molecular pathology of kidney diseases dr. k.w. chan
TRANSCRIPT

Molecular Pathology ofMolecular Pathology ofKidney DiseasesKidney Diseases
Dr. K.W. ChanDr. K.W. Chan

1. Hereditary Kidney 1. Hereditary Kidney DiseasesDiseases
•Adult polycystic diseaseAdult polycystic disease•Infantile polycystic diseaseInfantile polycystic disease•Alport syndromeAlport syndrome

1. Hereditary Kidney 1. Hereditary Kidney DiseasesDiseases
•[Adult polycystic disease][Adult polycystic disease]•Infantile polycystic diseaseInfantile polycystic disease•Alport syndromeAlport syndrome

Characteristics of APKDCharacteristics of APKD
• Gene frequency 1/1000.Gene frequency 1/1000.• Autosomal dominant.Autosomal dominant.• Symptoms onset in middle age.Symptoms onset in middle age.• Large polycystic kidneys.Large polycystic kidneys.• >50% end stage renal failure.>50% end stage renal failure.• A disorder affecting multiple organ A disorder affecting multiple organ
systems.systems.• Genetically heterogeneous.Genetically heterogeneous.

Localization of APKD Localization of APKD genesgenes
• 1986 (Reeders et al)1986 (Reeders et al)– A gene locus of APKD (now called PKD1) was shoA gene locus of APKD (now called PKD1) was sho
wn to be closely linked to the wn to be closely linked to the -globin locus on -globin locus on 16p.16p.
• 1988 (Kimberling)1988 (Kimberling)– Genetic heterogeniety of APKD was discovered.Genetic heterogeniety of APKD was discovered.
• 1992 (Peters et al)1992 (Peters et al)– The PKD2 locus was localized to 4q21-23.The PKD2 locus was localized to 4q21-23.

• 1994 (EPKDC)1994 (EPKDC)– PKD1 identified to be a gene encoding a 14-kb trPKD1 identified to be a gene encoding a 14-kb tr
anscriptanscript– encoding a 4,302 a.a. protein called polycystin-1encoding a 4,302 a.a. protein called polycystin-1
• 1996 (Mochizuki et al)1996 (Mochizuki et al)– PKD2 was cloned and polycystin-2 characterizedPKD2 was cloned and polycystin-2 characterized
• 1997 (Ariza et al)1997 (Ariza et al)– described a 2-generation Spanish family with Pdescribed a 2-generation Spanish family with P
KD in which linkage to the PKD1 and PKD2 loci wKD in which linkage to the PKD1 and PKD2 loci was excluded -> evidence of PKD3as excluded -> evidence of PKD3

Localization of PKD1Localization of PKD1
• A Portuguese family with both APKD aA Portuguese family with both APKD and TSCnd TSC– Father normal karyotypeFather normal karyotype– Mother 46,XX t(16;22)(p13.3;q11.21), suffeMother 46,XX t(16;22)(p13.3;q11.21), suffe
rs APKDrs APKD– Son 45,XY 16pter-p13.3 and 22pter-q11.2Son 45,XY 16pter-p13.3 and 22pter-q11.2
1, suffers TSC and APKD1, suffers TSC and APKD– The breakpoint at 16p13.3 has disrupted The breakpoint at 16p13.3 has disrupted
the the PKD1PKD1

Genetic Heterogeneity of Genetic Heterogeneity of APKDAPKD
Gene Involved Location Proportion
PKD1 16p13.3 80-90%
PKD2 4q21-23 10-15%
PKD3 not mapped rare

Characteristics of Characteristics of PKD1PKD1
• Located on 16p13.3Located on 16p13.3• ~52kb genomic DNA, 14kb mRNA, 46 exons, ~52kb genomic DNA, 14kb mRNA, 46 exons,
4,302 a.a.4,302 a.a.• Encodes for polycystin 1, an integral membEncodes for polycystin 1, an integral memb
rane glycoproteinrane glycoprotein• 70% duplication on 16p13.1 - the HG area70% duplication on 16p13.1 - the HG area
– HG-A 21 kbHG-A 21 kb– HG-B 17 kbHG-B 17 kb– HG-C 8.5 kbHG-C 8.5 kb

Characteristics of Characteristics of PKD2PKD2
• Located on 4q21-23.Located on 4q21-23.• Encodes a 4 kb mRNA, 968 a.a. Encodes a 4 kb mRNA, 968 a.a.
product.product.• Encodes for polycystin 2, an integral Encodes for polycystin 2, an integral
membrane glycoprotein.membrane glycoprotein.• Polycystin 1 and 2 function together Polycystin 1 and 2 function together
as part of a multi-component as part of a multi-component membrane-spanning complex involved membrane-spanning complex involved in cell-cell or cell-matrix interactions.in cell-cell or cell-matrix interactions.

Polycystin 2Polycystin 2
• Polycystin 2 has six transmembrane sPolycystin 2 has six transmembrane spans with intracellular amino- and carpans with intracellular amino- and carboxyl-termini.boxyl-termini.
• It has amino acid similarity with PKD1It has amino acid similarity with PKD1, and the family of voltage-activated c, and the family of voltage-activated calcium (and sodium) channelsalcium (and sodium) channels
• It contains a calcium-binding domain.It contains a calcium-binding domain.

Aims of genetic studyAims of genetic study
• Early diagnosis, including prenatal anEarly diagnosis, including prenatal and presymptomatic diagnosis.d presymptomatic diagnosis.
• Select embyro by “test tube baby”Select embyro by “test tube baby”• Correlate between phenotype and geCorrelate between phenotype and ge
notype.notype.• Understand mechanisms involved in cUnderstand mechanisms involved in c
yst formation and other associated leyst formation and other associated lesions of APKD at the molecular level.sions of APKD at the molecular level.

Problems in the study of Problems in the study of PKD1PKD1
• Only about 2.5 kb out of the 14 kb Only about 2.5 kb out of the 14 kb transcript is not duplicated.transcript is not duplicated.
• Mutations affecting the duplicated Mutations affecting the duplicated part are difficult to determine.part are difficult to determine.
• No “hot spot” mutations.No “hot spot” mutations.

Diagnosis by ImagingDiagnosis by Imaging
• UltrasonographyUltrasonography• Intravenous urographyIntravenous urography• Urogram with bolus intravenous nephUrogram with bolus intravenous neph
rotomographyrotomography• Computerized tomographyComputerized tomography
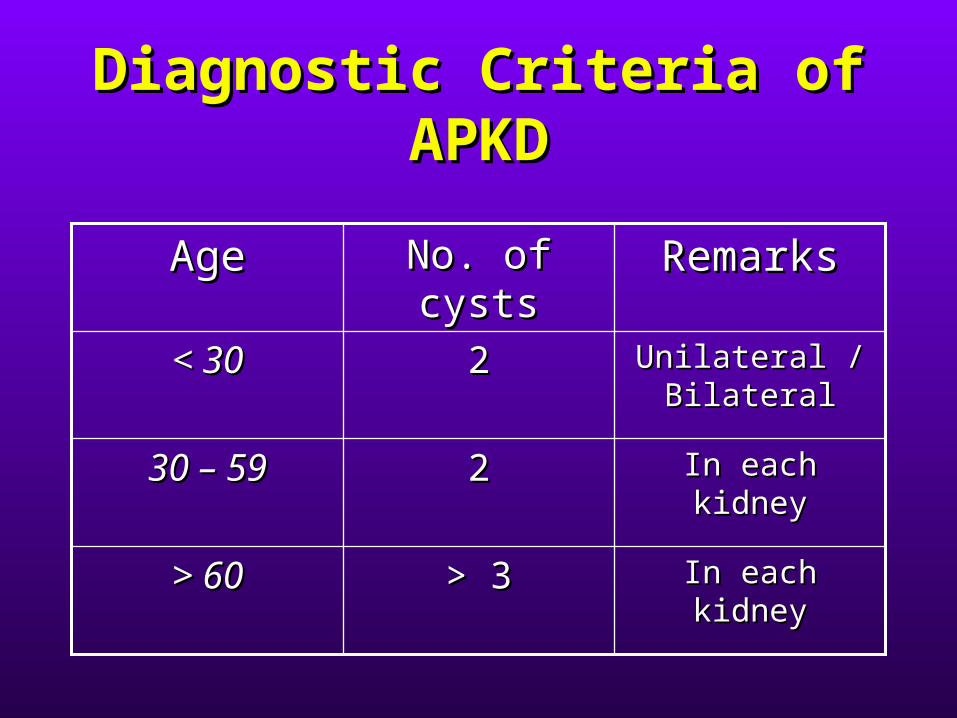
Diagnostic Criteria of Diagnostic Criteria of APKDAPKD
In each kidneyIn each kidney> 3> 3> 60> 60
In each kidneyIn each kidney2230 – 5930 – 59
Unilateral / Unilateral / BilateralBilateral
22< 30< 30
RemarksRemarksNo. of cystsNo. of cystsAgeAge

Strategies for genetic Strategies for genetic studystudy
• Genetic linkage study for early diagnosisGenetic linkage study for early diagnosis– Microsatellite studies in APKD kindredsMicrosatellite studies in APKD kindreds
• Mutation analysisMutation analysis– SequencingSequencing
• Only about 2.5 kb out of the 14 kb tranOnly about 2.5 kb out of the 14 kb transcript is not duplicated.script is not duplicated.
• Mutations affecting the duplicated parMutations affecting the duplicated part are difficult to determine.t are difficult to determine.
• No “hot spot” mutations.No “hot spot” mutations.

Mutation IdentifiedMutation IdentifiedMutation Type Mutation Method Paper Published
Deletion Exon 31-34, 35-46Exon 39, 44
PCRSSCP
Eur PKD Consortium 94Peral et al. 96
Insertion Novel aa SSCP Peral et al. 96
Missense Glu Asp2 C T
SSCPHA
Peral et al. 96Rossetti et al. 96
Nonsense GlnStopCysStopArg, GluStopC TC T
HASSCPSSCPHAHA
Turco et al. 95Neophytou et al. 96Peral et al. 96Rossetti et al. 96Turco et al. 96
Splicing Error Exon 44Intron 43
HA Eur PKD Consortium 94Peral et al. 95
Translocation Exon 15 CA Eur PKD Consortium 94

Microsatellite Microsatellite PolymorphismPolymorphism
• Variation in the number of dinucleotidVariation in the number of dinucleotides within (AC)n or other simple sequees within (AC)n or other simple sequence repeatsnce repeats

Microsatellite Markers for Microsatellite Markers for PKD1PKD1
Marker Locus Repeat Type Heterozygosity
SM7 D16S283 (AC)n 76.8%
CW2 D16S663 (AC)n 83%
AC2.5 D16S291 GGG(GT)nGAT 79%
SM6 D16S665 (TG)10C(CG)10TGC(GT)3 69%
KG8 (AC)n 56%
D16S521 D16S521 (AC)n 76.8%
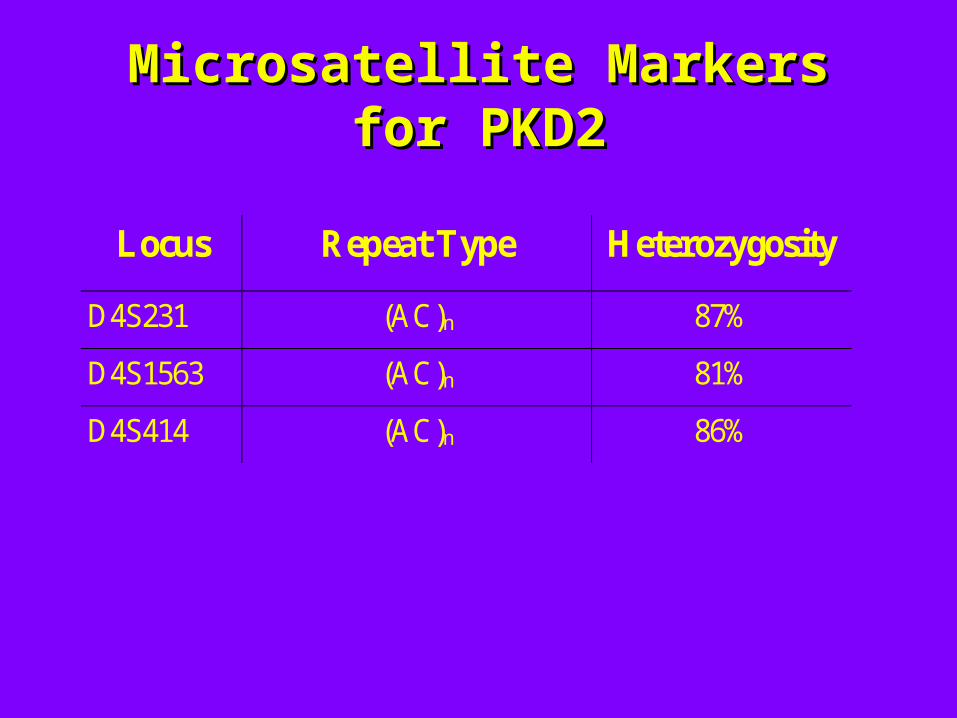
Microsatellite Markers for Microsatellite Markers for PKD2PKD2
Locus Repeat Type Heterozygosity
D4S231 (AC)n 87%
D4S1563 (AC)n 81%
D4S414 (AC)n 86%

Scope of workScope of work
• Characterization of markers in HK PopCharacterization of markers in HK Population (a sample of 63 unrelated adululation (a sample of 63 unrelated adults)ts)– 6 PKD1 markers, 3 PKD2 markers6 PKD1 markers, 3 PKD2 markers
• Microsatellite haplotyping in 5 APKD fMicrosatellite haplotyping in 5 APKD families with a total of 42 members.amilies with a total of 42 members.

MethodMethod
• 20 ml of peripheral blood20 ml of peripheral blood• DNA extracted and purifiedDNA extracted and purified• PCR amplification of each of the 9 PCR amplification of each of the 9
microsatellite markersmicrosatellite markers• Southern blotSouthern blot
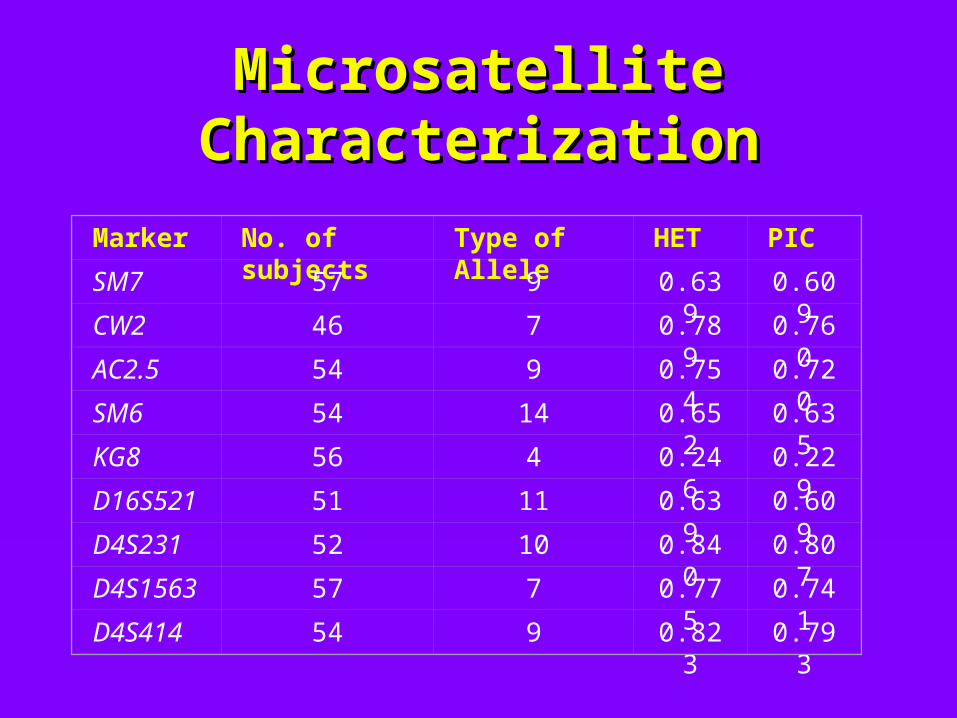
Microsatellite Microsatellite CharacterizationCharacterization
Marker No. of subjects Type of Allele HET PIC
SM7 57 9 0.639 0.609
CW2 46 7 0.789 0.760
AC2.5 54 9 0.754 0.720
SM6 54 14 0.652 0.635
KG8 56 4 0.246 0.229
D16S521 51 11 0.639 0.609
D4S231 52 10 0.840 0.807
D4S1563 57 7 0.775 0.741
D4S414 54 9 0.823 0.793
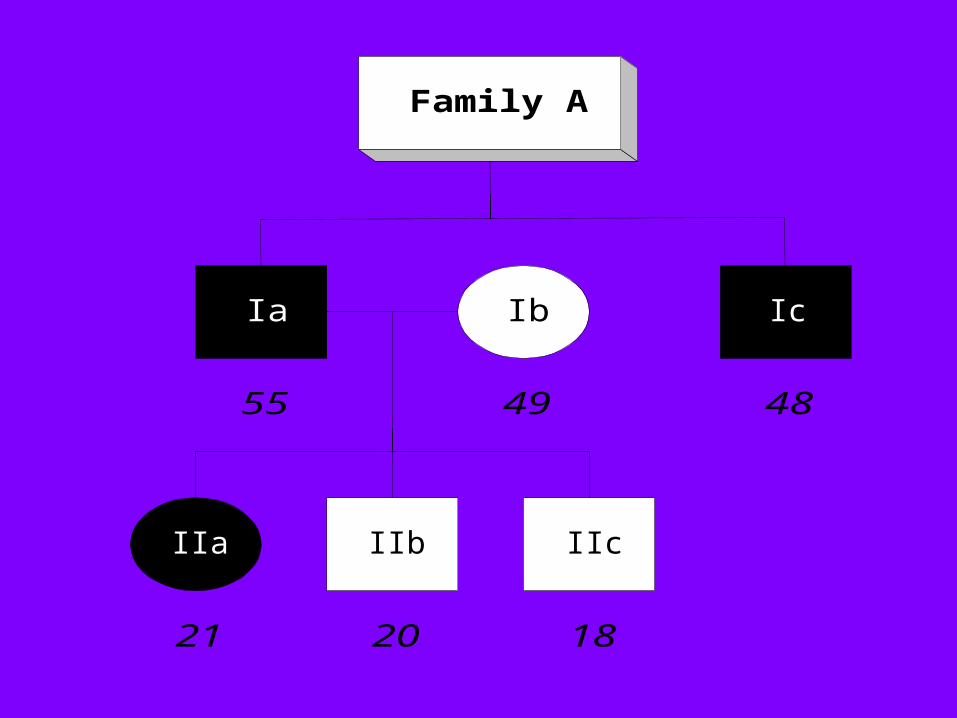
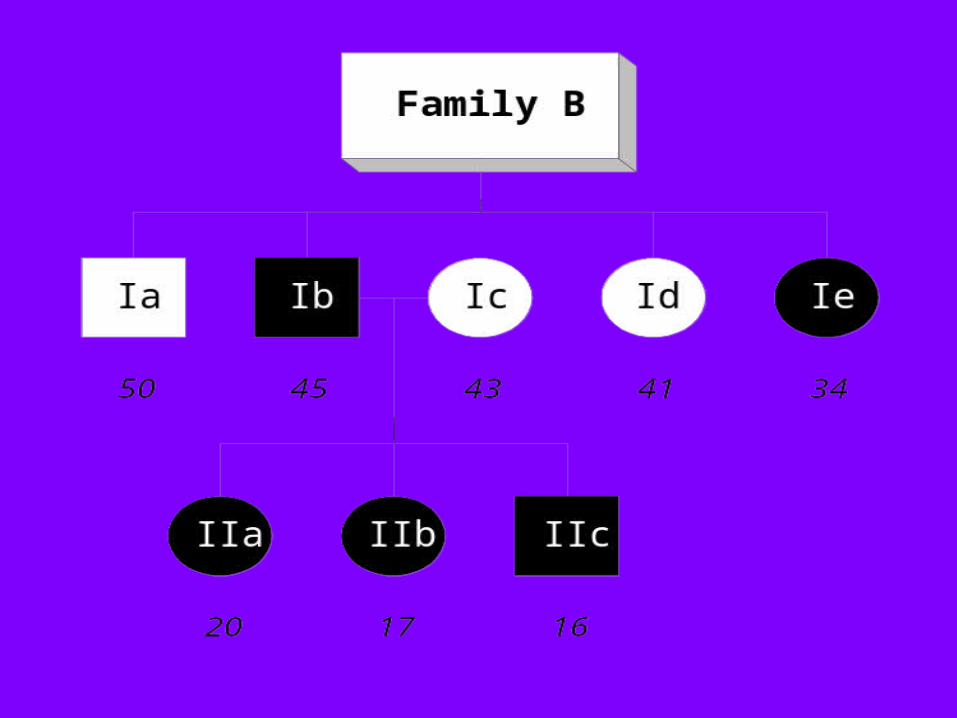
Family A
IIb IIc
Ic
IIa
Ia Ib
55
182021
4849

PKD1PKD1 Markers in Family A Markers in Family A

PKD2PKD2 Markers in Family A Markers in Family A
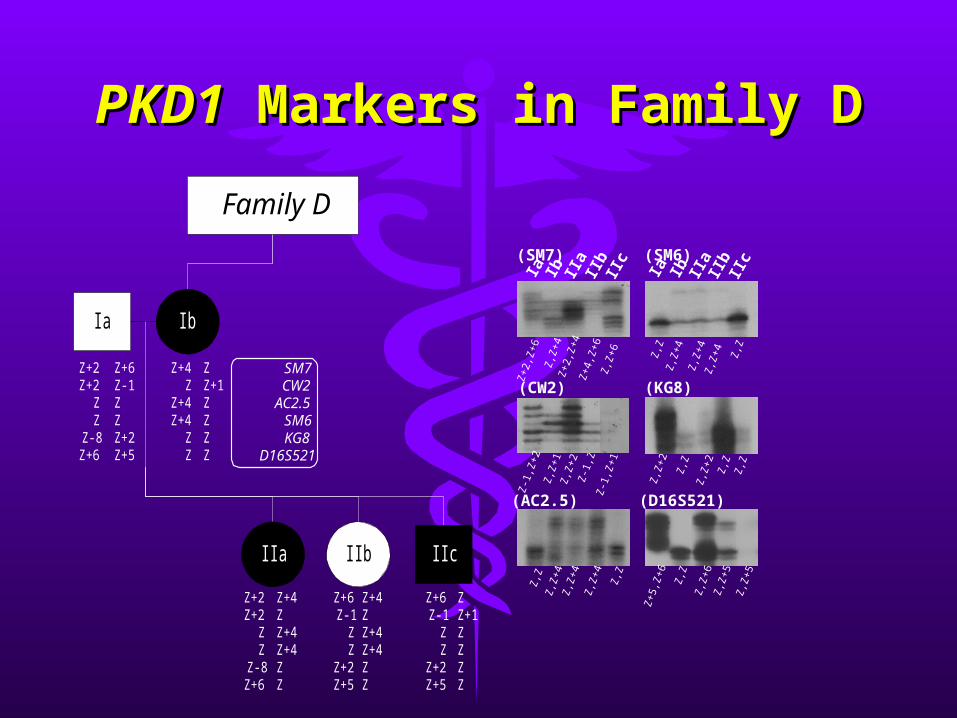
PKD1PKD1 Markers in Family D Markers in Family D
Ia
Ib II
aII
bII
c(SM7)
Ia
Ib II
aII
bII
c(SM6)
Z,Z
+4
Z+4
,Z+6
Z+2
,Z+6
Z,Z
+6
Z+2
,Z+4
Z,Z
+4
Z,Z
+4
Z,Z
Z,Z
Z,Z
+4
Z-1
,Z
(CW2)
(AC2.5)
Z,Z
+4
Z,Z
+4Z,Z
Z,Z
Z,Z
+4
(KG8)
Z,Z
Z,Z
Z,Z
+2
Z,Z
Z,Z
+2
(D16S521)
Z,Z
Z,Z
+5
Z+5
,Z+6
Z,Z
+5
Z,Z
+6
IIa
Z+2Z+2
ZZ
Z-8Z+6
Family D
SM7CW2
AC2.5SM6KG8
D16S521
Z+4Z
Z+4Z+4
ZZ
Z+6Z-1
ZZ
Z+2Z+5
Z+6Z-1
ZZ
Z+2Z+5
ZZ+1ZZZZ
Z+4ZZ+4Z+4ZZ
IIc
IbIa
Z+4ZZ+4Z+4ZZ
Z+2Z+2
ZZ
Z-8Z+6
Z+6Z-1ZZZ+2Z+5
ZZ+1ZZZZ
IIb
Z-1
,Z+2
Z,Z
+1Z
,Z+2
Z-1
,Z+1
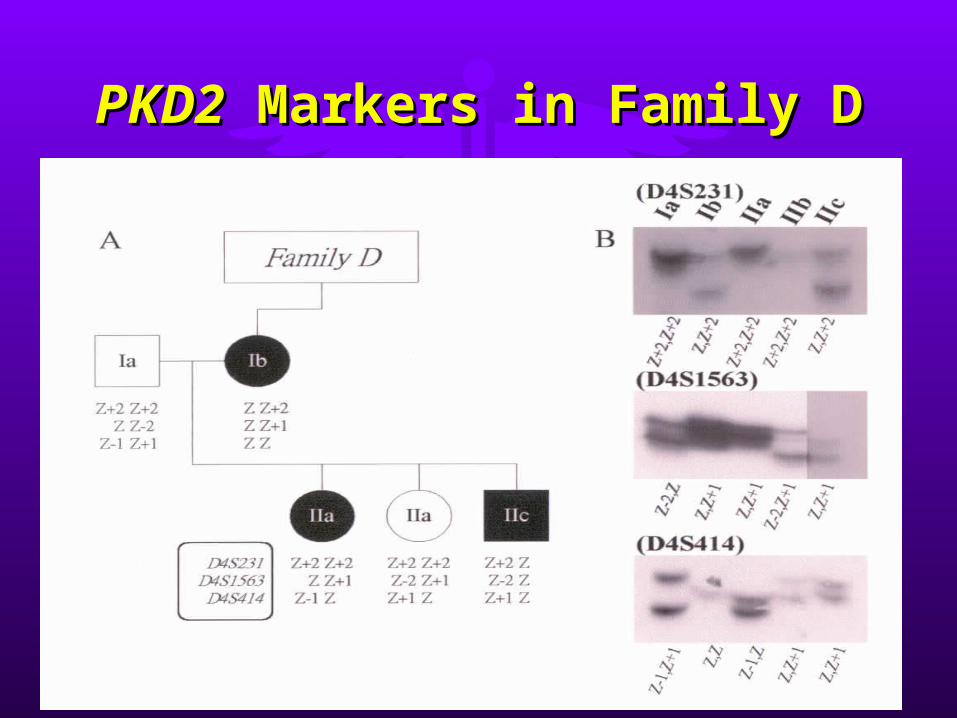
PKD2PKD2 Markers in Family D Markers in Family D
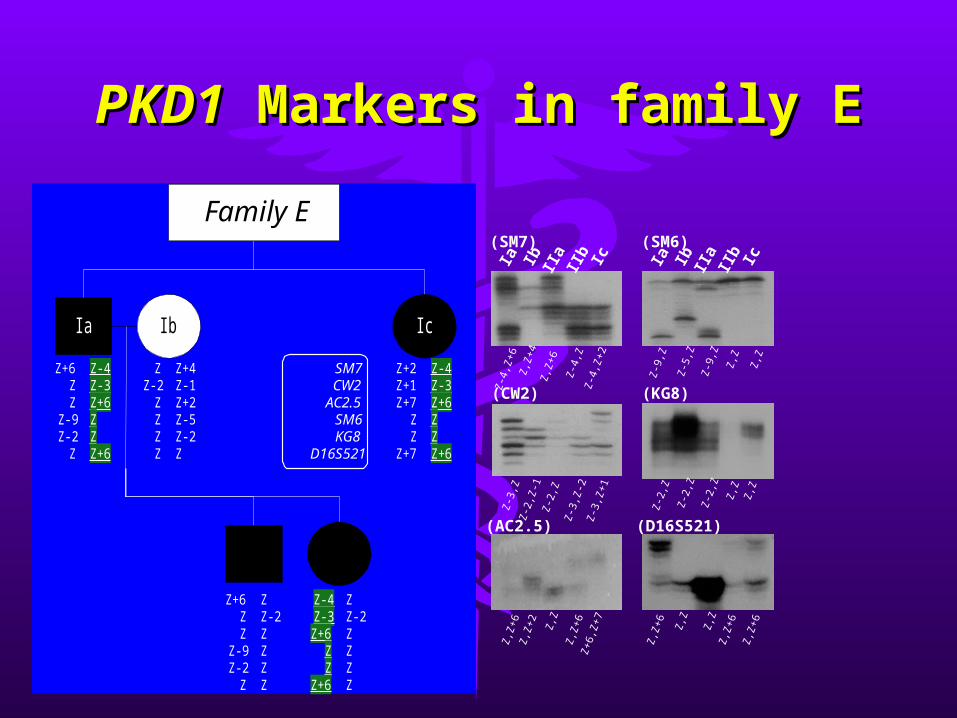
PKD1PKD1 Markers in family E Markers in family E
Ia Ib IIa
IIb Ic
(SM6)
Ia Ib IIa
IIb Ic
(SM7)
Z,Z
+4
Z-4
,Z
Z-4
,Z+6
Z-4
,Z+2
Z,Z
+6
Z-5
,Z
Z,Z
Z-9
,Z
Z,Z
Z-9
,Z
(CW2) (KG8)
Z-3
,Z
Z-3
,Z+1
Z-2
,Z
Z,Z
Z-2
,Z
Z,Z
Z-2
,Z
(AC2.5) (D16S521)Z
,Z+2
Z,Z
+6
Z,Z
+6
Z+6
,Z+7Z,Z
Z,Z
Z,Z
+6
Z,Z
+6
Z,Z
+6Z,Z
IIa
Z+6ZZ
Z-9Z-2
Z
Family E
SM7CW2
AC2.5SM6KG8
D16S521
Ia
Z+6ZZ
Z-9Z-2
Z
ZZ-2
ZZZZ
Z+2Z+1Z+7
ZZ
Z+7
Z-4Z-3Z+6
ZZ
Z+6
Z-4Z-3Z+6ZZZ+6
Z-4Z-3Z+6ZZZ+6
Z+4Z-1Z+2Z-5Z-2Z
ZZ-2ZZZZ
ZZ-2ZZZZ
Ib Ic
IIb
Z-2
,Z-1
Z
-2,Z
Z
-3,Z
-2

Results of Genetic Results of Genetic DiagnosisDiagnosis
NDNDNDND5522EE
----5522DD
----++++++181833CC
----++++8822BB
--++6622AA
ADPKD2ADPKD2ADPKD1ADPKD1No. of No. of MemberMember
GeneratiGenerationon
FamilyFamily

PKD3 - Is there one?PKD3 - Is there one?
• 2001 (Pei et al)– evidence of bilineal disease and trans-
heterozygotes in a large family of ADPKD– 28/48 members affected– SSCA screened for and found a PKD2
mutation (2152delA; L736X) in 12 affected pedigree members
– linkage analysis with markers at the PKD1 locus, found significant LOD scores (13.0).

PKD3 - Is there one?PKD3 - Is there one?
• 2001 (Pei et al)2001 (Pei et al)– evidence of bilineal disease and evidence of bilineal disease and
trans-heterozygotes in a large family trans-heterozygotes in a large family of ADPKDof ADPKD
– 28/48 members affected28/48 members affected– in 2/48, who had severe disease, in 2/48, who had severe disease,
evidence of trans-heterozygotesevidence of trans-heterozygotes

1. Hereditary Kidney 1. Hereditary Kidney DiseasesDiseases
•Adult Polycystic DiseaseAdult Polycystic Disease•[Infantile Polycystic Disease][Infantile Polycystic Disease]•Alport SyndromeAlport Syndrome

Infantile Polycystic Kidney Infantile Polycystic Kidney DiseaseDisease
• Autosomal recessive.Autosomal recessive.• Usually incompatible Usually incompatible
with life.with life.• Early antenatal Early antenatal
diagnosis for diagnosis for termination of termination of pregnancy is pregnancy is desirable.desirable.

Infantile Polycystic Infantile Polycystic DiseaseDisease
• In infantile PKD, the liver is always In infantile PKD, the liver is always affected.affected.
• The abnormal bile ducts in the liver The abnormal bile ducts in the liver are accompanied by periductal are accompanied by periductal fibrosis. Hence called congenital fibrosis. Hence called congenital hepatic fibrosis.hepatic fibrosis.

Localization of Localization of PKHD1PKHD1
• 19941994– Gene locus in 6p12.1-p21.Gene locus in 6p12.1-p21.
• 2002 (Ward et al)2002 (Ward et al)– PKHD1 PKHD1 cloned– 16kb transcript, 4,074 a.a. receptor-
like protein called fibrocystin– Missense and truncating mutations
identified in 14 probands

Genetics of PKHDGenetics of PKHD
• Compound heterozygosity.Compound heterozygosity.• Double truncating mutations – Double truncating mutations –
severe disease.severe disease.• Some families with mild disease Some families with mild disease
show compound heterozygosity for show compound heterozygosity for a missense and a truncating a missense and a truncating mutation.mutation.

1. Hereditary Kidney 1. Hereditary Kidney DiseasesDiseases
•Adult Polycystic DiseaseAdult Polycystic Disease•Infantile Polycystic DiseaseInfantile Polycystic Disease•[Alport Syndrome][Alport Syndrome]

Alport SyndromeAlport Syndrome
• A hereditary disorder of basement mA hereditary disorder of basement membrane collagen characterized clinicembrane collagen characterized clinically by hematuria, progressive renal faally by hematuria, progressive renal failure, and, frequently, neurosensory hilure, and, frequently, neurosensory hearing loss and ocular abnormalities.earing loss and ocular abnormalities.

Alport SyndromeAlport Syndrome
• Genetically heterogeneous.Genetically heterogeneous.• In the majority of cases, the In the majority of cases, the
disease is inherited as an X-linked disease is inherited as an X-linked trait, but an autosomal recessive trait, but an autosomal recessive form and also an autosomal form and also an autosomal dominant form exist.dominant form exist.

Alport SyndromeAlport Syndrome
• X-link recessive: mutations of X-link recessive: mutations of COL4A5 COL4A5 on Xqon Xq22.22.
• X-link recessive associated with diffuse leioX-link recessive associated with diffuse leiomyomatosis: mutations of myomatosis: mutations of COL4A5COL4A5 and and COLCOL4A64A6..
• Autosomal recessive: mutations of Autosomal recessive: mutations of COL4A3COL4A3 and and COL4A4 COL4A4 on 2q.on 2q.
• Autosomal dominant : no mutation in any oAutosomal dominant : no mutation in any of the COL4 genes.f the COL4 genes.

Prenatal Diagnosis of Prenatal Diagnosis of ASAS
• X-link recessive: by genetic linkage X-link recessive: by genetic linkage analysis using polymorphic analysis using polymorphic markers in and around markers in and around COL4A5.COL4A5.
• Autosomal recessive: by genetic Autosomal recessive: by genetic linkage analysis using polymorphic linkage analysis using polymorphic markers in and around markers in and around COL4A3 COL4A3 andand COL4A4. COL4A4.

Fabry diseaseFabry disease

Fabry diseaseFabry disease
The story begins…The story begins…

Fabry diseaseFabry disease
• X-linked recessive inborn error of X-linked recessive inborn error of glycosphingolipid catabolism that results glycosphingolipid catabolism that results from the deficient activity of the from the deficient activity of the lysosomal enzyme α-galactosidase A (EC lysosomal enzyme α-galactosidase A (EC 3.2.1.22).3.2.1.22).
• Accumulation of glycosphingolipid Accumulation of glycosphingolipid substrates in the vascular endothelium substrates in the vascular endothelium causing occlusive microvascular diseases causing occlusive microvascular diseases mainly affecting the kidney, the heart, mainly affecting the kidney, the heart, peripheral nerves and the brain.peripheral nerves and the brain.

F/50 Southern ChineseF/50 Southern Chinese
• Asymptomatic proteinuria Asymptomatic proteinuria • 24 hour urine protein 0.6 g24 hour urine protein 0.6 g• No skin or corneal lesionsNo skin or corneal lesions• Normal renal function testsNormal renal function tests

Family historyFamily history
• Mother died of kidney diseaseMother died of kidney disease• 4 siblings4 siblings
– one younger brother (age 44) in end one younger brother (age 44) in end stage renal failure since age of 35stage renal failure since age of 35
– one younger brother (age 41) has one younger brother (age 41) has cardiomyopathy since age of 33, not cardiomyopathy since age of 33, not in renal failurein renal failure

Renal Biopsy DiagnosisRenal Biopsy Diagnosis
• Consistent with heterozygous Consistent with heterozygous Fabry diseaseFabry disease

Diagnosis ConfirmedDiagnosis Confirmed
• Consistent withConsistent withheterozygous Fabry diseaseheterozygous Fabry disease
• Low serum α-galactosidase A Low serum α-galactosidase A activity at 5.2nmol/ml.hr activity at 5.2nmol/ml.hr – (normal range: 8.8-14.5nmol/ml.hr) (normal range: 8.8-14.5nmol/ml.hr)
• Low serum α-galactosidase A Low serum α-galactosidase A activity in both brothersactivity in both brothers (both at (both at <0.5nmol/ml.hr)<0.5nmol/ml.hr)

Genetic studies of 2 Genetic studies of 2 affected brothersaffected brothers
• Direct DNA sequencing of all 9 Direct DNA sequencing of all 9 exons of α-galactosidase geneexons of α-galactosidase gene
• New primers are designed for this New primers are designed for this studystudy

Genetic studies of 2 Genetic studies of 2 affected brothersaffected brothers
• Direct DNA sequencing of all 9 exons of Direct DNA sequencing of all 9 exons of α-galactosidase geneα-galactosidase gene
• ResultsResults– single T-to-C transition in codon 14 of exon single T-to-C transition in codon 14 of exon
11– missense mutation predicting a leucine to missense mutation predicting a leucine to
proline substitution (L14P)proline substitution (L14P)– same mutation in both brotherssame mutation in both brothers– other exon sequences are normalother exon sequences are normal
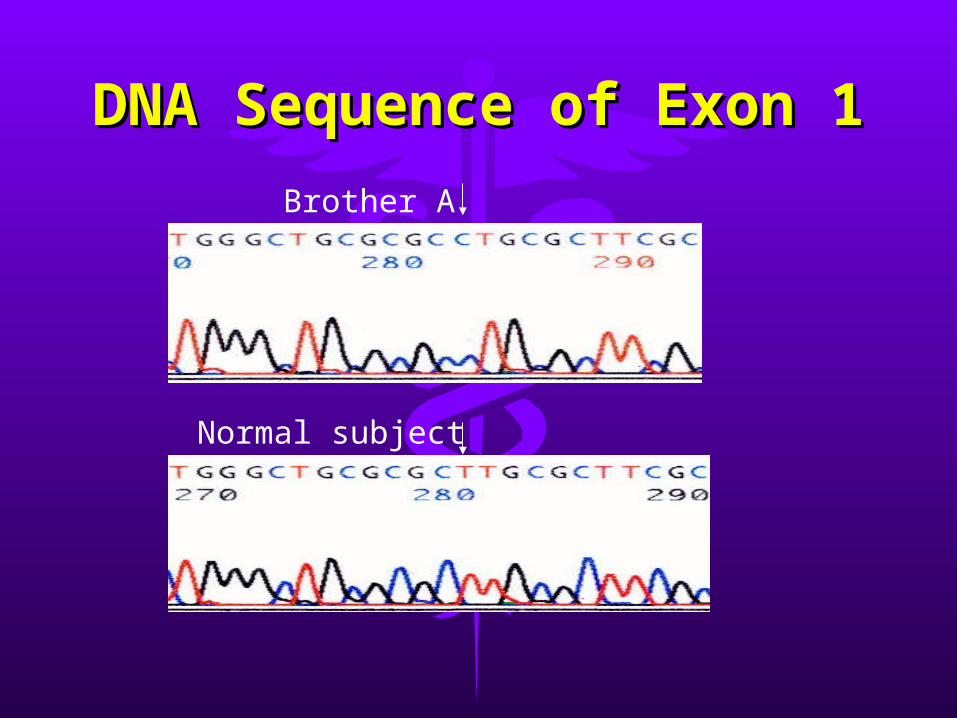
DNA Sequence of Exon DNA Sequence of Exon 11
Brother A
Normal subject
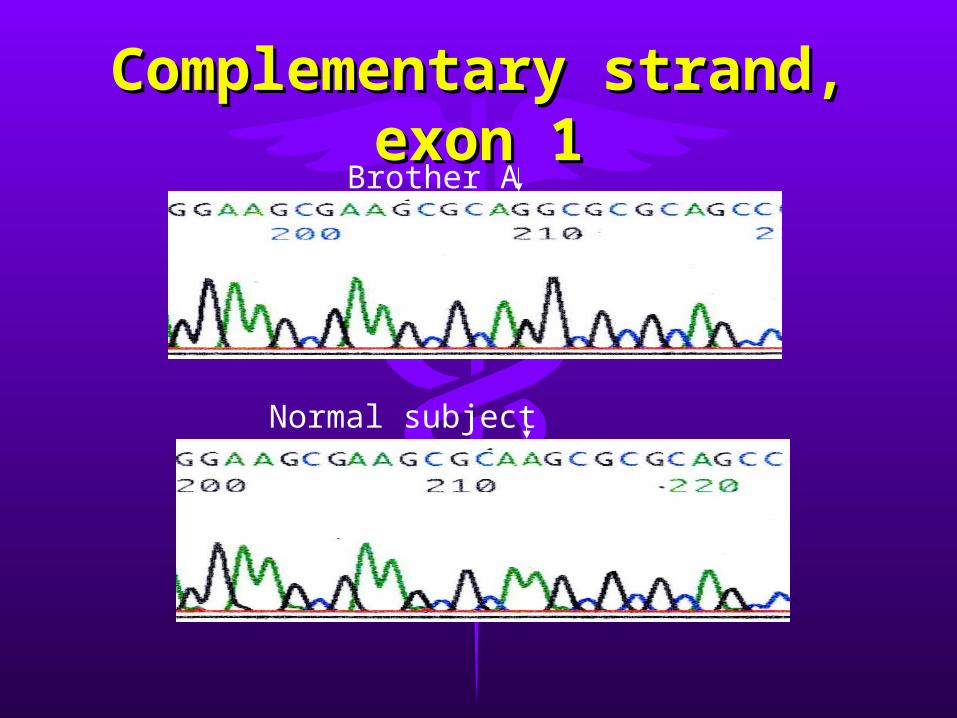
Complementary strand, Complementary strand, exon 1exon 1Brother A
Normal subject

Significance of the Significance of the studystudy
• Successful design of new primers Successful design of new primers for direct sequencing studies of the for direct sequencing studies of the Fabry disease geneFabry disease gene
• A novel mutation is documentedA novel mutation is documented• The two male patients have the The two male patients have the
same genetic mutations despite same genetic mutations despite different phenotypic different phenotypic manifestationsmanifestations

2. Tumors of the Kidney2. Tumors of the Kidney
•Renal Cell CarcinomaRenal Cell Carcinoma•Wilms’ tumor (nephroblastomWilms’ tumor (nephroblastoma)a)

2. Tumors of the Kidney2. Tumors of the Kidney
•[Renal Cell Carcinoma][Renal Cell Carcinoma]•Wilms’ tumor (nephroblastomWilms’ tumor (nephroblastoma)a)

Renal Cell Renal Cell CarcinomaCarcinomaRenal Cell Renal Cell CarcinomaCarcinoma
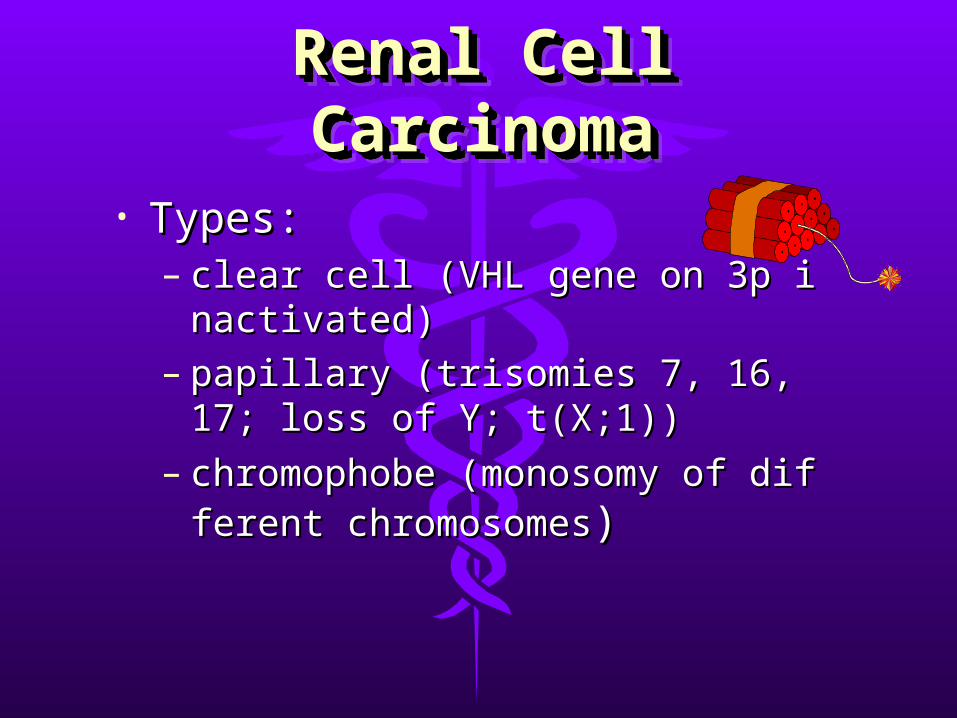
• Types (histological with a genetic Types (histological with a genetic basis):basis):– clear cellclear cell– papillarypapillary– chromophobechromophobe– collecting duct carcinomacollecting duct carcinoma
Renal Cell Renal Cell CarcinomaCarcinomaRenal Cell Renal Cell CarcinomaCarcinoma
• Types:Types:– clear cell (VHL gene on 3p inactivateclear cell (VHL gene on 3p inactivate
d)d)– papillary (trisomies 7, 16, 17; loss of papillary (trisomies 7, 16, 17; loss of
Y; t(X;1))Y; t(X;1))– chromophobe (monosomy of differchromophobe (monosomy of differ
ent chromosomesent chromosomes))

Nephroblastoma (Wilms’ tuNephroblastoma (Wilms’ tumor)mor)
Nephroblastoma (Wilms’ tuNephroblastoma (Wilms’ tumor)mor)
• 20% of malignant childhood tum20% of malignant childhood tumorsors
• highest incidence at age 3highest incidence at age 3
• abdomincal massabdomincal mass• hematuria, pain, feverhematuria, pain, fever

NephroblastomaNephroblastomaNephroblastomaNephroblastoma
• Deletions or mutations of WT1 Deletions or mutations of WT1 or WT2 genesor WT2 genes
• Both WT1 and WT2 genes are Both WT1 and WT2 genes are on the short arm of on the short arm of chromosome 11, but are chromosome 11, but are distinctly different genes.distinctly different genes.

WT1WT1• WT1 gene is encoded by 10 exons, resulting in
messenger RNA subject to a complex pattern of alternative splicing.
• WT1 gene encodes a zinc finger transcription factor, which binds to GC-rich sequences and functions as a transcriptional activator or repressor for many growth factor genes.
• WT 1 protein is mainly expressed in developing kidney, testis, and ovary, indicating that it is involved in the differentiation of genitourinary tissues, all thought to be the sites of origin of Wilms’ tumor.
• The point mutation of WT1 results in Denys-Drash syndrome.

WT1WT1
High level WT1 expression in leukemia High level WT1 expression in leukemia cells is linked to a poor prognosis.cells is linked to a poor prognosis.
A correlated expression between WT1 A correlated expression between WT1 and mdr-1 in vincristine resistant cells and mdr-1 in vincristine resistant cells indicates a close relation with multi-indicates a close relation with multi-drug resistance and is a promising drug resistance and is a promising diagnostic marker for chemoresistance diagnostic marker for chemoresistance in hematologic malignancies.in hematologic malignancies.

CystinuriaCystinuria
• BackgroundBackground– A hereditary disorder of cystine and A hereditary disorder of cystine and
dibasic amino acid transport across dibasic amino acid transport across the luminal membrane of renal the luminal membrane of renal proximal tubule and small intestine.proximal tubule and small intestine.
– Cystine is of low solubility and Cystine is of low solubility and forms urinary stones in sufferers of forms urinary stones in sufferers of cystinuria.cystinuria.

Localization of Cystinuria Localization of Cystinuria GenesGenes
• 1992 (Bertran et al)1992 (Bertran et al)– rBAT (SLC3A1) rBAT (SLC3A1) on 2p (type I cystinuria).on 2p (type I cystinuria).
• 1999 (Feliubadalo et al)1999 (Feliubadalo et al)– SLC7A9SLC7A9 on 19q (non-type I). on 19q (non-type I).
• SLC7A9 is the transmembrane channel SLC7A9 is the transmembrane channel mediating the uptake of these a.a.mediating the uptake of these a.a.
• rBAT is a smaller protein.rBAT is a smaller protein.• rBAT forms a heterodimeric complex with the rBAT forms a heterodimeric complex with the
channel and is critical for its targeting to the channel and is critical for its targeting to the luminal membrane.luminal membrane.

• Hereditary Kidney DiseasesHereditary Kidney Diseases– Adult polycystic diseaseAdult polycystic disease– Infantile polycystic diseaseInfantile polycystic disease– Alport syndromeAlport syndrome– Fabry diseaseFabry disease– Tubular transport - CystinuriaTubular transport - Cystinuria
• Tumours of the KidneyTumours of the Kidney– Renal Cell CarcinomaRenal Cell Carcinoma– Wilms’ tumor (nephroblastoma)Wilms’ tumor (nephroblastoma)

~~ the endthe end ~~