next generation sequencing presentation
DESCRIPTION
This presentation is the summary of the methods of Next generation sequencing. This also includes the applications that have been recently publishedTRANSCRIPT

Emerging Science: The Next-Generation Sequencing (NGS) Technology
Kalyan Kumar Pasumarthy

Its cheaper• $2700 million vs $1.5 million
The Power of Next-Generation SequencingSanger’s di-deoxy method vs NGS
Its faster• 13 years vs 5 months
Various applications• whole genome sequencing• targeted genomic resequencing• metagenomics• transcriptomics• chromatin analysis• DNA-protein binding analysis

Next-Generation Sequencing Platforms
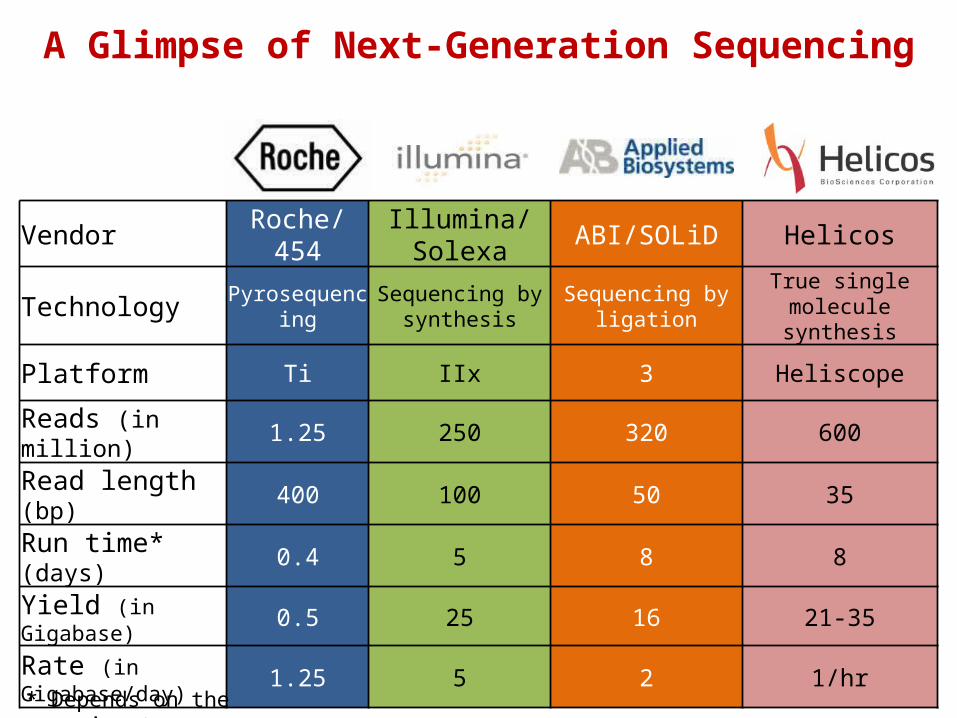
Vendor Roche/454 Illumina/Solexa ABI/SOLiD Helicos
Technology Pyrosequencing Sequencing by synthesis
Sequencing by ligation
True single molecule synthesis
Platform Ti IIx 3 Heliscope
Reads (in million) 1.25 250 320 600
Read length (bp) 400 100 50 35
Run time* (days) 0.4 5 8 8
Yield (in Gigabase) 0.5 25 16 21-35
Rate (in Gigabase/day)
1.25 5 2 1/hr
A Glimpse of Next-Generation Sequencing
* Depends on the experiment
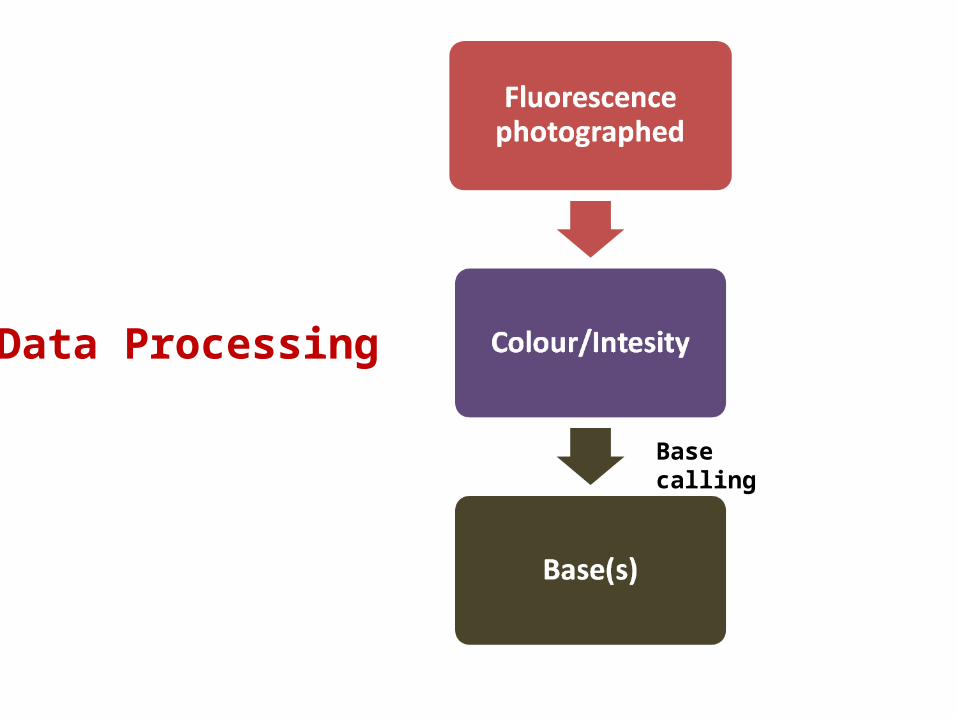
Data Processing
Base calling

Roche/454 – GS FLX• Clonal amplification by emulsion PCR
• Bead deposition on picotitre plates

• Pyrosequencing technology
A T G C A T
Roche/454 – GS FLX

Illumina/Solexa – Genome Analyzer• Clonal amplification by bridge PCR isothermally
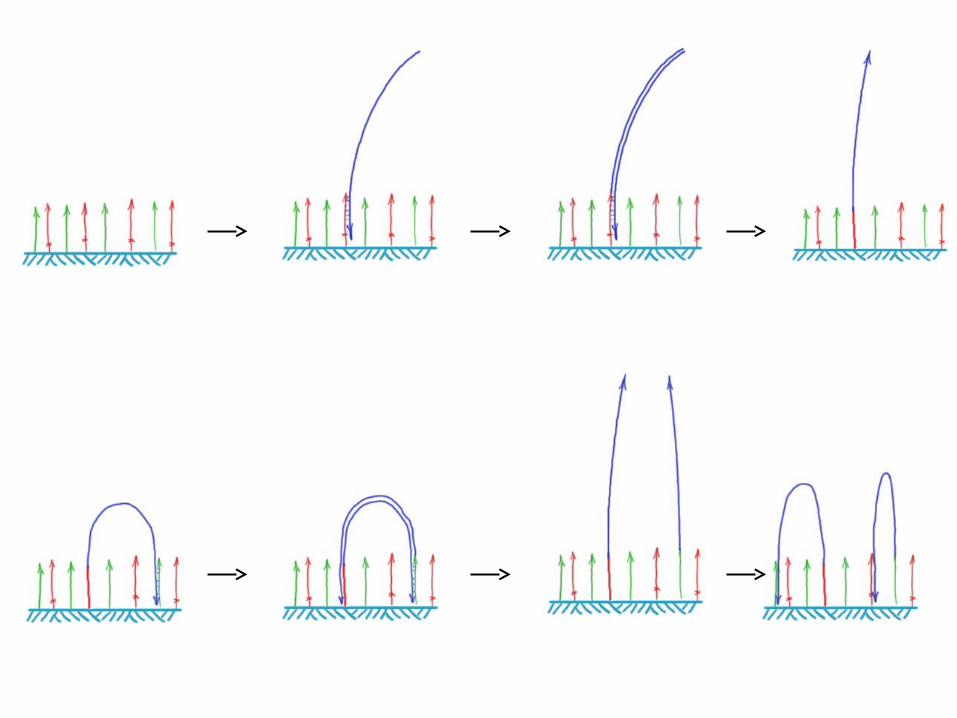
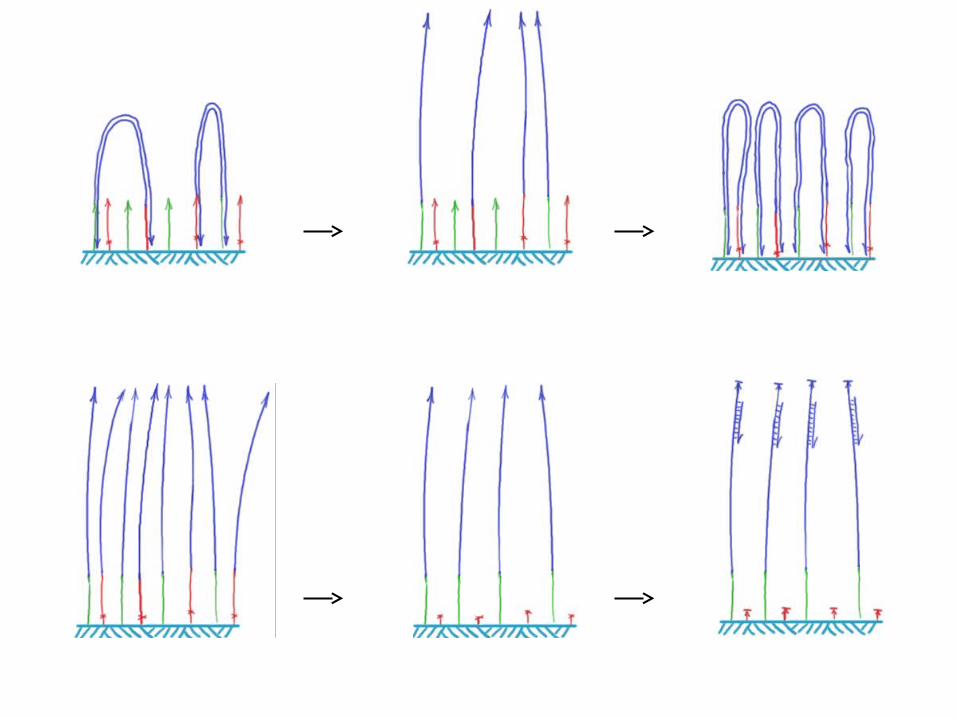
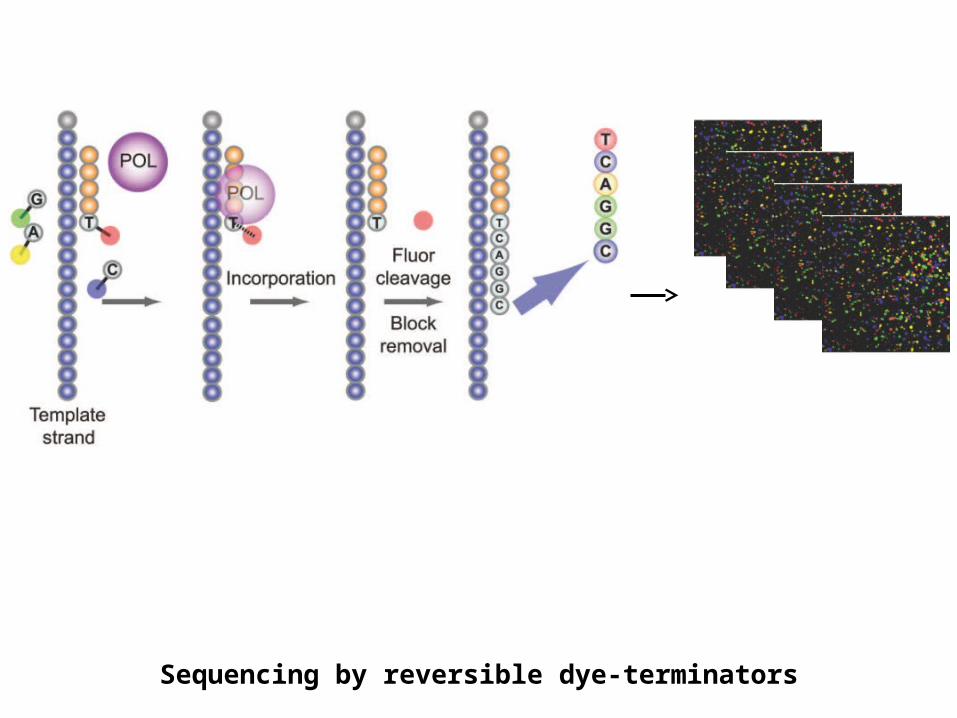
Sequencing by reversible dye-terminators

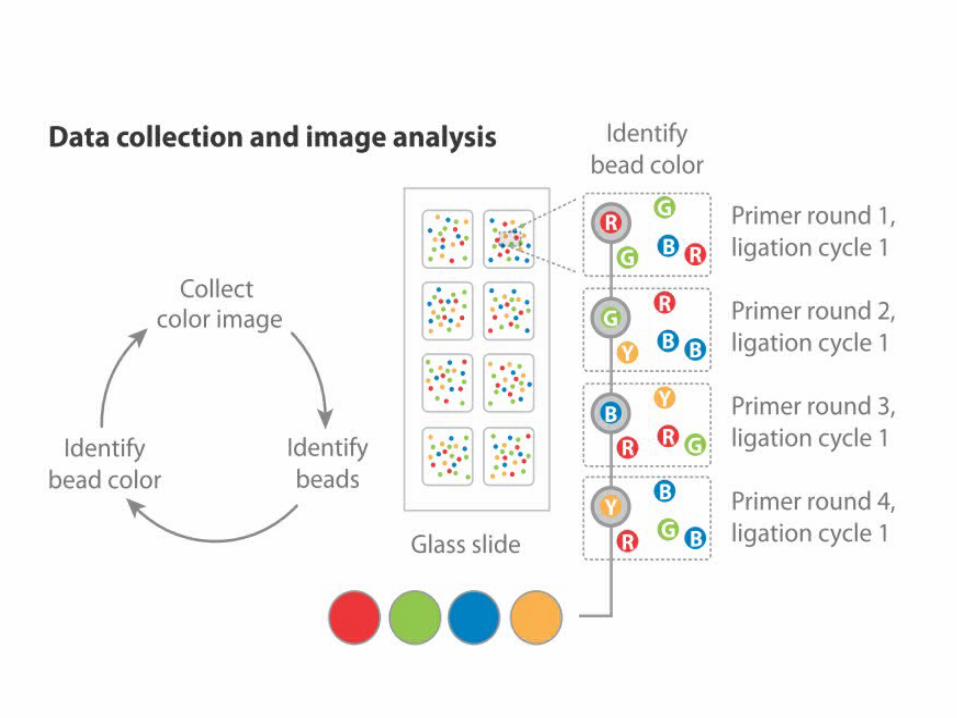
ABi- SOLiD (Supported Oligonucleotide Ligation and Detection)
• Clonal amplification by emulsion PCR
• Sequencing by ligation
• Template is probed by fluorescent tagged di-nucleotide probe


Primer n
Primer n-1 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
Primer n-2
Primer n-3
Primer n-4
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18


Helicos - Heliscope
• No amplification
• tSMS technology – True Single Molecular Synthesis
AAAA
TTTTTT
TACGAT
AAAA
TTTTTT
TACGGC
AAAA
TTTTTT
TCCATG
A A A
AAAA
TTTTTT
TACGAT
AAAA
TTTTTT
TACGGC
AAAA
TTTTTT
TCCATGA
AAAAA
Template preparationTemplate bound to
adapters on the flowcellAddition of a fluorescent
labelled base

AAAA
TTTTTT
TACGGT
AAAA
TTTTTT
TACTGC
AAAA
TTTTTT
TCCATGA
AAAA
TTTTTT
TACGGT
AAAA
TTTTTT
TACTGC
AAAA
TTTTTT
TCCATGA G
Fluorescent label removed
Fluorescent label removed
AAAA
TTTTTT
TACGGT
AAAA
TTTTTT
TACTGC
AAAA
TTTTTT
TCCATG
G G G
A
Probe with another labelled base
AAAA
TTTTTT
TACGGT
AAAA
TTTTTT
TACTGC
AAAA
TTTTTT
TCCATGA G
Polymerase adds the labelled base
AAAA
TTTTTT
TACGGT
AAAA
TTTTTT
TACTGC
AAAA
TTTTTT
TCCATGA G
C C C
Probe with another labelled base
AAAA
TTTTTT
TACGGT
AAAA
TTTTTT
TACTGC
AAAA
TTTTTT
TCCATGA G
C CC
Polymerase adds the labelled base
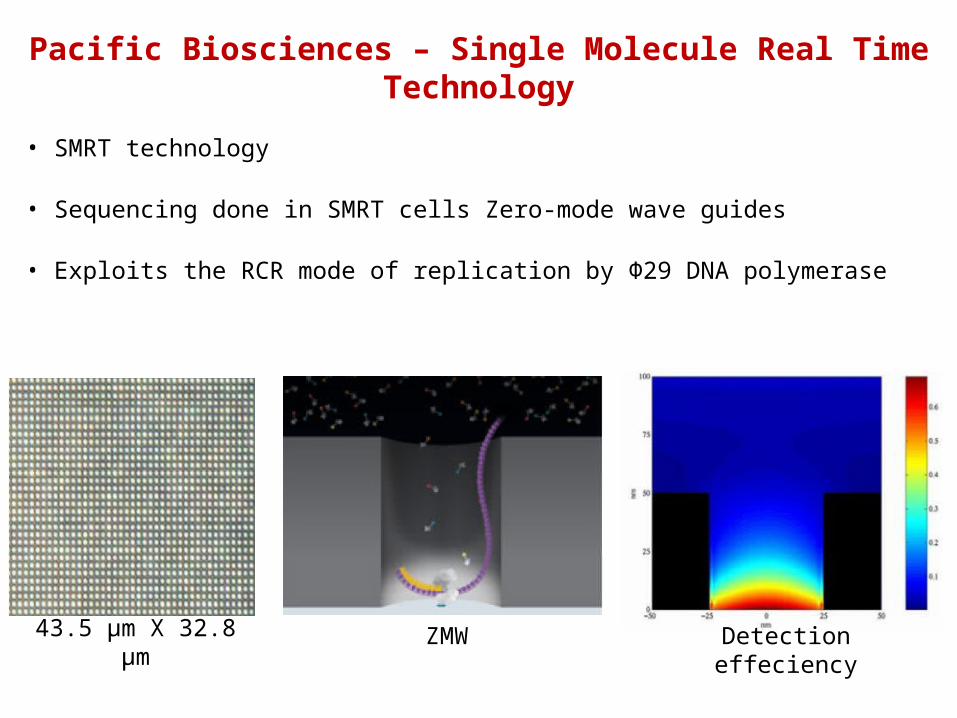
Pacific Biosciences – Single Molecule Real Time Technology
• SMRT technology
• Sequencing done in SMRT cells Zero-mode wave guides
• Exploits the RCR mode of replication by Φ29 DNA polymerase
43.5 µm X 32.8 µm ZMW Detection effeciency

Bases are individually labelled at γP
Pacific Biosciences – Single Molecule Real Time Technology

Various applications
• Whole genome sequencing
• Targeted genomic resequencing
• Metagenomics
• Transcriptomics
• Chromatin analysis
• DNA-protein binding analysis

Whole Genome Sequencing
• Best method at present is Roche/454 GS FLX
• Helps in mapping genomic structural variation : insertions, deletions & rearrangements
•1000 genome project aimed at characterising and cataloging of human genetic variation
• Long reads• Mycoplasma genetalium 96% coverage and 99.9% accuracy (Marguiles, 2005)• James Watson at 7.4 fold redundancy(Wheeler, 2008)• AML complete genome sequecing identified 8 non-synonymous mutations in comparision to normal skin genome of effecter person (Ley, 2008)

Targeted Genomic Resequencing
• To identify the genetic variation in genomic subregions
• Helps in identifying polymorphisms and mutations in genes implicated in cancer and in regions where whole-genome association studies have implicated in disease
• SNPs were catalogued in a region of 134 kb from 79 people (Yeager, 2008)
• 1000 mutations were identified in 23 genes by sequencing 623 genes from 180 cancer samples (Ding, 2008)

Metagenomics
• Study of genetic material recovered directly from environmental samples
• Microbial diversity from environmental and clinical samples can be studied
• Marine Biodiversity (Huber 2007)
• Soil Biodiversity (Urich, 2008)
• Oral cavity: 22 phyla with 19,000 phylotypes (Keijser, 2008)
• Gut metagenome from lean and obese twins identified that obesity is associated with phylum level changes in microbiota, reduced bacterial diversity and altered representation of bacterial genes and metabolic pathways (Turnbaugh, 2009)

Transcriptomics
• RNA analysis by sequencing: RNA-seq
• Qualitative and quantitative analysis is not limited by the knowledge of genome
• Helps in distinguishing RNA isoforms, allelic expression, alternative splicing events, demarcation of exon-intron boundaries
• Identification of transcripts from unidentified genes/pseudo genes
• Overlapping genes were identified in yeast (Nagalakshmi, 2008)
• Small RNAprofiling in Arabidopsis (Lister, 2008), Maize (Nobuta, 2008), Tomato (Moxon, 2008) Medicago ( Szittya, 2008), Rice under abiotic stress conditions (Zhou, 2009)

Mapping of DNA-Protein interactions
• Chromatin immuno-precipitation is followed by sequencing
• Advantageous over ChIP-chip
• Not limited by the genome knowledge
• Human neuro restrictive silencing factor (NRSF) binding sites identified (Johnson, 2007)
• STAT1 (Robertson, 2007)
• Histone modification and Chromatin accessibility can be assayed at genome level
• H2A.Z histone genome wide positioning in yeast (Albert, 2007)
• Chromatin accessibility in C. elegans (Johnson, 2006)

How does this Technique help us?
• In addressing the transcriptome (ESTs) during stress conditions• small RNA profiling under various conditions
• Abiotic stress• Biotic stress• Tomato• Rice• Cotton
•To examine the chromatin status in varying physiological conditions
• Analysis of the microbiota of the root and soil
• Analysis of the transcriptional activation of various genes by DNA binding proteins
• viral proteins that bind DNA and show transcriptional activation• genes modulated by various stress responsive transcription factors

Limitations
• Cost
• Error rate (Higher than Sanger sequencing ~ 1%)
• Short-read length
• Lack of effective computational algorithms
• Expertise in India is very meagre