograft mouse model
(Lavinsky et al., 1998, Girault et al., 2003).

General Introduction
23
Furthermore, activation of transcription factors (NF-κB and AP-1) promote ER binding
to specific gene promoters, have also been associated with endocrine resistance (Zhou et
al., 2007).
1.3.6.2 Loss of ER and activation of growth factor receptor
pathways
Another possible mechanism for resistance to endocrine therapy is the loss of ER
expression which occurs in 20% of patients treated with anti-hormonal therapy
(Encarnacion et al., 1993, Gutierrez et al., 2005). Upregulation of growth factor receptor
signalling pathways provides alternative escape pathways for proliferation and survival
of tumour cells which are no longer driven by estrogen (Osborne and Schiff, 2011).
Overexpression of HER2, as well as excessive EGFR, lead to improper activation of ER,
and consequently tamoxifen insensitivity (Campbell et al., 2001, Hutcheson et al., 2003,
Knowlden et al., 2003).
Loss of PR is another mechanism for resistance to endocrine therapy that occurs even
more frequently than ER and leads to more aggressive tumours (Brankovic-Magic et al.,
2002, Martinez et al., 2006). PR loss is associated with upregulation of the
phosphoinositide 3-kinase (PI3K)/AKT and p42/44 mitogen-activated kinases (MAPK)
pathways which also downregulate PR and ER expression (Arpino et al., 2005, Cui et al.,
2005).
1.3.6.3 Cell cycle signalling regulators
The third category of pathways implicated in tamoxifen resistance involves cell cycle
regulatory proteins. Overexpression of the positive regulators Myc, cyclins E1 (CCNE1),
and cyclin D1 (CCND1) have been involved in mediating tamoxifen resistance in patients
(Kenny et al., 1999, Stendahl et al., 2004). However, lack of subgroup stratification for
patients with CCND1 amplified tumours makes it difficult to determine the role of
CCND1 in endocrine resistance (Lundgren et al., 2012).

General Introduction
24
Inactivation of the retinoblastoma (RB) pathway is also thought to lead to tamoxifen
resistance in cell lines and xenograft models (Bosco et al., 2007, Lehn et al., 2011). High
expression of the cell cycle regulator p27 has been found to predict response to tamoxifen,
whereas exclusive cytoplasmic expression of p21 has been associated with tamoxifen
resistance (Perez-Tenorio et al., 2006, Chu et al., 2008, Stendahl et al., 2010).
In addition to overexpression of positive regulators and RB inactivation, upregulation of
downstream signalling pathways (PI3K/AKT, and MAPK), and activation of some
transcription factors (NF-κB), have also been shown to mediate cell survival and
contribute to endocrine resistance (Ali and Coombes, 2002).

General Introduction
25
1.4 Molecular pathogenesis of breast cancer
Genome instability is one of the hallmarks that characterize the pathogenesis of breast
cancer (Hanahan and Weinberg, 2011, Hainaut and Plymoth, 2013). Genetic changes that
drive and sustain cancer growth and metastasis are mainly categorized into two key
classes: 1) loss of function of tumour suppressor genes and 2) gain of function of
oncogenes.
1.4.1 Oncogenes and tumour suppressor genes
An oncogene is a mutated and overexpressed gene that alone, or in collaboration with
other changes, promotes cellular transformation, growth, and invasion. In contrast, a
tumour suppressor gene under normal conditions counteracts cell growth or other
processes that may increase invasive and metastatic potential and whose loss of function
promotes malignancy (Zhu et al., 2015).
The most frequently activated and best characterized oncogenes in breast cancer are
ERBB2 (erythroblasts leukemia viral oncogene homolog 2) (Shih et al., 2015), P1K3CA
(phosphoatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha) (Ibrahim et al.,
2015), MYC (Nadal et al., 2015), and CCND1 (Long et al., 2015).
The most frequently altered tumour suppressor genes in breast cancer are the tumour
suppressor protein p53 gene (TP53) (Evangelisti et al., 2015), the breast cancer
susceptibility genes 1 and 2 (BRCA1 and BRCA2) (Zghair et al., 2015), and the
retinoblastoma gene (RB1) (Witkiewicz and Knudsen, 2014).
Undoubtedly, many more oncogenes and tumour suppressor genes contribute to breast
carcinogenesis. Given the heterogeneity of breast cancer, a better understanding of
genetic lesions that drive tumourigenesis in the mammary gland will lead to
improvements in the clinical management of breast cancer patients.

General Introduction
26
1.4.2 The “case” of the 11q13-q14 amplicon in breast Cancer
DNA amplification is a common non-random cellular event in cancer that often involves
many genes and large segments of DNA defined as “amplicons”. Chromosome locus
11q13-q14 is amplified in some human malignancies including 15% of breast cancers
(Karlseder et al., 1994, Ormandy et al., 2003, Lundgren et al., 2008). The high frequency
of this particular amplification suggests that the region contains oncogenes that contribute
to positive selection for cell proliferation and survival. The four distinct core regions
within 11q13-q14 can be amplified independently or concurrently in different
combinations (Karlseder et al., 1994).
The four cores of 11q13-q14 amplicon are arranged unevenly from centromere to
telomere, and their boundaries and compositions are summarized in Figure 1-6
(Wilkerson and Reis-Filho, 2013). Although the region harbours several genes with
known or suspected oncogenic potential, the multipart structure of the amplicon has
hindered the determination of specific gene function and this remains a complex and
continuing process (Ormandy et al., 2003).
Breast cancers harbour amplification of all cores in this region (Courjal et al., 1996,
Schraml et al., 2003). Interestingly, there is a frequent concurrent amplification of 8p11.2-
p12 related to the 11q13-q14 amplicon that could confer additional advantages to cancer
cells (Courjal and Theillet, 1997, Bautista et al., 1998).
Expression profile landscape by Curtis et al. (2012) suggests a separate amplicon from
11q13.5-11q14 centred around C11orf67 and spanning PAK1-GAB2. The presence of 13
genes (Table 1-1) in this oncogenic cluster makes it more challenging to distinguish the
driver in the IntClust 2 breast cancer subtype. Several studies provide strong evidence
that these genes are important oncogenic elements. Potential drivers of the 11q13
amplicon in epithelial tumours are discussed in the following sections.

General Introduction
27
Figure 1-6 Detailed schematic diagram of the 11q13-q14 amplicon
Chromosome 11 ideogram showning in detail the 11q13-q14 amplicon with the
four cores highlighted green. Genes contained within each core are shown in
boxes (with the start and end points of each core), with the proposed driver
C11orf67 highlighted in red. Adapted from Wilkerson and Reis-Filho (2013).

General Introduction
28
Table 1-1 Potential oncogenes residing in the 11q13.5-q14 cis-acting
amplicon
Gene
Function
PAK1 p21 protein (Cdc42/Rac)-
activated kinase 1
Cytoskeleton organization, migration,
survival and nuclear signalling
(Bostner et al., 2007)
AQP11 Aquaporin 11 Membrane channel (water, ions)
(Okada et al., 2008)
CLNS1A Chloride channel, nucleotide-
sensitive, 1A
cytoskeleton organization,
ribonucleoprotein synthesis (Scandella
et al., 2000)
RSF1 Remodeling and spacing factor 1 Chromatin Remodeler (Yang et al.,
2014)
C11ORF67
(AAMDC)
Adipogenesis associated,
Mth938 domain containing
Unknown, putative anti-apoptotic
protein, regulation of (fat) cell
differentiation
INTS4 Integrator complex subunit 4
a multiprotein mediator of small
nuclear RNA processing associates
with RNA polymerase II
THRSP Thyroid hormone responsive
Transcriptional activator, putative role
in controlling tumour lipid metabolism
(Wu et al., 2012)
NDUFC2 NADH dehydrogenase
(ubiquinone) 1, subcomplex
Accessory subunit of the
mitochondrial membrane respiratory
chain NADH dehydrogenase
(Complex I) (Igci et al., 2015)
KCTD14
Potassium channel
tetramerization domain
containing 14
Unknown
ALG8 Alpha-1,3-glucosyltransferase
Catalyzes the addition of a glucose
residue to the lipid-linked
oligosaccharide precursor
(Remminghorst et al., 2009)
KCTD21
Potassium channel
tetramerization domain
containing 21
Unknown
USP35 Ubiquitin specific peptidase 35 Human ubiquitin-specific protease
(Liu et al., 2015)
GAB2 GRB2-associated binding
protein 2
Activator of phosphatidylinositol-3
kinase (Herrera Abreu et al., 2011)

General Introduction
29
1.4.2.1 CCND1
CCND1 encodes the cyclin D1 cell cycle protein, which forms an active complex with
CDK4 and is responsible for the cell cycle progression through G1-S phases. Its
overexpression increases proliferation of cancer cells. Cyclin D1 has been found to be
amplified in 10-20% of breast cancer cases (Bostner et al., 2007, Lundgren et al., 2012,
Tobin and Bergh, 2012) (see section 3.6.3 for more details).
1.4.2.2 EMSY
EMSY encodes a putative oncogenic product interacting with the transactivation domain
of BRCA2. Overexpression of EMSY was found to induce chromosomal instability in
normal human breast epithelial cells similar to that reported in BRCA2-deficient cells
(Benusiglio et al., 2005, Raouf et al., 2005). EMSY is amplified in 13% of sporadic breast
cancers, 18% of high-grade ovarian cancers, and is associated with a poor prognosis
(Hughes-Davies et al., 2003). There is also a strong association between EMSY gene
amplification and overexpression (Rodriguez et al., 2004, Brown et al., 2006).
1.4.2.3 PAK1
The gene encoding p-21-activated kinase 1 (PAK1) has been reported to be amplified in
breast (Bekri et al., 1997) and ovarian cancer (Schraml et al., 2003, Brown et al., 2008).
PAK1 regulates cytoskeletal structure, motility and mitosis of normal mammary
epithelial cells (Vadlamudi et al., 2000, Li et al., 2002, Vadlamudi et al., 2005). In breast
cancer, nuclear expression of PAK1 predicts resistance to tamoxifen therapy, while
cytoplasmic levels correlate with recurrence rate and mortality (Bostner et al., 2007,
Bostner et al., 2010). It was also found that PAK1 gene regulates anchorage-independent
growth and invasiveness of human breast cancer cells and is closely associated with the
invasive phenotypes of breast cancer cells and tumour grades (Vadlamudi et al., 2000,
Wang et al., 2002, Menard et al., 2005).

General Introduction
30
1.4.2.4 AQP11
Aquaporins (AQPs) are a family of channel-forming membrane proteins that facilitate the
transport of water and some low-molecular-weight solutes (Agre, 2006). AQPs are
expressed in various tumours (Mobasheri et al., 2005), and AQP expression often
correlates with tumour grade (Verkman et al., 2008, Ribatti et al., 2014), metastasis and
local invasion (Hu and Verkman, 2006).
1.4.2.5 RSF1
Remodeling and spacing factor 1 (RSF1) is a family member of a chromatin remodeling
complex that has been shown to play an essential role in transcriptional regulation, cell
cycle progression and carcinogenesis (Vignali et al., 2000, Wolffe, 2001, Sheu et al.,
2010). Increasing evidence suggests that the RSF1 gene is amplified and overexpressed
in ovarian cancer (Shih Ie et al., 2005) and in breast cancer (Mao et al., 2006).
Elevated levels of RSF1 are correlated with poor prognosis (Liang et al., 2012). RSF1
knockdown reduces proliferation of ovarian cancer cells in vitro (Sheu et al., 2008). RSF1
was also found to be upregulated in paclitaxel-resistant ovarian cancer cell lines (Choi et
al., 2009).
1.4.2.6 GAB2
GRB2-associated binding protein 2 (GAB2) is a scaffolding adaptor protein responsible
for the transduction of extracellular signals through some receptor tyrosine kinases, such
as HER2. GAB2 is overexpressed in breast cancer cell lines and primary tumours (Bekri
et al., 1997, Ormandy et al., 2003, Bentires-Alj et al., 2006). Moreover, it is implicated
in the metastatic spread of both breast cancer and melanoma (Ke et al., 2007, Horst et al.,
2009). Overexpression of GAB2 in mammary epithelial cells lead to marked changes in
cytoskeletal organization, maturation of cell matrix and cell motility (Herrera Abreu et
al., 2011).

General Introduction
31
1.4.2.7 C11orf67
Human C11orf67 is also known as Adipogenesis associated Mth938 domain containing
(AAMDC), encoding a hypothetical protein of unknown function. C11orf67 is located in
the centre of the 11q13-14 locus and is one of the novel genes overexpressed and
amplified by copy number in the ER+ subgroup of breast cancer. This subgroup is
characterized by poor prognosis (Curtis et al., 2012).
Studies of the overexpression of the murine homolog of AAMDC show that it can induce
adipogenesis in murine preadipocytes, suggesting a potential role in lipid metabolism (Ma
et al., 2012). Moreover, C11orf67 was found to be one of the novel key regulators of the
NF-B pathway in the 293T embryonic kidney cells (Gewurz et al., 2012). In addition,
C11orf67 was found to be overexpressed in some breast cancer cell lines (Kwek et al.,
2009), but the biological function of the protein has not yet been investigated in human
tumours.

General Introduction
32
1.5 Statement of aims
C11orf67 is a novel gene that is amplified and overexpressed in a subgroup of HR+ breast
cancer that show resistance to endocrine therapy resulting in poor patient outcomes.
However, the biological function of this protein has not been previously investigated in
human tumours. I hypothesize that C11orf67 is a new hormone-dependent oncogene
amplified and overexpressed in a subset of HR+ breast cancers associated with poor
outcome, and its expression is necessary to maintain high proliferation in the IntClust 2
tumours. The following four aims were developed to test this hypothesis.
Aim 1. Demonstrate the amplification and overexpression of C11orf67
In chapter 2, we analyse the protein structure of C11orf67 and its pattern of expression
in breast cancer cell lines and breast cancer tissues. We test the amplification of
C11orf67 in breast cancer and its possible correlation with poor outcomes.
(A) Test the transcriptional and protein expression levels of C11orf67 in
breast cancer cell lines and cellular localization of the C11orf67 protein.
(B) Immunohistochemical analysis of commercially available tissue
microarrays, and correlate the overexpression with tumour grade.
(C) Fluorescence in situ hybridization of HR+ breast cancer tissues, and
correlate the amplification with LN metastasis.
Aim 2. Investigate the molecular function of C11orf67 in HR+ breast cancers
In chapter 3, we test the functional consequences of C11orf67 knockdown and gain
of function in breast cancer cell lines. Moreover, we identify candidate signalling
pathways that are affected by changes in C11orf67 gene expression.
(A) Determine the capacity of C11orf67 knockdown to control
tumourigenesis and tumour progression.

General Introduction
33
(B) Investigate the effect of C11orf67 overexpression on drug sensitivity.
(C) Map the signalling pathways affected by perturbation of C11orf67
expression.
Aim 3. Investigate the possible mechanisms of regulation of C11orf67 and the
intracellular binding partners of C11orf67
In chapter 4, we explore the effect of hormonal treatment on the expression levels of
C11orf67, along with the different effect of tamoxifen in cases of high or low
C11orf67 expression. We also investigate possible cross-talk between C11orf67 and
NF-κB in breast cancer. Finally, we screen for the binding partners of C11orf67.
(A) Define the molecular cues that control the expression of C11orf67 at
11q13.5-q14.
(B) Explore the effect of the molecular control of C11orf67 on the endocrine
resistance of the HR+ breast cancer subtypes.
(C) Investigate potential binding partners of C11orf67 by yeast two-hybrid
screens.

Chapter 2:
C11orf67 as a Novel
Biomarker in Breast Cancer

C11orf67 as a Novel Biomarker in Breast Cancer
35
2.1 Introduction
Breast cancer is one of the leading causes of cancer-related deaths in Australia and
worldwide (AIHW, 2014). In recent years, genomic profiling of breast cancers has
allowed researchers to develop novel predictive biomarkers and guide targeted therapy.
In this chapter, we take advantage of the cancer genome project and other databases to
describe C11orf67 as a novel target in hormone receptor positive (HR+) breast cancer.
We propose that C11orf67 is a predictive biomarker that could help define a group of
poor prognosis ER+ tumours at risk of relapse.
Oncogenic activation in breast cancer is often the result of the loss, rearrangement or
amplification of particular regions of the genome (Luo et al., 2006). There is a
considerable interest to identify the genes in which chromosomal abnormality occurs
since they are likely to determine the behavior of the tumour (Gillett et al., 1994). The
chromosome region (11q13.5-q14) in chromosome 11 is amplified in almost 10% of
breast cancer and it is associated with very poor clinical outcome (Borg et al., 1991, Hui
et al., 1998, Ormandy et al., 2003). Gene(s) within this amplicon may contribute to the
clinical aggressiveness of the tumour.
For decades, breast cancer has been classified at the molecular level into five intrinsic
subtypes; luminal A, luminal B, HER2+, basal-like and normal-like (Perou et al., 2000).
However, gene expression data does not directly inform about potential drivers that lead
to essentially different breast cancer disease (Turner et al., 2010, Wilkerson and Reis-
Filho, 2013).
To search candidate driver oncogenes, the recent sequencing of the breast cancer genome
has provided new insights about the genes that are actually causative of the disease. By
integrating both genomic aberrations, such as copy number amplifications, and gene
expression data, the cancer genome project has defined novel subgroups of breast cancer
with very important consequences for therapy (Curtis et al., 2012). One of these
functional subgroups is characterized by copy number amplification and overexpression
in a cluster of potential drivers in the 11q13.5 chromosomal region, namely Intercluster

C11orf67 as a Novel Biomarker in Breast Cancer
36
2 (IntClust 2) subtype. This IntClust 2 subtype is a HR+ breast cancer associated with
poor prognosis and possibly resistance to endocrine therapy (Curtis et al., 2012).
Therefore, it is crucial to analyse the key drivers of this subgroup to develop novel
biomarkers that predict the poor outcome and ultimately improve survival for these
patients.
Within the 11q13.5-q14 oncogenic cluster we observed the selective overexpression of
C11orf67 in the IntClust 2 subtype (Figure 2-1). C11orf67 (also known as adipogenesis
Figure 2-1 Selective overexpression of C11orf67 in breast cancer subtypes
Boxplots of log2 expression of C11orf67 in (A) the Intercluster subtypes, and
(B) the Intrinsic subtypes of breast cancer. IntClust 2 and luminal subtypes
are indicated with red boxes. Figure modified from Curtis et al. (2012).

C11orf67 as a Novel Biomarker in Breast Cancer
37
associated Mth938 domain containing, AAMDC) encodes a hypothetical protein of
unknown function. Conserved domain analyses suggest that C11orf67 has a very similar
folding to a predicted protein (Mth938) from Methanobacterium thermoautotrophicum
which has suggested lateral gene transfer from bacteria to mammalian cells (Das et al.,
2001). However, the biological role of C11orf67 protein in human tumours has not been
studied previously.
In this chapter, we first characterized the C11orf67 gene organisation and the domain
structure of the protein encoded by C11orf67. Second, we developed antibody based
assays to determine the subcellular localization of C11orf67 in mammalian cells. Third,
to understand the extent and the significance of C11orf67 in the prognosis of breast
cancer, we examined a series of breast cancer tissues for the presence of the C11orf67
gene amplification. Lastly, we investigated the expression of a C11orf67 protein by IHC
in breast cancer tissues.
This study demonstrated the amplification and overexpression of C11orf67 in HR+ breast
cancer tissues and breast cancer cell lines over normal tissues. Our results are the first to
highlight the prognostic value of C11orf67 as a novel biomarker in HR+ breast cancer.
Our work suggests that C11orf67 could be used clinically to identify ER+ breast cancer
patients belonging to this IntClust 2 subtype associated with very poor prognosis.

C11orf67 as a Novel Biomarker in Breast Cancer
38
2.2 Materials and Methods
2.2.1 Cell lines and cell culture
All breast cell lines were purchased from American Type Culture Collection (ATCC,
Manassas, VA) and cultured in complete growth medium as described by the company.
All media and supplements were purchased from Invitrogen, Waverley, Australia, unless
stated otherwise.
Briefly, human breast cancer cell lines T47D, ZR751, SKBR3 and BT474 cells were
cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) and
1% Penicillin-streptomycin (Pen/Strep). HEK293T and MDAMB-231 cells were cultured
in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% FBS and 1%
Pen/Strep. MCF7 cells were cultured in Minimum Essential Medium (MEM)
supplemented with 1.5 mg/ml sodium bicarbonate, 0.1 mM non-essential amino acids
(NEAA), 1 mM sodium pyruvate, 10% FBS and 1% Pen/Strep. MCF12A cells were
cultured in DMEM/F12 (1:1) containing 20 ng/ml epidermal growth factor (EGF)
(Sigma-Aldrich, St Louis, MO, USA), 100 ng/ml cholera toxin (Sigma-Aldrich, St Louis,
MO, USA), 0.01 mg/ml insulin, 500 ng/ml hydrocortisone (Sigma-Aldrich, St Louis, MO,
USA), 5% horse serum, and 1% Pen/Strep. Human mammary epithelial cells (HuMEC)
were cultured in HuMEC media, supplemented with 1% bovine pituitary extract, and
1%Pen/Strep. All the cells were incubated in 5% CO2-containing humidified atmosphere
at 37°C. Once the cells reached 60-70% confluency, they were trypsinised with 0.25%
trypsin-ethylenediaminetetraacetic acid (EDTA) and passaged accordingly.
2.2.2 RNA extraction
Total RNA was extracted from breast cancer cells using TRIzol reagent according to the
manufacturer’s protocol (Qiagen, Hilden, Germany). Briefly, cells were homogenized
using 1 ml TRIzol reagent per one well of the six-well plate. The homogenized samples
were incubated for 5 minutes at room temperature to permit complete dissociation of

C11orf67 as a Novel Biomarker in Breast Cancer
39
nucleoprotein complexes. 0.2 ml chloroform per 1 ml TRIzoL reagent was added to the
homogenized samples followed by vigorous shaking for 15 seconds. After 2-3 minutes
incubation at room temperature, samples were centrifuged at 12,000 g for 15 minutes at
4°C. After centrifugation, the samples separated into three phases, and the aqueous phase
was transferred to a new tube. For RNA precipitation, 0.5 ml isopropyl alcohol was added
to the aqueous phase. The samples were incubated for 10 minutes at room temperature
and centrifuged at 12,000 g for 10 minutes at 4°C. The supernatant was discarded, and
the RNA gel-like pellet was washed once with 200 μl 70% ethanol, vortexed and
centrifuged at 7,500 g for 5 minutes at 4°C. Ethanol was aspirated, and the RNA pellet
was air-dried, dissolved in RNase-free water and stored at -80°C until required.
2.2.3 Reverse transcription and cDNA synthesis
Up to 3 μg of total RNA was reverse transcribed to complimentary DNA (cDNA) using
the High Capacity cDNA Kit (Invitrogen, Waverley, Australia) according to the
manufacturer’s protocol, the samples were kept on ice for the next step or stored at -20°C
2.2.4 Real-time Polymerase Chain Reaction
Quantitative real-time Plymerase chain reaction (qRT-PCR) was carried out to detect
levels of C11orf67 mRNA expression using Taqman probes (Applied Biosystems,
Scoresby, Australia) for C11orf67 (FAM/MGB #4331182) and the housekeeping gene
GAPDH (FAM/MGB #4333764F). Data were analyzed using the comparative ΔΔ Ct
method (ABPrism software, Applied Biosystems) using GAPDH as an internal
normalization control (Livak and Schmittgen, 2001).
2.2.5 Total protein extraction and western blotting
For protein extraction, cells were washed twice with PBS and lysed in ice-cold lysis buffer
(2% sodium dodecyl sulfate (SDS), 125 mmol/L Tris/HCL pH 6.8). Lysates were

C11orf67 as a Novel Biomarker in Breast Cancer
40
centrifuged at 12,000 g for 30 minutes at 4°C; the supernatant was stored in -80°C until
assayed.
Protein concentration was measured with the Bio-Rad DC Protein Assay kit (BioRad
Laboratory, Hercules, CA) according to the manufacturer protocol, with various
concentration of Bovine Serum Albumin (BSA) (Sigma-Aldrich, St Louis, MO, USA)
prepared in the lysis buffer as standards for this assay.
For western blotting, cell lysates (20 μg) was mixed with Laemmli sample buffer (BioRad
Laboratory, Hercules, CA) and dithiothreitol (DTT) (Sigma-Aldrich, St Louis, MO,
USA). The samples were then denatured by boiling at 95°C for 5 minutes. Samples were
loaded onto 4-15% pre-cast Mini gels (BioRad Laboratory, Hercules, CA). Proteins were
then electroblotted to nitrocellulose membranes by using Trans-Blot turbo transfer system
(BioRad Laboratory, Hercules, CA). and blocked for 1 hour at room temperature in Tris-
buffered saline containing Tween (TBS-T) buffer (50 m M Tris – HCL, pH 7.5, 150 m M
NaCl, 0.1% Tween-20), containing 5% non-fat milk. After blocking, blots were incubated
overnight at 4°C with the required primary antibodies: rabbit anti-C11orf67 polyclonal
antibody 1:250 (Santa Cruz Biotechnology, Texas, USA), and mouse anti-α tubulin
antibody 1:2000 (Sigma-Aldrich, St Louis, MO, USA). The next day, membranes were
washed three times with TBS-T, and then incubated with horseradish peroxidase-
conjugated secondary Anti-rabbit and anti-mouse respectively (1:10,000) for one hour at
room temperature. After three more washes with TBS-T, proteins were visualized by
enzymatic Chemiluminescence Detection Kit (Millipore, Victoria, Australia).
2.2.6 Tissue Microarrays
Commercially available tissue microarrays (TMAs) (Biomax, Rockville, MD, USA)
containing Formalin-fixed, Paraffin-embedded (FFPE) tissues from 75 breast cancer
patients, with two representative cores 1.5 mm diameter from each tumour were used in
this study.

C11orf67 as a Novel Biomarker in Breast Cancer
41
Immunohistochemistry (IHC) was performed at Royal Perth Hospital by Mr Nathan Acott
on Ventana Benchmark Ultra, using Ventana optiView detection system (760-700).
Antigen retrieval was performed on deparaffinized sections by boiling them at 99°C in
citrate buffer (10 mM sodium citrate buffer, pH 6.0) for 16 minutes using board Ventana
Benchmark Ultra. Endogenous peroxidase was blocked with 3% H2O2 followed by
incubation with rabbit polyclonal anti-C11orf67 antibody (Santa Cruz Biotechnology,
Texas, USA) diluted at 1:100 in DAKO diluent (S0809) for 32 minutes at 36°C, followed
by the EnVision+ System (DAKO, Carpinteria, CA) using the peroxidase method.
Scoring was performed by breast pathologist Dr Jeremy Parry, intensity score ranging
from 0 to 2+ to evaluate C11orf67 immunoreactivity in tumour samples.
2.2.7 Immunocytochemistry
For immunocytochemistry (ICC), VitroView LSAB IHC Kit, Rabbit IgG (Genecopoeia,
Rockville, MD, USA) was used according the manufacturer’s protocol. Briefly, cells
were fixed with 10% paraformaldehyde (PFA) in Phosphate Buffered Saline (PBS) for
20 minutes, permeabilized in 0.025% Triton X-100, and incubated with normal goat
serum for 30 minutes to block non-specific binding. Rabbit polyclonal anti-C11orf67
antibody (Santa Cruz Biotechnology, Texas, USA) was added to the cells at 1:100 dilution
in 10% goat serum in PBS and incubated overnight at 4°C. Peroxidase blocking was
performed by incubation of cells with 0.3% hydrogen peroxide in PBS for 10 minutes at
room temperature; then cells were incubated with the biotinylated anti-rabbit secondary
antibody for 30 minutes at room temperature. Detection was finally performed by
incubating cells with Streptavidin-Horseradish Peroxidase (HRP) for 30 minutes at room
temperature, followed by DAB solution for signal development. Positive C11orf67 cells
were detected under light microscope Olympus IX71 (Singapore).
2.2.8 Fluorescence microscopy
T47D cells were cultured on coverslips until 70% confluent, then transiently transfected
using Lipofectamine-2000 reagent (Invitrogen, Waverley, Australia) with C11orf67

C11orf67 as a Novel Biomarker in Breast Cancer
42
isoform_2 cDNA fused to an N-terminal 3xFLAG epitope (GenScript, NJ, USA)
according to the manufacturer's protocol. After 36 hour incubation, cells were fixed with
4% PFA in PBS at room temperature for 20 minutes, permeabilized with 0.5% Triton-X
in PBS at 4°C for 10 minutes and blocked with 2% BSA in PBS at 4°C for 24 hours. Cells
were incubated with mouse monoclonal Anti-Flag antibody 1:1600 (Cell Signaling, Gold
Coast, QLD, Australia) overnight, then goat anti-mouse secondary Alexa Fluor 488-
conjutated antibody (1:500) for 1 hour in the dark . To visualize nuclei, cells were
incubated with 1:5000 dilution of Hoechst nuclear stain (Sigma-Aldrich, St Louis, MO,
USA) at room temperature for 5 minutes. Coverslips were mounted on slides, preserved
using Fluorosave and examined with the Nikon Fluorescence microscope with
microscope camera (DS-Fi2).
2.2.9 Fluorescence In Situ Hybridization
Fluorescence in situ hybridization (FISH) was conducted in collaboration with the
department of Biology and Genetics, Medical University, Poland by Dr Iwona Kardas.
FISH protocol was performed using commercially available probes (Empire Genomics,
Buffalo, NY) labelled red for C11orf67 gene, and green pericentromeric region of
chromosome 11 (5-Fluorescein dUTP). After deparaffinization and rehydration, slides
were soaked in pre-treatment solution (Histology FISH Accessory Kit, Dako) and heated
in a microwave oven for 10 minutes, followed by 17 minutes Pepsin digestion (room
temperature, RTU Pepsin solution, Histology FISH Accessory Kit, Dako). Subsequently,
slides were dehydrated and 10 μl of probe working solution was applied to each slide.
After co-denaturation at 78℃ for 5 minutes, slides were placed in a humid chamber at 37
°C overnight. The next day residual probe was washed out and Fluorescence Mounting
Medium (Histology FISH Accessory Kit, Dako) was applied. After 30 minutes, sections
were observed under a fluorescence microscope (ZEISS, Germany). Digital images were
recorded using the integrated camera (AxioCamMRc, ZEISS) and analysed using Isis
Fluorescence Imagining (MetaSystems, Germany). For each sample at least 40 nuclei
were counted. Cells with a ratio of gene specific probe (C11orf67) to control probe
(pericentromeric region of chromosome 11) ≥ 2.0 were considered to have amplification.

C11orf67 as a Novel Biomarker in Breast Cancer
43
2.2.10 Statistical analysis
Each experiment was repeated at least three times. Data are presented as mean ± the
standard deviation (SD) of each experiment. Data analysis was performed using
GraphPad Prism. Student’s t-test was used to calculate the numerical data. Chi-square test
was utilised for comparisons of categorical data. Statistical tests were two-sided, and p
values less than 0.05 were considered statistically significant.

C11orf67 as a Novel Biomarker in Breast Cancer
44
2.3 Results
2.3.1 Bioinformatic and structural analyses of C11orf67
Bioinformatic analyses were performed using BLAST, Ensembl and NCBI databases
(www.ncbi.nlm.nih.gov/protein/NP_078960.1). These analyses showed that human
C11orf67 is located on chromosome 11, and the corresponding open reading frame (ORF)
encodes a hypothetical protein of 122 amino acids (aa) with a predicted MW of 13.3 kilo
Dalton (kD).
Nucleotide sequence conservation analysis of C11orf67 indicated that the gene is
conserved across different species including human, chimpanzee, rhesus monkey, cow,
dog, rat, mouse, chicken, zebrafish, and even bacteria (Figure 2-2A). Furthermore, the
resulting protein domain structure was found highly similar to that of MTH938 domain
from Methanobacterium, suggesting lateral gene transfer from bacteria to eukaryotic cells
(Das et al., 2001) (Figure 2-2B). The crystal structure of the MTH938 domain reveals a
unique tertiary fold consisting of three β-sheets and three α-helices. The β-sheets from
each monomer comprise 111 aa and associate to form a dimer with a cleft in the
interphase, which potentially bind double-stranded nucleic acid (Das et al., 2001).
2.3.2 Identification of the full-length cDNA sequence and
spliced transcripts of C11orf67.
To obtain the cDNA sequences of the C11orf67 gene, we used GeneBank
(www.ncbi.nlm.nih.gov/gene/28971) and UCSC genome browser databases. C11orf67
was mapped to human chromosome 11, and its 6 exons spanned 75.6 Kbps. UCSC
Genome Browser analysis of C11orf67 cDNA sequences revealed the existence of three
alternative spliced variants (Figure 2-3A). The homology between these three C11orf67
transcripts resides mainly in their NH2-terminal domain, whereas the amino COOH-
terminal region is much more divergent. The C11orf67 isoforms differ in their nucleotide
sequence, position in chromosome 11 and number of coding exons; thereby, these

C11orf67 as a Novel Biomarker in Breast Cancer
45
isoforms may encode protein products of distinct MW and aa sequence (Table 2-1).The
length of the splicing transcript 1 was 264 bp (includes exon 1, 2, and 6), transcript 2 was
366 bp (includes exon 1, 2 and 3), transcript 3 was 441 bp (includes exon 1, 2, 4 and 5).

C11orf67 as a Novel Biomarker in Breast Cancer
46
Figure 2-2 Phylogenetic analysis and structural similarity of C11orf67
(A) Phylogenetic tree analysis of C11orf67. (B) Structural similarity between
C11orf67 and MTH938 domain from Methanobacterium, modelled using
PyMol software. 1IHN and 2AB1 indicate the X-ray structure of the gene
product.

C11orf67 as a Novel Biomarker in Breast Cancer
47
Figure 2-3 Alternative splicing of the C11orf67 gene
(A) Sketch map of the C11orf67 alternative splicing model. The boxes show
the exons and their sizes. (B) cDNA sequence and predicted aa sequence of
human C11orf67 Isoform_2. Shared aa sequence between the three isoforms
are underlined. * indicates a stop codon.

C11orf67 as a Novel Biomarker in Breast Cancer
48
Table 2-1 Characteristics of the C11orf67 isoforms
C11orf67
isoform
Position
Chr11
Number
of aa
Coding
exon
count
Mw
(kD)
Isoform_1 77,553,543-
77,629,145 88 3 9.57
Isoform_2 77,553,543-
77,583,361 122 3 13.33
Isoform_3 77,553,543-
77,611,732 147 4 16.37
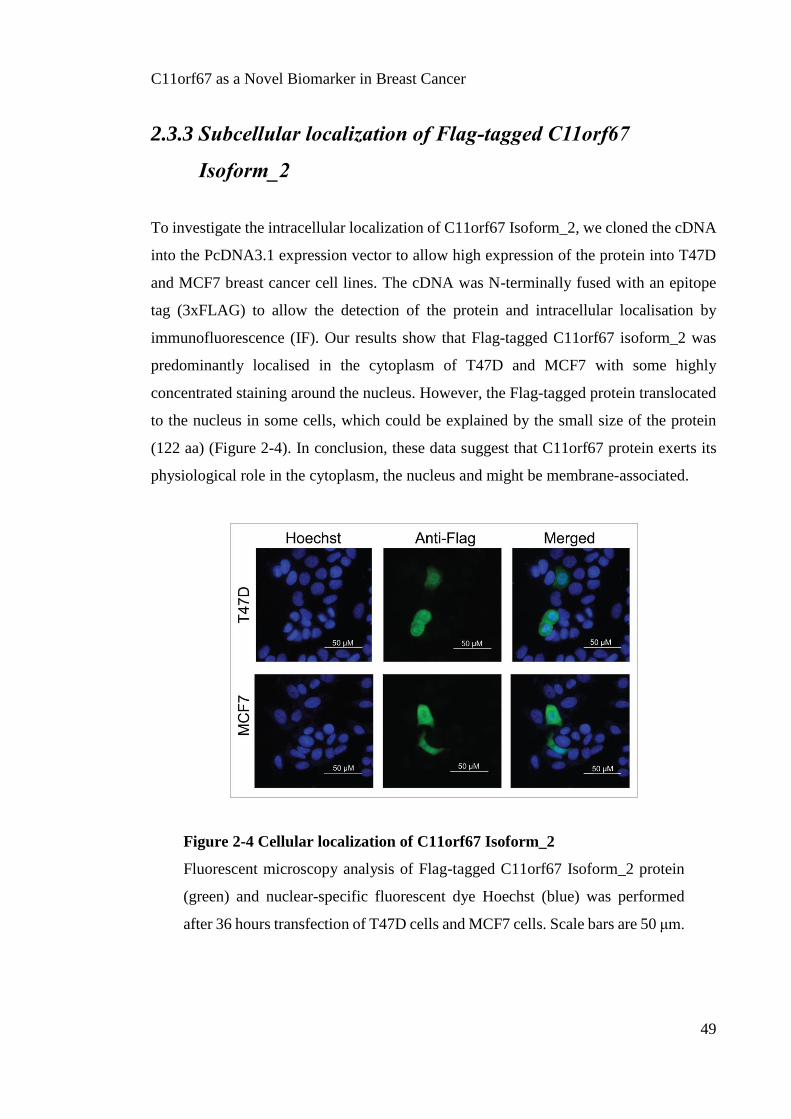
C11orf67 as a Novel Biomarker in Breast Cancer
49
2.3.3 Subcellular localization of Flag-tagged C11orf67
Isoform_2
To investigate the intracellular localization of C11orf67 Isoform_2, we cloned the cDNA
into the PcDNA3.1 expression vector to allow high expression of the protein into T47D
and MCF7 breast cancer cell lines. The cDNA was N-terminally fused with an epitope
tag (3xFLAG) to allow the detection of the protein and intracellular localisation by
immunofluorescence (IF). Our results show that Flag-tagged C11orf67 isoform_2 was
predominantly localised in the cytoplasm of T47D and MCF7 with some highly
concentrated staining around the nucleus. However, the Flag-tagged protein translocated
to the nucleus in some cells, which could be explained by the small size of the protein
(122 aa) (Figure 2-4). In conclusion, these data suggest that C11orf67 protein exerts its
physiological role in the cytoplasm, the nucleus and might be membrane-associated.
Figure 2-4 Cellular localization of C11orf67 Isoform_2
Fluorescent microscopy analysis of Flag-tagged C11orf67 Isoform_2 protein
(green) and nuclear-specific fluorescent dye Hoechst (blue) was performed
after 36 hours transfection of T47D cells and MCF7 cells. Scale bars are 50 μm.

C11orf67 as a Novel Biomarker in Breast Cancer
50
2.3.4 The pattern of C11orf67 gene alteration in cancer
After analyzing the genomic organisation and protein structure of C11orf67 we next
investigated whether C11orf67 was amplified and overexpressed in different tumour
types. In addition, we were interested to explore if its expression is of any prognostic
value in different tumours.
In order to achieve these objectives, we first investigated the pattern of C11orf67
alteration in a large-scale cancer genomic datasets with collaboration with Piotr
Kozlowski, utilizing cBioPortal for Cancer Genomics (http://www.cbioportal.org/public-
portal/). Our results show that C11orf67 tends to be amplified in a variety of cancers,
most predominantly in breast invasive carcinoma (~25% of cases), followed by ovarian
carcinoma (15% of cases) (Figure 2-5 A). In addition, a strong correlation between the
copy number amplification (CNAs) of C11orf67 and mRNA overexpression was
validated by examination of the Cancer Genome Atlas database. This correlation was also
described by Curtis et al in the context of the IntClust 2 subtype.
To assess the prognostic value of C11orf67 overexpression, we used the PPISURV
PORTAL tool to query the prognostic value of C11orf67 expression levels in a published
dataset of patients with different types of cancers. As expected from being an oncogenic
driver, patients with low C11orf67 expression had a significantly higher survival
probability than patients with high C11orf67 expression in breast cancer (p=0.0117),
ovarian cancer (p=0.00659), and lung non-small cell lung cancer (NSCLC) (p=0.0109)
(Figure 2-5B). This data support the notion that overexpression of C11orf67 has a
prognostic value in breast and other cancers.

C11orf67 as a Novel Biomarker in Breast Cancer
51
Figure 2-5 Alteration frequency and survival probability of C11orf67 in
different cancers
(A) Alteration frequency of C11orf67 (y-axis) in various cancer sequencing
studies (x-axis). Results are obtained via cBioPortal for cancer genomics.
Mutations are illustrated in green, deletions in blue, amplifications in red, and
multiple alterations in grey. Individual cancer types are shown along x-axis.
(B) Kaplan-Meier survival curves for patients with breast cancer, ovarian
cancer, and non-small cell lung cancer expressing different levels of C11orf67
(high C11orf67 expression: red line; low C11orf67 expression: green line).
Low C11orf67 expression correlates with higher survival probability in breast
cancer patients (p=0.0117), ovarian cancer patients (p=0.00659), and lung
cancer patients (p=0.0109).

C11orf67 as a Novel Biomarker in Breast Cancer
52
2.3.5 C11orf67 expression profile in breast cell lines
C11orf67 was found to be amplified and overexpressed in IntClust 2 breast cancer
subtype. This subgroup includes HR+ patients characterised with poor clinical outcome
and tendency to relapse (Curtis et al., 2012). Next, we asked the question whether
C11orf67 would be overexpressed in representative ER+ breast cancer cell lines. We
screened a panel of 8 tumourigenic and non-tumourigenic breast cell lines using a
C11orf67 antibody by western blot. Breast cell lines used were representative of luminal,
Her2+, and TNBC. Normal-like cell lines were used in our analyses as negative controls.
(Table 2-2).
First, qRT-PCR was performed to analyse the mRNA level of C11orf67 in the different
cell lines tested. Our results demonstrated that C11orf67 was overexpressed in several
breast cancer cell lines relative to the non-transformed breast epithelial cell line MCF12A.
The highest expression of C11orf67 was detected in the ER+ luminal T47D cell line
(p<0.0001), followed by the luminal cell lines BT474, MCF7 and ZR751 (p<0.0001).
C11orf67 was also found to be upregulated in the ER-, HER2+ cell line SKBR3 (p<0.001),
and in the basal cell line MDA-MB231 (p<0.0001) relative to MCF12A cells, but in less
extent that in the luminal lines. Normal-like HUMEC cells did not show any significant
difference in the expression of C11orf67 compared to MCF12A cells (Figure 2-6A).
Collectively, these data indicated that luminal T47D cells showed the highest levels of
C11orf67 mRNA.
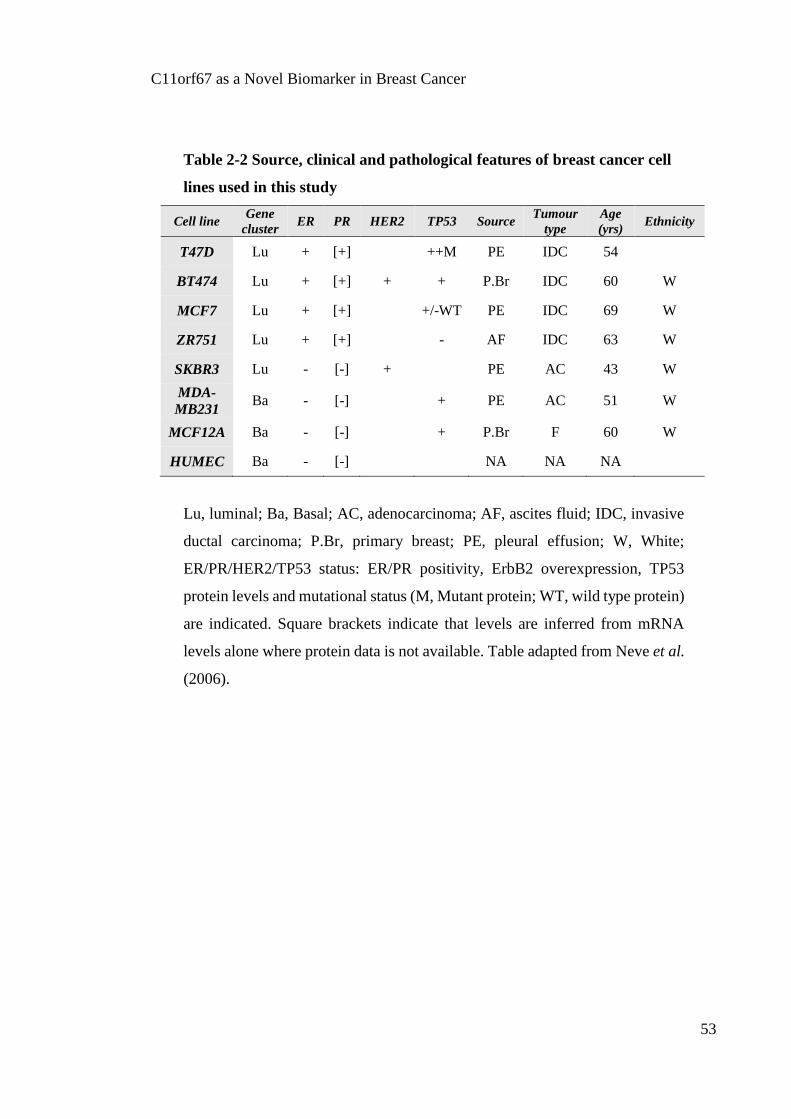
C11orf67 as a Novel Biomarker in Breast Cancer
53
Table 2-2 Source, clinical and pathological features of breast cancer cell
lines used in this study
Cell line Gene
cluster ER PR HER2 TP53 Source
Tumour
type
Age
(yrs) Ethnicity
T47D Lu + [+] ++M PE IDC 54
BT474 Lu + [+] + + P.Br IDC 60 W
MCF7 Lu + [+] +/-WT PE IDC 69 W
ZR751 Lu + [+] - AF IDC 63 W
SKBR3 Lu - [-] + PE AC 43 W
MDA-
MB231 Ba - [-] + PE AC 51 W
MCF12A Ba - [-] + P.Br F 60 W
HUMEC Ba - [-] NA NA NA
Lu, luminal; Ba, Basal; AC, adenocarcinoma; AF, ascites fluid; IDC, invasive
ductal carcinoma; P.Br, primary breast; PE, pleural effusion; W, White;
ER/PR/HER2/TP53 status: ER/PR positivity, ErbB2 overexpression, TP53
protein levels and mutational status (M, Mutant protein; WT, wild type protein)
are indicated. Square brackets indicate that levels are inferred from mRNA
levels alone where protein data is not available. Table adapted from Neve et al.
(2006).

C11orf67 as a Novel Biomarker in Breast Cancer
54
At the protein level, we first confirmed the specificity of the polyclonal anti-C11orf67
antibody by western blot analysis. We generated stable MCF7 cells transfected with a
C11orf67 Isoform_2 lentiviral vector as our positive control. We found that the anti-
C11orf67 antibody recognized two protein bands of a MW of 13 kD and 16 kD,
corresponding to C11orf67 Isoform_2 and Isoform_3, respectively. The western blot
analysis did not reveal any bands corresponding to the C11orf67 isoform_1 of a MW of
9 kD. Positive control cells showed an increase in the intensity of the band at the MW of
13 kD, corresponding to the expected C11orf67 Isoform_2, which indicates specificity of
the antibody.
Western blot analyses of the ER+ and ER- breast cancer cell lines showed a similar trend
of expression to that of the qRT-PCR analyses. Consistent with the transcriptional data,
MCF12A and HUMEC cells did not show detectable C11orf67 protein expression under
the same experimental conditions (Figure 2-6B).
To confirm the specific binding of the anti-C11orf67 antibody in cells, we tested the
expression pattern of C11orf67 protein in the high- and the low-expressing cells by ICC.
We stained the T47D luminal cell line that showed the highest levels of endogenous
C11orf67 mRNA, and the MCF12A normal-like cell line carrying the lowest levels of
expression. T47D cells showed cytoplasmic as well as peri-nuclear staining of the
C11orf67 protein, while MCF12A cells showed hardly detectable levels of C11orf67
staining both in the cytoplasm and nucleus (Figure 2-6C).
These data collectively validate the specificity of the anti-C11orf67 antibody and allowed
us to use this reagent in patient’s samples.

C11orf67 as a Novel Biomarker in Breast Cancer
55
Figure 2-6 Expression pattern of C11orf67 in breast cell lines
(A) Transcript levels of C11orf67 were determined in the indicated cell lines
by qRT-PCR and normalized to GAPDH. The data are normalized to T47D
cells. (B) Protein expression levels were determined by western blot, equal
loading of lanes was confirmed by expression of -tubulin. (C) C11orf67
protein localization was determined by ICC in T47D and MCF12A cells (***
p<0.0001, ** p<0.001).

C11orf67 as a Novel Biomarker in Breast Cancer
56
2.3.6 C11orf67 is overexpressed in HR+ and high-grade breast
cancer
We next examined the pattern and intensity of C11orf67 protein expression in breast
cancer specimens using a commercial breast cancer TMA by IHC staining. The breast
cancer TMA contained 3 cases of normal adjacent breast tissue (NAT), 3 cases of
fibroadenoma (benign breast tumours) and 69 cases of a malignant breast tumour (in
duplicate blocks). Information about the age, ER, PR, Androgen receptor (AR), p53,
Ki67, HER2, pathological type, TNM and grade of each patient sample was available.
Scoring was performed by breast pathologist Dr Jeremy Parry. All sections were divided
into negative (-), intermediate (+) and high (++) C11orf67 expression groups. A positive
reaction was defined as discrete localization of the chromogen in the cytoplasm, nucleus,
and membrane of stained sections (Figure 2-7A).
We found that C11orf67 was mainly localized in the cytoplasm, although it also showed
perinuclear localization in some breast cancer samples as well as membranous and
nuclear expression in other samples (Figure 2-7B)
Staining results for all cases are shown in Table 2-3. In 75 breast cores, the positive
expression rate of C11orf67 protein was 64%. Of these, 89.6% were malignant breast
cancer samples. It was observed that all 3 sections of NAT and 2 out of 3 cases of benign
breast conditions showed moderate (+) C11orf67 expression in luminal ductal epithelial
cells, either cytoplasmic or both cytoplasmic and nuclear. This observation suggests that
C11orf67 is weakly expressed in normal breast ductal epithelium (Figure 2-6A middle
panel).
Next, we investigated whether there is any correlation between the expression of
C11orf67 and available clinical/pathological information such as hormonal status and
grade. Our results indeed showed that in intermediate (+) and high (++) expression
groups, 48.6% and 53.8% of patients respectively were ER+, whereas only 14.8% of the
low expression group were ER+. This difference was statistically significant, p=0.0054

C11orf67 as a Novel Biomarker in Breast Cancer
57
(Figure 2-7C). This finding validated the bioinformatic analyses of genomic databases
and confirms that C11orf67 is predominantly overexpressed in the HR+ subgroups of
breast cancer.
Moreover, we investigated the correlation between C11orf67 expression and the PR status
of the samples. Although not all the ER+ tumours were PR+, we were still able to find a
significant correlation between the expression of C11orf67 and PR status of sections.
Analyses of TMA staining revealed that 53.8% of patients were PR+ in the high
expression group and 37.1% in the intermediate expression group, as compared to only
22.2% in the low expression group. This difference was also statistically significant,
p=0.0443 (Figure 2-7D), yet not as high as the correlation with ER+ tumours.
Tumour grade is a score that determines how abnormal the cancer cells and the tumour
tissue appear under a microscope. It is also an indicator of how quickly a tumour is likely
to grow and spread. Grades 1 and 2, well and moderately differentiated respectively, tend
to grow and spread at a slower rate than poorly and undifferentiated tumours (grades 3
and 4, respectively). Importantly, we were able to correlate high C11orf67 expression
with poorly differentiated high tumour grade (grade 3), p=0.0386 (Figure 2-7E). While
46.2% of high C11orf67 expression group was grade 3, only 13.6% and 14.8% of low
and intermediate expression groups respectively were grade 3.
No significant correlation was found between C11orf67 expression and any of the other
parameters available from the stained TMA sections, including Ki67, p53, Her2, AR, or
tumour stage (Table 2-3). It is worth mentioning that commercial TMA sections do not
allow access to response to treatment, follow-up or 5-year survival information of the
patient samples used in the assay.
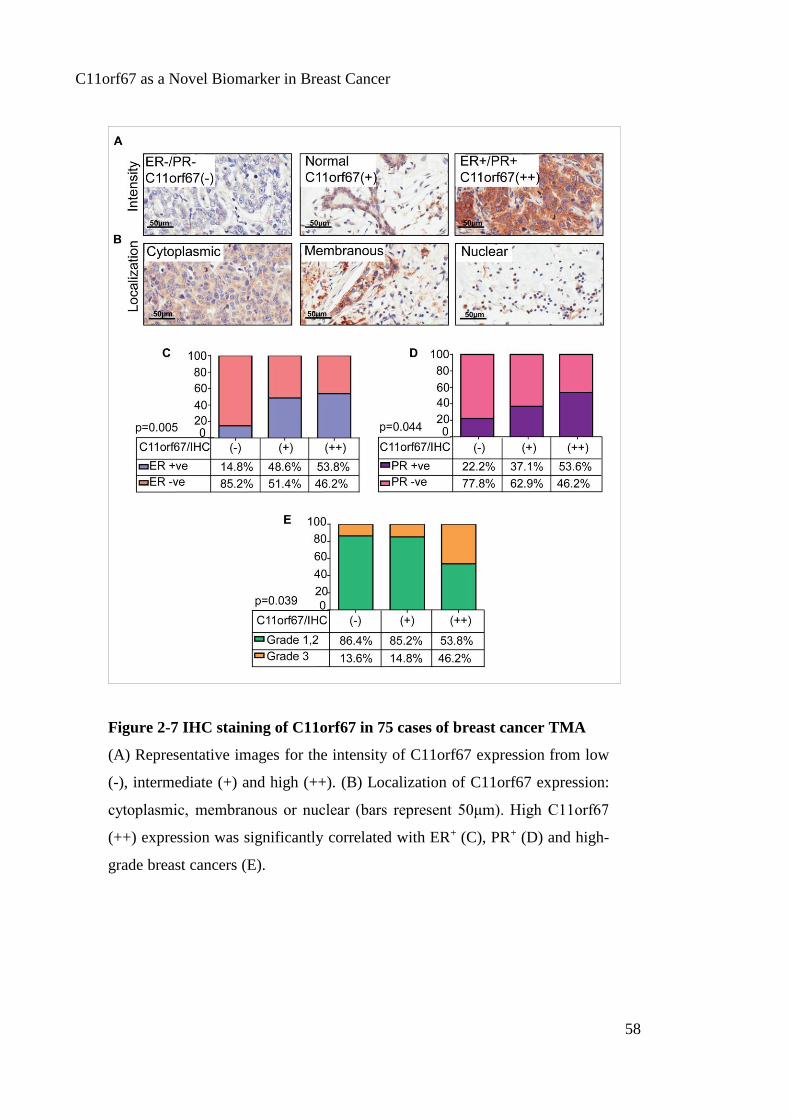
C11orf67 as a Novel Biomarker in Breast Cancer
58
Figure 2-7 IHC staining of C11orf67 in 75 cases of breast cancer TMA
(A) Representative images for the intensity of C11orf67 expression from low
(-), intermediate (+) and high (++). (B) Localization of C11orf67 expression:
cytoplasmic, membranous or nuclear (bars represent 50μm). High C11orf67
(++) expression was significantly correlated with ER+ (C), PR+ (D) and high-
grade breast cancers (E).

C11orf67 as a Novel Biomarker in Breast Cancer
59
Table 2-3 C11orf67 expression in 75 cases of breast cancer patients
a indicates unavailable information, ** p<0.01, * p<0.05.
Characteristics Total
No.
Low
(-)
No (%)
Intermediate
(+)
No (%)
High
(++)
No (%)
P
value
Type
Benign
Malignant
75 1 (3.7)
26 (96.3)
5 (14.3)
30 (85.7)
0 (0)
13 (100) 0.9423
Age
<=50
>50
75 15 (55.6)
12 (44.4)
21 (60)
14 (40)
7 (53.8)
6 (46.2) 0.9930
ER status
Positive
Negative
75 4 (14.8)
23 (85.2)
17 (48.6)
18 (51.4)
7 (53.8)
6 (46.2)
0.0054
**
PR status
Positive
Negative
75 6 (22.2)
21 (77.8)
13 (37.1)
22 (62.9)
7 (53.8)
6 (46.2)
0.0443
*
AR status
Positive
Negative
75 8 (29.6)
19 (70.4)
13 (37.1)
22 (62.9)
4 (30.8)
9 (69.2) 0.8171
Her2
Positive
Negative
75 2 (7.4)
25 (92.6)
7 (20)
28 (80)
3 (23.1)
10 (76.9) 0.1483
P53
Positive
Negative
75 19 (70.4)
8 (29.6)
22 (62.9)
13 (37.1)
8 (61.5)
5 (38.5) 0.5242
Ki67
Positive
Negative
75 11 (40.7)
16 (59.3)
16 (45.7)
19 (54.3)
3 (23.1)
10 (76.9) 0.4230
Lymph node
metastases
Positive
Negative
67a 6 (23.1)
20 (76.9)
7 (25)
21 (75)
4 (30.8)
9 (69.2) 0.6213
Grade
Low grade 1,2
High grade ≥ 3
62a 19 (86.4)
3 (13.6)
23 (85.2)
4 (14.8)
7 (53.8)
6 (46.2)
0.0386
*

C11orf67 as a Novel Biomarker in Breast Cancer
60
2.3.7 Copy number amplification of C11orf67 in HR+ breast
cancer
Our bioinformatic analyses revealed that C11orf67 is overexpressed and amplified in the
IntClust 2 subtype of breast cancer. In the previous section, we confirmed the
overexpression of C11or67 in breast cancer TMA. In this section, we experimentaly
confirmed the amplification of C11orf67 in breast cancer and we correlated the extent of
the amplification with the protein overexpression. Unfortunately, it was incompatible to
test C11orf67 amplification by FISH in the same TMA settings due to damaged nuclei or
multi-layer aggregates of tumour cells that all led to unsuccessful hybridization of tumour
blocks. Alternatively, to validate the amplification of C11orf67 in clinical specimens, we
conducted FISH analysis on 32 available ER+ breast cancer cases through our
collaboration with the pathology department, Gdansk University Hospital, Poland.
FISH was performed using detection probes for C11orf67 and Chromosome 11
centromere (CEN-11, empire genomics). Breast cancer samples were evaluated
microscopically by their C11orf67/CEN-11 ratios with a ratio ≥2 considered as positive
amplification (Figure 2-8A).
Our data has validated the amplification of C11orf67 in 9 ductal carcinomas (all ER+) out
of 32 cases tested (28%) with amplification indexes varying between 2.2 and 4.7. We also
had available information about the grade of the tumour and the TNM status.
While there was no significant difference in tumour grade between positive amplification
cases (33%) versus amplification negative cases (22%) p=0.08, we were able to
significantly correlate the cases with amplification to the LN metastases. 78% of the
tumours with C11orf67 amplification presented LN metastases over 26% of the negative
amplification group p<0.0001, keeping with expected aggressive pathobiology (Figure 2-
8B).
However, it is important to note that the statistical power of this analysis is limited
because of the small number of cases, lack of normal-like cases, and unavailable data

C11orf67 as a Novel Biomarker in Breast Cancer
61
about response to treatment or resistance. In the near future we are planing to conduct a
much extensive study in a larger available TMA of 1000 cases.
In summary, in this chapter we have validated for the first time the overexpression and
the amplification of C11orf67 in HR+ breast cancer. As expected, a general positive
correlation between C11orf67 overexpression and ER and PR expression, tumour grade
and LN involvement. Bioinformatic analyses revealed that C11orf67 indeed had
prognosis value not only in breast cancer but also in other tumours, particularly ovarian
and lung cancer. Lastly we also experimentally validated the existence of C11orf67
different isoforms in luminal cells and revealed the intracellular distribution of isoform 2
in luminal cells. Collectively our data suggest that C11orf67 could be a novel biomarker
to identify HR+ breast cancers with very poor prognosis.

C11orf67 as a Novel Biomarker in Breast Cancer
62
Figure 2-8 C11orf67 amplification detected by FISH in ER+ breast cancer
(A) Representative images of breast tumours positive and negative for the
C11orf67 amplification as assessed by FISH. Red dots represent C11orf67
FISH detection probe, and green dots represent Chromosome 11 centromere.
(B) Summary of the data analysis of FISH performed on 32 ER+ breast cancer
patients.

C11orf67 as a Novel Biomarker in Breast Cancer
63
2.1 Discussion
Breast cancer is a genetically heterogeneous disease with multiple genetic alterations
influencing its initiation and progression. Copy number amplification of several
oncogenic drivers at 11q13.5-q14 is a hallmark chromosomal aberration in the IntClust 2
subtype of breast cancer. These group of cancers are characterized by poor prognosis and
disease recurrence after treatment (Curtis et al., 2012). The identification of new
prognostic markers that stratify patients with poor prognosis, is very important to improve
the overall survival of ER+ breast cancer.
There are several established putative oncogenes harboured in the 11q13 region,
including MEN1, CCND1, FGF3, EMS1, PAK1, RSF1 and GAB2 (Shih Ie et al., 2005).
However, no previous reports have attempted to characterize the amplification of
C11orf67 gene as a significant candidate oncogene featuring amplification and
overexpression in this chromosome region.
The tertiary structure of the human AAMDC (C11orf67) protein that is higly homologus
to the bacterial protein MTH938 from Methanobacterium. This unique tertiary structure
of the protein has suggested that the gene has invaded the eukaryotic genomes by lateral
gene transfer (Das et al., 2001). Similar to the murine homologue of AAMDC (LI2),
human C11orf67 is highly conserved in human, mouse, rat, cow, chicken, and zebrafish
(Ma et al., 2012).
Bioinformatics analysis of subcellular localization suggests that human C11orf67 protein
is distributed in the cytoplasm (http://www.uniprot.org/uniprot/Q9H7C9). Our study
confirmed that flag-fused C11orf67 Isoform_2 protein is located in the cytoplasm with
some condensations around the nucleus in T47D and MCF7 breast cancer cell lines,
suggesting that C11orf67 protein has a physiological role in the cytoplasm and the
nucleus. This finding is not inconsistant with the study performed by Ma et al. (2012),
which showed that the murine homologue of AAMDC protein follows cytoplasmic
distribution in fibroblast cells (Ma et al., 2012).

C11orf67 as a Novel Biomarker in Breast Cancer
64
In this chapter we described the existence of three different isoforms of C11orf67 and we
hypothesised that these transcript variants are originated by alternative splicing occurring
at the C-terminus. Spliced variants of C11orf67 produces multiple mature mRNAs and
protein isoforms with distinct structural and functional properties, which could contribute
to the oncogenic potential of the C11orf67 protein and its ability to influence several
aspects of tumour formation, progression and resistance to therapy.
To the best of our knowledge, only one other study had investigated the mRNA
expression levels of C11orf67 in a number of breast cancer cell lines, together with
expression levels of other genes in the same locus (Kwek et al., 2009). However, this
study neither investigated the protein expression levels of C11orf67 in the tested cell lines
nor correlated the mRNA expression levels to the HR expression status of those cell lines.
Here we reported the expression of human C11orf67 at both the mRNA and protein levels
in different breast cancer lines with variable HR status, and we correlated the high
overexpression of C11orf67 in HR+ breast cell lines compared to the normal-like breast
cells.
A significant number of reports have detected the amplification of chromosome 11q13.5
in several types of human cancer, and most of them have linked the presence of the
amplicon with a worse clinical outcome and shorter overall survival (Ormandy et al.,
2003, Hui et al., 1998, Borg et al., 1991). These findings suggest that the 11q13.5
amplicon may contain important potential drivers that could participate to clinical
aggressiveness of certain tumour subtypes.
In the current study, we performed a detailed genomic analysis on a novel gene located
in the 11q13.5 amplicon that has not been studied before in the context of breast cancer.
The selective overexpression and copy number amplification of C11orf67 in IntClust 2
ER+ subgroup of breast cancer that is associated with low survival, makes C11orf67 an
attractive potential target which could serve to distinguish those ER+ tumours at risk of
relapse from those of low risk (Curtis et al., 2012, Stephens et al., 2012).

C11orf67 as a Novel Biomarker in Breast Cancer
65
We foud that C11orf67 protein expression in the breast cancer TMA significantly
correlated with positive ER and PR status and high tumour grade. On the other hand,
amplification analyses in a different cohort of ER+ patients by FISH revealed another
correlation with LN metastases. However, the statistical power of this analysis is limited
because of the small number of cases in the gene-amplified subgroups. However, these
observations suggest the predictive prognostic power of C11orf67 overexpression in ER+
patients, and strongly argue that C11orf67 amplification could represent a driver lesion
in breast carcinogenesis.
Choi et al. found that C11orf67 was amplified and overexpressed in high-grade ovarian
serous carcinoma, but this study focused more on neighbouring genes with higher
correlation between DNA and transcript copy number (e.g. RSF-1, INTS4, CLNS1A,
ALG8, GAB2, and PAK1) (Choi et al., 2009).
These findings are consistent with other studies that proved the involvement of 11q13
oncogenic targets in the prognosis of breast cancer and other epithelial cancers. CCND1
is one of the most important drivers in 11q13 chromosome locus and had been examined
by a significant number of studies. Lundgren et al. and others have found that gene
amplification of CCND1 is linked to poor clinical prognosis, detrimental response to
tamoxifen treatment, and is associated with high tumour grade (Roy et al., 2010,
Lundgren et al., 2012). Also, high CCND1 expression by IHC defines a subgroup of ER+
patients with a high risk of relapse and death (Beca et al., 2015).
Another important potential driver located in the same amplicon is PAK1 (p21-activated
kinase 1), which has been studied extensively in the context of breast cancer.
Experimental studies have recently shown increasing evidence that the overexpression of
PAK1 is associated with decreased tamoxifen sensitivity and poor clinical outcome in
breast cancer, particularly in postmenopausal women (Holm et al., 2006, Bostner et al.,
2007, Bostner et al., 2010).
RSF-1 (remodelling and spacing factor) is the neighbouring gene to C11orf67, and may
be coregulated with C11orf67 (this is discussed in more detail in chapter 4). RSF-1 is also

C11orf67 as a Novel Biomarker in Breast Cancer
66
an important biomarker that predicts prognosis in breast and other epithelial cancers. In
gastric adenocarcinoma and colorectal carcinoma, RSF-1 overexpression is known to
predict an unfavorable prognosis and poor overall survival (Hu et al., 2012, Liu et al.,
2012). High expression levels of RSF-1 have also been associated with aggressive
phenotypes and poor clinical outcome in breast cancer patients and paclitaxel resistance
in ovarian cancer patients (Choi et al., 2009, Ren et al., 2014).
In conclusion, there are many reports suggesting that other candidate genes in the 11q13
amplicon may contribute to tumour development and poor clinical outcome in breast
cancer. However, this is the first report that has clearly established C11orf67 as one of
the novel biomarkers in 11q13.5 in breast cancer that can stratify patients and identify
those patients with a higher risk of poor clinical outcome.
In the future, it will be of interest to conduct C11orf67 staining of FFPE tissues from
biopsies of patients with HR+ breast cancer, along with clinical follow-up of those
patients. This will be a necessary translational step to introduce C11orf67 staining to the
clinic. Also, generation of cell lines derived from patient samples that show amplification
of C11orf67 is a significant step to having a model that can be used in the development
of a routine clinical test in the future.

Chapter 3:
Functional Consequences of
C11orf67 Gene Alteration

Functional Consequences of C11orf67 Gene Alteration
68
3.1 Introduction
Despite increasing efforts towards developing new advances in surgery and novel
therapeutic agents, breast cancer remains a leading cause of cancer-related death in
women worldwide (Calaf et al., 2015). Identification of novel functional biomarkers is
essential for the developent of novel targeted therapies for specific breast cancer subtypes,
specially those characterised with poor prognosis (IntClust 2 subtype) (Nakshatri and
Badve, 2007).
In the previous chapters, we have demonstrated that C11orf67 is both amplified and
overexpressed in ER+ breast cancers and is associated with poor prognosis. These patients
usually develop highly proliferative tumours, which have a high incidence of metastases
and become resistant to endocrine therapy and chemotherapy (Curtis et al., 2012). As
C11orf67 is a fundamentally uncharacterised gene, its role as an oncogenic driver in
breast cancer has not been previously investigated.
To assess the role of C11orf67 in breast cancer tumourigenesis and tumour progression,
in this chapter we artificially altered the endogenous levels of C11orf67 in ER+ breast
cancer cells, by either loss of function (RNAi-mediated knockdown) in cell lines
expressing high levels of the gene, or by gain of function (exogenous cDNA
overexpression) in cell lines carrying very low levels of C11orf67. We hypothesized that
this alteration of C11orf67 expression would affect proliferation, tumourigenicity and
other aspects of tumour progression such as resistance to drugs. Indeed, our data revealed
that knockdown of C11orf67 was able to arrest proliferation and induced cellular
senescence of ER+ breast cancer cells. Furthermore, a C11orf67 gain of function
increased the sensitivity of ER+ breast cancer cells to antifolate chemotherapeutic agents.
Tumourigenesis is caused by genetic alterations that activate oncogenes or inactivate
tumour suppressor genes. Genes involved in tumourigenesis are usually associated with
aberrant cellular proliferation, apoptosis, angiogenesis, and cellular senescence (Hainaut
and Plymoth, 2013). Senescence is a state of irreversible and permanent growth arrest
that occurs in response to either aging (replicative senescence), stress (stress-induced

Functional Consequences of C11orf67 Gene Alteration
69
senescence), or mediation of an oncogenic factor (oncogene-induced senescence)
(Collado et al., 2007). Triggering the senescence of tumour cells may contribute to
successful cancer therapy (Lleonart et al., 2009).
Activation of the tumour suppressor p53 or RB pathways is usually essential for the
induction of the senescence program (Lowe et al., 2004). However, cell cycle arrest and
the inhibition of the AKT signalling pathway are also possible mechanisms that induce
senescence, and participant proteins of these pathways are used as essential markers for
the identification of senescent cells (Kuilman et al., 2010).
The phosphoinositide 3 kinase/Akt/mammalian target of rapamycin (PI3K/Akt/mTOR)
pathway plays a substantial role in tumour cell growth, proliferation, and has been
associated with resistance to endocrine therapy (Paplomata and O'Regan, 2014). A
simplified schematic representation of the PI3K/Akt/mTOR pathway is illustrated in
Figure 3-1.
Figure 3-1 Illustration of the PI3K/Akt/mTOR pathway
Adapted from Holmes (2011).

Functional Consequences of C11orf67 Gene Alteration
70
Growth fator stimulation leads to activation of tyrosine kinase receptors, whichs induce
the recruitment and activation of different proteins such as PI3K (Cantley, 2002). PI3K
phosphorylates phosphatidylinositol 4,5 bisphosphate, or PIP2, to phosphatidylinositol
3,4,4-triphosphate, or PIP3. This leads to the phosphorylation of Akt in two residues,
threonine 308 and serine 473, after which it is fully activated. Phosphorylated Akt then
phosphorylates several targets in the nucleus and cytoplasm, including mTOR. mTOR
induces cellular growth and proliferation through its effect on the downstream molecules
S6K1 (40S ribosomal protein S6 kinase 1), and 4EBP1 (eukaryotic initiation factor 4E
binding protein) (Kenerson et al., 2002, Dowling et al., 2010, Holz, 2012). Indeed,
rapamycin, the inhibitor of mTOR, is currently used for the treatment of HR+ breast
cancer (Ciruelos Gil, 2014). Furthermore, experimental studies have found an interaction
between Akt activity and the response to chemotherapy in breast and ovarian cancer (Page
et al., 2000, Stal et al., 2003).
In this chapter, we analyzed the effect of C11orf67 knockdown and overexpression on
the progression of breast cancer cells, to elucidate the mechanisms and molecular
pathways by which C11orf67 may induce cellular senescence and resistance to therapy.

Functional Consequences of C11orf67 Gene Alteration
71
3.2 Materials and Methods
3.2.1 Reagents and Antibiotics
Estradiol valerate, peanut oil, Methotrexate, 5-Fluorouracil , and Taxol were purchased
from Sigma-Aldrich, St Louis, MO, USA.
For IF and western blotting, primary antibodies specific to Ki-67, caspase-3, p-AKT
(Serine 473) , p-AKT (Threonine 308), p-mTOR (Serine 2448), p-MAPK p42/44,
CDK4, CDK6, CCND1, and CCNE1 were purchased from Cell Signaling, Gold Coast,
QLD, Australia.
3.2.2 Lentiviral C11orf67 shRNA infection
Knockdown of C11orf67 expression was performed using Mission TRC human
shRNA/pLKO.1 clone sets (Sigma-Aldrich, St Louis, MO, USA). This system allows
efficient intracellular delivery and stable expression of the small hairpin RNA (shRNA)
sequences specific to the C11orf67 mRNA (Table 3-1). Lentiviruses were produced by
transfecting HEK293T packaging cells at 80% confluency with the following packaging
plasmids: 1.54 μg VSV-G Envelop (vesicular stomatitis virus G), 1.1 μg RSV-Rev, 2.88
μg Gag-pol, and 4.5 μg of each pLKO.1/shRNA plasmid, using Lipofectamine-2000
reagent (Invitrogen, Waverley, Australia) according to the manufacturer’s protocol. An
empty pLKO.1 lentiviral vector was used as a negative control.
Recipient (host) T47D cells were seeded at 1.5×105 cells per 10 cm diameter tissue culture
dish and incubated for 24 hours at 37°C, and 5% CO2 atmosphere. Host cells were then
transfected 3 times every 8 hours with culture media containing viral particles collected
48, 56 and 72 hours post-infection. Viral particles were filtered through a 0.22 μm sterile
filter unit (Thermo Scientific, USA), and treated with 8 μg/ml polybrene (Sigma-Aldrich,
St Louis, MO, USA) before being added to the host cells. Stable T47D cells were selected

Functional Consequences of C11orf67 Gene Alteration
72
with 5 µg/ml puromycin (Invitrogen, Waverley, Australia) for 5 days before functional
assays were performed.
3.2.3 Lentiviral C11orf67 cDNA infection
The gain of function of C11orf67 was performed using C11orf67 Isoform_2 cDNA
pLV105 lentiviral plasmid (Genecopoeia, Rockville, MD, USA). MCF7 cells were
transfected with virus harvested from HEK293T cells, as described above. An empty
pLV105 lentiviral vector was used as a negative control.
Table 3-1 Nucleotide sequences and exon number of C11orf67-specific
shRNAs
Nucleotides involved in base pairing to C11orf67 transcript is shown in red.
C11orf67
shRNA Sequence & Location (Exon no.)
sh1 5'CCGGAGGCTCTAATACAACCTATAACTCGAGTTATA
GGTTGTATTAGAGCCTTTTTTG (Exon 1)
sh2 5'CGGCAGATGTGAAGGAAGTTGTTGCTCGAGCAACA
ACTTCCTTCACATCTGTTTTTG (Exon 2)
sh5 5'CCGGGTGTACAGACTCTTGTGATTGCTCGAGCAATC
ACAAGAGTCTGTACACTTTTTG (Exon 2)

Functional Consequences of C11orf67 Gene Alteration
73
3.2.4 MTT cell viability assays
Evaluation of cell proliferation and cell cytotoxicity was performed using MTT
colorimetric assay (Sigma-Aldrich, St Louis, MO, USA). Either T47D or MCF7 breast
cancer cell lines were transduced with the corresponding lentiviral vectors (C11orf67
shRNAs for knockdown, or C11orf67 cDNA for overexpression) or empty vector
lentiviral control. Transduced cells were seeded at a density of 1000 cells/well in 96-well
flat-bottom culture plates in triplicate for each condition.
For proliferation assays, cells were incubated for 0, 24, 48, 72, or 96 hours and the
percentage of proliferating cells was measured according to the MTT manufacturer’s
instructions. Briefly, 100 μl of filter sterilized 3-(4, 5-Dimethylthiazol-2-yl)-2, 5-
Diphenyltetrazolium Bromide (MTT) solution (final concentration 0.5 mg/ml in complete
media) was added to the media in each well. Following a 1 hour incubation period with
MTT, media were removed and the blue formazan crystals trapped in cells were dissolved
in sterile dimethyl sulfoxide (DMSO) (50 μl/well) (Sigma-Aldrich, St Louis, MO, USA)
by incubating at 37ºC for 5 minutes. The absorbance was measured at 570 nm using a
microplate spectrophotometer (Labsystems Multiskan RC). Background absorbance was
measured at 690 nm. The results were represented as mean ± SE of three independent
experiments. Results were normalized to values obtained immediately after seeding of
the cells (day=0).
For cytotoxicity assays, transduced cells were treated with various concentrations of
indicated drugs (methotrexate: 0.5, 1.0, and 5.0 μM; 5-fluorouracil: 0.5, 5.0, and 10.0 μM;
taxol: 1, 10, 20, 30, and 40 nM) for 48 or 72 hours. The percentage of viable cells in each
treatment group was normalized to that of vehicle control treated cells.
3.2.5 Immunofluorescence
2x104 T47D cells transduced with C11orf67 shRNAs or empty lentiviral vector were
seeded on coverslips and incubated overnight at 37°C to allow the cells to attach. Cells
were fixed with 4% PFA in PBS for 20 minutes, permeabilized with 0.5% Triton X-100

Functional Consequences of C11orf67 Gene Alteration
74
in PBS for 10 minutes at 4°C, blocked with 4% BSA in PBS for 60 minutes at room
temperature, incubated with the primary antibody (1:400) diluted in blocking solution
overnight at 4°C, and finally incubated with a secondary Alexa Fluor 488-conjutated
antibody (1:1000) for 1 hour in the dark. Nuclei were stained blue with Hoechst nuclear
stain (1:5000) (Sigma-Aldrich, St Louis, MO, USA), and coverslips were mounted in
Fluoromount aqueous mounting medium (Sigma-Aldrich, St Louis, MO, USA). The
percentage of positive cells was assessed by counting the percentage of green fluorescent
cells in 9 fields of views using a fluorescent Olympus IX71 microscope (Singapore). Data
were normalised to empty vector control cells.
3.2.6 Anchorage-independent colony formation assays
Anchorage-independent growth was assessed with soft agar colony formation assays.
T47D cells (10000 cells/well) were seeded in triplicate in six-well plates, in 0.48%
agarose (Sea Plaque Low-Melt Agarose, Sigma-Aldrich, St Louis, MO, USA) over a solid
layer of 0.8% agarose in RPMI medium with 20% FBS. 0.5 ml of growth media was
added to each well and gently replaced every 3 days. Plates were then incubated in 5%
CO2 humidified conditions at 37°C for 4 weeks.
The resulting colonies were fixed and stained with 4% PFA-0.01% crystal violet in PBS
for 5 minutes, washed in PBS, and then counted by Leika light microscope in 12 random
fields. Results were expressed as a percentage of colony count ± SE from three
independent experiments relative to the empty vector control.
3.2.7 In Vitro cell migration assay
To assess migratory capacity of T47D knockdown cells, a CytoSelect 24-well cell
migration assay (Cell Biolabs, San Diego, USA) was used according to the
manufacturer’s instructions. Briefly, the transwell polycarbonate membrane filter inserts
with 8-µm pore size were rehydrated and placed in 24-well tissue culture plates. T47D
cells transduced with either C11orf67 shRNAs or empty vector control were serum
starved overnight and then resuspended into serum free media (SFM). 1x 106 cells/well

Functional Consequences of C11orf67 Gene Alteration
75
from serum-free cell suspension were added to the top chambers of the transwells, and
10% serum-containing RPMI media was added to bottom chambers, which served as a
chemoattractant. The plates were incubated at 37°C to allow the cells to migrate. After
24 hours, the culture media was carefully aspirated, and the non-migrating cells were
removed from the top chamber of the membrane using a cotton swab without causing any
punctures to the polycarbonate layer. To visualise the migrating cells, the inserts were
incubated with 400 μl cell stain solution (Cell Biolabs, San Diego, USA) for 10 minutes
at room temperature. Pictures were captured using a digital camera attached to a cell
culture inverted Leika light microscope to quantitate the number of cells that had passed
through the porous membrane in 12 random fields. Results were expressed as average
values ± SE relatively to the empty vector control cells.
3.2.8 TUNEL assay
The terminal deoxy-transferase (TdT)-mediated deoxyuridine triphosphate (dUTP) nick-
end labeling (TUNEL) assay was used to detect the extent of DNA fragmentation caused
by apoptosis. The in Situ cell death detection kit (Roche, Castle Hill, Australia) was used
to detect the percentage of apoptotic cells in T47D cells transduced with either the
C11orf67 shRNAs or with empty vector control by flow cytometry according to the
manufacturer’s instructions.
Briefly, T47D cells were fixed in 2% PFA in PBS for 60 minutes at room temperature
and then permeabilized in 0.1% Triton X-100 in 0.1% sodium citrate for 2 minutes on
ice. Cells were incubated with 50 μl of freshly prepared TUNEL reaction mixture
according to the kit’s instruction for 60 minutes at 37°C in the dark. Fluorescein-
conjugated dUTP incorporated in nucleotide polymers was detected and quantified using
flow cytometry. As a positive control for apoptosis, T47D cells were treated with 20 nM
taxol for 72 hours before the experiment and treated the same way as the treatment groups.
Approximately 10,000 cells per sample were acquired and quantified using Cell-Quest
software (Becton-Dickinson) for the analysis of cell apoptosis (Bao et al., 2014).

Functional Consequences of C11orf67 Gene Alteration
76
3.2.9 In vivo tumourigenicity assays
All animal experiments were approved under and performed in accordance with the
animal ethics protocol RA 3/100/1159, University of Western Australia. 5x106 T47D
cells stably transduced with either pLKO.1 empty lentiviral vector or C11orf67 shRNAs
2 (sh2) were injected subcutaneously (sc) in the right flank of 5 weeks old female BALB/c
Foxn1/Arcmouse (nude) mice (Animal Resources Centre, WA, Australia). Prior to
inoculation, cells were resuspended (1 in 1) in a solution of SFM and BD Matrigel High
Concentration (BD Bioscience, North Ryde, NSW, Australia). A total of 100 μl of cell
suspension was slowly injected using a 26-gauge needle sc in the flank of nude mice. 1μg
of estradiol valerate in 100 μl peanut oil (Sigma-Aldrich, St Louis, MO, USA) was
injected sc into the back of the mice every 4 days to allow the estradiol-dependent T47D
cells to proliferate (Sartorius et al., 2003). Width and length of tumours were measured
every 3-4 days by a digital caliper, and tumour volumes were calculated using the
modified ellipsoid formula; V= [(width2) x ½ x length]. Animals bearing tumours
>800mm3 were humanely sacrificed, and tumours were dissected and extracted. Tumour
tissues were fixed in 4% PFA in PBS, washed 3 times with PBS and incubated in 70%
ethanol for up to one week. The tumours were embedded in paraffin and 5 µm sections
were made by Cell Central (University of Western Australia).
3.2.10 Immunohistochemistry of tumour sections
C11orf67 IHC staining was performed on FFPE tissue biopsies from mice. Tissue
sections were deparaffinized with xylene and rehydrated with 100% and then 75%
ethanol. Sections were then subjected to heat-induced antigen retrieval in 10 mM Sodium
citrate buffer, pH 6.0, using a microwave oven at high power for 10 minutes before the
power was lowered for another 10 minutes. Following that, the tissue sections were
cooled to room temperature in the same buffer and rinsed with distilled water.
Endogenous peroxidase activity was blocked by adding 3% hydrogen peroxide solution
(Sigma-Aldrich, St Louis, MO, USA) in methanol for 30 minutes at 4°C. The tissue
sections were then washed with distilled water before they were incubated with 1% BSA

Functional Consequences of C11orf67 Gene Alteration
77
in PBS for 1 hour at room temperature to block non-specific protein binding. The anti-
C11orf67 primary antibody (Santa Cruz Biotechnology, Texas, USA) was added
overnight at 4°C (1:100). Immunodetection was performed by incubation with
biotinylated goat anti-rabbit secondary antibody (GeneCopeia, Maryland, United States)
for one hour followed by incubation with streptavidin-horseradish peroxidase
(Genecopoeia, Rockville, MD, USA) at room temperature for 30 minutes. Sections were
then incubated with DAB solution (Genecopoeia, Rockville, MD, USA) for 5 minutes
until signal development and counterstained with Haematoxylin for 1 minute. After
rinsing with distilled water and then tap water, the sections were dehydrated through
increasing concentrations of ethanol (70%, 95% and 100%) and then xylene. Finally,
mounting of the coverslips was done using Acrymount IHC mounting media (McKinney,
Texas, USA).
3.2.11 Senescence-associated β-galactosidase staining
A senescence cellular staining kit (Sigma-Aldrich, St Louis, MO, USA ) was utilized for
the detection of senescent T47D cells according to the manufacturer's instructions.
Briefly, stable T47D cell lines transduced with either C11orf67 shRNAs or with empty
vector cells were seeded in a sex-well plate at a density of 4x105 cells/well. The cells were
washed with PBS and fixed in the kit’s fixative solution before staining with the X-gal
solution. After 24 hours of incubation at 37°C, cells were observed under the microscope
and the percentage of positive cells was calculated by counting the blue-stained cells
(senescent cells) versus total cells in at least 12 random microscopic fields.
3.2.12 Illumina microarray analysis
3.2.12.1 RNA preparation
For microarray gene expression analysis, total RNA was extracted using Trizol
(Invitrogen Life Technologies, Carlsbad, USA) and purified using RNeasy columns
(Qiagen, Valencia, USA) according to the manufacturers’ protocol. After processing with
DNase digestion, RNA samples were quantified and aliquoted and stored at -80°C until

Functional Consequences of C11orf67 Gene Alteration
78
use. For quality control, RNA purity and integrity were evaluated by denaturing gel
electrophoresis, OD 260/280 ratio, and analyzed on Agilent 2100 Bioanalyzer (Agilent
Technologies, Palo Alto, USA).
3.2.12.2 Labelling and Purification
Total RNA was amplified and purified using the Ambion Illumina RNA Amplification
kit (Ambion, Austin, USA) to yield biotinylated cRNA according to the manufacturer’s
instructions. Gene expression analysis were carried by Macrogen (Seoul Rep, Korea).
Briefly, 550 ng of total RNA was reverse-transcribed to cDNA using a T7 oligo (dT)
primer. Second-strand cDNA was synthesized, in vitro transcribed, and labeled with
biotin-NTP. After purification, the cRNA was quantified using the ND-1000
Spectrophotometer (NanoDrop, Wilmington, USA).
3.2.12.3 Hybridization and data export
750 ng of labeled cRNA samples were hybridized to each human HT-12 expression v.4
bead array for 16-18 h at 58°C, according to the manufacturer's instructions (Illumina,
Inc., San Diego, USA). Detection of array signal was carried out using Amersham
fluorolink streptavidin-Cy3 (GE Healthcare Bio-Sciences, Little Chalfont, UK) following
the bead array manual. Arrays were scanned with an Illumina bead array Reader confocal
scanner according to the manufacturer's instructions.
3.2.12.4 Raw data preparation and statistical analyses
The quality of hybridization and overall chip performance were monitored by visual
inspection of both internal quality control checks and the raw scanned data. Raw data
were extracted using the software provided by the manufacturer (Illumina GenomeStudio
v2011.1 (Gene Expression Module v1.9.0)). Array probes were transformed by logarithm
and normalized by quantile method. Statistical significance of the expression data was
determined using Independent t-test and fold change in which the null hypothesis was

Functional Consequences of C11orf67 Gene Alteration
79
that no difference exists among groups. False discovery rate (FDR) was controlled by
adjusting p-value using Benjamini-Hochberg algorithm. For a differentially expressed
genes (DEG) set, Hierarchical cluster analysis was performed using complete linkage and
Euclidean distance as a measure of similarity. Gene-Enrichment and Functional
Annotation analysis for significant probe list was performed using DAVID
(http://david.abcc.ncifcrf.gov/home.jsp). All data analysis and visualization of
differentially expressed genes were (www.r-project.org).
3.2.13 Statistical analysis
Statistical significance was detected using one-way ANOVA to compare differences
between mean values in cell viability, colony formation, cell migration and invasion
assays in shRNA-transduced samples relative to empty vector control group. Multiple
group comparisons were analyzed with Dunnet multiple comparison tests. Statistical
analyzes were performed using Prism software, p < 0.05 was considered to be statistically
significant. All data are represented as mean value ± SE of at least triplicate experiments.

Functional Consequences of C11orf67 Gene Alteration
80
3.3 Results
In previous chapters, we demonstrated that C11orf67 is a novel prognostic biomarker that
is overexpressed and amplified in ER+ breast cancer. However, the precise role C11orf67
in the tumourigenesis and tumour progression in breast cancer cell lines has not been
investigated before.
In this chapter, we hypothesized that C11orf67 has a role in breast carcinogenesis and
influences the response of breast cancer cells to different therapeutic agents. To
experimentally address this, we first identified suitable cellular model systems expressing
C11orf67 (Section 2.3.5). We selected T47D breast cancer cell line to be used in the
functional characterization of C11orf67 knockdown showing highest levels of C11orf67
expression, and MCF7 cells in the C11orf67 cDNA overexpression expressing lower
levels of the gene.
In the following sections, we will take advantage of in vitro and in vivo tumour biology
assays to analyse the consequences of the genetic manipulation of C11orf67 expression
to address the role of C11orf67 in breast cancer tumourigenesis.
3.3.1 Functional consequences of C11orf67 ablation in T47D
cells
3.3.1.1 Knockdown of C11orf67 by shRNAs
For stable knockdown of C11orf67 expression in T47D cells, we used the pLKO.1
lentiviral vector. This system allows efficient intracellular delivery and stable expression
of shRNA sequences to target gene of interest in mammalian cells. shRNA sequences
were designed to specifically targeting C11orf67 mRNA at the first and the second exons.
To account for potential off-target effects, we used three different shRNA constructs
specific to C11orf67 transcript in all the functional studies, shRNA1 (sh1), shRNA2

Functional Consequences of C11orf67 Gene Alteration
81
(sh2), and shRNA5 (sh5). As a negative control, we used pLKO.1 empty vector and
untransfected T47D (mock) cells.
Lentiviral particles were generated by co-transfection of lentiviral constructs with
packaging constructs in the recipient HEK293T cells, and supernatants were used to infect
the host lines. After transfection, the transduced cells were analyzed for quantitative
changes in C11orf67 expression by qRT-PCR, western blot, and ICC analyses (Figure 3-
2).
We found that C11orf67 sh1 and sh2 constructs yielded a significant knockdown of
C11orf67 expression at the mRNA level (90% and 99% respectively), p<0.0001. sh5
produced an incomplete yet significant downregulation of C11orf67 expression (30-50%)
as compared to the empty vector control, p<0.001 (Figure 3-2A). Western blot analysis
confirmed the existence of at least two isoforms of human C11orf67 in T47D cells.
Isoform_2 revealed a MW of 13 kD, and was completely downregulated with the sh2
construct, while Isoform_3 with a MW of 16 kD showed partial downregulation (Figure
3-2B).
We also confirmed the cytoplasmic and perinuclear downregulation of C11orf67
expression by sh2 at the protein level by ICC. Our results confirmed the intracellular
compartmentalization studies of the protein conducted in section 2.3.3 (Figure 3-2C).

Functional Consequences of C11orf67 Gene Alteration
82
Figure 3-2 Knockdown of C11orf67 by shRNAs in T47D cells
(A) C11orf67 mRNA expression analysis by qRT-PCR in T47D cells
transduced with mock control, empty vector, or specific C11orf67 shRNAs
(sh1, sh2 and sh5) (*** p<0.0001, ** p<0.001). (B) Western blot analysis of
the indicated cell lines blotted with an anti-C11orf67 antibody (Santa Cruz); -
tubulin was used as a loading control. MW markers are shown on the right. (C)
ICC staining of T47D cells transduced with empty vector or C11orf67 sh2
constructs.

Functional Consequences of C11orf67 Gene Alteration
83
3.3.1.2 C11orf67 ablation reduces the proliferative potential
of T47D
After C11orf67 downregulation by shRNAs was validated at the mRNA and the protein
level, we secondly investigated whether C11orf67 depletion had an effect on the
proliferation rates of the ER+ breast T47D cells by MTT cell viability assays.
T47D cells transduced with either empty vector, or C11orf67 shRNAs (sh1, sh2, and sh5)
were seeded at the same density in triplicate for each experimental point, then cell
proliferation was monitored over time for a total period of 96 hours. We observed that
T47D cells expressing C11orf67 shRNAs (sh1, sh2, sh5) exhibited a significant reduction
in cell growth relative to the empty vector cells at 48 hours, 72 hours, and 96 hours.
(p<0.0001) (Figure 3-3A).
To further confirm this finding, we quantitated the expression of the proliferation marker
Ki-67 in the shRNA cells by IF (Figure 3-3B). shRNA-transduced cells showed a
significant reduction in the percentage of cells exhibiting nuclear positivity of Ki-67 as
compared with the empty vector control cells (Figure 3-3C). C11orf67 sh1 and sh2 cells
showed about 70% and 85% drop in the expression of Ki-67 protein, respectively
(p<0.0001), while sh5 cells had 40% less Ki-67 expression relative to that of the empty
vector control (p=0.0001).
Overall, the above results indicate that C11orf67 is necessary for the proliferation of
T47D cells.

Functional Consequences of C11orf67 Gene Alteration
84
Figure 3-3 C11orf67 knockdown decreases cellular proliferation
(A) MTT cell proliferation analysis of T47D cells transduced with either empty
vector or C11orf67 shRNAs. (B) Representative IF images of Ki-67 positive
cells in the indicated cell lines (green), nuclei were labelled with Hoechst
nuclear stain (blue). (C) Percentage of Ki-67 positive cells in C11orf67 shRNA
cells normalized to the empty vector control (*** p<0.0001, ** p<0.001).

Functional Consequences of C11orf67 Gene Alteration
85
3.3.1.3 C11orf67 knockdown decreases tumourigenic
phenotype of T47D cells in vitro
As many oncogenic drivers induce tumourigenic behaviour, we next investigated whether
the loss of C11orf67 expression could affect the ability of T47D cells to grow in an
anchorage-independent fashion and to form colonies in soft agar, which monitors
tumourigenic capacity in vitro. To this end, T47D cells transduced with either the
C11orf67 shRNAs or control empty vector were overlaid over a solid layer of agar at a
clonogenic density of 1x104 cells per well in a six-well plate, and the cells were allowed
to grow for three weeks until visible colonies were formed.
Consistent with the results obtained from the proliferation assays, we observed a
significant decrease in the colony forming efficiency upon C11orf67 knockdown in T47D
cells (Figure 3-4A). Our results show that both mock treated and empty vector transduced
cells formed abundant foci in soft-agar while C11orf67 depletion by sh1 and sh2 caused
a dramatic and significant reduction in the number of colonies formed relative to controls
(75% and 83% reduction respectively, p<0.0001). sh5 cells demonstrated 60% decrease
in the percentage of the number of colonies, p=0.001 (Figure 3-4B). In addition to colony
number the size of the colonies was also affected by the C11orf67 shRNAs with an overall
trend similar to that of the colony scoring experiment (Figure 3-4C). Thus, our data shows
that colony forming ability is inversely correlated with the silencing efficiency achieved
with the various lentiviral vectors (lower efficiency of C11orf67 sh5).
In summary, these experiments demonstrated that C11orf67 was necessary for the
formation and proliferation of colonies in anchorage-independent assays, suggesting that
C11orf67 is playing a role in tumour formation.

Functional Consequences of C11orf67 Gene Alteration
86
Figure 3-4 C11orf67 knockdown decreases cellular colony formation
(A) Images of stained soft agar colonies from cells transduced with the
C11orf67 shRNAs or with empty vector control and incubated for 4 weeks. (B)
Percentage of colony formation normalised to the empty vector control cells.
(C) Percentage of colony size relative to the empty vector control cells (***
p<0.0001, ** p<0.001).

Functional Consequences of C11orf67 Gene Alteration
87
3.3.1.4 C11orf67 promotes migratory behaviour in ER+ breast
cancer cells
We also hypothesized that the knockdown of C11orf67 might impair the migratory
behaviour of ER+ breast cancer cells. To investigate this, we conducted a CytoSelect
migration assay to quantitate the migration capacity of T47D cells transduced with the
empty vector and the C11orf67 shRNAs. Importantly, we found that C11orf67
knockdown had a potent inhibitory effect on T47D cell migration (Figure 3-5A).
C11orf67 sh1 and sh2 significantly suppressed cell migration by more than 90% as
compared with the empty vector control (p<0.0001). Incomplete knockdown of C11orf67
by sh5 suppressed cell migration to a lesser extent (75%, p<0.0001) (Figure 3-5B).
Figure 3-5 C11orf67 knockdown decreases cellular migration
(A) Representative images of migratory T47D cells transduced with C11orf67
shRNAs or the empty vector control. (B) Quantification of migratory cells in
each condition normalised to the empty vector control cells (*** p<0.0001).

Functional Consequences of C11orf67 Gene Alteration
88
3.3.1.5 Downregulation of C11orf67 expression inhibits
tumour growth in a xenograft model
The finding that the C11orf67 knockdown strongly inhibited proliferation, tumour
formation and migration in vitro, prompted us to investigate the effect of C11orf67
knockdown in a tumourigenic model in vivo. In our in vivo experimental design, we used
cells transduced with the empty vector as a control and the T47D transfected with the
C11orf67 sh2 since these cells showed the most potent repression of C11orf67 gene
transcripts and protein. Cells were implanted into the posterior flank of nude mice, and
tumour growth was monitored every other day. Our results show that the knockdown of
C11orf67 by sh2 significantly reduced the tumour growth in mice bearing T47D sh2 cells
compared to the empty vector control (p< 0.0001) (Figure 3-6A). C11orf67 sh2 mediated
inhibition of tumour growth was evident in most of the animals at several time points
post-injection (Figure 3-6B).
Examination of the tumour sections by IHC staining using an anti-C11orf67 antibody
demonstrated a higher density of tightly packed tumour cells in the empty vector control
cells. In contrast, C11orf67 sh2 tumours showed small islands of tumour cells (Figure 3-
6C).
In summary, our in vivo analysis indicated that the tumour suppressive functions of the
C11orf67 shRNA weakened the aggressiveness of T47D cells and decreased tumour cell
proliferation in an animal model of breast cancer, suggesting an important role of this
oncogene in promoting breast cancer in vivo.

Functional Consequences of C11orf67 Gene Alteration
89
Figure 3-6 C11orf67 knockdown inhibits in vivo tumour growth
(A) Representative images of tumour sections collected at day 10 post-injection
of cells. (B) Tumour volume measurements at day 3 and day 10 post-injection
(N= 8 animals per group) (*** p<0.0001, ** p<0.001). (C) C11orf67 IHC
staining of empty vector and C11orf67 sh2 in mice tumour sections at day 10
post-injection of cells.

Functional Consequences of C11orf67 Gene Alteration
90
3.3.1.6 C11orf67 knockdown is not involved in cellular
apoptosis
Since C11orf67 knockdown caused growth suppression of ER+ T47D breast cancer cells,
we further explored the underlying molecular mechanisms related to the proliferation
inhibition. Suppression of expression of many oncogenic drivers has been attributed to
programmed cell death or apoptosis (Yu et al., 2007, Al Dhaheri et al., 2013). Thus, we
analyzed whether the C11orf67 knockdown induced apoptosis of T47D cells.
To quantitate cellular apoptosis in response to C11orf67 knockdown, we first scored the
levels of the active caspase-3 by IF using an anti-cleaved caspase-3 antibody. Caspase-3
is a protease from the caspase family (cysteinyl aspartate-specific proteases) that is
implicated with the initiation of cellular death and therefore is a well suited read-out in
an apoptosis assay (Nicholson et al., 1995). C11orf67 shRNA cells showed a very small,
yet significant percentage of caspase-3 positive cells relative to that of the empty vector
control. The highest percentage of positive apoptotic cells were found in the C11orf67
knockdown sh1 (8%) compared to the empty vector control cells (p=0.0006), while sh2
and sh5 showed only 3% and 4 % of caspase-3 positive cells respectively (p<0.05) (Figure
3-7A). The low percentage of apoptotic cells in case of C11orf67 knockdown, may
indicate that apoptosis is not the primary mechanism of the decreased cellular
proliferation induced by C11orf67 knockdown.
We also investigated later stages of apoptosis in the C11orf67 knockdown by analyzing
DNA fragmentation by TUNEL assay. The TUNEL reaction preferentially labels DNA
strand breaks generated during apoptosis, by TdT (Labat-Moleur et al., 1998, Negoescu
et al., 1998). Fluorescein labels incorporated in nucleotide polymers are detected and
quantified by flow cytometry.
Flow cytometric analysis of apoptosis by TUNEL assay confirmed a minor yet significant
increase in the percentage of TUNEL positive cells in C11orf67 sh1 knockdown
compared to the empty vector control (p<0.05). Insignificant changes in the percentage

Functional Consequences of C11orf67 Gene Alteration
91
of TUNEL positive cells were observed with C11orf67 sh1 or sh5 when compared to the
empty vector control, p>0.05 (Figure 3-7B). Collectively, these results suggest that the
C11orf67 knockdown does not induce cancer cell death predominantly through induction
of cellular apoptosis.
Figure 3-7 C11orf67 knockdown is not associated with cellular apoptosis
(A) Representative IF images of anti-cleaved caspase-3 antibody positive cells
(green) in the indicated cell lines, nuclei were labeled with Hoechst (blue),
(quantification of positive cells is indicated on the right panel). (B) Flow
cytometry histograms (Cell-Quest software) of TUNEL positive cells
compared to the empty vector control (quantification is indicated on the right
panel) (** p<0.001, * p<0.05).

Functional Consequences of C11orf67 Gene Alteration
92
3.3.1.7 C11orf67 knockdown induces cellular senescence
To explore other possible mechanisms underlying C11orf67 knockdown-induced
decrease in cell proliferation, we also investigated cellular senescence. For this purpose,
we stained the cells with β-galactosidase (-gal), a specific marker used to identify
senescent cells which are characterised by increased lysosomal β-gal (Dimri et al.,
1995).
We found that transduction of T47D cells with C11orf67 shRNAs significantly increased
the number of cells showing β-gal activity as compared with the T47D empty vector cells
(Figure 3-8A). In addition we found that in C11orf67 knockdown cells the percentage of
β-gal positive staining cells peaked at 24 hours with a value of approximately 34% for
sh1 (p<0.0001), 50% for sh2 (p<0.0001), and 18% for sh5 (p=0.003) compared to less
than 5% in the empty vector (Figure 3-8A). In conclusion, these results support the notion
that C11orf67 depletion induces arrest in cell growth via induction of cellular senescence.
3.3.1.8 C11orf67 knockdown deactivates AKT/mTOR and
MAPK pathways
In the search for the molecular pathways altered by C11orf67, we first analysed the
Akt/mTOR and MAPK pathways since they have been shown to control proliferation and
cellular senescence (Young et al., 2009, Kennedy et al., 2011, Xu et al., 2014).
Since all phosphorylated residues are known to regulate cell proliferation and senescence
(Park et al., 2013), we analyzed the levels of phosphorylated forms of different regulatory
proteins participating in the Akt/mTOR and MAPK pathways in C11orf67 shRNA cells
and control cells by western blot using specific phospho-antibodies. We found that
C11orf67 knockdown by sh2 reduced the activity of p-Akt at both serine 473 and
threonine 308, and also reduced the p-MAPK (Thr202/Tyr204). On the other hand,
C11orf67 sh1 decreased the p-Akt levels at threonine 308, but not at serine 473 or the p-
MAPK, a result that could be explained by the incomplete knockdown of the C11orf67

Functional Consequences of C11orf67 Gene Alteration
93
protein accomplished by sh1. We also tested the effect of C11orf67 depletion on the
phosphorylated mTOR, which is a downstream target of Akt. Our results show that
C11orf67 knockdown by sh1, sh2 and sh5 resulted in decreased phosphorylation of
mTOR protein at serine 2448 (Figure 3-9). Altogether, these data suggest that C11orf67
knockdown might decrease the activity of the Akt/mTOR and MAPK pathways.
Figure 3-8 C11orf67 knockdown induces cellular senescence
(A) Representative images of β-gal staining of T47D cells treated with empty
vector control or C11orf67 shRNAs. (B) Quantification of the percentage of
positive cells (blue) in C11orf67 shRNA cells compared to the empty vector
control (*** p<0.0001, ** p<0.001).

Functional Consequences of C11orf67 Gene Alteration
94
Figure 3-9 C11orf67 knockdown deactivates Akt/mTOR and MAPK
pathways
Western blot analysis of p-Akt, p-mTOR and p-MAPK proteins in the
C11orf67 shRNA T47D cells and empty vector control, α-tubulin was used as
a loading control. MW markers are indicated on the right.

Functional Consequences of C11orf67 Gene Alteration
95
3.3.1.9 Identification of genes and biological pathways
downstream of C11orf67
To identify genes and biological pathways downstream from C11orf67, we used global
transcriptome analyses. We determined the genes differentially regulated by the C11orf67
knockdown (sh1 and sh2) relative to the empty vector control cells by Illumina gene
expression arrays. Statistical analyses of three independent biological replicates were
conducted by t-test analyses (p<0.05). C11orf67 knockdown achieved by sh1 and sh2
constructs co-regulated 40 genes that are differentially regulated by the two shRNA
constructs relative to control cells (Figure 3-10A).
In order to investigate the biological relevance of these genes, we performed a STRING
network analyses (Jensen et al., 2009). Interestingly, one of the network nodes was highly
enriched in genes with known function in cell cycle regulation such as Cyclin-dependent
kinases, CDK4 and CDK6 which play a key role in regulating cell cycle progression and
regulation of cellular senescence (Anders et al., 2011), which is in agreement with our
functional studies (Figure 3-10B).
These network analyses also revealed a metabolic node, comprising several biosynthetic
enzymes, notably the methylenetetrahydrofolate dehydrogenase 1-like (MTHFD1L) that
protein, may link the mitochondria and the cytoplasm in the mammalian model of one-
carbon folate metabolism (Vazquez et al., 2013) (Figure 3-10C).
For subsequent validation, we performed qRT-PCR and western blot analyses for 16
important genes that were differentially regulated by C11orf67 sh1 and sh2 knockdown
in T47D cells (p<0.001) (Figure 3-10D).
In conclusion, these gene expression assays indicated that C11orf67 regulates genes
involved in cell cycle regulation and metabolism. The link between C11orf67 and
metabolic control (one-carbon folate) is particularly exciting and could explain the
induction of cellular senescence by depletion of fundamental metabolic pathways
required for cancer proliferation.

96
Fig
ure
3-1
0 E
xp
ress
ion
pro
fili
ng o
f C
11orf
67 k
nock
dow
n c
ells
(A)
Hie
rarc
hic
al c
lust
erin
g o
utl
inin
g t
he
40
-gen
e si
gnat
ure
of
targ
ets
dif
fere
nti
ally
reg
ula
ted i
n T
47
D c
ells
by b
oth
C11orf
67
sh1 a
nd s
h2
rela
tive
to e
mpty
vec
tor
contr
ol
cell
s. (
B a
nd C
) In
tera
ctio
n n
etw
ork
(S
TR
ING
) in
dic
atin
g t
arget
s dif
fere
nti
ally
reg
ula
ted b
y s
h1 (
bla
ck f
onts
)
and s
h1 a
nd
sh2 (
red f
onts
) re
lati
ve
to c
ontr
ol.
(D
) qR
T-P
CR
and w
este
rn b
lot
val
idat
ion o
f 16 t
arget
s dif
fere
nti
ally
reg
ula
ted i
n s
h1 a
nd s
h2
rela
tive
to e
mpty
vec
tor
contr
ol.

Functional Consequences of C11orf67 Gene Alteration
97
3.3.2 Functional analyses of C11orf67 overexpression in breast
cell lines
In parallel to the functional ablation studies, we analyzed the consequences of C11orf67
gain of function in breast cancer cells. We used MCF7 cells as a cellular model system of
tumour mammary cells that are ER+, PR+ and features low basal levels of C11orf67
mRNA transcript compared to that of T47D cells. We used a lentiviral construct to stably
overexpress C11orf67 in MCF7 cells. The pLV105 lentiviral vector containing the human
cDNA of C11orf67 Isoform_2 (Genecopoeia, Rockville, MD, USA) was utilized in this
assay. This system allows constitutive and stable expression of C11orf67 protein. Cells
were lentivirally transduced with either pLV105 empty vector or with a pLV105-
C11orf67 construct to yield stable overexpression of the protein. Stably transduced MCF7
cells were analyzed for C11orf67 expression at transcript and protein levels. qRT-PCR
and western blot analyses showed that lentiviral-mediated overexpression of C11orf67
yielded approximately 15-fold increase in the mRNA and protein levels, respectively, as
compared to the empty vector control or the un-transduced MCF7 (Mock) cells, p<0.0001
(Figure 3-11A and B).
To determine whether C11orf67 overexpression is sufficient per se to induce
tumourigenicity in MCF7 cells, we analyzed cell proliferation rate of MCF7/C11orf67
cells compared to the empty vector control and mock cells by MTT cell proliferation
assay. Our results revealed that ectopic C11orf67 overexpression was not associated with
significant increase in MCF7 cell proliferation (Figure 3-11C). Indeed, there were no
evident differences in terms of growth morphology, size, and number of outgrowths
detected between C11orf67 cDNA-transduced cells relative to controls. This could be
explained by the fact that other factors in addition to C11orf67 might be required to induce
tumourigenicity. It could also be possible that other isoforms, in addition to isoform_2,
might be responsible for the oncogenic potential of C11orf67. Moreover, results in
chapter 4 demonstrate that C11orf67 isoform_2 overexpression induces tamoxifen
resistance, and thus the overexpression of the cDNA appears not to change proliferation
but rather a resistance to certain forms of therapy.

Functional Consequences of C11orf67 Gene Alteration
98
Figure 3-11 C11orf67 overexpression does not increase cellular proliferation
(A) qRT-PCR analyses of C11orf67 mRNA levels in the C11orf67/MCF7,
empty vector control, or mock cells. (B) Western blot analyses of the indicated
cells blotted with anti-C11orf67 antibody, α-tubulin was used as a loading
control. (MW markers are shown on the right). (C) MTT cell proliferation
analysis of indicated cells, cell viability was monitored over 96 hours. Results
are plotted as fold increase with respect to day 0.

Functional Consequences of C11orf67 Gene Alteration
99
3.3.2.1 C11orf67 activates genes involved in metabolism
Our cDNA microarray data analyses revealed that C11orf67 knockdown lead to the
downregulation of many cell cycle regulation related genes and genes involved in
metabolism and biosynthesis, in addition to genes associated with senescence. In this
section, we explored the effect of C11orf67 overexpression on these targets identified in
the knockdown experiments.
We first used western blot analyses to score the expression and/or activation state of 8
proteins that were affected by the C11orf67 knockdown: p-Akt ser473, p-Akt threo308,
p-mTOR ser2448, p-MAPK p42/44, CDK4, CDK6, CCNE1, and CCND1 (Figure 3-
12A). Our western blot data showed that only CCND1 protein was upregulated in the
C11orf67 overexpressing cells relative to empty vector control cells. However it is
important to note that the cells tested here are MCF7 cells, which are different from T47D
cells used in the knockdown analyses.
We next performed qRT-PCR analyses to detect the expression level of some of the
targets of the C11orf67 shRNAs identified by gene expression microarrays. Here again
we found that C11orf67 overexpression in MCF7 leads to upregulation of genes involved
in metabolic control, particularly one-carbon folate metabolism and phospholipase
activity (MTHFD1L, ALDH1L2, PPARGC1A, PLCE1), p<0.001 (Figure 3-12B).
Overall these results suggest that C11orf67 isoform_2 altered the expression of the cell
cycle regulator CCND1 in MCF7 cells and also induced enzymes involved in metabolic
control, reinforcing a role of the gene in controlling metabolic pathways.

Functional Consequences of C11orf67 Gene Alteration
100
Figure 3-12 Changes in gene/protein expression in response to C11orf67
overexpression
(A) Western blot analyses of several signalling pathways affecting proliferation
and cell cycle control in MCF7 cells overexpressing C11orf67 cDNA relative
to control cells, this has been validated twice in two independent experiments.
(B) qRT-PCR analyses of genes involved in metabolism in case of C11orf67
overexpression and empty vector MCF7 (* p<0.01).

Functional Consequences of C11orf67 Gene Alteration
101
3.3.2.2 C11orf67 overexpression increases sensitivity to anti-
metabolites
Significant upregulation of genes involved in one-carbon folate metabolism raised the
hypothesis that C11orf67 overexpression could increase ER+ breast cancer cell sensitivity
to folate antagonists, such as methotrexate (MTX) and 5-fluorouracil (5-Fu).
To evaluate the role of C11orf67 conferring sensitivity to these drugs, we took advantage
of MCF7 cells stably expressing C11orf67 cDNA isoform_2 and control empty vector
cells. These lines were seeded in 96-well plates and challenged to increasing
concentrations of drugs. Cell viability assays were carried out to determine the percentage
of surviving cells by MTT assays.
We found that cells overexpressing the C11orf67 cDNA had significantly enhanced the
sensitivity of MCF7 cells to the anti-tumour effect of 5-Fu and MTX at a time and dosage-
dependent manner compared to the empty vector control cells (p<0.001), an effect that
did not occur with non-antimetabolite drugs such as taxol (p>0.05) (Figure 3-13).

Functional Consequences of C11orf67 Gene Alteration
102
Figure 3-13 C11orf67 overexpression increases the sensitivity to one-
carbon folate antagonists
MCF7 cells transfected with C11orf67 cDNA vector, or the empty vector
control were treated for 72 hours with different doses of (A) methotrexate, (B)
5-fluorouracil, and (C) taxol. Percentage of cell cytotoxicity was assessed by
MTT assay (** p<0.001).

Functional Consequences of C11orf67 Gene Alteration
103
3.4 Discussion
An important question addressed in this chapter is how C11orf67 deregulation is
mechanistically implicated in breast cancer, and which cellular processes are deregulated
on its depletion. So far, no studies have addressed the role of C11orf67 in breast cancer
tumourigenesis and progression. In this chapter, we mounted evidence that C11orf67 is
necessary for breast cancer tumourigenesis and progression. We are the first to report that
efficient C11orf67 knockdown results in decreased proliferation, tumour formation and
migration of ER+ breast cancer cells in vitro, and also tumour formation in a xenograft
model in vivo. In addition, we were able to demonstrate that apoptosis was not the main
mechanism by which downregulation of C11orf67 expression inhibited cellular growth
and proliferation. In contrast, deregulation of C11orf67 expression caused high levels of
induction of cellular senescence.
Several reports demonstrated that cellular senescence functions as a potent tumour
suppressor mechanism that is essential for preventing unregulated growth and malignant
transformation (Campisi and d’Adda di Fagagna, 2007). We thus tested the potential role
of C11orf67 in regulating cellular lifespan. A senescent phenotype was readily observed
in ER+ breast cancer cells, T47D following the inhibition of C11orf67 expression. This
result suggests that cellular senescence induced by C11orf67 knockdown might be the
mechanism underlying its potential promoting malignant transformation and sustained
cell growth effects. Moreover, cellular senescence induced by loss of function of
oncogenes has been considered as a therapeutic approach and has been verified by
previous studies (Zou et al., 2002, Lleonart et al., 2009, Li et al., 2012).
We were also able to demonstrate a potential link between C11orf67 depletion and
senescence induction through decreased activity of phosphorylated AKT/mTOR proteins.
However, AKT/mTOR pathway is complicated and contradicting reports in the literature
argue the ability of AKT/mTOR pathway to induce senescence. Some reports have
pointed out that activation of the AKT pathway induces senescence in mouse embryonic
fibroblasts and prostate, esophageal, and colon cancer cells (Oyama et al., 2007,

Functional Consequences of C11orf67 Gene Alteration
104
Majumder et al., 2008, Nogueira et al., 2008, Park et al., 2013). Other reports have
concluded that AKT/mTOR activity is downregulated in senescence , and can antagonize
senescence (Courtois-Cox et al., 2006, Young et al., 2009, Kennedy et al., 2011, Xu et
al., 2014). In this study, we show that C11orf67 knockdown impairs the Akt activity and
induce a senescencent phenotype in ER+ breast cancer cells. Thus, our results point out
towards the second hypothesis.
The notion that AKT/mTOR is associated with resistance to endocrine therapy in breast
cancer (Paplomata and O'Regan, 2014) is of special interest as it supports the idea of
stratifying breast cancer patients and predicting those with poor prognosis and endocrine
treatment resistance. Mounting evidence indicates that the combination of mTOR
inhibitors with the hormonal therapy can overcome the resistance and improve the
outcome of HR+ breast cancer patients (Baselga et al., 2012, Bachelot et al., 2014).
Furthermore, strategies to downregulate C11orf67 in endocrine resistant breast cancer in
combination with mTOR inhibitor might improve the response to endocrine therapy and
consequently the outcome of patients, a hypothesis that needs to be further validated.
To confirm the involvement of PI3K/Akt/mTOR in C11orf67 induced senescence, more
checkpoints in the pathway require further analyses. For instance, the PI3K the upstream
effector of AKT, which phosphorylate PIP2, to PIP3. On the other hand, we showed that
phosphorylated mTOR protein is downregulated in response to C11orf67 knockdown;
however, the intermediate proteins (TSC1/2 and Rheb-GTP), the other subunits of the
mTORC1 complex (Raptor, PRAS40, and mLST8), and the mTORC2 complex have not
been checked to be affected or not in response to C11orf67 depletion and required to be
dissected for the full understanding of the AKT/mTOR pathway involvement in the
cellular senescence induced by C11orf67 knockdown.
We also investigated whether alteration of C11orf67 expression in breast cancer cell lines
induces changes in the transcriptomic landscape of these cells. To our knowledge, this is
the first report conducted a genome-wide expression microarray analyses to identify
differentially regulated genes linked to C11orf67 knockdown in T47D cells, we identified
40 genes to be commonly co-regulated by both shRN1 and shRNA2 constructs specific

Functional Consequences of C11orf67 Gene Alteration
105
to C11orf67 transcript. The fact that these genes are regulated in these shRNAs in 6
independent transfections give high confidence that these targets are bona fide
downstream genes of C11orf67, and not the result of off-targets of the shRNAs. To date,
we have validated the regulation of 16 out of 40 genes by qRT-PCR and western blot
analyses.
Moreover, to gain insights into the biological relevance of the global transcriptional
changes induced by C11orf67 downregulation, we performed network pathway analysis.
This analyses resulted in the novel identification of a cell cycle control network and
metabolism network regulated by C11orf67 knockdown, suggesting an important role of
C11orf67 in the regulation of these processes.
It was not surprising that we found a cell cycle gene network regulated by C11orf67
knockdown, taking in consideration the results obtained in our functional studies, which
revealed that C11orf67 depletion in breast cancer cell lines decreased cellular
proliferation, and also in light of the global transcriptome changes conducted by the
cancer genome project in the context of the InClus2 subtype (Curtis et al., 2012).
Moreover, cell cycle regulation might provide another explanation for the gain of
senescence induced by C11orf67 knockdown. Cellular senescence caused by C11orf67
knockdown could be explained by the downregulation of many cell cycle regulation-
related genes (CCNE1, CCND1, CDK4, CDK6, PPP4R4, CEBPG, and PPARGC1). It is
important to note that suppression of most of these genes was usually found to be
associated with cell cycle arrest, cellular senescence or both as discussed below.
CDK4 and CDK6 are positive regulators of cell cycle entry, and inhibit senescence in
melanoma cells (Anders et al., 2011). CEBPG is a growth-promoting transcription factor
and its depletion induced senescence in lung tumour cells and primary mouse fibroblasts
(Huggins et al., 2013). Ppp4R4 (Phosphoprotein phosphatase 4) has been implicated in
DNA damage checkpoint signalling, NF-κB activation, and mTOR pathway regulation
(Zhou et al., 2002, Cohen et al., 2005, Nakada et al., 2008). PPARGC1 (peroxisome
proliferator-activated receptor gamma, coactivator 1) plays a critical role in the

Functional Consequences of C11orf67 Gene Alteration
106
transcriptional control of mitochondrial biogenesis and respiratory function (Scarpulla,
2011).
In addition to the cell cycle gene network found in the pathway analyses, we also found
a biosynthesis and a metabolism gene network that is deregulated in response to C11orf67
knockdown. Indeed, C11orf67 depletion led to remarkable lower expression of the
mitochondrial glycine biosynthetic pathway and also downregulation of mitochondrial
folate enzymes that include serine hydroxymethyltransferase (SHMT2),
methylenetetrahydrofolate dehydrogenase (MTHFD (Scarpulla, 2011, Vazquez et al.,
2013). The downregulation of these mitochondrial enzymes correlated with decreased
proliferation caused by C11orf67 ablation. The expression levels of the above mentioned
mitochondrial enzymes contribute to the sensitivity or the resistance of a tumour cell to
antifolate cancer therapy (MTX and 5-FU) (Vazquez et al., 2013), where there is a strong
correlation between the sensitivity to MTX treatment and increased expression of
mitochondrial enzyme MTHFD1L (Sorich et al., 2008).
Consequently, we raised the hypothesis that overexpression of C11orf67 in cancer cells
will upregulate the expression of mitochondrial one-carbon pathway, and glycine
consumption that render those cells to be more potently inhibited by MTX, whereas cells
with lower levels of C11orf67 would not be as sensitive. In this chapter, we are the first
to provide evidence that the anti-metabolites MTX, and 5-FU might show selectivity to
tumours showing higher levels of C11orf67 with the subsequent upregulation of the
mitochondrial one-carbon pathway components. These data support the hypothesis that
MTX, 5-FU and potentially other inhibitors of thymidylate and purine biosynthesis could
be used to target ER+ breast cancers with high expression levels of C11orf67.
In summary, our data demonstrates that C11orf67 has a novel oncogenic role in ER+
breast cancer and is required for breast cancer cell proliferation and survival. Further, we
identified the induction of cellular senescence as a potential mechanism underlying
C11orf67 malignant transformation effect. In addition, our results suggest that one-carbon
metabolism antagonists might increase the response of cancer cells overexpressing
C11orf67 which increase the dependence of cancer cells on this metabolic pathway.

Chapter 4:
Regulation and Binding
Partners of C11orf67

Regulation and Binding Partners of C11orf67
108
4.1 Introduction
To define the significance of C11orf67 in the initiation and progression of ER+ breast
cancer, it is essential to identify both its transcriptional regulation mechanisms and its
molecular interactions with putative proteins and cofactors. This information will also be
important at the cellular level to potentially understand the mechanisms of response to
therapy mediated by C11orf67, as well as the identification of more effective treatments
for tumours expressing this novel biomarker.
Approximately 75% of breast cancers are ER+, and different anti-hormonal based
treatments exert their action through various mechanisms to antagonize tumour
proliferation stimulated by estrogen (Zhang et al., 2013). One of the accepted models of
ER function is via binding of estrogen to ER to form the estrogen/ER complex, which
binds specifically to ERE of the DNA. Binding of the ERE initiates gene transcription
either directly, or indirectly through the interaction with other transcription factors such
as NF-κB (Thomas and Gustafsson, 2011, Baumgarten and Frasor, 2012).
Current anti-estrogen approaches include the use of selective estrogen receptor
modulators (SERM), such as tamoxifen, which antagonize ER activity by competing with
estrogen for ER binding. Although tamoxifen is very efficient in the treatment of ER+
breast cancer, resistance and tumour relapse occurs in 30% to 40% of patients who
showed an initial response to tamoxifen (Chia et al., 2005). Indeed, it is challenging to
find definitive methodology identifying ER+ patients that are more likely to benefit from
tamoxifen from those who are not (Jung et al., 2013).
The activity of the transcription factor NF-κB is thought to deliver means for tumour cells
to escape the antagonistic effect of tamoxifen, by either downregulating ER protein
expression, or by enhancing ER activity in a ligand-independent manner (Biswas et al.,
2000, Van Laere et al., 2007). Different inflammatory cytokines, such as tumour necrosis
factor α (TNFα), activate NF-κ B proteins to form dimers that bind to NF-κB response
elements of the DNA (Hoffmann et al., 2003, Chen and Greene, 2004, Sun, 2011).

Regulation and Binding Partners of C11orf67
109
Activation of NF-κ B occurs through either the canonical pathway or the alternative
pathway (explained in Figure 4-1) (Sen and Smale, 2010).
Figure 4-1 Canonical and alternative pathways of NF-κB activation
In the canonical pathway, cellular activation leads to phosphorylation of the
inhibitor of NF-κB α-protein (IκBα) by a macromolecular complex containing
an inhibitor of NF-κB kinase 1 and 2 (IKKα and IKKβ) and others.
Phosphorylation of IκBα stimulates the rapid ubiquitylation and degradation of
this cytoplasmic inhibitor by the 26S proteasome complex, which leads to
liberation and rapid translocation of NF-κB dimers (p65 and p50) into the
nucleus. In the non-canonical pathway, RelB/NF-κB2 dimer undergoes
inducible proteolytic processing of the NF-κB2 gene product, p100. (Ko et al.,
2010, Sun, 2011). Adapted from Dolcet et al. (2005).

Regulation and Binding Partners of C11orf67
110
In this chapter, we used bioinformatic analyses and different experimental approaches to
understand novel regulatory mechanisms that govern the expression of C11orf67 and
neighboring genes in the oncogenic cluster at 11q13.5-q14. We are the first to report that
C11orf67 and other genes of the cluster are co-regulated by a group of transcription
factors inducing proliferation including ER, Myc and NF-κB. The results of our analyses
suggest that transcriptional upregulation of C11orf67 by tamoxifen underlies the
development of resistance to endocrine therapy in these patients. Therefore, the C11orf67
expression could represent a novel biomarker for predicting tamoxifen resistance, which
occurs in 30% of patients.
Moreover, we also utilized yeast two-hybrid (Y2H) to identify the direct interaction
partners of C11orf67 protein, as an important step toward systematically defining the
C11orf67 protein function. Two independent Y2H screens revealed RABGAP1L (Ras-
related in brain GTPase-activating protein 1-like) (Mitra et al., 2011) as a very high
confidence prey physically interacting with C11orf67. RABGAP1L is a critical cell
signalling molecule known to be involved in a variety of pivotal cellular processes
including intracellular vesicular transport and membrane trafficking (Spiegel et al.,
2014). The outcomes of this research have revealed C11orf67 and other interacting
partners as a new signalling axis defining tamoxifen resistance in ER+ tumours.

Regulation and Binding Partners of C11orf67
111
4.2 Materials and Methods
4.2.1 Reagents and Antibodies
17β-Estradiol, Tamoxifen, Progesterone, Mifepristone, Prolactin and Oxytocin were
purchased from Sigma-Aldrich, St Louis, MO, USA. NF-κB inhibitor was purchased
from Calbiochem, Kilsyth, Victoria, Australia. TNFα was a kind gift from Professor
Juliana Hamzah (University of Western Australia).
For western blotting, primary antibodies specific to p-IKKα/β, IκBα , p-p65, NFκB2
p100/p52, and c-Myc were purchased from Cell Signaling, Gold Coast, QLD, Australia,
and were used at 1:1000. The anti-p65 antibody was purchased from Santa Cruz
Biotechnology, Texas, USA (1:500). Anti-RABGAP1L was purchased from Proteintech,
Chicago, USA, and was used for immunohistochemistry (1:400), and western bloltting
(1:100).
4.2.2 Cell Culture
Human breast cancer T47D, MCF7, and SKBR3 cell lines were routinely grown in the
appropriate media (Section 2.2.1). To evaluate the effect of hormonal signals, cells were
cultured in phenol red-free media supplemented with 5% dextran coated, charcoal-
stripped serum (CSS) (Invitrogen, Waverley, Australia) for 3 to 5 days, before being
incubated with either the indicated hormones or the antihormonal agent (Perillo et al.,
2000). 4x105 cells were seeded in a six-well plate and grown until they reached 60-70%
confluency. Cells were then treated with the indicated concentrations of hormones, drug
or vehicle control (mock) for 48 to 72 hours. Cells were trypsinized, and total RNA was
collected with the RNeasy Kit and converted to cDNA by reverse transcription (Sections
2.2.2, and 2.2.3). qRT-PCR analysis of human C11orf67 was performed with a PCR
Taqman Gene Expression Assay, using human GAPDH as a housekeeping gene (Section
2.2.4).

Regulation and Binding Partners of C11orf67
112
4.2.3 siRNA Transfection
The sequences of siRNA oligos specific for C11orf67 (Sigma-Aldrich, St Louis, MO,
USA) were as follows: 5’-ACUUGGGAUUGGAGAGAAA-3’ and 5’-
UUUCUCUCCAAUCCCAAGU-3’. The sequences of negative universal control siRNA
(Sigma-Aldrich, St Louis, MO, USA) were as follows: 5’-
UUCUCCGAACGUGUCACGUTT-3’ and 5’-ACGUGACACGUUCGGAGAATT-3’.
T47D cells were cultured in six-well plates until they reached 50-60% confluency. Cells
were transfected with C11orf67 siRNA or control siRNA according to the manufacturer's
protocol. Briefly, 1 μl (2 μg) of the siRNAs oligos was added to 99 μl of Opti-MEM
Reduced-Serum Medium (Invitrogen, Waverley, Australia) and mixed gently. At the
same time, 4 μl of Lipofectamine-2000 (Invitrogen, Waverley, Australia) was added to
96 μl of Opti-MEM and left at room temperature for 5 minutes. The media including the
siRNA was added gently to the media including the Lipofectamine and mixed by
pipetting. After 20 minutes, 200 μl of the mixture was added to each well and made up to
a final volume of 500 μl with Opti-MEM, and the plates were incubated at 37 °C and 5%
CO2 for 24 hours. C11orf67 mRNA levels were detected by qRT-PCR, and MTT
cytotoxicity assays were performed on transiently transfected cells up to 72 hours after
transfection (Section 3.2.4).
4.2.4 Rat AAMDC qRT-PCR
Pregnant rats (24 days of gestation) and control female rats were humanely sacrificed
under Ethics Approval number RA/3/100/1318 of the University of Western Australia.
Embryos were collected for a separate study while the mammary gland and
retroperitoneal fat pads were collected under aseptic conditions and snap frozen. RNA
was extracted from the collected fat pads using Qiagen RNA extraction kit (Hilden,
Germany) and processed as stated previously (Section 2.2.2). Rat AAMDC mRNA
expression levels were detected using TaqMan primers and probes (Applied Biosystems,
Scoresby, Australia), using rat GAPDH as a housekeeping gene.

Regulation and Binding Partners of C11orf67
113
4.2.5 Chromatin immunoprecipitation assay
Chromatin immunoprecipitation assay (ChIP) was performed as described by Yang et al.
(2014). Briefly, T47D cells were either maintained in their basal media or stimulated with
10 ng/ml TNF-α for 4 hours. Cells were fixed, sonicated, before being incubated with the
anti-p65 (Santa Cruz Biotechnology, Texas, USA) antibody for 16 hours at 4°C (1
mg/reaction). Chromatin was precipitated using Dynabeads Protein A (Invitrogen,
Waverley, Australia) according to the manifucturer’s protocol. DNA was extracted with
phenol/chloroform, precipitated with ethanol and analyzed by PCR using primers
flanking the predicted p65 binding sites. The immunoprecipitated sample DNA was
compared with the input DNA to determine the percent of the immunoprecipitated
product. The primers used to amplify a distinct region of the C11orf67 promoter with
putative NF-κB binding sites (determined using Transcription Factor software) were: 5’-
CCTCTGGTCCACTTGGGATA-3’ and 5’-GTGATCTCAGTGGCAAGCAA-3’, with
the following conditions: Cycle 1, 3 minutes at 95°C; Cycle 2, 30 sec at 95°C; Cycle 3,
30 sec at 60°C; Cycle 4, 1 minutes at 72°C; then repeat cycles 2 to 4 35 times followed
by a final step of 10 minutes at 72°C. The amplified products were analysed by
electrophoresis on a 2% agarose gel and visualized by ethidium bromide staining under
UV light.
4.2.6 Luciferase assay
Stable T47D cells that had been transfected with C11orf67 shRNAs or empty vector (see
section 3.2.3) were then transfected with the NF-κB–Luc reporter plasmid (a kind gift
from Dr. Xavier Dolcet, University of Lleida, Spain). Transfection was performed using
Lipofectamine-2000 transfection reagent according to manufacturer’s instructions. Cells
were lysed using Luciferase Reporter Assay System (Promega, Madison, WI, USA)
according to the manufacturer’s instructions. Luciferase activity was measured using the
EnVision 2012 Multilabel Reader, PerkinElmer (Waltham, MA, USA)

Regulation and Binding Partners of C11orf67
114
4.2.7 Yeast Two-Hybrid Analysis
Yeast two-hybrid screening was performed by Hybrigenics Services, S.A.S., Paris,
France (http://www.hybrigenics-services.com).
The coding sequence for Homo sapiens C11orf67 (GenBank accession number gi:
34328078) was PCR-amplified and cloned into the plasmid pB27 as a C-terminal fusion
to LexA (N-LexA-C11orf67-C), and into pB66 as a C-terminal fusion to Gal4 DNA-
binding domain (N-Gal4-C11orf67-C). The constructs were verified by sequencing and
used as bait to screen a random-primed cDNA library from human breast tumour cells
(T47D, MDA468, MCF7, and BT20) cloned into pP6. pB27, pB66 and pP6 derive from
the original pBTM116 (Vojtek and Hollenberg, 1995), pAS2ΔΔ (Fromont-Racine et al.,
1997), and pGADGH (Bartel et al., 1993) plasmids, respectively.
For the LexA bait construct, we screened 85 million clones (8-fold the complexity of the
library) using a mating method with YHGX13 (Y187 ade2-101:loxP-kanMX-loxP, matα)
and L40ΔGal4 (mata) yeast strains as previously described (Fromont-Racine et al., 1997).
A total of 16 His+ colonies were selected on a medium lacking tryptophan, leucine, and
histidine.
For the Gal4 construct, 68 million clones (6-fold the complexity of the library) were
screened using the same mating approach with HGX13 (Y187 ade2-101::loxP-kanMX-
loxP, matα) and CG1945 (mata) yeast strains. A total of 53 His+ colonies were selected
on a medium deficient in tryptophan, leucine, and histidine. PCR was used to amplify the
prey fragments of the positive colonies and then were sequenced at their 5’ and 3’ ends.
The NCBI GenBank database was used to identify the resulting sequence for the
corresponding interacting proteins. A Predicted Biological Score for each interaction was
attributed (Formstecher et al., 2005).

Regulation and Binding Partners of C11orf67
115
4.2.8 Statistical analysis
Statistical data are presented as the mean ± SD of three individual experiments performed
in triplicate. Statistical analysis was carried out using the Student’s t-test or one-way
ANOVA, and the level of significance was established at a P value of <0.05.

Regulation and Binding Partners of C11orf67
116
4.3 Results
4.3.1 11q13.5-q14 locus is enriched in transcription factor
binding sites
13 genes mapping in 11q13.5-q14 are found to be amplified in the IntClust 2 subtype of
breast cancer (Curtis et al., 2012). We have described the validation of the C11orf67
amplification by FISH and described the oncogenic potential of this protein in ER+ breast
cancer in previous chapters. However, the transcriptional regulation of C11orf67 and
other oncogenic drivers at 11q13.15-q14 has not been investigated before.
A key factor in determining the transcriptional expression of the 11q13.5-q14 genes is the
binding of sequence-specific DNA binding proteins (i.e. transcription factors, TFs) to
regulatory regions of these genes (i.e. promoters and enhancers) within a few hundred
nucleotides from the transcription start site (TSS). In addition, TFs can also bind
enhancers which act via chromatin loops many Kbps away from their binding site. In fact,
examination of ENCODE TF data in MCF7 cells demonstrates that chromatin
interactions can link many oncogenes in the 11q13.5-q14 amplicon even located more
than 500 Kbps away. Whilst these TFs show sequence-specific DNA binding properties,
they also bind more than one sequence, which makes it difficult to identify ultimately all
functional binding sites in a given cell line.
To determine the relevant TF binding sites, we first employed the transcription factor
binding site (TFBS) search program to scan sequences for putative binding sites in the
whole 11q13 amplicon, and particularly in the RSF1-C11orf67-INTS4 region. Outcomes
from TF analysis demonstrated a high-density of binding sites for ER, PR, p65 and Myc
in the promoter and other regulatory regions along 11q13.5-q14. A sketch map of the TF
binding sites and their relative position in the 11q13.5-q14 locus is illustrated in Figure
4-2A.

Regulation and Binding Partners of C11orf67
117
Interestingly, we found 4 out of 13 p65 binding sites in the C11orf67 promoter region are
a consensus with high-score motifs GGGAGTTCCC, GGGGGCCTCC, GGGACTTCT
and GGGAAATTCC. Furthermore, we observed that the RSF1 gene promoter sequence
(the gene located adjacently to C11orf67 start site) also contains several NF-κB binding
sites, which suggests possible co-regulation with the C11orf67 gene.
We next investigated the possibility of concomitant co-regulation of the C11orf67 gene
with the other genes in the amplicon. Interestingly, we found that C11orf67 is
significantly co-amplified with other distal genes in the locus, particularly the proximal
genes, AQP11, CLNS1A, RSF1 and INTS4 (Cancer Genome Atlas, 2012), with the
highest probability of co-amplification with the neighboring genes in the amplicon.
Collectively, these results suggest the possibility of chromatin interactions within the
11q13.5-q14 locus, and that the expression of the C11orf67 gene and other genes residing
in this amplicon might be regulated by ER, PR, p65, and Myc TFs.
4.3.2 Estrogen modulates C11orf67 gene expression.
In previous chapters, we have found that C11orf67 is overexpressed in ER+ breast
cancers. Consistently, here we have determined its gene promoter contains ER binding
sites, yet the exact transcriptional effect of estrogen on C11orf67 gene expression has not
previously been investigated. With this aim in mind, we chose ER+ T47D cells which are
ER+, PR+ and show high levels of C11orf67 mRNA expression as a model to understand
the transcriptional regulation of C11orf67 in hormone sensitive luminal breast cancers.
To study hormonal cues in gene expression in general two types of media are commonly
used: Phenol red-containing (red) medium supplemented with FCS, which is the standard
medium for optimum cellular growth, and Phenol red-free (white) medium supplemented
with CSS, which is used to analyse the expression of hormone-dependent genes, since
phenol red is known to activate ER-dependent gene regulation. For all the hormonal
assays, T47D cells were cultured in white media for 72 hours, to abolish the uncontrolled
effect of exogenous estrogens on gene expression.

Regulation and Binding Partners of C11orf67
118
Figure 4-2 Transcription factor binding sites of the 11q13.5-q14 amplicon
(A) Sketch map representing the ENCODE chromatin interaction data of the
11q13.5-q14.5 amplicon, indicating the TF-binding sites and their relative
position, with an enlarged image of the RSF1-C11orf67-INTS4 region with
putative NF-κB binding sites. (B) Co-amplification of C11orf67 with the other
immediate neigboring oncogenes at 11q13.5-q14.5 in breast cancer (analyses
performed by Dr. A. Woo at the Harry Perkins institute using Cancer Genome
Atlas input data).

Regulation and Binding Partners of C11orf67
119
We first investigated whether changing the culture media from red to white could affect
the expression levels of C11orf67 mRNA. To this end, T47D cells were grown in red
media, and switched to white media for 48 hours, after which C11orf67 mRNA levels
were analyzed. Importantly, we found that C11orf67 mRNA levels increased by at least
two-fold (p<0.0001) when the cells were cultured in white media, which suggests that
removing the estrogens from the growing media increases the C11orf67 mRNA levels
(Figure 4-3A).
To validate these findings, T47D cells were treated with increasing physiological
concentrations of 17β-Estradiol (E2), ranging from 1nM to 100 nM, for 48 hours before
conducting a qRT-PCR analysis to quantitate C11orf67 mRNA levels. Consistent with
our previous finding, we noticed a two-fold downregulation (p<0.0001) of C11orf67
mRNA levels in response to treatment of increasing doses of E2, compared to the mock
control cells (DMSO treated) (Figure 4-3B).
Tamoxifen (Tam) is an anti-estrogen drug that is most commonly used for the treatment
of ER+ breast cancer. To explore the effect of Tam on C11orf67 mRNA levels, T47D
cells were treated with increasing concentrations of Tam (0.5 μM, 1, and 5 μM) for 48
hours, before C11orf67 mRNA levels were measured and compared to that of the mock
control cells. Our findings did not demonstrate any significant changes in transcriptional
activity of C11orf67 upon treatment with any dose of Tam (Figure 4-3C).

Regulation and Binding Partners of C11orf67
120
Figure 4-3 C11orf67 is regulated by Estrogen
qRT-PCR analysis of C11orf67 mRNA levels in T47D cells in response to
different hormonal growing conditions: Phenol red-free media (A), Estradiol
treatment (B), and Tamoxifen treatment (C) (*** p<0.0001).

Regulation and Binding Partners of C11orf67
121
4.3.3 Estrogen downregulates C11orf67 expression in an ER-
dependent manner
We next investigated whether E2 exerts its action on C11orf67 expression in a way
dependent on ER expression. To this end, we treated the ER+ line T47D with gradually
increasing doses of the specific ER antagonist, Tam (0.5, 1, and 5 μM), to 1 nM of E2.
Interestingly, C11orf67 mRNA downregulation observed with E2 alone was gradually
abolished by adding Tam to the growing media, until it reached significantly higher levels
of expression of C11orf67 in response to 5μM Tam (as compared to the mock-treated
cells) (p<0.0001) (Figure 4-4A).
Moreover, we tested the same approach (E2 and Tam treatment) in an ER- cell line,
SKBR3. As expected from being hormone-independent cells, neither E2 nor Tam led to
a significant difference in the mRNA levels of C11orf67 expression in SKBR3 cells. It is
worth noting that 5μM Tam in the presence of 1nM E2 resulted in a slight yet significant
increase in C11orf67 mRNA levels compared to the mock-treated cells (p<0.05) (Figure
4-4B). Collectively, these data suggest that the effect of E2 on C11orf67 is mediated
through ER and is rescued by the anti-estrogen drug tamoxifen.
4.3.4 Tamoxifen upregulates expression of multiple oncogenes
in the 11q13.5-q14 cluster
Based on the previous finding of possible co-regulation and chromatin interactions among
multiple oncogenes residing at 11q13.5-q14, we next explored the effect of 5μM Tam in
combination with 1nM E2 on the expression levels of all 13 genes in the amplicon.
Strikingly, we detected significant upregulation of all 13 genes in the amplicon upon
treatment with Tam (p<0.001) as assessed by qRT-PCR. In addition, our results indicated
that 8 out of 13 genes in the amplicon were significantly transcriptionally downregulated
by E2 (p<0.01).

Regulation and Binding Partners of C11orf67
122
These results suggest that exposure to anti-estrogens might have an undesirable effect
by activating multiple oncogenes in Chromosome 11 and subsequently the IntClust 2
subtype of breast cancer.
Figure 4-4 Estrogen regulates C11orf67 expression in an ER dependent
manner
Effect of Estradiol (E2), Tamoxifen (TAM), or both on the mRNA levels of
C11orf67 in ER+ cell line; T47D (A) and ER- cell line; SKBR3 (B) (***
p<0.0001, * p<0.05)

123
Fig
ure
4-5
Eff
ect
of
estr
ad
iol
an
d t
am
oxif
en o
n t
he
exp
ress
ion
of
mu
ltip
le o
nco
gen
es a
t 11q
13.5
-q14
Eff
ect
of
1nM
est
radio
l al
one
or
in c
om
bin
atio
n w
ith 5
µM
tam
oxif
en o
n t
he
mR
NA
lev
els
of
the
13 g
enes
loca
ted i
n t
he
onco
gen
ic c
lust
er
11q13.5
-q14. V
alues
are
norm
aliz
ed t
o t
he
expre
ssio
n l
evel
s of
the
untr
eate
d c
ells
(M
ock
contr
ol)
(** p
<0.0
01, * p
<0.0
1)

Regulation and Binding Partners of C11orf67
124
4.3.5 Aberrant expression of C11orf67 alters the sensitivity to
tamoxifen
Previously, we have demonstrated that tamoxifen treatment results in upregulation of
genes in the oncogenic cluster 11q13.5-q14 in ER+ cell lines. To further validate this
observation and establish the role of C11orf67 in antiestrogen resistance, we examined
the effect of aberrant expression of C11orf67 on the response to tamoxifen treatment.
To this aim, we generated T47D cells with lower levels of C11orf67 by transient
transfection with siRNA oligos specific for C11orf67. We validated the successful
knockdown by qRT-PCR analysis of C11orf67 mRNA levels. siRNA transfection
resulted in 60% knockdown of C11orf67 mRNA levels compared to the negative control
siRNA-transfected cells. It is worth noting that we did not use C11orf67 shRNAs as an
alternative for the knockdown in this experiment, since they resulted in extensive
decreased proliferation of cells (Section 3.3.1.2), which masked the effect of Tam
treatment. To complement this study, we also generated MCF7 cells with higher levels of
C11orf67 by stable cDNA lentiviral plasmid (Section 3.2.3).
Our results indicate that T47D cells transfected with C11orf67 siRNA were more
sensitive to Tam treatment than T47D transfected with control siRNA (p<0.001) (Figure
4-6A). Reciprocally, MCF7 cells lentivirally expressing C11orf67 cDNA were more
resistant to Tam treatment compared to the MCF7/empty vector cells (p<0.0001) (Figure
4-6B).
Collectively, these results suggest that Tam treatment upregulates the expression of
C11orf67 and other genes, resulting in increased resistance of these cells to Tam. This
finding explains why anti-estrogen treatment might not be the proper approach for
treating this aggressive ER+ breast cancer subtype.

Regulation and Binding Partners of C11orf67
125
Figure 4-6 Changes in C11orf67 mRNA levels alter the sensitivity of T47D
cells to tamoxifen
(A) qRT-PCR analysis of C11orf67 mRNA expression in T47D/control siRNA
and T47D/C11orf67 siRNA cells (left panel), indicated cells were treated with
different concentrations of Tam for 48 hours and cell viability was measured
by MTT assay (right panel). (B) qRT-PCR analysis of C11orf67 mRNA
expression in MCF7/empty vector and MCF7/C11orf67 cells (left panel),
indicated cells were treated with different concentrations of Tam for 48 hours
and cell viability was measured by MTT assay (right panel) (*** p<0.0001, **
p<0.001).

Regulation and Binding Partners of C11orf67
126
4.3.6 C11orf67 is upregulated in pregnancy
Progesterone Receptor (PR) is one of the transcription factors that has high frequency of
predicted binding sites in promoter region of C11orf67, but whether it has any
transcriptional regulation of the expression of C11orf67 has not yet been investigated.
We next studied the effect of Progesterone (PG) and other pregnancy hormones (Prolactin
and Oxytocin) on the expression levels of C11orf67.
Our results show that physiological doses of PG treatment (100 nM) in T47D cells for 48
hours led to significant activation of C11orf67 mRNA levels by approximately 1.7 fold
(p<0.0001). This effect was rescued by treating the cells with the anti-PG compound
Mifepristone (Mif) at 1 nM, 10 nM and 100 nM (p<0.001, p>0.5 and p<0.001
respectively). 100 nM Mifepristone alone did not significantly alter the expression level
of C11orf67 (Figure 4-7A).
Similarly, physiological doses of other pregnancy hormones such as Prolactin and
Oxytocin, which are known to increase during pregnancy, resulted in significant
upregulation of C11orf67 mRNA levels (p<0.01) (Figure 4-7B).
The physiological responsiveness to hormones in vivo was confirmed by analyzing the
C11orf67 homolog in rats (rat AAMDC) mRNA expression in the mammary fat pads from
pregnant and control rats. Fat pads from pregnant rats (in which PG levels peak) exhibited
3-6 folds higher levels of AAMDC mRNA relative to non-pregnant rats (p<0.0001). While
no changes in expression were observed in non-mammary (retroperitoneal) fat pads
between the two groups (Figure 4-7C).
Collectively, these results suggest that pregnancy, and pregnancy hormones upregulate
the expression of C11orf67 (or rat AAMDC) at the transcriptional level. The opposing
effects of E2 and PG on C11orf67 gene expression could be due to differential
associations with co-repressors and co-activators .
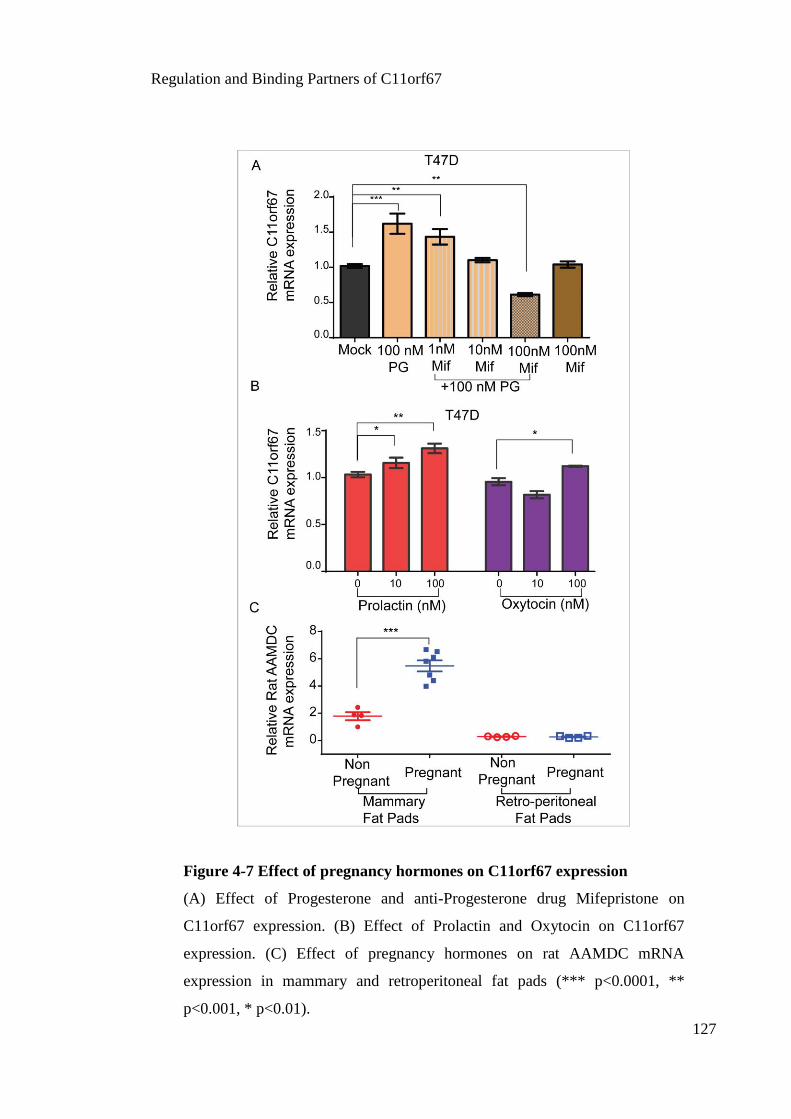
Regulation and Binding Partners of C11orf67
127
Figure 4-7 Effect of pregnancy hormones on C11orf67 expression
(A) Effect of Progesterone and anti-Progesterone drug Mifepristone on
C11orf67 expression. (B) Effect of Prolactin and Oxytocin on C11orf67
expression. (C) Effect of pregnancy hormones on rat AAMDC mRNA
expression in mammary and retroperitoneal fat pads (*** p<0.0001, **
p<0.001, * p<0.01).

Regulation and Binding Partners of C11orf67
128
4.3.7 NF-κB regulates the expression of C11orf67
Our previous TF search analysis revealed the transcription factor p65 (from the NF-κB
family) has strong consensus sites mapping in the regulatory regions of C11orf67, as well
as the neighboring gene RSF1. In previous sections, we showed that the C11orf67
promoter sequence contains 13 p65 binding sites, four of which are high consensus sites,
suggesting that p65 might regulate C11orf67 mRNA expression at the transcriptional
level. p65 is a multifunctional transcription factor that elicits its physiological function
by regulating target gene expression upon NF-κB activation. To determine whether NF-
κB transcriptionally controls the expression of C11orf67 in ER+ cells, we analyzed the
effect of either activation or inhibition of NF-κB pathway on the transcriptional levels of
C11orf67.
First, we tested the effect of TNFα on the transcriptional levels of C11orf67 mRNA in
T47D cells. Stimulation of T47D cells with 10 ng/ml and 100 ng/ml TNFα for 4 hours
significantly increased C11orf67 mRNA levels by 3 and 5 fold, respectively (p<0.001),
as compared with untreated (mock) cells (Figure 4-8A).
Second, we used the NF-κB inhibitor, APQ [6Amino-4(4-phenoxyphenylethylamino)
quinazoline], a cell-permeable quinazoline compound that acts as a potent inhibitor of
NF-κB transcriptional activation by inhibiting NF-κB translocation into the nucleus. APQ
treatment of T47D cells at 10 μM and 100 μM for 48 hours resulted in a significant dose-
dependent downregulation of C11orf67 mRNA levels by approximately 0.4 and 0.2 fold,
respectively, as compared with the untreated control (p<0.0001) (Figure 4-8B).
To physically validate the binding of p65 to the κB binding site in the C11orf67 promoter
in vivo, we performed a ChIP assay in T47D cells. The binding of p65 to the C11orf67
promoter was detected in the basal conditions (no TNFα), and this binding was
significantly higher upon stimulation with 10 ng/ml TNFα for 4 hours. Protein-DNA
complexes were immunoprecipitated with specific anti-p65 antibody. The PCR
quantification of the 177 bp ChIP product was performed with specific primers
encompassing the p65 consensus binding sites in the C11orf67 proximal promoter region.

Regulation and Binding Partners of C11orf67
129
Upon stimulation with TNFα, nuclear p65 binding to the human C11orf67 gene promoter
increased relative to the basal levels (Figure 4-8C).
ChIP results suggest the direct binding of the p65 transcription factor to the C11orf67
proximal promoter. This enhanced binding is consistent with the activation of C11orf67
mRNA expression induced by TNFα. In summary, these results suggest that p65 regulates
C11orf67 transcription by directly binding to the C11orf67 promoter in a TNFα
dependent fashion.
Figure 4-8 C11orf67 is regulated by NF-κB
C11orf67 mRNA levels were analysed by qRTPCR in T47D cells after being
treated with 10 ng/ml and 100 ng/ml of TNFα (A) and with 1µM and 10µM
NF-κB inhibitor APQ (B) for 48 hours. (*** p<0.0001, ** p<0.001). (C) ChIP
analysis of p65 in presence or absence of 10 ng/ml TNFα treatment for 4 hours.

Regulation and Binding Partners of C11orf67
130
4.3.8 C11orf67 acts as a regulator of NF-κB transcriptional
activity
To determine the molecular function of C11orf67 we first searched the genome-wide
siRNA phenotypic screening databases and found that C11orf67 was identified as one of
the 155 targets regulating NF-κB activity in 293T cells (Gewurz et al., 2012). To further
investigate this role in breast cancer cell lines, we performed a luciferase assay using an
NF-κB response element-derived reporter (pNF-κB–Luc) to monitor NF-κB activity.
Reducing C11orf67 using three independent shRNAs having satisfactory knockdown
efficiency to reduce levels of C11orf67 (see section 3.3.1.1), resulted in a significant
decrease in the activity of NF-κB reporter at both the constitutive (basal) levels or after
TNFα stimulation, p<0.001 (Figure 4-9A).
We also tested the effect of C11orf67 knockdown on the mRNA levels of some important
genes downstream the NF-κB pathway; c-Myc, RSF1, and Vascular endothelial growth
factor A (VEGFA). qRT-PCR analysis confirmed that C11orf67 knockdown was
accompanied with a significant downregulation of c-Myc, RSF1 and VEGFA transcripts
(p<0.001), which also follows the same trend of C11orf67 downregulation by the
different shRNAs (Figure 4-9B).
To identify the exact mechanisms of regulation of the endogenous NF-κB by C11orf67,
whole cell lysates were prepared from T47D cells transfected with C11orf67 shRNAs or
empty vector control. Western blotting was performed using specific antibodies against
checkpoints in the NF-κB signalling pathway (Figure 4-9C). Western blot results
indicated that C11orf67 knockdown led to blocked phosphorylation of IKKα/β at Ser
176/180, and also downregulated c-Myc expression at the protein level. However, no
significant changes were observed in IκBα, p65, p-p65, NF-κB2 (p100/p50) protein
expression (Figure 4-9C). Collectively, these data suggest that C11orf67 is an essential
upstream signalling factor controlling NF-κB activity and consequently downstream
targets.

Regulation and Binding Partners of C11orf67
131
Figure 4-9 Effect of C11orf67 knockdown on NF-κB activity
(A) Luciferase NF-κB reporter activity was assessed in T47D transfected with
C11orf67 shRNAs or empty vector control. (B) qRT-PCR analysis of genes
downstream NF-κB pathway in the indicated cell lines (** p<0.001). (C)
Western blot analyses of proteins of the NF-κB pathway in knockdown cells
and empty vector T47D, α-tubulin was used as a loading control. (MW markers
are indicated on the right).

Regulation and Binding Partners of C11orf67
132
4.3.9 RABGAP1L as a binding partner of C11orf67
The previous sections in this chapter have described transcriptional regulation of
C11orf67. Moreover, the identification of novel post-transcriptional protein-protein
interactions involving C11orf67 represents a critical step towards the elucidation of its
biochemical function.
To identify the direct binding partners of C11orf67 we conducted a Yeast two-hybrid
(Y2H) screen. Briefly, Y2H it is a technique conducted in yeast that takes advantage of
the properties of the galactose gene activating the transcription factor (GAL4) or other
similar modular TFs such as LexA, of the yeast Saccharomyces cerevisiae (S. cerevisiae)
(Fields and Song, 1989). The GAL4 protein consists of two separable domains: the N-
terminal domain and the C-terminal domain. The N-terminal domain, also called DNA
binding domain (DBD), recognizes and binds to specific sequences in the DNA upstream
of a promoter. These sequences are termed upstream activation sequences or UAS. The
C-terminal domain (activating domain, AD) stimulates transcription by binding RNA
polymerase. By fusing a protein to each of these domains, one can detect if two proteins
interact by transformation into S. cerevisiae. Provided that the two proteins fused to the
two separable domains interact, the reconstituted GAL4 protein activates transcription of
one or more reporter genes that enable a colour reaction or growth on specific media. The
protein “X” that is fused to the BD of GAL4 is termed the ‘bait’, whilst the second protein,
“Y”, fused to the AD of GAL4 is termed the ‘prey’ (Figure 4-10A) (Fields and Song,
1989, Phizicky and Fields, 1995, Topcu and Borden, 2000, Lentze and Auerbach, 2008).
In our case the screen was performed with the bait plasmids pB27 (N-LexA-bait-C fusion)
and pB66 (N-GAL4-bait-C fusion), and obtained 14 positive clones out of 75 million
interactions tested with a 7-fold coverage of the luminal breast cancer cDNA library. Our
results revealed RABGAP1L as a very high-confidence prey (score A), given the number
(7/14) of independent and overlapping clones obtained from the screen.
In addition to RABGAP1L, only two preys have been identified with a significant score:
SF3B1 (score C, good confidence interaction), and COPG (score D, moderate confidence

Regulation and Binding Partners of C11orf67
133
with a high possibility of false-positive interaction). SF3B1, is the splicing factor 3B
subunit 1 protein and is involved in RNA splicing particularly prespliceosome formation
(Will et al., 2002, Maguire et al., 2015), COPG, is the coatomer protein subunit gamma,
and is one of seven protein coatomers that coat vesicles as they bud from the Golgi
complex (Futatsumori et al., 2000). In the further analysis we will be focusing on
RABGAP1L as it has a very high confidence interaction (score A) obtained from the Y2H
screen.
4.3.10 RABGAP1L expression co-localises with C11orf67 in
ER+ breast cancers
The Y2H screen revealed RABGAP1L as a very high-confidence binding partner of
C11orf67. We next asked whether RABGAP1L follows the same pattern of expression
in breast cancer to that of C11orf67. To this aim, we took advantage of our breast cancer
TMA and performed IHC using anti-RABGAP1L and anti-C11orf67 specific antibodies
to assess co-localisation in the breast tumours. Our IHC staining revealed that
RABGAP1L follows a cytoplasmic, perinuclear and membrane-associated pattern of
expression very similar to that of C11orf67 (Figure 4-11A). With regards to the intensity
of staining, RABGAP1L showed weak expression (-) in the ER-/PR- breast cancer
patients, intermediate expression (+) in normal and benign breast tissues, and high
expression (++) in ER+/PR+ breast sections.
Moreover, we also investigated the effect of C11orf67 gene knockdown by shRNAs (see
Section 3.3.1) on both the mRNA and protein expression levels of RABGAP1L.
Interestingly, the C11orf67 knockdown by sh1 and sh2, but not control vector, led to
significant downregulation of RABGAP1L at the transcript and the protein level,
following the same trend of C11orf67 downregulation (Figure 4-11B). These results
suggest that C11orf67 and its binding partner RABGAP1L might participate the same
intracellular pathway. Further analysis might be required to understand the detailed
interaction and existence of the 2 proteins.

Regulation and Binding Partners of C11orf67
134
Figure 4-10 Y2H screen reveals RABGAP1L and SF3B1 as binding partners
of C11orf67
(A) Schematic illustration of the two-hybrid-based protein-protein interactions
in the yeast nucleus. Modified from James et al. (1996). (B) Y2H screen
parameters for proteins interacting with C11orf67 (Score A: very high
confidence in the interaction, Score C: good confidence in the interaction).
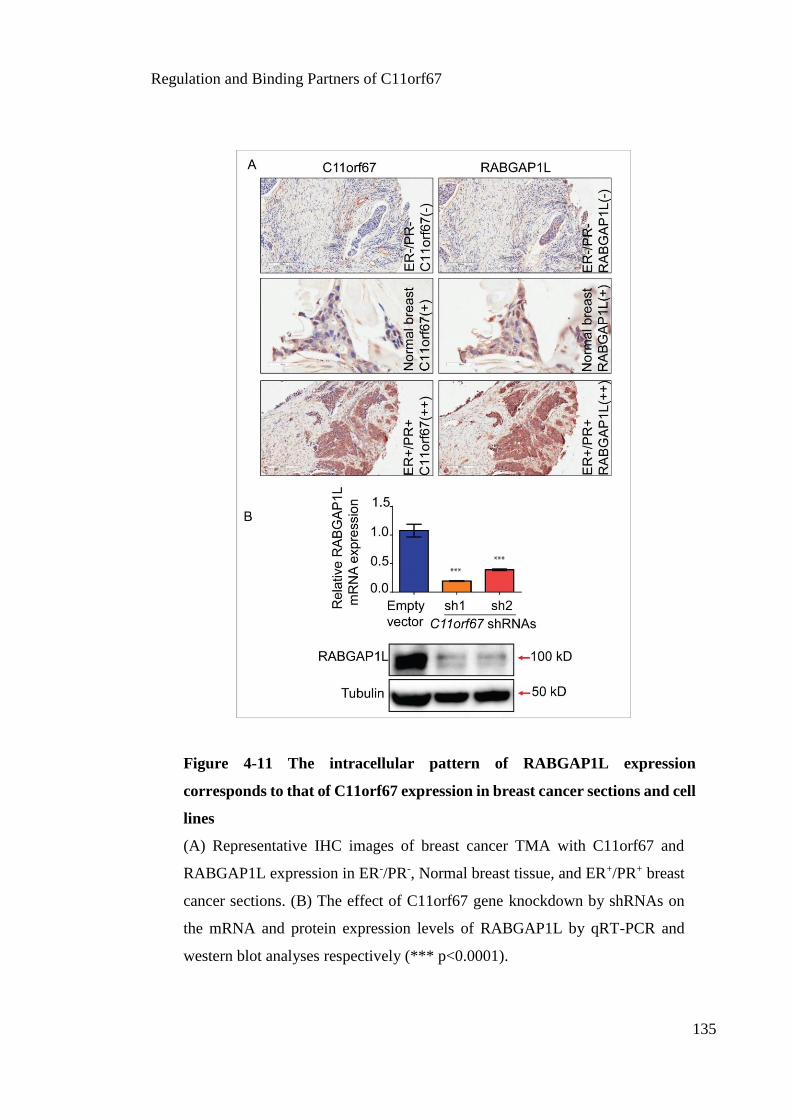
Regulation and Binding Partners of C11orf67
135
Figure 4-11 The intracellular pattern of RABGAP1L expression
corresponds to that of C11orf67 expression in breast cancer sections and cell
lines
(A) Representative IHC images of breast cancer TMA with C11orf67 and
RABGAP1L expression in ER-/PR-, Normal breast tissue, and ER+/PR+ breast
cancer sections. (B) The effect of C11orf67 gene knockdown by shRNAs on
the mRNA and protein expression levels of RABGAP1L by qRT-PCR and
western blot analyses respectively (*** p<0.0001).

Regulation and Binding Partners of C11orf67
136
4.4 Discussion
C11orf67 has previously been identified as a novel candidate gene located at the heart of
an oncogenic cluster in 11q13.5-q14 that is amplified and overexpressed in an ER+
subgroup of breast cancer (Curtis et al., 2012). The IntClust 2 subtype is characterized by
poor prognosis and resistance to endocrine therapy.
In previous chapters, we proposed C11orf67 as a novel biomarker in ER+ breast cancer
that may have a role in predicting poor prognosis. We also identified the oncogenic
potential of C11orf67 and its significance as a therapeutic target in ER+ breast cancer. In
this chapter, we delineated the regulation mechanisms leading to C11orf67 upregulation
and its involvement in the development of the endocrine resistance to tamoxifen treatment
in ER+ breast cancer. We also evaluated potential protein-protein interactions involving
C11orf67 and other signalling molecules which may be reponsible for its biochemical
and biological role.
One of the major clinical obstacles for the management of the IntClust 2 subtype is the
failure of tamoxifen to prevent early relapse in a percentage of patients receiving
treatment, which results in a poor clinical outcome. Moreover, lack of predictors of
tamoxifen response makes it difficult to distinguish those patients who will not respond
to tamoxifen treatment.
The 11q13.5-q14 amplicon comprises C11orf67 and 12 other potential oncogenes.
Among them, amplification of PAK1 has been well associated with poor response to
tamoxifen in ER+ breast cancer. It has also been suggested that therapies targeting PAK1
expression and activity represent a novel strategy to increase endocrine response in breast
cancer (Kilker and Planas-Silva, 2006, Jirstrom et al., 2005, Bostner et al., 2007, Holm et
al., 2006).
In agreement with these findings, we are the first to highlight the role of the C11orf67
gene in tamoxifen resistance in breast cancer. The exact response to tamoxifen treatment
varied among breast cancer cell lines with high or low C11orf67 expression. C11orf67

Regulation and Binding Partners of C11orf67
137
overexpression reduced the antitumour effect of tamoxifen in MCF7 cells, whereas
siRNA depletion of C11orf67 combined with tamoxifen increased its antitumour effects.
These results provide a novel explanation for cellular mechanisms of tamoxifen
resistance. In summary, our findings suggest C11orf67 may provide a potential novel
targeted therapy to increase the response to endocrine treatment in poor prognosis breast
cancer subtype.
It has been reported that amplification of 11q13.5-q14 might occur individually or
simultaneously, resulting in co-amplification of multiple oncogenes (Karlseder et al.,
1994, Bekri et al., 1997, Courjal et al., 1997). In line with these studies, we are the first
to report that the C11orf67 promoter region shares transcriptional factor binding sites that
are distributed over the 11q13.5-q14 cluster as a whole. We also demonstrated that the
transcriptional regulation of this oncogenic cluster includes hormonal binding sites, p65
and Myc binding sites and the possibility of variable chromatin interactions occurring in
different regions of the cluster. This finding suggests that this oncogenic cluster might be
co-regulated leading to co-amplification of potential oncogenes including C11orf67.
Despite the fact that the 11q13.5-q14 locus might be coregulated in ER+ poor prognosis
breast cancer, no previous studies have investigated the effect of tamoxifen on the gene
expression of the whole cluster. We were the first to test this novel hypothesis, and found
that treating ER+ breast cancer cells with tamoxifen leads to remarkable upregulation of
all the genes (including C11orf67) residing in the 11q cluster. This provides an
explanation for the potential adverse effect of treating this category of patients with the
anti-estrogen drug tamoxifen.
The molecular mechanisms involved in the development of resistance to tamoxifen are
poorly understood. However, growing evidence indicates that the NF-κB signalling
pathway plays an important role by regulating the expression of genes critical for cell
survival (deGraffenried et al., 2004, Oida et al., 2014, Kastrati et al., 2015). Direct
binding of the p65 component to the RSF1 gene promoter is one of the possible
mechanisms by which NF-κB elicits its role in resistance to therapy of cancer cells (Yang
et al., 2014). Thus, we invesigated the binding of p65 in the C11orf67 locus using ChIP.

Regulation and Binding Partners of C11orf67
138
Our data support a direct binding of p65 to the proximal promoter of C11orf67 in ER+
T47D breast cancer cells. Moreover, activating the NF-κB pathway by inflammatory
mediators (TNFα) or inhibiting its translocation into the nucleus resulted in significant
proportional changes in the expression of the C11orf67 transcript.
Both the direct binding of NF-κB to the C11orf67 promoter and the changes in
transcriptional levels of C11orf67 validate the possible role of NF-κB in regulating the
expression of C11orf67. This could be the underlying mechanism of developing
endocrine resistance. Additionally, these data also suggest that C11orf67 and RSF1 genes
are more likely to be co-regulated under the control of NF-κB.
A genome-wide siRNA screen for the regulators of NF-κB activity highlighted C11orf67
as one of 133 genes that if transiently knocked down by siRNA led to a significant
suppression of NF-κB activity in HEK 293T cells (Gewurz et al., 2012). However, this
study did not identify any mechanistic pathways underlying the effect of C11orf67 in
regulating NF-κB pathway, and findings in this embryonic cell line may not be applicable
to breast cancer. However, this finding added another dimension to our understanding of
the potential relation between C11orf67 and NF-κB, and motivated us to investigate the
effect of C11orf67 knockdown on NF-κB activity.
The outcomes of our experimental approach were consistent with the findings of Gewurz
(2012) and also answered additional questions. We showed that C11orf67 knockdown
dramatically decreased NF-κB reporter activity at both the basal level and after
stimulation with TNFα. We also showed downregulation of genes downstream of the NF-
κB pathway. Additionally, we pointed to one possible mechanism behind the decreased
activity of NF-κB, an impaired phosphorylation of IKKα and β levels associated with
C11orf67 knockdown. IKKα and β proteins are responsible for phosphorylation of the
inhibitor proteins stimulating its degradation by the proteasome, thus blocking
phosphorylation of IKKα and β proteins keeps p65 in the inactive form in combination
with IκB. Similar findings were observed after the RSF1 knockdown in ovarian cancer
cells by short hairpin RNAs, which suppressed the transcriptional activity of NF-κB
(Yang et al., 2014).

Regulation and Binding Partners of C11orf67
139
In summary, our preliminary data endorse C11orf67 as an essential, upstream signalling
factor controlling NF-κB activity and consequently downstream targets associated with
proliferation and resistance to anti-oestrogens and some forms of chemotherapy. On the
other hand, since C11orf67 and possibly other loci in 11p13.5 are regulated by p65, our
data provide evidence that C11orf67 acts in a feed-forward loop to amplify NF-κB
activation.
One of our striking findings is the novel discovery of RABGAP1L as a very high-
confidence prey by Y2H to be the binding partner of C11orf67, given the number (7/14)
of independent and overlapping clones obtained from the screen.
Ras-like proteins in brain (Rab) is a family of small GTPases that control the vesicular
trafficking that ensures appropriate binding of vesicles from their site of synthesis, with
the proper acceptor compartment and without any uncontrolled interactions or mixing.
Dysregulation of vesicular trafficking leads to diverse pathological consequences (Mitra
et al., 2011). In the cytosol, Rab proteins are kept in the inactive form by constant binding
with Guanine diphosphate (GDP). The Rab GTPase-activating proteins (RABGAP)
accelerate the intrinsic GTPase activity to promote the formation of the inactive GDP-
bound form. RABGAP proteins are “master organizers” that alternate the switches
between an inactive GDP-bound and an active Guanine triphosphate (GTP)-bound form
(Novick and Zerial, 1997, Mitra et al., 2011).
RABGAP1L, also known as HHL or KIAA0471, and is located in the 1q25.1 (Kim et al.,
2013). It is a signal transduction protein that regulates Rab GTPases and controls critical
pathways including vesicular trafficking, cell signalling, protein stability and cell death
(Spiegel et al., 2014). RABGAP1L encodes for a 298 aa protein, which is a GTPase-
activating protein, containing a putative phosphotyrosine-binding domain that binds
phospholipids and cell receptors, suggesting it is the tyrosine-kinase target in signalling
transduction (Roberti et al., 2009).

Regulation and Binding Partners of C11orf67
140
Importantly, RABGAP1L was found to be upregulated in human esophageal and oral
squamous cell carcinomas, and its downregulation was detected in Alzheimer’s disease
(Sharma et al., 2003, Arora et al., 2005).
In mammalian cells, Rab family include about 70 putative members, some with
particularly exciting roles. For instance, Rab 31 protein was found to be overexpressed in
IntClust 2 tumours (Curtis et al., 2012) and was also known to be one of the proteins
controlling NF-κB activity (Gewurz et al., 2012). Additionally, Rab 28 was found to
regulate p65 nuclear transport (Jiang et al., 2013), which could explain molecular
mechanisms by which C11orf67 control the NF-κB activity, resulting in transcriptional
activation of gene programs supporting proliferation and metabolic reprogramming.
In summary, our data demonstrates the important role of C11orf67 as a novel molecule
in tamoxifen resistance induced by the activation of the NF-κB pathway. Our findings
provide a potential mechanism of tamoxifen resistance in ER+ breast cancer subtype. It
can be predicted that targeting C11orf67 and other genes in the 11q13.5-q14 amplicon
can increase the efficiency of endocrine therapy in patients with C11orf67 overexpressing
breast cancer. In addition, tamoxifen should be used with caution in patients with
C11orf67 overexpression.

Chapter 5:
General Discussion

General Discussion
142
Breast cancer is one of the most prevalent malignancies in women around the world.
Although the relative survival from breast cancer is improving, the number of deaths from
this disease is likely to rise by the end of 2015 (AIHW, 2014). Breast cancer heterogeneity
is a major problem that gives rise to different clinical outcome and response to treatment
despite the same clinical diagnosis and prognostic profiles (Zhang et al., 2014).
Over the past few years, the introduction of advanced genomic platforms has added a new
dimension to the landscape of breast cancer and resulted in the identification of new
molecular subtypes. This integrative clustering approach has revealed the IntClust 2
subtype as a novel HR+ tumour subtype associated with high proliferation and very poor
survival. IntClust 2 is characterized by the overexpression of a cluster of candidate
oncogenic drivers in the 11q13.5-q14 locus.
Amplification of the 11q13.5-q14 amplicon has been detected in some types of human
cancer, and is associated with resistance to therapy and poor clinical prognosis (Hui et
al., 1997, Hui et al., 1998, Ormandy et al., 2003, Bostner et al., 2007). However, the
molecular mechanisms accounting for the poor response to treatment and the unfavorable
clinical outcomes in these patients are not understood. In the present study, we performed
functional genomic assays on the C11orf67 locus located in the centre of 11q13.5-q14
amplicon for its roles in tumourigenesis and the development of drug resistance in ER+
breast cancer.
C11orf67 encodes a hypothetical 122 aa protein of unknown function. It has been
previously shown that the protein structure is highly homologous to the bacterial protein
MTH938 (Das et al., 2001), and we have validated the existence of at least three different
oncogenic isoforms derived from C-terminal differential splicing. Importantly, the
function and regulation of C11orf67 is still largely unexplored, particularly in the breast
cancer field.
In the present study, we first assessed C11orf67 expression in clinical breast cancer
specimens and the potential prognostic role of C11orf67 in ER+ breast cancer patients.
Furthermore, we utilized model breast cancer cell lines to investigate the molecular

General Discussion
143
mechanisms of C11orf67 in promoting resistance to endocrine therapy, and proposed an
alternative approach for treating patients with C11orf67 overexpression.
C11orf67 is a novel prognostic biomarker in breast cancer
Incorporation of novel biomarkers that can be assessed in large patient cohorts into
routine clinical practice is very important towards prognostic improvement over the
traditional clinicopathological markers (ER, PR, HER2). In the current study, C11orf67
overexpression at the protein level (as assessed by IHC) appeared to be a strong
prognostic factor associated with HR+ status and the high grade breast cancer sections.
On the other hand, our preliminary data has validated the copy number amplification of
C11orf67 in ER+ breast tumours with a significant correlation with LN metastases. This
finding provides support to the notion that C11orf67 could be used as a novel biomarker
able to stratify breast cancer patients and identify those patients with a higher risk of
tumour recurrence and endocrine resistance.
However, in the near future larger cohorts of breast cancer patients are required for the
validation of the prognostic value of C11orf67. Further collaboration is planned to
validate the amplification and the overexpression of C11orf67 in clinical specimens by
performing IHC and FISH on invasive breast carcinomas from large Western Australian
patient cohorts. Both overexpression and amplification of C11orf67 need to be integrated
in the same patient cohort and then correlated with the risk of recurrence, resistance to
therapy and 5 year survival of patients. Significant correlations are imperative prior to the
development of clinical tests that can be implemented as prognostic biomarker of ER+
breast cancer patients with risk of recurrence and poor outcome.
C11orf67 is a novel oncogene in breast cancer
It is often unclear whether a given molecular alteration is a driver or a passenger in the
process of tumourigenesis and tumour progression. In the case of a driver, the expectation
is that tumour cells harbouring a given alteration are dependent on such alteration for the
origin and/or maintenance of the malignant phenotype (Koch, 2014). Several lines of

General Discussion
144
evidence presented in this thesis indicate that C11orf67 expression is a driver of some
aspects of ER+ breast cancer malignancy. However, it is presently unknown whether
C11orf67 functionally collaborates with some other flanking oncogenes. Clarification
will require combinatorial knockdowns of those genes. Current work ongoing in the
laboratory is implementing a knockdown multiplexing technology based on the
CRISPR/Cas9 system, to carry out multiple loss-of-function mutations in C11orf67 and
the flanking oncogenes and address their effect in the cell death phenotype.
C11orf67 is overexpressed in ER+ breast cancer cell lines, and as expected from being an
oncogene, has low expression levels in normal mammary epithelial cells. IHC was
conducted to evaluate C11orf67 expression in breast cancer specimens. High C11orf67
expression was mainly found in ER+/PR+ breast cancers compared to the normal breast
tissues or benign breast lesions.
More importantly, we demonstrated that silencing of C11orf67 expression in ER+ breast
cancer cell line by shRNA resulted in a significant decrease in the proliferation, colony
formation, in vitro migration, and in vivo tumourigenic potential of breast cancer cells.
Further supporting a causative role of C11orf67 in breast carcinogenesis, we showed that
C11orf67 depletion resulted in cellular senescence, which was evident by positive
staining of the β-gal assay.
C11orf67 is a potential therapeutic target in tamoxifen resistant breast tumours
ER+ breast cancers comprise 70% of all breast cancer cases, and these tumours exhibit a
relatively better prognosis than the ER- breast cancers. However, 30% of ER+ cases show
resistance to hormonal therapy and overall poor outcome (Goldhirsch et al., 2011).
Accordingly, the ER status is not the only factor that controls hormonal responsiveness.
Comprehensive understanding of the molecular signature profiles and identification of
novel predictors of tamoxifen resistance are essential for identifying patients who are
more likely to respond to tamoxifen treatment, and therefore designing novel therapeutic
strategies to improve the clinical outcome.

General Discussion
145
The effects of tamoxifen treatment were distinct between breast cancer cell lines with
high or low C11orf67 expression. We found that C11orf67 isoform_2 overexpression
increased the resistance to tamoxifen in ER+ breast cancer cell lines, whereas the siRNA-
mediated knockdown of the gene increased sensitivity. Furthermore, our molecular
studies provide a basis for potential mechanisms underlying tamoxifen resistance in ER+
breast cancer. Some proposed mechanisms for C11orf67-induced tamoxifen resistance
are discussed below, taking into consideration some gaps of knowledge and missing
information that were not fulfilled in our time frame, which will need to be explored in
further studies.
Loss or phosphorylation of ER
The ER status of breast cancer provides the primary target for endocrine therapy.
However, varying degrees of loss of ER signalling have been detected in about 20% of
tamoxifen-resistant breast cancer (Gutierrez et al., 2005). This phenomenon is not
contradictory with our results showing that C11orf67 is highly expressed in ER+ breast
cancer patients. However, further studies are required to determine if C11orf67
overexpression indeed correlates with ER loss in breast cancer patients treated with
tamoxifen.
Several pathways have been involved in tamoxifen resistance, including activation of
Akt/mTOR, MAPK, and NF-κB pathways. The activation of these proliferative pathways
induces the phosphorylation of ER, resulting in altered response to tamoxifen (Schiff et
al., 2004, Levin and Pietras, 2008, Santen et al., 2009). For instance, phosphorylation of
ERα at Serine 118 by p-MAPK, p-IKKα, or p-mTOR, increases ER activity and decreases
ER affinity to tamoxifen (Vendrell et al., 2005, de Leeuw et al., 2011). All the kinases
mentioned above were shown to be downregulated in the C11orf67 knockdown, and
could explain a possible mechanism of tamoxifen resistance. However, the
phosphorylated ER at S118 is an important checkpoint to be further tested in case of
C11orf67 depletion.

General Discussion
146
Growth factor receptor signalling pathways
Activation of proliferative pathways (EGFR and HER2) leads to the formation of ER-
independent transcriptional activity by either downregulating ER protein or by enhancing
ER activity in a ligand-independent manner, allowing tumour cells to escape the
inhibitory action of tamoxifen. Tamoxifen resistance, however, can also occur indirectly
by activation of other signalling pathways. In this study we found that the expression
levels of phosphorylated p42/44 MAPK, a downstream target of EGFR, decreased in the
case C11orf67 knockdown, an effect that could be associated with the reported
EGFR/HER2-induced tamoxifen resistance in ER+ tumours (Arpino et al., 2008,
Chakraborty et al., 2010). We plan to investigate the overexpression and/or the activation
state of EGFR/HER2 signalling pathways in the case of C11orf67 overexpression or
knockdown in the future.
Akt/mTOR pathway
The Akt/mTOR pathway can directly alter the sensitivity to tamoxifen leading to
tamoxifen resistance in ER+ breast cancer (Baselga et al., 2012, Cavazzoni et al., 2012).
ER+ breast cancer patients with high expression levels of p-Akt (Serine473) and p-mTOR
(Serine2448) exhibit an increased recurrence rate and poor outcome when they receive
tamoxifen treatment (Kirkegaard et al., 2005). However, the factors that induce the
activation of Akt/mTor signalling remain unknown. We are the first to show that
C11orf67 knockdown inhibits the phosphorylation of Akt/mTor proteins, suggesting that
a C11orf67-based target therapy is more likely to improve the response to tamoxifen in
ER+ breast cancer patients. It will also be interesting to define further the effect of mTOR
inhibitors combined with C11orf67 knockdown to increase the antitumour effect of
tamoxifen.
Cell cycle regulators and Myc
The expression and activity of the cyclins and cyclin-dependent kinases proteins have
been shown to have an impact on tamoxifen response. Increased expression of CCND1

General Discussion
147
has been reported in breast cancer cells in association with the development of tamoxifen
resistance, and with restored sensitivity in case of CCND1 depletion (Kilker et al., 2004).
Also, several studies have identified a significant correlation between CCND1
overexpression and poor response to tamoxifen treatment (Stendahl et al., 2004, Rudas et
al., 2008). This finding could be of particular interest, as it explains why C11orf67
knockdown cells showed lower levels of CCND1, while C11orf67 overexpression led to
upregulation of CCND1.
Furthermore, CCNE1 overexpression has been shown to induce resistance to anti-
estrogen therapy (Dhillon and Mudryj, 2002), and is considered one of the most critical
markers that predict endocrine resistance in ER+ breast cancer (Viedma-Rodriguez et al.,
2014). CCNE1 was found in our gene expression array to be downregulated in C11orf67
knockdown cells; these results offer another possible mechanism of tamoxifen resistance
mediated by C11orf67.
Another important marker for tamoxifen resistance is Myc, which is a nuclear
transcription factor that modulates the regulators of cell cycle progression (Nicholson et
al., 2005). We found that Myc was transcriptionally downregulated in the C11orf67
knockdown. Mounting evidence suggests that the overexpression of Myc is associated
with tamoxifen resistance in ER+ breast cancer patients. Myc activation occurs in
response to activation of the upstream mitogenic signalling pathways or the NF-κB
pathway that are both downregulated in C11orf67 ablation.
NF-κB activation
There is strong evidence in the literature that NF-κB activation is associated with
resistance to endocrine therapy (deGraffenried et al., 2004, Zhou et al., 2007). The
mechanisms of NF-κB activation are complex and crosstalk between multiple signalling
pathways is involved (Zhou et al., 2005, Van Laere et al., 2007, Godwin et al., 2013,
Zhang et al., 2013). Activation of Akt/mTor and MAPK pathways can in turn activate
NF-kB pathway (Sanchez-Perez et al., 2002, Crowley et al., 2005). A previous study
conducting a genome-wide screen in HEK 293T cells identified C11orf67 as one of the

General Discussion
148
targets that regulate NF-κB activity. In the current study, we confirmed this finding in the
context of the breast cancer cells, and also demonstrated that NF-κB regulated the
expression of C11orf67 by direct binding of p65 to the promoter of C11orf67. Our data
provide evidence that C11orf67, and possibly the 11q13.5-q14 amplicon, act as a
feedback loop to amplify NF-κB activation (Figure 5-1).
Figure 5-1 Proposed mechanisms underlying C11orf67-induce tamoxifen
resistance in breast cancer
Red dotted arrows indicate downstream direct effects that have been validated
in the study. Black dotted arrows and question marks indicate proposed effects
that have not been validated in the study.

General Discussion
149
Tamoxifen might be detrimental to ER+ patients with 11q13.5-q14 amplification
All the mechanisms mentioned above explain the potential role of C11orf67 in promoting
resistance to tamoxifen. On the other hand, we were able to demonstrate that treating ER+
breast cancer cells with a combination of estrogen and tamoxifen enhanced the expression
levels of almost all the genes residing in the 11q13.5q14 amplicon. This novel finding
suggests that the exposure to tamoxifen might have a detrimental effect on IntClust 2
patients through the activation of multiple oncogenes in 11q13.5-q14 amplicon. Indeed,
exposure to tamoxifen might represent a feedback loop that leads to more upregulation of
the expression levels of oncogenes, more resistance to tamoxifen, and so on.
Antimetabolites are a better choice for C11orf67 overexpression
Metabolic reprogramming of cancer cells is one of the hallmarks of cancer that is thought
to be essential to trigger rapid cancer cell proliferation and thus it is associated with poor
survival in breast cancer (Vazquez et al., 2013). Metabolic reprogramming of cancer cells
is expressed as increased nutrient uptake, upregulated consumption of glycine, and
increased expression levels of folate mitochondrial enzymes (SHMT2, MTHFD2, and
MTHFD1L) (Nilsson et al., 2014). The dependence of cancer cells on nucleotide
metabolism forms the basis that these cancer cells are potentially more inhibited by
chemotherapeutic agents targeting these metabolic enzymes (Jain et al., 2012) .
In the current study, we utilized genome-wide and targeted gene expression analyses to
study the expression patterns of metabolic enzymes in the case of C11orf67 knockdown
or overexpressing cancer cells. In particular, we demonstrated that enzymes of the
mitochondrial folate metabolic pathway are highly upregulated in cancer cells
overexpressing C11orf67, and markedly downregulated in C11orf67-depleted cancer
cells. Further, we provide evidence to show that clinically approved inhibitors of
thymidylate and purine biosynthesis (MTX and 5-FU) show selectivity to C11orf67-
overexpressing MCF7 cells. These findings were also consistent with other studies which
found a strong correlation between MTX sensitivity and high expression of folate
mitochondrial enzymes (Vazquez et al., 2013).

General Discussion
150
Our work suggests that C11orf67 overexpression in breast cancer is one of the underlying
reasons for the concomitant expression of folate metabolism-associated genes, which is
also are targets of Myc (Zeller et al., 2003). This highlights a novel pathway connecting
C11orf67, Myc, and metabolic enzymes in the responsiveness to MTX. Consequently,
overexpression of C11orf67 in breast cancer cells might provide a basis of stratifying the
patients that are more likely to benefit from MTX or 5-FU therapy; these tumours could
be more dependent on folate metabolism.
Concluding remarks
Outcomes from the present study may have several biological, clinical and prognostic
implications in the field of ER+ breast cancer.
First, the current results provide a potential explanation of how the 11q13.5-q14
amplification in the IntClust 2 subtype of breast cancer contributes to shorter overall
survival as compared to the other inter cluster subtypes. It is likely that tumours with
increased DNA copy number of C11orf67 overexpress C11orf67 protein that renders the
de novo tamoxifen resistance. Thus, they are likely related to poor clinical outcomes in
those patients.
Second, the C11orf67 knockdown sensitized breast cancer cells with high C11orf67
expression to the anti-cancer effects of tamoxifen. This result might have translational
consequences for the development of new, targeted cancer therapies to enhance the
sensitivity to tamoxifen. This could be achieved by inactivating the C11orf67 gene or by
interrupting the interaction with its potential binding partners, such as the RABGAP1L
protein in cancer cells.
Third, we found that C11orf67 was required for cell proliferation and survival in T47D
cells, indicating that cells with high expression of C11orf67 may be molecularly
“addicted” to C11orf67 expression.

General Discussion
151
Fourth, C11orf67 expression in ER+ breast cancer specimens could be used as a surrogate
marker alone or in combination with other candidate genes in the 11q13.5-q14 amplicon
(e.g. RSF1, INTS4) to predict treatment response to tamoxifen. To this end, future cohort
studies are required to validate the usefulness of C11orf67 immunoreactivity as a
potential prognostic test for ER+ breast cancer, or for other cancer types with C11orf67
amplification and overexpression.
There are several possible mechanisms that could explain how C11orf67 overexpression
contributes to tumour cell survival and growth in the presence of tamoxifen. First, an
upregulated C11orf67 might modulate the transcriptional activity of a set of genes that
participate in tamoxifen resistance. In fact, based on comparison of gene profiles between
C11orf67 knockdown and control T47D cells, we found that C11orf67 expression was
associated with changes in the expression of CCND1, CCNE1, and Myc. These gene
products have been previously reported to be involved in developing tamoxifen resistance
(Span et al., 2003, Butt et al., 2005).
Furthermore, using interaction network analysis, it appears that several major molecular
hubs were identified in this network, including NF-κB, MAPK, and Akt/mTOR. These
pathways have been suggested to participate in the development of tamoxifen resistance
in cancer cells. Further studies are required to demonstrate the detailed mechanisms of
how the C11orf67/NF-κB complex contributes to tumour development through its
downstream mediators.
While we have proposed the model outlined above, there are alternative interpretations to
consider. First, the shRNA approach used in this study suggests that C11orf67 is the main
gene within the 11q13.5-q14 amplicon responsible for tamoxifen resistance. However,
other gene(s) within the 11q13.5 amplicon might also play a role in the aggressive
behaviour of 11q13.5-amplified carcinomas. For example, PAK1 and Rsf-1 genes have
been shown to be associated with cancer cell proliferation and resistance to endocrine and
chemotherapy in breast and ovarian cancers (Holm et al., 2006, Mao et al., 2006, Bostner
et al., 2007, Keilty et al., 2013, Yang et al., 2014).

General Discussion
152
In summary, our data highlights an important role of C11orf67 as a novel marker in
tamoxifen resistant breast cancers. Our findings provide a possible mechanism of
tamoxifen-resistance in ER+ breast cancers by regulation of several pathways such as
Akt/mTOR and NF-κB. We propose that targeting C11orf67 and its downstream
signalling pathways could increase the efficacy of endocrine therapy in patients with
C11orf67-overexpressing breast cancer. In addition, tamoxifen should be used with
caution in patients presented with C11orf67 overexpression, as it leads to the upregulation
of the genes residing in the oncogenic cluster 11q13.5-q14. Instead, folate antagonists
might present a better choice for this subtype, as C11orf67 overexpression might increase
the dependence on folate metabolism.

Chapter 6:
References

References
154
ADAMI, H. O., SIGNORELLO, L. B. & TRICHOPOULOS, D. 1998. Towards an
understanding of breast cancer etiology. Semin Cancer Biol, 8, 255-62.
AGRE, P. 2006. The aquaporin water channels. Proc Am Thorac Soc, 3, 5-13.
AIHW 2014. Australian Institute of Health and Welfare - Breast cancer in Australia: an
overview. Cancer series no. 78. Cat. no. CAN 75. Canberra: AIHW.
AL DHAHERI, Y., ATTOUB, S., ARAFAT, K., ABUQAMAR, S., EID, A., AL
FARESI, N. & IRATNI, R. 2013. Salinomycin induces apoptosis and
senescence in breast cancer: upregulation of p21, downregulation of survivin
and histone H3 and H4 hyperacetylation. Biochim Biophys Acta, 1830, 3121-35.
ALI, S. & COOMBES, R. C. 2002. Endocrine-responsive breast cancer and strategies
for combating resistance. Nat Rev Cancer, 2, 101-12.
ALI, S. H., O'DONNELL, A. L., BALU, D., POHL, M. B., SEYLER, M. J.,
MOHAMED, S., MOUSA, S. & DANDONA, P. 2000. Estrogen receptor-alpha
in the inhibition of cancer growth and angiogenesis. Cancer Res, 60, 7094-8.
ALLRED, D. C., BROWN, P. & MEDINA, D. 2004. The origins of estrogen receptor
alpha-positive and estrogen receptor alpha-negative human breast cancer. Breast
Cancer Res, 6, 240-5.
ALTUCCI, L., ADDEO, R., CICATIELLO, L., DAUVOIS, S., PARKER, M. G.,
TRUSS, M., BEATO, M., SICA, V., BRESCIANI, F. & WEISZ, A. 1996.
17beta-Estradiol induces cyclin D1 gene transcription, p36D1-p34cdk4 complex
activation and p105Rb phosphorylation during mitogenic stimulation of G(1)-
arrested human breast cancer cells. Oncogene, 12, 2315-24.
ANDERS, L., KE, N., HYDBRING, P., CHOI, Y. J., WIDLUND, H. R., CHICK, J. M.,
ZHAI, H., VIDAL, M., GYGI, S. P., BRAUN, P. & SICINSKI, P. 2011. A
systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to
senescence suppression in cancer cells. Cancer Cell, 20, 620-34.
ANDERSON, E. 2002. The role of oestrogen and progesterone receptors in human
mammary development and tumorigenesis. Breast Cancer Res, 4, 197-201.
ANZICK, S. L., KONONEN, J., WALKER, R. L., AZORSA, D. O., TANNER, M. M.,
GUAN, X. Y., SAUTER, G., KALLIONIEMI, O. P., TRENT, J. M. &
MELTZER, P. S. 1997. AIB1, a steroid receptor coactivator amplified in breast
and ovarian cancer. Science, 277, 965-8.

References
155
ARORA, S., MATTA, A., SHUKLA, N. K., DEO, S. V. & RALHAN, R. 2005.
Identification of differentially expressed genes in oral squamous cell carcinoma.
Mol Carcinog, 42, 97-108.
ARPINO, G., DE ANGELIS, C., GIULIANO, M., GIORDANO, A., FALATO, C., DE
LAURENTIIS, M. & DE PLACIDO, S. 2009. Molecular mechanism and
clinical implications of endocrine therapy resistance in breast cancer. Oncology,
77 Suppl 1, 23-37.
ARPINO, G., WEISS, H., LEE, A. V., SCHIFF, R., DE PLACIDO, S., OSBORNE, C.
K. & ELLEDGE, R. M. 2005. Estrogen receptor-positive, progesterone receptor-
negative breast cancer: association with growth factor receptor expression and
tamoxifen resistance. J Natl Cancer Inst, 97, 1254-61.
ARPINO, G., WIECHMANN, L., OSBORNE, C. K. & SCHIFF, R. 2008. Crosstalk
between the estrogen receptor and the HER tyrosine kinase receptor family:
molecular mechanism and clinical implications for endocrine therapy resistance.
Endocr Rev, 29, 217-33.
BACHELOT, T., MCCOOL, R., DUFFY, S., GLANVILLE, J., VARLEY, D.,
FLEETWOOD, K., ZHANG, J. & JERUSALEM, G. 2014. Comparative
efficacy of everolimus plus exemestane versus fulvestrant for hormone-receptor-
positive advanced breast cancer following progression/recurrence after
endocrine therapy: a network meta-analysis. Breast Cancer Res Treat, 143, 125-
33.
BAE, S. Y., KIM, S., LEE, J. H., LEE, H. C., LEE, S. K., KIL, W. H., KIM, S. W.,
LEE, J. E. & NAM, S. J. 2015. Poor prognosis of single hormone receptor-
positive breast cancer: similar outcome as triple-negative breast cancer. BMC
Cancer, 15, 138.
BAO, A., LI, Y., TONG, Y., ZHENG, H., WU, W. & WEI, C. 2014. 1,25-
Dihydroxyvitamin D(3) and cisplatin synergistically induce apoptosis and cell
cycle arrest in gastric cancer cells. Int J Mol Med, 33, 1177-84.
BARROS, F. F., POWE, D. G., ELLIS, I. O. & GREEN, A. R. 2010. Understanding the
HER family in breast cancer: interaction with ligands, dimerization and
treatments. Histopathology, 56, 560-72.
BARTEL, P., CHIEN, C. T., STERNGLANZ, R. & FIELDS, S. 1993. Elimination of
false positives that arise in using the two-hybrid system. Biotechniques, 14, 920-
4.

References
156
BASELGA, J., CAMPONE, M., PICCART, M., BURRIS, H. A., 3RD, RUGO, H. S.,
SAHMOUD, T., NOGUCHI, S., GNANT, M., PRITCHARD, K. I., LEBRUN,
F., BECK, J. T., ITO, Y., YARDLEY, D., DELEU, I., PEREZ, A.,
BACHELOT, T., VITTORI, L., XU, Z., MUKHOPADHYAY, P., LEBWOHL,
D. & HORTOBAGYI, G. N. 2012. Everolimus in postmenopausal hormone-
receptor-positive advanced breast cancer. N Engl J Med, 366, 520-9.
BAUMGARTEN, S. C. & FRASOR, J. 2012. Minireview: Inflammation: an instigator
of more aggressive estrogen receptor (ER) positive breast cancers. Mol
Endocrinol, 26, 360-71.
BAUTISTA, S., VALLES, H., WALKER, R. L., ANZICK, S., ZEILLINGER, R.,
MELTZER, P. & THEILLET, C. 1998. In breast cancer, amplification of the
steroid receptor coactivator gene AIB1 is correlated with estrogen and
progesterone receptor positivity. Clin Cancer Res, 4, 2925-9.
BEAN, L. A., IANOV, L. & FOSTER, T. C. 2014. Estrogen receptors, the
hippocampus, and memory. Neuroscientist, 20, 534-45.
BECA, F., PEREIRA, M., CAMESELLE-TEIJEIRO, J. F., MARTINS, D. &
SCHMITT, F. 2015. Altered PPP2R2A and Cyclin D1 expression defines a
subgroup of aggressive luminal-like breast cancer. BMC Cancer, 15, 285.
BEKRI, S., ADELAIDE, J., MERSCHER, S., GROSGEORGE, J., CAROLI-BOSC, F.,
PERUCCA-LOSTANLEN, D., KELLEY, P. M., PEBUSQUE, M. J.,
THEILLET, C., BIRNBAUM, D. & GAUDRAY, P. 1997. Detailed map of a
region commonly amplified at 11q13-->q14 in human breast carcinoma.
Cytogenet Cell Genet, 79, 125-31.
BENTIRES-ALJ, M., GIL, S. G., CHAN, R., WANG, Z. C., WANG, Y., IMANAKA,
N., HARRIS, L. N., RICHARDSON, A., NEEL, B. G. & GU, H. 2006. A role
for the scaffolding adapter GAB2 in breast cancer. Nat Med, 12, 114-21.
BENUSIGLIO, P. R., LESUEUR, F., LUCCARINI, C., MCINTOSH, J., LUBEN, R.
N., SMITH, P., DUNNING, A., EASTON, D. F., PONDER, B. A. &
PHAROAH, P. D. 2005. Common variation in EMSY and risk of breast and
ovarian cancer: a case-control study using HapMap tagging SNPs. BMC Cancer,
5, 81.
BISWAS, D. K., CRUZ, A. P., GANSBERGER, E. & PARDEE, A. B. 2000. Epidermal
growth factor-induced nuclear factor kappa B activation: A major pathway of
cell-cycle progression in estrogen-receptor negative breast cancer cells. Proc
Natl Acad Sci U S A, 97, 8542-7.

References
157
BOCCHINFUSO, W. P. & KORACH, K. S. 1997. Mammary gland development and
tumorigenesis in estrogen receptor knockout mice. J Mammary Gland Biol
Neoplasia, 2, 323-34.
BORG, A., SIGURDSSON, H., CLARK, G. M., FERNO, M., FUQUA, S. A.,
OLSSON, H., KILLANDER, D. & MCGURIE, W. L. 1991. Association of
INT2/HST1 coamplification in primary breast cancer with hormone-dependent
phenotype and poor prognosis. Br J Cancer, 63, 136-42.
BORG, A., TANDON, A. K., SIGURDSSON, H., CLARK, G. M., FERNO, M.,
FUQUA, S. A., KILLANDER, D. & MCGUIRE, W. L. 1990. HER-2/neu
amplification predicts poor survival in node-positive breast cancer. Cancer Res,
50, 4332-7.
BOSCO, E. E., WANG, Y., XU, H., ZILFOU, J. T., KNUDSEN, K. E., ARONOW, B.
J., LOWE, S. W. & KNUDSEN, E. S. 2007. The retinoblastoma tumor
suppressor modifies the therapeutic response of breast cancer. J Clin Invest, 117,
218-28.
BOSTNER, J., AHNSTROM WALTERSSON, M., FORNANDER, T., SKOOG, L.,
NORDENSKJOLD, B. & STAL, O. 2007. Amplification of CCND1 and PAK1
as predictors of recurrence and tamoxifen resistance in postmenopausal breast
cancer. Oncogene, 26, 6997-7005.
BOSTNER, J., SKOOG, L., FORNANDER, T., NORDENSKJOLD, B. & STAL, O.
2010. Estrogen receptor-alpha phosphorylation at serine 305, nuclear p21-
activated kinase 1 expression, and response to tamoxifen in postmenopausal
breast cancer. Clin Cancer Res, 16, 1624-33.
BRANKOVIC-MAGIC, M., JANKOVIC, R., NESKOVIC-KONSTANTINOVIC, Z. &
NIKOLIC-VUKOSAVLJEVIC, D. 2002. Progesterone receptor status of breast
cancer metastases. J Cancer Res Clin Oncol, 128, 55-60.
BRISKEN, C. & O'MALLEY, B. 2010. Hormone action in the mammary gland. Cold
Spring Harb Perspect Biol, 2, a003178.
BROWN, L. A., IRVING, J., PARKER, R., KIM, H., PRESS, J. Z., LONGACRE, T.
A., CHIA, S., MAGLIOCCO, A., MAKRETSOV, N., GILKS, B., POLLACK,
J. & HUNTSMAN, D. 2006. Amplification of EMSY, a novel oncogene on
11q13, in high grade ovarian surface epithelial carcinomas. Gynecol Oncol, 100,
264-70.
BROWN, L. A., KALLOGER, S. E., MILLER, M. A., SHIH IE, M., MCKINNEY, S.
E., SANTOS, J. L., SWENERTON, K., SPELLMAN, P. T., GRAY, J., GILKS,

References
158
C. B. & HUNTSMAN, D. G. 2008. Amplification of 11q13 in ovarian
carcinoma. Genes Chromosomes Cancer, 47, 481-9.
BROWN, P. H. & LIPPMAN, S. M. 2000. Chemoprevention of breast cancer. Breast
Cancer Res Treat, 62, 1-17.
BUTT, A. J., MCNEIL, C. M., MUSGROVE, E. A. & SUTHERLAND, R. L. 2005.
Downstream targets of growth factor and oestrogen signalling and endocrine
resistance: the potential roles of c-Myc, cyclin D1 and cyclin E. Endocr Relat
Cancer, 12 Suppl 1, S47-59.
CALAF, G. M., ZEPEDA, A. B., CASTILLO, R. L., FIGUEROA, C. A., ARIAS, C.,
FIGUEROA, E. & FARIAS, J. G. 2015. Molecular aspects of breast cancer
resistance to drugs (Review). Int J Oncol, 47, 437-45.
CAMPBELL, R. A., BHAT-NAKSHATRI, P., PATEL, N. M., CONSTANTINIDOU,
D., ALI, S. & NAKSHATRI, H. 2001. Phosphatidylinositol 3-kinase/AKT-
mediated activation of estrogen receptor alpha: a new model for anti-estrogen
resistance. J Biol Chem, 276, 9817-24.
CANCER GENOME ATLAS, N. 2012. Comprehensive molecular portraits of human
breast tumours. Nature, 490, 61-70.
CANTLEY, L. C. 2002. The phosphoinositide 3-kinase pathway. Science, 296, 1655-7.
CAVAZZONI, A., BONELLI, M. A., FUMAROLA, C., LA MONICA, S., AIROUD,
K., BERTONI, R., ALFIERI, R. R., GALETTI, M., TRAMONTI, S.,
GALVANI, E., HARRIS, A. L., MARTIN, L. A., ANDREIS, D., BOTTINI, A.,
GENERALI, D. & PETRONINI, P. G. 2012. Overcoming acquired resistance to
letrozole by targeting the PI3K/AKT/mTOR pathway in breast cancer cell
clones. Cancer Lett, 323, 77-87.
CAZZANIGA, M. E., MUSTACCHI, G., PRONZATO, P., DE MATTEIS, A., DI
COSTANZO, F. & FLORIANI, I. 2006. Adjuvant systemic treatment of early
breast cancer: the NORA study. Ann Oncol, 17, 1386-92.
CHAKRABORTY, A. K., WELSH, A. & DIGIOVANNA, M. P. 2010. Co-targeting the
insulin-like growth factor I receptor enhances growth-inhibitory and pro-
apoptotic effects of anti-estrogens in human breast cancer cell lines. Breast
Cancer Res Treat, 120, 327-35.
CHEN, L. F. & GREENE, W. C. 2004. Shaping the nuclear action of NF-kappaB. Nat
Rev Mol Cell Biol, 5, 392-401.

References
159
CHESKIS, B. J., GREGER, J. G., NAGPAL, S. & FREEDMAN, L. P. 2007. Signaling
by estrogens. J Cell Physiol, 213, 610-7.
CHIA, S., BRYCE, C. & GELMON, K. 2005. The 2000 EBCTCG overview: a
widening gap. Lancet, 365, 1665-6.
CHOI, J. H., SHEU, J. J., GUAN, B., JINAWATH, N., MARKOWSKI, P., WANG, T.
L. & SHIH IE, M. 2009. Functional analysis of 11q13.5 amplicon identifies Rsf-
1 (HBXAP) as a gene involved in paclitaxel resistance in ovarian cancer. Cancer
Res, 69, 1407-15.
CHU, I. M., HENGST, L. & SLINGERLAND, J. M. 2008. The Cdk inhibitor p27 in
human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev
Cancer, 8, 253-67.
CIANFROCCA, M. & GOLDSTEIN, L. J. 2004. Prognostic and predictive factors in
early-stage breast cancer. Oncologist, 9, 606-16.
CIARDIELLO, F. & TORTORA, G. 2008. EGFR antagonists in cancer treatment. N
Engl J Med, 358, 1160-74.
CIRUELOS GIL, E. M. 2014. Targeting the PI3K/AKT/mTOR pathway in estrogen
receptor-positive breast cancer. Cancer Treat Rev, 40, 862-71.
CLARKE, M., COLLINS, R., DARBY, S., DAVIES, C., ELPHINSTONE, P., EVANS,
V., GODWIN, J., GRAY, R., HICKS, C., JAMES, S., MACKINNON, E.,
MCGALE, P., MCHUGH, T., PETO, R., TAYLOR, C. & WANG, Y. 2005.
Effects of radiotherapy and of differences in the extent of surgery for early
breast cancer on local recurrence and 15-year survival: an overview of the
randomised trials. Lancet, 366, 2087-106.
COHEN, P. T., PHILP, A. & VAZQUEZ-MARTIN, C. 2005. Protein phosphatase 4--
from obscurity to vital functions. FEBS Lett, 579, 3278-86.
COLLADO, M., BLASCO, M. A. & SERRANO, M. 2007. Cellular senescence in
cancer and aging. Cell, 130, 223-33.
COURJAL, F., CUNY, M., SIMONY-LAFONTAINE, J., LOUASON, G., SPEISER,
P., ZEILLINGER, R., RODRIGUEZ, C. & THEILLET, C. 1997. Mapping of
DNA amplifications at 15 chromosomal localizations in 1875 breast tumors:
definition of phenotypic groups. Cancer Res, 57, 4360-7.
COURJAL, F., LOUASON, G., SPEISER, P., KATSAROS, D., ZEILLINGER, R. &
THEILLET, C. 1996. Cyclin gene amplification and overexpression in breast

References
160
and ovarian cancers: evidence for the selection of cyclin D1 in breast and cyclin
E in ovarian tumors. Int J Cancer, 69, 247-53.
COURJAL, F. & THEILLET, C. 1997. Comparative genomic hybridization analysis of
breast tumors with predetermined profiles of DNA amplification. Cancer Res,
57, 4368-77.
COURTOIS-COX, S., GENTHER WILLIAMS, S. M., RECZEK, E. E., JOHNSON, B.
W., MCGILLICUDDY, L. T., JOHANNESSEN, C. M., HOLLSTEIN, P. E.,
MACCOLLIN, M. & CICHOWSKI, K. 2006. A negative feedback signaling
network underlies oncogene-induced senescence. Cancer Cell, 10, 459-72.
COUSE, J. F. & KORACH, K. S. 1999. Estrogen receptor null mice: what have we
learned and where will they lead us? Endocr Rev, 20, 358-417.
CREIGHTON, C. J. 2012. The molecular profile of luminal B breast cancer. Biologics,
6, 289-97.
CROWLEY, J. E., TREML, L. S., STADANLICK, J. E., CARPENTER, E. &
CANCRO, M. P. 2005. Homeostatic niche specification among naive and
activated B cells: a growing role for the BLyS family of receptors and ligands.
Semin Immunol, 17, 193-9.
CUI, X., SCHIFF, R., ARPINO, G., OSBORNE, C. K. & LEE, A. V. 2005. Biology of
progesterone receptor loss in breast cancer and its implications for endocrine
therapy. J Clin Oncol, 23, 7721-35.
CURTIS, C., SHAH, S. P., CHIN, S. F., TURASHVILI, G., RUEDA, O. M.,
DUNNING, M. J., SPEED, D., LYNCH, A. G., SAMARAJIWA, S., YUAN,
Y., GRAF, S., HA, G., HAFFARI, G., BASHASHATI, A., RUSSELL, R.,
MCKINNEY, S., GROUP, M., LANGEROD, A., GREEN, A.,
PROVENZANO, E., WISHART, G., PINDER, S., WATSON, P.,
MARKOWETZ, F., MURPHY, L., ELLIS, I., PURUSHOTHAM, A.,
BORRESEN-DALE, A. L., BRENTON, J. D., TAVARE, S., CALDAS, C. &
APARICIO, S. 2012. The genomic and transcriptomic architecture of 2,000
breast tumours reveals novel subgroups. Nature, 486, 346-52.
DARBY, S., MCGALE, P., CORREA, C., TAYLOR, C., ARRIAGADA, R.,
CLARKE, M., CUTTER, D., DAVIES, C., EWERTZ, M., GODWIN, J.,
GRAY, R., PIERCE, L., WHELAN, T., WANG, Y. & PETO, R. 2011. Effect of
radiotherapy after breast-conserving surgery on 10-year recurrence and 15-year
breast cancer death: meta-analysis of individual patient data for 10,801 women
in 17 randomised trials. Lancet, 378, 1707-16.

References
161
DAS, K., XIAO, R., WAHLBERG, E., HSU, F., ARROWSMITH, C. H.,
MONTELIONE, G. T. & ARNOLD, E. 2001. X-ray crystal structure of
MTH938 from Methanobacterium thermoautotrophicum at 2.2 A resolution
reveals a novel tertiary protein fold. Proteins, 45, 486-8.
DAUVOIS, S., DANIELIAN, P. S., WHITE, R. & PARKER, M. G. 1992. Antiestrogen
ICI 164,384 reduces cellular estrogen receptor content by increasing its turnover.
Proc Natl Acad Sci U S A, 89, 4037-41.
DAVIES, C., GODWIN, J., GRAY, R., CLARKE, M., CUTTER, D., DARBY, S.,
MCGALE, P., PAN, H. C., TAYLOR, C., WANG, Y. C., DOWSETT, M.,
INGLE, J. & PETO, R. 2011. Relevance of breast cancer hormone receptors and
other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of
randomised trials. Lancet, 378, 771-84.
DE LEEUW, R., NEEFJES, J. & MICHALIDES, R. 2011. A role for estrogen receptor
phosphorylation in the resistance to tamoxifen. Int J Breast Cancer, 2011,
232435.
DEGRAFFENRIED, L. A., CHANDRASEKAR, B., FRIEDRICHS, W. E., DONZIS,
E., SILVA, J., HIDALGO, M., FREEMAN, J. W. & WEISS, G. R. 2004. NF-
kappa B inhibition markedly enhances sensitivity of resistant breast cancer
tumor cells to tamoxifen. Ann Oncol, 15, 885-90.
DEL BARCO, S., CIRUELOS, E., TUSQUETS, I., RUIZ, M. & BARNADAS, A.
2013. SEOM clinical guidelines for the systemic treatment of early breast cancer
2013. Clin Transl Oncol, 15, 1011-7.
DHILLON, N. K. & MUDRYJ, M. 2002. Ectopic expression of cyclin E in estrogen
responsive cells abrogates antiestrogen mediated growth arrest. Oncogene, 21,
4626-34.
DIMRI, G. P., LEE, X., BASILE, G., ACOSTA, M., SCOTT, G., ROSKELLEY, C.,
MEDRANO, E. E., LINSKENS, M., RUBELJ, I., PEREIRA-SMITH, O. & ET
AL. 1995. A biomarker that identifies senescent human cells in culture and in
aging skin in vivo. Proc Natl Acad Sci U S A, 92, 9363-7.
DOLCET, X., LLOBET, D., PALLARES, J. & MATIAS-GUIU, X. 2005. NF-kB in
development and progression of human cancer. Virchows Arch, 446, 475-82.
DOWLING, R. J., TOPISIROVIC, I., FONSECA, B. D. & SONENBERG, N. 2010.
Dissecting the role of mTOR: lessons from mTOR inhibitors. Biochim Biophys
Acta, 1804, 433-9.

References
162
DOWSETT, M., SESTAK, I., LOPEZ-KNOWLES, E., SIDHU, K., DUNBIER, A. K.,
COWENS, J. W., FERREE, S., STORHOFF, J., SCHAPER, C. & CUZICK, J.
2013. Comparison of PAM50 risk of recurrence score with oncotype DX and
IHC4 for predicting risk of distant recurrence after endocrine therapy. J Clin
Oncol, 31, 2783-90.
DUBIK, D., DEMBINSKI, T. C. & SHIU, R. P. 1987. Stimulation of c-myc oncogene
expression associated with estrogen-induced proliferation of human breast
cancer cells. Cancer Res, 47, 6517-21.
EARLY BREAST CANCER TRIALISTS' COLLABORATIVE, G. 2005. Effects of
chemotherapy and hormonal therapy for early breast cancer on recurrence and
15-year survival: an overview of the randomised trials. Lancet, 365, 1687-717.
ENCARNACION, C. A., CIOCCA, D. R., MCGUIRE, W. L., CLARK, G. M.,
FUQUA, S. A. & OSBORNE, C. K. 1993. Measurement of steroid hormone
receptors in breast cancer patients on tamoxifen. Breast Cancer Res Treat, 26,
237-46.
ENMARK, E., PELTO-HUIKKO, M., GRANDIEN, K., LAGERCRANTZ, S.,
LAGERCRANTZ, J., FRIED, G., NORDENSKJOLD, M. & GUSTAFSSON, J.
A. 1997. Human estrogen receptor beta-gene structure, chromosomal
localization, and expression pattern. J Clin Endocrinol Metab, 82, 4258-65.
EVANGELISTI, C., DE BIASE, D., KURELAC, I., CECCARELLI, C., PROKISCH,
H., MEITINGER, T., CARIA, P., VANNI, R., ROMEO, G., TALLINI, G.,
GASPARRE, G. & BONORA, E. 2015. A mutation screening of oncogenes,
tumor suppressor gene TP53 and nuclear encoded mitochondrial complex I
genes in oncocytic thyroid tumors. BMC Cancer, 15, 157.
FEDELE, P., CALVANI, N., MARINO, A., ORLANDO, L., SCHIAVONE, P.,
QUARANTA, A. & CINIERI, S. 2012. Targeted agents to reverse resistance to
endocrine therapy in metastatic breast cancer: where are we now and where are
we going? Crit Rev Oncol Hematol, 84, 243-51.
FIELDS, S. & SONG, O. 1989. A novel genetic system to detect protein-protein
interactions. Nature, 340, 245-6.
FISHER, B., ANDERSON, S., BRYANT, J., MARGOLESE, R. G., DEUTSCH, M.,
FISHER, E. R., JEONG, J. H. & WOLMARK, N. 2002. Twenty-year follow-up
of a randomized trial comparing total mastectomy, lumpectomy, and
lumpectomy plus irradiation for the treatment of invasive breast cancer. N Engl J
Med, 347, 1233-41.

References
163
FLEMING, F. J., HILL, A. D., MCDERMOTT, E. W., O'HIGGINS, N. J. & YOUNG,
L. S. 2004. Differential recruitment of coregulator proteins steroid receptor
coactivator-1 and silencing mediator for retinoid and thyroid receptors to the
estrogen receptor-estrogen response element by beta-estradiol and 4-
hydroxytamoxifen in human breast cancer. J Clin Endocrinol Metab, 89, 375-
83.
FLOTOTTO, T., NIEDERACHER, D., HOHMANN, D., HEIMERZHEIM, T., DALL,
P., DJAHANSOUZI, S., BENDER, H. G. & HANSTEIN, B. 2004. Molecular
mechanism of estrogen receptor (ER)alpha-specific, estradiol-dependent
expression of the progesterone receptor (PR) B-isoform. J Steroid Biochem Mol
Biol, 88, 131-42.
FONG, Y., EVANS, J., BROOK, D., KENKRE, J., JARVIS, P. & GOWER-THOMAS,
K. 2015. The Nottingham Prognostic Index: five- and ten-year data for all-cause
survival within a screened population. Ann R Coll Surg Engl, 97, 137-9.
FORMSTECHER, E., ARESTA, S., COLLURA, V., HAMBURGER, A., MEIL, A.,
TREHIN, A., REVERDY, C., BETIN, V., MAIRE, S., BRUN, C., JACQ, B.,
ARPIN, M., BELLAICHE, Y., BELLUSCI, S., BENAROCH, P., BORNENS,
M., CHANET, R., CHAVRIER, P., DELATTRE, O., DOYE, V., FEHON, R.,
FAYE, G., GALLI, T., GIRAULT, J. A., GOUD, B., DE GUNZBURG, J.,
JOHANNES, L., JUNIER, M. P., MIROUSE, V., MUKHERJEE, A.,
PAPADOPOULO, D., PEREZ, F., PLESSIS, A., ROSSE, C., SAULE, S.,
STOPPA-LYONNET, D., VINCENT, A., WHITE, M., LEGRAIN, P.,
WOJCIK, J., CAMONIS, J. & DAVIET, L. 2005. Protein interaction mapping: a
Drosophila case study. Genome Res, 15, 376-84.
FRASOR, J., DANES, J. M., KOMM, B., CHANG, K. C., LYTTLE, C. R. &
KATZENELLENBOGEN, B. S. 2003. Profiling of estrogen up- and down-
regulated gene expression in human breast cancer cells: insights into gene
networks and pathways underlying estrogenic control of proliferation and cell
phenotype. Endocrinology, 144, 4562-74.
FROMONT-RACINE, M., RAIN, J. C. & LEGRAIN, P. 1997. Toward a functional
analysis of the yeast genome through exhaustive two-hybrid screens. Nat Genet,
16, 277-82.
FUTATSUMORI, M., KASAI, K., TAKATSU, H., SHIN, H. W. & NAKAYAMA, K.
2000. Identification and characterization of novel isoforms of COP I subunits. J
Biochem, 128, 793-801.
GEWURZ, B. E., TOWFIC, F., MAR, J. C., SHINNERS, N. P., TAKASAKI, K.,
ZHAO, B., CAHIR-MCFARLAND, E. D., QUACKENBUSH, J., XAVIER, R.

References
164
J. & KIEFF, E. 2012. Genome-wide siRNA screen for mediators of NF-kappaB
activation. Proc Natl Acad Sci U S A, 109, 2467-72.
GHERGHE, M., BORDEA, C. & BLIDARU, A. 2015. Sentinel lymph node biopsy
(SLNB) vs. axillary lymph node dissection (ALND) in the current surgical
treatment of early stage breast cancer. J Med Life, 8, 176-80.
GIAMPAGLIA, M., CHIURI, V. E., TINELLI, A., DE LAURENTIIS, M.,
SILVESTRIS, N. & LORUSSO, V. 2010. Lapatinib in breast cancer: clinical
experiences and future perspectives. Cancer Treat Rev, 36 Suppl 3, S72-9.
GIANNI, L., DAFNI, U., GELBER, R. D., AZAMBUJA, E., MUEHLBAUER, S.,
GOLDHIRSCH, A., UNTCH, M., SMITH, I., BASELGA, J., JACKISCH, C.,
CAMERON, D., MANO, M., PEDRINI, J. L., VERONESI, A., MENDIOLA,
C., PLUZANSKA, A., SEMIGLAZOV, V., VRDOLJAK, E., ECKART, M. J.,
SHEN, Z., SKIADOPOULOS, G., PROCTER, M., PRITCHARD, K. I.,
PICCART-GEBHART, M. J., BELL, R. & HERCEPTIN ADJUVANT TRIAL
STUDY, T. 2011. Treatment with trastuzumab for 1 year after adjuvant
chemotherapy in patients with HER2-positive early breast cancer: a 4-year
follow-up of a randomised controlled trial. Lancet Oncol, 12, 236-44.
GILLETT, C., FANTL, V., SMITH, R., FISHER, C., BARTEK, J., DICKSON, C.,
BARNES, D. & PETERS, G. 1994. Amplification and overexpression of cyclin
D1 in breast cancer detected by immunohistochemical staining. Cancer Res, 54,
1812-7.
GINES, J., SABATER, E., MARTORELL, C., GRAU, M., MONROY, M. &
CASADO, M. A. 2011. Efficacy of taxanes as adjuvant treatment of breast
cancer: a review and meta-analysis of randomised clinical trials. Clin Transl
Oncol, 13, 485-98.
GIRAULT, I., LEREBOURS, F., AMARIR, S., TOZLU, S., TUBIANA-HULIN, M.,
LIDEREAU, R. & BIECHE, I. 2003. Expression analysis of estrogen receptor
alpha coregulators in breast carcinoma: evidence that NCOR1 expression is
predictive of the response to tamoxifen. Clin Cancer Res, 9, 1259-66.
GODWIN, P., BAIRD, A. M., HEAVEY, S., BARR, M. P., O'BYRNE, K. J. &
GATELY, K. 2013. Targeting nuclear factor-kappa B to overcome resistance to
chemotherapy. Front Oncol, 3, 120.
GOLDHIRSCH, A., WOOD, W. C., COATES, A. S., GELBER, R. D.,
THURLIMANN, B., SENN, H. J. & PANEL, M. 2011. Strategies for subtypes--
dealing with the diversity of breast cancer: highlights of the St. Gallen
International Expert Consensus on the Primary Therapy of Early Breast Cancer
2011. Ann Oncol, 22, 1736-47.

References
165
GOSS, P. E., INGLE, J. N., MARTINO, S., ROBERT, N. J., MUSS, H. B., PICCART,
M. J., CASTIGLIONE, M., TU, D., SHEPHERD, L. E., PRITCHARD, K. I.,
LIVINGSTON, R. B., DAVIDSON, N. E., NORTON, L., PEREZ, E. A.,
ABRAMS, J. S., THERASSE, P., PALMER, M. J. & PATER, J. L. 2003. A
randomized trial of letrozole in postmenopausal women after five years of
tamoxifen therapy for early-stage breast cancer. The New England journal of
medicine, 349, 1793-802.
GREENBERG, S., STOPECK, A. & RUGO, H. S. 2011. Systemic treatment of early
breast cancer--a biological perspective. J Surg Oncol, 103, 619-26.
GRIFFITHS, C. L. & OLIN, J. L. 2012. Triple negative breast cancer: a brief review of
its characteristics and treatment options. J Pharm Pract, 25, 319-23.
GUIU, S., MICHIELS, S., ANDRE, F., CORTES, J., DENKERT, C., DI LEO, A.,
HENNESSY, B. T., SORLIE, T., SOTIRIOU, C., TURNER, N., VAN DE
VIJVER, M., VIALE, G., LOI, S. & REIS-FILHO, J. S. 2012. Molecular
subclasses of breast cancer: how do we define them? The IMPAKT 2012
Working Group Statement. Ann Oncol, 23, 2997-3006.
GUSTAFSSON, J. A. & WARNER, M. 2000. Estrogen receptor beta in the breast: role
in estrogen responsiveness and development of breast cancer. J Steroid Biochem
Mol Biol, 74, 245-8.
GUTIERREZ, M. C., DETRE, S., JOHNSTON, S., MOHSIN, S. K., SHOU, J.,
ALLRED, D. C., SCHIFF, R., OSBORNE, C. K. & DOWSETT, M. 2005.
Molecular changes in tamoxifen-resistant breast cancer: relationship between
estrogen receptor, HER-2, and p38 mitogen-activated protein kinase. J Clin
Oncol, 23, 2469-76.
HABASHY, H. O., POWE, D. G., ABDEL-FATAH, T. M., GEE, J. M., NICHOLSON,
R. I., GREEN, A. R., RAKHA, E. A. & ELLIS, I. O. 2012. A review of the
biological and clinical characteristics of luminal-like oestrogen receptor-positive
breast cancer. Histopathology, 60, 854-63.
HAINAUT, P. & PLYMOTH, A. 2013. Targeting the hallmarks of cancer: towards a
rational approach to next-generation cancer therapy. Curr Opin Oncol, 25, 50-1.
HAN, T., LIU, Z., LI, H., XIE, W., ZHANG, R., ZHU, L., GUO, F., HAN, Y., SHENG,
Y. & XIE, X. 2015. High expression of UBD correlates with epirubicin
resistance and indicates poor prognosis in triple-negative breast cancer. Onco
Targets Ther, 8, 1643-9.

References
166
HANAHAN, D. & WEINBERG, R. A. 2011. Hallmarks of cancer: the next generation.
Cell, 144, 646-74.
HARARI, D. & YARDEN, Y. 2000. Molecular mechanisms underlying ErbB2/HER2
action in breast cancer. Oncogene, 19, 6102-14.
HELDRING, N., PIKE, A., ANDERSSON, S., MATTHEWS, J., CHENG, G.,
HARTMAN, J., TUJAGUE, M., STROM, A., TREUTER, E., WARNER, M. &
GUSTAFSSON, J. A. 2007. Estrogen receptors: how do they signal and what
are their targets. Physiol Rev, 87, 905-31.
HERRERA ABREU, M. T., HUGHES, W. E., MELE, K., LYONS, R. J.,
RICKWOOD, D., BROWNE, B. C., BENNETT, H. L., VALLOTTON, P.,
BRUMMER, T. & DALY, R. J. 2011. Gab2 regulates cytoskeletal organization
and migration of mammary epithelial cells by modulating RhoA activation. Mol
Biol Cell, 22, 105-16.
HERSCHKOWITZ, J. I., SIMIN, K., WEIGMAN, V. J., MIKAELIAN, I., USARY, J.,
HU, Z., RASMUSSEN, K. E., JONES, L. P., ASSEFNIA, S.,
CHANDRASEKHARAN, S., BACKLUND, M. G., YIN, Y., KHRAMTSOV,
A. I., BASTEIN, R., QUACKENBUSH, J., GLAZER, R. I., BROWN, P. H.,
GREEN, J. E., KOPELOVICH, L., FURTH, P. A., PALAZZO, J. P.,
OLOPADE, O. I., BERNARD, P. S., CHURCHILL, G. A., VAN DYKE, T. &
PEROU, C. M. 2007. Identification of conserved gene expression features
between murine mammary carcinoma models and human breast tumors.
Genome Biol, 8, R76.
HERVOUET, E., CARTRON, P. F., JOUVENOT, M. & DELAGE-MOURROUX, R.
2013. Epigenetic regulation of estrogen signaling in breast cancer. Epigenetics,
8, 237-45.
HOFFMANN, A., LEUNG, T. H. & BALTIMORE, D. 2003. Genetic analysis of NF-
kappaB/Rel transcription factors defines functional specificities. EMBO J, 22,
5530-9.
HOLM, C., RAYALA, S., JIRSTROM, K., STAL, O., KUMAR, R. & LANDBERG,
G. 2006. Association between Pak1 expression and subcellular localization and
tamoxifen resistance in breast cancer patients. J Natl Cancer Inst, 98, 671-80.
HOLMES, D. 2011. PI3K pathway inhibitors approach junction. Nat Rev Drug Discov,
10, 563-4.
HOLZ, M. K. 2012. The role of S6K1 in ER-positive breast cancer. Cell Cycle, 11,
3159-65.

References
167
HORST, B., GRUVBERGER-SAAL, S. K., HOPKINS, B. D., BORDONE, L., YANG,
Y., CHERNOFF, K. A., UZOMA, I., SCHWIPPER, V., LIEBAU, J., NOWAK,
N. J., BRUNNER, G., OWENS, D., RIMM, D. L., PARSONS, R. & CELEBI, J.
T. 2009. Gab2-mediated signaling promotes melanoma metastasis. Am J Pathol,
174, 1524-33.
HOVEY, R. C., TROTT, J. F. & VONDERHAAR, B. K. 2002. Establishing a
framework for the functional mammary gland: from endocrinology to
morphology. J Mammary Gland Biol Neoplasia, 7, 17-38.
HU, B. S., YU, H. F., ZHAO, G. & ZHA, T. Z. 2012. High RSF-1 expression correlates
with poor prognosis in patients with gastric adenocarcinoma. Int J Clin Exp
Pathol, 5, 668-73.
HU, J. & VERKMAN, A. S. 2006. Increased migration and metastatic potential of
tumor cells expressing aquaporin water channels. FASEB J, 20, 1892-4.
HU, Z., FAN, C., OH, D. S., MARRON, J. S., HE, X., QAQISH, B. F., LIVASY, C.,
CAREY, L. A., REYNOLDS, E., DRESSLER, L., NOBEL, A., PARKER, J.,
EWEND, M. G., SAWYER, L. R., WU, J., LIU, Y., NANDA, R.,
TRETIAKOVA, M., RUIZ ORRICO, A., DREHER, D., PALAZZO, J. P.,
PERREARD, L., NELSON, E., MONE, M., HANSEN, H., MULLINS, M.,
QUACKENBUSH, J. F., ELLIS, M. J., OLOPADE, O. I., BERNARD, P. S. &
PEROU, C. M. 2006. The molecular portraits of breast tumors are conserved
across microarray platforms. BMC Genomics, 7, 96.
HUDIS, C. A. 2007. Trastuzumab--mechanism of action and use in clinical practice. N
Engl J Med, 357, 39-51.
HUGGINS, C. J., MALIK, R., LEE, S., SALOTTI, J., THOMAS, S., MARTIN, N.,
QUINONES, O. A., ALVORD, W. G., OLANICH, M. E., KELLER, J. R. &
JOHNSON, P. F. 2013. C/EBPgamma suppresses senescence and inflammatory
gene expression by heterodimerizing with C/EBPbeta. Mol Cell Biol, 33, 3242-
58.
HUGHES-DAVIES, L., HUNTSMAN, D., RUAS, M., FUKS, F., BYE, J., CHIN, S. F.,
MILNER, J., BROWN, L. A., HSU, F., GILKS, B., NIELSEN, T., SCHULZER,
M., CHIA, S., RAGAZ, J., CAHN, A., LINGER, L., OZDAG, H., CATTANEO,
E., JORDANOVA, E. S., SCHUURING, E., YU, D. S., VENKITARAMAN,
A., PONDER, B., DOHERTY, A., APARICIO, S., BENTLEY, D., THEILLET,
C., PONTING, C. P., CALDAS, C. & KOUZARIDES, T. 2003. EMSY links the
BRCA2 pathway to sporadic breast and ovarian cancer. Cell, 115, 523-35.
HUI, R., BALL, J. R., MACMILLAN, R. D., KENNY, F. S., PRALL, O. W.,
CAMPBELL, D. H., CORNISH, A. L., MCCLELLAND, R. A., DALY, R. J.,

References
168
FORBES, J. F., BLAMEY, R. W., MUSGROVE, E. A., ROBERTSON, J. F.,
NICHOLSON, R. I. & SUTHERLAND, R. L. 1998. EMS1 gene expression in
primary breast cancer: relationship to cyclin D1 and oestrogen receptor
expression and patient survival. Oncogene, 17, 1053-9.
HUI, R., CAMPBELL, D. H., LEE, C. S., MCCAUL, K., HORSFALL, D. J.,
MUSGROVE, E. A., DALY, R. J., SESHADRI, R. & SUTHERLAND, R. L.
1997. EMS1 amplification can occur independently of CCND1 or INT-2
amplification at 11q13 and may identify different phenotypes in primary breast
cancer. Oncogene, 15, 1617-23.
HUTCHESON, I. R., KNOWLDEN, J. M., MADDEN, T. A., BARROW, D., GEE, J.
M., WAKELING, A. E. & NICHOLSON, R. I. 2003. Oestrogen receptor-
mediated modulation of the EGFR/MAPK pathway in tamoxifen-resistant MCF-
7 cells. Breast Cancer Res Treat, 81, 81-93.
IBRAHIM, E. M., KAZKAZ, G. A., AL-MANSOUR, M. M. & AL-FOHEIDI, M. E.
2015. The predictive and prognostic role of phosphatase phosphoinositol-3 (PI3)
kinase (PIK3CA) mutation in HER2-positive breast cancer receiving HER2-
targeted therapy: a meta-analysis. Breast Cancer Res Treat, 152, 463-76.
IGCI, Y. Z., BOZGEYIK, E., BORAZAN, E., PALA, E., SUNER, A., ULASLI, M.,
GURSES, S. A., YUMRUTAS, O., BALIK, A. A. & IGCI, M. 2015. Expression
profiling of SCN8A and NDUFC2 genes in colorectal carcinoma. Exp Oncol,
37, 77-80.
JAIN, M., NILSSON, R., SHARMA, S., MADHUSUDHAN, N., KITAMI, T.,
SOUZA, A. L., KAFRI, R., KIRSCHNER, M. W., CLISH, C. B. & MOOTHA,
V. K. 2012. Metabolite profiling identifies a key role for glycine in rapid cancer
cell proliferation. Science, 336, 1040-4.
JAMES, P., HALLADAY, J. & CRAIG, E. A. 1996. Genomic libraries and a host strain
designed for highly efficient two-hybrid selection in yeast. Genetics, 144, 1425-
36.
JEMAL, A., BRAY, F., CENTER, M. M., FERLAY, J., WARD, E. & FORMAN, D.
2011. Global cancer statistics. CA Cancer J Clin, 61, 69-90.
JENSEN, E. V. 1962. On the mechanism of estrogen action. Perspect Biol Med, 6, 47-
59.
JENSEN, E. V., SUZUKI, T., KAWASHIMA, T., STUMPF, W. E., JUNGBLUT, P.
W. & DESOMBRE, E. R. 1968. A two-step mechanism for the interaction of
estradiol with rat uterus. Proc Natl Acad Sci U S A, 59, 632-8.

References
169
JENSEN, L. J., KUHN, M., STARK, M., CHAFFRON, S., CREEVEY, C., MULLER,
J., DOERKS, T., JULIEN, P., ROTH, A., SIMONOVIC, M., BORK, P. & VON
MERING, C. 2009. STRING 8--a global view on proteins and their functional
interactions in 630 organisms. Nucleic Acids Res, 37, D412-6.
JIANG, J., QI, Y. X., ZHANG, P., GU, W. T., YAN, Z. Q., SHEN, B. R., YAO, Q. P.,
KONG, H., CHIEN, S. & JIANG, Z. L. 2013. Involvement of Rab28 in NF-
kappaB nuclear transport in endothelial cells. PLoS One, 8, e56076.
JIAO, Q., WU, A., SHAO, G., PENG, H., WANG, M., JI, S., LIU, P. & ZHANG, J.
2014. The latest progress in research on triple negative breast cancer (TNBC):
risk factors, possible therapeutic targets and prognostic markers. J Thorac Dis,
6, 1329-35.
JORDAN, V. C. 2004. Selective estrogen receptor modulation: concept and
consequences in cancer. Cancer Cell, 5, 207-13.
JUNG, Y., ABDEL-FATAH, T. M., CHAN, S. Y., NOLAN, C. C., GREEN, A. R.,
ELLIS, I. O., LI, L., HUANG, B., LU, J., XU, B., CHEN, L., MA, R. Z.,
ZHANG, M., WANG, J., WU, Z., ZHU, T., PERRY, J. K., LOBIE, P. E. &
LIU, D. X. 2013. SHON is a novel estrogen-regulated oncogene in mammary
carcinoma that predicts patient response to endocrine therapy. Cancer Res, 73,
6951-62.
KALIDAS, M. & BROWN, P. 2005. Aromatase inhibitors for the treatment and
prevention of breast cancer. Clinical breast cancer, 6, 27-37.
KARLSEDER, J., ZEILLINGER, R., SCHNEEBERGER, C., CZERWENKA, K.,
SPEISER, P., KUBISTA, E., BIRNBAUM, D., GAUDRAY, P. & THEILLET,
C. 1994. Patterns of DNA amplification at band q13 of chromosome 11 in
human breast cancer. Genes Chromosomes Cancer, 9, 42-8.
KASTRATI, I., CANESTRARI, E. & FRASOR, J. 2015. PHLDA1 expression is
controlled by an estrogen receptor-NFkappaB-miR-181 regulatory loop and is
essential for formation of ER+ mammospheres. Oncogene, 34, 2309-16.
KAWALEC, P., LOPUCH, S. & MIKRUT, A. 2015. Effectiveness of targeted therapy
in patients with previously untreated metastatic breast cancer: a systematic
review and meta-analysis. Clin Breast Cancer, 15, 90-100 e1.
KE, Y., WU, D., PRINCEN, F., NGUYEN, T., PANG, Y., LESPERANCE, J.,
MULLER, W. J., OSHIMA, R. G. & FENG, G. S. 2007. Role of Gab2 in
mammary tumorigenesis and metastasis. Oncogene, 26, 4951-60.

References
170
KEILTY, D., BUCHANAN, M., NTAPOLIAS, K., ALEYNIKOVA, O., TU, D., LI,
X., SHEPHERD, L., BRAMWELL, V. & BASIK, M. 2013. RSF1 and not
cyclin D1 gene amplification may predict lack of benefit from adjuvant
tamoxifen in high-risk pre-menopausal women in the MA.12 randomized
clinical trial. PLoS One, 8, e81740.
KENERSON, H. L., AICHER, L. D., TRUE, L. D. & YEUNG, R. S. 2002. Activated
mammalian target of rapamycin pathway in the pathogenesis of tuberous
sclerosis complex renal tumors. Cancer Res, 62, 5645-50.
KENNEDY, A. L., MORTON, J. P., MANOHARAN, I., NELSON, D. M.,
JAMIESON, N. B., PAWLIKOWSKI, J. S., MCBRYAN, T., DOYLE, B.,
MCKAY, C., OIEN, K. A., ENDERS, G. H., ZHANG, R., SANSOM, O. J. &
ADAMS, P. D. 2011. Activation of the PIK3CA/AKT pathway suppresses
senescence induced by an activated RAS oncogene to promote tumorigenesis.
Mol Cell, 42, 36-49.
KENNY, F. S., HUI, R., MUSGROVE, E. A., GEE, J. M., BLAMEY, R. W.,
NICHOLSON, R. I., SUTHERLAND, R. L. & ROBERTSON, J. F. 1999.
Overexpression of cyclin D1 messenger RNA predicts for poor prognosis in
estrogen receptor-positive breast cancer. Clin Cancer Res, 5, 2069-76.
KILKER, R. L., HARTL, M. W., RUTHERFORD, T. M. & PLANAS-SILVA, M. D.
2004. Cyclin D1 expression is dependent on estrogen receptor function in
tamoxifen-resistant breast cancer cells. J Steroid Biochem Mol Biol, 92, 63-71.
KIM, H. S., PARK, I., CHO, H. J., GWAK, G., YANG, K., BAE, B. N., KIM, K. W.,
HAN, S., KIM, H. J. & KIM, Y. D. 2012. Analysis of the potent prognostic
factors in luminal-type breast cancer. J Breast Cancer, 15, 401-6.
KIM, J. H., JUNG, S. H., BAE, J. S., LEE, H. S., YIM, S. H., PARK, S. Y., BANG, S.
Y., HU, H. J., SHIN, H. D., BAE, S. C. & CHUNG, Y. J. 2013. Deletion
variants of RABGAP1L, 10q21.3, and C4 are associated with the risk of
systemic lupus erythematosus in Korean women. Arthritis Rheum, 65, 1055-63.
KIRKEGAARD, T., WITTON, C. J., MCGLYNN, L. M., TOVEY, S. M., DUNNE, B.,
LYON, A. & BARTLETT, J. M. 2005. AKT activation predicts outcome in
breast cancer patients treated with tamoxifen. J Pathol, 207, 139-46.
KLEIN-HITPASS, L., RYFFEL, G. U., HEITLINGER, E. & CATO, A. C. 1988. A 13
bp palindrome is a functional estrogen responsive element and interacts
specifically with estrogen receptor. Nucleic Acids Res, 16, 647-63.

References
171
KNOWLDEN, J. M., HUTCHESON, I. R., JONES, H. E., MADDEN, T., GEE, J. M.,
HARPER, M. E., BARROW, D., WAKELING, A. E. & NICHOLSON, R. I.
2003. Elevated levels of epidermal growth factor receptor/c-erbB2 heterodimers
mediate an autocrine growth regulatory pathway in tamoxifen-resistant MCF-7
cells. Endocrinology, 144, 1032-44.
KO, B. S., CHANG, T. C., CHEN, C. H., LIU, C. C., KUO, C. C., HSU, C., SHEN, Y.
C., SHEN, T. L., GOLUBOVSKAYA, V. M., CHANG, C. C., SHYUE, S. K. &
LIOU, J. Y. 2010. Bortezomib suppresses focal adhesion kinase expression via
interrupting nuclear factor-kappa B. Life Sci, 86, 199-206.
KOCH, L. 2014. Cancer genomics: Non-coding mutations in the driver seat. Nat Rev
Genet, 15, 574-5.
KOK, M. & LINN, S. C. 2010. Gene expression profiles of the oestrogen receptor in
breast cancer. Neth J Med, 68, 291-302.
KUILMAN, T., MICHALOGLOU, C., MOOI, W. J. & PEEPER, D. S. 2010. The
essence of senescence. Genes Dev, 24, 2463-79.
KUIPER, G. G., CARLSSON, B., GRANDIEN, K., ENMARK, E., HAGGBLAD, J.,
NILSSON, S. & GUSTAFSSON, J. A. 1997. Comparison of the ligand binding
specificity and transcript tissue distribution of estrogen receptors alpha and beta.
Endocrinology, 138, 863-70.
KUIPER, G. G., ENMARK, E., PELTO-HUIKKO, M., NILSSON, S. &
GUSTAFSSON, J. A. 1996. Cloning of a novel receptor expressed in rat
prostate and ovary. Proc Natl Acad Sci U S A, 93, 5925-30.
KUMAR, V., GREEN, S., STACK, G., BERRY, M., JIN, J. R. & CHAMBON, P.
1987. Functional domains of the human estrogen receptor. Cell, 51, 941-51.
KUSHNER, P. J., AGARD, D. A., GREENE, G. L., SCANLAN, T. S., SHIAU, A. K.,
UHT, R. M. & WEBB, P. 2000. Estrogen receptor pathways to AP-1. J Steroid
Biochem Mol Biol, 74, 311-7.
KWAN, M. L., HAQUE, R., LEE, V. S., JOANIE CHUNG, W. L., AVILA, C. C.,
CLANCY, H. A., QUINN, V. P. & KUSHI, L. H. 2012. Validation of AJCC
TNM staging for breast tumors diagnosed before 2004 in cancer registries.
Cancer Causes Control, 23, 1587-91.
KWEK, S. S., ROY, R., ZHOU, H., CLIMENT, J., MARTINEZ-CLIMENT, J. A.,
FRIDLYAND, J. & ALBERTSON, D. G. 2009. Co-amplified genes at 8p12 and

References
172
11q13 in breast tumors cooperate with two major pathways in oncogenesis.
Oncogene, 28, 1892-903.
LABAT-MOLEUR, F., GUILLERMET, C., LORIMIER, P., ROBERT, C.,
LANTUEJOUL, S., BRAMBILLA, E. & NEGOESCU, A. 1998. TUNEL
apoptotic cell detection in tissue sections: critical evaluation and improvement. J
Histochem Cytochem, 46, 327-34.
LANFRANCHI, A. 2014. Normal breast physiology: the reasons hormonal
contraceptives and induced abortion increase breast-cancer risk. Issues Law
Med, 29, 135-46.
LAVINSKY, R. M., JEPSEN, K., HEINZEL, T., TORCHIA, J., MULLEN, T. M.,
SCHIFF, R., DEL-RIO, A. L., RICOTE, M., NGO, S., GEMSCH, J.,
HILSENBECK, S. G., OSBORNE, C. K., GLASS, C. K., ROSENFELD, M. G.
& ROSE, D. W. 1998. Diverse signaling pathways modulate nuclear receptor
recruitment of N-CoR and SMRT complexes. Proc Natl Acad Sci U S A, 95,
2920-5.
LEE, H., JIANG, F., WANG, Q., NICOSIA, S. V., YANG, J., SU, B. & BAI, W. 2000.
MEKK1 activation of human estrogen receptor alpha and stimulation of the
agonistic activity of 4-hydroxytamoxifen in endometrial and ovarian cancer
cells. Mol Endocrinol, 14, 1882-96.
LEHN, S., FERNO, M., JIRSTROM, K., RYDEN, L. & LANDBERG, G. 2011. A non-
functional retinoblastoma tumor suppressor (RB) pathway in premenopausal
breast cancer is associated with resistance to tamoxifen. Cell Cycle, 10, 956-62.
LENTZE, N. & AUERBACH, D. 2008. The yeast two-hybrid system and its role in
drug discovery. Expert Opin Ther Targets, 12, 505-15.
LEVIN, E. R. 1999. Cellular Functions of the Plasma Membrane Estrogen Receptor.
Trends Endocrinol Metab, 10, 374-377.
LEVIN, E. R. & PIETRAS, R. J. 2008. Estrogen receptors outside the nucleus in breast
cancer. Breast Cancer Res Treat, 108, 351-61.
LI, F., ADAM, L., VADLAMUDI, R. K., ZHOU, H., SEN, S., CHERNOFF, J.,
MANDAL, M. & KUMAR, R. 2002. p21-activated kinase 1 interacts with and
phosphorylates histone H3 in breast cancer cells. EMBO Rep, 3, 767-73.
LI, Z., WANG, N., FANG, J., HUANG, J., TIAN, F., LI, C. & XIE, F. 2012. Role of
PKC-ERK signaling in tamoxifen-induced apoptosis and tamoxifen resistance in
human breast cancer cells. Oncol Rep, 27, 1879-86.

References
173
LIANG, P. I., WU, L. C., SHEU, J. J., WU, T. F., SHEN, K. H., WANG, Y. H., WU,
W. R., SHIUE, Y. L., HUANG, H. Y., HSU, H. P., CHEN, Y. H., CHEN, L. T.,
LI, C. F. & LIAO, A. C. 2012. Rsf-1/HBXAP overexpression is independent of
gene amplification and is associated with poor outcome in patients with urinary
bladder urothelial carcinoma. J Clin Pathol, 65, 802-7.
LIN, P. L., HAO, Y., XIE, J., LI, N., OHASHI, E., KOO, V. & WU, E. Q. 2015. Real-
world effectiveness of everolimus-based therapy versus endocrine monotherapy
and chemotherapy in patients of HR+/HER2- breast cancer with liver metastasis
in the USA. Expert Opin Pharmacother, 1-11.
LIU, C., WANG, L., CHEN, W., ZHAO, S., YIN, C., LIN, Y., JIANG, A. & ZHANG,
P. 2015. USP35 activated by miR let-7a inhibits cell proliferation and NF-
kappaB activation through stabilization of ABIN-2. Oncotarget.
LIU, S., DONG, Q. & WANG, E. 2012. Rsf-1 overexpression correlates with poor
prognosis and cell proliferation in colon cancer. Tumour Biol, 33, 1485-91.
LIVAK, K. J. & SCHMITTGEN, T. D. 2001. Analysis of relative gene expression data
using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods,
25, 402-8.
LLEONART, M. E., ARTERO-CASTRO, A. & KONDOH, H. 2009. Senescence
induction; a possible cancer therapy. Mol Cancer, 8, 3.
LONG, J., OU, C., XIA, H., ZHU, Y. & LIU, D. 2015. MiR-503 inhibited cell
proliferation of human breast cancer cells by suppressing CCND1 expression.
Tumour Biol.
LOWE, S. W., CEPERO, E. & EVAN, G. 2004. Intrinsic tumour suppression. Nature,
432, 307-15.
LUNDGREN, K., BROWN, M., PINEDA, S., CUZICK, J., SALTER, J., ZABAGLO,
L., HOWELL, A., DOWSETT, M., LANDBERG, G. & TRANS, A. I. 2012.
Effects of cyclin D1 gene amplification and protein expression on time to
recurrence in postmenopausal breast cancer patients treated with anastrozole or
tamoxifen: a TransATAC study. Breast Cancer Res, 14, R57.
LUNDGREN, K., HOLM, K., NORDENSKJOLD, B., BORG, A. & LANDBERG, G.
2008. Gene products of chromosome 11q and their association with CCND1
gene amplification and tamoxifen resistance in premenopausal breast cancer.
Breast Cancer Res, 10, R81.

References
174
LUO, M. L., SHEN, X. M., ZHANG, Y., WEI, F., XU, X., CAI, Y., ZHANG, X., SUN,
Y. T., ZHAN, Q. M., WU, M. & WANG, M. R. 2006. Amplification and
overexpression of CTTN (EMS1) contribute to the metastasis of esophageal
squamous cell carcinoma by promoting cell migration and anoikis resistance.
Cancer Res, 66, 11690-9.
LUPORSI, E., ANDRE, F., SPYRATOS, F., MARTIN, P. M., JACQUEMIER, J.,
PENAULT-LLORCA, F., TUBIANA-MATHIEU, N., SIGAL-ZAFRANI, B.,
ARNOULD, L., GOMPEL, A., EGELE, C., POULET, B., CLOUGH, K. B.,
CROUET, H., FOURQUET, A., LEFRANC, J. P., MATHELIN, C., ROUYER,
N., SERIN, D., SPIELMANN, M., HAUGH, M., CHENARD, M. P., BRAIN,
E., DE CREMOUX, P. & BELLOCQ, J. P. 2012. Ki-67: level of evidence and
methodological considerations for its role in the clinical management of breast
cancer: analytical and critical review. Breast Cancer Res Treat, 132, 895-915.
MA, X., DING, W., WANG, J., WU, G., ZHANG, H., YIN, J., ZHOU, L. & LI, D.
2012. LOC66273 isoform 2, a novel protein highly expressed in white adipose
tissue, induces adipogenesis in 3T3-L1 cells. J Nutr, 142, 448-55.
MACGREGOR, J. I. & JORDAN, V. C. 1998. Basic guide to the mechanisms of
antiestrogen action. Pharmacol Rev, 50, 151-96.
MAGUIRE, S. L., LEONIDOU, A., WAI, P., MARCHIO, C., NG, C. K., SAPINO, A.,
SALOMON, A. V., REIS-FILHO, J. S., WEIGELT, B. & NATRAJAN, R. C.
2015. SF3B1 mutations constitute a novel therapeutic target in breast cancer. J
Pathol, 235, 571-80.
MAJUMDER, P. K., GRISANZIO, C., O'CONNELL, F., BARRY, M., BRITO, J. M.,
XU, Q., GUNEY, I., BERGER, R., HERMAN, P., BIKOFF, R., FEDELE, G.,
BAEK, W. K., WANG, S., ELLWOOD-YEN, K., WU, H., SAWYERS, C. L.,
SIGNORETTI, S., HAHN, W. C., LODA, M. & SELLERS, W. R. 2008. A
prostatic intraepithelial neoplasia-dependent p27 Kip1 checkpoint induces
senescence and inhibits cell proliferation and cancer progression. Cancer Cell,
14, 146-55.
MALHOTRA, G. K., ZHAO, X., BAND, H. & BAND, V. 2010. Histological,
molecular and functional subtypes of breast cancers. Cancer Biol Ther, 10, 955-
60.
MAO, T. L., HSU, C. Y., YEN, M. J., GILKS, B., SHEU, J. J., GABRIELSON, E.,
VANG, R., COPE, L., KURMAN, R. J., WANG, T. L. & SHIH IE, M. 2006.
Expression of Rsf-1, a chromatin-remodeling gene, in ovarian and breast
carcinoma. Hum Pathol, 37, 1169-75.

References
175
MARTINEZ, S. R., YOUNG, S. E., GIULIANO, A. E. & BILCHIK, A. J. 2006. The
utility of estrogen receptor, progesterone receptor, and Her-2/neu status to
predict survival in patients undergoing hepatic resection for breast cancer
metastases. Am J Surg, 191, 281-3.
MCGUIRE, W. L. 1975. Endocrine therapy of breast cancer. Annu Rev Med, 26, 353-
63.
MCKENNA, N. J., LANZ, R. B. & O'MALLEY, B. W. 1999. Nuclear receptor
coregulators: cellular and molecular biology. Endocr Rev, 20, 321-44.
MENARD, R. E., JOVANOVSKI, A. P. & MATTINGLY, R. R. 2005. Active p21-
activated kinase 1 rescues MCF10A breast epithelial cells from undergoing
anoikis. Neoplasia, 7, 638-45.
MENASCE, L. P., WHITE, G. R., HARRISON, C. J. & BOYLE, J. M. 1993.
Localization of the estrogen receptor locus (ESR) to chromosome 6q25.1 by
FISH and a simple post-FISH banding technique. Genomics, 17, 263-5.
MILLS, D., GORDON, E. J., CASANO, A., LAHTI, S. M., NGUYEN, T., PRESTON,
A., TONDRE, J., WU, K., YANASE, T., CHAN, H., CHIA, D., ESFANDIARI,
M., HIMMEL, T. & LOVE, S. M. 2011. The physiology of the normal human
breast: an exploratory study. J Physiol Biochem, 67, 621-7.
MITRA, S., CHENG, K. W. & MILLS, G. B. 2011. Rab GTPases implicated in
inherited and acquired disorders. Semin Cell Dev Biol, 22, 57-68.
MOBASHERI, A., AIRLEY, R., HEWITT, S. M. & MARPLES, D. 2005.
Heterogeneous expression of the aquaporin 1 (AQP1) water channel in tumors
of the prostate, breast, ovary, colon and lung: a study using high density multiple
human tumor tissue microarrays. Int J Oncol, 26, 1149-58.
NADAL, R., SALIDO, M., NONELL, L., RODRIGUEZ-RIVERA, M.,
PUIGDECANET, E., GARCIA-PUCHE, J. L., MACIA, M., COROMINAS, J.
M., SERRANO, M. J., LORENTE, J. A. & SOLE, F. 2015. Combined analysis
of copy number alterations by single-nucleotide polymorphism array and MYC
status in non-metastatic breast cancer patients: comparison according to the
circulating tumor cell status. Tumour Biol, 36, 711-8.
NAKADA, S., CHEN, G. I., GINGRAS, A. C. & DUROCHER, D. 2008. PP4 is a
gamma H2AX phosphatase required for recovery from the DNA damage
checkpoint. EMBO Rep, 9, 1019-26.

References
176
NAKSHATRI, H. & BADVE, S. 2007. FOXA1 as a therapeutic target for breast cancer.
Expert Opin Ther Targets, 11, 507-14.
NEGOESCU, A., GUILLERMET, C., LORIMIER, P., BRAMBILLA, E. & LABAT-
MOLEUR, F. 1998. Importance of DNA fragmentation in apoptosis with regard
to TUNEL specificity. Biomed Pharmacother, 52, 252-8.
NEVE, R. M., CHIN, K., FRIDLYAND, J., YEH, J., BAEHNER, F. L., FEVR, T.,
CLARK, L., BAYANI, N., COPPE, J. P., TONG, F., SPEED, T., SPELLMAN,
P. T., DEVRIES, S., LAPUK, A., WANG, N. J., KUO, W. L., STILWELL, J.
L., PINKEL, D., ALBERTSON, D. G., WALDMAN, F. M., MCCORMICK, F.,
DICKSON, R. B., JOHNSON, M. D., LIPPMAN, M., ETHIER, S., GAZDAR,
A. & GRAY, J. W. 2006. A collection of breast cancer cell lines for the study of
functionally distinct cancer subtypes. Cancer Cell, 10, 515-27.
NEVILLE, M. C., MCFADDEN, T. B. & FORSYTH, I. 2002. Hormonal regulation of
mammary differentiation and milk secretion. J Mammary Gland Biol Neoplasia,
7, 49-66.
NICHOLSON, D. W., ALI, A., THORNBERRY, N. A., VAILLANCOURT, J. P.,
DING, C. K., GALLANT, M., GAREAU, Y., GRIFFIN, P. R., LABELLE, M.,
LAZEBNIK, Y. A. & ET AL. 1995. Identification and inhibition of the
ICE/CED-3 protease necessary for mammalian apoptosis. Nature, 376, 37-43.
NICHOLSON, R. I., HUTCHESON, I. R., HISCOX, S. E., KNOWLDEN, J. M.,
GILES, M., BARROW, D. & GEE, J. M. 2005. Growth factor signalling and
resistance to selective oestrogen receptor modulators and pure anti-oestrogens:
the use of anti-growth factor therapies to treat or delay endocrine resistance in
breast cancer. Endocr Relat Cancer, 12 Suppl 1, S29-36.
NIELSEN, T. O., PARKER, J. S., LEUNG, S., VODUC, D., EBBERT, M., VICKERY,
T., DAVIES, S. R., SNIDER, J., STIJLEMAN, I. J., REED, J., CHEANG, M.
C., MARDIS, E. R., PEROU, C. M., BERNARD, P. S. & ELLIS, M. J. 2010. A
comparison of PAM50 intrinsic subtyping with immunohistochemistry and
clinical prognostic factors in tamoxifen-treated estrogen receptor-positive breast
cancer. Clin Cancer Res, 16, 5222-32.
NILSSON, R., JAIN, M., MADHUSUDHAN, N., SHEPPARD, N. G.,
STRITTMATTER, L., KAMPF, C., HUANG, J., ASPLUND, A. & MOOTHA,
V. K. 2014. Metabolic enzyme expression highlights a key role for MTHFD2
and the mitochondrial folate pathway in cancer. Nat Commun, 5, 3128.
NOGUEIRA, V., PARK, Y., CHEN, C. C., XU, P. Z., CHEN, M. L., TONIC, I.,
UNTERMAN, T. & HAY, N. 2008. Akt determines replicative senescence and

References
177
oxidative or oncogenic premature senescence and sensitizes cells to oxidative
apoptosis. Cancer Cell, 14, 458-70.
NOVICK, P. & ZERIAL, M. 1997. The diversity of Rab proteins in vesicle transport.
Curr Opin Cell Biol, 9, 496-504.
OIDA, K., MATSUDA, A., JUNG, K., XIA, Y., JANG, H., AMAGAI, Y., AHN, G.,
NISHIKAWA, S., ISHIZAKA, S., JENSEN-JAROLIM, E., MATSUDA, H. &
TANAKA, A. 2014. Nuclear factor-kB plays a critical role in both intrinsic and
acquired resistance against endocrine therapy in human breast cancer cells. Sci
Rep, 4, 4057.
OKADA, S., MISAKA, T., TANAKA, Y., MATSUMOTO, I., ISHIBASHI, K.,
SASAKI, S. & ABE, K. 2008. Aquaporin-11 knockout mice and polycystic
kidney disease animals share a common mechanism of cyst formation. FASEB J,
22, 3672-84.
ORMANDY, C. J., MUSGROVE, E. A., HUI, R., DALY, R. J. & SUTHERLAND, R.
L. 2003. Cyclin D1, EMS1 and 11q13 amplification in breast cancer. Breast
cancer research and treatment, 78, 323-35.
OSBORNE, C. K., BARDOU, V., HOPP, T. A., CHAMNESS, G. C., HILSENBECK,
S. G., FUQUA, S. A., WONG, J., ALLRED, D. C., CLARK, G. M. & SCHIFF,
R. 2003. Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-
2/neu in tamoxifen resistance in breast cancer. J Natl Cancer Inst, 95, 353-61.
OSBORNE, C. K. & SCHIFF, R. 2011. Mechanisms of endocrine resistance in breast
cancer. Annu Rev Med, 62, 233-47.
OYAMA, K., OKAWA, T., NAKAGAWA, H., TAKAOKA, M., ANDL, C. D., KIM,
S. H., KLEIN-SZANTO, A., DIEHL, J. A., HERLYN, M., EL-DEIRY, W. &
RUSTGI, A. K. 2007. AKT induces senescence in primary esophageal epithelial
cells but is permissive for differentiation as revealed in organotypic culture.
Oncogene, 26, 2353-64.
PAGE, C., LIN, H. J., JIN, Y., CASTLE, V. P., NUNEZ, G., HUANG, M. & LIN, J.
2000. Overexpression of Akt/AKT can modulate chemotherapy-induced
apoptosis. Anticancer Res, 20, 407-16.
PAPLOMATA, E. & O'REGAN, R. 2014. The PI3K/AKT/mTOR pathway in breast
cancer: targets, trials and biomarkers. Ther Adv Med Oncol, 6, 154-66.

References
178
PARK, J. H., KIM, J. J. & BAE, Y. S. 2013. Involvement of PI3K-AKT-mTOR
pathway in protein kinase CKII inhibition-mediated senescence in human colon
cancer cells. Biochem Biophys Res Commun, 433, 420-5.
PARKER, J. S., MULLINS, M., CHEANG, M. C., LEUNG, S., VODUC, D.,
VICKERY, T., DAVIES, S., FAURON, C., HE, X., HU, Z., QUACKENBUSH,
J. F., STIJLEMAN, I. J., PALAZZO, J., MARRON, J. S., NOBEL, A. B.,
MARDIS, E., NIELSEN, T. O., ELLIS, M. J., PEROU, C. M. & BERNARD, P.
S. 2009. Supervised risk predictor of breast cancer based on intrinsic subtypes. J
Clin Oncol, 27, 1160-7.
PARKER, M. G. 1993. Steroid and related receptors. Curr Opin Cell Biol, 5, 499-504.
PEREZ-TENORIO, G., BERGLUND, F., ESGUERRA MERCA, A.,
NORDENSKJOLD, B., RUTQVIST, L. E., SKOOG, L. & STAL, O. 2006.
Cytoplasmic p21WAF1/CIP1 correlates with Akt activation and poor response
to tamoxifen in breast cancer. Int J Oncol, 28, 1031-42.
PERILLO, B., SASSO, A., ABBONDANZA, C. & PALUMBO, G. 2000. 17beta-
estradiol inhibits apoptosis in MCF-7 cells, inducing bcl-2 expression via two
estrogen-responsive elements present in the coding sequence. Mol Cell Biol, 20,
2890-901.
PEROU, C. M., SORLIE, T., EISEN, M. B., VAN DE RIJN, M., JEFFREY, S. S.,
REES, C. A., POLLACK, J. R., ROSS, D. T., JOHNSEN, H., AKSLEN, L. A.,
FLUGE, O., PERGAMENSCHIKOV, A., WILLIAMS, C., ZHU, S. X.,
LONNING, P. E., BORRESEN-DALE, A. L., BROWN, P. O. & BOTSTEIN,
D. 2000. Molecular portraits of human breast tumours. Nature, 406, 747-52.
PHIZICKY, E. M. & FIELDS, S. 1995. Protein-protein interactions: methods for
detection and analysis. Microbiol Rev, 59, 94-123.
PICCART-GEBHART, M. J., PROCTER, M., LEYLAND-JONES, B.,
GOLDHIRSCH, A., UNTCH, M., SMITH, I., GIANNI, L., BASELGA, J.,
BELL, R., JACKISCH, C., CAMERON, D., DOWSETT, M., BARRIOS, C. H.,
STEGER, G., HUANG, C. S., ANDERSSON, M., INBAR, M., LICHINITSER,
M., LANG, I., NITZ, U., IWATA, H., THOMSSEN, C., LOHRISCH, C.,
SUTER, T. M., RUSCHOFF, J., SUTO, T., GREATOREX, V., WARD, C.,
STRAEHLE, C., MCFADDEN, E., DOLCI, M. S., GELBER, R. D. &
HERCEPTIN ADJUVANT TRIAL STUDY, T. 2005. Trastuzumab after
adjuvant chemotherapy in HER2-positive breast cancer. N Engl J Med, 353,
1659-72.
PRAT, A., PARKER, J. S., KARGINOVA, O., FAN, C., LIVASY, C.,
HERSCHKOWITZ, J. I., HE, X. & PEROU, C. M. 2010. Phenotypic and

References
179
molecular characterization of the claudin-low intrinsic subtype of breast cancer.
Breast Cancer Res, 12, R68.
PRAT, A. & PEROU, C. M. 2011. Deconstructing the molecular portraits of breast
cancer. Mol Oncol, 5, 5-23.
RAKHA, E. A., EL-SAYED, M. E., LEE, A. H., ELSTON, C. W., GRAINGE, M. J.,
HODI, Z., BLAMEY, R. W. & ELLIS, I. O. 2008a. Prognostic significance of
Nottingham histologic grade in invasive breast carcinoma. J Clin Oncol, 26,
3153-8.
RAKHA, E. A., EL-SAYED, M. E., POWE, D. G., GREEN, A. R., HABASHY, H.,
GRAINGE, M. J., ROBERTSON, J. F., BLAMEY, R., GEE, J., NICHOLSON,
R. I., LEE, A. H. & ELLIS, I. O. 2008b. Invasive lobular carcinoma of the
breast: response to hormonal therapy and outcomes. Eur J Cancer, 44, 73-83.
RAOUF, A., BROWN, L., VRCELJ, N., TO, K., KWOK, W., HUNTSMAN, D. &
EAVES, C. J. 2005. Genomic instability of human mammary epithelial cells
overexpressing a truncated form of EMSY. J Natl Cancer Inst, 97, 1302-6.
RAZANDI, M., PEDRAM, A., PARK, S. T. & LEVIN, E. R. 2003. Proximal events in
signaling by plasma membrane estrogen receptors. J Biol Chem, 278, 2701-12.
REDMOND, A. M., BANE, F. T., STAFFORD, A. T., MCILROY, M., DILLON, M.
F., CROTTY, T. B., HILL, A. D. & YOUNG, L. S. 2009. Coassociation of
estrogen receptor and p160 proteins predicts resistance to endocrine treatment;
SRC-1 is an independent predictor of breast cancer recurrence. Clin Cancer Res,
15, 2098-106.
REIS-FILHO, J. S. & PUSZTAI, L. 2011. Gene expression profiling in breast cancer:
classification, prognostication, and prediction. Lancet, 378, 1812-23.
REMMINGHORST, U., HAY, I. D. & REHM, B. H. 2009. Molecular characterization
of Alg8, a putative glycosyltransferase, involved in alginate polymerisation. J
Biotechnol, 140, 176-83.
REN, J., CHEN, Q. C., JIN, F., WU, H. Z., HE, M., ZHAO, L., YU, Z. J., YAO, W. F.,
MI, X. Y., WANG, E. H. & WEI, M. J. 2014. Overexpression of Rsf-1
correlates with pathological type, p53 status and survival in primary breast
cancer. Int J Clin Exp Pathol, 7, 5595-608.
RIBATTI, D., RANIERI, G., ANNESE, T. & NICO, B. 2014. Aquaporins in cancer.
Biochim Biophys Acta, 1840, 1550-3.

References
180
ROBERTI, M. C., LA STARZA, R., SURACE, C., SIRLETO, P., PINTO, R. M.,
PIERINI, V., CRESCENZI, B., MECUCCI, C. & ANGIONI, A. 2009.
RABGAP1L gene rearrangement resulting from a der(Y)t(Y;1)(q12;q25) in
acute myeloid leukemia arising in a child with Klinefelter syndrome. Virchows
Arch, 454, 311-6.
ROCHEFORT, H., PLATET, N., HAYASHIDO, Y., DEROCQ, D., LUCAS, A.,
CUNAT, S. & GARCIA, M. 1998. Estrogen receptor mediated inhibition of
cancer cell invasion and motility: an overview. J Steroid Biochem Mol Biol, 65,
163-8.
RODRIGUEZ, C., HUGHES-DAVIES, L., VALLES, H., ORSETTI, B., CUNY, M.,
URSULE, L., KOUZARIDES, T. & THEILLET, C. 2004. Amplification of the
BRCA2 pathway gene EMSY in sporadic breast cancer is related to negative
outcome. Clin Cancer Res, 10, 5785-91.
ROMOND, E. H., PEREZ, E. A., BRYANT, J., SUMAN, V. J., GEYER, C. E., JR.,
DAVIDSON, N. E., TAN-CHIU, E., MARTINO, S., PAIK, S., KAUFMAN, P.
A., SWAIN, S. M., PISANSKY, T. M., FEHRENBACHER, L., KUTTEH, L.
A., VOGEL, V. G., VISSCHER, D. W., YOTHERS, G., JENKINS, R. B.,
BROWN, A. M., DAKHIL, S. R., MAMOUNAS, E. P., LINGLE, W. L.,
KLEIN, P. M., INGLE, J. N. & WOLMARK, N. 2005. Trastuzumab plus
adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J
Med, 353, 1673-84.
ROY, P. G., PRATT, N., PURDIE, C. A., BAKER, L., ASHFIELD, A., QUINLAN, P.
& THOMPSON, A. M. 2010. High CCND1 amplification identifies a group of
poor prognosis women with estrogen receptor positive breast cancer. Int J
Cancer, 127, 355-60.
RUDAS, M., LEHNERT, M., HUYNH, A., JAKESZ, R., SINGER, C., LAX, S.,
SCHIPPINGER, W., DIETZE, O., GREIL, R., STIGLBAUER, W., KWASNY,
W., GRILL, R., STIERER, M., GNANT, M. F., FILIPITS, M., AUSTRIAN, B.
& COLORECTAL CANCER STUDY, G. 2008. Cyclin D1 expression in breast
cancer patients receiving adjuvant tamoxifen-based therapy. Clin Cancer Res,
14, 1767-74.
RUSSO, I. H. & RUSSO, J. 1998. Role of hormones in mammary cancer initiation and
progression. J Mammary Gland Biol Neoplasia, 3, 49-61.
SAJI, S. & KIMURA-TSUCHIYA, R. 2015. Combination of molecular-targeted drugs
with endocrine therapy for hormone-resistant breast cancer. Int J Clin Oncol, 20,
268-72.

References
181
SANCHEZ-PEREZ, I., BENITAH, S. A., MARTINEZ-GOMARIZ, M., LACAL, J. C.
& PERONA, R. 2002. Cell stress and MEKK1-mediated c-Jun activation
modulate NFkappaB activity and cell viability. Mol Biol Cell, 13, 2933-45.
SANTARIUS, T., SHIPLEY, J., BREWER, D., STRATTON, M. R. & COOPER, C. S.
2010. A census of amplified and overexpressed human cancer genes. Nat Rev
Cancer, 10, 59-64.
SANTEN, R. J., FAN, P., ZHANG, Z., BAO, Y., SONG, R. X. & YUE, W. 2009.
Estrogen signals via an extra-nuclear pathway involving IGF-1R and EGFR in
tamoxifen-sensitive and -resistant breast cancer cells. Steroids, 74, 586-94.
SANTOS, C., SANZ-PAMPLONA, R., NADAL, E., GRASSELLI, J., PERNAS, S.,
DIENSTMANN, R., MORENO, V., TABERNERO, J. & SALAZAR, R. 2015a.
Intrinsic cancer subtypes--next steps into personalized medicine. Cell Oncol
(Dordr), 38, 3-16.
SANTOS, M., CORREIA-GOMES, C., MARCOS, R., SANTOS, A., A, D. E. M.,
LOPES, C. & DIAS-PEREIRA, P. 2015b. Value of the Nottingham Histological
Grading Parameters and Nottingham Prognostic Index in Canine Mammary
Carcinoma. Anticancer Res, 35, 4219-27.
SARTORIUS, C. A., SHEN, T. & HORWITZ, K. B. 2003. Progesterone receptors A
and B differentially affect the growth of estrogen-dependent human breast tumor
xenografts. Breast Cancer Res Treat, 79, 287-99.
SCANDELLA, E., NAGL, U. O., OEHL, B., BERGMANN, F., GSCHWENTNER, M.,
FURST, J., SCHMARDA, A., RITTER, M., WALDEGGER, S., LANG, F.,
DEETJEN, P. & PAULMICHL, M. 2000. The promoter for constitutive
expression of the human ICln gene CLNS1A. J Biol Chem, 275, 15613-20.
SCARPULLA, R. C. 2011. Metabolic control of mitochondrial biogenesis through the
PGC-1 family regulatory network. Biochim Biophys Acta, 1813, 1269-78.
SCHIFF, R., MASSARWEH, S. A., SHOU, J., BHARWANI, L., MOHSIN, S. K. &
OSBORNE, C. K. 2004. Cross-talk between estrogen receptor and growth factor
pathways as a molecular target for overcoming endocrine resistance. Clin
Cancer Res, 10, 331S-6S.
SCHMITT, F. 2009. HER2+ breast cancer: how to evaluate? Adv Ther, 26 Suppl 1, S1-
8.
SCHRAML, P., SCHWERDTFEGER, G., BURKHALTER, F., RAGGI, A.,
SCHMIDT, D., RUFFALO, T., KING, W., WILBER, K., MIHATSCH, M. J. &

References
182
MOCH, H. 2003. Combined array comparative genomic hybridization and tissue
microarray analysis suggest PAK1 at 11q13.5-q14 as a critical oncogene target
in ovarian carcinoma. Am J Pathol, 163, 985-92.
SCHWABE, J. W., CHAPMAN, L., FINCH, J. T., RHODES, D. & NEUHAUS, D.
1993. DNA recognition by the oestrogen receptor: from solution to the crystal.
Structure, 1, 187-204.
SEN, R. & SMALE, S. T. 2010. Selectivity of the NF-{kappa}B response. Cold Spring
Harb Perspect Biol, 2, a000257.
SENKUS, E., KYRIAKIDES, S., PENAULT-LLORCA, F., POORTMANS, P.,
THOMPSON, A., ZACKRISSON, S. & CARDOSO, F. 2013. Primary breast
cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-
up. Ann Oncol, 24 Suppl 6, vi7-23.
SHARMA, R., SAMANTARAY, S., SHUKLA, N. K. & RALHAN, R. 2003.
Transcriptional gene expression profile of human esophageal squamous cell
carcinoma. Genomics, 81, 481-8.
SHASTRY, M. & YARDLEY, D. A. 2013. Updates in the treatment of basal/triple-
negative breast cancer. Curr Opin Obstet Gynecol, 25, 40-8.
SHEU, J. J., CHOI, J. H., YILDIZ, I., TSAI, F. J., SHAUL, Y., WANG, T. L. & SHIH
IE, M. 2008. The roles of human sucrose nonfermenting protein 2 homologue in
the tumor-promoting functions of Rsf-1. Cancer Res, 68, 4050-7.
SHEU, J. J., GUAN, B., CHOI, J. H., LIN, A., LEE, C. H., HSIAO, Y. T., WANG, T.
L., TSAI, F. J. & SHIH IE, M. 2010. Rsf-1, a chromatin remodeling protein,
induces DNA damage and promotes genomic instability. J Biol Chem, 285,
38260-9.
SHIH IE, M., SHEU, J. J., SANTILLAN, A., NAKAYAMA, K., YEN, M. J.,
BRISTOW, R. E., VANG, R., PARMIGIANI, G., KURMAN, R. J., TROPE, C.
G., DAVIDSON, B. & WANG, T. L. 2005. Amplification of a chromatin
remodeling gene, Rsf-1/HBXAP, in ovarian carcinoma. Proc Natl Acad Sci U S
A, 102, 14004-9.
SHIH, J., BASHIR, B., GUSTAFSON, K. S., ANDRAKE, M., DUNBRACK, R. L.,
GOLDSTEIN, L. J. & BOUMBER, Y. 2015. Cancer Signature Investigation:
ERBB2 (HER2)-Activating Mutation and Amplification-Positive Breast
Carcinoma Mimicking Lung Primary. J Natl Compr Canc Netw, 13, 947-52.

References
183
SHOU, J., MASSARWEH, S., OSBORNE, C. K., WAKELING, A. E., ALI, S.,
WEISS, H. & SCHIFF, R. 2004. Mechanisms of tamoxifen resistance: increased
estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J
Natl Cancer Inst, 96, 926-35.
SKIBINSKI, A. & KUPERWASSER, C. 2015. The origin of breast tumor
heterogeneity. Oncogene.
SLAMON, D. J., CLARK, G. M., WONG, S. G., LEVIN, W. J., ULLRICH, A. &
MCGUIRE, W. L. 1987. Human breast cancer: correlation of relapse and
survival with amplification of the HER-2/neu oncogene. Science, 235, 177-82.
SOBIN, L. H. 2003. TNM: evolution and relation to other prognostic factors. Semin
Surg Oncol, 21, 3-7.
SORICH, M. J., POTTIER, N., PEI, D., YANG, W., KAGER, L., STOCCO, G.,
CHENG, C., PANETTA, J. C., PUI, C. H., RELLING, M. V., CHEOK, M. H.
& EVANS, W. E. 2008. In vivo response to methotrexate forecasts outcome of
acute lymphoblastic leukemia and has a distinct gene expression profile. PLoS
Med, 5, e83.
SORLIE, T., TIBSHIRANI, R., PARKER, J., HASTIE, T., MARRON, J. S., NOBEL,
A., DENG, S., JOHNSEN, H., PESICH, R., GEISLER, S., DEMETER, J.,
PEROU, C. M., LONNING, P. E., BROWN, P. O., BORRESEN-DALE, A. L.
& BOTSTEIN, D. 2003. Repeated observation of breast tumor subtypes in
independent gene expression data sets. Proc Natl Acad Sci U S A, 100, 8418-23.
SPAN, P. N., TJAN-HEIJNEN, V. C., MANDERS, P., BEEX, L. V. & SWEEP, C. G.
2003. Cyclin-E is a strong predictor of endocrine therapy failure in human breast
cancer. Oncogene, 22, 4898-904.
SPIEGEL, J., CROMM, P. M., ITZEN, A., GOODY, R. S., GROSSMANN, T. N. &
WALDMANN, H. 2014. Direct targeting of Rab-GTPase-effector interactions.
Angew Chem Int Ed Engl, 53, 2498-503.
STAL, O., PEREZ-TENORIO, G., AKERBERG, L., OLSSON, B., NORDENSKJOLD,
B., SKOOG, L. & RUTQVIST, L. E. 2003. Akt kinases in breast cancer and the
results of adjuvant therapy. Breast Cancer Res, 5, R37-44.
STENDAHL, M., KRONBLAD, A., RYDEN, L., EMDIN, S., BENGTSSON, N. O. &
LANDBERG, G. 2004. Cyclin D1 overexpression is a negative predictive factor
for tamoxifen response in postmenopausal breast cancer patients. Br J Cancer,
90, 1942-8.

References
184
STENDAHL, M., NILSSON, S., WIGERUP, C., JIRSTROM, K., JONSSON, P. E.,
STAL, O. & LANDBERG, G. 2010. p27Kip1 is a predictive factor for
tamoxifen treatment response but not a prognostic marker in premenopausal
breast cancer patients. Int J Cancer, 127, 2851-8.
STEPHENS, P. J., TARPEY, P. S., DAVIES, H., VAN LOO, P., GREENMAN, C.,
WEDGE, D. C., NIK-ZAINAL, S., MARTIN, S., VARELA, I., BIGNELL, G.
R., YATES, L. R., PAPAEMMANUIL, E., BEARE, D., BUTLER, A.,
CHEVERTON, A., GAMBLE, J., HINTON, J., JIA, M., JAYAKUMAR, A.,
JONES, D., LATIMER, C., LAU, K. W., MCLAREN, S., MCBRIDE, D. J.,
MENZIES, A., MUDIE, L., RAINE, K., RAD, R., CHAPMAN, M. S.,
TEAGUE, J., EASTON, D., LANGEROD, A., LEE, M. T., SHEN, C. Y., TEE,
B. T., HUIMIN, B. W., BROEKS, A., VARGAS, A. C., TURASHVILI, G.,
MARTENS, J., FATIMA, A., MIRON, P., CHIN, S. F., THOMAS, G.,
BOYAULT, S., MARIANI, O., LAKHANI, S. R., VAN DE VIJVER, M., VAN
'T VEER, L., FOEKENS, J., DESMEDT, C., SOTIRIOU, C., TUTT, A.,
CALDAS, C., REIS-FILHO, J. S., APARICIO, S. A., SALOMON, A. V.,
BORRESEN-DALE, A. L., RICHARDSON, A. L., CAMPBELL, P. J.,
FUTREAL, P. A. & STRATTON, M. R. 2012. The landscape of cancer genes
and mutational processes in breast cancer. Nature, 486, 400-4.
SUKEL, M. P., VAN DE POLL-FRANSE, L. V., NIEUWENHUIJZEN, G. A.,
VREUGDENHIL, G., HERINGS, R. M., COEBERGH, J. W. & VOOGD, A. C.
2008. Substantial increase in the use of adjuvant systemic treatment for early
stage breast cancer reflects changes in guidelines in the period 1990-2006 in the
southeastern Netherlands. Eur J Cancer, 44, 1846-54.
SUN, S. C. 2011. Non-canonical NF-kappaB signaling pathway. Cell Res, 21, 71-85.
SWAMINATHAN, G., VARGHESE, B. & FUCHS, S. Y. 2008. Regulation of
prolactin receptor levels and activity in breast cancer. J Mammary Gland Biol
Neoplasia, 13, 81-91.
THOMAS, C. & GUSTAFSSON, J. A. 2011. The different roles of ER subtypes in
cancer biology and therapy. Nat Rev Cancer, 11, 597-608.
TOBIN, N. P. & BERGH, J. 2012. Analysis of Cyclin D1 in Breast Cancer: A Call to
Arms. Curr Breast Cancer Rep, 4, 171-173.
TOLANEY, S. M., BARRY, W. T., DANG, C. T., YARDLEY, D. A., MOY, B.,
MARCOM, P. K., ALBAIN, K. S., RUGO, H. S., ELLIS, M., SHAPIRA, I.,
WOLFF, A. C., CAREY, L. A., OVERMOYER, B. A., PARTRIDGE, A. H.,
GUO, H., HUDIS, C. A., KROP, I. E., BURSTEIN, H. J. & WINER, E. P. 2015.
Adjuvant paclitaxel and trastuzumab for node-negative, HER2-positive breast
cancer. N Engl J Med, 372, 134-41.

References
185
TOPCU, Z. & BORDEN, K. L. 2000. The yeast two-hybrid system and its
pharmaceutical significance. Pharm Res, 17, 1049-55.
TORRE, L. A., BRAY, F., SIEGEL, R. L., FERLAY, J., LORTET-TIEULENT, J. &
JEMAL, A. 2015. Global cancer statistics, 2012. CA Cancer J Clin, 65, 87-108.
TURNER, N., PEARSON, A., SHARPE, R., LAMBROS, M., GEYER, F., LOPEZ-
GARCIA, M. A., NATRAJAN, R., MARCHIO, C., IORNS, E., MACKAY, A.,
GILLETT, C., GRIGORIADIS, A., TUTT, A., REIS-FILHO, J. S. &
ASHWORTH, A. 2010. FGFR1 amplification drives endocrine therapy
resistance and is a therapeutic target in breast cancer. Cancer Res, 70, 2085-94.
UTSUMI, T., KOBAYASHI, N. & HANADA, H. 2007. Recent perspectives of
endocrine therapy for breast cancer. Breast Cancer, 14, 194-9.
VADLAMUDI, R. K., ADAM, L., WANG, R. A., MANDAL, M., NGUYEN, D.,
SAHIN, A., CHERNOFF, J., HUNG, M. C. & KUMAR, R. 2000. Regulatable
expression of p21-activated kinase-1 promotes anchorage-independent growth
and abnormal organization of mitotic spindles in human epithelial breast cancer
cells. J Biol Chem, 275, 36238-44.
VADLAMUDI, R. K., BARNES, C. J., RAYALA, S., LI, F., BALASENTHIL, S.,
MARCUS, S., GOODSON, H. V., SAHIN, A. A. & KUMAR, R. 2005. p21-
activated kinase 1 regulates microtubule dynamics by phosphorylating tubulin
cofactor B. Mol Cell Biol, 25, 3726-36.
VAN LAERE, S. J., VAN DER AUWERA, I., VAN DEN EYNDEN, G. G., VAN
DAM, P., VAN MARCK, E. A., VERMEULEN, P. B. & DIRIX, L. Y. 2007.
NF-kappaB activation in inflammatory breast cancer is associated with
oestrogen receptor downregulation, secondary to EGFR and/or ErbB2
overexpression and MAPK hyperactivation. Br J Cancer, 97, 659-69.
VAZQUEZ, A., TEDESCHI, P. M. & BERTINO, J. R. 2013. Overexpression of the
mitochondrial folate and glycine-serine pathway: a new determinant of
methotrexate selectivity in tumors. Cancer Res, 73, 478-82.
VENDRELL, J. A., BIECHE, I., DESMETZ, C., BADIA, E., TOZLU, S., NGUYEN,
C., NICOLAS, J. C., LIDEREAU, R. & COHEN, P. A. 2005. Molecular
changes associated with the agonist activity of hydroxy-tamoxifen and the
hyper-response to estradiol in hydroxy-tamoxifen-resistant breast cancer cell
lines. Endocr Relat Cancer, 12, 75-92.
VERKMAN, A. S., HARA-CHIKUMA, M. & PAPADOPOULOS, M. C. 2008.
Aquaporins--new players in cancer biology. J Mol Med (Berl), 86, 523-9.

References
186
VERONESI, U., CASCINELLI, N., MARIANI, L., GRECO, M., SACCOZZI, R.,
LUINI, A., AGUILAR, M. & MARUBINI, E. 2002. Twenty-year follow-up of a
randomized study comparing breast-conserving surgery with radical mastectomy
for early breast cancer. N Engl J Med, 347, 1227-32.
VERONESI, U., LUINI, A., DEL VECCHIO, M., GRECO, M., GALIMBERTI, V.,
MERSON, M., RILKE, F., SACCHINI, V., SACCOZZI, R., SAVIO, T. & ET
AL. 1993. Radiotherapy after breast-preserving surgery in women with localized
cancer of the breast. N Engl J Med, 328, 1587-91.
VIEDMA-RODRIGUEZ, R., BAIZA-GUTMAN, L., SALAMANCA-GOMEZ, F.,
DIAZ-ZARAGOZA, M., MARTINEZ-HERNANDEZ, G., RUIZ ESPARZA-
GARRIDO, R., VELAZQUEZ-FLORES, M. A. & ARENAS-ARANDA, D.
2014. Mechanisms associated with resistance to tamoxifen in estrogen receptor-
positive breast cancer (review). Oncol Rep, 32, 3-15.
VIGNALI, M., HASSAN, A. H., NEELY, K. E. & WORKMAN, J. L. 2000. ATP-
dependent chromatin-remodeling complexes. Mol Cell Biol, 20, 1899-910.
VISVADER, J. E. 2009. Keeping abreast of the mammary epithelial hierarchy and
breast tumorigenesis. Genes Dev, 23, 2563-77.
VISVADER, J. E. 2011. Cells of origin in cancer. Nature, 469, 314-22.
VOJTEK, A. B. & HOLLENBERG, S. M. 1995. Ras-Raf interaction: two-hybrid
analysis. Methods Enzymol, 255, 331-42.
WANG, R. A., MAZUMDAR, A., VADLAMUDI, R. K. & KUMAR, R. 2002. P21-
activated kinase-1 phosphorylates and transactivates estrogen receptor-alpha and
promotes hyperplasia in mammary epithelium. EMBO J, 21, 5437-47.
WEIGELT, B., GEYER, F. C. & REIS-FILHO, J. S. 2010. Histological types of breast
cancer: how special are they? Mol Oncol, 4, 192-208.
WILKERSON, P. M. & REIS-FILHO, J. S. 2013. The 11q13-q14 amplicon:
clinicopathological correlations and potential drivers. Genes Chromosomes
Cancer, 52, 333-55.
WILL, C. L., URLAUB, H., ACHSEL, T., GENTZEL, M., WILM, M. &
LUHRMANN, R. 2002. Characterization of novel SF3b and 17S U2 snRNP
proteins, including a human Prp5p homologue and an SF3b DEAD-box protein.
EMBO J, 21, 4978-88.

References
187
WITKIEWICZ, A. K. & KNUDSEN, E. S. 2014. Retinoblastoma tumor suppressor
pathway in breast cancer: prognosis, precision medicine, and therapeutic
interventions. Breast Cancer Res, 16, 207.
WOLFFE, A. P. 2001. Chromatin remodeling: why it is important in cancer. Oncogene,
20, 2988-90.
WU, J., LI, J. & MEN, X. L. 2012. [The role of Thrsp in the regulation of lipogenesis].
Sheng Li Ke Xue Jin Zhan, 43, 393-7.
XIE, J., HAO, Y., LI, N., LIN, P. L., OHASHI, E., KOO, V., SIGNOROVITCH, J. E.,
WU, E. Q. & YARDLEY, D. A. 2015. Comparative effectiveness of
everolimus-based therapy versus endocrine monotherapy among
postmenopausal women with HR+/HER2- metastatic breast cancer: a
retrospective chart review in community oncology practices in the US. Curr
Med Res Opin, 31, 1095-103.
XU, X. & CHEN, J. 2009. One-carbon metabolism and breast cancer: an
epidemiological perspective. J Genet Genomics, 36, 203-14.
XU, X., LU, Z., QIANG, W., VIDIMAR, V., KONG, B., KIM, J. J. & WEI, J. J. 2014.
Inactivation of AKT induces cellular senescence in uterine leiomyoma.
Endocrinology, 155, 1510-9.
YANG, Y. I., AHN, J. H., LEE, K. T., SHIH IE, M. & CHOI, J. H. 2014. RSF1 is a
positive regulator of NF-kappaB-induced gene expression required for ovarian
cancer chemoresistance. Cancer Res, 74, 2258-69.
YERUSHALMI, R., WOODS, R., RAVDIN, P. M., HAYES, M. M. & GELMON, K.
A. 2010. Ki67 in breast cancer: prognostic and predictive potential. Lancet
Oncol, 11, 174-83.
YOUNG, A. R., NARITA, M., FERREIRA, M., KIRSCHNER, K., SADAIE, M.,
DAROT, J. F., TAVARE, S., ARAKAWA, S., SHIMIZU, S., WATT, F. M. &
NARITA, M. 2009. Autophagy mediates the mitotic senescence transition.
Genes Dev, 23, 798-803.
YU, J. H., CUI, Q., JIANG, Y. Y., YANG, W., TASHIRO, S., ONODERA, S. &
IKEJIMA, T. 2007. Pseudolaric acid B induces apoptosis, senescence, and
mitotic arrest in human breast cancer MCF-7. Acta Pharmacol Sin, 28, 1975-83.
ZELLER, K. I., JEGGA, A. G., ARONOW, B. J., O'DONNELL, K. A. & DANG, C. V.
2003. An integrated database of genes responsive to the Myc oncogenic

References
188
transcription factor: identification of direct genomic targets. Genome Biol, 4,
R69.
ZGHAIR, A. N., SINHA, D. K., KASSIM, A., ALFAHAM, M. & SHARMA, A. K.
2015. Differential Gene Expression of BRCA1,ERBB2 and TP53 biomarkers
between Human Breast Tissue and Peripheral Blood Samples of Breast Cancer.
Anticancer Agents Med Chem.
ZHANG, M. H., MAN, H. T., ZHAO, X. D., DONG, N. & MA, S. L. 2014. Estrogen
receptor-positive breast cancer molecular signatures and therapeutic potentials
(Review). Biomed Rep, 2, 41-52.
ZHANG, X., MU, X., HUANG, O., XIE, Z., JIANG, M., GENG, M. & SHEN, K.
2013. Luminal breast cancer cell lines overexpressing ZNF703 are resistant to
tamoxifen through activation of Akt/mTOR signaling. PLoS One, 8, e72053.
ZHENG, J., ALSAADI, T., BLAICHMAN, J., XIE, X., OMEROGLU, A.,
METERISSIAN, S. & MESUROLLE, B. 2013. Invasive ductal carcinoma of the
breast: correlation between tumor grade determined by ultrasound-guided core
biopsy and surgical pathology. AJR Am J Roentgenol, 200, W71-4.
ZHOU, G., MIHINDUKULASURIYA, K. A., MACCORKLE-CHOSNEK, R. A.,
VAN HOOSER, A., HU, M. C., BRINKLEY, B. R. & TAN, T. H. 2002. Protein
phosphatase 4 is involved in tumor necrosis factor-alpha-induced activation of c-
Jun N-terminal kinase. J Biol Chem, 277, 6391-8.
ZHOU, Y., EPPENBERGER-CASTORI, S., EPPENBERGER, U. & BENZ, C. C.
2005. The NFkappaB pathway and endocrine-resistant breast cancer. Endocr
Relat Cancer, 12 Suppl 1, S37-46.
ZHOU, Y., YAU, C., GRAY, J. W., CHEW, K., DAIRKEE, S. H., MOORE, D. H.,
EPPENBERGER, U., EPPENBERGER-CASTORI, S. & BENZ, C. C. 2007.
Enhanced NF kappa B and AP-1 transcriptional activity associated with
antiestrogen resistant breast cancer. BMC Cancer, 7, 59.
ZHU, K., LIU, Q., ZHOU, Y., TAO, C., ZHAO, Z., SUN, J. & XU, H. 2015.
Oncogenes and tumor suppressor genes: comparative genomics and network
perspectives. BMC Genomics, 16 Suppl 7, S8.
ZOU, X., RAY, D., AZIYU, A., CHRISTOV, K., BOIKO, A. D., GUDKOV, A. V. &
KIYOKAWA, H. 2002. Cdk4 disruption renders primary mouse cells resistant
to oncogenic transformation, leading to Arf/p53-independent senescence. Genes
Dev, 16, 2923-34.