observations on the biochemistry and … · measured by this method were 174,000, ... agglutination...
TRANSCRIPT

OBSERVATIONS ON THE BIOCHEMISTRY AND CLINICAL SIGNIFICANCE OF THE
RHESUS ANTIGENS AND ANTIBODIES
being a thesis submitted for the
Degree of Doctor of Philosophy
in the Faculty of Medicine
University of London
by
Elizabeth Jean Folkerd, BSc.
Medical Research Council's Experimental Haematology Research Unit,
Wright-Flemming Institute of Microbiology, St. Mary's Hospital Medical School,
London
1978

1
ABSTRACT
Current ideas on the biochemistry of the Rhesus antigens were
investigated by techniques involving the use of enzymes, radiation and
detergents.
The action of phospholipase A2 and phospholipase C on erythrocyte
membranes resulted in the degradation of membrane phospholipids and a
decline in Rh (D) activity, thus indicating the involvement of phospholipid
molecules with D antigen activity. There was some evidence that coating
red cell membranes with anti-D before phospholipase treatment protected
the D antigen from enzymic attack.
Measurement of the percentage of biological activity surviving
increasing doses of radiation can be used to measure the molecular weights
of proteins in situ. The molecular weights of the D, C, c and e antigens
measured by this method were 174,000, 191,000, 194,000 and 221,000 daltons
respectively. The molecular arrangement of the Rh antigens at the membrane
surface was discussed in the light of these results.
Rh (D) activity could be regained in membrane material solubilized
by sodium deoxycholate after removal of detergent and reaggregation of
the lipid and protein molecules. Further experiments demonstrated for
the first time the stability of the D antigen-antibody complex in detergent.
The latter observation encouraged attempts to purify the D antigen by
absorbing the deoxycholate-solubilized proteins on a solid phase anti-IgG
column, but this was not successful.

2
The clinical significance of the Rh antigens was studied with respect
to haemolytic disease of the newborn. Erythrophagocytosis, agglutination
and complement binding were all considered as possible mechanisms of
erythrocyte removal in Rh and ABO haemolytic disease. The relationship
between the number of molecules of anti-D and anti-A bound to red cells
and the extent of phagocytosis by monocytes and polymorphonuclear leucocytes
was investigated in vitro. Physiological serum concentrations of IgG
inhibited the observed erythrocyte ingestion. Nevertheless it was
concluded that erythrophagocytosis is the most likely mechanism of
red cell removal in haemolytic disease and various suggestions were made
concerning how the observed in vitro inhibition of ingestion by IgG might
be overcome in vivo in the spleen.

3
PREFACE
The biochemistry of the ABO blood group substances has been
successfully investigated (for review see Walkins, 1966). The information
gained from such studies can be used to further the understanding of the
genetic control of blood group antigens. In contrast, the biochemistry
of the Rh antigens is still uncertain and the suggested mechanisms of
genetic control purely speculative. The aim of the investigations
described in this study was to further the current understanding of the
physiology and biochemistry of the Rh antigens.
Chapter I is an introduction to the experiments concerning the
biochemistry of the Rhesus antigens that follow. The history and current
knowledge of the subject are considered and a series of experiments
formulated to further this knowledge.
In the second chapter the lipid dependence of the Rh (D) antigen is
examined by the use of phospholipase enzymes. Phospholipase A2 is known
to cleave the fatty acyl ester bond at the C-2 position of the glycerol
backbone of most phospholipids. Phospholipase C catalyses the hydrolysis
of phospholipids to diglycerides and phosphorylated amines. Red cell
membranes were exposed to phospholipase A2 and C and the Rh activity of
the degraded membranes investigated.
Chapter III is concerned with the measurement of the molecular size
of the Rh D, C, c and e antigens by radiation inactivation, a technique
involving the bombardment of red cell membranes with high energy electrons.
In general terms the dose of radiation required to destroy the biological
activity of a molecule will be inversely proportional to the size of the

4
molecule. Large molecules will present a larger target area to the
ionizing particle than will a smaller molecule and therefore a smaller
dose of radiation would be required to inactivate a large molecule than
a small molecule. The structure of the Rh antigens at a molecular level
is discussed on the basis of the results obtained in this study.
The next chapter, (IV) describes experiments designed to isolate and
purify the Rh (D) antigen after solubilization with the bile salt sodium
deoxycholate. Lorusso and Green (1975) found that Rh (D) antigen activity
could be restored in the material solubilized from Rh positive red cell
membranes after the removal of bile salt. Experiments were carried out
on similar lines except that the Rh positive red cell membranes were treated
with 125I-labelled anti-D before solubilization and the radioactive label
was used as a marker for the D antigen in subsequent purification procedures.
Finally, the experiments in chapter V are concerned with the
physiological significance of the antigen-antibody reaction in vivo with
special reference to haemolytic disease of the newborn. The experiments
were principally designed to investigate the possibility of the involvement
of erythrophagocytosis in the destruction of antibody-coated red cells in
ABO and Rh haemolytic disease. The in vivo factors which may modify the
results observed from phagocytic experiments conducted in vitro are considered.

ACKNOWLEDGEMENTS
I am indebted to all of the staff at the MRC Experimental Haematology
Unit, in particular I would like to thank Professor N.C. Hughes-Jones for
his excellent tutorship and patience, and Professor P.L. Mollison for his
helpful discussions.
I would also like to thank the staff in the department of
Radiotherapeutics at Addenbrookes Hospital, Cambridge for their assistance
and the use of the linear accelerator and Dr. J.C. Ellory for his
guidance during the radiation inactivation experiments.
I am very grateful to Mrs. Eileen Law for her excellent typing
and to my husband, David for his help and encouragement.
5

CONTENTS
TITLE
ABSTRACT
PREFACE
ILLUSTRATIONS
TABLES
CHAPTER I : THE BIOCHEMISTRY OF THE Rh ANTIGENS
1) History
2) Nomenclature
3) Early research into the chemistry of the Rh(D) antigen
4) Early evidence for a sialic acid structure
5) Early evidence for a protein structure
6) Recent experiments on Rh(D) antigen biochemistry
7) Rh null
8) Stereochemistry
9) Conclusions and speculations
CHAPTER II : INVESTIGATIONS INTO THE EFFECT OF PHOSPHOLIPASES
ON THE Rh(D) ANTIGEN
I) INTRODUCTION
1) The role of phospholipids in the red cell membrane
2) Experiments demonstrating the involvement of
phospholipids with the Rh(D) antigen
3) Phospholipases
a) General observations
b) The action of phospholipases on red cell membranes
i) Chemical
ii) Physical
c) The use of phospholipases in demonstrating the 46 phospholipid requirement for biological activity
d) Some conclusions from studies involving phospholipases 46
6
page
1
3
17
20
22
22
23
25
26
28
31
33
34
36
39
39
39
41
43
43
43
43
45

II)
B)
SECTION I
page
49
49
49 49
49
50 50
51
51
51
52
52
53
53
54
55
55
55
55
58
58
58
62
A) MATERIALS AND METHODS
1)
2)
3)
4)
5)
6)
7)
8)
9)
10)
11)
12)
13)
RESULTS
Enzymes
Red cells
Preparation of stroma
125I-labelled anti-D
Measurement of phospholipase A2 activity
Incubation of stroma with phospholipase A2
Incubation of stroma with phospholipase C
Assessment of Rh(D) antigen activity remaining
Analysis of membrane phospholipids
Measurement of the action of phospholipase A2
on antigen-antibody complex
Detection of solubilized D antigen after the
action of phospholipase A2
Measurement of the effect of phospholipase A2
and phospholipase C on intact red cells
Detection of protease activity in phospholipase
preparations
1)
2)
3)
4)
5)
Measurement of phospholipase A2 activity
Adtion of phospholipase A2 on stroma
a) The effect on Rh(D) antigen activity
b) The effect on phosphatidylcholine
Incubation of phospholipase A2 with the antigen-
antibody complex
Detection of solubilized D antigen after the
action of phospholipase A2
Action of phospholipase C on stroma
7

6) The effect of phospholipase A2 and C on
intact red cells
7) Detection of protease activity in
phospholipase preparations
C) DISCUSSION
III) SECTION II
A) INTRODUCTION
1) Phospholipid metabolism in the human red cell
membrane
a) Chemical reactions
b) movement of phospholipid molecules
c) Phospholipid exchange between red cells
and plasma
2) Conclusions
B) MATERIALS AND METHODS
1) Solutions
2) Attempts to restore Rh(D) activity with plasma
3) Attempts to restore Rh(D) activity by incubation
with linoleic acid in the presence of coenzyme A
and ATP
4) Attempts to restore Rh(D) activity with sonicated 73 lecithin
5) Experiments involving 32P-labelled phospholipids 73
a) Labelling whole blood with 32P 73
b) Lipid extraction from plasma 74
c) Lipid extraction from red cells 74
d) Attempts to restore Rh(D) activity to 74 phospholipase A2-treated stroma with sonicated 32P-labelled lipid extract from red cells and
plasma
8 page
62
62
65
68
68
68
68
69
70
70
71
71
72
72

9 page
C) RESULTS 75
1) Attempts to restore Rh activity with plasma 75
2) Attempts to restore Rh activity with linoleic acid, 76
Co A and ATP
3) Attempts to restore Rh activity with sonicated lecithin 76
4) Experiments involving 32P-labelled phospholipids 78
a) Labelling and lipid extraction 78
b) Attempts to restore Rh(D) activity with 78
sonicated 32P-labelled lipid extract from whole
red cells and from plasma
D) DISCUSSION 80
CHAPTER III : THE RADIATION INACTIVATION OF THE RHESUS ANTIGENS 83
I) INTRODUCTION 83
1) Direct effects 83
2) Indirect effects 83
3) Target theory 83
4) Dependence on radiation source 87
5) Calculation of molecular size from survival curves 87
6) Criticisms of target theory
90
7) The significance of molecular size determinations
92
II) SECTION I 93
A) MATERIALS AND METHODS 93
1) Red cells 93
2) Preparation of red cell membranes 93
3) Preparation of 125I-labelled anti-D 93
4) Anti-A 94

' 5) Radiation procedure
6) Measurement of Rh(D) antigen activity
7) Measurement of acetylcholinesterase activity
8) Measurement of A antigen activity
10 page
94
94
95
95
B) RESULTS 95
1) Rh(D) antigen activity 95
2) A antigen activity 97
3) Acetylcholinesterase activity 97
4) Temperature changes 97
C) DISCUSSION 99
102
102
103
103
103
105
103
103
104
10Z
10/
105
10:;
106
106
SECTION II
A) INTRODUCTION
B) MATERIALS AND METHODS
1) Antibodies
i) Anti-A
ii) Anti-D
iii) Anti-C
iv) Anti-c
v) Anti-e
2) Treatment of red cells with papain
3) Irradiation of red cell membranes
4) Spectrophotometric measurement of antigenic activity
a) Optimum antibody concentration determination
b) Calibration curves
5) Antigenic activity of irradiated membranes
6) Measurement of the remaining A antigen activity
after irradiation

11
page
C) RESULTS 106
1) Optimum antibody dilutions 106
2) Calibration curves 109
3) Antigenic activity after irradiation 109
a) D antigen 109
b) C antigen 109
c) c antigen 11';
d) e antigen 115
e) A antigen 115
D) DISCUSSION 115 1) Speculations on the molecular structure of the Rh antigens 120
2) Conclusions 128
CHAPTER IV : THE SOLUBILIZATION OF RED CELL MEMBRANES USING 130
SODIUM DEOXYCHOLATE
I INTRODUCTION 130
1) General methods of membrane disruption 130
2) Bile salts 134 3) Isolation of the D antigen 135
4) Proposed experiments 137
II MATERIALS AND METHODS 137
1) Preparation of red cell membranes 137
2) Anti-D 138
3) Preparation of 125I-labelled anti-D 138
4) Solubilizing buffer 138

page
5) Polyacrylamide gel electropheresis 139
a) Solutions 139
b) Preparation of 7.3% polyacrylamide gels 139
c) Gel electropheresis 139
d) Destaining gels 140
6) Preparation of Biobeads SM-2 140
7) The action of sodium deoxycholate on the Rh(D) antigen 141
a) Solubilizing red cell membranes with sodium 141
deoxycholate
b) Determination of the Rh activity of reaggregated 141
proteins
c) Separation of solubilized proteins with ultrafilters 141
8) The action of sodium deoxycholate on the Rh(D) antigen- 142
antibody complex
a) Treatment of red cells with 125I-labelled anti-D 1;2
followed by solubilization with sodium deoxycholate
b) Separation of solubilized proteins on Sepharose 4B
142
c) Measurement of the amount of combined antibody and
142 antigen after treatment with sodium deoxycholate
9) Attempts to purify the Rh(D) antigen
143
a) Separation of solubilized proteins on Sepharose 4B 143 followed by affinity chromatography on S-CNBr-anti-IgG
i) Purification of IgG anti-IgG 143
ii) Activating the S-CNBr 143
iii) Coupling the protein to S-CNBr 144
iv) Separating the solubilized proteins 144
12

13 page
b) The use of buffers with acid pH to split the D antigen- 145 antibody complex
i) The effect of pH on the stability of the D antigen 145 antibody complex
ii) The elution of deoxycholate-treated anti-D from 145
S-CNBr-anti-IgG at various pH values
c) The separation of solubilized proteins on S-CNBr- 146 anti-IgG
III) RESULTS 147
1) The action of sodium deoxycholate on the Rh(D) antigen 147
a) Solubilizing red cell membranes with sodium deoxycholate 147
b) Determination of the Rh(D) antigen activity of 147 reaggregated proteins
c) Separation of solubilized proteins with ultrafilters 151
2) The action of sodium deoxycholate on the Rh(D) antigen- 151 antibody complex
a) Treatment of red cells with 125I-labelled anti-D 151 followed by solubilization with sodium deoxycholate
b) Separating solubilized proteins on Sepharose 4B 153
c) Measurement of the amount of bound and free antibody 153 after treatment with sodium deoxycholate
3) Attempts to purify the Rh(D) antigen 158
a) Separation of solubilized proteins on Sepharose 4B followed 158
by purification on S-CNBr-anti-IgG
b) The use of buffers with acid pH to split the D antigen- 161 antibody complex
i) The effect of pH on the stability of the D antigen- 161 antibody complex
ii) The elution of solubilized anti-D from S-CNBr-anti- 161
IgG at various pH values
c) Separation of solubilized proteins on S-CNBr-anti-IgG 161

page
IV) DISCUSSION 169
CHAPTER V : OBSERVATIONS ON THE ROLE OF LEUCOCYTES IN THE 174
DESTRUCTION OF ANTIBODY-COATED RED CELLS IN
HAEMOLYTIC DISEASE OF THE NEWBORN
I) INTRODUCTION 174
II) MATERIALS AND METHODS 178 1) Red cells
170 2) White cells 178 3) Antibodies 178
a) IgG anti-D 178
b) IgG anti-A 179
c) Horse anti-human IgG 179
4) Tissue culture medium 179
5) IgM myeloma protein and IgG preparation 180
6) Experimental techniques 180
a) Dextran sedimentation 180
b) Opsonizing red cells with IgG antibodies 180
c) Preparation of leucocyte monolayers 180
d) Incubation of red cells with leucocyte monolayers 181
e) fixing and staining the coverslips 181
f) Microscopic examination 181
14

page
7) Experiments 182
a) Measurement of bound 125
I-labelled anti-D 182
or anti-A
125 b) Measurement of bound antibody using I-labelled 182
anti-IgG
c) Measurement of the effect of plasma constituents 182
on erythrophagocytosis
i) Measurement of phagocytosis in plasma 182 and serum
ii) Measurement of phagocytosis in the 183
presence of IgM
iii) Measurement of phagocytosis in the 183 presence of various concentrations of IgG
d) Red cell agglutination at low levels of bound 183 antibody and the effects of agglutination on
phagocytosis
i) Red cell agglutination at low levels of
183 antibody sensitization in plasma
ii) Measurement of the phagocytosis of
184 agglutinated red cells in serum
e) The effect of complement on erythrophagocytosis 184
III) RESULTS 185
1) Recovery of white cells
185
2) Microscopic examination 185
3) Measurement of the phagocytosis of red cells opsonized 186
with IgG antibodies
a) Ingestion of red cells coated with anti-D 186
b) Ingestion of red cells coated with anti-A 186
15

page
4) The effect of plasma constituents on erythrophagocytosis 187
a) Measurement of phagocytosis in plasma and serum 187
b) Measurement of phagocytosis in the presence of IgM 187
c) Measurement of phagocytosis in the presence of
195 various concentrations of IgG
5) Red cell agglutination at low levels of bound antibody 193
and the effects of agglutinated red cells on phagocytosis
a) Red cell agglutination in plasma at low levels of 193
IgG antibody sensitization
b) Measurement of the phagocytosis of agglutinated 196
red cells in serum
6) The effect of complement on erythrophagocytosis 196
197
1) The response of leucocyte monolayers to IgG anti-D and IgG 197
anti-A opsonized erythrocytes
2) Comparison of the amount of antibody on red cells in ABO 200 and Rh haemolytic disease and that required to induce
phagocytosis in peripheral blood leucocytes
3) The inhibition of erythrophagocytosis by serum IgG 200
4) Agglutination of red cells sensitized with IgG anti-D 201 and anti-A
5) The effect of complement 202
6) Speculations on the possible mechanism of red cell 203 destruction in haemolytic disease of the newborn
16
IV) DISCUSSION
CHAPTERS II, III, IV AND V SUMMARY AND CONCLUSIONS 207
REFERENCES 213

ILLUSTRATIONS
Page (a) The action of p-chloromercuribenzoate 29
(b) The chemical structure of a phospholipid molecule
A possible genetic pathway for the production of 32 Rh and LW antigens
(a) Common red cell phospholipids 40 (b) Red cell total and individual phospholipids
of adult blood
The positions on the phospholipid molecule at which 42
the various phospholipases attack
The enzymic activity of each of the phospholipase 56
A2 preparations
The decline in Rh(D) activity of red cell membranes 57 in response to various amounts of 3 preparations of
phospholipase A2
The degradation of phosphatidylcholine in response 59
to various amounts of 3 preparations of phospholipase
A2
The relationship between the amount of bound anti-D 60 and lecithin levels in stroma after treatment with
phospholipase A2
The decline in Rh(D) antigen activity of red cell 61 membranes after treatment with phospholipase C
The degradation of phosphatidylcholine in stroma after 63
treatment with phospholipase C
The relationship between the amount of bound anti-D 64 and lecithin levels in stroma after treatment with
phospholipase C
Fig III-1 (a) The decline in Rh activity of red cell membranes 96
after treatment with increasing doses of ionizing
radiations
(b) The decline in red cell membrane acetylcholin-
esterase activity after treatment with increasing
doses of ionizing radiations
1'7
Fig I-1
Fig 1-2
Fig II-1
Fig 11-2
Fig 11-3
Fig II-4
Fig 11-5
Fig 11-6
Fig 11-7
Fig 11-8
Fig 11-9

18
Page Fig III-2 The absorbance of red cell suspensions in the 107
presence of various antibody dilutions
Fig III-3 An example of a calibration curve prepared to show 108
the effect of adding various amounts of red cell
membranes to a specific antibody dilution before adding
red cells and measuring the absorption at 600 nm
Fig III-4 The decline in Rh(D) activity of red cell membranes 111
as measured by the binding of anti-D by spectro-
photometric technique, after treatment with
increasing doses of ionizing radiation
Fig 111-5 The decline in Rh(C) activity of red cell membranes, 112
as measured by the binding of anti-C by the spectro-
photometric technique, after treatment with increasing
doses of ionizing radiation
Fig 111-6 The decline in Rh(c) activity of red cell membranes, 115 as measured by the binding of anti-c by the spectro-
photometric method, after treatment with increasing
doses of ionizing radiation
Fig III-7 The decline in Rh(e) activity of red cell membranes, 114 as measured by the binding of anti-e by the spectro-
photometric technique, after treatment with increasing
doses of ionizing radiation
Fig 111-8 The conjugated operon model for the Rh antigens 116
Fig 111-9 Speculations on the molecular structure of the Rh 122 antigens
Fig IV-1 Red cell membranes separated on 7.3% SDS-polyacrylamide 131
gels stained for protein and glycoprotein
Fig IV-2 A possible arrangement for the major erythrocyte 135 membrane polypeptides
Fig IV-3 The actions of sodium deoxycholate on red cell membranes 148
Fig IV-4 The separation of deoxycholate solubilized proteins 152 on Sepharose 4B
Fig IV-5 The elution of the D antigen-antibody complex on 156 Sepharose 4B

19
page
Fig IV-6 The elution of the D antigen-antibody complex from 157 S-CNBr-anti-IgG
Fig IV-7 Polyacrylamide gels demonstrating the elution of the 159 D antigen-antibody complex from S-CNBr-anti-IgG
Fig IV-8 Polyacrylamide gels demonstrating the elution of 162 deoxycholate solubilized IgG from S-CNBr-anti-IgG
Fig IV-9 The elution of deoxycholate solubilized proteins on 163 S-CNBr-anti-IgG
Fig IV-10 Polyacrylamide gels demonstrating the elution of 164 deoxycholate solubilized proteins on S-CNBr-anti-IgG
Fig IV-11 Polyacrylamide gels demonstrating the elution of 166 deoxycholate solubilized proteins at various pH values
on S-CNBr-anti-IgG
Fig IV-12 Separation of deoxycholate solubilized proteins on 167
S-CNBr-anti-IgG
Fig IV-13 Polyacrylamide gels demonstrating the elution of
168 deoxycholate solubilized proteins at pH 3.0 from
S-CNBr-anti-IgG
Fig V-1
Fig V-2
(a) The relationship between the amount of 125I-
labelled anti-D bound to red cells and the degree
to which they were subsequently ingested by monocytes
(b) The relationship between the amount of anti-D
bound to red cells and the degree to which they were
subsequently ingested by monocytes (bound anti-D
measured with an 125I-labelled anti-IgG)
(a) The relationship between the amount of 125I-
labelled anti-A bound to red cells and the degree to
which they were subsequently ingested by monocytes
and neutrophils
(b) The relationship between the amount of anti-A
bound to red cells and the degree to which they were
subsequently ingested by monocytes and neutrophils
(bound anti-A measured with an 125I-labelled anti-IgG)
189
191

20 TABLES
page Table I-1 Terminology of the Rh antigens 24
Table 1-2 The published data on the molecular weight
of the Rh antigens 3'5
Table II-1 The action of various examples of phospholipases
on intact red cells 44
32 Table 11-2 The incorporation of inorganic P into red cell i
and plasma lipid
77
Table 11-3 The number of pmoles of labelled lipid incorporated79
into various phospholipids in the phospholipase A-
treated membranes
Table III-1 Individual values taken from 9 experiments of the 98
molecular size of the Rh(D) antigen
Table 111-2 The D37 and molecular size of the Rh antigens 110 as estimated from radiation inactivation data
Table 111-3 The available data on the molecular weight of the 124
Rh antigens according to Abraham and Bakerman
Table 111-4 The possible molecular weights of complexes of 126
specified Rh genotypes computated from the data
of Abraham and Bakerman compared with similar
measurements computated from radiation inactivation
data
Table IV-1 Determination of the Rh(D) activity of 149
reaggregated proteins
Table IV-2 Estimation of the amount of labelled anti-D
remaining bound to red cell membranes after
treatment with sodium deoxycholate and
reaggregation of the solubilized proteins
after dialysis
150
Table IV-3 Measurement of the amount of combined antibody 155 and antigen after treatment with sodium deoxycholate
and separation on Sepharose 4B

21
Table IV-4 The effect of pH on the stability of the D antigen- 160
antibody complex
Table V-1 The inhibitory effect of various amounts of IgG 192
on the ingestion of opsonized red cells
Table V-2 The agglutination, in plasma of red cells coated 194
with anti-D
Table V-3 The agglutination, in plasma of red cells coated 195
with anti-A
Table V-4 Comparison of the amount of antibody on red cells 199
in ABO and Rh haemolytic disease of the newborn
with that necessary to induce ingestion by
leucocytes in vitro

22 CHAPTER I
BIOCHEMISTRY OF THE Rh ANTIGEN
History
As a consequence of Landsteiner's discovery of the ABO blood groups,
at the beginning of the twentieth century, transfusions of blood between
humans became safer because donors could be selected after ABO grouping.
However, as more and more transfusions were given it became obvious that
there were intragroup haemolytic transfusion reactions. These occurred
mainly in subjects who had been previously transfused and presumably
became immunized by the transfusion of an antigen not present on their
own red cells.
In 1939 Levine and Stetson reported the case of a woman who had
just given birth to a stillborn child. This woman had a severe haemolytic
reaction to the transfusion of her husband's blood even though she had
never previously had a blood transfusion. The woman's serum agglutinated
the cells of 80 out of 104 ABO compatible donors. Levine and Stetson
suggested that the mother had become immunized by her foetus which had
inherited the antigen responsible from the father. The authors did
not name the antigen.
One year later, following some work by Wiener on the M and N antigens
Landsteiner and Wiener (1940) reported that when rabbits were immunized
with blood from the monkey Macacus rhesus, the resulting antibodies
agglutinated the red cells of 85 percent of Caucasians. Wiener and
Peters (1940) demonstrated that an apparent similar antibody to that
produced by the rabbits was present in the serum of certain patients who
had experienced incompatible transfusion reactions following the
transfusion of blood of the correct ABO group.

In 1941 Levine et al. showed that Erythroblastosis foetalis was
the result of Rh incompatibility between mother and foetus. Much later
(Levine, 1961) it was realized that the rabbit anti-Rh and human anti-Rh
are not the same. The rabbit antibody is now widely called anti-LW
after Landsteiner and Wiener.
Nomenclature
Very soon it became apparent that the Rh blood group system was
not simple. By 1944 the British (Race et al., 1944) had four antisera
and had defined seven alleles. Fisher (1944) noticed that two of the
four antisera were antithetical and he suggested that the antigens
(and genes) corresponding to these two antibodies, were 'allelic' and
called then C and c. The remaining two antisera were not antithetical.
He called their corresponding antigens D and E and proposed the existence
of their 'allelic' forms d and e. The Rh complex could then be made
in eight different ways:
CDe, cDE, cde, cDe, cdE, Cde, CDE and CdE.
Every individual would inherit one of the above gene complexes
from each parent.
Later on Fisher realized that in the English population some gene
complexes occurred more frequently than others:
CDe, cde, cDE 12% or over
cDe, cdE, Cde and CDE 3%
CdE very rare
Fisher (1946, 1947 and 1953) suggested that the rarer combinations
were the result of crossing-over from the more common heterozygotes.
For example a crossing-over between D and E in cDE/cde produces
cdE and cDe. He also suggested
23

TABLE I-1
TERMINOLOGY OF Rh ANTIGENS
NUMERICAL CDE Rh-Hr NUMERICAL CDE Rh-Hr
Rh1 D Rho Rh18 Hr
Rh2 C rh1 Rh19 hrs
Rh3 E rh11 Rh20 VS, es
Rh4 lir1 Rh21 CG
Rh5 hr11 Rh22 CE
Rh6 f, ce hr Rh23 Wiel, Dw
Rh7 Gs Ce rh'1 Rh24 ET
rh Rh8 Cw wl Rh25 LW
Rh9 CX re Rh26 'Deal'
Rh10 V, ces hry Rh27 cE
Rh11 Ew rhW2 Rh28 hr
H
Rh12 G rhG Rh29 'Total Rh'
RhA a Rh13 Rh30 GO
Rh14 RhB Rh31 hrB
RhC
Rh15 Rh32 determined by R N
RhD 0 Har
Rh16 Rh33 determined by R
Rh17 Hro Rh34 Bas
‘11

25
that C lies between D and E because the frequency ratio of cdE to the
heterozygote CDE/cde is larger than the ratios of Cde to CDe/cde (cross over
between C and D) and of CDE to CDe/cDE (cross over between C and E).
Fisher's ideas were critised by Wiener who believed that a multiple
allelic gene determines the production of an entire Rh antigen (agglutinogen).
He developed the Rh-Hr system of nomenclature shown in Table 1. As the
discovery of more antibodies and antigens increases the complexity of the
Rhesus system, neither the CDE nor the Rh-Hr systems are really adequate and
eventually a numerical system similar to that proposed by Rosenfield (1973)
will have to be adopted. The three systems are shown in Table I-1. Since
the CDE system is more easily appreciated than any of the others it will be
used throughout the following chapters.
Early Iesearch into the chemistry of the Rh(D) antigen
Soon after the discovery of the Rh complex, research on the biochemical
structure of the antigen began. Much of this early work was limited to looking
for substances which would inhibit the reaction between the D antigen and its
corresponding antibody, and which consequently may be chemically-similar in
some way to the antigen. This method of investigating antigenic structure
has many disadvantages, principally because such substances may inhibit the
antigen-antibody reaction by destroying the binding sites in a nonspecific
manner rather than by reacting with the antibody directly.
In 1946 Calvin found the Rh activity to be confined to an ether-soluble
fraction of stroma, probably lipoprotein in nature. This work was repeated
and extended by Moskowitz et al. (1950a) who found that the fraction containing
Rh activity was water-soluble. He called it elinin, and suggested that it may
be a protein or a simple molecule attached to the protein portion of elinin

2G
because the Rh factor was unstable under conditions in which proteins are
denatured. Moskowitz was unable to demonstrate the production of antibodies
in rabbits, guinea pigs or humans in response to Rh elinin.
In 1951 Howe suggested that the influenza virus receptor, the Rh and the
ABO antigens were part of a complex. At the same time Moskowitz and Treffers
(1950b) and Morgan and Watkins (1951) described the destruction of Rh (D)
antigen by periodate and Bigley et al. (1958) demonstrated inhibition of Rh (D)
antibody by eluates of erythrocytes treated with mumps virus or periodate. tt tt Makela et al. (1959) could not repeat the above results with mumps virus, due,
according to Dodd et al. (1964) not to differences in the haemagglutinating
capacity of their virus, but to its lack of enzymic activity on urinary
mucoprotein inhibitor. This is related to the neuraminidase activity of the
virus.
Hackel (1958) reported that some ribonucleic acid derivatives e.g. adenylic
acid and uridylic acid inhibited anti-D sera. One year later Boyd (1959)
reported that anti-D sera were inhibited weakly but specifically by L-glucose,
L-mannose, or D-glucose. The effect was not observed with other monosaccharides
or with the nucleic acid derivatives that Hackel used. Bogoch (1958) found
that when brain gangliosides were injected into rabbits antibodies to the
ganglioside were produced. He suggested that this might be a molecule suited
to function at membrane surfaces.
Early evidence for a sialic acid structure
In 1960 Dodd et al. reported that N-acetyl neuraminic acid and certain
related compounds inhibit anti-D. This was confirmed by Boyd and Reeves (1961)
who also noted that colominic acid, a polymer of N-acetyl neuraminic acid

27
produced by certain strains of E. Coli, was also inhibitory. On trying to
repeat this work Johnson and McCluer (1961) could only find a slight inhibition
by crude, impure, sialic acid preparations. To account for the failure of
Johnson and McCluer (1961) to repeat his results, Dodd et al.(1963)emphasized
that the inhibition by N-acetyl neuraminic acid is dependent on the amount of
antibody, time of incubation and reversibility of reaction. Yokoyama et al.
(1963) could not repeat the production of ganglioside antibodies by intravenous
injection of ganglioside solution described by Bogoch (1958) unless the gang-
liosides were in complete Freund adjuvant and injected with foreign proteins.
Dodd et al. (1964) was able to demonstrate antibodies to gangliosides and
Rh (D) on injecting ganglioside in complete Freund adjuvant. Rh antibody
and antibody to the ganglioside appeared eleven days after the first injection.
The maximum titre of the anti-ganglioside being 32 and 256 for trypsinized
Rh (D) positive red cells. The latter was specific for the D antigen. The
results were limited due to the small amount of available ganglioside and if
the red cells were not trypsinized the titre was much lower (1:8 being the
highest). In the same year (1964) Johnson and Dodd also reported a specific
inhibition of anti-D by human urinary mucoprotein which is about 9.1% sialic
acid. However, a maximum agglutinating dose of anti -D was not inhibited by
the mucoprotein unless the cells were treated with a 1% solution of trypsin.
A saline suspension of red cells treated with trypsin will agglutinate
in the presence of incomplete antibody. This effect is not limited to trypsin-
treated red cells. Treatment of red cells with bromelin, ficin, papain and
neuraminidase all produce this effect. Bromelin, ficin and papain are all
thiol proteases with a sulphydryl group at the active site which requires
activation by a reagent e.g. cysteine, which frees the sulphydryl. In
contrast, trypsin is a serine protease with a very reactive serine residue

28
at the active site. Neuraminidase is not a protease; however, it shares
with the four proteases the ability to effect release of sialic acid from the
treated cells (Prager and Fletcher, 1966). There is a progression in N-acetyl
neuraminic acid release and agglutination as a function of neuraminidase
concentration. The amount of agglutination for a given quantity of N-acetyl
neuraminic acid released varies with the genotype of the cell. The release
of N-acetyl neuraminic acid is consistent with the decrease in negative charge
on the red cells which occurs during the first ten minutes of incubation with
trypsin (Prager and Fletcher, 1962) and may lead to sufficiently decreased
repulsion between sensitized cells to permit agglutination to occur. The
agglutination may be a thermodynamic process (Prager and Fletcher, 1962).
Enzyme treated cells have a greater capacity for binding globulins and there
may also be less bound water when antibody and antigen interact. The release
of the bound water would result in an increase in entropy and provide a driving
force for agglutination.
Springer and Tegtmeyer (1964) reported a specific inhibition of anti-D by
extracts from twigs of angiospermous plants. The extracts were not inactivated
by boiling or ethanol treatment and the authors concluded that it was likely
that the material was different physically and chemically from the Rh (D)
antigen.
Early evidence for a protein structure
Various researchers have found evidence leading to the conclusion that
the Rh antigen is, at least in part, protein in nature, and that one or more
disulphide bonds and one or more free sulphydryl groups are required for activity.
As previously mentioned, Moskowitz et al. (1950) studied ether-extracted stroma
and recovered a heat-sensitive material with Rh activity which was probably
protein in nature although the Rh activity could be detected only in lipid-rich
fractions.

(a) Reversible action of parachloromercuribenzoate
1. R-SH + C1HgC6H4COOH
RSHgC6H4COOH + HC1
PCMB SH-MB
2. RSHgC6H4COOH + Rt-SH \ R-SH RISHgC6H4COOH
e.g. 2-Mercaptoethanol
(b) Chemical structure of a phospholipid
0
CH2-0-C-R1 0 tt
R2-C-0-CH
0
CH2-0-P-O-BASE
Fig I-1 (a) The action of p-chloromercuribenzoate
(b) The chemical structure of a phospholipid molecule
29

30
Green (1965) noticed that Rh antigenic activity was lost following
treatment with certain sulphydryl reagents. Failure to bind antibody
following p-chloromercuribenzoate treatment was reversible suggesting that
major structural changes were not necessary to bring about the loss of activity.
(illustrated in Fig I-la).
In 1968 Green found that the Rh (D) antigen activity of human erythrocyte
membranes was lost following extraction with 100% 1-butanol, but could be
regenerated by the addition of the membrane extract or of a chloroform-methanol
extract of human plasma. Studies on this extract indicate that the active
component was phosphatidylcholine (shown ii Fig1-16) and that it must contain
unsaturated fatty acids. The same year (1968) Weicker reported that the Rh
factor was a small molecular weight peptide which was liberated from the
erythrocyte membrane by hemolysis of Rh (D) cells in water. The peptide was
free of lipids but contained a small amount of xylose. In later reports
(1971 and 1973) the molecular weight of the peptide was found to be 5,000-
e 6,000 daltons with a, peptide contint of 96%. There were fourteen different
amino acids and a small amount of liquid phosphorus, not due to phosphatidyl-
choline. However, Weicker was unable to establish the antigenicity of the
protein by the haemagglutination inhibition test or by any of the usual tests.
He used the 'Schultz-Dales technique to prove antigenicity (the antigen-antibody
reaction causes the release of an anaphylatoxin which triggers the contraction
of uterus muscle segments in the guinea pig). Weicker's results have not been
substantiated as yet and doubts have been raised by at least one group of
workers (Fisher et al., 1970).
Green (1972) published more experiments on red cell membrane lipids and Rh
antigen activity. He had previously (1968) found that antigen activity was
abolished after extraction with 100% 1-butanol, but could be regenerated to about
50% of the unextracted membrane activity by the addition of certain lipids.

31
He found that phospholipids were the only class of lipid that would result
in regeneration of antigenic activity. The binding of aqueous sonicated
phospholipids was associated with the best regeneration. Labelled
phosphatidylcholine showed little binding to the membranes, and large amounts
of unlabelled phosphatidylcholine only slightly depressed the binding of the
labelled phospholipid, which suggests that the binding of phospholipid is
mainly hydrophobic. Green concluded that Rh (D) antigenic activity is
dependent on the presence of bound phospholipids containing at least one
unsaturated fatty acid with neither the polar nor the nonpolar portion of the
molecule alone satisfying this requirement.
Recent experiments on Rh (D) antigen Biochemistry
Floyd Green working in Buffalo, New York, and Abraham and Bakerman from
Virginia, USA are responsible for much of the most recent work on the bio-
chemistry of the Rh antigen.
Abraham and Bakerman (1975a) claim to have isolated the Rh (D) antigen,
and also the c (1974), C (1975b) and E (1976) antigens by solubilising red cell
stroma with EDTA followed by dialysis against saline and ultrafiltration.
They detected Rh (D) activity in the fraction of molecular weight 10,000-20,000
daltons. After purifying the fraction by iso-electric focusing they injected
it into guinea pigs and obtained a high titre anti-D. The molecular weight
estimations for the c, C and E antigens were: 20,000-30,000; 50,000-100,000
and 50,000-100,000 respectively.
Lorusso and Green (1975) used detergents in an attempt to isolate the
antigen. Freeze-dried stroma was treated with deoxycholate and spun at high
speed. The solubilised proteins in the supernatant were put through Biobeads SPS-2
SUBSTANCE 1
32
'REGULATOR' GENES X°X° N
'REGULATOR'
GENE X'
SUBSTANCE 2 Rhnull No Rh or LW antigens
CDE
GENES
N NO CDE GENES
(---/---)
N V
Rh ANTIGENS Rh null No Rh or LW antigens
LW GENE NN NO LW GENE
(1w/lw)
V Rh and LW ANTIGENS Rh but no LW antigens
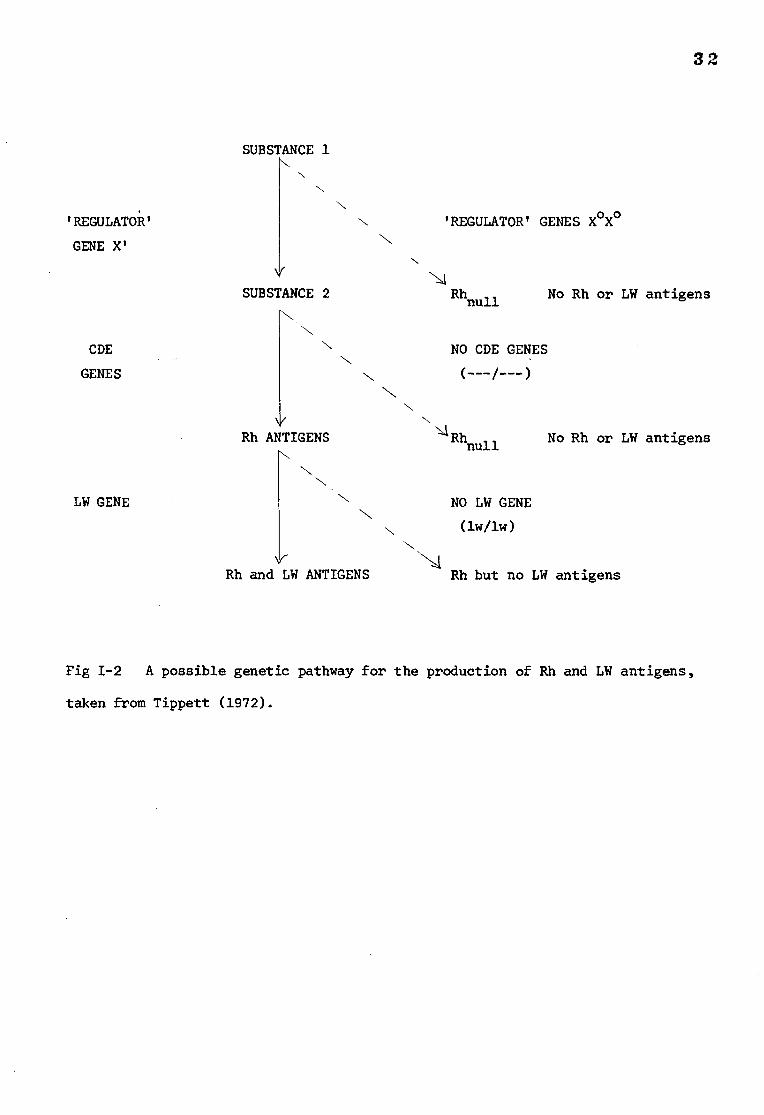
Fig 1-2 A possible genetic pathway for the production of Rh and LW antigens,
taken from Tippett (1972).

33
to remove the detergent and then dialysed against buffered water containing
magnesium ions. After several days a precipitate formed which was found to
have Rh (D) antigen activity. Attempts to partially purify the antigenic
material indicated a molecular weight of less than 300,000 daltons.
Rhnull
Rhnul1 red cells do not express the CDE antigens. There are two
conditions in which Rhnull
can occur. Firstly by the action of a 'regulator'
gene, not part of the Rh complex locus, which blocks the synthesis of Rh
antigens when present in double dose. The parents of some individuals
affected in this way (who only have a single dose of the repressor gene) show
depressed Rh antigen activity. Alternatively, and more rarely, the Rhnuil
condition can result from the action of amorph genes which are a part of the
Rh complex locus. In this case no Rh antigens are produced. A possible
genetic pathway for the biosynthesis of Rh and LW antigens has been suggested
by Tippett (1972) and is shown in Fig 1-2.
Individuals with Rhnull
cells often have anaemia, shortened red cell
survival, stomatocytosis, increased red cell fragility, mild spherocytosis and
a raised reticulocyte count. Rhnuil cells also may have abnormalities in the
MNSsU system (Schmidt and Vos, 1967 and Schmidt et al., 1967). There is no
evidence for linkage between the structural gene loci of Rh and MNSsU.
Schmidt et al. (.1967) concluded that there was a possibility that "the
aberration is one of sequential action of genes controlling shared terminal
sugar(s) giving various specificities depending on the precursor substance".
Lauf and Joiner (1976) studied K+ influx and 3H- ouabain binding in
Rhnull
cells as compared to normal Rh positive red cells. Rhnull
cells appear
to have a membrane defect. The tendency to hemolyse suggested a defect in cell

34
volume regulation. Their findings were consistent with the idea that Rhnull
cells have more Na+K+ pumps.
Smith et al. (1973) investigated the lipid-protein interactions in
normal Rh positive cells and Rhnull cells. They found that the fluorescence
intensity of the membrane bound probe 1-anilino naphthalene 8 sulphonate (ANS)
and the labelling of sulphydryl groups with N-1- (14c) -ethyl maleimide increased
after treatment of normal q Rh(D) positive erythrocyte membranes with
phospholipase A2. In contrast treatment of 0 Rhnull erythrocyte membranes
with phospholipase A2 did not result in increased fluorescence intensity or
an increase in sulphydryl group labelling. Smith et al. (1973) concluded that
hydrophobic bonding between n-fatty acid side chains on lipids and nonpolar
regions of al etric proteins is necessary for maintaining the structure of
the Rh (D) membrane. The red cell membrane of Rhnull
individuals are not
noticably deficient in any of the major proteins visible after polyacrylamide
gel electropheresis (unpublished observations by N.C. Hughes-Jones) neither
are the lipids noticably abnormal (Sturgeon, 1970). These observations
suggest that the Rhnull condition results in the altered properties of a
molecule or molecules rather than the complete deletion of one of the membrane
proteins, and this alteration results in the apparent membrane defect.
Stereochemistry
Nicolson et al. (1971), using Rh positive red cells sensitized with 1251-
labelled anti-D, lysed and stained with ferritin conjugated anti-human gamma
globulin, found that the Rh (D) antigenic sites appeared to be molecularly
dispersed on the membrane surface in a random two-dimensional array. In a
similar study using gold anti-IgG reagent Romano (1975) also found that the two
dimensional distribution of sites was random. Following papain-treatment of

35
Antigen
TABLE 1-2
Authors Molecular Weight
1 D 5-6,000 Weicker et al. (1968)
2 D 10,000-20,000 Abraham and Bakerman (1975a)
3 c 20,000-30,000 IT TT (1974)
4 E 50,000-100,000 IT 11 (1976)
5 C 50,000-100,000 tt ti (1975b)
6 D 300,000 Lorusso and Green (1965)
7 D 174,000+10,000 Folkerd et al. (1977)
TABLE 1-2 The published data on the molecular weight of the Rh antigens.

36
Rh positive cells a clustered distribution of sites was evident. This was
also seen in -D- cells without enzyme treatment; these cells have more D sites
than normally found on Rh positive red cells. Romano's results also indicated
that D and c antigen sites may be located close together on R2R2 red cells.
Conclusions and speculations
Investigations into the biochemistry of the Rh antigen have been
summerized briefly in the previous pages. The information which has been
gained is surprisingly limited and probably reflects not only the complexity
of the antigen but also a lack of sensitive tools with which to probe membrane
surfaces.
It would be very interesting to know how antigens and antibodies react
at a molecular level, to compare their mode of action with those of enzymes and
substrates, to understand their significance, and their role, if any, in
membrane structure and biochemistry.
The limited data on the molecular weight of the substance or substances
on which the Rh antigens are expressed is shown in Table 1-2. However it is
always difficult to evaluate information on the molecular weight of substances
extracted from biological membranes because isolation procedures and the
molecular environment of the purified molecule can lead either to polymerisation
and aggregation or to fragmentation of the original molecule leading to
anomolous results. Information regarding the type of molecule on which the
D antigen is carried favours a protein structure which interacts with
phospholipid molecules for full expression of antigenic activity. The membrane
defect in Rhnull cells suggests that expression of the Rh antigens is a
prerequisite for the normal maintenance of the integrity of the red cell
membrane.

37
Whether all the antigens in the Rh complex can be represented on one
molecule or whether there are different molecules for each one remains to be
seen. Perhaps the simplest way of visualising the Rh complex would be to
imagine each antigen, whose synthesis would be directed by the CDE genes, as
a specific sequence of amino acids in a polypeptide chain, which is orientated,
as a result of the tertiary and quarternary structure of the protein to expose
the antigens at the exterior surface of the membrane. The folding of the
polypeptide chain would be influenced by the constituent amino acids and would
be subtly altered by the composition of the antigenic complex dictated by the
CDE genes. Therefore the chains of amino acids and their spatial orientation
at the membrane surface would be different for each phenotype and would react
with antibodies with corresponding specificity. Interactions with surrounding
lipid or protein molecules are probably also important if not essential in the
expression of full antigenic activity.
In an attempt of investigate the physiology and biochemistry of the Rh
antigens, experiments were undertaken with the following aims and methods:
1. To investigate the phospholipid requirement for antigenic activity.
In recent years phospholipases have been used as a tool for investigating red
cell membranes and it was considered that their use in investigating the effect
of phospholipids on Rh (0) antigen activity might provide some interesting
information.
2. To determine the molecular size of the D antigen so that it might be
possible to identify the membrane protein on which the antigen is carried. The
molecular size was measured by radiation inactivation. This method does not
require purification of the active molecule and in combination with more
conventional techniques can provide information regarding the physical state of
the molecule in its normal environment.

3. To isolate and purify the D antigen after the initial treatment of
the red cell membrane with sodium deoxycholate.
4. To examine the physiological significance of the D antigen-antibody
reaction in vivo with special reference to haemolytic disease of the newborn.
The experimental detail and results are described in the following
chapters.
38

39
CHAPTER II
INVESTIGATIONS INTO THE EFFECT OF PHOSPHOLIPASES ON THE Rh (D)
ANTIGEN
INTRODUCTION
The role of phospholipids in the red cell membrane
The human red cell membrane is composed of approximately 50% w/w
protein, 40% w/w lipid and 10% w/w carbohydrate. The lipid fraction can
be subdivided into phospholipids and neutral lipids - mainly cholesterol
and glycolipids. The structures of some of the commonly occurring membrane
lipids are shown in Fig II-1 with the normal values for the various phospholipids
found in red cells and plasma. Since phospholipids have both acid and basic
groups they behave as zwitterions and because they have both hydrophilic and
hydrophobic groups they are somewhat soluble in both water and fats which
suggests that they may be suitable as structural materials in the cell.
Most modern theories of membrane structure, for example that of Singer and
Nicolson (1972) propose a major role for phospholipids in the membrane.
The bulk of the membrane lipids are thought to form a discontinuous bilayer
in which the globular membrane proteins are embedded. Both lipids and
proteins are arranged with polar groups facing into the aqueous phase and
nonpolar groups in the hydrophobic membrane interior. It is quite probable
that there are interactions between the lipids and proteins in the membrane.
The presence of phospholipids on the exterior of the cell membrane and
their known involvement in the activities of certain enzymes on the cell
surface (Jurtshuk et al., 1961) prompted Green (1968) to investigate the
effect of phospholipid extraction on the Rh (D) antigen.

Fig II-1
(a) Common red cell phospholipids
1 0
CH -0-C- R 2
9 CH2 - 0 - P - 0 - CH2CH2N(CH3)3 0-
2 1
CH - 0 - C - R2
CH2 - 0 - P - 0 - 0 - CH2CH2NH3
PHOSPHATIDYLCHOLINE PHOSPHATIDYLETHANOLAMINE
(Lecithin)
2 CH - NH - C - R
0 NH
CH2 - 0 - P - 0 - CH2 - CH3
CH2 - 0 - P - 0 - CH2CH2N(CH3)3 0- co 3 0-
PHOSPHATIDYLSERINE SPHINGOMYELIN
(b) Red cell total and individual phospholipids of adult blood
(including standard deviations)
TOTAL PHOSPHATIDYL PHOSPHATIDYL PHOSPHATIDYL SPHINGOMYELIN
PHOSPHOLIPID ETHANOLAMINE SERINE
(pmoles x 10-10 / cell)
CHOLINE
RED CELLS 4.11 1.18 0.62 1.24 1.00
+ S.D. 0.37 0.13 0.06 0.14 0.10
TOTAL PHOSPHATIDYL PHOSPHATIDYL SPHINGOMYELIN LYSOLECITHIN
PHOSPHOLIPID ETHANOLAMINE CHOLINE
(pmoles /
PLASMA 2.88 0.13 1.90 0.53 0.21
+ S.D. 0.69 0.02 0.52 0.12 0.11
9 CH - 0 - C - R
40
9 CH - 0 - C - R
0 CH -0-C- R 2 1
LH -0-C- R
CH(OH) - CH = CH - (CH2)12CH3 0 rr
Taken from Hurter et al. (1970)

41
Experiments demonstrating the involvement of phospholipids with the
Rh (D) antigen
In 1968 Green reported that Rh antigen activity was abolished after
treatment of erythrocyte membranes with 1-butanol, but could be restored
by the addition of the butanol extract or of a chloroform-methanol extract
of human plasma. His studies indicated that the active component of the
lipid extract was phosphatidylcholine (lecithin). A later paper (Green, 1972)
established that the best regeneration of Rh activity was associated with
the addition of aqueous, sonicated lecithin and his results led him to the
conclusion that Rh antigen activity was dependent on the presence of bound
phospholipid, containing at least one unsaturated fatty acid.
Leddy et al. (1970) investigated the effect of lipid extraction
using butanol on various red cell antigens. They demonstrated a loss of
binding capacity for anti-D, anti-C, anti-E, anti-c, anti-e and various
unspecified IgG autoantibodies. Blood group A, B and H antigens were
unaffected.
Additional evidence for the lipid requirement of the Rh (D) antigen comes
from the experiments of Weicker et al. (1973). Weicker has isolated a membrane
protein with D antigen activity as measured by the Schultz-Dale test. He
failed to demonstrate any antigen-antibody reaction i.e. muscle contraction,
after incubation of the muscle with phospholipase A2 indicating that the
phospholipid requirement of the purified membrane protein is presumably
satisfied by phospholipids from the uterus muscle. If the purified membrane
protein was recombined with phosphatides of synthetic lecithins containing
oleic or linoleic acid at the C-2 position on the glycerol backbone of the
molecule, a positive Schultz-Dale reaction was obtained even after treatment
of the muscle segments with phospholipase A2.
R2 - C - - CH
PHOSPHOLIPASE A2
PHOSPHOLIPASE D
0
- 0 - P - 0 - BASE
1 0
PHOSPHOLIPASE C
CH2
PHOSPHOLIPASE Al
1 2 CH2 - 0 - C - R1
42
0
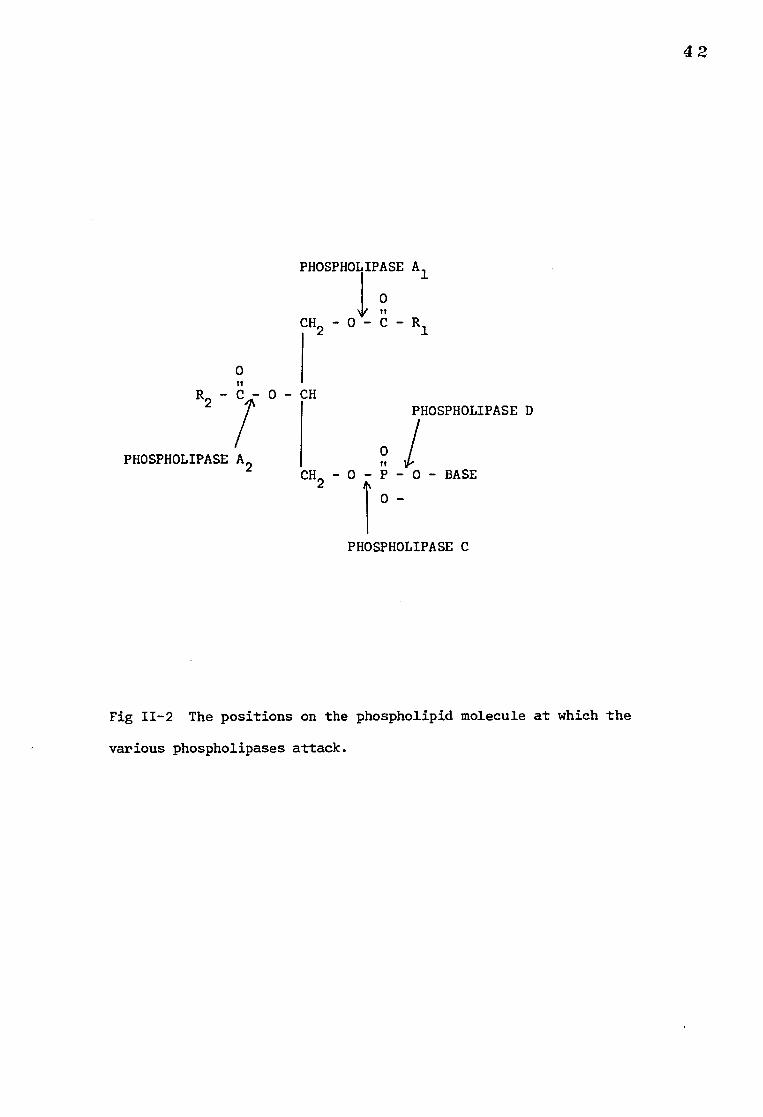
Fig 11-2 The positions on the phospholipid molecule at which the
various phospholipases attack.

43 Phospholipases
a) General observations
The positions at which the various groups of phospholipases attack
phospholipid molecules are shown in Fig. 11-2. Phospholipase A2 (PLA2)
and phospholipase C(PLC) are the two most commonly used classes of this
enzyme. PLA2 cleaves the fatty acyl ester bond on the C-2 position of the
glycerol backbone of most phospholipids except sphingomyelin, in which this
linkage is replaced by a peptide bond. PLC catalyses the hydrolysis of
phospholipids to diglycerides and water-soluble phosphorylated amines.
However as will be shown later, the amounts and types of products released on
treatment of membranes with enzymes from different sources vary as a result of
the substrate specificity and purity of the enzyme concerned.
In general phospholipases require calcium ions for activity, are heat
stable and have constrained tertiary structures due to many disulphide bonds
(Tsao et al., 1975). Although most are active as monomers with a molecular
weight around 15,000, the phospholipase A2 from rattlesnakes (Crotalus
adamanteus and Crotalus atrox) and from bel venom (Apix mellifica) are dimers
(Wells, 1971 and Tsao et al., 1975).
b) The action of Phospholipases on red cell membranes
i) chemical
The phospholipids of intact red cell membranes are much more resistant
to the action of phospholipases than those in the isolated membranes (Ibrahim
and Thompson, 1965). Depending on the enzyme used, intact red cell membranes
may undergo: (a) haemolysis and phospholipid attack, or (b) no haemolysis
but phospholipid attack, or (c) no haemolysis and no phospholipid attack.

etl R:t1
1:AW171..
ENZYME SOURCE DEGRADATION OF HAEMOLYSIS AUTHORS
PHOSPHOLIPIDS
PLA2 Pancreas No No Zwaal et al. (1975)
PLA2 C. adamanteus It It tl
PLC B. cereus TI Vt ft
PLD Cabbage
PLA2
Whole sea snake venom Yes No Ibrahim & Thompson. (1965)
PLA2 Cobra venom Condrea et al. (1970) — —
PLA2 Bee venom Zwaal et al. (1975)
PLA2 Naja Naja Gul & Smith (1974)
PLC C. welchii Yes Zwaal et al. (1975)
Table II-I: The action of various examples of phospholipases
on intact red cells.

45
On the other hand isolated red cell membranes normally undergo phospholipid
attack in the presence of phospholipases quite readily. On average approxi-
mately 70% of the membrane phospholipids are degraded by the action of
phospholipase A2 (Kahlenberg and Banjo, 1972) or phospholipase C (Glaser et al.,
1970 and Finean et al., 1971) on isolated red cell stroma.
The reactions of a few examples of phospholipases with intact cells are
shown in Table II-1. Gut and Smith (1974) reported that with phospholipase
A2 from Naja Naja venom haemolysis did occur if bovine serum albumin was added.
They suggested that this was caused either by removal of cleaved fatty acids
by albumin leading to a weaker membrane which was unable to contain haemoglobin
or possibly by removal of cleaved lysophosphoglycerides by albumin.
ii) Physical
Glaser et al. (1970) found that membranes treated with PLC (B.cereus)
remained intact, although shrunken, and contained some nodules. In agreement
with this are the findings of Finean et al. (1971) who found a 45-55% decrease
in the surface area of ghosts after PLC (c. Welchii) and Colman et al. (1970)
and Colley et al. (1973) who reported the appearance of black dots in ghosts
after PLC treatment and decided that they were composed predominantly of
diglycerides formed as a product of the hydrolysis of phospholipids.
The circular dichroism spectrum, reflecting the average conformation of
the proteins in the membrane, was unaltered after treatment with PLC (Glaser
et al., 1970) but the same authors showed that the proton magnetic resonance
spectra demonstrated a changed physical state of the fatty acid chains of the
phospholipids in the membrane, about three quarters of the fatty acid chains
became much more mobile than in the untreated membrane.

46
With intact cells Zwaal et al. (1975) noticed that the action of
phospholipases resulted in an increase in osmotic fragility, but not always
lysis. Allan et al. (1975) using PLC (C.perfringens) on intact cells found
that up to 30% of the membrane phospholipids could be broken down without
significant cell lysis but there was a morphological change in the erythrocytes
resulting in spherical cells containing internal membrane vescicles.
Verkleij et al. (1973) found that the action of PLA2 did not change the
freeze-etch morphology of intact cells but incubation of ghosts with the
same enzyme resulted in a complete reorganization of the membranes.
c) The use of phospholipases in demonstrating the phospholipid
requirement for biological function
The action of phospholipases on red cell membranes has been linked with
loss in activity of various membrane-associated biologically active processes.
For example, Kahlenberg and Banjo (1972) reported that PLA2 treatment of human
erythrocyte membranes produced a decrease in glucose uptake activity of 75%.
In contrast, hydrolysis of approximately 64% and 46% of the membrane
phospholipid phosphodiester linkages by PLC and PID respectively resulted in
only a 25% decrease in D-glucose uptake activity. Acetylcholinesterase
activity was not affected but there was a decrease in the activity of the
erythrocyte membrane (Na2+
+K+)-activated ATPase which was also more marked
after phospholipase A than after PLC or PLD. In addition, Coleman and Bramley
(1975) found that human erythrocyte (Ca2+, Mg
2+).•-, ATPase activity is lost as
a result of treatment with PLC (C. welchii) and could be reactivated by a
mixed lipid preparation.
d) Some conclusions from studies involving Phospholipases
The information gained from experiments involving the action of
phospholipases on cell membranes up until the present time is limited and open
to criticism.

47
It was accepted that, because phospholipase C attacked phospholipids in
isolated membrane preparations, this was evidence that phospholipid molecules
were present on the surface of the membrane, orientated with the polar heads
facing outwards and the hydrophobic fatty acid side chains on the inside of the
membrane. However, the action of phospholipases on intact red cells showed
that the situation was not as simple as this and illustrates that the membranes
in red cell ghosts are by no means identical to those in the intact cell.
Considering the subtle variations in substrate specificity demonstrated by
phospholipases, the difference in the reaction of these enzymes from differing
sources to the phospholipids in intact red cells was thought to reflect an
asymetric distribution of phospholipids in the membrane (Zwaal et al., 1973)
but Martin et al. (1975) and Taguchi and Ikezawa (1976) have found that the
susceptibility of phospholipids to attack by phospholipases is a function of
many variables, the most critical is probably the accessibility of the enzyme
to that part of the phospholipid molecule which satisfies its individual
substrate requirement. Evidence for the asymetry of phospholipids in
erythrocyte membranes must come from other studies.
The observations that, even under optimal conditions, up until now, a
maximum of only 70% of the membrane phospholipids are degraded by phospholipase
enzymes suggested to Glaser (1970) that 25-30% of the phospholipids are in a
physical state different from the remainder of the lipids, and suggested that
they are involved in a more tightly coupled interactionwith membrane proteins.
In agreement with this are the findings of Marinetti et al. (1973). The
results of these authors, obtained from experiments using cross-linking agents,
indicated that 20% of the total membrane phospholipids are closely associated
with proteins.

4 3
The information obtained regarding the phospholipid requirement for the
activity of various membrane-associated enzymes, for example (Ca2+ Mg2+) ATPase
(Coleman and Bramley, 1975) and glucose uptake (Kahlenberg and Banjo, 1972)
was very interesting and prompted these investigations into the phospholipid
requirement of the Rhesus D antigen in isolated membranes and intact cells.
Initially several experiments were designed to investigate the Rh (D)
activity of Rh positive red cell membranes after treatment with various
phospholipase preparations and these experiments are described in section 1.
Rh activity was found to be decreased after phospholipase treatment and
therefore it was decided to attempt to restore Rh (D) activity to the treated
membranes. The experimental techniques utilised for this purpose are
described in section 2.

49 SECTION 1
THE ACTION OF PHOSPHOLIPASE A2 AND C ON THE Rh (D) ANTIGEN
A) METHODS AND MATERIALS
1) Enzymes
Purified preparations of phospholipase A2 (EC 3.1.1.4) from Vipera
russellii (Koch-Light), Beal venom (Sigma) and Crotalu: terr. terr.
(Boehringer, Marnheim) were used. The purity of the preparatiohc was
examined by electropheresis on cellulose acetate paper using barbiturate
buffer, pH 8.9. The Viper and Bee preparations showed only a single band,
, The Crotalus preparation showed a mjaor protein band and an additional minor
band. The phospholipase C (EC 3.1.4.3) was a purified preparation from
Clostridium perfringens (Koch-Light).
Phospholipase A2 from Naja Naja (Koch-Light Laboratories) was used
in some experiments involving intact red cells.
2) Red Cells
Group 0 Rh positive (CDe/cDE) and Rh negative (cde/cde) erythrocytes
were used. The erythrocytes were stored in acid-citrate-dextrose solution
at 4°C for up to 3 weeks before using.
3) Preparation of stroma
Red cell stroma was prepared by the method of Dodge et al. (1963) using
20 mOsm phosphate buffer pH 8.0. The stroma was made up to the original
volume of packed cells with 20 mOsm phosphate buffer and stored at 4°C.
Penicillin (100 pg/m1) and streptomycin (100 pg/m1) were added to prevent
bacterial growth.

50
4) 125I-labelled anti-D
An IgG preparation known to contain anti-D activity was preoxidised and
labelled with by by the iodine monochloride method as described by McFarlane
(1958). The labelled antibody was purified by absorbing on Rh positive red
cells and eluting with ether by a modification of the method of Rubin (1963),
described in the following paragraph.
The antibody preparation was diluted to 30 ml with 1% bovine serum
albumin in saline. The solution was spun at 3,000 rpm for 10 minutes.
The supernatant was removed and incubated at 37°C with an excess of packed,
washed Rh positive red cells for 15 minutes. The mixture was spun at 3,000
rpm for 10 minutes and the supernatant removed. The cells were washed five
times with an equal volume of ice-cold saline before lysing and preparing
stroma from the cells by the method of Dodge et al. (1963). The red cell
membranes were resuspended in saline to a total volume of 30 ml. An equal
volume of diethyl ether was added and the mixture incubated at 37°C for 20 min.
The stroma was spun at 3,000 rpm for 10 minutes. The antibody in the saline
layer was carefully removed and tested for purity by a method based on that
described by Hughes-Jones (1967). The purity of the anti-D
preparation was estimated to be approximately 40% i.e. 40 pg anti-D/100 pg IgG
and the specific activity was 2,400 counts/minute/pg.
5) Measurement of phospholipase A2 activity
The enzyme activity of each of the phospholipase A2 preparations was
assessed by measuring the rate of release of fatty acid from egg lecithin
(Koch-Light Labs.) using the decolourisation of cresol red by fatty acid as
an indicator. Phospholipase A2 was added to 5 ml of a 0.02M glycine-NaOH
buffer, pH 9.2, containing phosphatidyl choline (0.7 mg), Tween 80 (20 pl)

51
calcium chloride (1 mg) and cresol red (0.02 mg). The change in absorbance
at 587 nm at room temperature (22°C) was measured at 5 minute intervals and
related to the enzyme activity in units (U) assuming that 1 unit of enzyme
was responsible for a change in absorbance of 0.11 per minute under the
defined conditions (Boehringer-Mannheim catalogue). The value for the
enzyme activity of the phospholipase C preparation (1.5U/mg) as stated in
the catalogue was accepted. One unit being defined as the amount of enzyme
required to liberate one micromole of inorganic phosphate from egg lecithin
per minute at 37°C, pH 7.3.
6) Incubation of stroma with phospholipase A2
Aliquots of stroma (0.1 ml) were added to 0.6 ml glycine (0.15M) NaCl
(0.08M) containing CaC12 (0.002M) pH 6.7. Phospholipase A2 was added in
amounts varying between 0 and 30 milliunits (m-units). After incubation
at 37°C for 10 minutes EDTA was added to a final concentration of 0.003 M and
the mixture was cooled to 0°C to stop the reaction. The amount of active D
antigen detectable on the stroma and the change in the membrane phospholipids
was then measured.
7) Incubation of stroma with phospholipase C
Aliquots of stroma (0.1 ml) were incubated at 37°C for one hour with
between 0 and 8 m-units of phospholipase C in 1 ml of a buffer containing
Tris-HCl (0.1 M), NaC1 (0.08 M) and CaCl2 (0.01 M) pH 7.4. The active D
antigen sites remaining and the change in the membrane phospholipids was
estimated.
8) Assessment of Rh (D) antigen activity remaining
Approximately 1 pg of purified 125I-labelled anti-D was added to each
stroma aliquot after the initial incubation with the enzymes. After incubation
at 37°C for a further 15 minutes, the stroma was ultracentrifuged at 150,000 g

52
for 20 minutes and the amount of radioactivity in the stromal precipitate
was estimated after resuspending the precipitate in clean plastic tubes.
Control samples of Rh positive and Rh negative stroma, not exposed to
enzymes, were treated in a similar way.
9) Analysis of the membrane phospholipids
The enzyme-treated stroma was mixed with 40 volumes of chloroform-
methanol (2:1) for one hour. After centrifuging 0.8 ml of distilled water
was added to the extract and the lower chloroform layer was withdrawn and
dried by heat evaporation.
The lipid was dissolved in 0.05 ml of chloroform and applied to Whatman
silica-impregnated paper (SG81). The constituent phospholipids were separated
by ascending paper chromatography in chloroform-methanol-water (65:25:4)
as the solvent system (Stott, 1972).
The chromatograms were stained with Dragendorff's reagent (Stott, 1972)
and the relative amounts of the phospholipids were estimated by the colo-ri-
metric method of Stott (1972). The amount of lipid phosphorus in the
lecithin spot was determined by the microanalytical technique described by
Kates (1972).
10) Measurement of the action of Phospholipase A2
on the antigen-antibody complex
125 I-labelled anti-D (0.3 pg) was incubated with red cell stroma (0.03 ml)
in 0.4 ml glycine (0.15 M) NaCl (0.08 M) pH 6.7 containing 0.002 M calcium
chloride at 37°C for 20 minutes.

53
10 m-units phospholipase A2 (Vipera russellii) was added and the mixtures
incubated a further 10 min before ultracentrifuging at 150,000 I for 20 min.
The supernatant was removed and the stroma resuspended in clear plastic tubes
and the bound radioactivity estimated in a Wallac gamma counter. Control
samples were treated in the same way omitting the phospholipase A2 addition.
11) Detection of solubilised D antigen after the action of phospholipase A2
Aliquots of stroma were incubated with various amounts of phospholipase A2
(Vipera russellii)as previously described. The stroma was spun at 150,000 .E
for 20 min and the supernatant removed. The supernatant was incubated with
125I-labelled anti-D at 37°C for 15 min. 0.1 ml washed Rh positive red cells
were added and incubated a further 15 min at 37°C. The mixture was
centrifuged and the supernatant removed. The cells were washed three times
and the radioactive content measured. Control samples were treated in the
same way omitting the phospholipase treatment.
12) Measurement of the effect of phospholipase A2 and phospholipase C
on intact cells
Rh positive red cells were washed three times in saline and 0.1 ml
aliquots were incubated with 0, 0.35, 3.5, 35, m-units phospholipase A2
(bee venom) or 0, 1.14, 11.4, 114, m-units phospholipase A2 (Naja Naja
Koch-Light Labs) in 0.6 ml glycine (0.15 M) Neel (0.08 M) containing CaC12
(0.002M) pH 6.7. After incubation for 10 min EDTA was added to a final
concentration of 0.003M. The remaining Rh (D) active antigen on the cells
was estimated using 125I-labelled anti-D.
The effect of phospholipase C on intact cells was estimated by incubating
0.1 ml aliquots of washed Rh positive red cells with 0, 0.5, 2, and 4 m-units
of phospholipase C (C. perfringens) for one hour at 37°C in 1 ml of a buffer

54
containing Tris-HCl (0.1M) NaC1 (0.08M) and CaC12 (0.01M) pH 7.4. The
remaining active D antigen sites were estimated using 125I-labelled anti-D.
For comparison the effect of similar amounts of phospholipase A2
and phospholipase C on stroma was measured simultaneously.
13) Detection of protease activity in phospholipase preparations*
The methods of Eagle (1937) and Northrop et al. (1948) using gelatin
and denatured haemoglobin respectively were used.
*The experiments concerning the detection of protease activity in
phospholipase were carried out by Miss V.A.M. Hunt.

55
B) RESULTS
1) Measurement of phospholipase A2 activity
For the purposes of these experiments, under the conditions defined
in methods, 1 unit of enzyme activity was assumed to be responsible for a
change in absorbance (A A) of 0.11 per minute (Boehringer-Mannheim catalogue).
This change (AA) is equivalent to the release of approximately 1 pmole of
fatty acid. The change in absorbance with time is shown in Fig 11-3 for
the three enzyme preparations. The activity was calculated from a line
drawn at a tangent to the curve through time 0. The E for the Vipera,
Bee and Crotalus preparations was 0.0125, 0.005 and 0.001 respectively.
2) The action of phospholipase A on stroma
a) The effect on Rh (D) antigen activity
The amount of 1251-labelled anti-D that bound to stroma treated with
increasing amounts of the phospholipase preparations is shown in Fig 11-4.
It can be seen that treatment of membranes with PLA2 brought about a fall in
the amount of 125I-labelled anti-D that could be bound to the stroma. The
number of D sites lost was dependent upon experimental conditions since it
was found that increasing the amount of enzyme (Vipera russellii) to 50 m-units
and the incubation time to 30 minutes resulted in a reduction in the uptake
of 125I-labelled anti-D by the stroma to about 10% of that of untreated stroma.
Large doses of the Crotalus preparation (12 m-units) were consistently required
to produce approximately the same loss of Rh activity as 2 m-units of the Bee
or Viper preparations. Doses in excess of 10 m-units of the Crotalus
preparation produced almost the same reduction in Rh activity as equivalent
doses of the other two enzyme preparations.
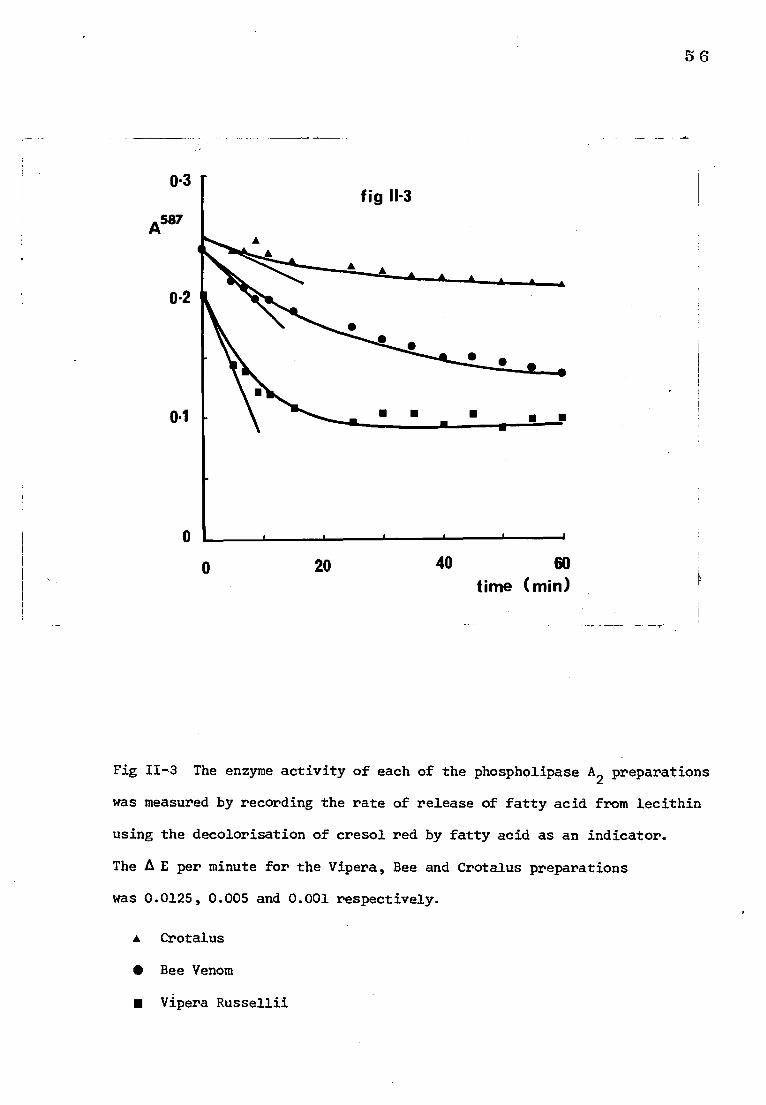
0.3
A587
0.2
0.1
0
56
0
20 40 60 time (min)
Fig 11-3 The enzyme activity of each of the phospholipase A2 preparations
was measured by recording the rate of release of fatty acid from lecithin
using the decolorisation of cresol red by fatty acid as an indicator.
The A E per minute for the Vipera, Bee and Crotalus preparations
was 0.0125, 0.005 and 0.001 respectively.
• Crotalus
• Bee Venom
■ Vipera Russellii
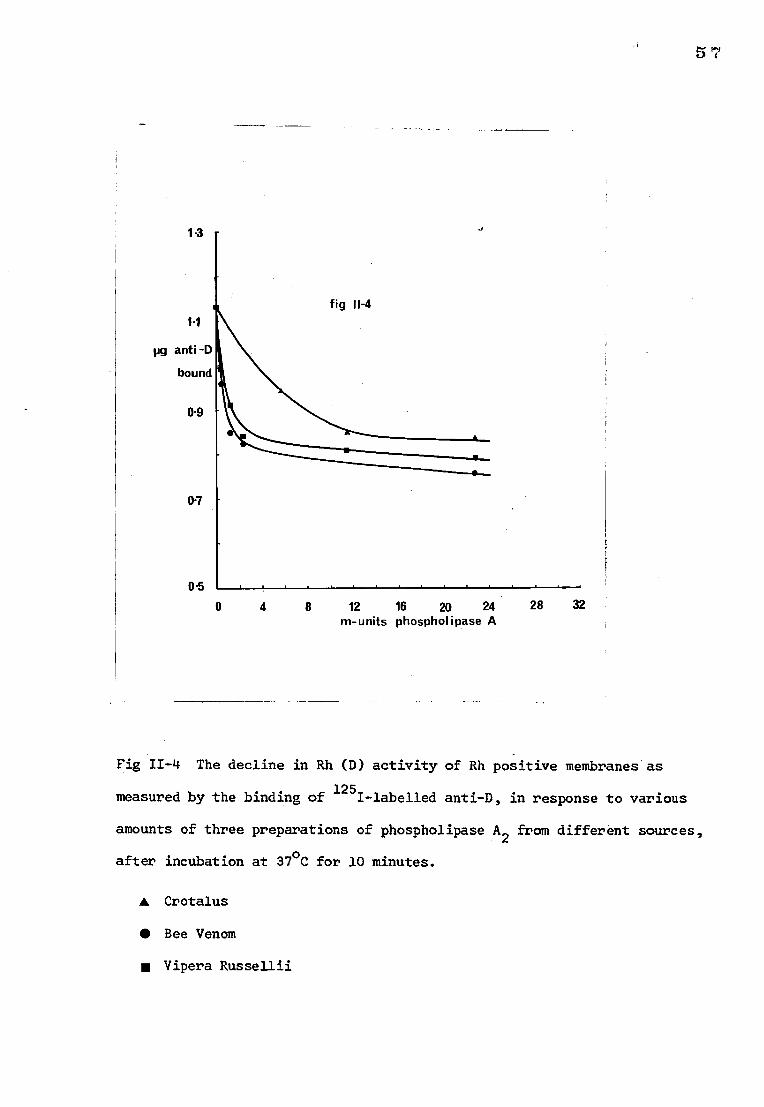
1.3
1.1
pg anti -D
bound
0.9
07
05
0 4 8 12 16 20 24
28
32 m-units phospholipase A
Fig II-4 The decline in Rh (D) activity of Rh positive membranes as
measured by the binding of 125I-labelled anti-D, in response to various
amounts of three preparations of phospholipase A2 from different sources,
after incubation at 37°C for 10 minutes.
• Crotalus
• Bee Venom
■ Vipera Russellii

5$
b) The effect on phosphatidylcholine
Chromatograms of the lipids extracted from phospholipase A2 treated
stroma showed a fall in colour intensity of both phosphatidylcholine and
phosphatidyl ethanolamine spots (phosphatidyl serine could not be distinguished
from phosphatidyl choline with the solvent system used). A lysophosphatidyl-
choline spot appeared.
The degradation of phosphatidylcholine as measured by the amount of
lecithin phosphorus remaining in the stroma at increasing doses of
phospholipase A2 from 0 to 30 m-units is shown in Fig. 11-5. Increasing
the amount of enzyme to 50 m-units and the incubation time to 30 min resulted
in complete loss of visible colour on the chromatogram in the position of the
phosphatidylcholine, phosphatidylserine and phosphatidylethanolamine spots.
The relationship between the decrease in Rh activity and fall in
stroma lecithin is shown in Fig. 11-6.
3. Incubation of Phospholipase A2 with the antigen-antibody complex
There was no difference in the amount of 125I-labelled anti-D on the
stroma which had been treated with phospholipase A2 and the control, untreated
stroma, suggesting that the D antigen is not inactivated by phospholipase A2
when it is bound to anti-D.
4. Detection of solubilised D antigen after the action of phospholipase A2
There was no evidence for the release of D antigen into the supernatant
after enzyme treatment as measured by the ability of the supernatant to
inhibit the uptake of purified 125I-labelled anti-D by intact red cells.
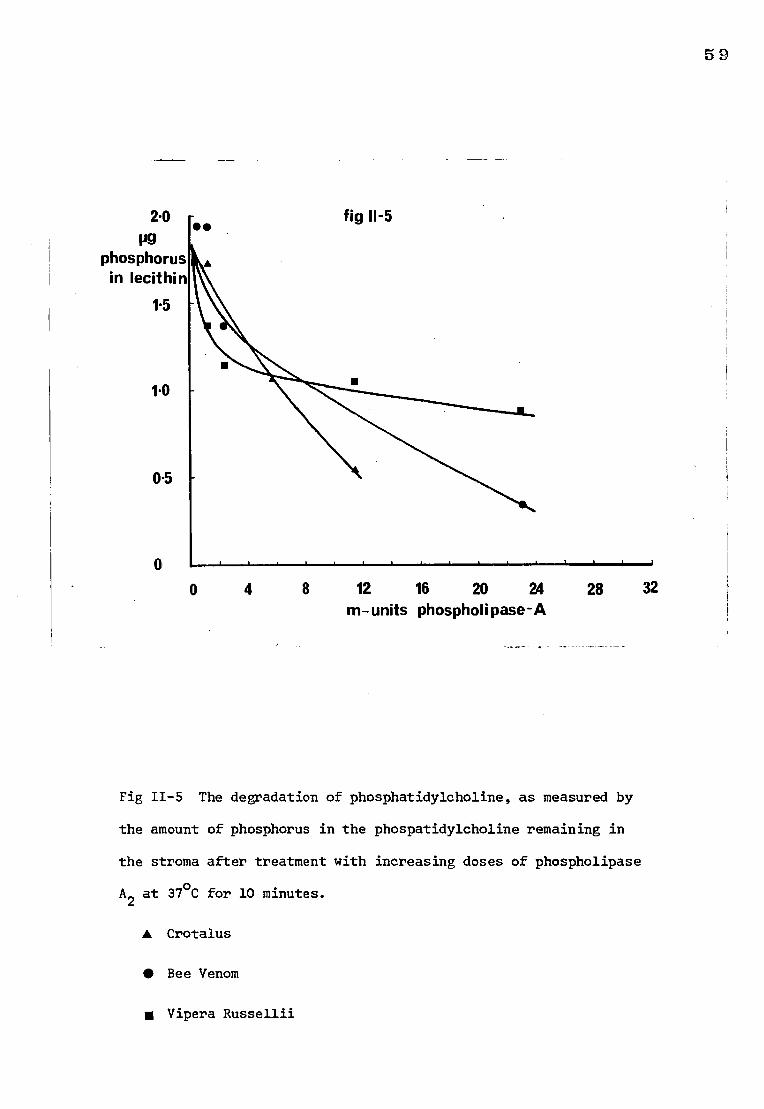
59
2.0
phosphorus in lecithin
1.5
1.0
0.5
0
0 4 8 12 16 20 24 28 32 m-units phospholipase-lk
Fig 11-5 The degradation of phosphatidylcholine, as measured by
the amount of phosphorus in the phospatidylcholine remaining in
the stroma after treatment with increasing doses of phospholipase
A2 at 37°C for 10 minutes.
A Crotalus
• Bee Venom
• Vipera Russellii
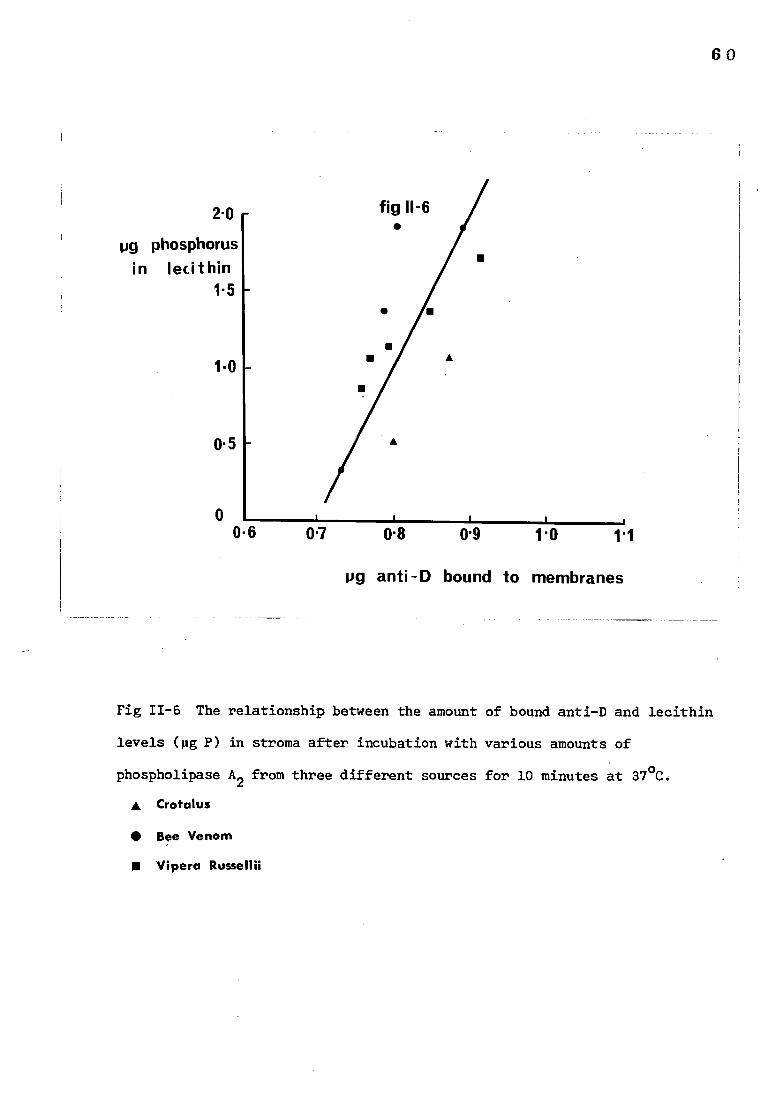
60
fig 11-6 • 2.0
pg phosphorus in lecithin
1.5
1.0
0.5
■
0 0.6 0.7 0.8 0.9 1.0 1.1
pg anti -D bound to membranes
Fig 11-6 The relationship between the amount of bound anti-D and lecithin
levels (pg P) in stroma after incubation with various amounts of
phospholipase A2 from three different sources for 10 minutes at 37°C.
• Crotalus
• Bee Venom
■ Vipera Russellii
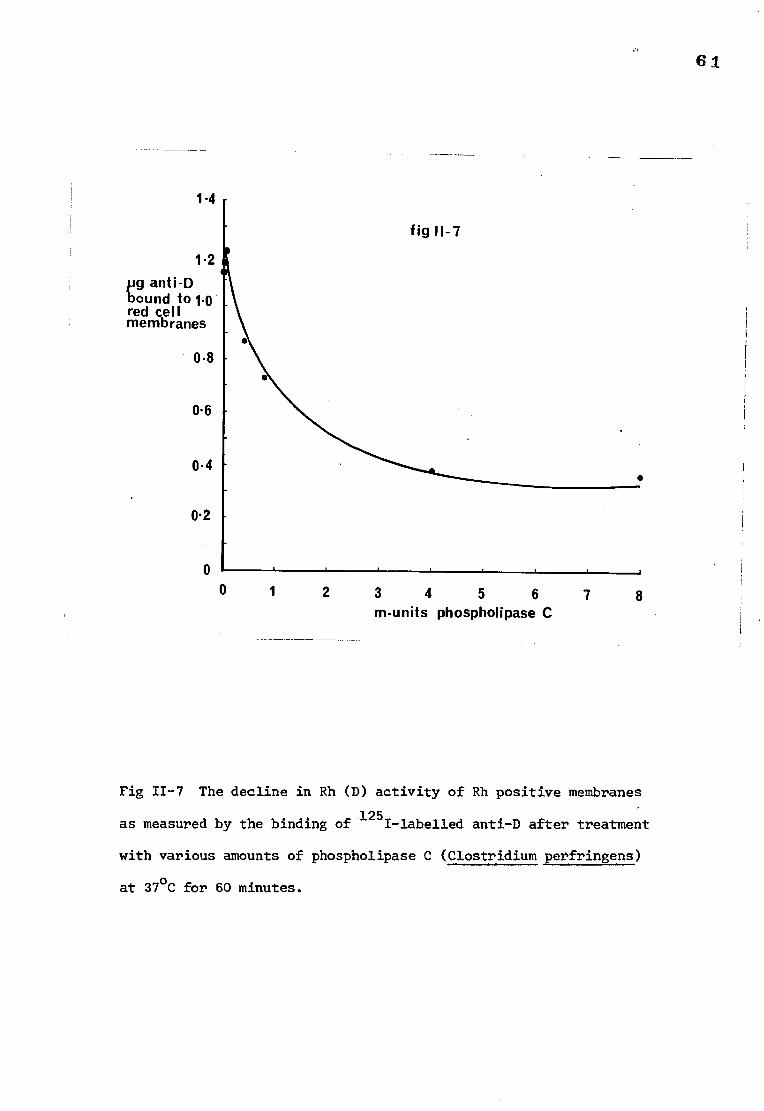
1-4
fig 11-7
1.2 pg anti-D bound to 1.0 red cell membranes
0.8
0.6
0.4
0.2
•
0
1
2 3 4 5 6
7 8 rn-units phospholipase C
Fig 11-7 The decline in Rh (D) activity of Rh positive membranes
as measured by the binding of 1251-labelled anti-D after treatment
with various amounts of phospholipase C (Clostridium perfringens)
at 37°C for 60 minutes.
61

62 5) Action of phospholipase C on stroma
Treatment with phospholipase C results in the conversion of phosphatidyl-
choline, phosphatidylserine and phosphatidylethanolamine to their respective
diglycerides. In stroma this conversion was accompanied by a fall in the
amount of 125I-labelled anti-D which would bind to the stroma. This fall
in bound 125I-labelled anti-D and the decrease in membrane phosphatidylcholine
are shown in Figs 11-7 and 11-8 and the relationship between them is shown in
Fig 11-9.
6) The effect of phospholipase A2 and phospholipase C on intact red cells
Phospholipase A2 (Naja Naja and Beed venom) did not cause a decline in
the Rh activity of intact red cells at the doses used. These doses, 0.35,
3.5 and 35 m-units of Bee venom phospholipase A2 and 1.14, 11.4 and 114 m-units
phospholipase A2 from Naja Naja led to the expected decline in the Rh (D)
activity of stroma. Chromatographic examination of the red cell phospholipids
after treatment with phospholipase A2 from either source did not show any
visible conversion of phosphatidylcholine to lysolecithin.
Incubation with phospholipase C (C. perfringens) resulted in red cell
lysis. Since this lysis rendered the red cell membranes as available to
phospholipase attack as stroma, the effect of this enzyme on whole cells could
not be ascertained.
7) Detection of protease activity in phospholipase preparations
No protease activity could be detected either by the gelatin or denatured
haemoglobin method in any of the phospholipase A2 or C preparations, using
concentrations of the preparations 200 times greater than that of the highest
concentration used on stroma. Trypsin was used as a control for both the
gelatin and the haemoglobin methods and could be detected at a dilution . of
1:100,000 (w/v); it was found that it was necessary to use a concentration of
trypsin 100 times greater than the detectable amount in order to reduce D activity when added to stroma.
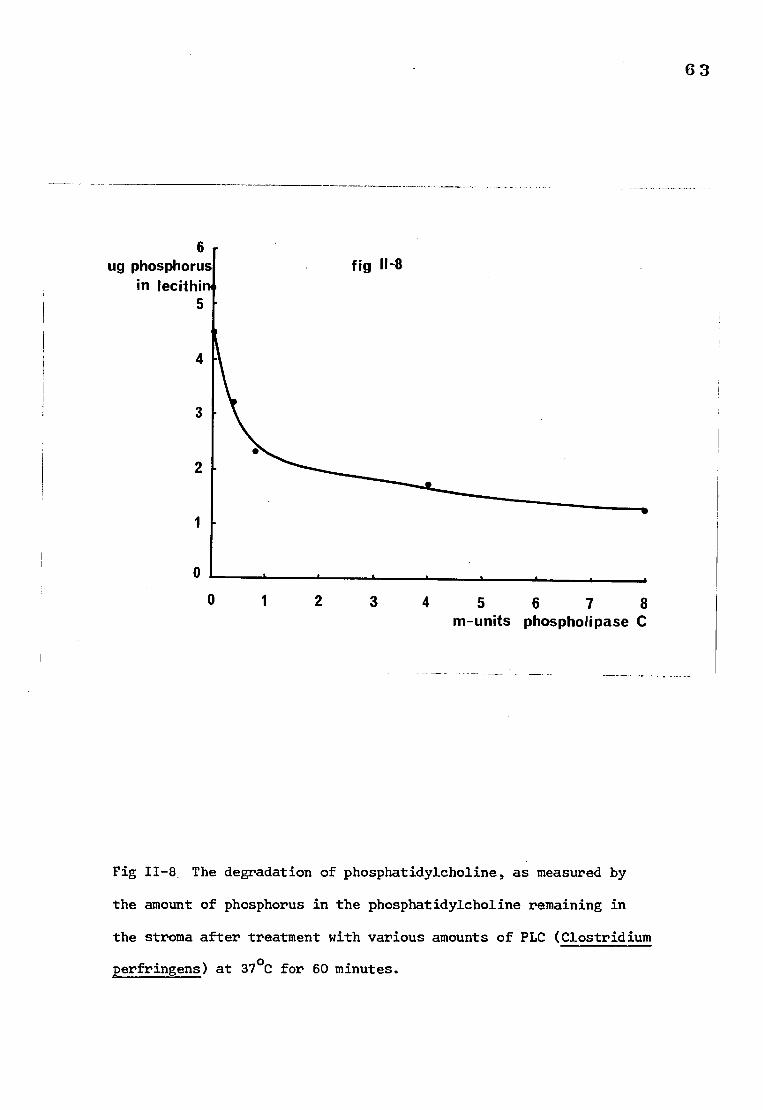
6 ug phosphorus
in lecithi 5
0
0
63
2 3 4 5 6 7 8 m-units phospholipase C
Fig 11-8 The degradation of phosphatidylcholine, as measured by
the amount of phosphorus in the phosphatidylcholine remaining in
the stroma after treatment with various amounts of PLC (Clostridium
perfringens) at 37°C for 60 minutes.
fig 11-8
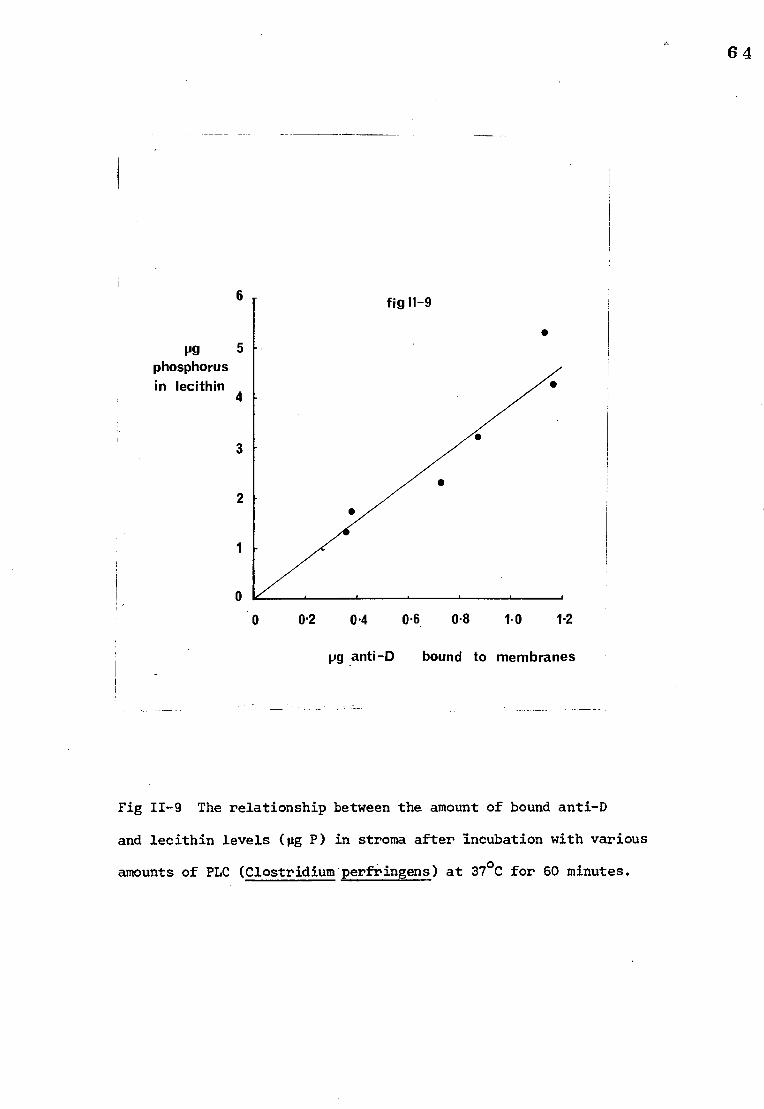
fig 11-9 6
• PO
phosphorus in lecithin
4
3
2
0 0.2 0.4 0.6 0.8 t 0 1.2
pg anti-D bound to membranes
Fig 11-9 The relationship between the amount of bound anti-D
and lecithin levels (pg P) in stroma after incubation with various
amounts of PLC (Clostridium perfringens) at 37°C for 60 minutes.
64

65
DISCUSSION
The three phospholipase A2 preparations used here brought about the
conversion of phosphatidylcholine, phosphatidylserine and phosphatidylethanol-
amine of the red cell membrane to their respective lyso-compounds and also
reduced the ability of the red cell membranes to bind with anti-D.
It is possible that another constituent in the phospholipase preparations
was responsible for the inactivation of the D antigen. Wolff and Springer
(l964) have shown that the D antigen is destroyed by proteolytic enzymes.
However, no protease activity could be detected in the preparations using
two methods which were both capable of detecting trypsin at a concentration
one hundred times less than that required to destroy D antigen activity.
Moreover, when the three phospholipase preparations from different sources
were used in the same concentrations, as judged by their action on lecithin,
they produced a similar degree of inactivation of the D antigen. It is unlikely
that the three different preparations each contained the same contaminant at
the same concentration.
The finding that phospholipase C destroyed D antigen activity also
suggests that it is the degradation of the phospholipids that is responsible
for the loss in antigenic activity.
When 125I-labelled anti-D was first combined with stroma and then
exposed to phospholipase A2 there was no release of the antibody. This
suggests that the D antigen remains intact under these conditions, due either
to the antigen-antibody bond maintaining the structure of the antigen site
intact or to the failure of the phospholipase to degrade the phosphiipids
associated with the antigen owing to steric hindrance by the antibody.

66
The results of this study confirm the findings of Green (1972) that
intact phospholipids are essential for the maintenance of Rh antigen activity.
Much of the evidence hitherto reported has suggested that the Rh antigen
is a protein-like substance (Green, 1965). It is possible that the tertiary
structure of this protein could be modified through a noncovalent bond with
a phospholipid molecule, and if the phospholipid is altered, for example, by
the action of phospholipase A2, then the antigenic site on the protein may
be incapable of binding antibody. Alternatively, the protein on which the
Rh antigen is carried may require a completely intact lipid bilayer for its
correct spatial presentation on the membrane surface. This latter hypothesis
would infere a rather generalized lipid requirement and would not be entirely
in keeping with the findings of Green (1972) that for Rh activity the
phospholipids in the membrane should have at least one unsaturated fatty acid
implying that the double bond confers on the molecule a specific property
that enables it to interact with the protein by some sort of weak intra-
molecular binding directly or by altering the phospholipid sterically and
thereby permitting some form of interaction that would not be otherwise
possible.
However the results of this study must be interpreted cautiously.
Doses of phospholipase A2 which resulted in loss of D antigen activity
in isolated membranes, failed to have any action on the D antigen activity
of intact red cells. This is in agreement with the studies of Prager et al.
(1963) using Russells viper venom, on the Rh activity of intact red cells.
They could demonstrate no consistent significant effect on Rh agglutination
reactions. In the present study, it was found that larger doses of bee venom
phospholipase A2 (70, 140 and 210 m-units) resulted in cell lysis.

67
Gul and Smith (1974) and Zwaal et al. (1975) have reported that Naja
Naja and bee venom phospholipase A2 respectively, both degrade phospholipids
in intact red cells without haemolysis. In the present study there was no
measurable phospholipid degradation although this could merely reflect a
lack of sensitivity in the technique employed to detect lysophospholipids.
Whether or not the membrane phospholipids were degraded it is evident
that the phospholipid molecules which are responsible for the maintenance of
Rh antigen activity are not present on the surface of the red cell in a
manner in which they can be as readily attacked by phospholipase A2 from
Naja Naja or bee venom, as they are in isolated membrane preparations.

68
SECTION II
ATTEMPTS TO REACTIVATE THE Rh ANTIGEN AFTER PHOSPHOLIPASE TREATMENT
INTRODUCTION
Phospholipid metabolism in the human red cell membrane
a) Chemical reactions
The manner in which phospholipids are attached to the red cell membrane
must be dictated by the manner in which the membrane is able to incorporate
these molecules and their precursors.
Mature human erythrocytes are not able to syntheK long-chain fatty
acids from acetate because they do not contain an acetyl CoA-carboxylase
which catalyses the synthesis of malonyl CoA (Pittman and Martin, 1966)
but they do have acid CoA-ligase and acyl-CoA:lysolecithin acyltransferase
and therefore are able to activate and incorporate long-chain fatty acids.
Mulder and Van Deenen (1965) demonstrated that erythrocytes free from
leucocytes and reticulocytes incorporated fatty acids into phosphoglycerides.
The incorporation in vitro of fatty acids into the intact erythrocyte was slow.
In 5 hours the uptake of linoleate from the medium amounted to about 1.5 pg
fatty acid per 0.5 ml rabbit red cells, which corresponds to about 0.6% of
the linoleic acid present in the phospholipids from rabbit erythrocytes.
The rate of incorporation was increased in lysed cells and addition of
coenzyme A (CoA) and adenosine 5'-triphosphate (ATP) promoted the fatty acid
uptake. Incubation of red cell membranes in Ringers solution supplemented
with lysolecithin resulted in a significant increase in the incorporation of
linoleic acid into the phospholipids. Mulder et al. (1965) demonstrated that
lysolecithin is converted by rabbit stroma into lecithin and glyceryl
phosphorylcholine. They showed that there were two separate reactions:

69 ATP CoA
1. lysolecithin + fatty acid >lecithin
a transacylation requiring ATP and CoA, and
2. lysolecithin glycerophosphorylcholine + fatty acid
under conditions favourable for fatty acid incorporation the first pathway
was predominant.
Hokin and Hokin (1961) showed the existence of phosphorylation
reactions in human erythrocyte membranes in which diglycerides are
phosphorylated to phosphatidic acid in the presence of ATP, and catalysed
by diglyceride kinase.
1, 2-diglyceride t ATP phosphatidic acid + ADP
It is probable that this is the pathway whereby radioactive phosphate
was incorporated into phosphatidic acid in the experiments of Mulder and
Van Deenen (1965).
b) Movement of phospholipid molecules
Phospholipids with unsaturated fatty acids have a low melting temperature,
and those with saturated fatty acids undergo a melting transition at a higher
temperature. Below the melting temperature the hydrocarbon chains are rigid
and above it they are free to move. Once the hydrocarbon chains have melted
it may become possible for individual phospholipid molecules to exchange with
their neighbours. According to Devaux and McConnel (1972) phospholipids
have an estimated neighbour exchange rate of less than 10-6 seconds.
Kornberg and McConnell (1971) found however, that the movement of a
phospholipid molecule from one side of a bilayer to the other ('flip-flop')
is very slow. The half-life for a spin-labelled phospholipid to 'flip-flop'
was found to be about 6 hours at 30°C.

70
c) Phospholipid exchange between red cells and plasma
Reed (1959) was able to demonstrate that erythrocyte phospholipids
(except phosphatidylserine) are in a dynamic state of exchange with the
plasma phosphatides. He found that red cell lecithin and sphingomyelin
exchanged with plasma lecithin and sphingomyelin at the rate of 10% per
24 hours. In addition normal cells, in vitro, incorporated lipid phosphorus
from labelled plasma over a 12 hour period at a rate equal to that in vivo.
Longer incubation times reduced the cells ability to exchange phospholipids.
The results of Farquahar and Ahrens (1963) also led them to conclude that
"exchange of fatty acids or of intact phospholipids must occur between mature
red cells and one or more precursor pools". Lovelock et al. (1960) incubated
whole blood, in vitro, with sodium (Me 14C) acetate. Approximately 3-5%
of the added carbon isotope was found in lipid fractions at equilibrium and
the half life for the exchange process was 6 hours.
Conclusions
It was decided to attempt reactivation of the Rh antigen by removing and
replacing or altering the membrane phospholipids damaged by phospholipase A2
In Green's experiments (1968) the phospholipids were removed from the membrane
and replaced by incubation of the butanol-treated membranes with appropriate
lipid extracts. However in the experiments in the present study the
phospholipids have been treated with phospholipase A2 which cleaves the bond
between the fatty acid at the C-2 position but leaves the lysophospholipid in
the membrane. Therefore either the molecule must be acylated or exchanged.
The following methods were used to achieve these aims.
1. Incubation with Plasma. It is possible that enough lecithin
molecules from plasma would exchange with the lysolecithin in the membranes
to reactivate the antigen. There is unlikely to be enough ATP or CoA in the

71
preparation to enable the membrane to incorporate fatty acids from plasma
into the C2 position of phospholipid molecules which had been attacked by
phospholipase A2.
2. Incubation with linoleic acid, ATP and coenzyme A.
Incubation of the membranes with a supply of fatty acid, ATP,
and coenzyme A may enable the membrane to incorporate the fatty acids
into the lysophospholipids formed as a result of enzyme action.
3. Incubation with sonicated lecithin.
If the phospholipid responsible for Rh activity is lecithin (Green,
1968) perhaps providing a medium rich in this substrate would promote
exchange between the lysophospholipids in the membrane and the incubation
medium so that enough lecithin could be incorporated into the membrane to
enable a measurable recovery of Rh (D) activity.
4. Incubation with 32P-labelled phospholipids.
Incubation with a labelled lipid extract from whole blood should
demonstrate whether or not exchange is occurring between the membranes
and the added lipid.
METHODS AND MATERIALS
The materials and methods described in Section 1 were used in
combination with those described below in an attempt to restore Rh (D)
activity in phospholipase treated red cell membranes.
1) Solutions
a) Hepes buffer, 4-(2-hydroxyethyl)-1-piperazine-ethansulphonic acid.
b) Ringer's solution (9g NaCl; 0.3g KCl; 0.25g CaC12. 2H20)
(0.2g NaHCO3; Distilled water to 1000 m1).

7 2
c) Scintillation fluid for a-counting
15g PPO (2, 5 Diphenyloxazole); 0.3g POPOP (1, 4 -Di 2-
(5-phenyloxazoly1)-benzene in 2i ..1 toluene).
d) Rhodamine 6g (Serle diagnostic laboratories)
A stock solution (1.6g/1000m1 H20) was diluted 1:100
with water before use.
2) Attempts to restore Rh activity with plasma
Aliquots of Rh positive stroma (0.2 ml) were treated with 5 m-units of
phospholipase A2 (Vipera russellii), then spun at 150,000 g for 20 min, washed
once and made up to 0.2 ml with phosphate buffered saline. The membranes
were then incubated overnight with 5.0 ml aliquots of fresh plasma obtained
from Rh negative donors. The stroma was spun at 150,000 £ for 30 min and
washed twice with phosphate buffered saline. Half of the resulting pellet
was tested for Rh (D) activity with 125I-labelled anti-D, and the phospholipids
in the remaining half were extracted with chloroform-methanol (2:1) as
described in Section I.
Control aliquots were treated similarly either omitting the phospholipase
treatment or the incubation with plasma.
3) Attempts to restore Rh (D) activity by incubation with linoleic acid
in the presence of coenzyme A and ATP
Aliquots of Rh positive stroma (0.2 ml) were treated with phospholipase A2
(5 m-units), spun down and washed then made up to 0.5 ml in Ringers solution
(containing 2% glucose) and incubated in the presence of 50 umoles ATP
(Sigma Labs.), 1 umole coenzyme A (Sigma Labs.) and 0.04 ml linoleic acid
(Sigma Labs.) overnight at 37°C in a shaking water bath. The stroma was then

73
spun at 150,000 g and washed before testing half for Rh (D) activity and
extracting the remainder with chloroform-methanol (2:1). The extracted
phospholipids were separated by ascending chromatography and examined visually
after staining as described in section 1.
4) Attempts to restore Rh (D) activity with sonicated lecithin
These experiments were carried out as described above for those with
plasma or linoleic acid except that the phospholipase treated membranes were
incubated for one hour at 37°C in 0.5 ml lecithin (Ex-egg lecithin 1g/10 ml
in ethanol) which had been sonicated at an amplitude setting of 2 microns
for 2 minutes in an MSE ultrasonic disintegrator to disperse the lecithin
into micelles. After incubation the membranes were split into two equal
aliquots and the phospholipids were extracted from one aliquot and the Rh (D)
activity investigated in the other aliquot.
5) Experiments involving 32P-labelled phospholipids
a) Labelling whole blood with 32P
Rh negative blood was drawn into EDTA and mixed. Approximately
50 pCiP(0.1 ml) was added to 2.0 ml 0.014 M Hepes buffer pH 7.3 containing
0.09 g glucose, 1 pg Penicillin and 1 pg streptomycin. 1.0 ml plasma and
1.0 ml red cells were added and incubated at 37°C for five hours. Assuming
the plasma contained 3 mg/100 ml inorganic phosphate (Documenta Geigy
scientific tables, 6th ed. page 542) the mixture would have 6 x 105 cpm/mgP.
After incubation the mixture was spun at 3,000 rpm for 10 minutes and the
lipids were extracted from the supernatant and from the cells as described
below.

7 4
b) Lipid extraction from plasma supernatant
Chloroform-methanol (2:1) (15 ml) was added, mixed and filtered through
Whatman No.1 qualitative filter paper. The top layer of the filtrate was
removed and the interface gently washed once (with the upper layer obtained
on mixing chloroform-methanol-water in the ratio 8:4:3). The bottom layer
was evaporated before estimating the amount of incorporated 32P and separating
the constituent phospholipids, in duplicate, chromatographically as described
in section 1. The individual phospholipid spots were visualised by staining
with Rhodamine 6G for 2 minutes, and the corresponding areas on unstained
duplicates were cut out and put in glass counting vessels with 4.0 ml
. scintillation fluid. The amount of 32P in the samples was estimated in a
Packard beta counter.
c) Lipid extraction from red cells
The cells were mixed for five minutes in 5 ml methanol. Chloroform
(5 ml) was added and mixed for a further 5 minutes. The mixture was
centrifuged at 3,000 rpm for 10 minutes and the supernatant was removed.
The extraction of the cells was repeated and the lipids from both extractions
pooled and evaporated to reduce the volume before estimating the 32P
incorporation in the manner described for the plasma lipids.
d) Attempts to restore Rh (D) activity to phospholipase A2 treated
stroma with sonicated 32P-labelled lipid extract from red cells
and plasma
Phospholipase A2 treated stroma was incubated overnight with 32P-labelled
lipids extracted either from cells or from plasma. Before incubation, the
extracted lipids were evaporated to dryness under vacuum, then dispersed by
sonicating for 3 minutes at an amplitude setting of 2 microns, in 2.5 ml
Hepes buffer (0.014 M pH 7.3). The cell lipid sample had 5,481 counts per

7 5
10 minutes per ml and was estimated to contain 0.7 pmoles phospholipid.
The plasma lipid sample had 5,580 counts per 10 minutes per ml and was
estimated to contain 0.52 pmoles phospholipid. The phospholipid
preparations (0.5 ml) were both added to enzyme-treated membranes and
incubated for 18 hours at 37°C. After incubation the membranes were
centrifuged at 35,000 rpm for 30 minutes and the supernatant was removed.
The sediment was resuspended to a total volume of 0.2 ml. Half was
extracted with chloroform-methanol (2:1) as described in section 1 and the
phospholipids were separated by ascending paper chromatography in chloroform-
methanol-water (65:25:4) according to the method of Stott (1972). The
phospholipids were stained in Dragondorff reagent and the individual lipid
spots, cut out, dried and counted in a Packard beta counter. The remaining
0.1 ml membranes was tested for Rh (D) antigen activity using 1251-labelled
anti-D as described in section 1. The results were compared with those of
control samples which had been treated with Phospholipase A2, but not incubated
with labelled lipid, and with untreated stroma.
RESULTS
1) Attempts to restore Rh activity with plasma
There was no evidence of any restoration of Rh activity in the
phospholipase treated membranes after overnight incubation with plasma.
Visual examination of the lipid chromatograms of the plasma and membranes
after incubation with plasma showed the presence of lecithin and lysolecithin
in both plasma and membranes. The presence of lecithin in the membranes
suggested that phospholipid from the plasma had been incorporated into the
membranes. Lysolecithin is a normal constituent of plasma, however the spot
corresponding to this phospholipid on the chromatograms was larger and brighter

7 6
than that on the chromatograms of untreated plasma, indicating that there
was more lysolecithin in the plasma after it had been incubated with
phospholipase-treated membranes. The extra lysolecithin in the plasma
after incubation could have been phospholipid that had exchanged from the
membranes. On the other hand, it could have been due to contaminating
phospholipase, since phospholipase activity could be demonstrated in the
progressive washings of phospholipase treated membranes. It was found
that at least three washes of the stroma were needed to remove the enzyme
activity and therefore it is probable that much of the excess lysolecithin
in the plasma after incubation with the membranes was an artifact caused by
the action of phospholipase A2 on plasma lecithin. In subsequent experiments
the membranes were washed at least three times before incubation with lipid
to minimise the effect of phospholipase on the added lipid.
2) Attempts to restore Rh activity with linoleic acid, coenzyme A and ATP
Rh (D) activity was not restored in the membranes after incubation with
linoleic acid, as judged by comparing the amount of 125I-labelled anti-D
bound to these membranes with untreated Rh positive membranes and Rh negative
membranes treated with phospholipase A2 and incubated with linoleic acid.
Furthermore, visual examination of chromatograms of the phospholipids
extracted from phospholipase treated stroma which had been incubated with
linoleic acid indicated that lecithin had not been synthesized under the
experimental conditions employed.
3) Attempts to restore Rh activity with sonicated lecithin
There was no evidence of regeneration of Rh activity in phospholipase A2
treated membranes which had been incubated with sonicated lecithin for one hour
at 37°C, although small amounts of lecithin could be seen in chromatograms of
the enzyme-treated membranes after incubation with lecithin.

TABLE 11-2
pg inorganic P Percentage of pg inorganic Percentage of
incorporated phospholipid incorporated lecithin
into molecules which into lecithin molecules which
phospholipids had taken up had taken up
inorganic P inorganic P
RED CELLS 3.2 2.5 1.4 3.7
PLASMA 2.6 2.8
1.9 3.2
Table 11-2: The incorporation of inorganic P into red cell and plasma lipid. The results were calculated by assuming
that there are 0.03 mg P/ml plasma which made the specific
activity of the labelling mixture 600 cmp/pg P, and using
the results of Hurter et al. (1970) for the number of pmoles
of phospholipids in red cells and plasma.
7 7

78
4) Experiments involving 32P-labelled phospholipids
a) Labelling and lipid extraction
An estimate of the amount of inorganic phosphorus incorporated into
red cell phospholipids and plasma phospholipids under the conditions defined
in methods is given in Table 11-2. The individual result for lecithin is
also given. Assuming that all of the phospholipids were removed from the
red cells and the plasma, and using the values obtained by Hurter et al. (1970)
for the number of pmoles of total phospholipid and the individual value for
lecithin (pmoles) in the red cell and the plasma (as given in Fig II-1) it
was possible to calculate the percentage of lipid molecules which had
incorporated inorganic phosphorus. In both red cells and plasma just under
3% of the phospholipid molecules had incorporated inorganic phosphorus, and
just over 3% of the lecithin molecules had been involved in exchange.
b) Attempts to restore Rh (D) activity with sonicated 32P-labelled
lipid extract from whole red cells and from plasma
Rh (D) activity was not restored after incubation with the 32P-labelled
lipid extracts. The amount of labelled lipid incorporation was investigated
by extracting the lipid from the samples of phospholipase A2 treated stroma
which had been incubated with labelled lipid from red cells or from plasma.
The extracted lipid was run on chromatograms and the spots representing the
individual phospholipids were cut out and counted in a Packard beta counter.
The counts were used to estimate the number of pmoles of certain phospholipids
which had been incorporated into the membranes (providing the extraction of
the lipids was 100% efficient, 4.11 pmoles of phospholipid should have been
extracted from 1 ml red cells. The fraction of the preparation which was
used in this attempt to restore Rh activity was calculated to contain 0.7 pmoles
of phospholipid). The individual results for the phospholipids investigated

TABLE 11-3
LABELLED LIPID
LABELLED LIPID
FROM RED CELLS
FROM PLASMA
units: pmoles of phospholipid
LIPID SPOTS ON PHOSPHOLIPASE CONTROL PHOSPHOLIPASE CONTROL
CHROMATOGRAM TREATED STROMA TREATED STROMA
ORIGIN 0.004 0.009
LYSOLECITHIN 0.012 0.009
SPHINGOMYELIN 0.024 0.010
LECITHIN 0.016 0.006 0.014 0.004
SOLVENT FRONT 0.005 -
The number of Table II-3:/gmoles of labelled lipid incorporated into various
phospholipids in the phospholipase A treated membranes. The results
were calculated by assuming that the red cell lipid had 0.7 gmoles
phospholipid and 7857 counts per 20 min per mole and the plasma lipid
had 0.52 pmoles phospholipid and 10730 counts per 20 min per gmole.

Bo
are shown in Table 11-3. Of the added labelled lipid, 7.4% was incorporated
into the phospholipids estimated in this experiment. The labelled plasma
lipid sample was estimated to contain 0.52 gmoles of phospholipids by similar
considerations to those outlined above for the red cell lipids. Of the
added lipids, 6.3% was incorporated into the phospholipids investigated in
the enzyme treated membranes. The individual results are given in Table 11-3.
The amount of these labelled lipids taken up by the membranes seems to be
independent of whether the lipid was originally obtained from red cells or
from plasma.
The incorporation of 32P-labelled lecithin into membranes which had not
been previously incubated with phospholipase A2 is shown in Table 11-3 for
comparison with the uptake in enzyme treated membranes. There was approximately
three times as much 32P-labelled lipid incorporated into the phospholipase
treated membranes compared with the untreated membranes.
DISCUSSION
The methods used in this section to replace or modify the membrane
phospholipids after phospholipase treatment all failed to restore Rh activity.
Incubation with plasma was probably unsuccessful because insufficient
lecithin was able to exchange in the time available. In addition to this
the amount of lecithin in the plasma that could exchange was reduced due to
attack by phospholipase A2 which had remained associated with the membranes
after washing.

81
Linoleic acid was not incorporated to a measurable extent into lysolecithin
in the phospholipase treated membranes even in the presence of coenzyme A
and ATP. The rate limiting factor in this reaction was almost certainly
the availability or activity of the enzymes responsible for its catalysis.
It is possible that either phospholipase A2 has a destructive action on these
enzymes or that they were lost in the preparation of the red cell membranes.
Ferber (1973) has reported that the activity of the enzymes involved in
phospholipid metabolism is reduced in haemoglobin-free membrane preparations.
He suggested that this inactivation is caused by the dissolution of relatively
tightly bound haemoglobin and other membrane proteins.
The egg lecithin also failed to restore Rh activity. Green (1968)
reported a 60% regeneration of D activity after incubation of butanol-treated
membranes with egg lecithin, although, he found that hydrogenated egg lecithin
had only a very small capacity to restore antigenicity. Thus it is possible
that the lecithin used in the present study contained too few unsaturated
fatty acids to fulfil the requirements of the D antigen. It is also possible
that lecithin is incorporated more rapidly into butanol-treated membranes,
in which the lipid has been removed, than phospholipase-treated membranes in
which the phospholipids have been converted into their corresponding lyso-
compounds.
The experiments with the 32P-labelled lipids showed that the phospholipid
molecules were exchanging with those in the membranes and that approximately
three times as much lecithin was exchanged in a given time in membranes which
had been treated with phospholipase A2 than in membranes which were untreated.

82
The failure of the techniques employed to reactivate Rh activity
after treatment with phospholipase A2 could be explained in several ways:
1. The normal phospholipid exchange process, although three times as
effective for lecithin in phospholipase-treated membranes than in untreated
membranes, was unable to compensate for the change in phospholipid composition
brought about by phospholipase treatment in the time available.
2. The presence of lysophospholipids in the red cell membrane had an
irreversible destructive effect on the Rh antigen.
3. The phospholipase enzymes attacked the D antigen by an activity other
than phospholipid degradation, for example, by the action of contaminating
proteases.
Considering these alternatives, it is quite possible that the normal
lipid exchange process was unable to replace the enzyme-altered phospholipids
with physiological amounts of their normal counterparts under the conditions
employed. One might expect a slight increase in Rh activity since lecithin
molecules were exchanging, although, it is likely that insufficient numbers
of phospholipid molecules were able to exchange in order to restore Rh activity
to a detectable level.
Lysophospholipids may be able to react chemically with the Rh antigen
inducing a conformational change which is not reversible by replacement of
the lyso compound. The possibility of attack by contaminant proteases
has been considered in the discussion at the end of section 1; however,
some other form of inactivating action cannot be entirely eliminated.

83
CHAPTER III
TIM RADTATIO7 r:ZACTIVATT_C'T afiTISIJS A.T7IGENS INTRODUCTION
The procedures which are commonly employed in the isolation of
pure protein samples from cell membranes often result in a denatured
protein and consequent loss of biological activity. Using the technique
described in this chapter it was possible to obtain information regarding
the size, in situ, of the molecule which exhibits Rh (D) antigenic activity
without extracting the substance from its membrane environment.
Macromolecules in the path of an ionizing radiation tend to lose
electrons and become biologically inactive. The way in which the change
in biological activity in a fixed volume of tissue and at various radiation
doses, can be related to the size of the protein concerned is explained in
the following brief account of the actions of radiation on biological matter.
Radiation alters matter directly by interactions between the ionizing
particles and macromolecules leading to energy transfer between the substances.
Indirect effects are caused by the production of free radicals, formed in the
paths of ionizing particles which diffuse readily and react with macromolecules
outside the path of the radiation.
Direct effects
An ionizing radiation dissipates energy by transferring a part of its
kinetic energy to any molecule in its path. With 15 meV electrons, as used
in this study, as a radiation source, the energy transfer, or 'primary event'
raises the free energy of the electrons in the affected atom. When the energy

84
transferred is greater than the binding energy of the electron, the electron
will be released. If smaller amounts of energy are transferred the electron
may be raised to a higher atomic energy level resulting in the production of
an 'excited' atom. The released electron in the case of the ionized molecule
may have sufficient energy to cause further secondary ionizations. However,
Ore and Larsen (1964) have reported that over 80% of primary events result in
only 1 or 2 ion pairs. The energy transferred to the ionized or excited
molecule is subsequently redistributed to lower the molecular free energy
of the molecule. In the case of ionized molecules, evidence from electron
spin resonance (ESR) studies indicates that the charge migrates from the
primary event until it reaches a site in the molecule where the free energy
of ionization is lowest. The migration of charge disrupts the secondary
structure of the molecule (Dertinger and Jung, 1970) and denatures the
molecule if it affects the active site or that part of the molecule which
is responsible for the biological activity of the molecule. ESR studies'
have shown that the charge migration is dependent on temperature. The
new energy site is called the 'primary lesion' and is usually very reactive.
At this stage the molecule may readily react with itself or neighbouring
molecules. If the reaction involves the active site, loss of biological
activity can result. Excited macromolecules may redistribute their charge
either by emitting a photon or by loss of less strongly bound electrons which
can also result in an inactivated molecule as a result of disruption of
secondary structure in the manner described for ionized molecules.
Irradiation of a tissue has a very complex and often lethal effect on
the metabolism of constituent cells. The experiments described in the
present study were restricted to examining the effect of high energy radiations
on the antigenic activity of biological membranes rather than relating the
effect of the inactivation of these antigens to the biochemistry of the membranes.

85
Indirect effects
Small molecules are affected by radiation in the same way as
macromolecules i.e. Energy is transferred and redistributed; however,
indirect effects on larger molecules may result from the diffusion of
small radicals particularly if they are produced in water or air.
Irradiation in water produces diffusing hydrogen and hydroxyl radicals
and irradiation in oxygen produces diffusing reactive oxygen radicals.
The indirect effects are a result of the interaction of small diffusible
radicals with biomolecules. Substances therefore tend to be more sensitive
to radiation in water or an oxygen atmosphere. For the purpose of
determining molecular size from radiation inactivation studies, the indirect
effects must be eliminated. In the present study this was achieved by
irradiating freeze-dried material in a nitrogen atmosphere. Under such
conditions free radicals are unable to diffuse and interaction of macro-
molecules with molecular oxygen is not possible.
Target theory
The dose of radiation required to produce chemical change in a given
proportion of the molecules of a substance by direct action is inversely
proportional to the size of the molecule (Lea, 1955). The molecule or
structure in which the ionization has to be produced is the 'target' and
the energy transfer is a 'hit'.
There are many actions of radiations on organisms which cannot be
interpreted by target theory, for example, in circumstances where indirect
effects are likely, or if the observed effect in a single cell is due to
changes in the surrounding tissues.

86
Target theory can be applied when the biological effect is believed
to be caused by a single ionization or 'hit'. The total number of hits
is proportional to the dose of radiation given. However, although the
total number of hits increases in proportion to the dose, the number of
targets hit increases more slowly, so that the yield plotted against the
dose gives a characteristic exponential shape. If, for example, one is
following the loss in activity of a particular enzyme, each dose increment
results in the inactivation of the same proportion of the number of molecules
which have remained active until that time. The number of active molecules
falls off in a geometrical progression and therefore the survival curve is
exponential. If it is assumed that the energy transfer by direct action
takes place in discrete highly localized packets on the target molecule
and the primary events are distributed randomly with respect to volume then
the number of hits(n) a target Suffers is related to the target size V and
can be described by a Poisson distribution:
P (n) = (VxD)n exp-VD
where D is the radiation dose in primary events per volume matter and P is
the probability of n hits. n or more hits inactivate the target, so the
fraction of the original number of molecules (N/No) which survive a dose D
will be:
n=1 (VD)n
N/No = exp-VD E
n0 n!
Irradiation survival curves are usually plotted as log10 N/No against dose.
For most molecules the survival curves are best described by a single hit
inactivation process (Kepner and Macey, 1968). For n=1 the above
relationship becomes:
N/No = exp-VD
On a semi-log plot this will give a linear inactivation curve of slope -V.

87
Dependence on radiation source
If, in a reaction where a single hit in a molecule is sufficient to
inactivate it, the ionizations were distributed at random, the survival
of a molecule would depend only on the number of ionizations produced per
unit volume in the tissue. In fact, ionizations are located along the
paths of ionizing particles. If the particles have a high linear energy
transfer (LET) the distance between successive primary events is very small
(< 1 R) and it is probable that several ionizations may take place when the
particle passes through the target. However in the case of high energy
electrons (as used in the present study) the LET is low. The electrons
are moving so fast that primary events occur at greater distances (2,000 A)
and are therefore distributed randomly with respect to the volume of the
target.
Calculation of molecular size from survival curves
Radiation doses are usually measured in units of rads. One rad can
be defined as an energy transfer of 100 ergs per gram of tissue. Target
theory, as explained in a previous paragraph, predicted that the fraction
N/No of active molecules which survive a radiation dose D (measured in
inactivating events /cm3 i ) is given by:
N = e -VD
N
The slope of the semi-log plot of N/N0 against D corresponds to V, the target
volume in cm3. The major problem is to relate the dose which is measured in
rads to the dose absorbed by the target molecules (in units of inactivating
events per cm 3). _). Therefore it is necessary to determine the energy transferred
in the primary event, this calculation involves determining the electron volts

88
per inactivating event (Q). In the present study the method developed
by Kepner and Macey (1968) was employed. They produced a calibration
curve relating the molecular weights of various enzymes as determined by
biochemical studies to the dose (in rads) required to achieve 37% survival
i.e. the D37. They obtained a linear relationship from which they were
able to obtain a value for Q of 66 electron volts per inactivating event.
If p is the target density and N Avogadro's number, then it is
possible to calculate the molecular weight from the target volume V from
the relationship:
molecular weight =pNV (1)
If ionizations are produced in the tissue singly and at random,
the dose needed to produce an average of one ionization per target of
1 volume V (i.e. the D37) would produce ionizations per unit volume in
the tissue
1 i.e. V = D
37
to convert the measured dose (in rads) to the dose in inactivating events
percmh3 the following relationship must be used:
(p (g/ml) 103 erg/g.rad
D37 (inactivating electron volts - X D37 Q (electron volts 1.6 x 10-12 erg/e (rads)
per inactivating event)
(taken from Kepner and Macey, 1968) (3)
(2)

using Q = 66 electron volts per inactivating event and substituting
equations 2 and 3 into equation 1 gives:
molecular weight =pNx
/ p (g/cm3) x 103 erg/g.rad
66 eV/inactivating 1.6 x 10-12 erg/eV event
8 9
D37 (rads)
1
which simplifies to give:
6.4 x 1011
molecular weight =
D37 (rads)
This relationship is independent of the density of the target and
can be used to relate the dose of radiation which corresponds to 37%
survival of the activity being measured to the size of the target in vivo
if one assumes that:
1. The radiation is sparsely ionizing i.e. the primary events are
randomly distributed with respect to volume.
2. A single 'hit' inactivates the target.
3. There are no indirect effects.
Irradiation inactivation measures the functional size of a molecule
rather than its true molecular weight; in most cases the terms are
synonymous, but on occasion the molecular size measurement may over-
or under estimate the molecular weight depending on the quarternary
structure of the molecule involved. This point is considered more
fully in a subsequent paragraph.
(4)

90
Investigators who have studied the radiation inactivation of
proteins of known molecular weight have found good correlation between
target size (as calculated above) and the known values for molecular weight.
In addition, inactivation curves (log surviving fraction against dose) are
normally linear indicating that for most protein molecules a hit anywhere
within the molecule will cause inactivation.
Non-linear inactivation curves can be analysed by other models,
for example, the 'multihit target model' (which assumes that inactivation
of the molecule requires multiple hits within the same target), or the
'multi-target model' (which assumes that the molecule contains multiple
targets each of which must be inactivated by a single hit for inactivation
of the whole molecule). The latter model has been applied to the IgM
haemolytic antibody by Rosse et al. (1967). They found that there was
at least 3 independent target areas in one IgM haemolytic antibody molecule.
Criticisms of target theory
1. Although survival curves of many molecular activities can be described
by a single hit inactivation process it is possible that almost linear
survival curves may also be obtained from more complex actions in which
a number of ionizations must be produced within the target. In addition
indirect actions of radiations lead to survival curves of similar shape
to those obtained from direct action and therefore the shape of the survival
curve alone does not show whether direct or indirect processes predominate
in inactivation. However, indirect action was limited in the present study
by irradiating in the dry state under nitrogen and so anomalous results due
to the action of free radicals should be minimal.

91
2. Much of the energy dissipated by radiation in tissue becomes
degraded to heat energy and heating denatures proteins. Therefore it
could be argued that loss of biological activity was due to heat. However,
most proteins have a threshold value for thermal inactivation and therefore
the shape of the survival curve would be altered i.e. there would be a
rapid loss of activity at the threshold temperature rather than a linear
survival curve, if activity loss was due to temperature rather than
ionization. The radiosensitivity of a material normally increases as a
function of temperature and so it is important that the heat generated
during irradiation is controlled and standardized for each experiment in
a series.
3. Target theory assumes that the target is inactivated by a hit if it
occurs in that part of the molecule which is sensitive to inactivation.
There are a number of instances in which this may not happen. A molecule
may lose energy by emitting a photon without any decline in biological
activity and furthermore, one can imagine, for example; in the case of
an enzyme, a lesion in the molecule far away from the active site which
would not affect enzyme activity. In addition there is some evidence
which suggests that the energy transferred at the primary event is sometimes
insufficient to cause an ionization (Johnson and Rymer, 1967; Rauth and
Simson, 1967). If, however, one assumes that the ratio of sensitive volume
to actual target volume is a constant for every target and the proportion of
primary events which inactivate is the same for all different targets then c
target theory will still apply. This reasoning is probably more true for
large molecules where energy may be transferred to a wider choice of locations
than small molecules where the choice is more likely to be restricted
(Davis and Pollard, 1952). These assumptions are substantiated by the
general agreement between irradiation inactivation molecular weight
estimations and those obtained by more conventional techniques (Pollard, 1959).

92
A more detailed criticism of target theory can be found in Lea (1955).
The significance of molecular size determinations
Throughout this chapter the term 'molecular size' rather than molecular
weight is used. This is because the method based on irradiation inactivation
determines the functional size of a molecule rather than its true molecular
weight. If the Rh antigen were composed of smaller protein molecules
which could be inactivated by a hit on any of the subunits, then the
molecular weight as measured by irradiation inactivation would be that
of the whole polymer. Insulin is an example of a molecule where the
large molecular weight as estimated by irradiation is related to the
polymeric form of the monomer (Setlow and Doyle, 1953). However, if a
direct hit on one particular subunit was necessary for inactivation then
the molecular weight of a complex would be that of the subunit as in the
case of catalase (Setlow, 1952). Therefore radiation inactivation data
taken with conventional molecular weight determination studies can reveal
information regarding the protein molecule as it exists and functions in
its normal molecular environment.
Irradiation inactivation as a technique for molecular size determination
has been subject to criticism for many years, principally because of the
assumptions which must be made before it can be applied. The most
convincing support for the validity of the method is the general agreement
between determinations by conventional methods and irradiation inactivation
(Pollard, 1959). To reject the technique merely because at this time one
is unable to understand its mechanism fully or explain its shortcomings
would be to reject the valuable information which can be revealed when the
method is applied in the appropriate circumstances.

93
SECTION 1
RADIATION INACTIVATION OF THE Rh (D) ANTIGEN
METHODS AND MATERIALS
1) Red cells
The red cells used in both section 1 and section 2 of this chapter
were Rh phenotyped. All the cells of probable genotype CDe/cDE and
cde/cde used were phenotyped at the North London Blood Transfusion Centre.
The other cells were phenotyped for C, c, E and e antigens by standard
techniques (Mollison, 1975). The D antigens were investigated using
125I-labelled anti-D and the data used in combination with Rh frequency
tables to determine the probable genotypes. For the experiments involving
the A antigen, outdated group A cells were obtained and tested with an
extract of Dolichlos biflorus (lectin-A1) and only red cells which
agglutinated with this reagent (A1 cells) were used.
2) Preparation of red cell membranes
Membranes were prepared from fresh or 21-day old normal, human, Rh (D)
positive blood by hypotonic lysis in 20 mosmol/1 phosphate buffer pH 8.0,
followed by at least 3 washes, by centrifugation (90,000 g, 20 min), in the
same buffer. The stroma was lyophilized and stored at -20°C until required.
3) Preparation of 125I-labelled anti-D
An IgG preparation containing high titre anti-D was labelled with 1251
by the iodine monochloride method and purified by absorption onto Rh positive
red cells followed by ether elution as described in chapter II, section 1.
The final product had a specific activity of 5,200 cts/min/pg and was
approximately 8% pure.

94
4) Anti-A
Prepared using ethanol fractionation followed by elution from DEAE
cellulose, kindly donated by Professor P.L. Mollison. The product was
98% pure IgG and had 60 pg anti-A per ml. This preparation was used as
a source of anti-A throughout the following chapters.
5) Radiation procedure
Stroma samples, normally 5 mg, were irradiated under nitrogen in
pyrex tubes using the linear accelerator at Addenbrooke's hospital,
Cambridge. Doses between 1 and 10 megarads were used at a rate of
1 megarad per min. The tubes were cooled by a jet of air at 0°C.
These precautions were taken to minimise 'non-specific' inactivation
by free radicals (Kepner and Macey, 1968; Ebert, 1973). The temperature
at the end of a 10 megarad irradiation was measured with a thermocouple
to ensure that any loss of activity could not be attributed to excessive
heat. Irradiated stroma was stored dry at -20°C until required for assay.
6) Measurement of Rh (D) antigen activity
The Rh (D) activity remaining in the stroma samples was measured
by an inhibition method using 125I-labelled anti-D. Each aliquot of
irradiated stroma was reconstituted in phosphate buffered saline (2.0 ml)
and sonicated at an amplitude setting of 2 microns for 2 minutes in an
MSE 100 watt ultrasonic disintegrator. The amount of active D antigen
on the stroma was measured by incubating 1 pg of purified 125I-labelled
anti-D with 0.4 ml aliquots of the stroma for 30 min at 37°C. Rh positive
red cells (0.05 ml) of the probable genotype CDe/cDE were added and the
mixtures incubated for a further 30 minutes at 37°C. The red cells were

95
washed three times in phosphate buffered saline and the amount of 1251._
labelled anti-D bound to the cells was estimated. The amount of D antigen
remaining in the irradiated stroma was determined from a calibration curve
prepared using varying amounts of non-irradiated stroma from the same batch
as used in the test.
7) Measurement of acetylcholinesterase activity
The activity of this enzyme after irradiation was measured by
Dr. J.C. Ellory by the col,irimetric method of Ellman et al. (1961).
8) Measurement of A antigen activity
The irradiated stroma was reconstituted in saline and sonicated
as described for the Rh (D) assay. Aliquots of stroma (0.4 ml) were
incubated with 6 pg anti-A for 30 min at 37°C. The stroma was then
centrifuged at 90,000 g for 20 min and the supernatant containing
unbound anti-A removed. Serial dilutions of the supernatant were made
and one drop of each dilution was mixed on a glass tile with one drop
of a 10% suspension of washed Al red cells. The end point of
haemagglutination was assessed visually after 5 min and compared with
that of a non-irradiated stroma control sample.
RESULTS
1) Rh (D) antigen activity
Stroma samples from 9 donors were irradiated as described in the
methods. The percentage of Rh (D) activity remaining was plotted on a
log scale against the dose of radiation. A straight line could be drawn
through the data indicating an exponential relationship. The D37 (megarads)
was estimated from a regression line calculated for each experiment and
substituted in equation 4, page 89
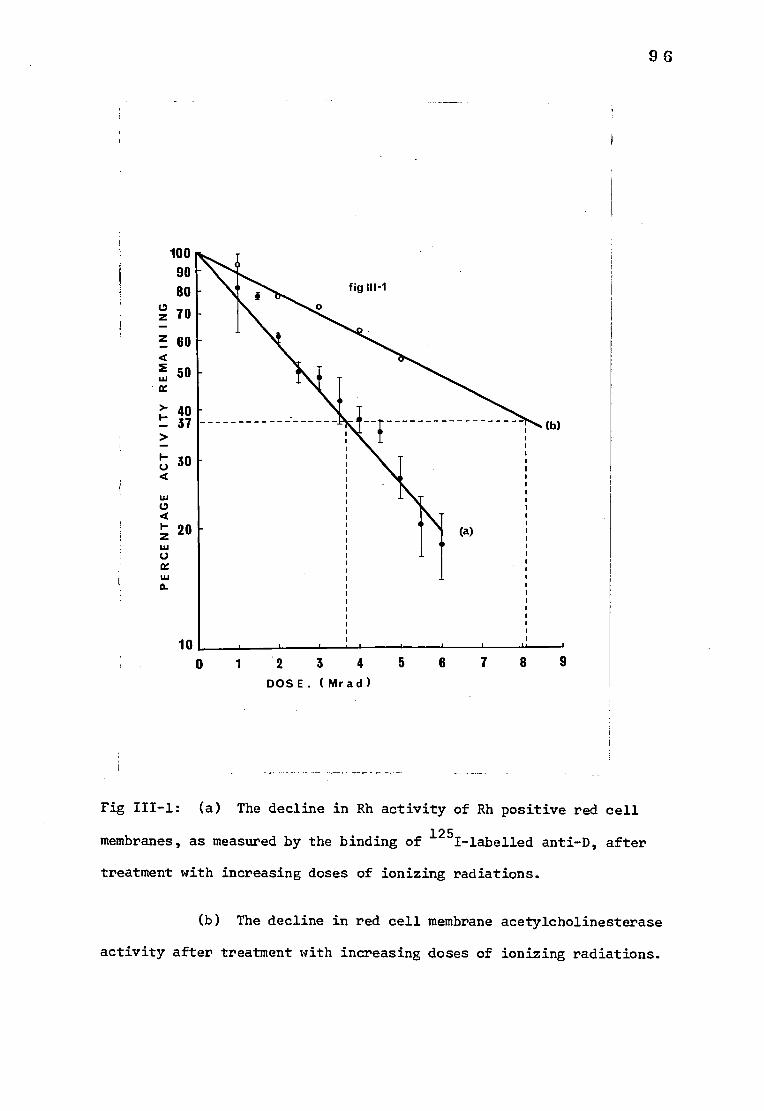
6 2 3 4
5 DOSE. ( Mr ad)
0 z 70
z 60
2 50
>- 40 37
30
100 90 80
0 a 2°
U
1 7 8 9
Fig III-1: (a) The decline in Rh activity of Rh positive red cell
membranes, as measured by the binding of 125I-labelled anti-D, after
treatment with increasing doses of ionizing radiations.
96
(b) The decline in red cell membrane acetylcholinesterase
activity after treatment with increasing doses of ionizing radiations.

97
The results are shown in Table III-1. The mean molecular size from
these 9 estimates was 174,000 4. 10,000 daltons. The data from all the
experiments was used to find the mean result at each radiation dose.
The means and the standard error of each mean are shown in Fig III-1.
2) A antigen activity
There was no measurable decrease in A antigen activity after
exposure to ionizing radiations of the doses used in the present study.
3) Acetylcholinesterase activity
Three experiments were carried out to determine the molecular size
of the membrane acetylcholinesterase. The average value obtained for
the esterase activity remaining after each irradiation dose is shown in
Fig and the estimate of the molecular size obtained from these
results was 79,000 daltons. This result compares well with the estimates
of 70,000-80,000 previously reported (Levinson and Ellory, 1974).
4) Temperature changes
The temperature after an ionizing dose of 10 megarads was found to
be 27-34°C. This temperature would have had no measurable effect on
D antigen activity, since, in a separate experiment, there was no loss
in Rh (D) activity on heating lyophilized stroma at 56°C for 15 minutes.
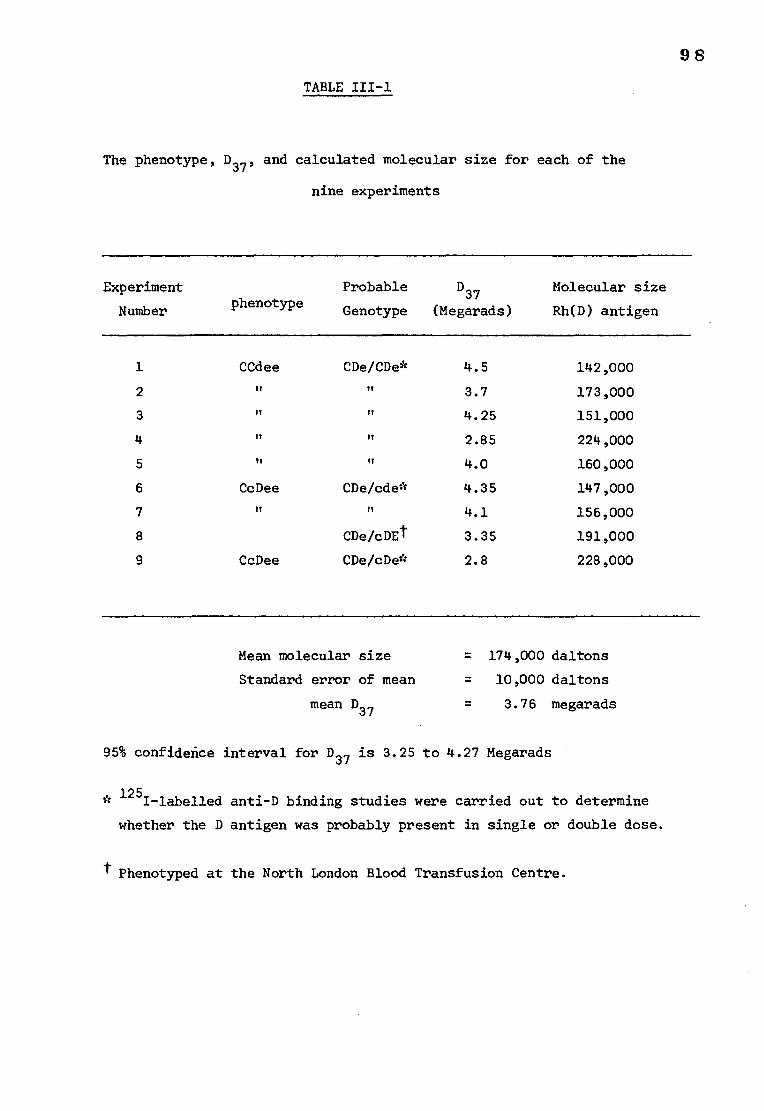
98
TABLE III-1
The phenotype, D37, and calculated molecular size for each of the
nine experiments
Experiment
Number phenotype Probable
Genotype
D37
(Megarads)
Molecular size
Rh(D) antigen
1 CCdee CDe/CDe* 4.5 142,000
2 II II 3.7 173,000
3 n n 4.25 151,000
4 n n 2.85 224,000
5 n ft 4.0 160,000
6 CcDee CDe/cde* 4.35 147,000
7 n n 4.1 156,000
8 CDe/cDEt 3.35 191,000
9 CcDee CDe/cDe* 2.8 228,000
Mean molecular size = 174,000 daltons
Standard error of mean = 10,000 daltons
mean D37 3.76 megarads
95% confidence interval for D37 is 3.25 to 4.27 Megarads
* 125I-labelled anti-D binding studies were carried out to determine
whether the D antigen was probably present in single or double dose.
t Phenotyped at the North London Blood Transfusion Centre.

99
DISCUSSION
The possibility that the loss in Rh activity during irradiation was
due to indirect factors such as heat or free radicals must be considered
The small rise in temperature recorded at the end of a 10 megarad
irradiation dose was not sufficient to explain the biological effects
produced, unless it was initially localized within a small number of atoms
in the tissues causing a large rise in temperature in these atoms.
However, the ionization of an atom is considered to result in a chemical
change in the affected molecule rather than create a 'hot spot' (Lea, 1955)
and so the temperature rise in dry tissues can probably be discounted as a
cause of reduction in Rh (D) activity. Inactivation of the antigen by free
radicals is unlikely since their mobility is restricted in lyophilized
preparations. In addition the membranes were irradiated under nitrogen
since some materials are more sensitive to radiation in the presence of
oxygen (Ebert, 1973).
Additional evidence that the decline in Rh activity was not a
'non-specific' effect comes from the experiments investigating the effect
of radiation on the A antigen. The red cell blood group antigens of the
ABO system are mainly oligosaccharides attached to ceramide (Stellner et al.,
1973; Hanfland and Egli, 1975; Anstee and Tanner, 1975) and hence would
have a molecular weight of the order of 2,000 daltons. Molecules of this
size would require a radiation dose of approximately 300 megarads to bring
about a significant reduction in antigen activity, a dose far higher than
that used in the present experiments. The stability of the A antigen at
radiation doses up to 10 megarads is consistant with a small molecular
weight and also indicates that there are no potent 'non-specific' destructive
agents present as a result of the ionizing radiation, although the presence of
a weaker destructive agent affecting protein but not carbohydrate antigens
cannot be eliminated.

100
The data from this irradiation inactivation study indicated that
the molecular size of the D antigen is approximately 174,000 daltons.
Conventional molecular weight studies on the D antigen are very limited.
Only two groups (Weickerand Metz, 1971; Abraham and Bakerman, 1974) have
claimed to have isolated the active antigen in recent years and both have
suggested a molecular weight in the range 10,000 to 20,000 for a purified
isolated preparation of the antigen. Lorusso and Green (1975) have studied
the solubilization of the antigen using deoxycholate. Their results
indicated that the proteins solubilized by deoxycholate regained Rh activity
after removal of detergent and dialysis against buffered water containing
magnesium ions. Microscopic examination of the solubilized proteins after
dialysis showed the formation of vesicles indicating that some form of
crude aggregation into 'membrane-like' structures may be a pre-requisite
for Rh activity. The solubilized proteins were also fractionated by
filtration and the results indicated a molecular weight of less than
300,000 daltons.
The work of Green (1972), Lorusso and Green (1975) and Weicker et al.
(1973) has indicated that the active site of the D antigen is situated on
a protein but that the conformation of the protein requires the presence
of phospholipid to maintain the antigenic activity. It is possible that
D activity is only expressed when a number of small protein molecules poly-
merise. If Rh (D) activity is lost only when a particular molecule in the
aggregate is hit then the molecular weight obtained by irradiation inactivation
would be that of the individual molecule rather than that of the aggregate.
Conversely, if Rh (D) activity was lost when any of the molecules in the
aggregate was hit then the molecular weight would be that of the aggregate
rather than the molecule which had been hit. Therefore it is possible that

1 0 1
the Rh antigen could be a subunit of molecular weight 10,000-20,000
daltons in an aggregate of total molecular weight 174,000 and that the
D antigen interacts with the other molecules in the aggregate such that
an ionization in any part of the aggregate will inactivate it. Alternatively,
the D antigen could be a single molecule of molecular weight 174,000 daltons.
There are a number of proteins in the red cell membrane of molecular weights
in the range 150,000-200,000 which could be the Rh antigen(Conrad and
Pennington, 1976). Whitely and Berg, (1974) have identified a protein
of 170,000 daltons on the outer surface of the cell by amidation.

102
SECTION II
RADIATION-INACTIVATION'OF THE Rh (D, C, c and e) ANTIGENS
INTRODUCTION
The successful radiation inactivation of the Rh (D) antigen led
naturally to the investigation of other Rh antigens. The antigenic
activity of the D antigen can readily be determined accurately using
high titre 1252-labelled antibody. It is however, very difficult to
obtain sufficient amounts of antibodies with Rh antigenic specificities
other than D, with a high enough titre to enable purified labelled antibody
preparations to be made. After several unsuccessful attempts to label
an IgG preparation (with anti-C specificity) with 1251 it was decided to
develop a different technique.
It is possible to measure the Rh antigenic activity of red cell
membranes by assessing visually the extent to which the membranes are
able to inhibit the agglutination of papain treated red cells in the
presence of an appropriate antibody. However, it is difficult to express
an antibody titre accurately in terms of the percentage activity surviving
radiation and the visual estimation of agglutination is rather subjective.
A less biased result can be obtained by using a spectrophotometer.
The agglutination of red cells causes the solution in which they are
suspended to become less dense thereby permitting the passage of more
light than one in which the cells are not agglutinated. The extent to
which the cells are clumped can therefore be determined in a spectrophotometer.

103
Hence the antigenic activity of stroma can be estimated by its ability
to inhibit the agglutination of red cells in the presence of a suitable
antibody.
The irradiation inactivation of the D, C, c and e antigens is
described in the following section. As a control the blood group
A activity was also measured after irradiation by a spectrophotometric
method.
METHODS AND MATERIALS
1) Antibodies
i) Anti-A, the purified preparation used was 98% pure IgG and had
60 pg anti-A per ml.
ii) Anti-D, an IgG preparation containing high titre anti-D;
(1600 by agglutination of papainised red cells). Used without further
purification.
iii) Anti-C, from Nordisk insulin laboratories, Denmark. The titre
was 128 by agglutination of papainised red cells. No anti-D activity
detectable at the dilution used. Used without further purification.
iv) Anti-c, a post mortem serum sample. The serum had weak anti-D
activity and therefore red cells of the probable genotype cde/cde were
used in all tests involving this antibody although the anti-D activity was
not measurable at the antibody dilution used in the tests. The serum was
used without further purification.

104
v) Anti-e, a post mortem serum sample with a titre of 8 by
the indirect antiglobulin test. The serum was used without further
purification
2) Treatment of red cells with papain
Two drops of washed, packed red cells were mixed with one drop of
0.1% papain and incubated at 37°C for 7 minutes. The cells were washed
twice with warm saline and kept at 0°C before use.
3) Irradiation of cell membranes
Red cell membranes of various phenotypes were prepared and irradiated
as described in section I.
4) Spectrophotometric measurement of antigenic activity
Papain treated red cells agglutinate in the presence of the
appropriate Rh antibodies. In the present study the antigenic activity
of irradiated red cell membranes was estimated by the ability of the
membranes to inhibit the agglutination of papainized red cells in the
presence of a fixed amount of antibody. The accuracy of this method for
estimating antigenic activity depends on determining the antibody concentration
which, under the experimental conditions used, will maximally agglutinate
the red cells of the chosen phenotype. If too much antibody is used a
small decrease in the antigenic activity of the membranes will not be
measurable and if too little antibody is used the range of values for
absorption over which agglutination takes place will be too small. A
dilution of papainized red cells which gave an absorption of approximately
0.7 in a volume of 1.0 ml (1 cm light path) was chosen and a suitable antibody
concentration determined as described below.

105
(a) Optimum antibody concentration determination
Various amounts (1.0, 0.5, 0.25, 0.125 and 0.0625 ml) of a trial
antibody dilution were each mixed with saline to make a total volume of 1.0 ml.
Papainized red cells (0.035 ml of a 1:100 dilution of packed cells), with
a double dose of the antigen where possible i.e. cde/cde cells for the c
and e antigens and CDe/CDe cells for the C antigen, were added to each
antibody dilution and rotated 'end over end' for 60 minutes. The cells
were then centrifuged at approximately 500 rpm for 5 minutes to encourage
agglutination and finally resuspended by rotation for a further 10 minutes.
The absorption of the cell suspensions at 600 nm were read immediatly in
a Unicam SP 600 spectrophotometer. The experiment was repeated with
different antibody dilutions until one was found which agglutinated the
red cells maximally with the largest antibody volume used i.e. 1.0 ml.
(b) Calibration curves
Red cell membranes which had been freeze-dried but not irradiated
were reconstituted in saline and sonicated at an amplitude setting of
2 microns in a MSE ultrasonic disintegrator (100 watts) for 2 minutes.
Aliquots of stroma between 0 and 0.2 ml were mixed with 1.0 ml samples of
the chosen antibody dilution and incubated 60 minutes at 37°C. The stroma
was then centrifuged at 35,000 rpm for 30 minutes. The supernatants were
removed and papainized red cells (0.035 ml of a 1:100 dilution) were added
and the mixtures rotated for 60 minutes at room temperature. The cells
were spun at approximately 500 rpm for 5 minutes and resuspended by rotation
for a further 10 minutes before reading the absorption at 600 nm. The
absorption of control samples with saline substituted for the antibody were
also measured. All experiments were performed in duplicate.

106
5. Antigenic activity of irradiated membranes
Each aliquot of irradiated membranes was reconstituted in 2.0 ml
phosphate buffered saline and sonicated at an amplitude setting of 2 microns
for 2 minutes. In general, 0.2 ml samples of stroma were incubated with
1.0 ml of the chosen antibody dilution as described for the calibration
curves. After incubation, and ultracentrifuging the antibody supernatants
were removed and papainized red cells added and rotated as previously described.
The absorption of the red cell suspensions was recorded and compared with
the appropriate calibration curve in order to enable an estimation to be
made of the percentage of the original antigen activity remaining after
irradiation. This technique was used for all the Rh antibodies tested.
6. Measurement of the remaining A antigen activity after irradiation
A antigen activity was measured by the technique described above
for the Rh antigens except that the red cells were not treated with papain
since group A red cells readily agglutinate in the presence of IgG anti-A
without enzyme treatment.
RESULTS
1) Optimum antibody dilutions
The effect of the antibody dilution on the absorption of light by
red cell suspensions can be seen in Fig 111-2 for various dilutions of
anti-D. The aim of this experiment was to determine the greatest dilution
of antibody which would maximally agglutinate the red cells of the chosen
phenotype. In the experiment illustrated in Fig 111-2 a 1:3200 dilution
of the anti-D preparation produced maximum red cell agglutination. Using
to much or too little antibody reduced the sensitivity of the technique.
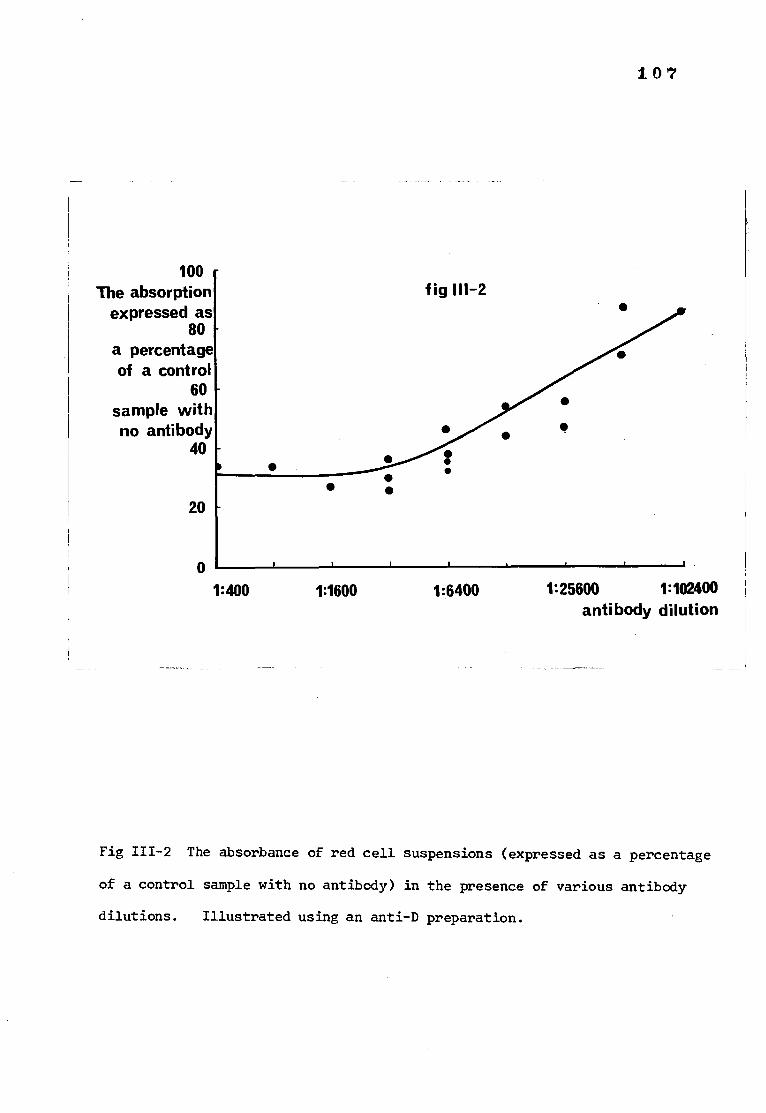
107
100 - The absorption
expressed as 80
a percentage of a control
60 sample with no antibody
40
fig 111-2
•
•
•
•
• •
•
• •
20
0 1:400 1:1600 1:6400 1:25600 1:102400
antibody dilution
Fig 111-2 The absorbance of red cell suspensions (expressed as a percentage
of a control sample with no antibody) in the presence of various antibody
dilutions. Illustrated using an anti-D preparation.
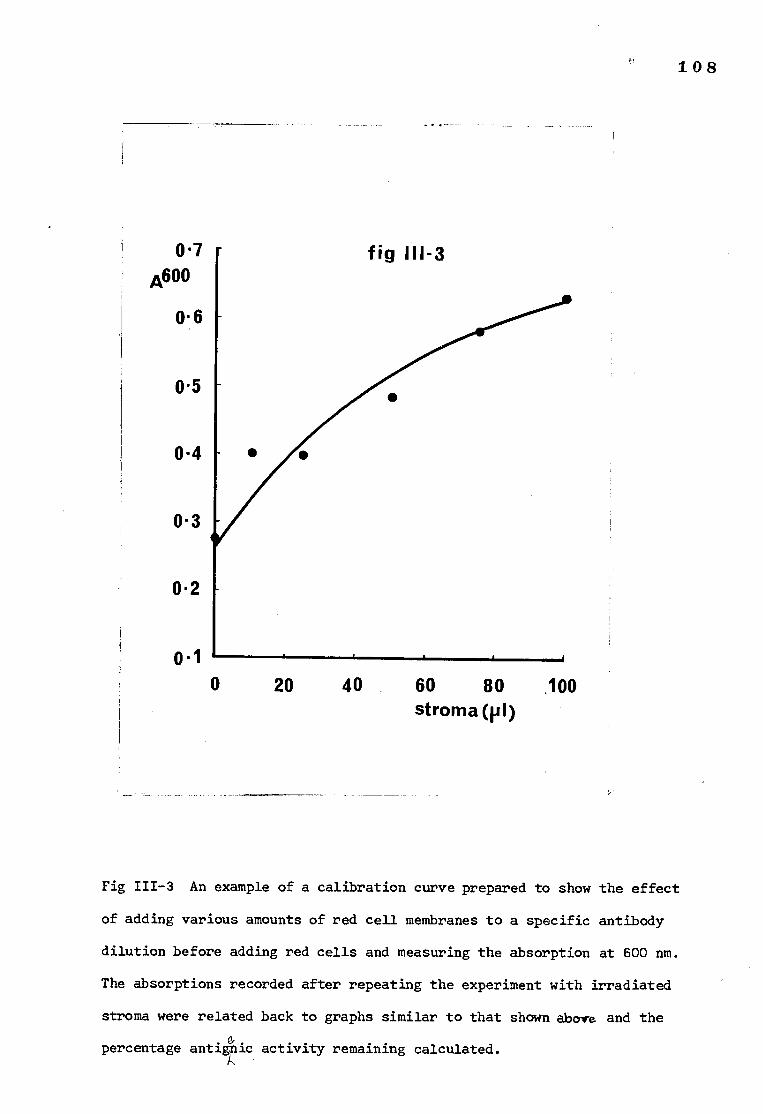
108
0.7 A600
0.6
0.5
0-4
0.3
0.2
0 ' 1
fig 111-3
•
0 20 40 60 80 .100 stroma(p1)
Fig 111-3 An example of a calibration curve prepared to show the effect
of adding various amounts of red cell membranes to a specific antibody
dilution before adding red cells and measuring the absorption at 600 nm.
The absorptions recorded after repeating the experiment with irradiated
stroma were related back to graphs similar to that shown above and the
percentage antignic activity remaining calculated. k

109
The optimal antibody dilutions, as determined by this method, for
the antibodies tested, were, for anti-D, 1:3200, for anti-C, 1:200, for
0 anti-c, 1:100, for anti, 1:40 and for anti-A, 1:16.
2) Calibration curves
Calibration curves and estimations of irradiated stroma were made
simultaneously using the same reagents so that experimental variations
could be minimised. A typical calibration curve, for anti-D, is shown
in Fig 111-3.
3) Antigenic activity after irradiation
a) D antigen The percentage of D activity remaining after irradiation
was plotted on a log scale against the dose of radiation.
For each experiment a regression line was calculated and the D37
obtained from each line is given in Table 111-2. The data was also
analysed by pooling all the results. The pooled regression data were
used to produce mean values of percentage survival for each radiation dose.
The line drawn from these means and the standard error of each mean (SEM)
are shown in Fig 111-4. The mean molecular size from Fig 111-4 was
183,000 daltons and the 95% confidence limits for the D37 (3.5) were
2.81-4.19.
b) C antigen Five estimates of the molecular size of the C antigen
were made by the method described for the D antigen. The individual
results are shown in Table 111-2. Using the pooled data, the means and
standard errors of the means for each radiation dose were calculated and
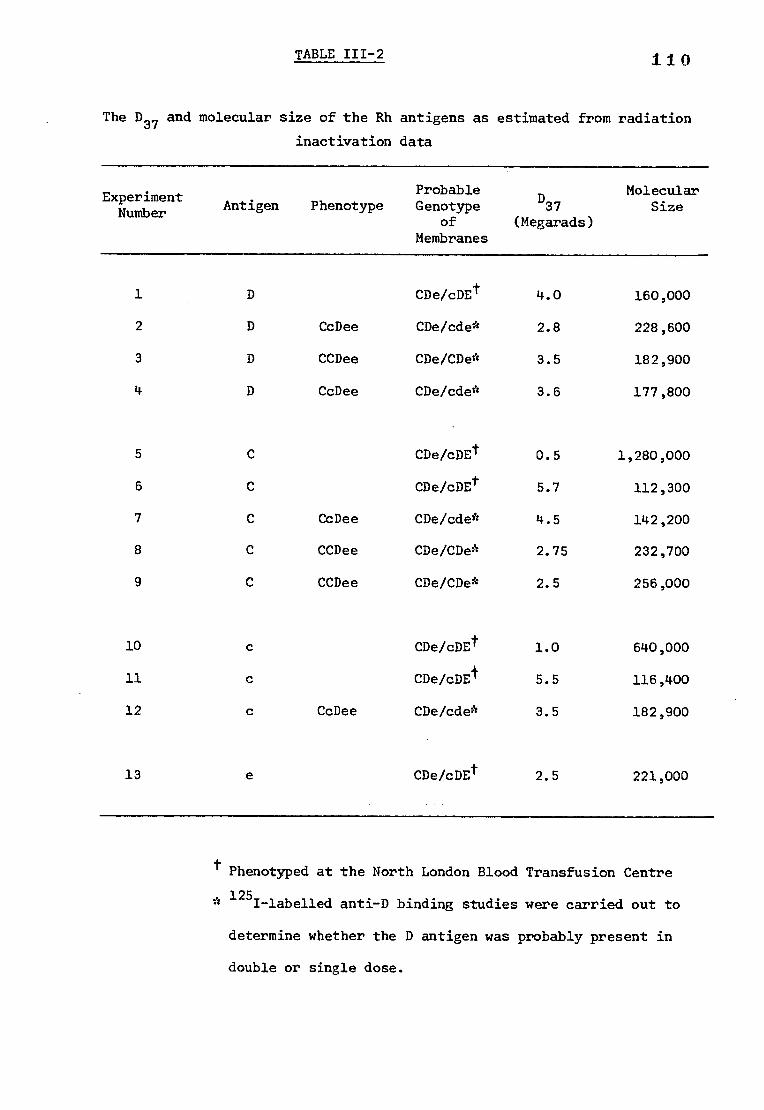
TABLE III-2 110
The D37 and molecular size of the Rh antigens as estimated from radiation
inactivation data
Experiment Number Antigen Phenotype
Probable Genotype
of Membranes
D37
(Megarads)
Molecular Size
1 D CDe/cDEt 4.0 160,000
2 D CcDee CDe/cde' 2.8 228,600
3 D CCDee CDe/CDe* 3.5 182,900
4 D CcDee CDe/cde' 3.6 177,800
5 C CDe/cDEt 0.5 1,280,000
6 C CDe/cDEt 5.7 112,300
7 C CcDee CDe/cde* 4.5 142,200
8 C CCDee CDe/CDe' 2.75 232,700
9 C CCDee CDe/CDe* 2.5 256,000
10 c CDe/cDEt 1.0 640,000
11 c CDe/cDEI 5.5 116,400
12 c CcDee CDe/cde* 3.5 182,900
13 a CDe/cDEt 2.5 221,000
Phenotyped at the North London Blood Transfusion Centre
* 125I-labelled anti-D binding studies were carried out to
determine whether the D antigen was probably present in
double or single dose.
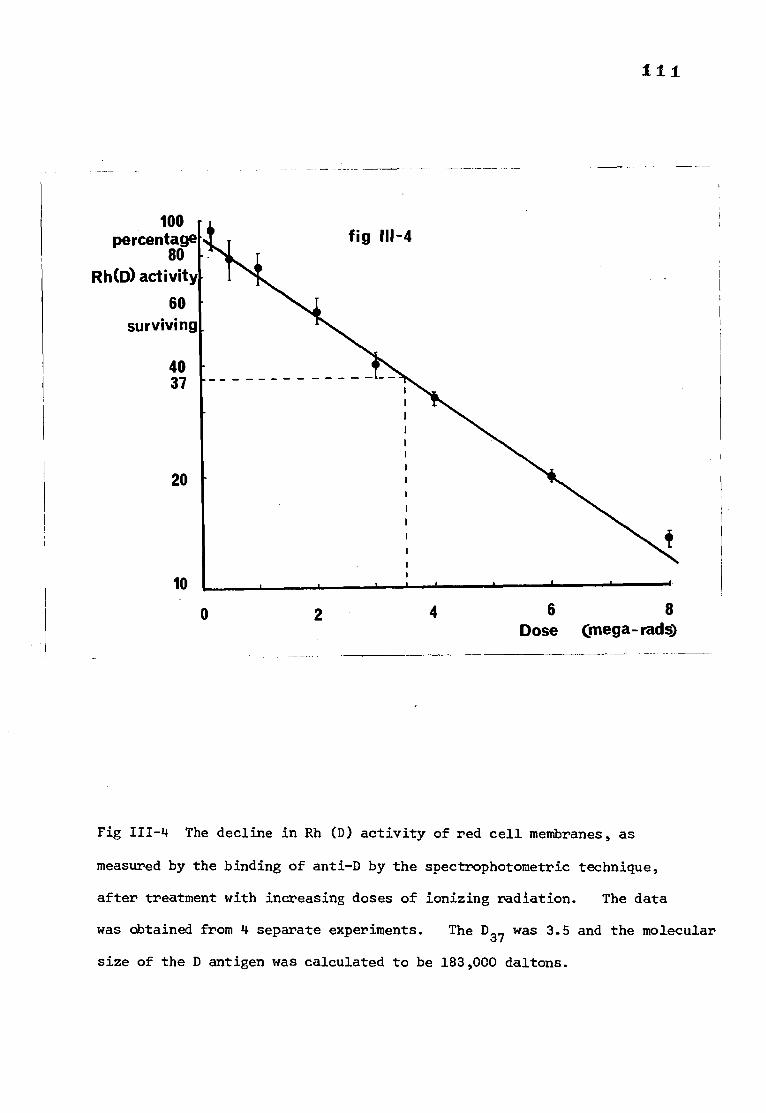
I 1 1
100 percentage
80 Rh (3) activity
60 surviving
40 37
20
10
0
2 4 6 8
Dose (mega- rads)
Fig 111-4 The decline in Rh (D) activity of red cell membranes, as
measured by the binding of anti-D by the spectrophotometric technique,
after treatment with increasing doses of ionizing radiation. The data
was obtained from 4 separate experiments. The D37 was 3.5 and the molecular
size of the D antigen was calculated to be 183,000 daltons.
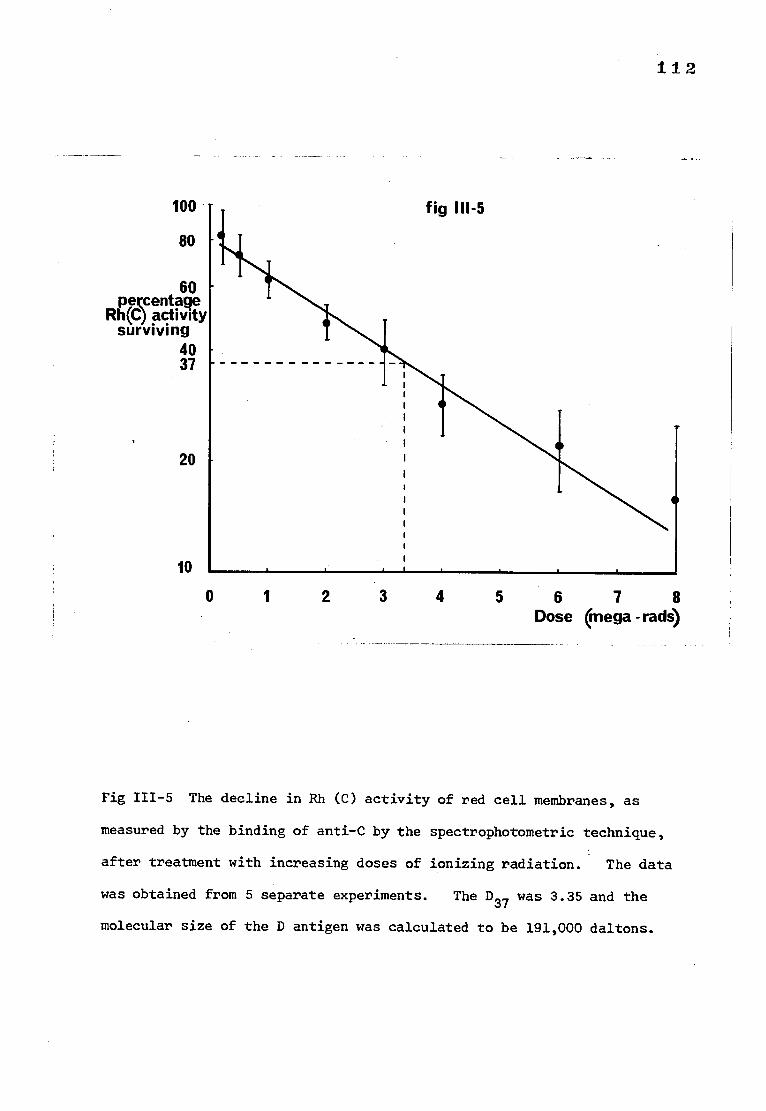
fig 111-5
112
100
80
60 percentage
Rh(C) activity surviving
40 37
20
10
0 1 2 4 5 6 7 8 Dose (mega -rads)
Fig 111-5 The decline in Rh (C) activity of red cell membranes, as
measured by the binding of anti-C by the spectrophotometric technique,
after treatment with increasing doses of ionizing radiation. The data
was obtained from 5 separate experiments. The D37 was 3.35 and the
molecular size of the D antigen was calculated to be 191,000 daltons.
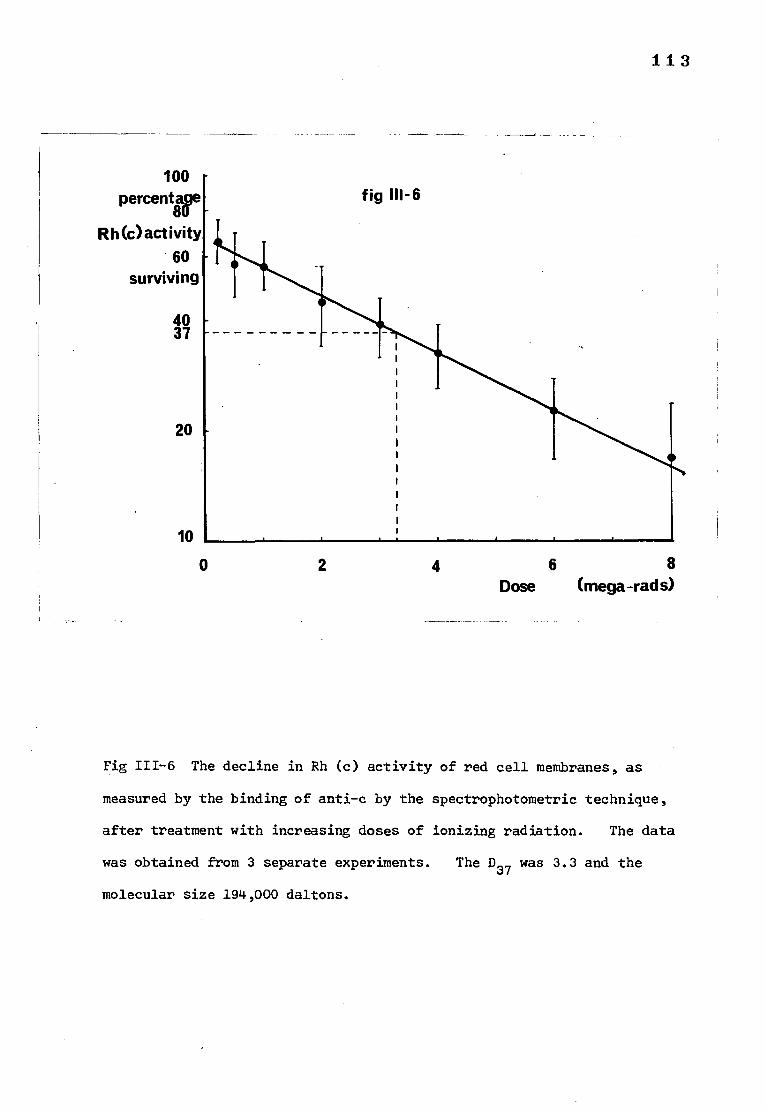
fig
113
100 - percenttoge
Rh(c)activity 60
surviving
40 37
20
10
0 2 4 6 8 Dose (mega-rads)
Fig 111-6 The decline in Rh (c) activity of red cell membranes, as
measured by the binding of anti-c by the spectrophotometric technique,
after treatment with increasing doses of ionizing radiation. The data
was obtained from 3 separate experiments. The D37
was 3.3 and the
molecular size 194,000 daltons.
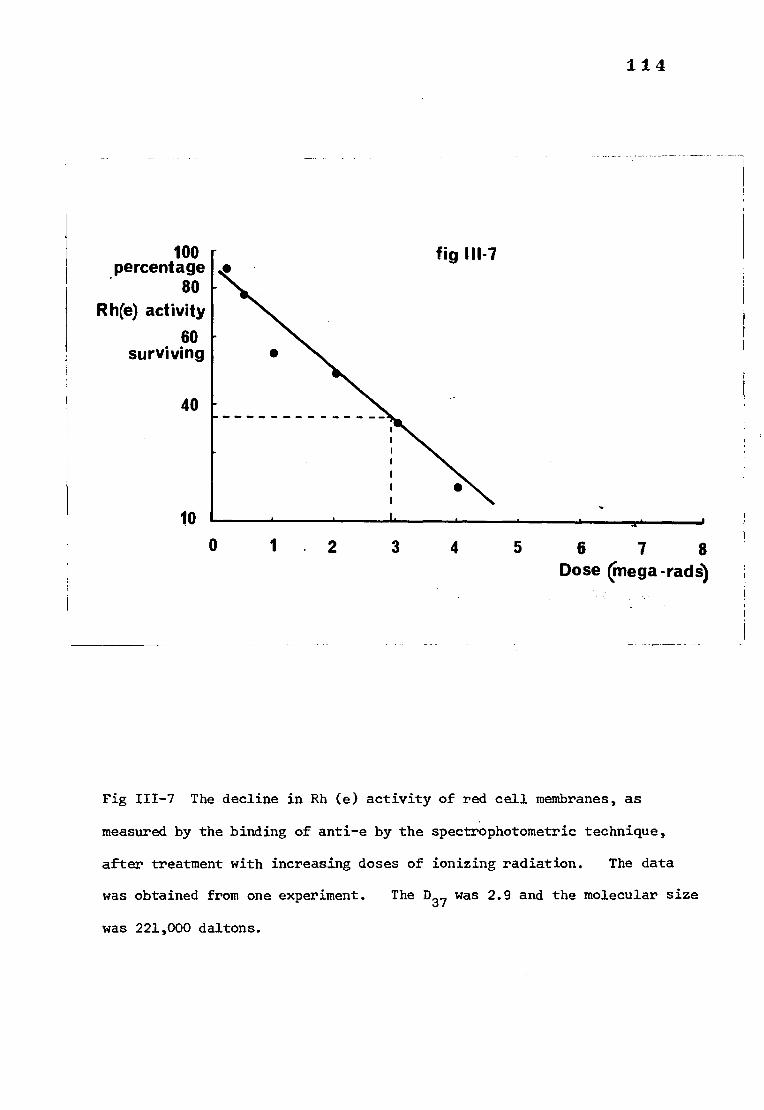
• fig 111-7
114
100 percentage
80 R h(e) activity
60 surviving
40
10 0 1 2 3 4 5 6 7 8
Dose mega -rads)
Fig 111-7 The decline in Rh (e) activity of red cell membranes, as
measured by the binding of anti-e by the spectrophotometric technique,
after treatment with increasing doses of ionizing radiation. The data
was obtained from one experiment. The D37 was 2.9 and the molecular size
was 221,000 daltons.

1 1 5
are shown in Fig 111-5. The molecular size of the C antigen as calculated
from the D37 (3.35) from Fig 111-5 was 191,000 daltons.
c) c antigen Three estimates of the irradiation inactivation of
the c antigen were made. The individual results are shown in Table 111-2.
Using the pooled data, the means and standard errors of the means for each
radiation dose were calculated and are shown in Fig 111-6. The D37 from
Fig 111-6 was 3.3 and the molecular size 194,000 daltons.
d) e antigen It was only possible to make one estimation of the
e antigen and therefore little importance can be attached to this
measurement. The result is given in Table III-2and the individual
result for each dose in Fig 111-7. The D37 was 2.9 and the molecular
size was 221,000 daltons.
e) A antigen There was no measurable decline in A antigen activity
after exposure to ionizing radiations of the doses used in the present study.
DISCUSSION
The reasons for assuming that indirect effects of the radiation were
negligible in these experiments were discussed in section 1. It does seem,
however, that the radiosensitivity of the red cell membranes was greater than
expected in one experiment concerning the C antigen (experiment number 5)
in which the D37 was only 0.5. Taking into account the general agreement
between the other C antigen estimations it seems likely that some other
factor was potentiating the direct action of the radiation in this
particular experiment.
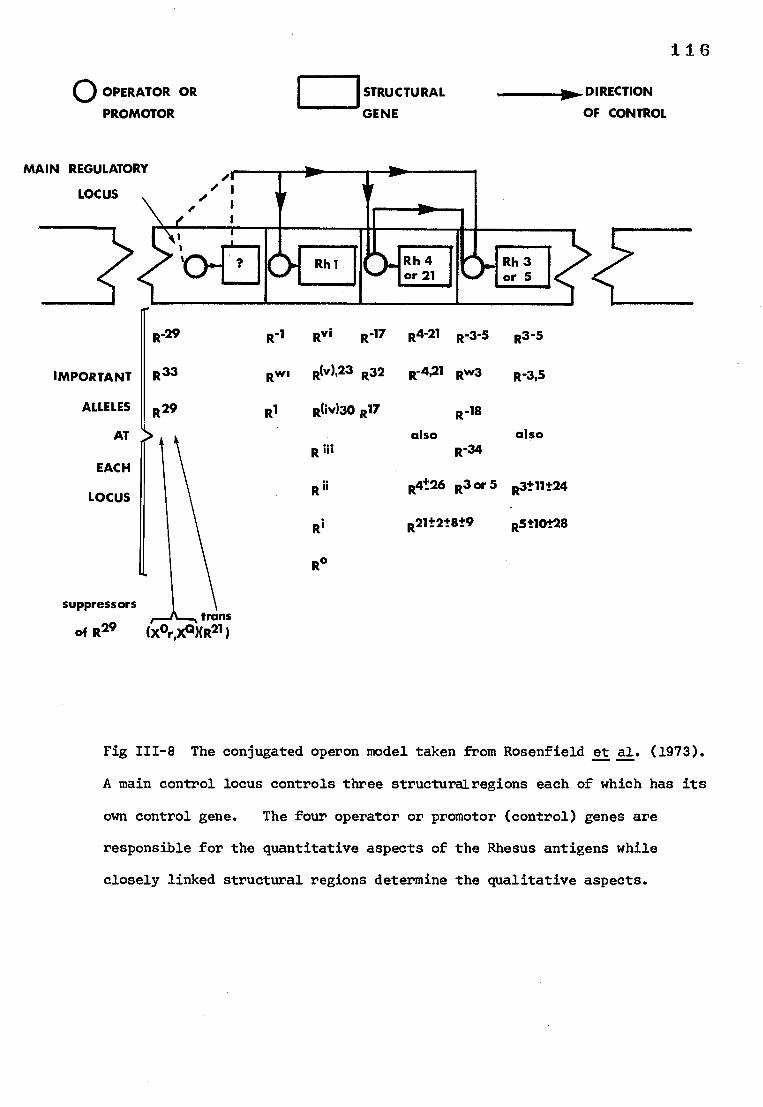
V
061or 21
YMMINEm.1■I
IC>, Rh3 or 5
MAIN REGULATORY
LOCUS
Rh 4
IMPORTANT
ALLELES
AT
EACH
LOCUS
116
O OPERATOR OR
PROMOTOR
STRUCTURAL ■1110... DIRECTION
GENE OF CONTROL
R-29 RA Rvi R-17 R4-21 R-3-5 R3-5
R33 Rwi Rliv)'23 R32 R-4,21 Rw3 R-315
R29 R1 01430 R17 R-18
also also R ili R-34
R ii R4t26 R3 or 5 R3±11t24
Ri
R°
21+2+ R - -8+9 R5t10t28
suppressors trans
of R29 (XOr,XQXR21 )
Fig 111-8 The conjugated operon model taken from Rosenfield et al. (1973).
A main control locus controls three structurairegions each of which has its
own control gene. The four operator or promotor (control) genes are
responsible for the quantitative aspects of the Rhesus antigens while
closely linked structural regions determine the qualitative aspects.

117
The D37 for the Rh (D) antigen obtained by measuring antigen activity
in the spectrophotometer compares well with that obtained using the labelled
antibody technique used in section 1. The D37 for the C and c antigens
are remarkably similar to that of the D antigen and fall within the 95%
confidence limits for the D37 of the D antigen as measured in section 2.
Even the D37 for the e antigen is just inside the 95% confidence limits for
the D antigen. Obviously the number of estimates is too small for any of
the antigens tested to state with confidence that the molecular size of
the D, C, c and e antigens is the same, as determined by irradiation
inhibition.
It was hoped that the determination of the molecular size of the Rh
antigens in situ would help to clarify the genetic pathway of the Rh antigens.
For many years there has been argument over whether the Rh antigens are
controlled by one gene with one site for mutation or 3 genes with three
sites for mutation. The conjugated operon model proposed by Rosenfield
et al. (1973) Shown in Fig 111-8, has explained the qualitative and
quantitative expression of the Rh antigens more thoroughly than any of the
previous attempts and it is proposed to use this model as a basis from which
to discuss the mechanism by which the genes express the Rh antigens at a
molecular level on the red cell surface.
The ultimate function of genes is to control the synthesis of proteins,
structural proteins or enzymes. The alternative alleles of the ABO blood
group system determine the synthesis of alternative enzymes which, in turn,
determine the addition of specific carbohydrates to a mucopolysaccharide
molecule (Watkins, 1966). It is difficult to imagine how a similar mechanism
could be used to express the complexity of the Rh antigens, and therefore for

1 1 8
the purposes of this discussion it will be assumed that the polypeptides
coded for by the 'CDE' genes are structural proteins. The genes responsible
for LW are not sited at the Rh locus (Tippett, 1972). However, since all
examples of Rhnull are LW negative and LW-positive subjects may become
phenotypically negative (Chown et al., 1971), it has been suggested
(Rosenfield et al., 1973) that the polypeptide responsible for the expression
of LW is only antigenic when bound to Rh. This idea must be accommodated
in the final expression of the Rh antigens. In addition, the Rh antigens
can be suppressed by independently segregating partially recessive genes
(X°r) (Levine et al., 1965) and when in double dose (X°r/X°r) produce Rhnull'
Rhnull can also be of the amorph type which might occur if the main
regulatory locus of Rh locus were permanently suppressing the action of
the 'CDE' genes.
Using the conjugated.operon model of Rosenfield et al. (1973) and
taking into account the restrictions cited above, various schemes can be
proposed to describe the production of the Rh antigens.
1) Each structural gene produces one polypeptide. If the gene products
do not react with each other or any other substance, then each would be
expected to be a discrete molecule.
2) Alternatively, the gene products from each chromosome could polymerise
in various ways such that there would be between 2 and 6 polypeptide chains
per polymer molecule, i.e. the C antigens from opposite chromosomes could
dimerise, likewise the D and the E antigens or, the C, D and E antigens from
each chromosome could form trimers or, all the antigens from both chromosomes
could form a hexamer. A major difficulty with this scheme is the 'd antigen'.
According to the conjugated operon theory of Rosenfield et al., 1973, the Rh

1 1 9
negative status (d) happens when the R1 control gene is 'off' and there
would be no structural product d. Therefore in a cell heterozygous for
the D antigen, D dimers (or CDE trimers) would be unable to form.
3) If the Rh system is governed by a complex locus rather than by the
conjugated operon model of Rosenfield et al. (1973), then it is possible
that the Rh antigens are all present on a single gene product. The
composition of the gene product would presumably vary according to the
Rh genotype.
The final expression of the Rh antigens may be dependent upon the
combination of the 'CDE' gene products with another (non-Rh) substance,
protein, carbohydrate or lipid which could act to orientate and anchor the
antigens in the red cell membrane such that their antigenic and membrane
functions can be accomplished. Such a substance could interact with single
polypeptides or polymers with equal ease and could be accommodated into any
of the basic schmes described above. The proposed interaction of the LW
antigens with the Rhesus antigens could be compared with the binding of a
non-Rh substance, but the bonding would be expected to be looser (i.e. non-
covalent) since LW antigenic activity can be lost temporarily without
affecting Rh expression (Chown et al., 1971).
Rh antigen activity could also be modified on the cell membrane.
Interactions with surrounding lipid and protein molecules may be a pre-
requisite for the expression of certain antigens. There is for example,
good evidence to suggest that Rh (D) antigen activity is dependent upon an
association with phospholipid molecules (Green, 1972; Hughes-Jones et al.,
1975).

120
In the case of Rhnull of the 'regulator' type i.e. er/X r, the X°r
gene might act in several ways. For example at the level of the messenger
RNA, either by controlling the amount of messenger RNA available or by
restricting its binding to ribosomes. In this manner it could switch the
main regulatory locus 'off' or stop the synthesis of the 'CDE' gene products.
Under such conditions the Rh antigens would not be formed, the LW antigens
would not be able to interact with them and would therefore not be expressed.
A further possibility is that the gene product of the X°r genes is an enzyme
which adds, for example, a carbohydrate onto the individual Rh antigen
polypeptides thereby preventing polymerisation or antigenic expression.
Speculations on the molecular structure of the Rh antigens
If the molecular size of each of the Rh antigens as determined by
radiation inactivation is approximately the same, this would suggest one
of the following possibilities:
1. Each Rh antigen is on a separate molecule of exactly the same molecular
weight (Approx. 200,000 daltons) shown in Fig 111-9 (a).
2. Each Rh antigen is part of a complex consisting of one molecule
containing one Rh antigen plus non-Rh material with a total molecular weight
of 200,000 daltons (Fig 111-9 (b)).
3. All the Rh antigens are subunits in the same complex with non-Rh
substances and the total molecular weight of the complete unit is
approximately 200,000 daltons (Fig 111-9 (c)).

121
Fig 111-9 Speculations on the molecular structure of the Rh antigens.
(a) Each antigen on a separate molecule of molecular weight approximately
200,000 daltons.
(b) Each antigen on a separate molecule with non-Rh material of total
molecular weight approximately 200,000 daltons.
(c) All the Rh antigens (expressed by a cell phenotype) are in a single
complex, with non-Rh material, of total molecular weight approximately
200,000 daltons.
(d) All the Rh antigens are on a single molecule of molecular weight
approximately 200,000 daltons.
(e) The Rh antigens are present as polymers with 2-6 polypeptide chains
per polymer molecule of molecular weight approximately 200,000 daltons.
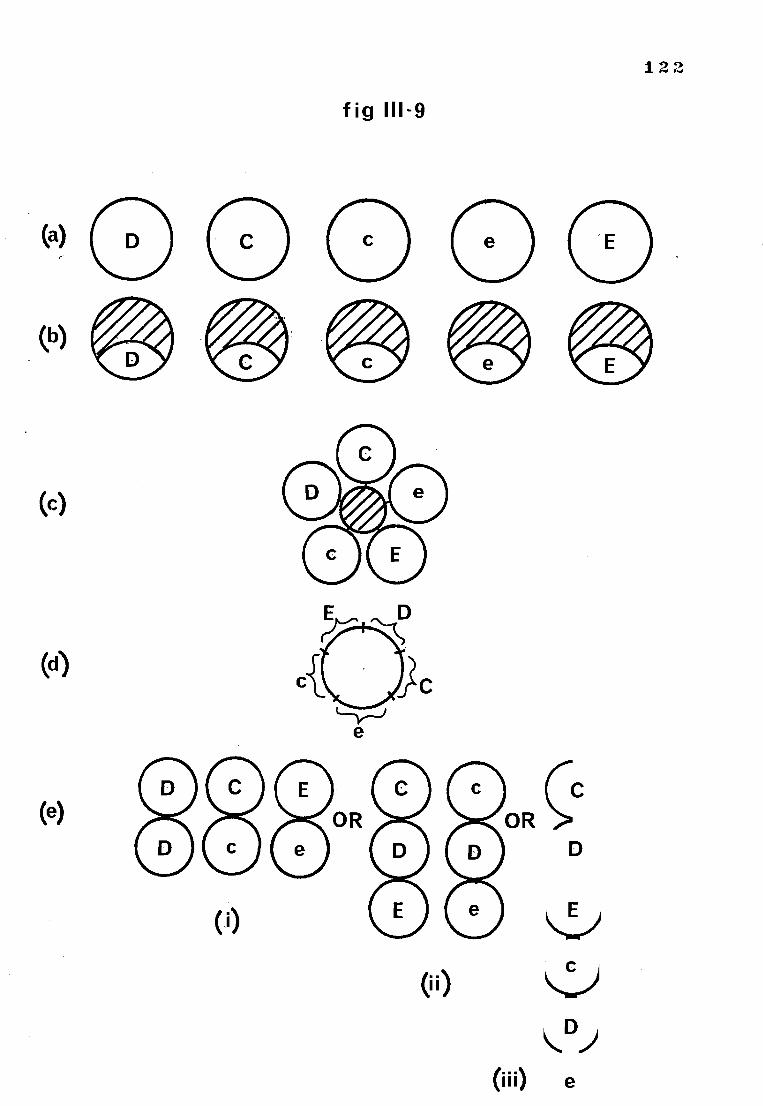
(c)
(a)
(e)
(i)
0i)
122
fig 111-9
e

123
4. All the Rh antigens are on the same molecule of molecular weight
200,000 daltons (Fig 111-9 (d)).
5. Alternatively, the Rh antigens expressed by each chromosome could be
present as polymers, with between 2 and 6 polypeptide chains per polymer
molecule, of molecular weight approximately 200,000 daltons (Fig 111-9 (e)).
However, if for example, the D antigen was present as a dimer on a red cell
membrane whose genotype expressed a 'double-dose' of the D antigen, the
size of the molecule as measured by radiation inactivation would be twice
that measured in the membrane of a cell whose genotype expressed only a
'single-dose' of the D antigen (where dimers would be unable to form).
The size of a 'CDE trimer' as measured by radiation inactivation would
similarly be smaller in a membrane with only a 'single-dose' of the D antigen.
Since there was no apparent decrease in the molecular weight of the D antigen
in membranes with only a 'single-dose' of D antigen it seems unlikely that
the D antigen exists as di- or trimers. In a hexamer of the same molecular
weight i.e. 200,000 daltons, the D antigen would account for a smaller
percentage of the total molecular weight and therefore the decrease in
molecular weight in 'single-dose' membranes may not be apparent, this also
applies to the complex shown in Fig 111-9 (c) and the single molecule in
Fig 111-9 (d).
Radiation inactivation measures the molecular size of a substance
in situ. Radiation studies used in combination with molecular weight
determinations by conventional biochemical techniques i.e. gel filtration
and ultracentrifugation, could provide information regarding the structure
of a molecule in situ. The Rh antigens must be isolated from the red cell
membranes and their size investigated before the data from the present study
can be fully appreciated.
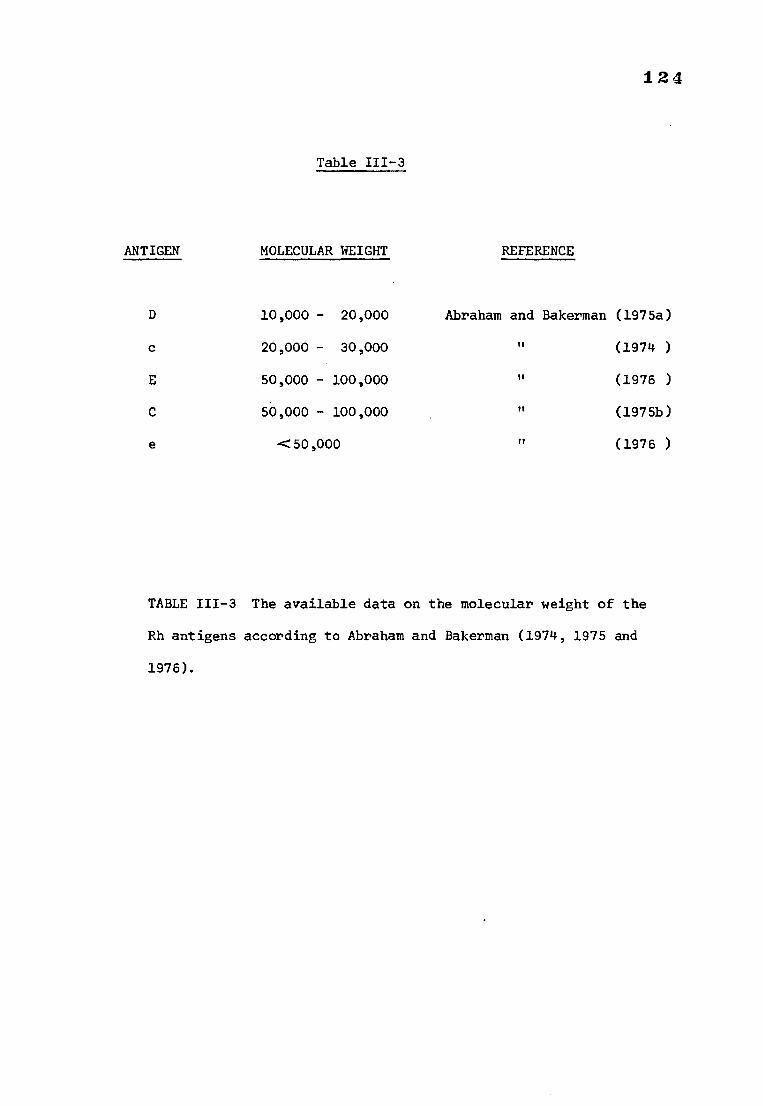
124
Table 111-3
ANTIGEN MOLECULAR WEIGHT REFERENCE
D 10,000 - 20,000
c 20,000 - 30,000
E 50,000 - 100,000
C 50,000 - 100,000
e <50,000
Abraham and Bakerman (1975a)
(1974 )
(1976 )
(1975b)
(1976 )
TABLE 111-3 The available data on the molecular weight of the
Rh antigens according to Abraham and Bakerman (1974, 1975 and
1976).

125
There is very little information regarding the size or biochemistry
of the C, c or e antigens in the literature. Abraham and Bakerman (1975b,
1974 and 1976) claim to have isolated the C, c and E antigens as well as
the D antigen. The molecular weight estimations of Abraham and Bakerman
(shown in Table 111-3) are all much lower than the values obtained in this
study. If both sets of data are correct it could follow that the Rh antigens
are small subunits within a larger structure which is inactivated by doses
of radiation that would be expected to inactivate molecules of the same size
as the complete Rh structure.
Suppose that the molecule associated with Rh activity in situ, is
composed exclusively of the material which expressed one copy of each of
the Rh antigens as defined by the phenotype of the cell from which it
originated. That is, either the Rh antigens as expressed in the cell
phenotype are all on one molecule of molecular weight 200,000 daltons
(Fig 111-9 (d)) or they are present as small subunits aggregated to form
a complex or polymer of molecular weight 200,000 daltons (Fig III-10 (5c)).
If the latter is true the combined molecular weight of the individual antigens
as proposed by Abraham and Bakerman, for a given red cell phenotype should be
approximately equal to the radiation inactivation data for the same phenotype.
Considering cells of the genotype CDe/cDe and assuming that the molecular
weight of each antigen is the middle value in the range given by Abraham and
Bakerman, then 75,000(C) + 15,000(D) + 25,000(e) + 25,000(c) + 15,000(D) +
75,000(E) = 230,000 which is very similar to the values obtained by irradiation
inactivation in the present study. The agreement between the molecular weight
estimation according to the data from Abraham and Bakerman and those from the
experiments in this chapter can be appreciated better if individual results
for particular phenotypes obtained from radiation experiments are compared
with those obtained by computation of the combined molecular weights as shown
in Table 111-4. Providing that the individual Rh antigens are inactivated
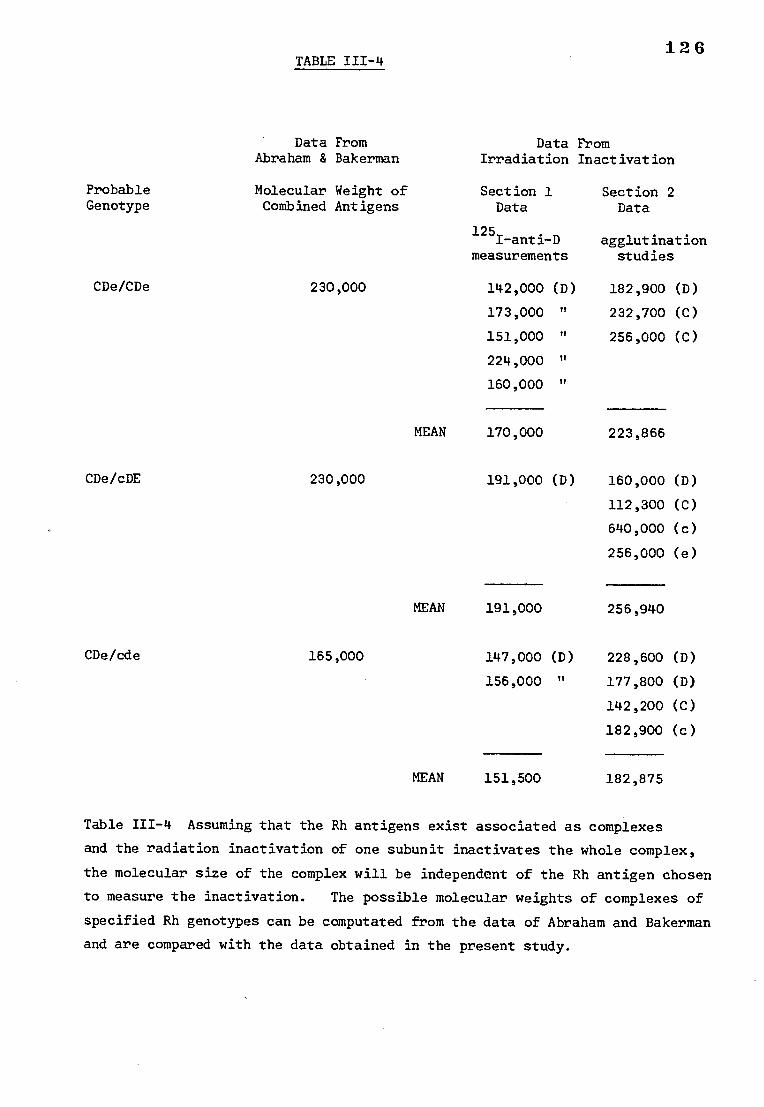
126 TABLE 111-4
Data From Data From Abraham & Bakerman Irradiation Inactivation
Probable Genotype
CDe/CDe
Molecular Weight of Combined Antigens
230,000
Section 1 Data
125I-anti-D measurements
142,000 (D)
173,000 "
Section 2 Data
agglutination studies
182,900 (D)
232,700 (C)
151,000 " 256,000 (C)
224,000 "
160,000 "
MEAN 170,000 223,866
CDe/cDE 230,000 191,000 (D) 160,000 (D)
112,300 (C)
640,000 (c)
256,000 (e)
MEAN 191,000 256,940
CDe/cde 165,000 147,000 (D) 228,600 (D)
156,000 " 177,800 (D)
142,200 (C)
182,900 (c)
MEAN 151,500 182,875
Table 111-4 Assuming that the Rh antigens exist associated as complexes
and the radiation inactivation of one subunit inactivates the whole complex,
the molecular size of the complex will be independent of the Rh antigen chosen
to measure the inactivation. The possible molecular weights of complexes of
specified Rh genotypes can be computated from the data of Abraham and Bakerman
and are compared with the data obtained in the present study.

127
according to the molecular size of the whole complex, the molecular weight
of an antigen complex corresponding to a particular phenotype will be the
same irrespective of which antigen is used to measure the radiation inactivation.
The data from each section have been grouped separately and the mean values
can be compared with the molecular weights calculated from the data of
Abraham and Bakerman. There is agreement between the data from two different
sources which is particularly noticeable when the molecular weights for the
CDe/cde cells are compared with those for the CDe/cDE and CDe/CDe cells.
This model for Rh antigenic structure can be criticized on several
counts. Firstly, it is difficult to account for the observed number of Rh
antigen sites per cell. If all the Rh antigens as expressed by the
phenotype are all present at the surface of the cell on the same molecule
then why are there more c sites than e sites (Hughes-Jones et al., 1971)
and why do some cells with a double-dose of D (e.g. CDe/cDE) have more sites
than others (e.g. CDe/CDe) (Rochna and Hughes-Jones, 1965). It could be
argued that the stereochemistry of the molecule is affected by the
constituent antigens and therefore not all the available antigen sites
within the molecule are exposed on the cell surface. Alternatively, as
suggested by Rosenfield, control genes determine the degree of expression
of the structural genes.
Secondly, there is the question of the LW antigens. If the LW
antigens are bound to the Rh antigens directly, and constitute an integral
part of the Rh antigen 'complex' then the molecule on which the LW antigens
are expressed would be expected to contribute to the molecular weight of the
Rh antigen 'complex' as measured by radiation inactivation. Since neither
the size nor chemistry of the LW antigens is known it is difficult to discuss
the relevance of this factor. However, LW antigen activity can be

128
temporarily lost from the red cell without affecting the Rn antigens
(Chown et al., 1971), and therefore it is unlikely that they are tightly
bound. If the LW antigen is on a unit linked to the Rh antigen complte
in a manner which has no influence on the expression of the Rh entigen
then it is unlikely that an ionization within the LW antigen would ,also
inactivate the Rh antigens and therefore the target size of the Rh antigen
'complex. would not include the LW antigen.
Conclusions
-Ineconclusien it must bgesaidethat there'are:really insufficient data
to propose a. model for the structure of the Rh antigete tttt a
The data of Abraham and Bakerman have not been verified by other workers
although Weicker (1971) has reported isolating a small peptide of molecular
weight 5,000-6,000 daltons with Rh (D) activity. Perhaps it ie peee;ib.le
to isolate amall fraction of an Rh 7,n-T-57,r which will
degree of biological activity, although in vivo the antigenic activity 1-c-uld , -7 ---
be associated with a larger molecule. Under such circumstances it would
not be valid to comparathe data of Abraham and Bakerman With that of the
present study. Until the Rh antigens can be isolated and characterised
intact from the red cell membrane speculations on pathway from 'genes to
antigens have limited value.
Irradiation inactivation has not been previously appliefl ,e
determination of the molecular weight of a red cell antigen. The techniquc
has mainly been confined to the study of enzymes (Levinson and Ellory, 1973;
Kepner and Macey, 1968) and nucleic acids (Ginoza, 1963) and therefore

129
validity of extending the technique to antigens could be questioned. The
sensitivity of antigenic activity to radiation obviously depends on what an
antigen is and what the sequence of events is when an antigen reacts with an
antibody. Therefore the true significance of the results of this study will
have to await not only the biochemical purification of the Rh antigens but
also the mechanism of antigen-antibody reactions.

tii 130
CHAPTER IV
THE SOLUBILIZATION OF RED CELL MEMBRANES USING SODIUM DEOXYCHOLATE
INTRODUCTION
General methods of membrane disruption
The red cell membrane is composed of a mixture of lipid, protein and
carbohydrate. There is usually approximately 40% w/w lipid, up to 10% w/w
carbohydrate and the balance protein. Between 20 and 40% of the membrane
protein from red cell ghosts can be released by changing the ionic strength
of the surrounding medium. The remaining protein is more firmly associated
with the membrane and can only be released by the action of protein
perturbants, for example; 6M guanidine HCl (Gwynne and Tanford, 1970);
acetic acid (Schubert, 1973); lithium diiodosalicylate (Marchesi and Andrews,
1971); or detergents, for example, sodium dodecyl sulphate (Kirkpatrick et al.,
1974), triton X-100 (Yu and Steck, 1973) and the bile salt sodium deoxycholate
(Philippot, 1971).
Sodium dodecyl sulphate (SDS) solubilizes red cell membranes. The
bonds between proteins and lipids or other proteins are broken and the
polypeptides can be separated on a semi-micro scale on SDS-polyacrlamidel gels
where they migrate according to their molecular weights. Red cell membranes,
as separated on a polyacrylamide gel, after treatment with SDS, are shown in
Fig IV-1. The bands are named according to the nomenclature suggested by
Steck (1972). If the gels are stained for carbohydrate instead of protein,
one major and several minor components are seen. Their molecular weights
cannot be ascertained from the gels because of the large proportion of
carbohydrate they carry (Bretscher, 1971a).
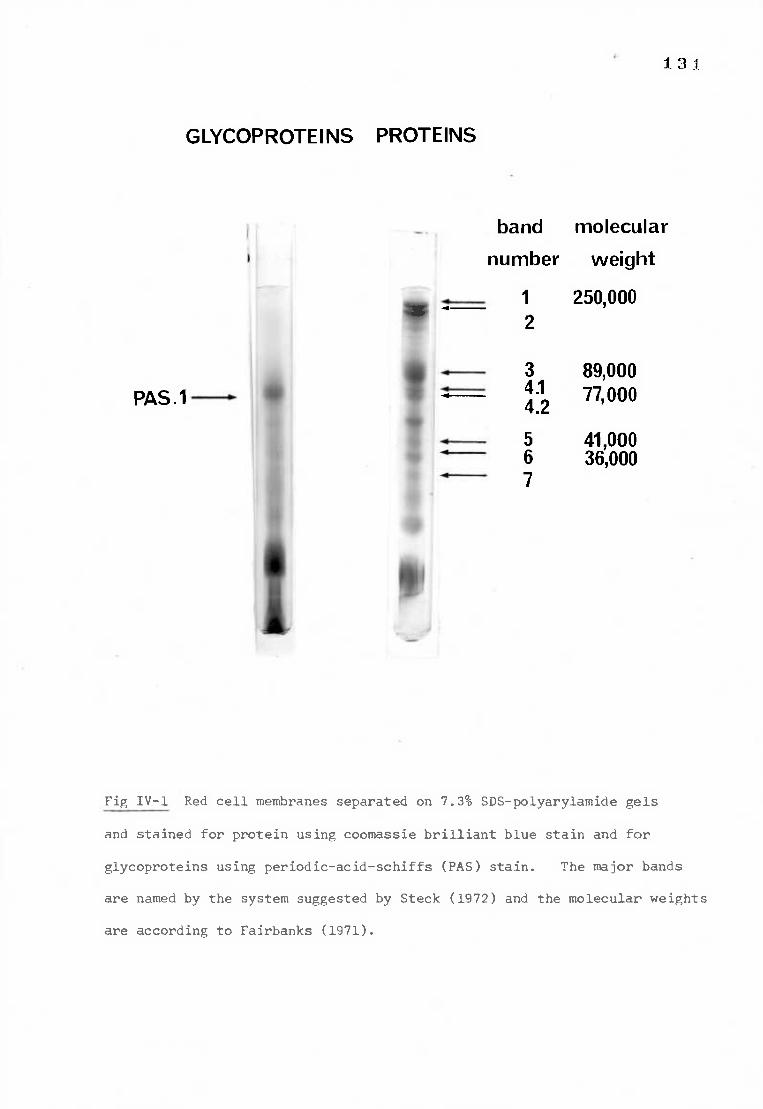
1 3 1
GLYCOPROTEINS PROTEINS
band molecular number weight
1 250,000 2
3 4.1 4.2
89,000 77,000
PAS.1
4
5 41,000 6 36,000 7
Fig IV-1 Red cell membranes separated on 7.3% SDS-polyarylamide gels
and stained for protein using coomassie brilliant blue stain and for
glycoproteins using periodic-acid-schiffs (PAS) stain. The major bands
are named by the system suggested by Steck (1972) and the molecular weights
are according to Fairbanks (1971).
z

132
Protein bands 1 and 2 account for about one third of all the membrane
protein, they are thought to be associated with the inner surface of the
cell membrane and can be released by low ionic strength media (Fairbanks
et al., 1971) or protein perturbants. Bands 1 and 2 together with band 5
may be identified with the fibrillar material observed at the inner surface
of red cell ghosts (Marchesi and Steers, 1968) and may be involved in the
maintenance of red cell shape.
The other major protein band, band 3, is often rather diffuse,
indicating a heterogeneous composition. Experiments involving proteolytic
enzymes (Bender et al., 1971) and chemical labelling (Berg, 1969; Bretcher,
1971band Phillips and Morrison, 1970) have shown that band 3 is associated
with both the inner and the outer surface of the membrane and it is thought
to extend across the bilayer. Band 3 has only been removed from the
membrane by using detergents, for example, triton X-100 (Yu and Steck, 1973).
According to Steck, band 3 behaves as a dimer in the membrane and is
associated with band 4.2 and band 6, both of which are tetramers in situ
(Yu and Steck, 1973).
Bands 4.1 and 4.2 can be removed from the membrane by most protein media
disruptants. Band 5 is easily released by low ionic strengthAand may be
associated with bands 1 and 2. Band 6 is glyceraldehyde 3 phosphate
dehydrogenase protomer; it can be released from the membrane in high ionic
strength media (Fairbanks et al., 1971) and is thought to associate with
band 3 at the cytoplasmic side of the membrane. Very little is known about
band 7; It is an intrinsic protein i.e. tightly bound within the membrane
and thought to occur on the cytoplasmic surface only.
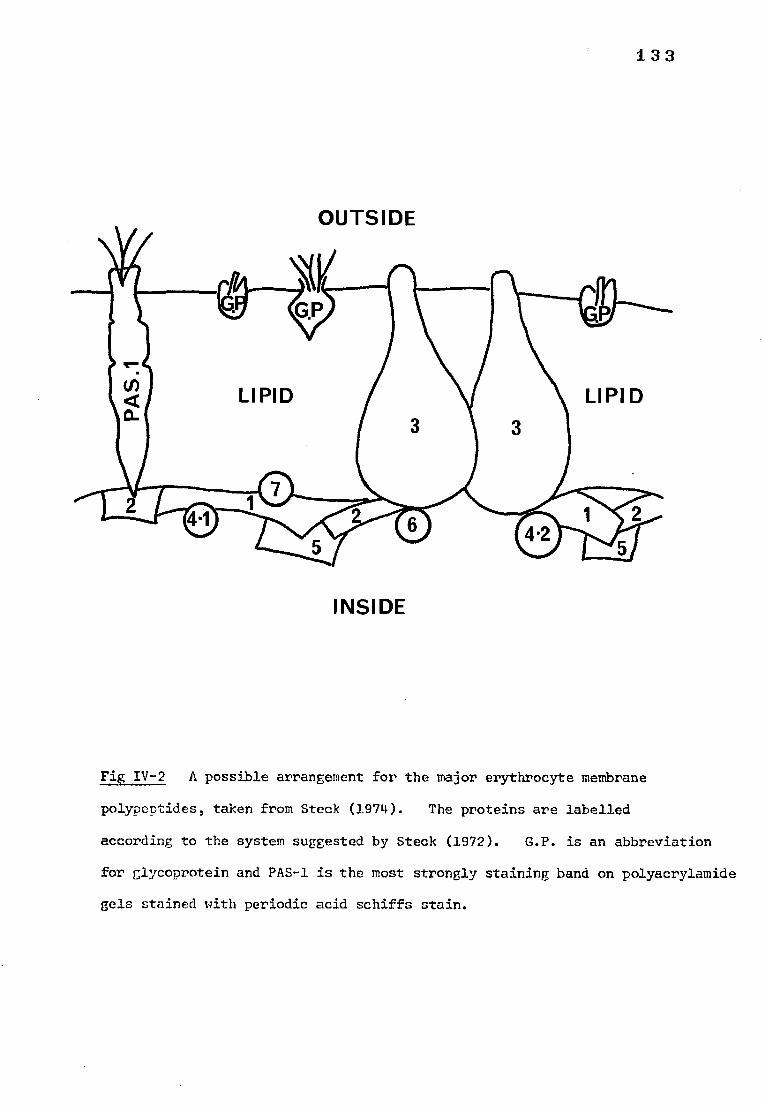
LIPID LIPID
133
OUTSIDE
INSIDE
Fig IV-2 A possible arrangement for the major erythrocyte membrane
polypeptides, taken from Steck (1974). The proteins are labelled
according to the system suggested by Steck (1972). G.P. is an abbreviation
for glycoprotein and PAS-1 is the most strongly staining band on polyacrylamide
gels stained with periodic acid schiffs stain.

134
At present, band 3 and the glycoprotein are thought to be the
only major membrane proteins occurring on the exterior of the red cell
membrane (Bretscher, 1971b; Phillips and Morrison, 1970), although
Whiteley and Berg (1974) have also found evidence that there may be a
protein of molecular weight 170,000 daltons also labelled by non-penetrating
membrane labels which would indicate an external location. The proposed
location of the various membrane proteins has been illustrated by Steck (1974)
as shown in Fig IV-2.
The aim of the present study was to isolate the membrane protein
responsible for the expression of Rh (D) activity. The identity of this
protein is unknown and therefore it is necessary to disrupt the entire
membrane and release all of the constituent polypeptides without denaturing
the antigen. SDS was thought to be unsuitable because it has been known
to irreversibly denature biologically active proteins. Yu and Steck (1973)
have found that the tertiary structure of proteins was maintained after
solubilization with triton X-100 which would make it a possible choice.
However in the present study sodium deoxycholate was used because Lorusso
and Green (1975) have reported that Rh activity could be restored to proteins
solubilized with this bile salt, indicating that the antigen was not denatured.
Bile salts
Bile salts are soluble amphiphiles i.e. they have polar and non-polar
regions which aggregate into micelles under certain conditions in water.
Bile salt micelles are smaller, more highly charged and different in
structure from detergent micelles. Deoxycholate solubilizes phospholipids
by incorporation into mixed micelles. The ratio of deoxycholate to lecithin
within the micelles is variable. Cholesterol can also be incorporated.

135
It has been reported (Makino et al., 1973) that deoxycholate binds to
proteins at specific sites e.g. with serum albumin, and to hydrophobic but not
hydrophilic proteins (Helenius and Simons, 1972). The critical micellar
concentration (CMC) of a bile salt is the critical solute concentration
in water above which colloidal aggregates or micelles spontaneously form
if the temperature is above the critical micellar temperature (CMT). The
CMT of the common bile salt conjugates is usually lower than 0°C, but at
high concentrations and low temperatures e.g. 4°C, solutions of deoxycholate
will form gels. Primary bile salt micelles aggregate hydrophobically back
to back. Secondary micelles consist of bands of completely lipophilic
regions held by hydrophobic interactions alternating with bands of
hydrophilic groups held together by hydrogen bonds. Disruption of these
hydrogen bonds by urea or increasing the temperature promotes the conversion
of secondary to primary micelles.
Above the CMC phospholipids begin to be removed from protein by
deoxycholate micelles. Cholesterol is then solubilized followed by the
residual protein and lipids (Kirkpatrick et al., 1974). The membrane is
thereby dissociated into its various components. The solubilisation is
often accompan'Ied by a loss of biological activity which is reversible
by removal of the bile salt, as, for example, in the case of (Nal--e)-ATPase
(Philippot, 1968).
Isolation of the D antigen
The isolation of the Rh (D) antigen has been reported by Weicker and
Metz (1971) and Abraham and Bakerman (1975a). Weicker obtained a low
molecular weight protein by dialysing haemolysed red cells against water.
He was unable to demonstrate D activity in the protein by conventional

1_ 3 6
methods but a positive reaction was obtained using the Schultz-Dale test.
This test depends on the antibody-antigen reaction causing the contraction
of uterine muscle-segments from guinea pigs. There have not been any
reports in the literature in support of his findings.
Abraham and Bakerman (1975a) claim to have isolated the D antigen by
solubilizing red cell stroma with EDTA followed by dialysis against saline
and ultracentrifugation. They detected Rh (D) activity in the fraction of
molecular weight 10,000 - 20,000 daltons. After purifying the fraction by
isoelectric focusing they injected it into guinea pigs and obtained a high
titre anti-D. This work has not been substantiated either. Both low ionic
strength and EDTA extraction would be expected to release bands 1, 2 and 5
from the membrane (Fairbanks et al., 1971; Bhakdi et al., 1974). However,
these bands are thought to be associated with the inner surface of the cell
membrane and have molecular weights higher than 20,000 daltons. It is
possible that the Rh activity is associated with other proteins running in
the same position as bands 1, 2 and 5 on polyacrylamide gels or they may
run too fast (as a result of a very small molecular weight) to be seen on
polyacrylamide gels under normal circumstances.
Lorusso and Green (1975) have reported Rh (D) activity in the proteins
solubilized by deoxycholate after removal of the bile salt by dialysis.
Between 55 and 70% of the membrane protein was solubilized. Bands 1 and 2
and some of the polypeptides in bands 3, 4 and 5 were not solubilized.
Electron photomicroscopic studies of the soluble proteins after dialysis
against magnesium ions showed membrane vesicular structure, indicating some
form of membrane reconstruction was taking place. Gel filtration of the
soluble proteins on sepharose 4B showed two peaks of approximately 200,000
and 10,000 daltons and ultrafiltration indicated that the molecular weight
of the protein with Rh activity was less than 300,000.

137
Proposed experiments
Initially it was decided to repeat the experiments of Lorusso and
Green (1975) concerning the solubilization of the Rh antigen using sodium
deoxycholate. Previous experience, for example, in the phospholipase
experiments, has shown that the D antigen is often protected when in
combination with anti-D and the antigen-antibody bond remains unaffected
by many outside factors. If this were the case in the presence of
deoxycholate then the use of a labelled antibody would act as a marker
for the antigen in purification procedures. Furthermore the antibody
would provide a means for the purification of the antigen by, for example,
affinity chromatography. If the soluble proteins were passed through
cyanogen bromide-activated sepharose 4B (S-CNBr) to which anti-human IgG
had been attached, then the IgG anti-D (and D antigen) would bind to the
anti-IgG and the remainder of the proteins would be eluted leaving the
pure antibody-antigen complex on the column.
The experiments described in this chapter describe how this initial
idea was developed and extended.
METHODS AND MATERIALS
1) Preparation of red cell membranes
Normal, human, 21-day old red cells were washed three times with
phosphate buffered saline and lysed in 20 volumes of 20 mosmol/1 phosphate
buffer pH 8.0. The membranes were centrifuged at 90,000 g for 30 minutes
and washed at least three times with 20 mosmo1/1 phosphate buffer pH 8.0.
The stroma was then freeze dried and stored dry at -20°C until required.

1 3 8
2) anti -D
IgG preparations containing high titre anti-D were used. The
amount of anti-D in the preparations was estimated by Dr. Hughes-Jones
at the WHO reference centre in 1974. It has been calculated that the
error of a single estimate is between 66 and 150% of the stated value
assuming 95% fiducial units (Hughes-Jones, 1974).
3) Preparation of 125I-labelled anti-D
An IgG preparation containing high titre anti-D wasIseparated on
CM-52 by the method of Frame and Mollison (1969). The peak containing
anti-D activity was concentrated and labelled by the iodine monochloride
method (McFarlane, 1958). Large amounts of 125I-labelled anti-D were
required for the experiments described in this chapter and therefore many
labelled antibody preparations were made. The specific activity of the
preparations was usually approximately 7,000 counts per minute per pg IgG
and the purity about 2%, i.e. 2% of the total IgG had anti-D activity.
4) Solubilizing buffer
Taken from Lorrusso and Green, (1975).
20 millimolar (mM) phosphate buffer pH 7.5
containing : 0.8% w/v sodium chloride
10 mM dithiothreitol
1 mM EDTA (sodium salt)
20% w/v glycerol
0.9% w/v sodiumdeoxycholate (Koch-Light laboratories)
The buffer forms a gel at temperatures below on on storage and therefore
was prepared freshly for each experiment.

139
5) Polyacrylamide gel electrophoresis
a) Solutions Buffer A : 0.22M sodium phosphate buffer pH 7.0
0.2 % w/v sodium dodecyl sulphate (SDS)
Bis acrylamide: 22.2% w/v acrylamide
0.6 % w/v Methylene bis acrylamide
Ammonium persulphate : 75 mg in 10 ml distilled water
prepared fresh when required.
Accelerator : Tetramethylethylenediamine (TEMED)
Buffer B : 4% w/v SDS
2% w/v B-mercaptoethanol (2-me)
in 0.01M phosphate buffer pH 7.0
Marker : 60% sucrose (w/v) mixed 9:1
0.05% (w/v) bromophenol blue (BPB))
Protein stain : 0.025% w/v Coomassie brilliant blue
5% glacial acetic acid (V/V)
50% methanol (v/v) ; 45% v/v distilled water
b) Preparation of 7.3% polyacrylamide gels. Buffer A (15 ml), bis
acrylamide (10 ml) and water (3.5 ml) were mixed and degassed. Ammonium
persulphate (1.5 ml) and three drops of TEMED were added and mixed. The
mixture was gently pipetted into glass tubes (1 x 12 cm), which had been
stoppered at one end, up to a height of 10 cm. Distilled water was slowly
layered onto the liquid surface and the gels were left to solidify.
c) Gel electropheresis. The stoppers were removed from the ends of the
tubes and the gels were fitted into a Shandon disc electrophecr) resis tank.
A 1:2 dilution in water of buffer A was made and used to fill the buffer
compartments. ¶he gels were pre-run in this buffer for 60 minutes at a

140
current of 8 milliamps (m-amps) per tube. Aliquots (approximately 50 pg
in 0.1 ml) of the proteins to be separated were mixed with 0.1 ml buffer
B in small glass test tubes. The tubes were incubated at 37°C for 30 minutes
then put in a boiling water bath for five minutes. After cooling, 0.04 ml
of the BPB-sucrose mixture was added and the proteins were gently applied
to the interface between gel and buffer using a syringe. Electrophoresis
was continued at a setting of 8 m-amps per tube until the BPB had travelled
approximately 7 cm. The gels were then removed from the tubes by
aspiration and stained in coomassie brilliant blue for 18 hours.
d) Destaining the gels. The gels were destained in a Shandon destainer
with 7% v/v acetic acid in the buffer compartments for approximately 2 hours.
After destaining the gels were kept in 7% acetic acid in stoppered tubes.
6) Preparation of Biobeads SM-2
Biobeads SM-2, a spherical macromolecular styrene-divinyl-benzene
corlymer, were obtained from Bio-Rad laboratories, Richmond, California
and used to remove detergent from the solublized membranes. Approximately
15 g Biobeads SM-2 were stirred with 100 ml methanol for 15 minutes. The
beads were collected on a scintered glass funnel and washed with a further
250 ml methanol. Water (500 ml) was added immediately and the beads poured
into a small (1 x 8 cm) column. The beads were washed slowly with 1000 ml
water before use. Solubilized proteins were normally eluted with 20 mM
phosphate pH 7.5 and therefore the column was equilibrated with this buffer
before adding the proteins.

141
7) The action of deoxycholate on Rh (D) antigen activity
a) Solubilizing red cell membranes with sodium deoxycholate
Red cell membranes were mixed with solubilizing buffer (normally 5 mg
membranes per ml buffer) at 4°C for up to 3 hours. The mixture was then
centrifuged at 90,000 g for 60 minutes. The supernatant containing the
solubilized membrane was put through a Biobead SM-2 column and eluted with
20 mM phosphate buffer pH 7.5. The eluate was dialysed against water
(buffered to pH 7.5 with tris base) containing 5 mM magnesium chloride,
for three days. Thei centrifuged at 90,000 g for 60 minutes and the Rh (D)
activity of the resultant pellet determined.
b) Determination of the Rh activity of reaggregated proteins
Saline (3.0 ml) was added to the reaggregated protein pellet and the
mixture was dispersed by sonication at an amplitude setting of 2 microns
for 2 minutes in a MSE ultrasonic disintegrator. Aliquots of the protein
preparation (0.05, 0.1 and 0.2 ml) were then incubated at 37°C for 30 minutes
with approximately 1 pg 125I-labelled anti-D. After incubation the mixtures
were spun at 35,000 rpm for 30 minutes, the supernatants, containing the
unbound antibody were removed and incubated with 0.05 ml Rh (D) positive
red cells at 37°C for 30 minutes. The red cells were washed three times
and the amount of 125I-labelled anti-D bound was estimated in a Wallac gamma
counter. Control samples of untreated membranes were also tested for Rh
activity in the same way, for comparison.
c) Separation of solubilized proteins with ultrafilters
Red cell membranes were solubilized as described but before passing
through the Biobeads SM-2 column the solution was put through a XM 100 filter,
a pressure of 15 psi was applied. Both the filtrate and the residue were put

142
through a Biobeads SM-2 column and dialysed for 3 days against tris buffered
water (pH 7.5) containing 5 mM MgCl2. The pellet of reaggregated proteins
obtained after dialysis was tested for Rh activity as described.
8. THE ACTION OF SODIUM DEOXYCHOLATE ON THE Rh (D) ANTIGEN-ANTIBODY COMPLEX
a) Treatment of red cells with 125I-labelled anti-D followed by
solubilization with Deoxycholate
Rh positive red cells (2.0 ml) and Rh (D) negative red cells (2.0 ml)
were each washed three times with phosphate buffered saline and incubated
20 minutes at 37°C with approximately 15 pg 125I-labelled anti-D. The
cells were washed three times with phosphate buffered saline and the amount
of bound 125
I was estimated. The cells were then lysed and the membranes
washed and freeze-dried. The stroma was solubilized and dialysed as
described above. The amount of 1251 in the pellet obtained from each
stroma sample after dialysis was determined.
b) Separation of solubilized proteins on sepharose 4B
A column containing sepharose 4B (Pharmacia, Sweden) was prepared
( 1 x 90 cm.) and equilibrated with 20 mM phosphate buffer pH 7.5 containing
0.8% NaCl and 40 mg/1 sodium deoxycholate. Solubilized proteins from
membranes which had been preincubated with 125I-labelled anti-D were added
to the column (without prior treatment with Biobeads SM-2) and eluted with
the buffer with which the column was equilibrated. The absorption at
280 nm and the amount of 1251 in the fractions was estimated.
c) Measurement of the amount of combined antibody and antigen after
treatment with sodium deoxycholate
This test was generally performed on the fractions of soluble protein

143
eluted from a sepharose 4B column. The fraction to be tested was divided
into three equal aliquots. Two of the aliquots were incubated at 56°C for
15 minutes. At 56°C the antibody-antigen complex would be expected to break,
releasing denatured antigen but active antibody. One of the 56°C treated
samples was then incubated with a suitable volume (usually 0.2 ml) of washed
Rh positive red cells and the other with an equal volume of washed Rh (D)
negative red cells. The remaining untreated aliquot was incubated with the
same volume of Rh positive red cells as used for the 56°C treated samples.
All were incubated at 37°C for 30 minutes, then the red cells were washed
and the amount of 125I-labelled anti-D bound to the cells was estimated in
a Wallac gamma counter. If 56°C treatment released the antibody which was
combined with D antigen then the amount of antibody available for reaction
with Rh positive red cells should be greater than in the sample which had
not been treated at 56°C. The sample treated at 56°C and incubated with Rh
negative red cells should give an estimate of the non-specific uptake of
1251 by the red cells.
9. ATTEMPTS TO PURIFY THE Rh (D) ANTIGEN
a) Separation of solubilized proteins on sepharose 4B followed by affinity
chromatography on S-CNBr-anti-IgG
(i) Purification of IgG anti-IgG
Horse serum (3.0 ml) containing 12 mg anti-human IgG per ml was put
on a small column (1.5 x 20 cm) containing an ion exchange cellulose (DE 52,
Whatman Ltd.). The IgG peak was eluted with 0.0175 M phosphate buffer
pH 6.5 and concentrated by vacuum dialysis to approximately 2.0 ml
(ii) Activating the S-CNBr
Cyanogen bromide activated sepharose 4B (2.0 g) was washed with 500 ml
10-3M hydrochloric acid on a scintered glass filter for 15 minutes to
activate it.

144
(iii) Coupling the protein to the S-CNBr
The purified anti-IgG (approximately 20 mg) was dialysed against
carbonate buffer pH 8.4 (100mM NaHCO3; 500 mM NaCl; NaOH to pH 8.4) for
3 hours. The protein was then added to the freshly activated S-CNBr and
rotated end over end for 16 hours at and and 1 hour at room temperature.
The gel and protein mixture was then spun for 5 minutes at 2,000 rpm.
The supernatant was removed and the gel washed four times with 10 ml
carbonate buffer pH 8.4. The gel was left for 2 hours with 10 ml 1 M
ethanolamine pH 8.0 to block the unreacted groups. The gel was washed
alternatively with 200 ml, three times each, of sodium acetate buffer
(0.1 M acetate; 1M NaC1, pH 4.0) and borate buffer (0.1 M borate, 1M NaCl,
pH 8.5) to remove non-covalently bound protein. The prepared gel was stored
at 4°C in C n saline containing 0.01% sodium azide. It was estimated, from the
absorbance at 280 nm of the washings, that approximately 17 mg protein
remained attached to the gel.
(iv) Separation of solubilized proteins
Rh positive stroma (100 ml) was incubated at 37°C for 30 minutes with
approximately 230 pg 125I-labelled anti-D and 1 mg unlabelled IgG anti-D in
5 ml bovine serum albumin (BSA) and 30 ml 0.3 M glycine; 0.8% NaCl. The
stroma was then centrifuged at 90,000 g and washed two,-times with saline
before freeze-drying. The stroma was solubilized in 100 ml deoxycholate
buffer (section 4 of Methods) at 4°C for 3 hours. The soluble proteins were
removed by centrifugation and dialysed against 20 mM phosphate buffer pH 8.0
overnight to remove the glycerol and some of the detergent. The proteins
were then freeze-dried because the volume was too large to apply to a column.
The lyophilised preparation was dissolved in water and dialysed against 20 mM
phosphate buffer pH 8.0 containing 0.8% NaCl and 40 mg/1 sodium deoxycholate,
before separating on sepharose 4B.

145
The fractions expected to contain the antibody-antigen complex
(12-18 inclusive, see results) were freeze-dried, dissolved in a small
volume of water and dialysed against saline for at least three hours
before separating on a S-CNBr-anti-IgG column. The proteins were
initially eluted with saline, and when most of the unbound protein had
been eluted the buffer was changed to 0.2 M glycine-HCl, pH 2.8. The
absorbance at 280 nm and the amount of 1251 in the eluted fractions was
estimated. Certain representative fractions were freeze-dried and
0 redissolved in approximately 0.3 ml distilled water before electroph1resing
on 7.3% polyacrylamide gels.
b) The use of buffers with acid pH to split the D antigen-antibody complex
(i) The effect of pH on the stability of the D antigen-anti-D complex
Red cell membranes (0.6 ml) were incubated for 30 minutes at 37°C
with approximately 10 pg 125I-labelled anti-D in 0.7 ml saline. Aliquots
(0.2 ml) of this anti-D-treated stroma were incubated with 1.0 ml volumes
of 0.3 M glycine-HC1 at pH 7.0, 5.0, 4.5, 4.0, 3.5 or 2.8 at 37°C for
60 minutes. The stroma was spun at 90,000 g for 30 minutes and the
supernatant was removed. The amount of 1251 in the supernatant and the
sediment was estimated after resuspending the sediment in clean tubes.
(ii) The elution of deoxycholate-treated anti-D from cyanogen bromide
activated sepharose 4B-anti-IgG at various pH values
This experiment was carried out to determine the pH at which anti-D is
eluted from anti-IgG. An IgG preparation containing high titre anti-D
(1.5 mg protein) was solubilized in 10 ml solubilizing buffer for 3 hours
at 4°C. The preparation was spun at 90,000 g for 30 minutes. The supernatant

146
was dialysed against saline before putting through a S-CNBr-anti-IgG column.
The unbound antibody was eluted with saline, then the bound antibody was
treated with 0.3 M glycine-HC1 pH 5.0, then pH 4.0 and finally with pH 2.8.
The absorbance at 280 nm of the eluates was recorded and then the fractions
eluted at each pH were freeze-dried separately and run on 7.3% polyacrylamide
gels.
c) The separation of solubilized proteins on S-CNBr-anti-IgG
(i) Rh positive stroma (10 ml) was incubated at 37°C for 60 minutes
with 46 pg 125I-labelled anti-D and 0.5 mg unlabelled IgG anti-D. The
stroma was spun at 90,000 &for 30 minutes, washed two times with water
and freeze-dried. Rh negative stroma (10 ml) was prepared and freeze-dried.
Each of the stroma samples were solubilized in 20 ml solubilizing buffer at
4oC for 3 hours. The soluble proteins were removed by centrifugation and
dialysed for 18 hours against saline to remove the glycerol and some of the
detergent. The dialysed soluble proteins were put on a S-CNBr-anti-IgG
column and eluted first with saline then with 0.3 M glycine-HC1, pH 5.0,
then pH 4.0 and finally pH 2.8. The fractions from each of the acid
elutions were pooled and freeze-dried then re-dissolved in approximately
0.2 ml water and electrophoresed on 7.3% polyacrylamide gels.
(ii) In further experiments aimed at verifying the results obtained
from the experiment described above (1), the labelled antibody was added to
red cells rather than membranes because this results in less non-specfic
uptake by the stroma. The solubilized proteins were put through Biobeads SM-2
prior to separating on CNBr-anti-IgG instead of dialysing overnight (which
could result in some reaggregation of the soluble proteins) and a fraction of
the solubilized proteins was tested to make sure that there was some antigen-
antibody complex in the proteins applied to the column.

1 4 7
RESULTS
1) THE ACTION OF SODIUM DEOXYCHOLATE ON THE Rh (D) ANTIGEN
a) Solubilizing red cell membranes with sodium deoxycholate
SDS-polyacrylamide gels of red cell membranes treated with deoxycholate
are shown in Fig IV— 3. Band 6 seems to have been solubilized completely,
together with at least some of the proteins running in the positions of all
the visible bands on untreated stroma. This is in contrast to the findings
of Lorusso and Green (1977) who found that bands 1 and 2 were not solubilized.
Gels of the reaggregated proteins after removal of the bile salt were rather
diffuse and the individual protein bands are difficult to see. The gel of
the unaggregated proteins is clearer, bands 1 and 2 were only just visible,
bands 3, 5, 6 and 7 are quite clear and there appears to be an additional
band between band 3 and band 5, probably due to a contaminant. Visual
examination of chromatograms of the chloroform-methanol (2:1) extract of the
reaggregated proteins showed the presence of lecithin and sphingomyelin
indicating that the proteins and lipids solublized by deoxycholate had
recombined on removal of the bile salt.
b) Determination of the Rh (D) activity of reaggregated proteins
The results from a typical experiment are shown in Table IV-1. The
uptake of 125I-labelled anti-D by three different volumes of the reaggregated
proteins is compared with the uptake by untreated membranes so that an
estimate could be made of the antigenic activity recovered after deoxycholate
treatment and dialysis. The uptake of 125I-labelled anti-D by the
reaggregated 'membranes' was compared with a calibration curve of the uptake
of anti-D by various amounts of untreated stroma. After compensating for
the volume tested the uptake of 125I-labelled anti-D was used to calculate
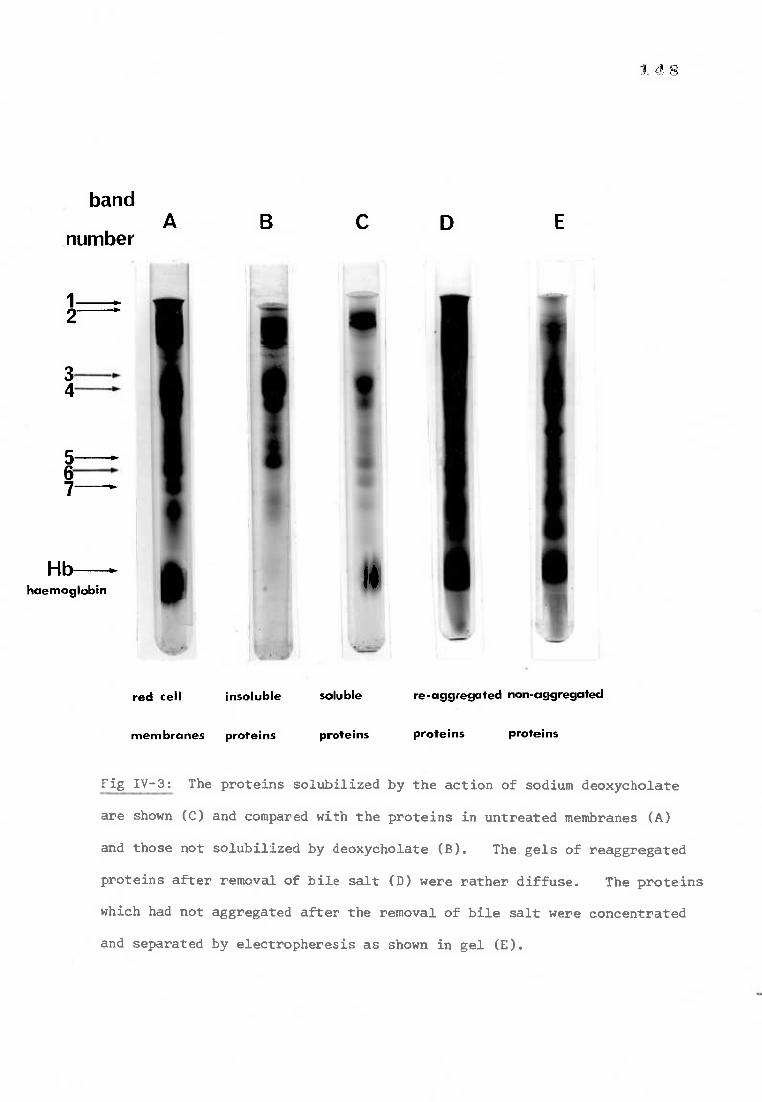
5— 6 7—
Hb---- haemoglobin
a
14
band
. 4 8
A number
red cell insoluble soluble re-aggregated non-aggregated
membranes proteins proteins proteins proteins
Fig IV-3: The proteins solubilized by the action of sodium deoxycholate
are shown (C) and compared with the proteins in untreated membranes (A)
and those not solubilized by deoxycholate (B). The gels of reaggregated
proteins after removal of bile salt (D) were rather diffuse. The proteins
which had not aggregated after the removal of bile salt were concentrated
and separated by electropheresis as shown in gel (E).

149
TABLE IV-1
Determination of the Rh (D) activity of reaggregated proteins
volume pg anti-D pg anti-D total
tested bound bound activity
(ml) reaggregated untreated recovered
proteins stroma %
0.05 0.10 0.19 6
0.10 0.23 0.24 12
0.20 0.28 0.29 13
In this experiment 20 ml of red cell membranes were solubilized and
reaggregated by dialysis. The pellet of reformed 'membranes' was
resuspended in 3.0 ml saline. The uptake of 1251-labelled anti-D by
the reaggregated 'membranes' was compared with a calibration curve of
the uptake of anti-D by various amounts of untreated stroma. After
compensating for the volume tested the values were used to calculate
the percentage of the original Rh activity which had been recovered.
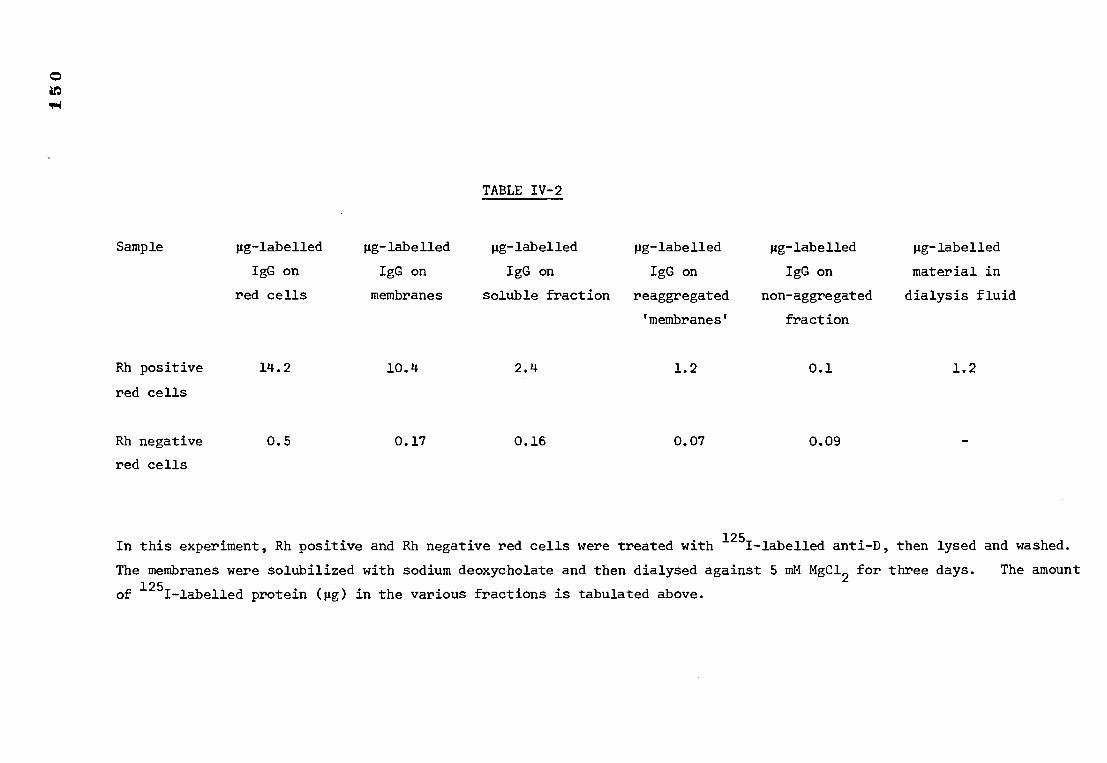
TABLE IV-2
pig-labelled µg-labelled pg-labelled pg-labelled pig-labelled pig-labelled
IgG on IgG on IgG on IgG on IgG on material in
red cells membranes soluble fraction reaggregated non-aggregated dialysis fluid
fraction
Sample
Rh positive
red cells
Rh negative
red cells
'membranes'
14.2 10.4 2.4 1.2
0.5 0.17 0.16 0.07
0.1 1.2
0.09
In this experiment, Rh positive and Rh negative red cells were treated with 125I-labelled anti-D, then lysed and washed.
The membranes were solubilized with sodium deoxycholate and then dialysed against 5 mM MgC12 for three days. The amount
of 125I-labelled protein (gg) in the various fractions is tabulated above.

1 5 1
the total amount of activity regained, expressed as a percentage of the
total activity of the membranes before treating with deoxycholate. In
the majority of experiments of this type up to 13% of the original Rh (D)
activity was regained.
c) Separation of solubilized proteins with ultrafilters
An XM-100 filter (Amicon, Diaflo filter) has a cut off point of
approximately 100,000 daltons. No Rh (D) activity, as measured by the
uptake of 125I-labelled anti-D could be detected in the protein which passed
the ultrafilter after dialysis. Approximately 3% of the original Rh (D)
activity of the membranes before treatment with deoxycholate was recovered
from the proteins which did not pass the filter.
2) ACTION OF SODIUM DEOXYCHOLATE ON THE Rh (D) ANTIGEN-ANTIBODY COMPLEX
a) Treatment of red cells with 1251-labelled anti-D followed by
solubilization with sodium deoxycholate
The results are shown in Table IV-2. After the preparation of stroma
there was only 0.17 pg labelled anti-D on the Rh negative membranes i.e. non-
specifically bound IgG (compared with 10.4pg) on the Rh positive membranes)
and therefore approximately 98% of the 125I-labelled IgG on the Rh positive
membranes could be expected to be anti-D. Approximately 23% of the counts
were released by solubilizing the membrane with deoxycholate and virtually
all the counts associated with the Rh negative membranes were released as
would be expected with nonspecifically bound molecules. Approximately half
of the counts on the solubilized Rh positive proteins were lost during dialysis.
It is difficult to account for the presence of labelled material in the
dialysis fluid although the action of proteases or deoxycholate on the
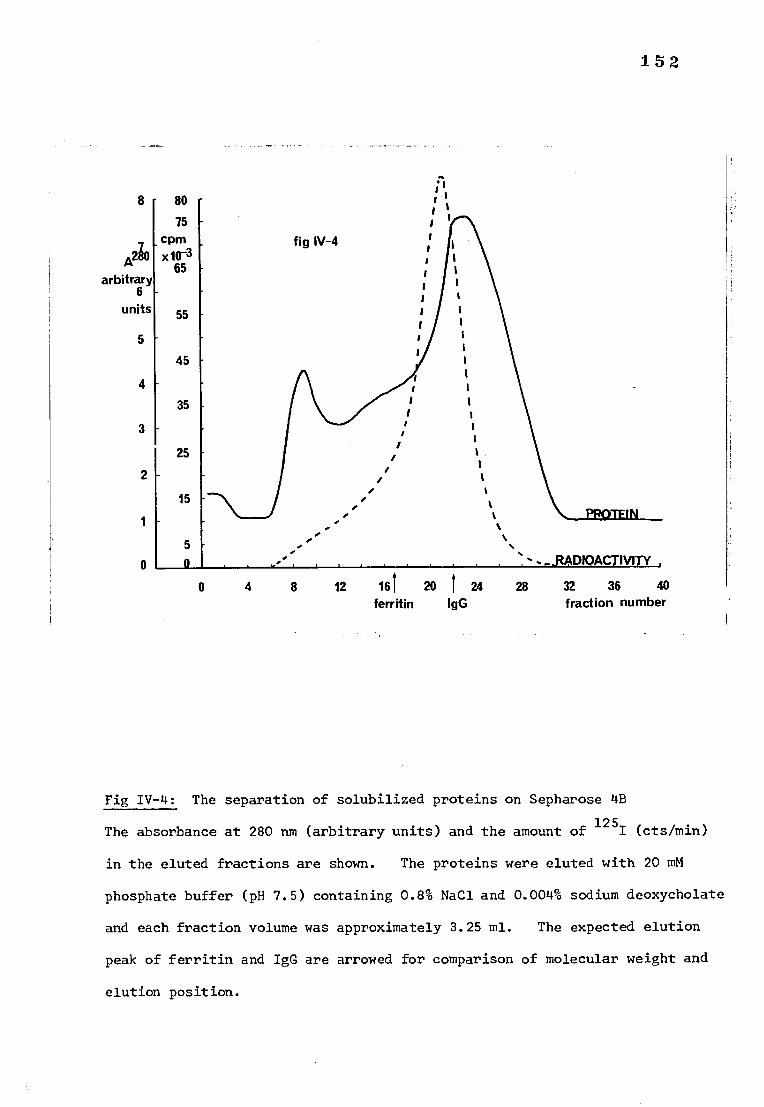
ferritin IgG fraction number
8
A230
arbitrary 6
units
5
4
3
0 4 8 12 161 20 1 24 28 32 36 40
152
Fig IV-4: The separation of solubilized proteins on Sepharose 4B
The absorbance at 280 nm (arbitrary units) and the amount of 125I (cts/min)
in the eluted fractions are shown. The proteins were eluted with 20 mM
phosphate buffer (pH 7.5) containing 0.8% NaCl and 0.004% sodium deoxycholate
and each fraction volume was approximately 3.25 ml. The expected elution
peak of ferritin and IgG are arrowed for comparison of molecular weight and
elution position.

1 53
antibody molecule may have resulted in the production of low M.W. fragments
which were able to pass through the dialysis sac. The remaining half of
the counts were associated with the reaggregated proteins. None of the
counts associated with the Rh negative sample were lost by dialysis;
they were distributed fairly equally between the aggregated proteins and
those which had not reaggregated indicating no particular affinity with
either protein population.
b) Separation of solubilized proteins on Sepharose 4B
The solubilized membrane proteins were fractionated on Sepharose 4B.
The absorbance at 280 nm and the radioactivity (cts./min.) of the
fractions eluted from the Sepharose 4B column are shown in Fig IV-4. The
elution positions of untreated IgG and ferritin on the same column are shown.
The peak of 125I-activity appears to be slightly earlier than the expected
position for IgG elution, indicating a higher molecular weight than normal,
either due to association with the D antigen or to association with deoxycholate
c) Measurement of the amount of bound and free antibody after treatment
with sodium deoxycholate
In one particular experiment involving this test, 185 mg stroma (pre-
treated with 125I-labelled anti-D) were solubilized in 30 ml of buffer.
The soluble proteins were removed and dialysed against 20 mM phosphate
buffer pH 7.5 containing 0.8% NaC1 and 0.004% deoxycholate, before separating
on Sepharose 4B. Fractions 8 to 20 inclusive were tested for bound antibody
and antigen as described in the methods. Prior treatment with Biobeads SM-2
was unnecessary since most of the deoxycholate is eluted later. Fractions 21
to 32 inclusive were treated with Biobeads SM-2 before carrying out the test.

154
The results are shown in Table IV-3. Fractions 28 to 32 are not included
because the red cells used to measure the available antibody lysed on
incubation in these fractions presumably because of high concentrations
of deoxycholate being eluted at this point on the column. There was no
evidence of appreciable amounts of antibody free or bound in any of the
later fractions (fractions up to No. 60 were tested). The amount of free
antibody and bound antibody (released by treatment at 56°C) in the fractions
eluted from Sepharose 4B are shown diagrammatically in Fignr-5. It can
be seen that the highest amount of bound antibody was eluted between
fractions 12 and 18. Molecules of molecular weight approximately 800,000
daltons would be expected to be eluted at this point under the experimental
conditions described. (Based on information gained by plotting log molecular
weight against elution volume for substances of known molecular weight
i.e. Ferritin and IgG). The largest amount of free anti-D recorded was
eluted with fractions of lower molecular weight. Free anti-D in fractions
10 to 18 may have been due to antibody dissociating from the antibody-antigen
complex. The peak of bound antibody presumably corresponds to the elution
of the Rh antigen-antibody complex. Little significance can be attached
to the expected molecular weight of proteins eluted at this point because
deoxycholate may be bound to the complex in unknown amounts thereby altering
the molecular weight.
Since the antigen-antibody complex appears to be eluted in a discrete
peak from Sepharose 4B, separation of this material could be used as a
partial purification of the antigen before passing down a column containing
cyanogen bromide-activated Sepharose 4B to which anti-IgG had been attached.
As a final purification the antigen-antibody complex should bind to the anti-
IgG and consequently remain bound to the column while the contaminating
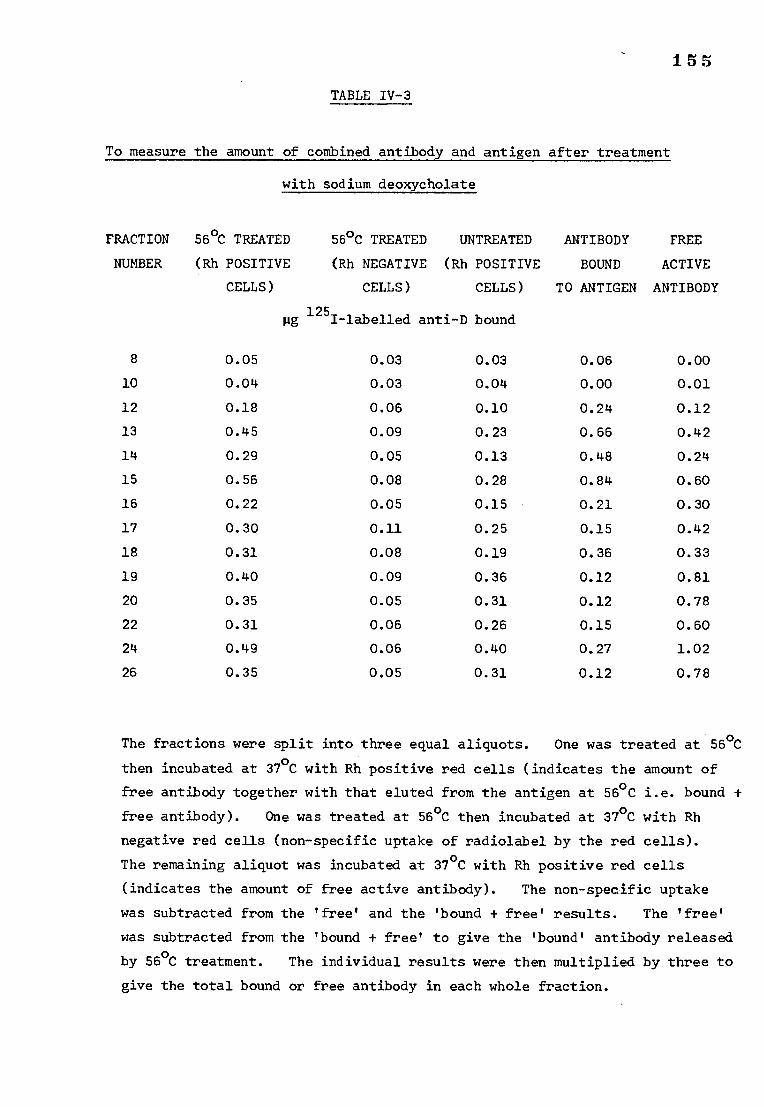
155
TABLE IV-3
To measure the amount of combined antibody and antigen after treatment
with sodium deoxycholate
FRACTION 56°C TREATED 56°C TREATED UNTREATED ANTIBODY FREE
NUMBER (Rh POSITIVE (Rh NEGATIVE (Rh POSITIVE BOUND ACTIVE
CELLS) CELLS) CELLS) TO ANTIGEN ANTIBODY
pg 1251-labelled anti-D bound
8 0.05 0.03 0.03 0.06 0.00
10 0.04 0.03 0.04 0.00 0.01
12 0.18 0.06 0.10 0.24 0.12
13 0.45 0.09 0.23 0.66 0.42
14 0.29 0.05 0.13 0.48 0.24
15 0.56 0.08 0.28 0.84 0.60
16 0.22 0.05 0.15 0.21 0.30
17 0.30 0.11 0.25 0.15 0.42
18 0.31 0.08 0.19 0.36 0.33
19 0.40 0.09 0.36 0.12 0.81
20 0.35 0.05 0.31 0.12 0.78
22 0.31 0.06 0.26 0.15 0.60
24 0.49 0.06 0.40 0.27 1.02
26 0.35 0.05 0.31 0.12 0.78
The fractions were split into three equal aliquots. One was treated at 56°C
then incubated at 37°C with Rh positive red cells (indicates the amount of
free antibody together with that eluted from the antigen at 56°C i.e. bound +
free antibody). One was treated at 56°C then incubated at 37°C with Rh
negative red cells (non-specific uptake of radiolabel by the red cells).
The remaining aliquot was incubated at 37°C with Rh positive red cells
(indicates the amount of free active antibody). The non-specific uptake
was subtracted from the 'free' and the 'bound + free' results. The 'free'
was subtracted from the 'bound + free' to give the 'bound' antibody released
by 56°C treatment. The individual results were then multiplied by three to
give the total bound or free antibody in each whole fraction.
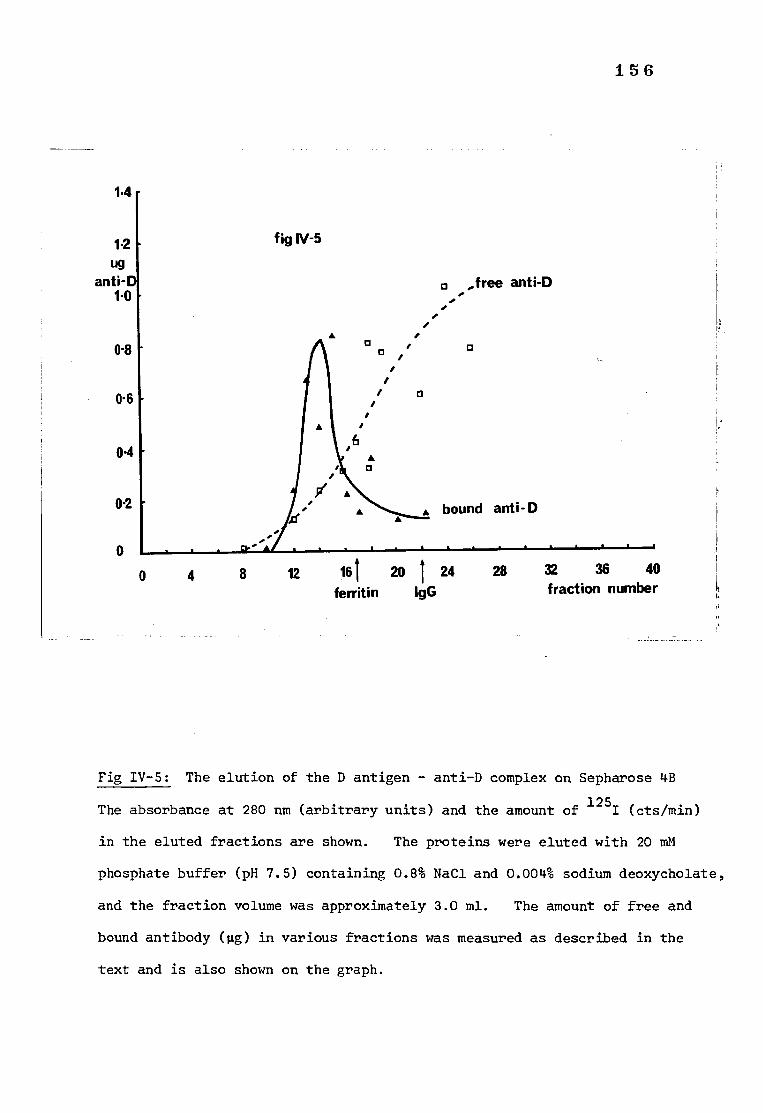
a a a •
i
0 ,free anti-D
• •
• a
• • bound anti-D
0 4 8 12 161 20 I 24 ferritin IgG
28 32 36 40 fraction number
•
fig IV-5
156
1.4
1.2 ug
anti- 1.0
0-8
0.6
04
02
Fig IV-5: The elution of the D antigen - anti-D complex on Sepharose 4B
The absorbance at 280 nm (arbitrary units) and the amount of 125I (cts/min)
in the eluted fractions are shown. The proteins were eluted with 20 mM
phosphate buffer (pH 7.5) containing 0.8% NaC1 and 0.004% sodium deoxycholate,
and the fraction volume was approximately 3.0 ml. The amount of free and
bound antibody (pg) in various fractions was measured as described in the
text and is also shown on the graph.
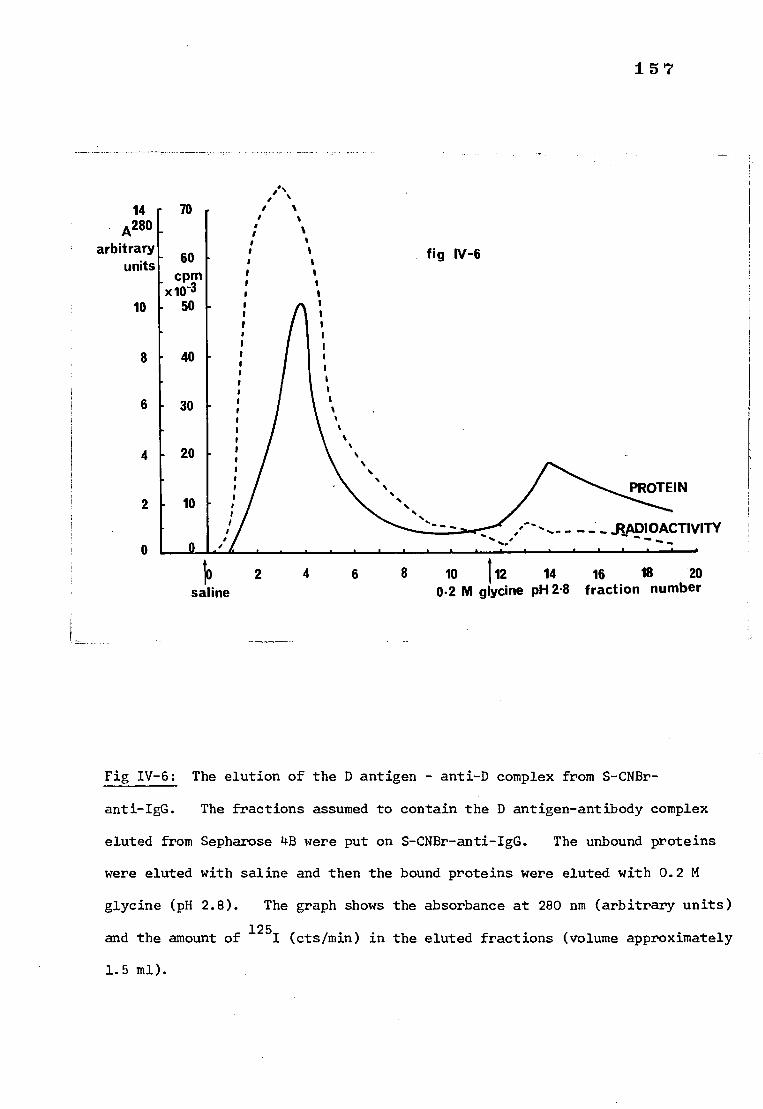
157
14 A280
arbitrary units
10
8
6
4
2
0
70
- 60 cpm
- x1W1 - 50
- 40
30
20
- 10
•
: / : / / / /
I
S
s s
. .
• ........
fig IV-6
..
/ ------- - ....
PROTEIN
- RADIOACTIVITY
10 2 4 6 8 10 112 14 16 18 20 saline 0.2 M glycine pH 2.8 fraction number
Fig IV-6: The elution of the D antigen - anti-D complex from S-CNBr-
anti-IgG. The fractions assumed to contain the D antigen-antibody complex
eluted from Sepharose 4B were put on S-CNBr-anti-IgG. The unbound proteins
were eluted with saline and then the bound proteins were eluted with 0.2 M
glycine (pH 2.8). The graph shows the absorbance at 280 nm (arbitrary units)
and the amount of 125
I (cts/min) in the eluted fractions (volume approximately
1.5 ml).

158
material is washed off. The antibody (and denatured antigen) could then
be eluted at low pH and it might be possible to recognise the extra band,
corresponding to the D antigen, when the acid eluate is separated on SDS
polyacrylamide gels.
3) ATTEMPTS TO PURIFY THE Rh (D) ANTIGEN
a) Separation of solubilized proteins on Sepharose 4B followed by
purification on S-CNBr-anti-IgG.
The separation of fractions 12 to 18 inclusive (eluted from Sepharose 4B
as described in the methods) on S-CNBr-anti-IgG is shown in Fig IV-6.
SDS polyacrylamide gels of various fractions are shown in Fig IV-7. The
proteins eluted at pH 2.8 from the S-CNBr-anti-IgG were indistinguishable
from reduced IgG after separation on SDS-polyacrylamide gels.
It was obviously necessary to try to develop a means of eluting the
D antigen and toward this end the possibility of breaking the D antigen-
anti-D bond at a higher pH than the anti-D-anti-IgG bond and thereby eluting
the antigen before the anti-D was investigated.
A further problem which emerged during this experiment was the overloading
of the Sepharose 4B column when 100 ml red cell membranes were solubilized and
separated. The separation was so bad that it is probable that the antigen-
antibody complex was more widely distributed in the eluted fractions than 'rc)
expected it-shTld have been and therefore only a small amount of the antigen-
antibody complex was purified on CNBr-anti-IgG. In the following experiments
the separation on Sepharose 4B was abandoned.
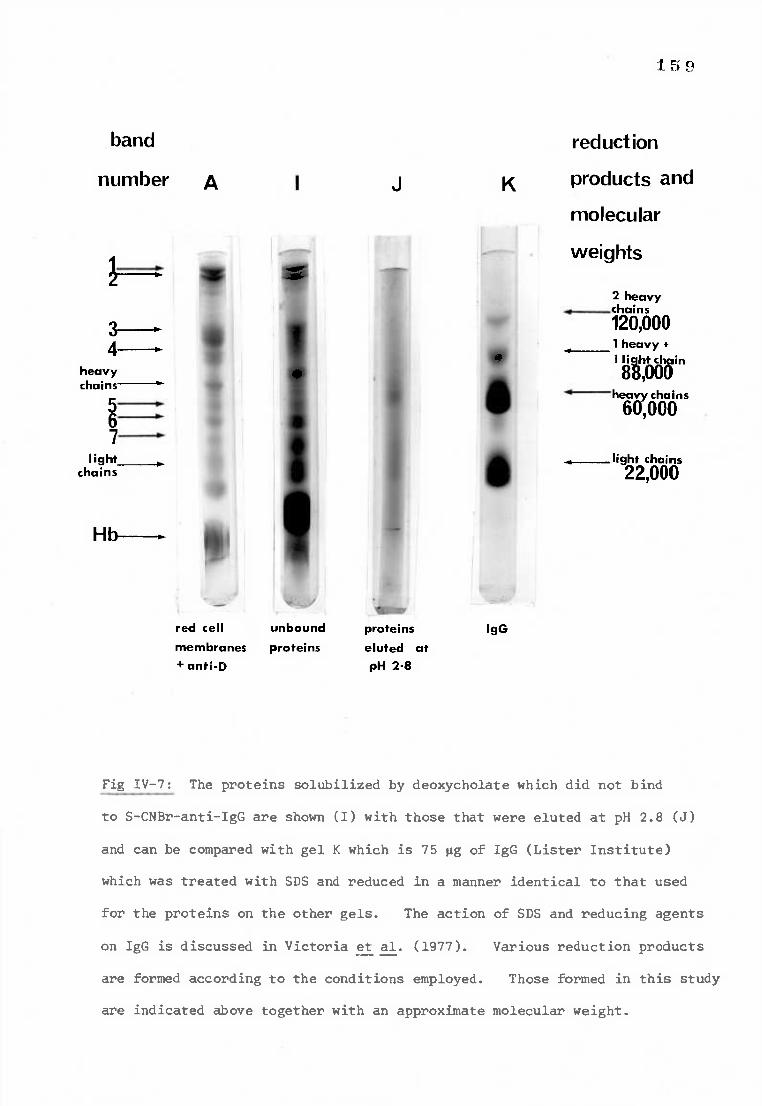
5
band reduction
number A I J K products and
molecular
weights 41
2 heavy chains 120,000 1 heavy +
3-- 4-11'
heavy chains
4 , gobh6in
O heavy chains
60,
light chains 22,000
5
6 7
light chains
red cell unbound proteins IgG
membranes proteins eluted at +anti-D pH 2.8
Fig IV-7: The proteins solubilized by deoxycholate which did not bind
to S-CNBr-anti-IgG are shown (I) with those that were eluted at pH 2.8 (J)
and can be compared with gel K which is 75 pg of IgG (Lister Institute)
which was treated with SDS and reduced in a manner identical to that used
for the proteins on the other gels. The action of SDS and reducing agents
on IgG is discussed in Victoria et al. (1977). Various reduction products
are formed according to the conditions employed. Those formed in this study
are indicated above together with an approximate molecular weight.

160
TABLE IV-4
The effect of pH on the stability of the D antigen-anti-D complex
pH of buffer mg antibody bound to stroma
after treatment at various
pH values
7.0 5.50
5.0 4.96
4.5 4.80
4.0 1.75
3.5 0.55
2.8 0.55
Rh positive red cell membranes were treated with 125I-labelled
anti-D and then incubated with glycine buffer at various pH values.
After ultracentrifugation the amount of 1251 remaining on the
membranes was estimated.

161
b) The use of buffers with acid pH to split the D antigen-antibody complex
(i) The effect of pH on the stability of the D antigen-anti-D complex
The amount of 125I-labelled anti-D remaining attached to red cell
stroma after incubation at various pH values is shown in Table IV-4.
It can be seen that the antibody-antigen complex begins to dissociate
at pH 4.0 with consequent loss of bound antibody.
(ii) The elution of solubilized anti-D from S-CNBr-anti-IgG
at various pH values
Anti-D alone (in solubilizing buffer) was eluted from S-CNBr-anti-IgG
at various pH values. SDS-polyacrylamide gels of the fractions eluted at
pH 5.0, 4.0 and 2.8 are shown in Fig IV-8. There was no evidence of the
elution of IgG at pH 5.0 and 4.0. It appears that the IgG anti-D-anti-IgG
bond is broken at pH 2.8. Considering the results of the previous experiment
(section b (i)) it is possible that the D antigen would elute from the anti-D
at a pH of about 4.0, while the anti-D would remain bound to the anti-IgG
until the pH was reduced to 2.8.
c) The separation of solublized proteins on S-CNBr-anti-IgG
(i) The elution of Rh positive and Rh negative soluble proteins from
S-CNBr-anti-IgG at various pH values is shown in Fig IV-9. SDS-polyacrylamide
gels of the eluates are shown in Fig IV-10. Comparing the patterns produced
by normal IgG with that produced by the eluates, there appears to be two
additional bands (arrowed) in the position expected for molecules of
approximate molecular weight 142,000 and 170,000 daltons. Some IgG was
eluted at pH 5.0 and 4.0, probably because the gel was overloaded and the
change in buffer caused any lightly bound IgG to be released.
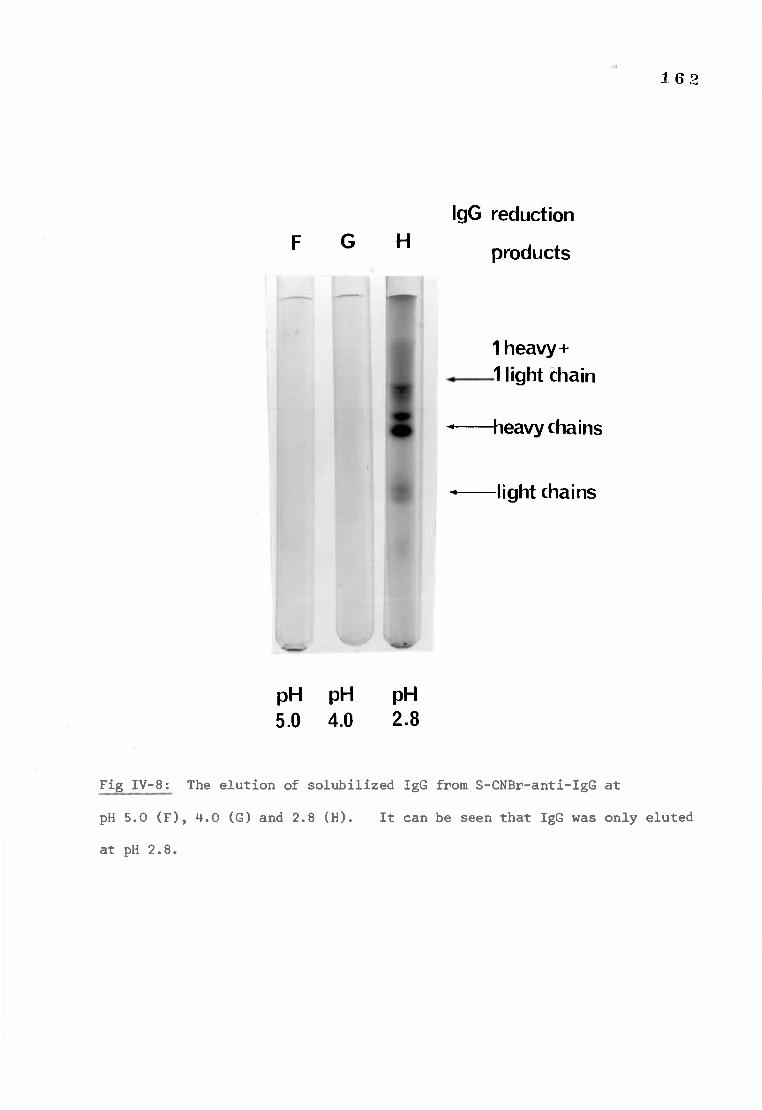
x. 62
IgG reduction F G H products
1 heavy+ 1 light chain
11 —heavy chains
—light chains
pH pH pH 5.0 4.0 2.8
Fig IV-8: The elution of solubilized IgG from S-CNBr-anti-IgG at
pH 5.0 (F), 4.0 (G) and 2.8 (H). It can be seen that IgG was only eluted
at pH 2.8.
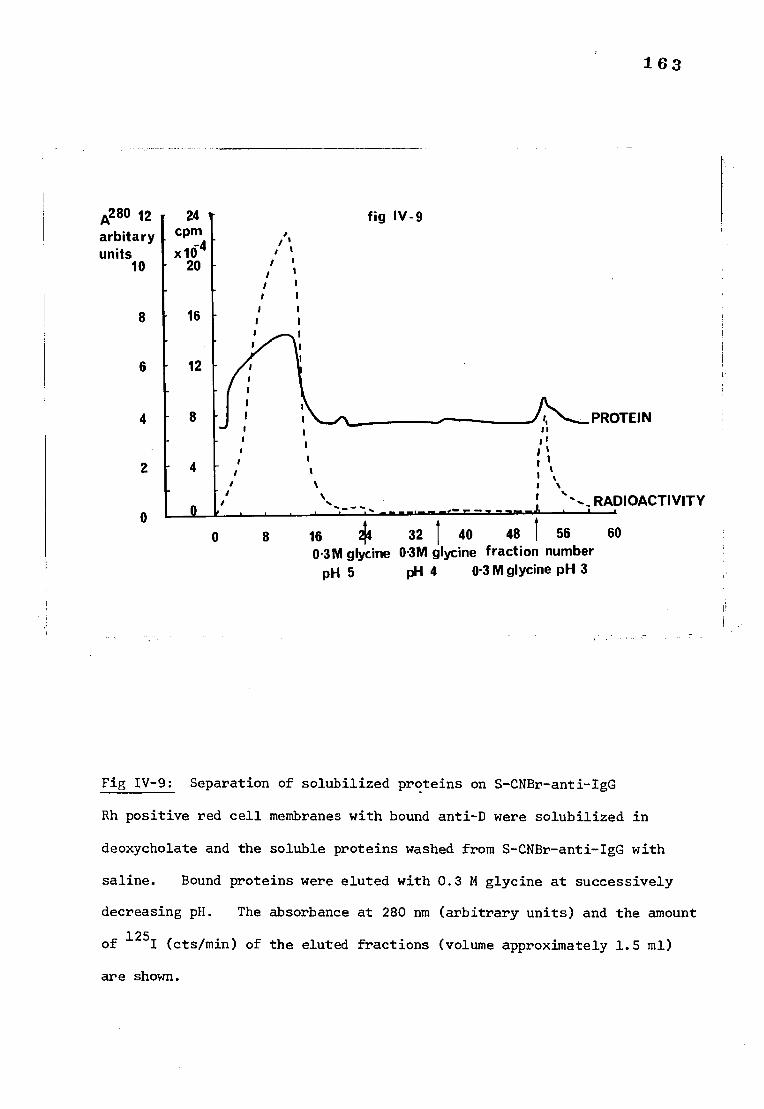
163
A280 12 24 arbitary cpm units x10 4
10 20
8 16
12
fig IV-9
4
2
0
PROTEIN 1 1
ti
. RADIOACTIVITY
0 8 16 214 32 1 40 48 1 56 60 01M glycine 01M glycine fraction number
pH 5 pH 4 0.3 M glycine pH 3
Fig IV-9: Separation of solubilized proteins on S-CNBr-anti-IgG
Rh positive red cell membranes with bound anti-D were solubilized in
deoxycholate and the soluble proteins washed from S-CNBr-anti-IgG with
saline. Bound proteins were eluted with 0.3 M glycine at successively
decreasing pH. The absorbance at 280 nm (arbitrary units) and the amount
of 125I (cts/min) of the eluted fractions (volume approximately 1.5 ml)
are shown.
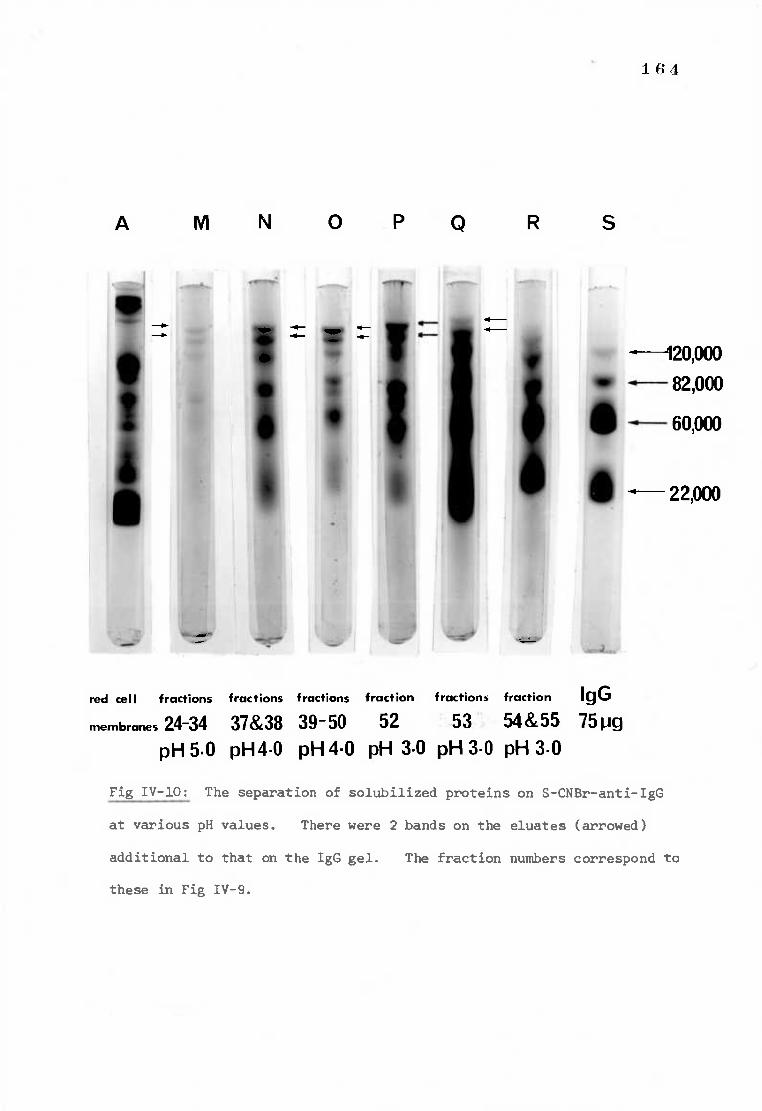
120,000 82,000
60,000
• 22,000
• •
•
1€4
A MN 0 PQ
red cell fractions fractions fractions fraction fractions fraction IgG membranes 24-34 37&38 39-50 52 53 54&55 75 pg
pH 5.0 pH 4.0 pH 4.0 pH 3.0 pH 3.0 pH 3.0
Fig IV-10: The separation of solubilized proteins on S-CNBr-anti-IgG
at various pH values. There were 2 bands on the eluates (arrowed)
additional to that on the IgG gel. The fraction numbers correspond to
these in Fig IV-9.

165
(ii) Before attempting to evaluate the significance of the result
obtained above it was necessary to repeat the experiment to determine whether
the additional protein bands at approximately 142,000 and 170,000 daltons
were a reproducible feature of the SDS-polyacrylamide gels of eluates from
S-CNBr-anti-IgG, or whether they were an artifact. Two similar experiments
were performed. One was abandoned due to technical difficulties. In the
other experiment the soluble protein fraction applied to the column was
tested for bound antigen and antibody by the technique described in the
methods section. The test for bound antibody indicated that 3.7% (8.0 pg)
of the labelled antibody which had been solubilized had remained in
combination with the antigen. Assuming that the same proportion of
unlabelled antibody had also remained bound (i.e. 3.7% or 160 pg) there must
have been approximately 168 pg of antibody bound to antigen in the sample
applied to the column. SDS-polyacrylamide gels of the fractions eluted at
various pH values from the S-CNBr-anti-IgG globulin column are shown in
Fig IV-11. The two additional bands (MW approximately 142,000 and 170,000)
were visible in gels V and W (and very faintly in U), although they were
much fainter than in the previous experiment. A comparison of the results
of this and the previous experiment is shown in Fig IV-12. Gel P from the
previous experiment showing the additional bands is compared with gel W from
this experiment and gel Z (150 pg IgG). The additional bands on gel P
(which are present very faintly on gel W) could correspond to two of the bands
on the IgG gel (Z) which represent whole IgG (2 heavy chains + 2 light chains)
and a reduction product of IgG in which 2 heavy chains and one light chain
(M.W. 142,000) are present (Victoria et al., 1977). These two lines are under the
only visible in the normal IgG preparation/experimental conditions employed,
when large amounts (150 pg) of IgG are separated on SDS-polyacrylamide gels
(and reduction is incomplete). It is still difficult to explain why the extra
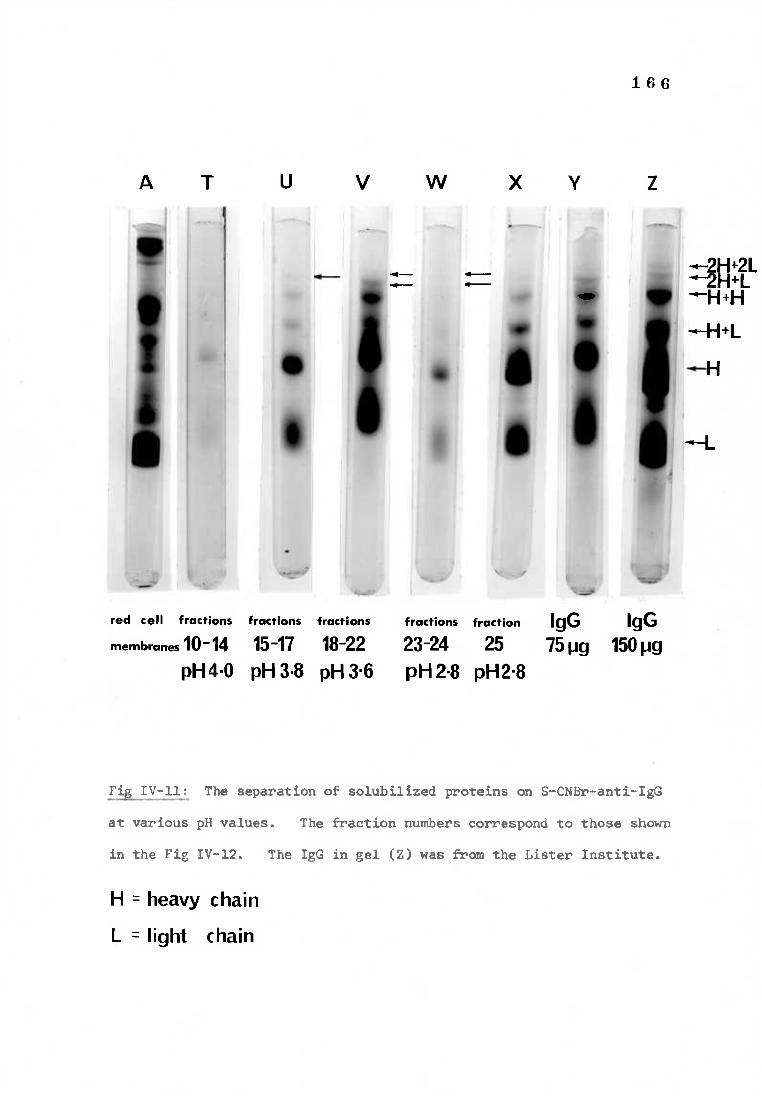
1 6 6
A T U V W X Y
• •
•
41--••• 4-
• --2H+2L —H+H
red cell fractions
membranes 10-14 pH SO
fractions fractions fractions fraction IgG IgG 15-17 18-22 23-24 25 75 pg 150 pg pH 3.8 pH 3.6 pH 2.8 pH2.8
Fig IV-11: The separation of solubilized proteins on S-CNBr-anti-IgG
at various pH values. The fraction numbers correspond to those shown
in the Fig IV-12. The IgG in gel (2) was from the Lister Institute.
H = heavy chain
L = light chain
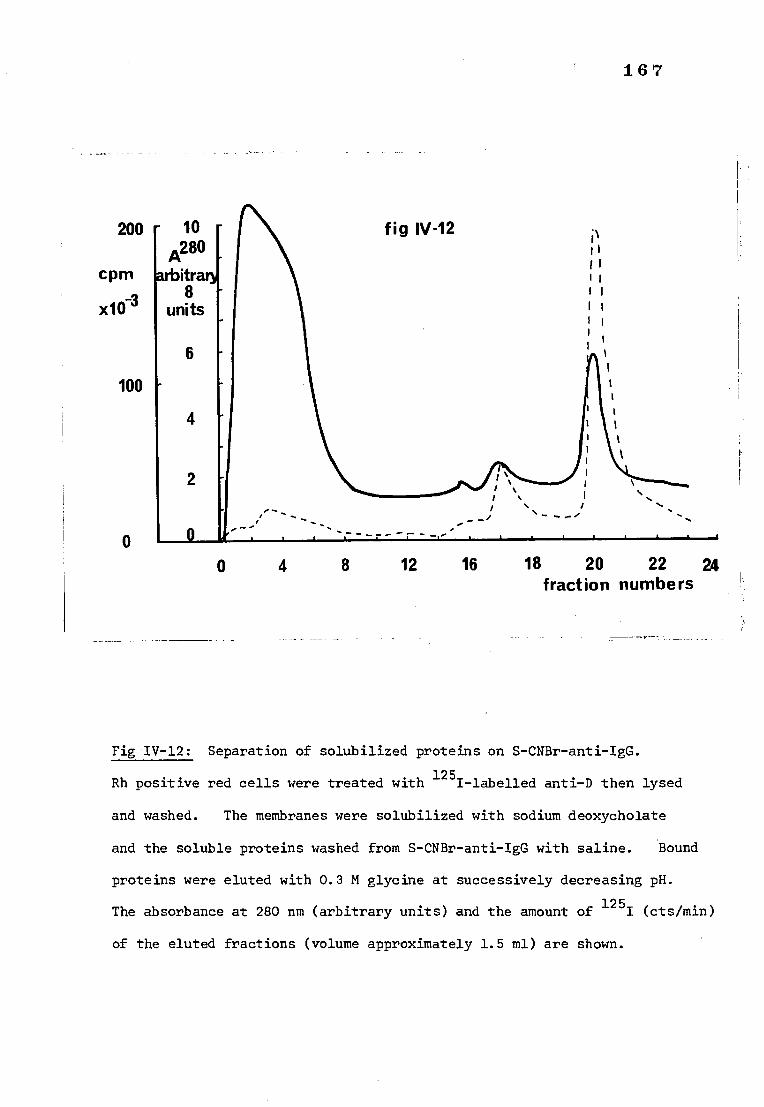
200
cpm
x10 3
100
0
fig IV-12
■
• •
•
10 A280
arbitrary 8
units
6
4
2
0 4 8 12 16 18 20 22 24 fraction numbers
1 6 7
Fig IV-12: Separation of solubilized proteins on S-CNBr-anti-IgG.
Rh positive red cells were treated with 125I-labelled anti-D then lysed
and washed. The membranes were solubilized with sodium deoxycholate
and the soluble proteins washed from S-CNBr-anti-IgG with saline. Bound
proteins were eluted with 0.3 M glycine at successively decreasing pH.
The absorbance at 280 nm (arbitrary units) and the amount of 1251 (cts/min)
of the eluted fractions (volume approximately 1.5 ml) are shown.
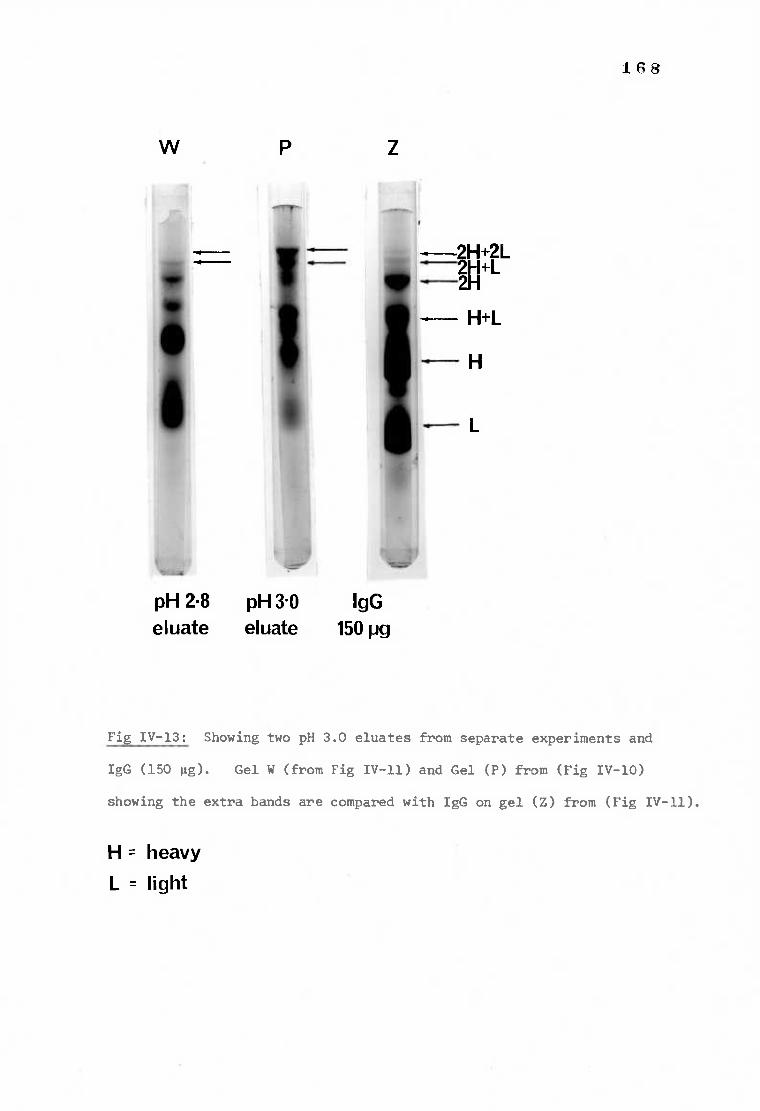
H+L
H
L
1 6 8
2H+LL
111.
•••••••■•■
pH 2.8 pH 3.0 IgG
eluate eluate 150 pg
Fig IV-13: Showing two pH 3.0 eluates from separate experiments and
IgG (150 mg). Gel W (from Fig IV-11) and Gel (P) from (Fig IV-10)
showing the extra bands are compared with IgG on gel (Z) from (Fig IV-11).
H = heavy L = light

169
bands are so pronounced in the gels from the first experiment (see gel M to R
Fig IV-10) compared to that in gel Z, when it is taken into account that there
was far less IgG (as assessed by the depth of protein staining) in gels M to
R than in gel Z. Furthermore, the incubation conditions in SDS and 2-
mercaptoethanol were identical for all the gels in this study. A possible
explanation could be that the pH or ionic strength of the eluate fractions
might influence the reduction of IgG. However there appeared to be no
constraint on the reduction of deoxycholate-treated IgG eluted from S-CNBr-
anti-IgG at pH 2.8 in the absence of membrane proteins (Fig IV-8). Therefore
either the IgG in the eluates was altered before treatment with SDS and
reducing agent, such that it could not easily be reduced, or, proteins other
than IgG, of molecular weight 142,000 and 170,000 daltons are present on
gels of the eluates.
DISCUSSION
The results of this study were in agreement with the findings of
Lorusso and Green (1975) in that the proteins solubilized by the action
of sodium deoxycholate regained Rh activity after the removal of detergent
and consequent reaggregation of the membrane lipids and proteins. Experiments
demonstrated that if Rh positive membranes were treated with 125I-labelled
anti-D before treatment with deoxycholate, a certain percentage of anti-D
remained in combination with the antigen after solubilization. Consequently,
the binding of labelled antibody could be used as a marker for the D antigen
in purification procedures. Estimation of the amount of 125I attached to
D antigen, as measured by the amount of active 125I-labelled anti-D which
could be released from the antigen by incubation at 56°C, was used to
demonstrate the existence of D antigen-antibody complex in fractions of

170
soluble proteins eluted from Sepharose 4B. The complex was eluted with
proteins of approximately 800,000 daltons. This value for the molecular
weight of the antigen-antibody complex is probably an overestimate because
it has been suggested (Helenius and Simons, 1972) that deoxycholate binds
to lipophilic and hydrophilic proteins with differing affinities. Therefore
unknown quantities of deoxycholate may have been bound to the antigen-antibody
complex thereby increasing its apparent molecular weight.
Attempts to purify the antigen-antibody complex by affinity chromato-
graphy were largely unsuccessful. The unattached proteins were washed
from a S-CNBr-anti-IgG column presumably leaving the D antigen-anti-D complex
attached to the anti-IgG. Treatment with buffers of decreasing pH should
have eluted first the antigen (at about pH 4.0) with progressively more
antibody as the pH decreased to 2.8. Comparison of the SDS-polyacrylamide
gels of the eluted fractions with the gels of pure IgG showed that in addition
to the protein bands present on the 'pure IgG' gel, there were two bands on
the gels of the eluted fractions running in the expected position of IgG
(2 heavy + 2 light chains, 170,000 daltons) and a reduction product of IgG
consisting of 2 heavy + 1 light chain, (142,000 daltons). These two bands
were not present on the gels of'pure IgG' reduced under identical conditions
unless very large amounts (150 Mg) were treated (and presumably, incompletely
reduced). Therefore either the IgG in the eluates has been altered before
treating with SDS and 2-mercaptoethanol such that it is not easily reduced,
or, alternatively, other (non-IgG) proteins of molecular weight 142,000 and
170,000 daltons are present on the gels of the eluates. Although it is
tempting to relate this result to that obtained by radiation inactivation
(174,000 daltons) caution must be used in evaluating its significance.

171
The regime utilized in this study for the identification of the
protein associated with Rh activity depended upon the resolution of proteins
on SDS-polyacrylamide gels. Although an extremely useful technique,
polyacrylamide gel electropheresis does have certain disadvantages. For
example, the bands visible on the gels after staining are not necessarily
individual polypeptides; they could be mixtures of polypeptides of the same
molecular weight, aggregates of low molecular weight peptides or even,
protein molecules which are not dissociated by the action of SDS. Therefore
if the D antigen ran on SDS-polyacrylamide gels in the position of one of the
IgG bands then it would not be distinguishable. The failure of the
experiments in the present study to demonstrate conclusively the presence
of the Rh (D) antigen could also be explained if there was insufficient
antigen protein applied to the gels, if the antigen was not a protein or if
it had a very small molecular weight (less than 10,000 daltons). It is
also concievable that the antigen-antibody complex was unable to bind to
the S-CNBr-anti-IgG and was therefore eluted with the other soluble proteins,
leaving the free anti-D bound to anti-IgG on the column.
Further experiments could be designed to investigate why the D antigen
was not identified in this study. However since it is unlikely that the
D antigen could be separated from anti-D without being inactivated by the
methods available at this time, it would perhaps be better to develop a
purification procedure which did not involve a denatured antigen as an end
product. The isolated antigen could then be tested for antigenic activity
and the whole isolation procedure would then be much more credible. For
example, if one can assume that the D antigen must be on the surface of the
membrane in order to fulfil its antigenic function then in theory it should
be possible to label the protein on which the antigen is carried by using a

272
non-penetrating radioactive label on the intact red cell. Providing that
antigenic activity is retained after labelling this should serve as a
possible marker for the antigen when the membrane is disrupted.
Solubilization of the membrane would still need a strong detergent, for
example, triton X-100 which Yu and Steck (1973) claim solubilizes without
disruption of the tertiary structure of proteins, or possibly deoxycholate,
using the regime utilised by Edwards (1977) in his investigations into the
glucose transport mechanisms in the red cell. The isolation of the antigen
from the other solubilized proteins remains a problem. It would be
interesting to pass the labelled solubilized proteins down Biobeads S-M2
followed by a S-CNBr column to which anti-D had been attached and then to
measure the radioactivity of the unbound protein. However, it is quite
possible that the D antigens would be unable to bind to anti-D in its
soluble state. If the antigen was unable to bind then perhaps various
molecular weight fractions of the soluble proteins could be reaggregated
by the removal of detergent and the Rh activity of each fraction investigated.
Considering the results of the radiation inactivation experiments
described in Chapter III, the Rhesus antigens may be small polypeptides
which aggregate or polymerize to form a complex and the various antigenic
activities associated with the Rhesus system may depend upon the integrity
of the whole complex. The success, therefore, of attempts to isolate the
D antigen by solubilizing the red cell membrane may depend on the effect of
the solubilizing agent on the quarternary structure of the Rhesus antigen
complex. The experiments of Lorusso and Green (1975) and those described
in the present study have shown that Rh antigenic activity can be restored
to deoxycholate solubilized protein molecules and therefore, providing the
demonstration of Rh activity does not depend upon the existence of large areas
of membrane structure, it may be possible to isolate the active components and
reaggregate them with concomitant restoration of Rh activity.

173
Since the completion of this study Lorusso et al. (1977) have
published more of their work concerning the solubilization of the Rh (D)
antigen using sodium deoxycholate. They have undertaken a similar series
of experiments to those described in the present study and their results are
very similar. They found that solubilization of Rh (D)-positive membrane -
14C anti-D complexes followed by chromatography on agarose columns
demonstrated that the complexes had a higher molecular weight than 14C-IgG
anti-D alone (an approximate molecular weight was not given) indicating that
the Rh (D) antigen and antibody remain associated in detergent. This is in
agreement with the results of the present study using 125I-labelled anti-D.

174
CHAPTER V
OBSERVATIONS ON THE ROLE OF LEUCOCYTES IN THE DESTRUCTION OF ANTIBODY-
COATED RED CELLS IN HAEMOLYTIC DISEASE OF THE NEWBORN
INTRODUCTION
Phagocytosis is the process by which certain cells are able to
transport particles from the extracellular environment into intracellular
vacuoles or phagosomes (Stossel, 1975).
Many of the cells which form a part of the reticuloendothelial system
in man are able to phagocytose foreign or unwanted particles for example,
human peripheral blood neutrophils are involved in the elimination of
bacteria from sites of skin injury. Monocytes and macrophages are also
capable of phagocytosis and the activity of these cells is not limited to
bacteria. Polystyrene particles (Al-Ibrahim et al., 1976), immune complexes
(Mantovani et al., 1972) and erythrocytes (Zipursky and Brown, 1974) are
known to be ingested in the appropriate conditions.
Various serum constituents have been found to increase the susceptibility
of particles to phagocytes. These constituents are called opsonins.
Immunoglobulins and the third component of complement (C3) bind to red cells
and can act as opsonins. IgG antibodies bind to red cell antigens by the
(Fab)2 fragments of the molecule and interact with receptors on the white
cell surface via the Fc fragment of the molecule (Berkin and Benacerraf, 1966;
Holm et al., 1974). The opsonised erythrocytes gather in this way around a
white cell to form a rosette (Spiegelberg, 1974). Erythrophagocytosis may be
a subsequent step in thig process. Ingested red cells are visible within
the white cells before losing colour as the haemoglobin disappears leaving

175
clear vacuoles which gradually diminish in size (Bonnin and Schwartz,
1954).
In man monocytes have receptors for IgG subclasses 1 and 3 (Huber and
Fudenberg, 1970; Holm et al., 1974). IgM is not cytophilic for monocytes
and rosettes will not form unless complement is present (Huber and Fudenberg,
1968).
The importance of phagocytic cells in combating infections caused by
pathogenic bacteria is reflected in the leucocytosis evident in these
conditions. There is evidence of erythrophagocytosis occurring naturally
in the spleen and liver in the removal of senescent erythrocytes from the
circulation (Kay, 1975) and in various haematological disorders, for example,
paroxysmal haemoglobinuria (Jordan et al., 1952) incompatible blood trans-
fusions (Hopkins, 1910; Mackay, 1939) and haemolytic disease of the newborn
(Abt, 1931; Cooper, 1950).
Haemolytic disease of the newborn occurs when foetal red cells, which
have antigens not present in the mother, pass across the placenta. The
mother may produce antibodies to these antigens if she has been previously
immunized and the IgG antibodies can cross the placenta resulting in the
destruction of foetal red cells.
Cases of haemolytic disease of the newborn due to ABO incompatibility
are usually quite mild and the amount of antibody on the foetal cells is often
less than 0.6 pg/ml cells (Romano et al., 1973). In contrast, Rh allo-
immunization usually produces a more severe haemolytic process than ABO
haemolytic disease, although in cases of comparable severity there is usually
more antibody on the cells in Rh haemolytic disease (Romano and Mollison,
1975). In the latter condition there is commonly between 0.4 and 18 pg IgG
anti-D bound / ml foetal cells (Hughes-Jones et al., 1967).

176
IgG Rh antibodies are destructive in vivo as is evident in infants
with haemolytic disease of the newborn. However, these antibodies cause
no visible damage to red cells in vitro. There is therefore an interaction
between antibody-coated cells and the reticuloendothelial system of the body,
which results in the removal of the affected cells from the circulation and
a characteristic haemolytic syndrome.
The mechanism by which red cells are destroyed in diseases like
haemolytic disease of the newborn has not been fully explained. There are
a number of possibilities.
1. Lysis by complement binding. This seems unlikely since the incomplete
Rh antibodies do not bind complement (with rare exceptions e.g. Ripley) and
the small amount of IgG anti-A on the red cells in haemolytic disease of the
newborn, usually less than 2 pg/ml cells, is insufficient to bind complement
at a detectable level (Romano and Mollison, 1975).
2. Agglutination. Romano and Mollison (1975) found that red cells coated
with as little as 200 molecules IgG anti-A per cell agglutinated spontaneously
when suspended in plasma and they suggested that IgG anti-A may cause red cell
destruction in haemolytic disease of the newborn by producing agglutination
in vivo leading to trapping of cells and metabolic change. This explanation,
however, is unlikely to explain the relatively severe haemolytic syndrome
observed in Rh haemolytic disease since red cells coated with IgG anti-D will
not agglutinate spontaneously in vitro, even in plasma, unless at least 50 pg
IgG anti-D is bound / ml cells (Romano and Mollison, 1975).

177
3. Phagocytosis. Phagocytes remove non-viable erythrocytes and foreign
particles from the circulation, and the extention of these activities to
antibody-coated cells seems reasonable. IgG antibodies are opsonizing and
therefore, red cells coated with IgG antibodies, such as those occurring in
haemolytic disease of the newborn, should be ingested by phagocytic cells
in the peripheral blood. The major problem with this explanation of red
cell removal is the fact that erythrophagocytosis in vitro is inhibited in
the presence of free IgG (Lobuglio et al., 1967 and Lay and Nussenzweig, 1968).
Any IgG in the white cell culture medium presumably competes with IgG bound
to erythrocytes for receptor sites on leucocytes and effectively prevents
binding and subsequent ingestion of the red cells.
4. Lysis by monocytes and lymphocytes. Holm et al. (1974) has described
the lysis of antibody-coated red cells in the presence of human monocytes.
Holm found that this process required IgG antibody of subclass 1 or 3, in
common with monocyte-mediated phagocytosis. Urbaniak (1976), using lymphocyte
preparations freed from monocytes and neutrophils by preincubation in plastic
containers, found that lymphocytes also brought about lysis of Rh-opsonized
red cells. In contrast, Poplack (1976) found that only monocytes were
cytotoxic for human red cells coated with red cell antibodies. In Poplack's
experiments the monocytes were removed magnetically after iron ingestion.
The lytic process is also inhibited by free IgG and it is probable that the
same receptor sites are involved in both erythrophagocytosis and red cell
lysis by monocytes.
The experiments described in this chapter were carried out to determine
whether red cells opsonized with IgG anti-D and IgG anti-A to the same extent
as those occurring in haemolytic disease of the newborn could be ingested by

178
peripheral blood leucocytes in vitro and whether the results would be
applicable to in vivo conditions by measuring the extent to which erythro-
phagocytosis is inhibited in the presence of physiological concentrations
of free IgG. It was hoped that the results might explain the observation
that in cases of ABO and Rh haemolytic disease in which comparable amounts
of antibody are bound to the red cells of the infant, antibodies of the ABO
system often result in a more severe disease (Romano and Mollison, 1975).
MATERIALS AND METHODS
1) Red cells
Normal adult human red cells, less than 14 days old were used.
When experiments involved the use of anti-D, group 0 red cells of the
probable genotype CDe/cDE were used. For experiments involving the use
of anti-A, group Al red cells were used.
2) White cells
Peripheral blood was taken from normal human donors, into heparin
immediately before an experiment was started, and mixed by inversion.
3) Antibodies
a) IgG anti-D The IgG preparation had 516 pg IgG anti-D/ml (assayed
by Dr. N.C. Hughes-Jones). An aliquot of this preparation was partially
purified and labelled with 125
I. The anti-D was separated on an ion exchange
cellulose (CM-52, Whatman) as described by Frame and Mollison (1969). The
IgG fraction containing anti-D was labelled with 125I by the iodine mono-
chloride method (McFarlane, 1958). One hundred pCi of 1251 and 0.1 ml of
ICl (0.4 mg I/m1 in 0.1 M HCL) were used for each 10 mg protein. The specific
activity of the newly labelled antibody was usually between 6,000 and 11,000
cts/ min/µg protein. Before use the labelled preparation was incubated at

179
37°C for 60 min with an equal volume of group 0 Rh negative washed red cells
to reduce the non-specific binding of labelled protein to the red cells.
The antibody concentration of the labelled preparation was determined by
a method based on the experiments described by Hughes-Jones (1967) for the
determination of the concentration and equilibrium constant of anti-D
preparations. The results were analysed according to the derivation of
the law of mass action derived by Scatchard (1949). A plot of the ratio
of bound antibody to free antigen against bound antibody was made. The
line obtained was extrapolated to the abscissa to give the concentration
of antibody in the labelled preparation. The antibody concentration of
the 125I-labelled anti-D was 46 gg IgG anti-D /ml and corresponded to a
purity of about 3%, i.e. 3 mg anti-D/100 mg IgG.
b) IgG anti-A The IgG preparation contained about 60 pig IgG anti-A/ml.
An aliquot was iodinated (with prior preoxidation) and the concentration
determined as described above for anti-D. The specific activity of the
labelled preparation was 8,000 cts/min/gg protein, the concentration was
approximately 8 pig anti-A/ml and the purity was about 1 mg anti-A/100 mg IgG.
c) Horse anti-human IgG 125I-labelled anti-human IgG was prepared
purified and calibrated by Mrs. B. Gardner by a modification of the method
of Rochna and Hughes-Jones (1965). The anti-IgG was used in 0.5 ml aliquots
(50 pig/0.5 ml, 625,325 cts/min/ aliquot). The amount of antibody bound was
measured by reference to a plot of the amount of 125I-labelled anti-IgG bound
to known amounts of 131I-labelled anti-D.
4) Tissue culture media
Hanks' balanced salt solution (HBSS) from Wellcome Laboratories Ltd.,
was used in all the experiments in combination with 30% foetal calf serum
(BDH Ltd.).

180
5) IgM myeloma protein and IgG preparations
A purified IgM myeloma protein was kindly donated by Dr. E.L. Romano.
The product had 0.15 mg protein/ml. Normal human IgG (150 mg protein/ml)
was obtained from the Lister Institute, Elstree, Herts.
6) Experimental Techniques
a) Dextran Sedimentation
One volume of heparinised whole blood was mixed by inversion with two
volumes of 3% dextran (M.W. 60,000-90,000) and allowed to sediment at room
temperature in plastic bottles for 30 minutes.
The red cell-depleted supernatant was removed and spun at 2,000 rpm
for 4 mins. The cells were washed once in Hanks' balanced salt solution
(HBSS) containing 20% foetal calf serum (FCS). The white cells were then
resuspended in HBSS containing 30% FCS to give an approximately 1:2 dilution
of the original volume of whole blood. The number of leucocytes and
erythrocytes remaining in this preparation was measured in a Coulter counter.
b) Opsonizing red cells with IgG antibodies
Red cells were washed 3 times in phosphate buffered saline (PBS).
Routinely, between 0 and 10 pg antibody was incubated with 0.1 ml packed
red cells at 37°C for 60 mins in a total volume of 1 ml. The cells were
then washed 3 times in PBS and made up to 1 ml in HBSS containing 30% FCS.
c) Preparation of leucocyte monolayers
After sedimenting and washing, aliquots of white cells, usually 0.5 or
1.0 ml were pipetted onto glass coverslips in petri-dishes. The dishes were
incubated at 37°C for 60 mins then the unattached cells were removed from the
coverslips by aspiration. The attached cells were gently washed once with
HBSS containing 20% FCS before adding red cells.

181
d) Incubation of red cells with leucocyte monolayers
Immediately after washing, 1 ml of opsonized red cells in HBSS was
pipetted onto the leucocytes and the cells were incubated together at 37°C
for 60 mins. The non-adherent cells were removed by gentle washing in HBSS
and the coverslips were air-dried.
e) Fixing and staining the coverslips
The coverslips were fixed in methanol for 10 mins then stained 30 mins
in Leishman's stain. The coverslips were then washed by immersion in
buffered water, pH 7.4 for 30 secs, air-dried, and mounted on slides.
f) Microscopic examination
The stained monolayers were examined for the presence of white cells
containing phagocytosed red cells under the microscope.
The microscope viewer was positioned at the top right-hand end of the
coverslip and fields were examined from right to left down the coverslip until
200-500 cells of the same type i.e. monocytes or neutrophils, had been counted.
In cases where few cells had adherred to the slide it was examined in a zig-zag
manner.
The percentage of a certain cell type containing phagocytosed erythrocytes
was recorded. Leucocytes with attached red cells were not considered to have
phagocytosed a red cell unless at least one could be seen inside the leucocyte.
White cells which could not be easily identified because of bad staining or
cellular damage were not counted.

182
EXPERIMENTAL PROCEDURE
a) Measurement of bound 125I-labelled anti-D or anti-A
Red cells were washed three times in PBS, then 0.1 ml aliquots were
incubated with between 0.09 and 1.8 pg labelled anti-D or between 0.1 and
20 pg labelled anti-A and 0.1 ml bovine serum albumin (BSA) in a total volume
of 1.0 ml at 37°C for 60 min. The non-specific uptake of labelled protein
by red cells was estimated by doing a similar experiment using group 0 Rh
negative red cells. The unbound radioactivity was removed after centri-
fugation. The red cells were washed three times in PBS, transferred to
clean tubes and the bound radioactivity was measured in a gamma counter.
The red cells were resuspended in 1.0 ml HBSS containing 30% FCS and incubated
with adult white cell monolayers as described previously.
125 b) Measurement of bound antibody using I-labelled anti-IgG
Red cells (0.1 ml aliquots) were incubated with unlabelled anti-D
at concentrations between 0.1 and 4.0 pg/ml in 1.0 ml total volume, or
unlabelled anti-A at concentrations between 0.06 and 12 pg/ml. To give an
estimation of the non-specific uptake 0.1 ml aliquots of group 0 Rh negative
cells were incubated with equivalent amounts of antibody. The red cells
were incubated 60 min at 37°C then washed three times and 0.05 ml aliquots
were incubated 15 min at 37°C with 125I-labelled anti-IgG, then washed three
times, lysed and transferred to clean tubes before estimating the bound
radioactivity in a gamma counter. The remaining cells were resuspended in
1.0 ml HBSS containing 30% FCS and incubated with adult white cell monolayers.
c) Measurement of the effect of plasma constituents on erythrophagocytosis
i) Measurement of phagocytosis in plasma and serum
Group 0 Rh positive red cells were washed three times in PBS and
0.1 ml aliquots were opsonized with 4 pg unlabelled anti-D/ml. After

183
washing, the erythrocytes were resuspended in 1.0 ml fresh autologous serum
or plasma and incubated with white cell monolayers.
ii) Measurement of phagocytosis in the presence of IgM
Washed red cells were opsonized with 4.0 gg unlabelled anti-D/ml.
After washing the cells were resuspended in HBSS containing 30% FCS and
various concentrations of IgM i.e. 0, 0.0150.12 and 0.5 mg/ml myeloma
protein. The red cells were incubated 60 min at 37°C with white cell
monolayers.
iii) Measurement of phagocytosis in the presence of various
concentrations of IgG
Group 0 Rh positive red cells were opsonized with 0.93 pg labelled
anti-D or 9.2 pg unlabelled anti-D. The cells incubated with labelled
antibody were counted after washing. The erythrocytes were resuspended
in HBSS containing FCS and various amounts (0, 7, 15, 30 or 45 gg of IgG).
d) Red cell agglutination at low levels of bound antibody and the effects
of agglutination on phagocytosis
i) Red cell agglutination at low levels of antibody sensitization
in plasma
Group 0 Rh positive and group Al red cells (0.1 ml aliquots) were
incubated at 37°C for 60 minutes with between 0 and 2.7 pg labelled IgG
anti-D or between 0 and 0.7 gg labelled IgG anti-A respectively.
The cells were washed and the bound radioactivity measured. Two
percent suspensions of the cells in PBS were prepared. One drop of red cells
was added to two drops of autologous plasma from various donors. The cells
were incubated for 60 minutes at 37°C in precipitin tubes and the agglutination

1 8 4
estimated microscopically. The reactions were recorded semi-quantitatively
as 4 (2 or 3 solid clumps), 3 (large clumps), 2 (medium clumps), 1 (small
clumps) and W (very small clumps) 31, 21 and 11 were used to describe
reactions between those defined above.
The experiment was repeated incubating the cells in saline instead of
plasma.
ii) Measurement of the phagocytosis of agglutinated red cells in serum
Red cells were agglutinated prior to incubating with white cell
monolayers. In the first experiment described the red cells were incubated
with saturating amounts of anti-D which caused their agglutination in serum,
and in the second experiment group A red cells were agglutinated by suspending
in serum from a group 0 donor (as a source of anti-A).
(a) Group 0 Rh positive red cells (0.1 ml) were opsonized with
9.3 pg unlabelled IgG anti-D in a total volume of 1 ml and incubated
with white cell monolayers at 37°C for 60 minutes in autologous serum.
(b) Group A Rh positive cells (0.1 ml) were opsonized with various
amounts of anti-D (usually between 2 and 10 pg unlabelled IgG anti-D)
in 1 ml total volume, and incubated with white cell monolayers in
group 0 serum.
e) The effect of complement on erythrophagocytosis
Group A Rh positive red cells were incubated with various amounts of
IgG anti-D or IgG anti-A in duplicate, washed three times in PBS then diluted
to make a 20% suspension in saline. Two volumes of fresh, autologous serum
(from defibrinated blood) were mixed with one volume of 20% cell suspension

185
and incubated at 37°C for 30 minutes. The cells were washed three times
in PBS and resuspended in 1 ml HBSS before pipetting onto white cell mono-
layers. Duplicate cell samples were tested for complement binding by mixing
one volume of .anti-B1C/A (anti-C3) and reading the agglutination
microscopically.
The percentage of monocytes and neutrophils with ingested red cells
was estimated from examination of the coverslips.
RESULTS
1) Recovery of white cells
Approximately 70% of the white cells and 5% of the red cells present
in the original whole blood sample were recovered in the supernatant after
dextran sedimentation. Assuming that all of the white cells adhered to the
coverslips there would be about 2 x 106 white cells per coverslip. Usually
either 0.1 or 0.05 ml (1.1 x 109 or 0.5 x 109) red cells were added and so
the number of available red cells should not limit the amount of erythro-
phagocytosis.
2) Microscopic examination
Providing that the staining of the coverslips was of good quality the
cell types were easily identifiable and white cells with enclosed erythrocytes
could be readily discerned. A technique of this type is open to criticism
because it is sometimes difficult to differentiate between attached and
internalized particles on a fixed preparation. However, examination of slides
prepared from white cell monolayers which had been incubated with untreated
red cells did not reveal any 'apparent' phagocytosis due to artifacts
resulting from the nature of the white cell preparation.

186
3) Measurement of the phagocytosis of red cells opsonized with IgG antibodies
a) Ingestion of red cells coated with anti-D
The relationship between the amount of 125I-labelled anti-D bound to
red cells and the degree to which they were subsequently ingested by monocytes
in leucocyte monolayers is shown in Fig V-1(a). There was very little
measurable ingestion of red cells coated with less than 1 pg anti-D/ml
(approximately 350 molecules / red cell). When there was between 1.0 and
7.5 pg/ml (350 to 2,600 molecules / red cell) the percentage of monocytes
which had ingested erythrocytes increased in a roughly linear manner.
Antibody opsonization in excess of 2,600 molecules / red cell did not
increase the percentage of ingesting monocytes above approximately 60%.
There was no evidence of erythrophagocytosis in the neutrophil population.
The results obtained when the amount of bound antibody was also measured
indirectly using labelled anti-IgG (shown in Fig V-1(b)) and were in good
agreement with those obtained when the amount of bound antibody was measured
using a labelled anti-D. There was an increase in the number of ingesting
monocytes when the amount of bound antibody was between 0.5 and 10 pg anti-D/ml
red cells up to a maximum of approximately 60% of the monocyte population.
b) Ingestion of red cells coated with anti-A
The results are shown in Fig V-2(a).
Red cells with less than 3 pg anti-A bound/ml cells (approximately
1,000 molecules/red cell) were not ingested by monocytes or neutrophils in
the experiments in this study. There was an increase in the number of
phagocytosing monocytes in response to red cells coated with between 3 and
10 pg anti-A/ml cells (corresponding to between 1,000 and 3,500 molecules/
red cell). Red cells coated with even larger amounts of antibody (20 to
140 pg/ml cells) did not induce further phagocytosis in the monocyte
population. There was an increase in the percentage of phagocytosing

187
neutrophils from about 6% when there was 16 gg anti-A/ml cells (5,500
molecules/red cell) to a maximum of about 30% at an opsonizing dose of 70 gg/ml
(24,200 molecules/red cell). There was very little ingestion of red cells
opsonized with less than 3,500 molecules anti-A per red cell by neutrophils.
The use of the labelled anti-IgG reagent was not really valid for
the experiments using anti-A because the anti-IgG preparation had been
calibrated using an IgG anti-D. However, the results (shown in Fig V-2(b))
compare very well with those obtained using a direct measurement. There
was ingestion of red cells coated with 1.2 gg anti-A/ml cells in about 4%
monocytes increasing to a maximum of just under 50% when the red cells were
opsonized with 15 gg anti-A/ml cells. There was very little ingestion of
red cells coated with less than 15 gg anti-A/ml cells by neutrophils.
4) Measurement of the effect of plasma constituents on erythrophagocytosis
a) Measurement of phagocytosis in plasma and serum
There was no visible phagocytosis of anti-D opsonized red cells
on incubation with white cell monolayers in the presence of plasma or serum.
There was more than 50% saturation of the D sites on the red cells with
anti-D and on incubation with white cells in tissue culture media free of
IgG there was pronounced ingestion of the red cells by monocytes.
b) Measurement of phagocytosis in the presence of IgM
The presence of IgM had no measurable effect on the phagocytosis of
anti-D opsonized erythrocytes by monocytes. When there was no IgM present
75% of the monocyte population had ingested red cells. In the presence of
0.015, 0.12 and 0.5 mg IgM myeloma protein there was 66, 70 and 65% of the
monocytes with ingested erythrocytes respectively, a slight decrease but
probably within the experimental error for the experiment.

188
Fig V-1(a) The relationship between the amount of 125I-labelled
anti-D bound to red cells and the degree to which they were subsequently
ingested by monocytes after incubation at 37°C for 60 minutes
Fig V-1(b) The percentage of monocytes with ingested red cells
after incubation at 37°C for 60 minutes with red cells which had been
coated with various amounts of IgG anti-D. The amount of bound anti-D
was measured using an 125I-labelled anti-IgG reagent
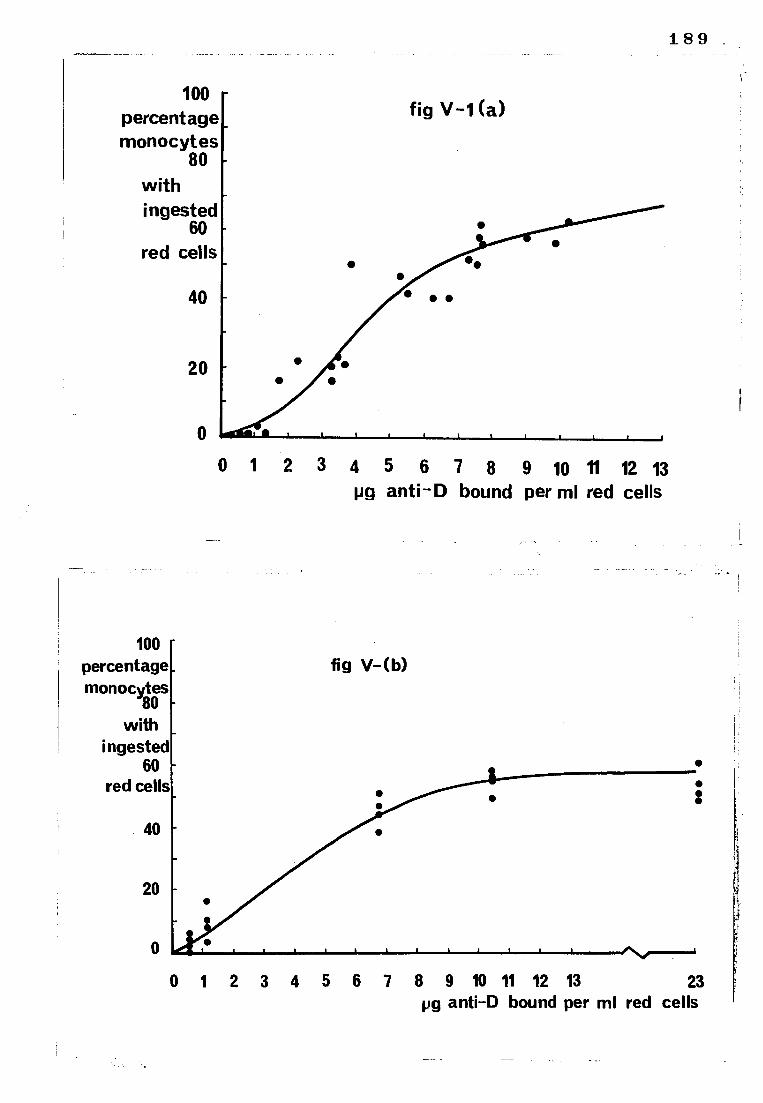
100 percentage monocytes
80
with
ingested 60
red cells
fig V-1 (a)
100 percentage monocytes
80 with
ingested 60
red cells
fig V- (b)
189
0 1 4 5 6 7 8 9 10 11 12 13 pg anti-D bound per ml red cells
• • • •
0 1 2 3 4 5 8 9 10 11 12 13 23 pg anti-D bound per ml red cells

190
Fig V-2(a) The relationship between the amount of 125I-labelled
anti-A bound to red cells and the degree to which they were subsequently
ingested by monocytes and neutrophils after incubation at 37°C for
60 minutes
Fig V-2(b) The percentage of monocytes and neutrophils with ingested
red cells after incubation at 37°C for 60 minutes with red cells which
had been coated with various amounts of IgG anti-A. The amount of
bound anti-A was measured using an 125I-labelled anti-IgG reagent
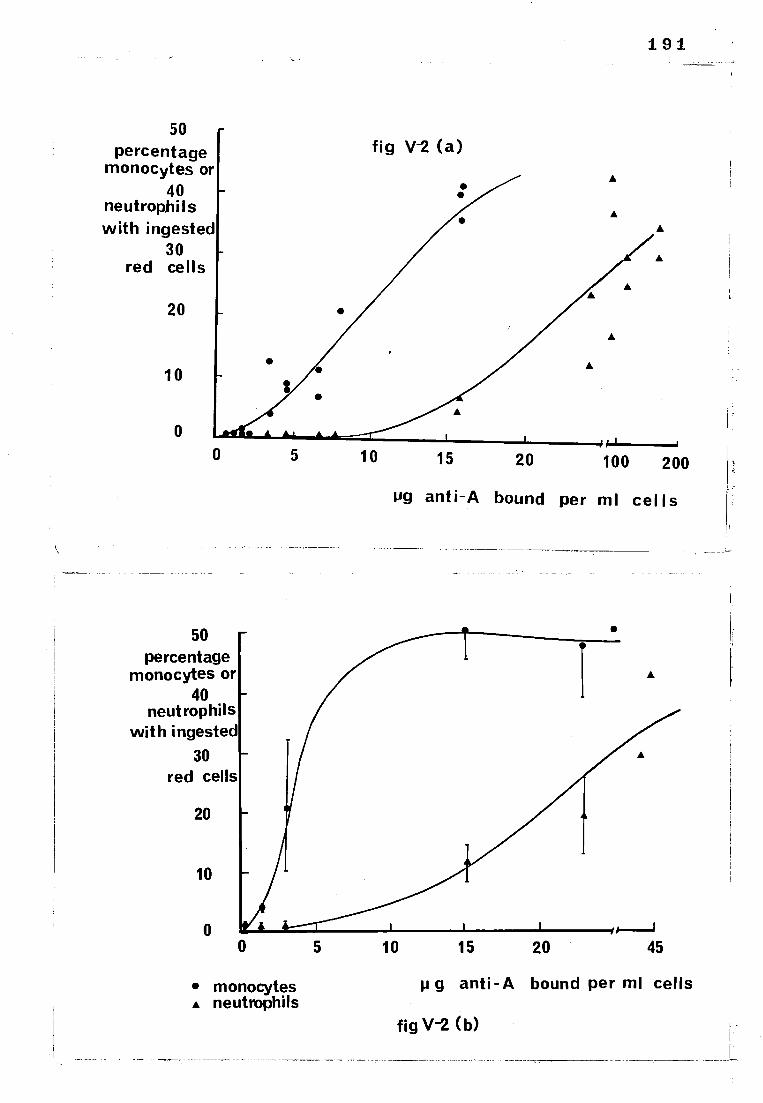
191
50 percentage
monocytes or 40
neutrophi Is with ingested
30 red cells
20
10
0 0 5 10 15 20 100 200
ug anti-A bound per ml cells
fig V-2 (a)
• •
•
50 percentage
monocytes or 40
neutrophils with ingested
30 red cells
20
10
0
• monocytes • neutrophils
5 10 15 20 45
p g anti-A bound per ml cells
fig V-2 (b)
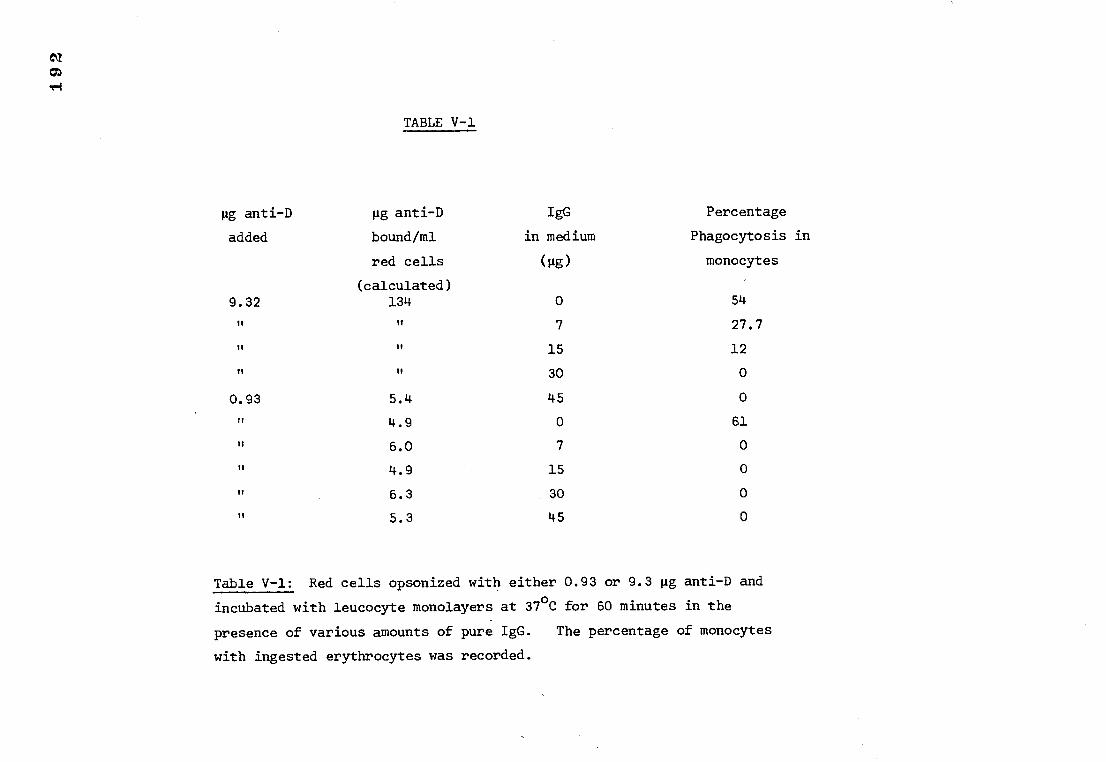
TABLE V-1
pg anti-D
added
pg anti-D
bound/ml
red cells
(calculated)
IgG
in medium
(pg)
Percentage
Phagocytosis in
monocytes
9.32 134 0 54
II TT 7 27.7
u 11 15 12
11 30 0
0.93 5.4 45 0
4.9 0 61
6.0 7 0
u 4.9 15 0
It 6.3 30 0
u 5.3 45 0
Table V-1: Red cells opsonized with either 0.93 or 9.3 pg anti-D and
incubated with leucocyte monolayers at 37°C for 60 minutes in the
presence of various amounts of pure IgG. The percentage of monocytes
with ingested erythrocytes was recorded.

193
c) Measurement of phagocytosis in the presence of various
concentrations of IgG
Red cells were opsonized with either 0.93 or 9.3 pg anti-D and
incubated with leucocyte monolayers in the presence of 0, 7, 15, 30, or
45 gg IgG. The results are shown in Table V-1. It appears that red cells
coated with almost saturating amounts of anti-D are able to overcome the
inhibitory effect of IgG to a small extent. However, red cells coated
with anti-D to the same extent as those found in haemolytic disease of the
newborn (i.e. between 0.5 and 17 pg/ml red cells) were not ingested in the
presence of IgG even at concentrations of IgG as low as 7 pg/ml. The
normal physiological level of IgG in the serum is between 6 and 12 mg/ml.
5) Red cell agglutination at low levels of bound antibody and the effects
of agglutinated red cells on phagocytosis
a) Red cell agglutination in plasma at low levels of IgG antibody
sensitization
Red cells coated with between approximately 500 and 2,700 molecules of
IgG anti-D per red cell or between 110 and 1,000 molecules IgG anti-A per
red cell formed clumps in plasma after 60 minutes incubation at 37°C. The
results are shown in Table V-2 and V-3. There appears to be a difference
in the extent to which the cells agglutinated in plasma from different donors.
If the incubation of the red cells in plasma was performed at room
temperature there was still agglutination although not as pronounced as after
a 37°C incubation using the same plasma sample. The red cells did not
agglutinate when the plasma was replaced by saline.
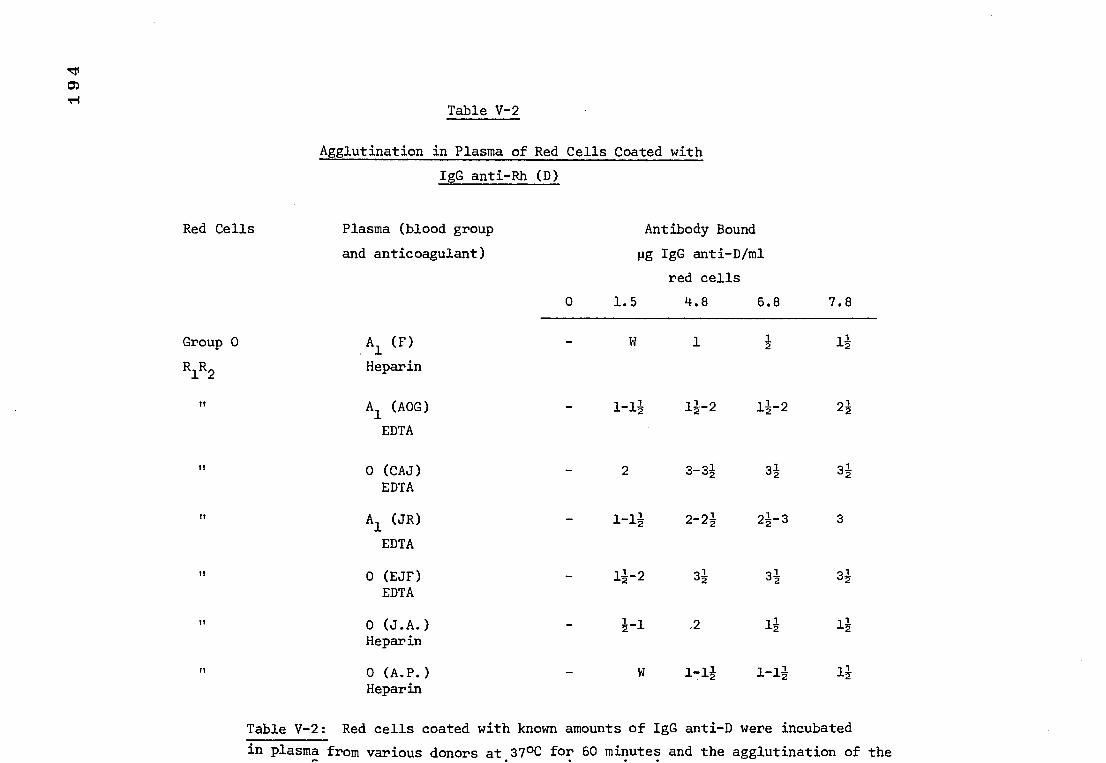
Table V-2
Agglutination in Plasma of Red Cells Coated with
IgG anti-Rh (D)
Red Cells Plasma (blood group
and anticoagulant)
0
Antibody Bound
mg IgG anti-D/ml
red cells
1.5 4.8 6.8 7.8
Group 0 Al (F) 2 11
R1R2 Heparin
Al (AOC) 1-11 11-2 11-2 21
EDTA
0 (CAJ) 2 3-3i 32 31 EDTA
Al (JR) 1-11 2-2a 21-3 3
EDTA
0 (EJF) 11-2 3i 31 31 EDTA
0 (J.A.) 1-1 ,2 11 11 Heparin
fl 0 (A.P.) 1-11 1-11 11 Heparin
Table V-2: Red cells coated with known amounts of IgG anti-D were incubated
in plasma from various donors at 370C for 60 minutes and the agglutination of the • •
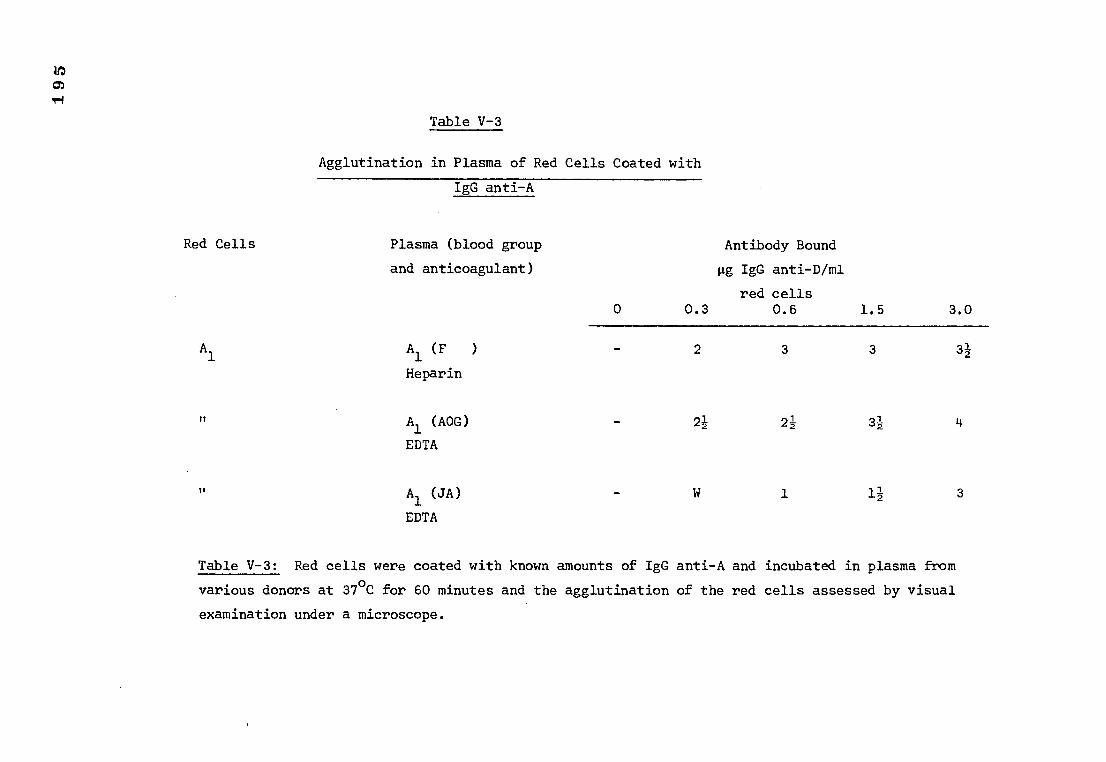
Table V-3
Agglutination in Plasma of Red Cells Coated with
IgG anti-A
Red Cells Plasma (blood group Antibody Bound
and anticoagulant)
pg IgG anti-D/ml
red cells 0 0.3 0.6 1.5 3.0
Al Al (F )
2 3 3
Heparin
11
Al (AOC) 21 21 32
EDTA
11
Al (JA) 1 12 3
EDTA
Table V-3: Red cells were coated with known amounts of IgG anti-A and incubated in plasma from
various donors at 37°C for 60 minutes and the agglutination of the red cells assessed by visual
examination under a microscope.

196
The agglutinates dispersed on shaking and did not reform fully after
standing for 1 hour at room temperature.
b) Measurement of the phagocytosis of agglutinated red cells
in serum
(i) Rh positive red cells opsonized with saturating amounts of
anti-D agglutinated readily in serum at 37°C. Neither the agglutinates
nor the free red cells were phagocytosed by the monocytes in a serum medium.
There did appear to be adherence, however, particularly between the
neutrophils and the agglutinated red cells, although this could merely
be an artifact resulting from the microscopic examination of a monolayer
of cells because agglutinated red cells tend to obscure the white cells in
these preparations.
(ii) Group A Rh positive cells agglutinated in group 0 serum
(containing anti-A). They were not phagocytosed however, even though
they had also been opsonized with anti-D.
6) The effect of complement on erythrophagocytosis
Red cells coated with anti-D did not bind complement in the experiments
described in the present study. Although IgG anti-D is not known to activate
complement, the experiments were carried out in case complement was bound at
levels which were not detectable in vitro but were nevertheless able to
potentiate ingestion. There was however no visible incase or decrease in
the number of monocytes which ingested erythrocytes incubated in fresh serum
compared with control red cells which had not been incubated in the presence
of complement components.

1 9 7
Red cells coated with IgG anti-A did not bind complement,
as measured by a positive reaction with an anti-B1C/A (anti-C3) serum,
until there was at least 15 pg anti-A bound / ml red cells. Even when
complement was bound there was no marked effect on the number of phagocytosing
monocytes or neutrophils although microscopic examination did reveal an
increase in the number of red cells attached to leucocytes.
DISCUSSION
The response of leucocyte monolayers to IgG anti-D and IgG anti-A
opsonized red cells
At least some of the cells of the reticuloendothelial system possess
a mechanism able to recognise antibody (or complement components) which
have combined with antigen. This recognition mechanism is visualised as
consisting of receptor sites, probably proteins, on the plasma membrane
which are able to interact with specific molecules (antibodies or complement
components) resulting in the binding of those molecules to the leucocyte
surface; subsequently, internalisation of the molecules and anything to
which they may be attached may take place.
The results of this study indicate that only monocytes have receptors
for IgG anti-D. In contrast, both monocytes and neutrophils have receptors
for IgG anti-A. It is possible that there are 2 types of binding sites on
IgG molecules, not always present at the same time, one reacting with
monocytes and the other with neutrophils. It has been reported by various
groups that IgG anti-D antibodies have failed to bind to neutrophils
(Lobuglio et al., 1967; Zipursky and Brown, 1974) and it has been inferred

198
from this that neutrophils do not have receptors for IgG. However, there
is evidence, apart from the present study, indicating that there are IgG
receptors on neutrophils although they may differ in certain aspects from
those on monocytes. Messner and Jelinek (1970) found that neutrophils can
ingest certain IgG opsonized bacteria and red cells. Ishizaka et al. (1970)
using radiolabelled immunoglobulins, have detected IgG molecules and IgG
receptors on the surface of neutrophils. In fact Ishizaka's findings
indicate that neutrophils bind IgG more firmly than monocytes.
There is some evidence which suggests that white cell receptors are
specific for IgG subclasses 1 and 3. Holm et al. (1974) tested several
human sera containing mainly IgG 1 or 3 subclasses of anti-D for monocyte
mediated haemolytic activity. Their results suggest that there are receptor
sites for IgG 1 and 3 on monocytes. The finding that IgG 1 and 3 myeloma
proteins were strong and equally efficient inhibitors of haemolysis of IgG
1 and 3 sensitized erythrocytes suggests that the receptors on human monocytes
for IgG 1 and 3 are identical. Similarly, other groups (Huber et al., 1971
and Abramson et al., 1970b) have shown cross-inhibition by IgG 1 and 3 of the
binding of these proteins to monocytes. Whether or not monocytes have
different receptors for IgG subclasses 2 and 4 is still not known. Receptors
for IgG 1 and 3 have been demonstrated on human neutrophils by Messner and
Jelinek (1970) using conventional techniques. It is probable therefore
that both monocytes and neutrophils have receptor sites for IgG antibodies,
and failure to detect IgG sites on neutrophils in some instances is a
consequence of the experimental technique employed.

Table V-4
Antibody
anti -D
Amount of Antibody on Amount of Antibody on
Red Cells in Haemolytic Red Cells to Induce
Disease Ingestion in vitro
0.4-18 pg/m1 red cells 1-7.5 pg/m1 red cells
(Hughes-Jones et al., 1967) (for ingestion by monocytes)
anti-A
0.25-3.5 pg/m1 red cells
3-10 mg/m1 red cells
(Romano et al., 1973)
(for ingestion by monocytes)
2 10 pg/m1 red cells
(for ingestion by neutrophils)

200
Comparison of the amount of antibody on red cells in ABO and Rh haemolytic
disease and that required to induce phagocytosis in peripheral blood
leucocytes
The amount of antibody normally found on red cells in ABO and Rh
haemolytic disease are compared in Table V-4 with the amount of antibody
on red cells required to initiate ingestion by leucocytes in vitro.
It can be seen that antibody coated red cells in Rh haemolytic disease
have levels of antibody opsonization which would induce ingestion by monocytes
(at least in vitro). The antibody coated red cells in ABO haemolytic
disease however would only be ingested by about 1% of peripheral blood
monocytes under the experimental conditions used in the present study.
If erythrophagocytosis is the mechanism of sensitized red cell
destruction in vivo then it is probable that the conditions in which the
reaction takes place are more favourable than those in the laboratory -
there would, for example, be a higher proportion of viable leucocytes than
in an isolated preparation maintained in tissue cultrue medium - therefore
it is quite likely that the levels of antibody sensitization found in Rh
and ABO haemolytic disease would allow their ingestion by macrophages
if such a process occurs in vivo.
The inhibition of erythrophagocytosis by serum IgG
The experiments in the present study are in agreement with those of
Lobuglio et al. (1967) and Lay and Nussenzweig (1968) who also found that
IgG inhibits the ingestion of antibody coated red cells. IgM has little
or no effect at physiological concentrations. Lobuglio et al. (1967) and

201
Holm et al. (1974) reported that it is the Fc, not the F(ab)2 fragment
of the IgG which is inhibitory, suggesting that IgG binds to monocytes
through the Fc fragment. Lay and Nussenzweig (1968) indicated that the
degree of inhibition was dependent on the amount of antibody on the red
cells and the concentration of IgG in the incubation medium. The findings
reported here suggest that the phagocytosis of red cells coated with anti-D
to the same extent as those in Rh haemolytic disease would be inhibited by
as little as 7 pg IgG/ml. Since the normal serum level of IgG is between
6 and 12 mg/ml, erythrophagocytosis should be inhibited in normal blood.
Lobuglio et al. (1967) found that although binding to monocytes was not
influenced by the concentration of red cells in saline, binding was
increased in direct proportion to the red cell concentration in serum
and at high haematocrit values serum had little inhibitory effect.
Lobuglio suggested that a high antibody coated red cell: macrophage ratio
(as may occur in the spleen) might allow the ingestion of weakly sensitized rt
red cells despite the high in vivo concentration of IgG.
Agglutination of red cells sensitized with IgG anti-D and anti-A
Red cells coated with between 500 and 2,700 molecules anti-D/ red cell
or between 110 and 1000 molecules anti-A/ red cell agglutinated in plasma
after 60 minutes incubation at 37°C in vitro. The results of Mollison and
Cutbush (1955) and Jandl et al. (1957) and Hughes-Jones et al. (1957)
indicate that red cells coated with Rh antibodies are removed from the
circulation in the spleen, appreciable destruction in the liver occurs only
when there is more than 20 pg antibody/ml red cells. If sensitized red
- cells agglutinate in vivo it could lead to trapping of cells, especially
in the spleen, overcoming the inhibitory effect of serum IgG by erythro-
concentration (Lobuglio et al, 1967) and resulting in a slow destruction
of the red cells by splenic macrophages.

202
Mollison (1972) has pointed out that following the administration of
IgG anti-A to group A subjects, spontaneous agglutination in whole blood
samples obtained from the recipients is invariably seen. Crawford et al.
(1953) reported that freshly drawn blood from an infant with ABO haemolytic
disease formed large agglutinates when the blood was examined on an opal
glass tile. In contrast, blood from infants with Rh haemolytic disease
may not agglutinate when mixed with serum unless the serum is mixed with 30%
albumin (Witebsky, 1952). In the present study red cells coated with
similar amounts of antibody to those occurring in Rh haemolytic disease
readily agglutinated in plasma at 37°C after 60 minutes incubation, although
the extent of agglutination was variable between subjects. The agglutination
of anti-A-coated red cells was evident at lower levels of anti-A sensitization
indicating that plasma is more effective at enhancing agglutination due to
anti-A than anti-D. This finding is in accordance with the results of
Romano and Mollison (1975). The same authors were unable to demonstrate
agglutination of anti-D-coated cells in plasma on a tile, at room temperature
unless there was at least 50 pg anti-D/ml red cells. In the present study
red cells coated with 5 pg anti-D/ml red cells agglutinated in plasma but
only after 60 minutes incubation at 37°C, suggesting that perhaps the
agglutination of anti-D-coated cells in plasma is temperature dependent.
The effect of complement
Spiegelberg et al. (1963) reported that erythrocytes and bacteria are
more rapidly ingested by the reticuloendothelial system of liver and spleen
in the presence of complement. In addition Lay and Nussenzweig (1968)
found that the adherence of red cells coated with antibody and complement
to leucocytes was not inhibited by the presence of IgG in the incubation
medium.

203
However Rh antibodies do not bind complement and it is not possible
to detect complement binding to red cells coated with small amounts of
anti-A. In the present study at least 15 gg anti-A/ml red cells was
bound before there was a positive reaction with the anti-61C/A serum, '
suggesting that complement is not involved directly in the leucocyte mediated
destruction of antibody-coated red cells in haemolytic disease of the newborn
unless complement coating is too low to detect in vitro but sufficient for
the initiation of in vivo red cell destruction.
Speculations on the possible mechanism of red cell destruction in
haemolytic disease of the newborn
The problem of how antibody-coated red cells are destroyed in haemolytic
disease remains unsolved. Phagocytosis remains the most probable mechanism
if the problem of inhibition by plasma IgG can be explained. Complement
binding has to be ruled out in the case of anti-D and is unlikely for low
concentrations of anti-A. It has been demonstrated in the present study
that red cells coated with small amounts of anti-D and anti-A will agglutinate
in plasma at 37°C. If agglutination occurs in vivo it may be followed by
filtration and erythrostasis as suggested by Jandl (1965) or it may
encourage erythroconcentration in the spleen thus raising the antibody-
coated red cell:macrophage ratio and overcoming the inhibition to ingestion
by serum IgG.
The possibility that erythrophagocytosis is a means of red cell
removal is supported by the recent experiments of Von Dem Borne et al. (1977).
The latter authors prepared a F(ab')2G anti-D by removing the Pc part of an
IgG anti-D by pepsin digestion. F(ab')2G anti-D-coated red cells did not
adhere to monocytes, nor were they eliminated at an accelerated rate on

204
injection in vivo into volunteers (in contrast to the rapid elimination
of IgG anti-D-coated cells under similar circuMstances). Von Dem Borne's
experiments indicate that the mechanism by which anti-D-coated red cells
are eliminated in vivo does not function if the Fc part of the immunoglobulin
molecule is not intact. It could be argued that complement binding will
not take place unless the Fc part of the IgG molecule is intact but complement
binding is unlikely particularly in the case of anti-D and therefore this
argument has little in its favour. The experiments of Von Dem Borne et al.
(1977) indicate that interactions between white cell receptors and the Fc
fragment of IgG bound to red cells are a likely mechanism of red cell
destruction in haemolytic disease.
There remains the question of whether red cell elimination is intra-
cellular. Lay and Nussenzweig (1968) and Lobuglio et al. (1967) have made
electron micrographs of rosettes on monocytes and neutrophils. Finger-like
cytoplasmic extensions were seen to extend from the leucocytes into the red
cells. The erythrocytes showed several points of adhesion to the white cells
and sometimes appeared deformed and fragmented. In addition, Lobuglio
et al. (1967) and Abramson et al. (1970a) found that bound red cells rapidly
change shape to become spherocytes and accompanied with this change there
was a shift in the osmotic fragility curve resulting in the cells becoming
sensitive to hypotonic lysis. Leucocytes may therefore destroy red cells
by ingestion and intracellular digestion or by attachment and extracellular
lysis. Spherocytes are not a notable feature of the peripheral blood of
neonates suffering from Rh haemolytic disease (although they are found in
infants suffering from ABO haemolytic disease). In addition rosette
formation was not commonly seen in the present study when white cells were
incubated with lightly sensitized red cells. It seems more likely that at
low levels of antibody sensitization red cells are ingested and the haemoglobin
metabolised intracellularly.

205
The observed in vitro inhibition of red cell ingestion by serum
IgG is still a mystery. There is one basic reaction:
F(abt)2-Fc + White cell > Fc-White cell
If there was only this one basic reaction and antibody bound to red cells
had to compete on equal terms with serum IgG for the white cell receptors
then it is unlikely that erythrophagocytosis could take place. Therefore
for the purposes of this discussion it is assumed that there are two
reactions which can be considered independently:
Red cell-IgG + White cell 'Red cell-IgG-White cell
Plasma IgG + White cell >IgG-White cell • • • • 2
If erythrophagocytosis is the method of red cell removal in vivo
then the equilibrium of reactions 1 and 2 must differ from that observed
in vitro. If the equilibrium of the reactions differs, then, either the
physiological conditions in which the reactions take place are dissimilar
or alternatively the reactants are not in the same physical form in both
situations.
Considering firstly that possibly the physiological conditions are
different, the temperature and media were identical (when the experiments
were carried out in serum or plasma) and therefore there must be something
about the spleen which is not simulated in vitro. It is difficult to
imagine how the tissue matrix could encourage the preferential binding of
antibody-coated red cells unless the Fc-receptors on splenic macrophages
differ from those on peripheral blood monocytes, for example splenic
macrophages are more mature than peripheral blood monocytes. Van Loghem
et al. (1977) compared the amount of IgG necessary to inhibit the adherence
of IgG anti-D-coated red cells to freshly collected monocytes with the

206
amount needed to inhibitcultivated (older) monocytes. Much more IgG was
needed to inhibit the more mature monocytes. Alternatively, perhaps
ingestion is aided by the flow of blood in the spleen. On a glass cover-
slip the media is stationary and there is equal opportunity for
the serum IgG and that attached to red cells to bind to leucocytes, since
there is much more serum IgG it will occupy virtually all of the white cell
receptors. In contrast, in the spleen, although the red cells are
concentrated, the plasma continually passes through the organ. As a result
the red cells coated with antibody remain in the same area as the macrophages
much longer than the plasma IgG which is continually moving. Perhaps the
probability of a macrophage Fc receptor colliding with an antibody-coated
red cell (which incidentally has its Fc receptor directed at approaching
objects) in preference to a moving serum IgG molecule is higher in vivo
than observed under stationary conditions experimentally.
Alternatively, IgG may exist in a form in vivo which differs from that
in which it is found in vitro. For example, IgG aggregates form readily
in vitro, it is just possible that the experiments in the present study
(and similar experiments condticted by other workers) have been measuring
the inhibition of red cell ingestion by aggregated IgG. Perhaps in vivo
the proportion of IgG aggregates in the spleen is small and the equilibrium
constant for the combination of free IgG with white cell receptors is much
smaller than that for aggregated IgG. Under such circumstances the turnover
of IgG molecules on the white cell would be much faster in vivo and
sensitized red cells would be able to compete more effectively for Fc
receptors than they would with the more strongly binding aggregated IgG.
Thus the inhibition of antibody-coated red cell ingestion by IgG in vitro
may be an artifact resulting from the artificial conditions under which
phagocytosis experiments are carried out.

207
SUMMARY AND CONCLUSIONS : CHAPTERS II, III, IV AND V
CHAPTER II
The action of phospholipase A2, from bee venom, Crotalus terr. terr.
and Vipera russellii, and phospholipase C (C. perfringens) on isolated
red cell membranes resulted in the degradation of membrane phospholipids
and a decline in Rh antigenic activity as measured by the amount of
1251-labelled anti-D that was able to bind to the membranes. These
results are consistent with the findings of Green (1972) that intact
phospholipids are essential for the maintenance of Rh antigen activity.
Phospholipase C (C. perfringens) lyses intact red cells. In
contrast phospholipase A2 from bee venom or Naja Naja did not lyse
intact red cells, neither did they cause a decline in Rh activity at
doses which reduced Rh activity in stroma, and there was not any
measurable phospholipid degradation either. This probably reflects
the insensitivity of the phospholipid detection technique together with
the relative inefficiency of these enzymes when attacking intact cells
compared to isolated membranes (Zwaal et al., 1975). The experiments
with intact red cells illustrate that the phospholipid molecules which
are responsible for the maintenance of Rh antigenic activity are not
present on the surface of the red cell in a manner in which they can
readily be attacked by phospholipase A2 from Naja Naja or bee venom.
In addition, the finding that phospholipase enzymes act differently with
intact red cells compared to isolated membrane preparations shows that
red cell stroma is not necessarily identical to its intact counterpart
in the red cell in conformation or reaction.

208
Attempts to reactivate Rh antigenic activity by removing and
replacing or resynthesizing the membrane phospholipids damaged by
phospholipase A2 all failed. Experiments using 32P-labelled
phospholipids demonstrated that there was exchange between the lipids
in the media and those in the membrane. Three times as much 32P-
labelled lecithin was incorporated into phospholipase A2-treated
membranes compared to untreated membranes. The failure to restore
Rh activity could be explained if either there was not enough replace-
ment of the enzyme altered lipid, or if the lysophospholipids had an
irreversible destructive action on the D antigen, alternatively it is
possible that there were contaminants in the phospholipase preparations
that attacked the D antigen by some activity other than phospholipid
degradation.
CHAPTER III
The molecular sizes of the Rh D, C, c and e antigens as determined
by irradiation inactivation were 174,000, 191,000, 194,000 and 221,000
daltons respectively. There were insufficient data for the C, c and e
antigens to determine accurately whether the molecular sizes of all the
Rh antigens were identical but they were certainly similar.
In general terms this similarity in molecular weight can be
interpreted in two ways. All the Rh antigens could be located on the
same molecule (or aggregated to form a complex of M.W. 200,000 daltons).
Alternatively, each antigen could be located on a separate molecule of
molecular weight 200,000 daltons.

209
Irradiation inactivation measures the molecular size of a target
in situ and therefore the true significance of the results cannot be
fully appreciated until the Rh antigens have been isolated from the
red cell membranes and investigated. Abraham and Bakerman (1975b,
1974 and 1976) claim to have isolated the C, c and E antigens as well
as the D antigen (1975a). The data from Abraham and Bakerman were
considered together with the findings of the present study and found
to be consistent with a molecular arrangement in which either each of
the Rh antigens, as expressed in the cell phenotype, are on one molecule
of molecular weight 200,000 daltons, or they are present as small
subunits aggregated to form a complex or polymer of molecular weight
200,000 daltons.
The findings of Abraham and Bakerman have not been verified by
other workers and until they are, or new evidence is published, any
proposed model must be speculative.
CHAPTER IV
Treatment of red cell membranes with the bile salt sodium deoxy-
cholate results in the release of protein and lipid molecules which are
able to reaggregate after the removal of deoxycholate. The lipoprotein
aggregates were able, under the experimental conditions employed, to
bind IgG anti-D. Up to 13% of the original antigenic activity, in terms
of the amount of 125
I-labelled anti-D taken up by the reaggregated
'membranes' was regained after dialysis. If the Rh (D) antigen was
treated with 125I-labelled anti-D before solubilizing, approximately 25%
of the counts were found in the soluble protein fractions and of this

210
approximately half were recovered in the reaggregated protein giving
a total recovery of about 10% of the original counts on the untreated
red cell membranes. Therefore, at least a percentage of the labelled
antibody remained in combination with the antigen after treatment with
sodium deoxycholate and consequently the binding of labelled antibody
was used as a marker for the D antigen in purification procedures.
The utilisation of this fact led to the demonstration that the D antigen-
antibody complex was eluted from Sepharose 4B at a position where
molecules of molecular weight 800,000 daltons would be expected. This
value is only approximate because it has been suggested (Helenius and
Simons, 1972) that deoxycholate binds to proteins with various degrees
of affinity and therefore it is not possible to calculate the amount
of deoxycholate bound to the antigen-antibody complex.
Attempts to purify the antigen by passing the solubilized proteins
down a column of cyanogen bromide-activated Sepharose 4B to which anti-
Ig G had been attached, and dissociating the antigen-antibody complex
bound to the anti-IgG by acid elution at successively decreasing pH
were inconclusive. Two bands in addition to those normally associated
with reduced IgG could be demonstrated on polyacrylamide gels in a
position of approximately 200,000 daltons. However bands in the same
position could be seen on polyacrylamide gels of IgG if large amounts
(150 pg) were electropheresed and therefore it could not be said
conclusively that the eluates from the S-CNBr-anti-IgG contained any
material other than IgG.

211
CHAPTER V
1) Receptors for IgG were shown to be present on monocytes by the
ability of these cells to bind and ingest erythrocytes coated with
IgG anti-D or anti-A in vitro.
2) Anti-A-coated red cells were ingested by neutrophils suggesting
the presence of an IgG receptor on these leucocytes, although, in
contrast, anti-D-coated red cells were not ingested by neutrophils
in the experiments described in this study.
3) Up to 60% of the monocyte population ingested red cells with
between 1 and 7.5 gg anti-D bound/ml red cells. In Rh haemolytic
disease red cells are normally coated with 0.4-18 pig anti-D/ml cells
(Hughes-Jones et al., 1967), and would therefore be ingested if the
same conditions prevailed in vivo as in vitro.
4) The minimum amount of bound anti-A required to opsonize erythrocytes
for ingestion by monocytes was 3 pg/m1 red cells and by neutrophils was
10 pg/m1 cells. In ABO haemolytic disease Romano et al. (1973) found
that there was normally between 0.25 and 3.5 pig/ml red cells. It is
therefore possible that lightly sensitized red cells could be ingested
in vivo particularly, if, as suggested by Von Loghem et al. (1977),
splenic macrophages have more Fc receptors than peripheral blood monocytes.
5) Normal serum levels of IgG inhibit the in vitro binding of red cells
sensitized with similar amounts of anti-A and anti-D to those found in
haemolytic disease of the newborn. Inhibition by IgG could be partially
overcome by increasing the amount of antibody bound to the red cells.

212
6) Red cells coated with as little as 520 molecules IgG anti-D or
110 molecules IgG anti-A formed small clumps in plasma after 60 minutes at
37°C which suggests that red cells in the neonate suffering from haemolytic
disease may agglutinate in vivo in areas of the circulation in which the
turbulence is not too great.
7) It seems unlikely that complement plays an active role in the
destruction of antibody sensitized red cells in haemolytic disease because
Rh antibodies rarely bind complement and neither does anti-A at the low
levels of antibody-coating normally found in affected infants. It is
however conceivable that complement binds to lightly sensitized red cells
at levels that cannot be detected in vitro but result in vivo in red cell
destruction.
8) In conclusion it is difficult considering the available evidence to
indicate, with confidence, the manner in which red cells are removed from
the circulation in haemolytic disease of the newborn. Erythrophagocytosis
remains the most likely possibility particularly in the light of the
experiments by Von Dem Borne et al. (1977) in which it was shown that the
Fc fragment of the IgG molecule must be intact if antibody-coated red cells
are to be removed from the circulation at an accelerated rate. The observed
inhibition of phagocytosis by serum IgG in vitro is difficult to explain
satisfactorily, however, it is not hard to appreciate that the physiological
conditions in the spleen under which the ingestion would occur might differ
considerably from the conditions simulated on a coverslip in the laboratory.
Thus it is proposed that the conditions prevailing in the spleen, physical
or biochemical, act such that the inhibition by serum IgG is overcome and
red cell ingestion may take place.

REFERENCES 213
Abraham, C.V. and Bakerman, S. (1974) Isolation and purification
of Rh(c) antigen. Biochim. Biophys. Acta., 365, 445.
Abraham, C.V. and Bakerman, S. (1975a) Isolation and purification
of the Rh (D) blood group receptor component from human erythrocyte
membrane. Clinica Chimica Acta, 60, 33.
Abraham, C.V. and Bakerman, S. (1975b) Isolation and purification
of Rh(C) antigen. Clinica Chimicia Acta, 59, 129.
Abraham, C.V. and Bakerman, S. (1976) Isolation of the Rh(E)
antigen. Biochim. Biophys. Acta, 420, 221.
Abramson, N., Lobuglio, A.F., Jandl, J.H. and Cotran, R.S. (1970a)
The interaction between human monocytes and red cells.
Binding Characteristics. J. Exp. Med., 132, 1191.
Abramson, N., Gelfland, E.W., Jandl, J.H. and Rosen, F.S. (1970b)
The interaction between human monocytes and red cells.
Specificity for IgG subclasses and IgG fragments.
J. Exp. Med., 132, 1207.
Abt, A.F. (1931) Mononuclear erythrophagocytosis in the blood
of a new-born infant. Am. J. Dis. Child., 42, 1364.
Al-Ibrahim, M.S., Chandra, R., Koshore, R., Valentine, F.T. and
Sherwood-Lawrence, H. (1976) A micromethod for evaluating the
phagocytic activity of human macrophages by ingestion of radio-labelled
polystyrene particles. J. Immunol. Methods, 10, 207.

214
Allan, D., Low, M.G., Finean, J.B. and Michell, R.M. (1975)
Changes in lipid metabolism and cell morphology following attack
by phospholipase C (Clostridium perfringens) on red cells or lymphocytes.
Biochim. Biophys. Acta, 413, 309.
Anstee, D.J. and Tanner, M.S. (1975) Separation of ABH, I, Ss
antigenic activity from the MN-active sialoglycoprotein of the
human erythrocyte membrane. Vox Sang., 29, 378.
Bender, W.W., Garan, H. and Berg, H.C. (1971) Proteins of the
human erythrocyte membrane as modified by pronase.
J. Mol. Biol., 58, 783.
Berg, H.C. (1969) Sulphanilic acid diazonium salt : A label for
the outside of the human erythrocyte membrane. Biochim. Biophys.
Acta, 183, 65.
Berken, A. and Bencerraf, B. (1966) Properties of antibodies cytophilic
for macrophages. J. Exp. Med., 123, 119.
Bhakdi, S., Knilfermann, H. and Hoelzl-Wallach, D.F. (1974)
Separation of EDTA-extractable erythroCyte membrane proteins by
isoelectric focussing linked to electrophoresis in sodium dodecyl
sulphate. Biochim. Biophys. Acta, 345, 448.
Bigley, N.J., Chandler, R.W. and Dodd, M.C. (1958) Inhibition of
Rho(D) antibody by eluates from Rho(D) erythrocytes treated with mumps
virus or the periodate ion. J. Immunol., 80, 85.

215
Bogoch, S. (1958) Studies on the structure of brain ganglioside.
Biochem. J., 68, 319.
Bonnin, J.A. and Schwartz, L. (1954) The combined study of agglutination,
hemolysis and erythrophagocytosis with special reference to acquired
hemolytic anemia. Blood, 9, 773.
Boyd, W.C., McMaster, M.M. and Waszczenko-Zacharaczenko, E. (1959)
Specific inhibition of anti-Rh serum by 'unnatural' sugars.
Nature, 184, 989.
Boyd, W.C. and Reeves, E. (1961) Specific inhibition of anti-D antibody
by colominic acid. Nature, 191, 511.
Bretscher, M.S. (1971a) Major human erythrocyte glycoprotein spans
the cell membrane. Nature (New Biol.), 231, 229.
Bretscher, M.S. (1971b) Human erythrocyte membranes : speeific labelling
of surface proteins. J. Mol. Biol., 58, 775.
Calvin, M., Evans, R.S., Behrendt, V. and Calvin, G. (1946)
Rh antigen and hapten. Nature of antigen and its isolation from
erythrocyte stroma. Proc. Soc. Exper. Biol. and Med., 61, 416.
Chown, B., Kaira, Ii., Lowen, B. and Lewis, M. (1971) Transient production
of anti-LW by LW-positive people. Transfusion, 11, 220.

216
Colley, C.M., Zwaal, R.F.A., Roelofen, B. and Van Deenen, L.L.M. (1973)
Lytic and non-lytic degradation of phospholipids in mammalian erythrocytes
by pure phospholipases. Biochim. Biophys. Acta, 307, 74.
Colman, R., Finean, J.B., Knutton, S. and Limbrick, A.R. (1970)
A structural study of the modification of erythrocyte ghosts by
phospholipase C. Biochim. Biophys. Acta, 219, 81.
Colman, R. and Bramley, T.A. (1975) Hydrolysis of erythrocyte membrane
phospholipids by a preparation of phospholipase C from clostridium
Welchii. Deactivation of (Ca2+, Mg2+)-ATPase and its reactivation by
added lipids. Biochim. Biophys. Acta, 382, 565.
Condrea, E., Barzilay, M. and Mager, J. (1970) Role of cobra venom
direct lytic factor and Ca2+ in promoting the activity of snake venom
phospholipase A. Biochim. Biophys. Acta, 210, 65.
Conrad, M.J. and Pennington, J.T. (1976) Resolution of erythrocyte
membrane proteins by two-dimensional electrophoresis.
J. Biol. Chem., 251, 253.
Cooper, M.B. (1950) Erythrophagocytosis in hemolytic disease of the
newborn: A report of twenty-five cases. Blood, 5, 678.
Crawford, H., Cutbush, M. and Mollison, P.L. (1953) Haemolytic disease
of the newborn due to anti-A. Blood, 8, 620.
Davis, M. and Pollard, E. (1952) The inact-ivation of dry penicillin
by deutron bombardment. Arch,Biochem. Biophys.,37 112

217
Dertinger, H. and Jung, M. (1970) Molecular Radiation Biology.
The English Universities Press, London.
Devaux, P. and McConnell, H.M. (1972) Lateral diffusion in spin-
labelled phosphatidylcholine multilayers. J. Amer. Chem. Soc., 94, 4475.
Dodd, M.C., Bigley, N.J. and Geyer, V.B. (1960) Specific inhibition
of Rho(D) antibody by sialic acids. Science, 132, 1398.
Dodd, M.C., Bigley, N.J. and Geyer, V.B. (1963) Further observations
on the serologic specificity of sialic acid for Rho(D) antibody.
J. Immunol., 90, 518.
Dodd, M.C., Bigley, N.J., Johnson, G.A. and McCluer, R.H. (1964)
Chemical aspects of inhibitors of Rho(D) antibody. Nature (London),
204, 549.
Dodge, J.T., Mitchell, C. and Hanahan, D.J. (1963) The preparation and
chemical characteristics of haemoglobin-free ghosts of human erythrocytes.
Arch. Biochim. Biophys., 100, 119.
Eagle, H. (1937) The coagulation of blood by snake venoms and its
physiological significance. J. exp. Med., 65, 613.
Ebert, M. (1973) Molecular Radiobiology. Br. Med. Bull., 29, 12.
Edwards, P.A.W. (1977) Reconstitution of glucose-transporting vesicles
from erythrocyte membranes disaggregated in detergent. Biochem. J.,
164, 125.

218
Ellman, G., Courtney, K.D., Andres, V. and Featherstone, R.M. (1961)
A new and rapid colorimetric determination of acetylcholinesterase
activity. Biochem. Pharmac., 7, 88.
Fairbanks, G., Steck, T.L., Wallach, D.F.H. (1971) Electrophoretic
analysis of the major polypeptides of the human erythrocyte membrane.
Biochem., 10, 2606.
Farquhar, J.W. and Ahrens Jr., E.H. (1963) Effects of dietary fats
on human erythrocyte fatty acid patterns. J. Clin. Invest., 42, 675.
Feber, E. (1973) Biological Membranes, Vol.2. Ed. D. Chapman and
D.F.H. Wallach. Academic Press.
Finean, J.B., Bramley, T.A. and Coleman, R. (1971) Lipid layer in
cell membranes. Nature (Lond.), 229, 114.
Fisher, B.D., Parker, C.W., Haynes, C.R. and Chaplin Jr., H. (1970)
Inhibition of erythrocyte antibody binding by a dialysate of
hemolysed red blood cells. Clinica. chimica. Acta, 30, 842.
Fisher, R.A., cited by Race, R.R. (1944) "An incomplete antibody
in human serum". Nature (Lond.), 153, 771.
Fisher, R.A. and Race, R.R. (1946) Rh gene frequencies in Britain.
Nature (Lond.), 157, 48.
Fisher, R.A. (1947) The Rhesus factor: a study in scientific method.
Am Science, 35, 95.

219
Fisher, R.A. (1953) Population genetics. Proc. Roy. Soc. B., 141, 510.
Folkerd, E.J., Ellory, J.C. and Hughes-Jones, N.C. (1977)
A molecular size determination of Rh (D) antigen by radiation
inactivation. Immunochemistry, 14, 529.
Frame, M. and Mollison, P.L. (1969) Charge differences between
human IgG isoantibodies associated with specificity.
Immunology, 16, 277.
Ginoza, W. (1963) Radiosensitive molecular weight of single-
stranded virus nucleic acids. Nature, 199, 453.
Glaser, M., Simpkins, H., Singer, S.J., Sheetz, M. and Chan, S.I.
(1970) On the interactions of lipids and proteins in the red
blood cell membrane. Proc. Nat. acad. Sci., 65, 721.
Green, F.A. (1965) Studies on the Rh(D) antigen. Vox Sang., 10, 32.
Green, F.A. (1968) Phospholipid requirement for Rh antigenic activity.
J. Biol. Chem., 243, 5519.
Green, F.A. (1972) Erythrocyte membrane lipids and Rh antigen activity.
J. Biol. Chem., 247, 881.

220
Gul, S. and Smith, A.D. (1974) Hemolysis of intact human erythrocytes
by purified cobra venom phospholipase A2 in the presence of albumin.and k
Gwynne, J.T. and Tanford, C. (1970) A polypeptide chain of very high
molecular weight from red blood cell membranes. J. Biol. Chem., 245,
3269.
Hackel, E., Smolker, R.E. and Fenske, S.A. (1958) Inhibition of
anti-Rh and anti-lutheran sera by ribonucleic acid derivatives.
Vox Sang. a, 402.
Hanfland, P. and Egli, H. (1975) Quantitative isolation and purification
of blood group-active glycosphingolipids from human B erythrocytes.
Vox Sang. 28, 438.
Helenius, A. and Simons, K. (1972) The binding of detergents to
lipophilic and hydrophilic proteins. J. Biol. Chem., 247, 3656.
Hokin, L.E. and Hokin, M.R. (1961) Diglyceride kinase and phosphatidic
acid phosphatase in erythrocyte membranes. Nature (Lond.), 189, 836.
Holm, G., Engwall, E., Hammarstrom, S. and Natvig, J.B. (1974)
Antibody-induced haemolytic activity of human blood monocytes.
Scand. J. Immunol., 3, 173.
Hopkins, J.G. (1910) Phagocytosis of red blood cells after transfusions.
Arch. Int. Med., 6, 270.
Ca2+. Biochim. Biophys. Acta, 367, 271.

221
Howe, C. (1951) The influenza virus receptor and blood group antigens
of human erythrocyte stroma. J. Immunol., 66, 9.
Huber, H., Fudenberg, H.H. (1968) Receptor sites of human monocytes
for IgG. Int. Arch. Allergy, 34, 18.
Huber, H., Fudenberg, H.H. (1970) The interaction of monocytes and
macrophages with immunoglobulins and complement. Ser. Haem., 3, 160.
Huber, H., Douglas, S.D., Nusbacher, J., Kochwa, S. and Rosenfield, R.E.
(1971) IgG subclass specificity of human monocyte receptor sites.
Nature (Lond.), 229, 419.
Hughes-Jones, N.C., Mollison, P.L. and Veall, N. (1957) Removal of
incompatible red cells by the spleen. Brit. J. Haematol., 3, 125.
Hughes-Jones, N.C. (1967) The estimation of the concentration and
equilibrium constant of anti-D. Immunology, 12, 565.
Hughes-Jones, N.C., Hughes, N.I.J. and Walker, W. (1967) The amount of
anti-D on red cells in haemolytic disease of the newborn.
Vox. Sang., 12, 279.
Hughes-Jones, N.C., Gardner, B. and Lincoln, P.J. (1971) Observations
of the number of available c, D and E antigen sites on red cells.
Vox Sang., 21, 210.
Hughes-Jones, N.C. (1974) Prophylaxe der Rhesus-Sensibilisierung;
The reproducability of the assay of anti-D. ed. Schneider, J. and Weitzel,
H., Die Medizinische Verlagsgesellschaft mbH. Marburg, p.113.

222
Hughes-Jones, N.C., Green, E.J. and Hunt,,V.A.M. (1975) Loss of Rh
antigen activity following the action of phospholipase A2 on red cell
stroma. Vox Sand. , 29, 184.
Ibrahim, S.A. and Thompson, R.H.S. (1965) Action of phospholipase A
on human red cell ghosts and intact erythrocytes. Biochim. Biophys.
Acta, 99, 331.
Ishizaka, K., Tomioka, H. and Ishizaka, T. (1970) Mechanisms of passive
sensitization 1. Presence of IgE and IgG molecules on human leukocytes.
J. Immunol., 105, 1459.
Jandl, J.H., Jones, A.R. and Castle, W.B. (1957)
The destruction of red
cells by antibodies in man. I. observations on the sequestration and
lysis of red cells altered by immune mechanisms. J. Clin. Invest.,
36, 1428.
Jandl, J.H. (1965) Mechanisms of antibody-induced red cell destruction
Series Haematologica, 9, 35.
Johnson, G.A. and McCluer, R.H. (1961) Relation of sialic acid to Rho(D)
antigen. Proc. soc. exp. Biol. and Med., 107, 692.
Johnson, H.M. and Dodd, M.C. (1964) Specific inhibition of anti-D by
human urinary mucoprotein. Nature, 203, 1082.
Johnson C.D. and Rymer T.B. (1967) Existancef-of collective excitation energy
losses from an electron beam passing throlugh biological materials. Nature 213 11445

223
Jordan Jr., W.S., Prouty, R.L., Heinle, R.W. and Dingle, J.H. (1952)
The mechanism of hemolysis in paroxysmal cold hemoglobin urea III.
Erythrophagocytosis and leukopenia. Blood, 7, 387.
Jurtshuk, Jr., P., Sekuzu, I. and Green, D.E. (1961) The interacIion
of the D(-) B- hydroxybutric apoenzyme with lecithin. Biochem Biophys.
Res Comm 6 76.
Kahlenberg, A. and Banjo, B. (1972) Involvement of phospholipids
in the D-glucose uptake activity of isolated human erythrocyte membranes.
J. Biol. Chem., 247, 1156.
Kates, M. (1972) Techniques of Lipidology. Isolation, analysis and
identification of lipids. (North Holland, Amsterdam, 1972).
Kay,kUiB. (1975) Mechanism of removal of senescent cells by human
macrophages in situ. Proc. Natl. Acad. Sci. USA, 72, 3521.
Kepner, G.R. and Macey, R.I. (1968) Membrane enzyme systems molecular
size determinations by radiation inactivation. Biochim. Biophys. Acta,
163, 188.
Kirkpatrick, F.H., Gordesky, S.E. and Marinette, G.V. (1974)
Differential solubilization of proteins, phospholipiariand cholesterol
of erythrocyte membranes by detergents. Biochim. Biophys. Acta, 345,
154.
Kornberg, R.D. and McConnel, H.M. (1971) Lateral diffusion of
phospholipids in a vesicle membrane. Proc. Natl. Acad. Sci., 68, 2564.

224
Landsteiner, K. and Wiener, A.S. (1940) An agglutinable factor in
human blood recognized by immune sera for rhesus blood.
Proc. soc. exp. Biol. N.Y., 43, 223.
Lauf, P.K. and Joiner, C.H. (1976) Potassium transport and ouabain
binding in human Rhnull red blood cells. Blood 48, 457.
Lay, W.H. and Nussenzweig, V. (1968) Receptors for complement on
leucocytes. J. Exp. Med., 128, 991.
Lea, D.E. (1955) Actions of radiations on living cells.
Cambridge University Press, London second edition.
Leddy, J.P., Whittemore, N.B. and Weed, J.I. (1970) Human erythrocyte
membranes : effect of lipid extraction on binding of IgG ('warm')
autoantibodies and Rh isoantibodies. Vox Sang., 19, 444.
Levine, P. and Stetson, R.E. (1939) An unusual case of intragroup
agglutination. J. Amer. Med. Ass., 113, 126.
Levine, P., Katzin, E.M. and Burnham, L. (1941) Isoimmunization in
pregnancy, its possible bearing on the etiology of erythroblastosis
fetalis. J. Amer. Med. Ass., 116, 825.
Levine, P., Vogel, P., Katzin, E.M. and Burnham, L. (1941)
Pathogenesis of erythroblastosis fetalis : statistical evidence.
Science, 94, 371.

225
Levine, P., Celano, M., Fenichel, R. and Singher, H. (1961)
A 'D-like' antigen in rhesus red blood cells and in Rh-positive
and Rh-negative red cells. Science, 133, 332.
Levine, P., Celano, M.J. Falkowski, F., Chambers, J., Hunter, Jr., O.B.
and English, C.T. (1965) A second example of ---/--- or Rhnull blood.
Transfusion, 5, 492.
Levinson, S.R. and Ellory, J.C. (1973) Molecular size of the
tetrodoxin binding site estimated by irradiation inactivation.
Nature New Biol., 245, 122.
Levinson, S.R. and Ellory, J.C. (1974) The molecular form of
acetylcholinesterase as determined by irradiation inactivation.
Biochem. J., 137, 123.
Lobuglio, A.F., Cotran, R.S. and Jandl, J.H. (1967) Red cells coated
with immunoglobin G : binding and sphering by mononuclear cells in man.
Science, 158, 1582.
Lorusso, D.J. and Green, F.A. (1975) Reconstitution of Rh(D) antigen
activity from human erythrocyte membranes solubilized by deoxycholate.
Science, 188, 66.
Lorusso, D.J. and Green, F.A. (1977) The Rh antigen system and
disaggregated human erythrocyte membranes. Immunochemistry, 14, 503.
Lovelock, J.E., James, A.T. and Rowe, C.E. (1960) The lipids of whole
blood 2. The exchange of lipids between the cellular constituents and
the lipoproteins of human blood. Biochem. J., 74, 137.

McFarlane, A.S. (1958) Efficient trace-labelling of proteins with 226
iodine. Nature (Lond.), 182, 53.
Mackay, I. (1939) Two cases of hematological interest-erythrophagocytosis
following blood transfusion. Brit. Med. J., 1, 494.
rt ” Makela, 0., Cantell, K. and Pentrinen, K. (1959) Failure of mumps
virus to inactivate agglutinogens M, N and Rho(D) of human red cells.
J. Immunol., 83, 127.
Makino, S., Reynolds, J.A. and Tanford, C. (1973) The binding of
deoxycholate and triton X-100 to proteins. J. Biol. Chem., 248, 4926.
Mantovani, B., Rabinovitch, M. and Nussenzweig , V. (1972)
Phagocytosis of immune complexes by macrophages. Different roles
of the macrophage receptor sites for complement (C3) and for immuno-
globulin (IgG). J. Exp. Med., 135, 780.
Marchesi, V.T. and Steers, Jr., E. (1968) Selective solubilization
of a protein component of the red cell membrane. Science, 159, 203.
Marchesi, V.T. and Andrews, E.P. (1971) Glycoproteins : isolation
from cell membranes with lithium diiodosalicylate. Science, 174, 1247.
Marinetti, G.V., Baumgarten, R., Sheeley, D. and Gordesky, S. (1973.)
Cross Linking of phospholipids to proteins in the erythrocyte membrane.
Biochim. Biophys. Res. Comm., 53, 302.
Martin, J.K., Luthra, M.G., Wells, M.A., Watts, R.P. and Hanahan, D.J. (1975)
Phospholipase A2 as a probe of phospholipid distribution in erythrocyte
membranes. Factors influencing the apparent specificity of the reaction.
Biochemistry, 14, 5400.

227
Messner, R.P. and Jelinek, J. (1970) Receptors for human TG-globulin
on human neutrophils. J. Clin. Invest., 49, 2165.
Mollison, P.L. and Cutbush, M. (1955) Use of isotope-labelled red
cells to demonstrate incompatibility in vivo. Lancet i, 1290.
Mollison, P.L. (1972) Blood Transfusion in Clinical Medicine
5th edition Blackwell Scientific Publications, Oxford.
Morgan, W.T.J. and Watkins, W.M. (1951) The inactivation of the blood
group receptors on the human erythrocyte surface by the periodate ion.
Brit. J. Exp. Path., 32, 34.
Moskowitz, M., Dandliker, W.B., Calvin, M. and Evans, R.S. (1950)
Studies on the antigens of human red cells 1. The separation from
human erythrocytes of a water soluble fraction containing the Rh, A
and B factors. J. Immunol., 65, 383.
Moskowitz, M. and Treffers, H.P. (1950) An agglutinin in normal sera
for periodate-treated red cells. Science, 111, 717.
Mulder, E. and Van Deenen, L.L.M. (1965) Metabolism of red cell lipids
III. Pathways for phospholipid renewal. Biochim. Biophys. Acta,
106, 348.
Mulder, E., Van Den Berg, J.W.O. and Van Deenen, L.L.M. (1965)
Metabolism of red cell lipids II conversions of lysophosphoglycerides.
Biochim. Biophys. Acta, 106, 118.

2 2
NiColson, G.L., Masouredis, S.P. and Singer, S.J. (1971) Quantitative
two-dimensional ultrastructural distribution of Rho(D) antigenic sites
on human erythrocyte membranes. Proc. Nat. Acad. Sci. USA, 68, 1416.
Northrop, J.H., Kunitz, M. and Herriott, R.M. (1948) Crystalline
Enzymes. Columbia University Press, New York.
Ore, A. and Larsen, A. (1964) Relative frequencies of ion clusters
containing various numbers of ion paris. Radiation Res., 21, 331.
Philippot, J. (1968) Reconstitution de l'activit6 de l'ATPase de
transport apres solubilization par le desoxycholate de sodium.
R6le des ions Na+ et k+ dans la structure du complexe Lipo-protelque
enzymatique. Bull. Soc. Chim. Biol., 50, 1481.
Philippot, J. (1971) Study of human red blood cell membrane using
sodium deoxycholate, I. mechanism of the solubilization.
Biochim. Biophys. Acta, 225, 201.
Phillips, D.R. and Morrison, M. (1970) The arrangement of proteins in
the human erythrocyte membrane. Biochim. Biophys. Res. Comm., 40, 284.
Pittman, J.G. and Martin, D.B. (1966) Fatty acid biosynthesis in human
erythrocytes : evidence in mature erythrocytes for an incomplete long chain
fatty acid synthesising system. J. Clin. Invest., 45, 165.
Pollard E.(1959) Radiation inactivation of enzymes, nucleic acids,
and phage particles. In Biophysical Science - A study program, Ed.
Oncley, J.L. New York: John Wiley & sons.

229
Poplack, D.G., Bannard, G.D., Holiman, B.J. and Blaese, R.M. (1976)
Monocyte-mediated antibody-dependent cellular cytotoxicity : A clinical
test of monocyte function. Blood, 48, 809.
Prager, M.D., Fletcher, M.A. (1962) Mechanism of the effect of trypsin
on specific hemagglutination with incomplete Rh antisera.
Proc. Soc. Exp. Biol. Med., 111, 722.
Prager, M.D., Hopkins, G. and Foster, M. (1963) The effect of
phosphatases and organic phosphates on Rh reactions. Vox Sang., 8, 410.
Prager, M.D. and Fletcher, M.A. (1966) The effect of enzymatic release
of sialic acid from human erythrocytes on Rh agglutinations.
J. Immunol., 97, 165.
Race, R.R., Taylor, G.L., Cappell, D.F. and McFarlane, M.N. (1944)
Recognition of a further common Rh genotype in man. Nature (Lond.),
153, 52.
Rauth, A.M. and Simson, J.A. (1964) The energy loss of electrons
in solids. Radiation Res., 22, 643.
Reed, C.F. (1959) Studies of in vivo and in vitro exchange of
erythrocyte and plasma phospholipids. J. Clin. Invest., 38, 1032.
Rochna, E. and Hughes-Jones, N.C. (1965) The use of purified 125I-labelled
anti-Y globulin in the determination of the number of D antigen sites on
red cells of different phenotypes. Vox Sang., 10, 675.

230
Romano, E.L., Hughes-Jones, N.C. and Mollison, P.L. (1973) Direct
antiglobulin reaction in ABO-haemolytic disease of the newborn.
Brit. Med. J., i, 524.
Romano, E.L. (1974) Interactions between red cell antigens and
corresponding antibodies, with special reference to ABO-haemolytic
disease. Studies with labelled antibodies and the electron microscope.
Ph.D. Thesis, London University.
Romano, E.L., Stolinski, C. and Hughes-Jones, N.C. (1975) Distribution
and mobility of the A, D and c antigens on human red cell membranes :
studies with a gold-labelled antiglobulin reagent. Brit. J. Haemat.,
30, 507.
Romano, E.L. and Mollison, P.L. (1975) Red cell destruction in vivo
by low concentrations of IgG anti-A. Brit. J. Haematol., 29, 121.
Rosenfield, R.E., Allen, Jr., F.H. and Rubinstein, P. (1973) Genetic
model for the Rh blood-group system. Proc. Nat. Acad. Sci. USA., 70,
1303.
Rosse, W.F. Rapp, H.J. and Borsos, T. (1967) Structural characteristics of
haemolytic antibodies as determined by the effects of ionizing radiation.
J. Immunol., 98 , 1190.
Rubin, M. (1963) Antibody elution from red blood cells. J. clin. Path.,
16, 70 •

231
Scatchard, G. (1949) The attractions of proteins for small molecules
and ions. Ann. N.Y. Acad. Sci., 51, 660.
Schmidt, P.J. and Vos, G.H. (1967) Multiple phenotypic abnormalities
associated with Rhnull (---/---). Vox Sang., 13, 18.
Schmidt, P.J., Lostumbo, M.M., English, C.T. and Hunter, 0.B. (1967)
Aberrant U blood group accompanying Rhnull'
Transfusion Philad.,
7, 33.
Schubert, D. (1973) Association of protein fractions and lipids from
human erythrocyte membranes, II. Hoppe-Seyler's Z. Physiol. Chem.,
354, 781.
Setlow, R.B. (1952) The radiation sensitivity of catalase as a function
of temperature. Pro. Natl. Acad. Sci. USA, 38, 166.
Setlow, R.B. and Doyle, B. (1953) The molecular weight of insulin
and the inactivation of insulin by fast charged particles.
Arch. Biochim. Biophys., 42, 83.
Singer, S.J. and Nicolson, G.L. (1972) The fluid mosaic model of the
structure of cell membranes. Science, 175, 720.
Smith, J.A., Lucus, Jr., F.V., Martin, A.P. Stenhauser, D.A. and Vorbeck,
M.L. (1973) Lipid-protein interactions of erythrocyte membranes :
comparison of normal 0, Rh(D) positive with the rare 0, Rhnuil.
Biochim. Biophys. Res. Comm., 54, 1015.

232
Spiegelberg, H.L., Miescher, P.A. and Benacerraf, B. (1963) Studies
on the role of complement in the immune clearance of Escherichia coli
and rat erythrocytes by the reticuloendothelial system in mice.
J. Immunol., 90, 751.
Spiegelberg, H.L. (1974) Biological activities of immunoglobulins
IX. Reaction with white blood cells-cytophilic antibodies.
Advances in Immunol., 19, 276.
Springer, G.F. and Tegtmeyer, H. (1964) Specific inhibition of anti-Rh
antibodies by extracts from twigs of angiospermous plants.
Nature (Lond.), 203, 298.
Steck, T.L. (1972) Cross-linking the major proteins of the isolated
erythrocyte membrane. J. Mol. Biol., 66, 295.
Steck, T.L. (1974) The organization of proteins in the human red
blood cell membrane. A review. J. cell Biol., 62, 1.
Stellner, K., Wantanabe, K. and Hakomori, S. (1973) Isolation and
characterization of glycosphingolipids with blood group H specificity
from membranes of human erythrocytes. Biochemistry, 12, 656.
Stossel, T.P. (1975) Phagocytosis : Recognition and Ingestion.
Seminars in Haematology, 12, 83.
Stott, A.W. (1972) A simple scanning method for the quantitative
estimation of serum lecithin. Clin. Chem. Acta, 37, 535.

233
Sturgeon, P. (1970) Haematological observations on the anaemia
associated with blood type Rhnull. Blood, 36, 310.
Taguchi, R. and Ikezawa, H. (1976) Studies on the hemolytic and
hydrolytic actions of phospholipases against mammalian erythrocyte
membranes. Arch. Biochim. Biophys., 173, 538.
Tippett, P. (1972) A present view of Rh. Pathologica, 64, 29.
Tsao, F.H.C., Keim, P.S. and Heinrikson, R.L. (1975) Crotalus
adamanteus phospholipase A2-a : subunit structure, NH2-terminal
sequence and homology with other phospholipases. Arch. Biochim. Biophys.
167, 706.
Urbaniak, S.J. (1976) Lymphoid cell dependent (K-cell) lysis of human
erythrocytes sensitized with rhesus alloantibodies.
Brit. J. Haemat., 33, 409.
Van Loghem, J.J., Van der Meulen, F.W., Fleer, A. ,Van der Hart, M.,
Van der Borne, Kr. A.E.G. and Engelfriet, C.P. (1976) The importance
of the FC receptor for red cell destruction under the influence of non-
complement-binding antibodies. Human blood groups 5th Int. Convoc.
Immunol., Buffalo, N.Y. pp.75.
Verkleijy.A.J., Zwaal, R.F.A., Roelofsen, B., Comfurius, P., Kastelijn, D.
and Van Deenen, L.L.M. (1973) The asymmetric distribution of phospho-
lipids in the human red cell membrane. A combined study using phospholipases
and freeze-etch electron microscopy. Biochim. Biophys. Acta, 323, 178.

234 Victoria, E.J., Mahan, L.C. and Masouredis, S.P. (1977) Immunoglobulin
G dissassembly during thermal denaturation in sodium dodecyl sulphate
solutions. Biochem., 16, 2566.
Von dem Borne, Kr., A.E.G., Beckers, D. and Engelfriet, C.P. (1977)
Mechanisms of red cell destruction mediated by non-complement binding
IgG antibodies : the essential role in vivo of the Fc part of IgG.
Brit. J. Haemat., 36, 485.
Watkins, W.M. (1966) Blood group substances. Science, 152, 172.
Weicker Von, H. (1968) Isolation of a small molecular peptide from
human erythrocyte membrane with Rh properties. Clin. Chem. Acta,
21, 513.
Weicker Von, H. and Metz, J. (1971) Isolierung eines kleinmolekularen
erythrocyten membran-proteins durch anti-D immunadsorption.
Z. Klin. Chem. Klin. Biochem., 9, 367.
Weicker Von, H., Schmid, H., Kropp, J., Helmstadter, V. and Ebert, W.
(1973) Die bedeutung von membranphospholipiden fur antigen-antikorper-
bindung im Rh-D-system. Z. Klin. Chem. Klin. Biochem., 11, 543.
Wells, M.A. (1971) Evidence that the phospholipase A2 of crotarlus
adamanteus venom are dimers. Biochemistry, 10, 4074.
Whiteley, N.M. and Berg, H.C. (1974) Amidation of the outer and inner
surfaces of the human erythrocyte membrane. J. Mol. Biol., 87, 541.
Wiener, A.S. and Peters, H.R. (1940) Hemolytic reactions following
transfusion of blood of the homologous group, with three cases in which
the same agglutinogen was responsible. Ann. Int. Med., 13, 2306.

235
Witebsky, E. (1954) The immunology of acquired haemolytic anaemia :
diagnostic and therapeutic considerations. Proceedings of the fourth
international congress of the international society of haematology.
Grune and Stratton, New York, p. 284.
Wolff, I. and Springer, G.F. (1964) Observations on Rh-active substances.
Proc. 10th Congr. Int. Blood Transfusion, Stockholm, p. 510.
Yokoyama, M., Trams, E.G. and Brady, R.O. (1963) Immunological studies
with gangliosides. J. Immunol., 90, 372.
Yu, J. and Steck, T.L. (1973) Selective solubilization, isolation
and characterization of a predominant polypeptide from human erythrocyte
membranes. J. cell. Biol., 59, Abstract No.748, p.374(a).
Zipursky, A. and Brown, E.J. (1974) The ingestion of IgG-sensitized
erythrocytes by abnormal neutrophils. Blood, 43, 737.
Zwaal, R.F.A., Roelofsen, B., Comfurius, P. and Van Deenen, L.L.M. (1975)
Organization of phospholipids in human red cell membranes as detected
by the action of various purified phospholipases. Biochim. Biophys. Acta,
406, 83.
Zwaal, R.F.A., Roelofsen, B. and Colley, C.M. (1973) Localization of
red cell membrane constituents. Biochim. Biophys. Acta, 300, 159.