on the interaction of the electron acceptor 2,6
TRANSCRIPT

TIE JOURNAL OF BIOLOQICAL CHEMISTRY Vol. 246, No. 2, Issue of January 25, pp. 286-293, 1971
Printed in U.S.A.
On the Interaction of the Electron Acceptor 2,6=Dichlorophenolindophenol with Bovine Milk Xanthine Oxidase*
(Received for publication, July 23, 1970)
H. L. GURTOO~ AND D. G. JOHN@
From the Department of Pharmacology, Yale University, New Haven, Connecticut 06510
SUMMARY
The electron acceptor 2,6-dichlorophenolindophenol (DCI) forms a spectrophotometrically and kinetically detectable complex with bovine milk xanthine oxidase. The visible absorption spectrum of complexed DC1 exhibits an absorp- tion maximum at 660 nm at pH 7.8 (E = 34.2 X lo3 M-’ cm-‘), a marked bathochromic shift from the visible absorption maximum at 608 nm characteristic of the free dye (E = 21.0 x lo3 M-’ cm-l). The bound dye can be partially removed from the enzyme by prolonged dialysis (24 hours), and totally removed by gel filtration, indicating a reversible rather than a covalent interaction. From the DC1 concentration depend- ence of the spectral change, the stoichiometry of the interac- tion is 2.08 DCI-binding sites per molecule of xanthine oxi- dase (assuming a molar extinction coefficient for the enzyme of 66 X lo3 M-I cm-l at 450 nm); and the dissociation con- stant of the DCI-enzyme complex is 1.3 x 1OmB M. The spectral shift characteristic of DCI-xanthine oxidase interac- tion is blocked by 2 mu arsenite, by 2 mu j-hydroxymercuri- benzoate, by the competitive inhibitor 2,4-diamino-6-hydroxy- s-triazine, 1.5 mu, and by the “stoichiometric” xanthine oxidase inhibitor, 4,6-dihydroxypyrazolo[3,4-djpyrimidine (4,6-diHPP), at levels equimolar with those of the enzyme. With the latter inhibitor, the DC1 spectral shift, although not present initially, develops concomitantly with reoxidation of the reduced enzyme molybdenum of the 4,6-diHPP-xanthine oxidase complex. In contrast to the results seen with the other xanthine oxidase inhibitors, the DCI-xanthine oxidase interaction is not blocked by 16 mu sodium cyanide. In kinetic studies, DCI, in addition to acting as an electron ac- ceptor, is also a noncompetitive inhibitor of the enzyme, with a Ki of 2.8 x lOTEM. Prior incubation with DC1 protects the enzyme from inhibition by arsenite, but not from inhibition by cyanide. On the basis of the spectrophotometric and kinetic data, it is proposed that the DCI-binding site is the molybdenum or molybdenum-sulfur component of the inter- nal electron transport chain of the enzyme.
* This investigation was supported by Grants CA-11334 and CA-10748 from the United States Public Health Service. and Grant PRA-58 from the American Cancer Society, Inc.
$ Present address, Miles Laboratories, Inc., Elkhart, Indiana 46514.
$ To whom reprint requests should be directed. Present ad- dress, Laboratory of Chemical Pharmacology, National Cancer Institute, National Institutes of Health, Bethesda,Maryland 20014.
2,6-Dichlorophenolindophenol and other indophenol dyes are frequently used as electron acceptors for enzyme-catalyzed oxi- dation reactions. With the enzyme bovine milk xanthine oxi- dase, the rate of electron transfer from conventional substrates to DCII approximates that for electron transfer to the physio- logical acceptor, molecular oxygen (1). Despite this similarity, the two processes differ fundamentally: electron transfer to oxy- gen takes place from the terminal components of the internal electron transport chain of the enzyme, iron and FAD (2, 3), whereas electron transfer to DC1 and other artificial acceptors (phenazine methosulfate; ferricyanide) takes place from a site preceding the flavin, presumably the enzyme-bound molybde- num (4-6).
DCI, in addition to its use as an electron acceptor in enzyme- catalyzed oxidations, is also a convenient indicator for nonenzy- mic oxidation-reduction reactions. With sulfhydryl compounds, however, the stoichiometry of oxidation by DC1 is complex; in titration studies with the thiols cysteine and glutathione, it was observed by Hadler, Erwin, and Lardy (7), and by Coffey and Hellerman (8), that oxidation of these compounds by DC1 is accompanied by a quantitatively significant side reaction, that of adduct formation between the thiol and the dye, yielding a covalently bound glutathione-DC1 or cysteine-DC1 conjugate. Furthermore, in subsequent studies, Coffey and Hellerman (9) showed that DC1 can bind tightly to certain sodium dodecyl sulfate-treated thiol-containing proteins; and Hadler, Alt, and Falcone (10) demonstrated interaction between DC1 and mito- chondrial proteins, which appeared to correlate with the thiol content of the mitochondria. Both groups of authors have sug- gested, on the basis of their previous studies of DC1 conjugation with glutathione and cysteine, that DCI-protein interaction in- volves oxidative addition of DC1 to enzyme sulfhydryl groups.
Our interest in DCI-protein interaction arose from studies on the site of electron transfer from the internal electron transport chain of xanthine oxidase to artificial acceptors (II), in the course of which it was noted that, as reported originally by Fridovich (12), marked inhibition of the rate of electron transfer from xanthine oxidase to DCI occurs as the acceptor concentration is raised. In studies of this phenomenon, described below, it was observed that this inhibition is accompanied by spectrophoto- metric evidence of DCI-xanthine oxidase binding, and that such
1 The abbreviations used are: DCI, 2,6-dichlorophenolindo- phenol; 4-HPP, 4-hydroxypyrazolo[3,4-dlpyrimidine; 4,6-diHPP, 4,6-dihydroxypyrazolo[3,4-dlpyrimidine; PMB, p-chloromercuri- benzoate; EPR, electron paramagnetic resonance.
286
by guest on April 13, 2018
http://ww
w.jbc.org/
Dow
nloaded from
Issue of January 25, 1971 H. L. Gurtoo and D. G. Johns 287
dye-enzyme interaction can be prevented by thiol reagents and other inhibitors known to bind at or near the substrate-binding site of the enzyme. St.udies of the stoichiometry of the inter- action indicate the presence of two DCI-binding sites per mole- cule of xanthine oxidase; the binding appears to differ, however, from the DCI-protein interaction described by the authors cited above in being reversible in type. A preliminary account of these studies has appeared (13).
METHODS
Bovine milk xanthine oxidase was purified by the procedure of Hart et al. (14), with the modification that the salicylate de- naturation step was omitted. The specific activity of the en- zyme preparations used in the present experiments varied be- tween 12 and 18 units per ml per Asso, as defined by Fridovich (15), and the AUK,. *A450 ratio varied between 5.7 and 6.3. For the kinetic and arsenite protection experiments, enzyme pur- chased from a commercial source was used (Seravac Labora- tories, code 5P) ; the latter preparation had been subjected to purification by the method of Gilbert and Bergel (16) and had a specific activity of 9.3 units per ml per Azso and a Atso:A~, ratio of 6.2. Enzyme preparations which were not used immediately were stored in 0.1 M potassium phosphate buffer, pH 7.8, con- taining 4 mu sodium salicylate, and 0.2 mM EDTA, and were dialyzed before use. 4,6-DiHPP-inhibited xanthine oxidase was prepared by a modification of previously published methods (6, 17), as follows. A solution containing xanthine oxidase (27 mpmoles), 4,8diHPP (2.7 pmoles), potassium phosphate buffer (pH 7.8, 0.5 mmole), and EDTA (0.2 pmole) in a total volume of 2.7 ml in a Thunberg-type anaerobic cuvette was subjected to deoxygenation and the absorption spectrum recorded. Sub- strate (either propionaldehyde (0.40 mmole) or xanthine (2.7 pmoles)) was then added from a side arm. The absorption spec- trum of the substrate-reduced enzyme was recorded, and the mixture incubated for 1 hour at 25”. The solution was then applied to a column of Sephadex G-25, 2 x 25 cm, and eluted with deoxygenated 0.1 M potassium phosphate buffer, pH 7.8, containing 0.2 MIM. EDTA. The effluent was monitored by means of a Uvicord absorptiometer; the peak containing the catalytically inactive xanthine oxidase-4,6-diHPP complex ap- peared immediately after the void volume.
Spectrophotometric assays at a single wave length were car- ried out using a Gilford model 2000 spectrophotometer equipped with a thermostated cell compartment; absorption spectra were obtained using an Aminco-Chance dual wave length split-beam recording spectrophotometer. Split cells with a total path length of 0.9 cm were obtained from Pyrocell Manufacturing Company, Inc., Westwood, New Jersey, and Thunberg-type anaerobic cuvettes were obtained from Hellma Cells, Inc., Jamaica, New York (codes 190 and 197). The sources of the reagents used have been indicated previously (18), with the ex- ception of sodium arsenite (Mallinckrodt Chemical Works) and p-chloromercuribenzoate (Eastman). Details of the individual assay methods are given in the legends for figures and tables; unless otherwise specified, all studies were carried out in 0.167 M
potassium phosphate buffer, pH 7.8, containing 66 pM EDTA. Nitrogen used in the anaerobic experiments was deoxygenated by the vanadous sulfate method of Meites and Meites (19). The molar extinction coefficients used in the spectrophotometric assay methods have previously been indicated (18); for the present studies, the molar extinction coefficient of DC1 at 608
0.05 - \
OL I I I I I 350 400 500 600 700 770
WAVELENGTH, “m
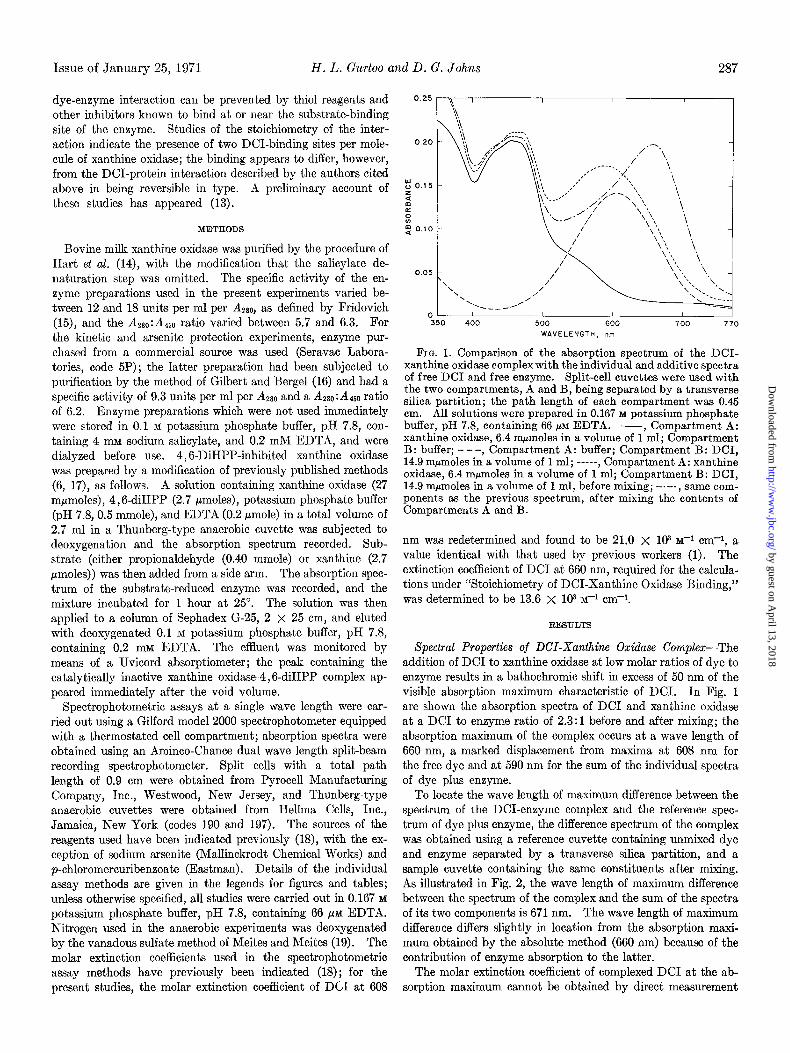
FIG. 1. Comparison of the absorption spectrum of the DCI- xanthine oxidase comnlex with the individual and additive snectra of free DC1 and free enzyme. Split-cell cuvettes were use2 with the two compartments, A and B, being separated by a transverse silica partition; the path length of each compartment was 0.45 cm. All solutions were prepared in 0.167 M potassium phosphate buffer, pH 7.8, containing 66 NM EDTA. -, Compartment A: xanthine oxidase, 6.4 mpmoles in a volume of 1 ml; Compartment B: buffer; - - -, Compartment A: buffer; Compartment B: DCI, 14.9 mpmoles in a volume of 1 ml; -----, Compartment A: xanthine oxidase, 6.4 mrmoles in a volume of 1 ml; Compartment B: DCI, 14.9 mfimoles in a volume of 1 ml, before mixing; -.-. , same com- ponents as the previous spectrum, after mixing the contents of Compartments A and B.
nm was redetermined and found to be 21.0 x lo3 I@ cm-l, a value identical with that used by previous workers (1). The extinction coefficient of DC1 at 660 nm, required for the calcula- tions under “Stoichiometry of DCI-Xanthine Oxidase Binding,” was determined to be 13.6 X lo3 M-’ cm-l.
RESULTS
Spectral Properties of DCI-Xanthine Oxidase Complex-The addition of DC1 to xanthine oxidase at low molar ratios of dye to enzyme results in a bathochromic shift in excess of 50 nm of the visible absorption maximum characteristic of DCI. In Fig. 1 are shown the absorption spectra of DC1 and xanthine oxidase at a DC1 to enzyme ratio of 2.3:1 before and after mixing; the absorption maximum of the complex occurs at a wave length of 660 nm, a marked displacement from maxima at 608 nm for the free dye and at 590 nm for the sum of the individual spectra of dye plus enzyme.
To locate the wave length of maximum difference between the spectrum of the DCI-enzyme complex and the reference spec- trum of dye plus enzyme, the difference spectrum of the complex was obtained using a reference cuvette containing unmixed dye and enzyme separated by a transverse silica partition, and a sample cuvette containing the same constituents after mixing. As illustrated in Fig. 2, the wave length of maximum difference between the spectrum of the complex and the sum of the spectra of its two components is 671 nm. The wave length of maximum difference differs slightly in location from the absorption maxi- mum obtained by the absolute method (660 nm) because of the contribution of enzyme absorption to the latter.
The molar extinction coefficient of complexed DC1 at the ab- sorption maximum cannot be obtained by direct measurement
by guest on April 13, 2018
http://ww
w.jbc.org/
Dow
nloaded from
288 Interaction of Z? ,6-Dichloroindophenol with Xanthine Oxidase Vol. 246, No. 2
I 1 I I
-O.’ I 350 400 500 600 700 770
WAVELENGTH, “m
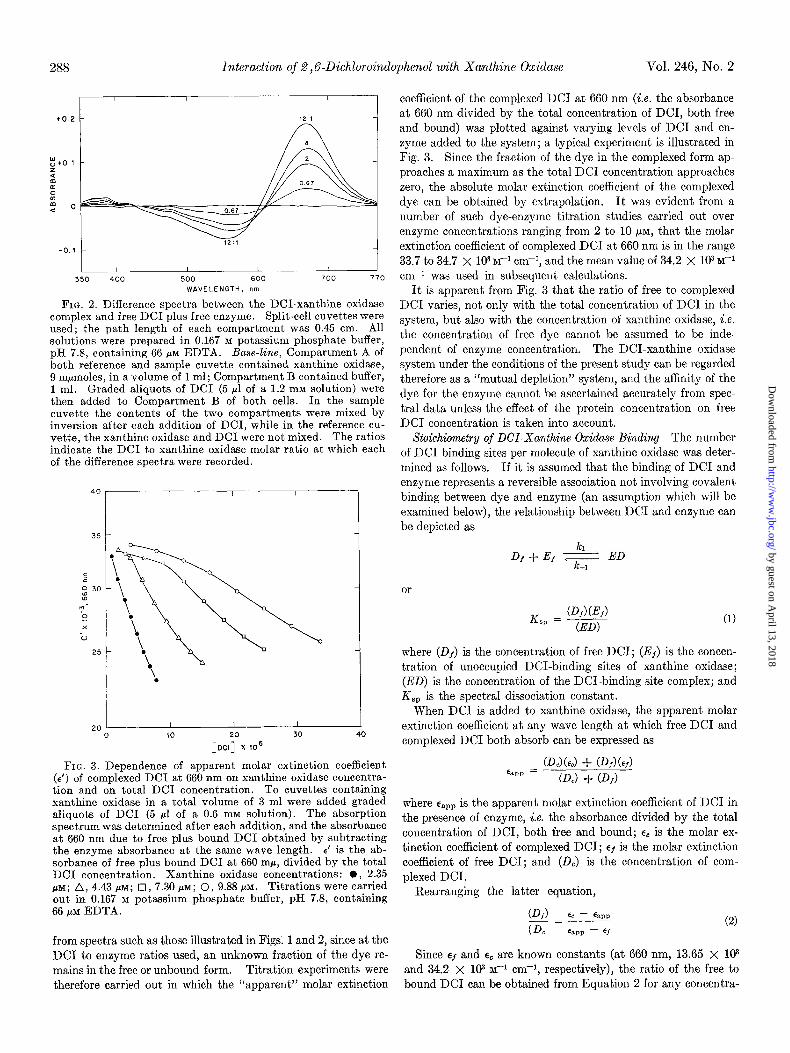
FIG. 2. Difference spectra between the DCI-xanthine oxidase complex and free DC1 plus free enzyme. Split-cell cuvettes were used; the path length of each compartment was 0.45 cm. All solutions were prepared in 0.167 M potassium phosphate buffer, pH 7.8, containing 66 PM EDTA. Base-line, Compartment A of both reference and sample cuvette contained xanthine oxidase, 9 mpmoles, in a volume of 1 ml; Compartment B contained buffer, 1 ml. Graded aliquots of DC1 (5 pl of a 1.2 mM solution) were then added to Compartment B of both cells. In the sample cuvette the contents of the two compartments were mixed by inversion after each addition of DCI, while in the reference cu- vette, the xanthine oxidase and DC1 were not mixed. The ratios indicate the DC1 to xanthine oxidase molar ratio at which each of the difference spectra were recorded.
r I / /
" 10 20 30 40
[DCI] X IO6
FIG. 3. Dependence of apparent molar extinction coefficient (E’) of complexed DC1 at 660 nm on xanthine oxidase concentra- tion and on total DC1 concentration. To cuvettes containing xanthine oxidase in a total volume of 3 ml were added graded aliquots of DC1 (5 ~1 of a 0.6 mM solution). The absorption spectrum was determined after each addition, and the absorbance at 660 nm due to free plus bound DC1 obtained by subtracting the enzyme absorbance at the same wave length. E’ is the ab- sorbance of free plus bound DC1 at 660 rnp, divided by the total DC1 concentration. Xanthine oxidase concentrations: 0, 2.35 PM; A, 4.43 PM; 0, 7.30 FM; 0, 9.88 PM. Titrations were carried out in 0.167 M potassium phosphate buffer, pH 7.8, containing 66 /.tM EDTA.
from spectra such as those illustrated in Figs. 1 and 2, since at the DC1 to enzyme ratios used, an unknown fraction of the dye re- mains in the free or unbound form. Titration experiments were therefore carried out. in which the “apparent” molar extinction
coefficient of the complexed DC1 at 660 nm (i.e. the absorbance at 660 nm divided by the total concentration of DCI, both free and bound) was plotted against varying levels of DC1 and en- zyme added to the system; a typical experiment is illustrated in Fig. 3. Since the fraction of the dye in the complexed form ap- proaches a maximum as the total DC1 concentration approaches zero, the absolute molar extinction coefficient of the complexed dye can be obtained by extrapolation. It was evident from a number of such dye-enzyme titration studies carried out over enzyme concentrations ranging from 2 to 10 PM, that the molar extinction coefficient of complexed DC1 at 660 nm is in the range 33.7 to 34.7 X lo3 M-l cm-i, and the mean value of 34.2 X 103 M-i cm-* was used in subsequent calculations.
It is apparent from Fig. 3 that the ratio of free to complexed DC1 varies, not only with the total concentration of DC1 in the system, but also with the concentration of xanthine oxidase, i.e. the concentration of free dye cannot be assumed to be inde- pendent of enzyme concentration. The DCI-xanthine oxidase system under the conditions of the present study can be regarded therefore as a “mutual depletion” system, and the affinity of the dye for the enzyme cannot be ascertained accurately from spec- tral data unless the effect of the protein concentration on free DC1 concentration is taken into account.
Stoichiometry of DCI-Xanthine Oxidase Binding-The number of DC1 binding sites per molecule of xanthine oxidase was deter- mined as follows. If it is assumed that the binding of DC1 and enzyme represents a reversible association not involving covalent binding between dye and enzyme (an assumption which will be examined below), the relationship between DC1 and enzyme can be depicted as
kl DI + Ef ; ED
k-1
or
K _ (D,)(E,) -~ w (ED)
where (Df) is the concentration of free DCI; (Ef) is the concen- tration of unoccupied DCI-binding sites of xanthine oxidase; (ED) is the concentration of the DCI-binding site complex; and K,, is the spectral dissociation constant.
When DC1 is added to xanthine oxidase, the apparent molar extinction coefficient at any wave length at which free DC1 and complexed DC1 both absorb can be expressed as
(D&c) + CD,) (~1) %pp =
CD,) + CD,)
where sapp is the apparent molar extinction coefficient of DC1 in the presence of enzyme, i.e. the absorbance divided by the total concentration of DCI, both free and bound; cc is the molar ex- tinction coefficient of complexed DCI; cf is the molar extinction coefficient of free DCI; and (DJ is the concentration of com- plexed DCI.
Rearranging the latter equation,
PI) EC - G*p _=~ (De
(2) %P - 6f
Since ef and ec are known constants (at 660 nm, 13.65 X lo3 and 34.2 x lo3 M+ cm+, respectively), the ratio of the free to bound DC1 can be obtained from Equation 2 for any concentra-
by guest on April 13, 2018
http://ww
w.jbc.org/
Dow
nloaded from
Issue of January 25, 1971 H. L. Gurtoo and D. G. Johns 289
tion of dye or enzyme by determining the apparent molar extinc- tion coefficient, espp, of the total DC1 present.
Since (DC) in Equation 2 is equal to (ED), Equation 1 can be rewritten as
K % -(Df)(Ej) = (E ) Cc - Cap,, -__ (DJ
/- %P - 9
(3)
It follows from Equation 3 that, irrespective of the total amount of enzyme in the solution, K,, = (Ef) when the total concentration of DC1 in the solution is at the level where (Df) = (DC). At any other fixed ratio of (Df) : (DC), i.e. when (Df)/ (DA = K,,
Ks, = &(Ef) (4)
The total concentration of DCI-binding sites (free plus oc- cupied sites) in the enzyme is proportional to the enzyme con- centration, i.e.
(ED) + (&) = m(XO)
or
(De) + 0%) = m(XO)
where m is the number of binding sites per molecule of enzyme and (X0) is the concentration of xanthine oxidase.
Rearranging the latter equation,
(DE) = m(X0) - (EI) (5)
From titration plots such as those shown in Fig. 3, the total concentration of DC1 necessary to give any fixed ratio of (DE) : (Df) can be determined, and from the latter data, the concen- tration of (De) can be ascertained. If (De) or (ED) is then plotted against (X0) for a number of different enzyme levels, the slope of the straight line obtained is equal to the number of DCI-binding sites per molecule of enzyme, and the intercept on the vertical axis = - (Ef). From Equation 4, the dissociation constant of the enzyme-DC1 complex, Ksp, can then be deter- mined.
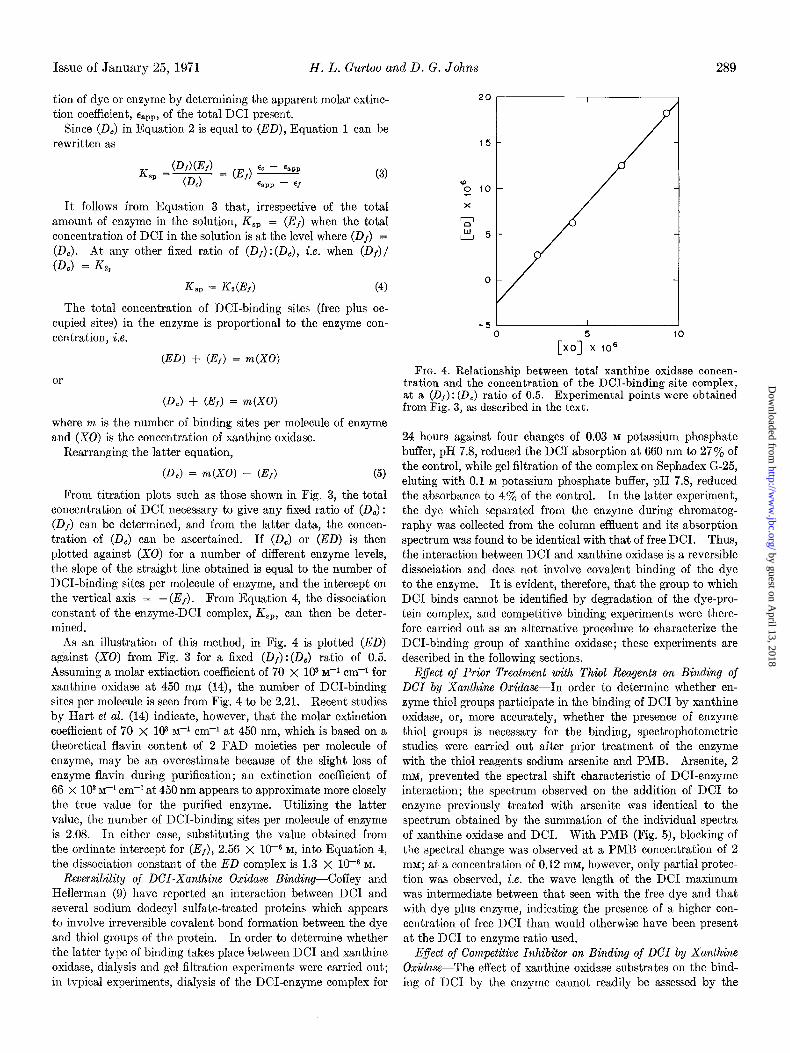
As an illustration of this method, in Fig. 4 is plotted (ED) against (X0) from Fig. 3 for a fixed (Df) :(Dc) ratio of 0.5. Assuming a molar extinction coefficient of 70 x lo3 I@ cm-+ for xanthine oxidase at 450 rnb (14), the number of DCI-binding sites per molecule is seen from Fig. 4 to be 2.21. Recent studies by Hart et al. (14) indicate, however, that the molar extinction coefficient of 70 x lo3 M+ cm-l at 450 nm, which is based on a theoretical flavin content of 2 FAD moieties per molecule of enzyme, may be an overestimate because of the slight loss of enzyme flavin during purification; an extinction coefficient of 66 X lo3 M-I cm-l at 450 nm appears to approximate more closely the true value for the purified enzyme. Utilizing the latter value, the number of DCI-binding sites per molecule of enzyme is 2.08. In either case, substituting the value obtained from the ordinate intercept for (Ef), 2.56 x 10m6 M, into Equation 4, the dissociation constant of the ED complex is 1.3 x 1OV M.
Reversibility of DCI-Xanthine Oxidase Binding-Coffey and Kellerman (9) have reported an interaction between DC1 and several sodium dodecyl sulfate-treated proteins which appears to involve irreversible covalent bond formation between the dye and thiol groups of the protein. In order to determine whether the latter type of binding takes place between DC1 and xanthine oxidase, dialysis and gel filtration experiments were carried out; in tvpical experiments, dialysis of the DCI-enzyme complex for
0
[xo15x 106 ‘O
FIG. 4. Relationship between total xanthine oxidase concen- tration and the concentration of the DCI-binding site complex, at a (Of): (DC) ratio of 0.5. Experimental points were obtained from Fig. 3, as described in the text.
24 hours against four changes of 0.03 M potassium phosphate buffer, pH 7.8, reduced the DC1 absorption at 660 nm to 27% of the control, while gel filtration of the complex on Sephadex G-25, eluting with 0.1 M potassium phosphate buffer, pH 7.8, reduced the absorbance to 4% of the control. In the latter experiment, the dye which separated from the enzyme during chromatog- raphy was collected from the column effluent and its absorption spectrum was found to be identical with that of free DCI. Thus, the interaction between DC1 and xanthine oxidase is a reversible dissociation and does not involve covalent binding of the dye to the enzyme. It is evident, therefore, that the group to which DC1 binds cannot be identified by degradation of the dye-pro- tein complex, and competitive binding experiments were there- fore carried out as an alternative procedure to characterize the DCI-binding group of xanthine oxidase; these experiments are described in the following sections.
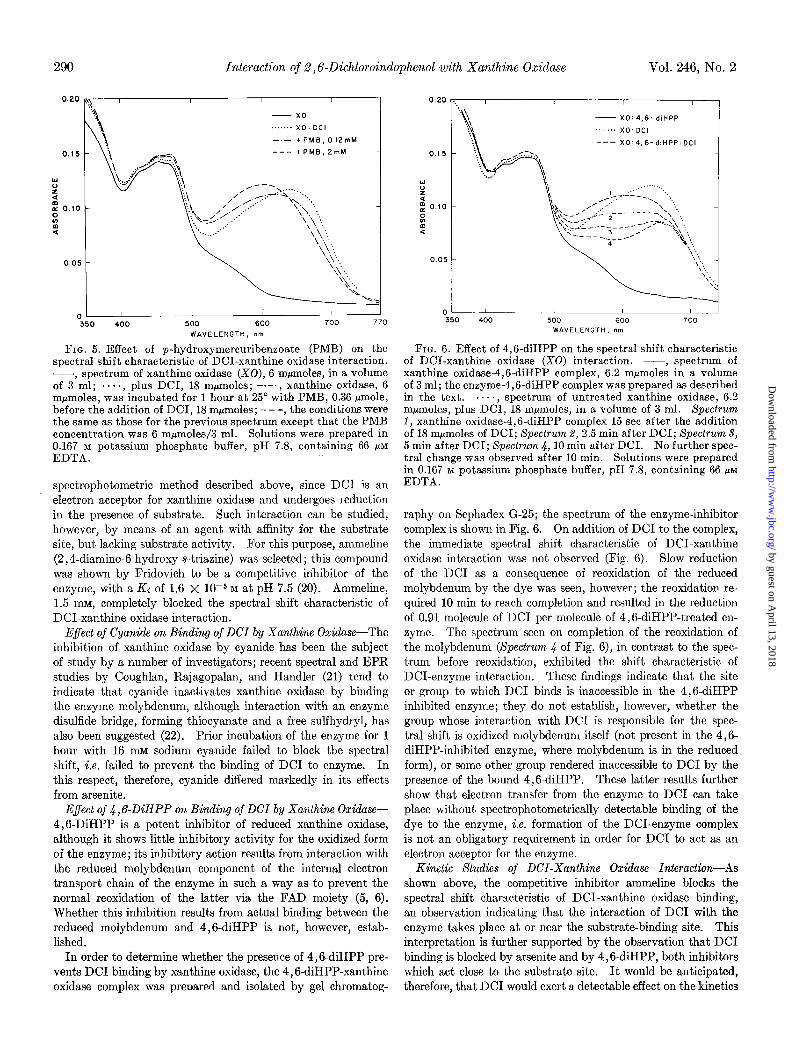
Effect of Prior Treatment with Thiol Reagents on Binding of DCI by Xanthine Oxidase-In order to determine whether en- zyme thiol groups participate in the binding of DC1 by xanthine oxidase, or, more accurately, whether the presence of enzyme thiol groups is necessary for the binding, spectrophotometric studies were carried out after prior treatment of the enzyme with the thiol reagents sodium arsenite and PMB. Arsenite, 2 mM, prevented the spectral shift characteristic of DCI-enzyme interaction; the spectrum observed on the addition of DC1 to enzyme previously treated with arsenite was identical to the spectrum obtained by the summation of the individual spectra of xanthine oxidase and DCI. With PMB (Fig. 5), blocking of the spectral change was observed at a PMB concentration of 2 mM; at a concentration of 0.12 mM, however, only partial protec- tion was observed, i.e. the wave length of the DC1 maximum was intermediate between that seen with the free dye and that with dye plus enzyme, indicating the presence of a higher con- centration of free DC1 than would otherwise have been present at the DC1 to enzyme ratio used.
Effect of Competitive Inhibitor on Binding of DCI by Xanthine Oxidase-The effect of xanthine oxidase substrates on the bind- ing of DC1 by the enzyme cannot readily be assessed by the
by guest on April 13, 2018
http://ww
w.jbc.org/
Dow
nloaded from
290 Interaction of 2,6-Dichloroindophenol with Xanthine Oxidase Vol. 246, No. 2
0.20 1. I I / /
- x0
. x0: DC,
-.- +PMB. 0.12mM
--- +PMB, 2mM
:
5
z 0.10 -
::
:
0.05 -
c-7 I 1 I
-350 400 500 600 700 770
WAVELENGTH, n”
FIG. 5. Effect of p-hydroxymercuribenzoate (PMB) on the spectral shift characteristic of DCI-xanthine oxidase interaction. -7 spectrum of xanthine oxidase (X0), 6 mpmolee, in a volume of 3 ml; ...., plus DCI, 18 mpmoles; -.-., xanthine oxidase, 6 mpmoles, was incubated for 1 hour at 25’ with PMB, 0.36 @mole, before the addition of DCI, 18 mpmoles; - - -, the conditions were the same as those for the previous spectrum except that the PMB concentration was 6 m&oles/3 ml: Solutions were prepared in 0.167 M potassium phosphate buffer, pH 7.8, containing 66 pM EDTA.
spectrophotometric method described above, since DC1 is an electron acceptor for xanthine oxidase and undergoes reduction in the presence of substrate. Such interaction can be studied, however, by means of an agent with affinity for the substrate site, but lacking substrate activity. For this purpose, ammeline (2,4-diamino-6hydroxy-s-triazine) was selected; this compound was shown by Fridovich to be a competitive inhibitor of the enzyme, with a Ki of 1.6 X 10e5 M at pH 7.5 (20). Ammeline, 1.5 mu, completely blocked the spectral shift characteristic of DCI-xanthine oxidase interaction.
E$ect of Cyanide on Binding of DCI by Xanthine Oxidase-The inhibition of xanthine oxidase by cyanide has been the subject of study by a number of investigators; recent spectral and EPR studies by Coughlan, Rajagopalan, and Handler (21) tend to indicate that cyanide inactivates xanthine oxidase by binding the enzyme molybdenum, although interaction with an enzyme disulfide bridge, forming thiocyanate and a free sulfhydryl, has also been suggested (22). Prior incubation of the enzyme for 1 hour with 16 lll~ sodium cyanide failed to block the spectral shift, i.e. failed to prevent the binding of DC1 to enzyme. In this respect, therefore, cyanide differed markedly in its effects from arsenite.
Effect of 4,GDiHPP on Binding of DCI by Xanthine Oxidase- 4,BDiHPP is a potent inhibitor of reduced xanthine oxidase, although it shows little inhibitory activity for the oxidized form of the enzyme; its inhibitory action results from interaction with the reduced molybdenum component of the internal electron transport chain of the enzyme in such a way as to prevent the normal reoxidation of the latter via the FAD moiety (5, 6). Whether this inhibition results from actual binding between the reduced molybdenum and 4,6-diHPP is not, however, estab- lished.
In order to determine whether the presence of 4,6-diHPP pre- vents DC1 binding by xanthine oxidase, the 4,6-diHPP-xanthine oxidase complex was preoared and isolated by gel chromatog-
0.20
0.1 5
z
5
“, 0.10
s m 4
0.05
0
, I I
~ X0:4,6-diHPP
. . . . . . . x0: DC,
--- X0:4,6-diHPP:DCI
I I I 1
) 400 500 600 700
WAVELENGTH, nm
FIG. 6. Effect of 4,6-diHPP on the spectral shift characteristic of DCI-xanthine oxidase (X0) interaction. -, spectrum of xanthine oxidase-4.6-diHPP comdex. 6.2 mumoles in a volume of 3 ml; the enzyme:4,6-diHPP complex was prepared as described in the text. .s.., spectrum of untreated xanthine oxidase, 6.2 mpmoles, plus DCI, 18 mpmoles, in a volume of 3 ml. Spectrum 1, xanthine oxidase-4,6-diHPP complex 15 set after the addition of 18 mpmoles of DCI’; Spectrum 2, 2:5 min after DCI; Spectrum 3, 5 min after DCI; Spectrum .J, 10 min after DCI. No further spec- tral change was observed after 10 min. Solutions were prepared in 0.167 M potassium phosphate buffer, pH 7.8, containing 66 PM EDTA.
raphy on Sephadex G-25; the spectrum of the enzyme-inhibitor complex is shown in Fig. 6. On addition of DC1 to the complex, the immediate spectral shift characteristic of DCI-xanthine oxidase interaction was not observed (Fig. 6). Slow reduction of the DC1 as a consequence of reoxidation of the reduced molybdenum by the dye was seen, however; the reoxidation re- quired 10 min to reach completion and resulted in the reduction of 0.91 molecule of DC1 per molecule of 4,6-diHPP-treated en zyme. The spectrum seen on completion of the reoxidation of the molybdenum (Spectrum 4 of Fig. 6), in contrast to the spec- trum before reoxidation, exhibited the shift characteristic of DCI-enzyme interaction. These findings indicate that the site or group to which DC1 binds is inaccessible in the 4,6-diHPP inhibited enzyme; they do not establish, however, whether the group whose interaction with DC1 is responsible for the spec- tral shift is oxidized molybdenum itself (not present in the 4,6- diHPP-inhibited enzyme, where molybdenum is in the reduced form), or some other group rendered inaccessible to DC1 by the presence of the bound 4,6-diHPP. These latter results further show that electron transfer from the enzyme to DC1 can take place without spectrophotometrically detectable binding of the dye to the enzyme, i.e. formation of the DCI-enzyme complex is not an obligatory requirement in order for DC1 to act as an electron acceptor for the enzyme.
Kinetic Studies of DCI-Xanthine Oxidase Interaction-As shown above, the competitive inhibitor ammeline blocks the spectral shift characteristic of DCI-xanthine oxidase binding, an observation indicating that the interaction of DC1 with the enzyme takes place at or near the substrate-binding site. This interpretation is further supported by the observation that DC1 binding is blocked by arsenite and by 4,6-diHPP, both inhibitors which act close to the substrate site. It would be anticipated, therefore, that DC1 would exert a detectable effect on the kinetics
by guest on April 13, 2018
http://ww
w.jbc.org/
Dow
nloaded from
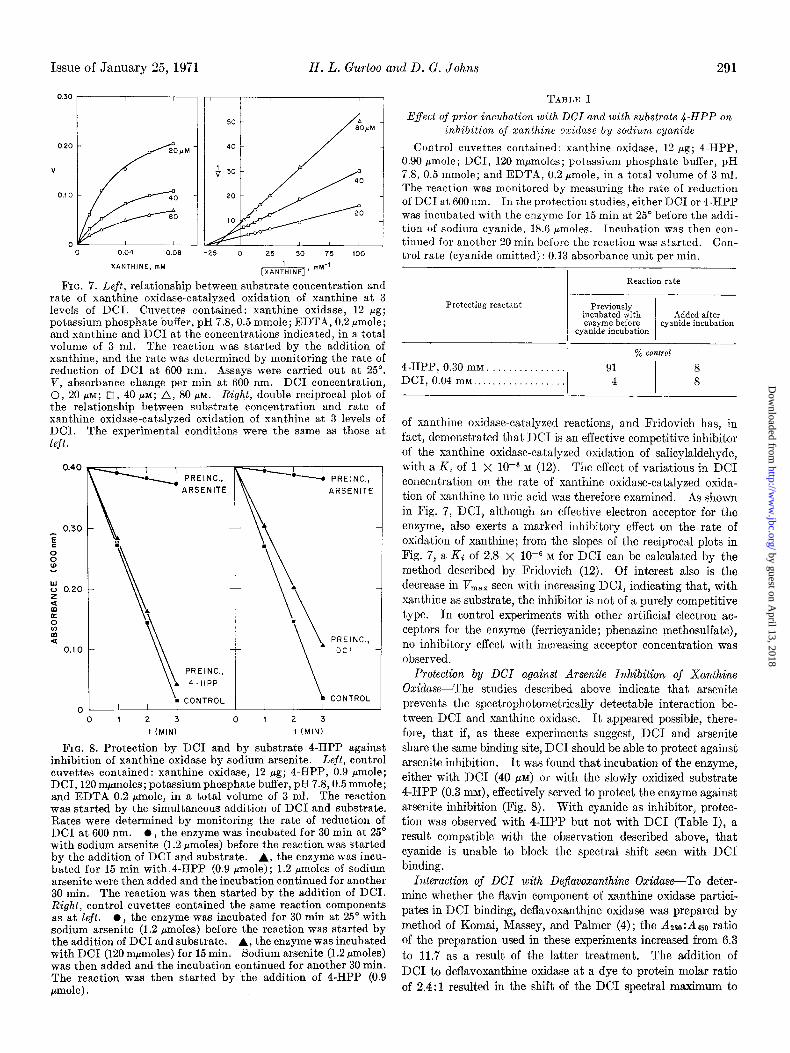
Issue of January 25, 1971 H. L. Gurtoo and D. G. Johns 291
O”O I------
0.20 2Oplr
"
0.1 0
E 40
60
0 AL 0 0.04 0.08 -25 0 25 50 75 100
XANTHINE. rnM [& ’ m”-’
FIG. 7. Left, relationship between substrate concentration and rate of xanthine oxidase-catalyzed oxidation of xanthine at 3 levels of DCI. Cuvettes contained: xanthine oxidase, 12 fig; potassium phosphate buffer, pH 7.8,0.5 mmole; EDTA, 0.2 pmole; and xanthine and DC1 at the concentrations indicated, in a total volume of 3 ml. The reaction was started by the addition of xanthine, and the rate was determined by monitoring the rate of reduction of DC1 at 600 nm. Assays were carried out at 25”. V, absorbance change per min at 600 nm. DC1 concentration, 0, 20 FM; q i, 40 PM; A, 80 PM. Right, double reciprocal plot of the relationship between substrate concentration and rate of xanthine oxidase-catalyzed oxidation of xanthine at 3 levels of DCI. The experimental conditions were the same as those at left.
0.40
0.30
P
8 z
i? 0.20
5 E
2 :
0.1 0
0
PREINC., ARSENITE
PREINC..
4-HPP
CONTROL
-.-m
l -• PREINC.,
ARSENITE
PREINC., DCI
CONTROL
0 1 2 3 0 1 2 3
I (MIN) t (MIN)
FIG. 8. Protection by DC1 and by substrate 4-HPP against inhibition of xanthine oxidase by sodium arsenite. Left, control cuvettes contained: xanthine oxidase, 12 pg; 4-HPP, 0.9 pmole; DCI, 120 mpmoles; potassium phosphate buffer, pH 7.8,0.5 mmole; and EDTA 0.2 pmole, in a total volume of 3 ml. The reaction was started by the simultaneous addition of DC1 and substrate. Rates were determined by monitoring the rate of reduction of DC1 at 600 nm. 0, the enzyme was incubated for 30 min at 25’ with sodium arsenite (1.2 rmoles) before the reaction was started by the addition of DC1 and substrate. A, the enzyme was incu- bated for 15 min with 4-HPP (0.9 pmole); 1.2 pmoles of sodium arsenite were then added and the incubation continued for another 30 min. The reaction was then started by the addition of DCI. Right, control cuvettes contained the same reaction components as at left. l , the enzyme was incubated for 30 min at 25” with sodium arsenite (1.2 pmoles) before the reaction was started by the addition of DC1 and substrate. A, the enzyme was incubated with DC1 (120 mpmoles) for 15 min. Sodium arsenite (1.2 pmoles) was then added and the incubation continued for another 30 min. The reaction was then started by the addition of 4-HPP (0.9 pmole) .
TABLE I
Effect of prior incubation with DCI and with substrate 4.HPP on inhibition of xanlhine oxidase by sodium cyanide
Control cuvettes contained: xanthine oxidase, 12 pg; 4-HPP, 0.90 Fmole; DCI, 120 mpmoles; potassium phosphate buffer, pH 7.8, 0.5 mmole; and EDTA, 0.2 pmole, in a total volume of 3 ml. The reaction was monitored by measuring the rate of reduction of DC1 at 600 nm. In the protection studies, either DC1 or 4-HPP was incubated with the enzyme for 15 min at 25” before the addi- tion of sodium cyanide, 18.6 +moles. Incubation was then con- tinued for another 20 min before the reaction was started. Con- trol rate (cyanide omitted) : 0.13 absorbance unit per min.
Protecting reactant
4-HPP, 0.30 mM .............. DCI, 0.04 rnM .................
I Reaction rate
Previously incubated with Added after enzyme before
cyanide incubation cyanide incubation
% co?ztrol 91
I
8 4 8
of xanthine oxidase-catalyzed reactions, and Fridovich has, in fact, demonstrated that DC1 is an effective competitive inhibitor
of the xanthine oxidase-catalyzed oxidation of salicylaldehyde, with a Ki of 1 X lop6 M (12). The effect of variations in DC1 concentration on the rate of xanthine oxidase-catalyzed oxida- tion of xanthine to uric acid was therefore examined. As shown in Fig. 7, DCI, although an effective electron acceptor for the enzyme, also exerts a marked inhibitory effect on the rate of oxidation of xanthine; from the slopes of the reciprocal plots in Fig. 7, a Ki of 2.8 x 10e6 M for DC1 can be calculated by the method described by Fridovich (12). Of interest also is the decrease in Vm,, seen with increasing DCI, indicating that, with xanthine as substrate, the inhibitor is not of a purely competitive
type. In control experiments with other artificial electron ac- ceptors for the enzyme (ferricyanide; phenazine methosulfate), no inhibitory effect with increasing acceptor concentration was observed.
Protection by DCI against Arsenite Inhibition of Xanthine Oxidase-The studies described above indicate that arsenite prevents the spectrophotometrically detectable interaction be- tween DC1 and xanthine oxidase. It appeared possible, there- fore, that if, as these experiments suggest, DC1 and arsenite share the same binding site, DC1 should be able to protect against arsenite inhibition. It was found that incubation of the enzyme, either with DC1 (40 ~11) or with the slowly oxidized substrate 4-HPP (0.3 m), effectively served to protect the enzyme against arsenite inhibition (Fig. 8). With cyanide as inhibitor, protec- tion was observed with 4-HPP but not with DC1 (Table I), a result compatible with the observation described above, that cyanide is unable to block the spectral shift seen with DC1 binding.
Interaction of DCI with Deflavozanthine Oxidase-To deter- mine whether the flavin component of xanthine oxidase partici- pates in DC1 binding, deflavoxanthine oxidase was prepared by method of Komai, Massey, and Palmer (4) ; the Azso: A450 ratio of the preparation used in these experiments increased from 6.3
to 11.7 as a result of the latter treatment. The addition of DC1 to deflavoxanthine oxidase at a dye to protein molar ratio
of 2.4:i resulted in the shift of the DC1 spectral maximum to
by guest on April 13, 2018
http://ww
w.jbc.org/
Dow
nloaded from

292 Interaction of 2 ,6-Dichloroindophenol with Xanthine Oxidase Vol. 246, No. 2
660 nm characteristic of DCI-xanthine oxidase complex forma- tion. It was concluded, therefore, that the presence of the en- zyme flavin is not obligatory for DCI-xanthine oxidase inter- action.
DISCUSSION
These studies indicate that the electron acceptor DC1 binds specifically and tightly to oxidized or noncycling xanthine oxi- dase. The number of DCI-binding sites per molecule of enzyme closely approximates 2 (2.21 or 2.08, using c450 values for xan- thine oxidase of 70 X lo3 and 66 x lo3 M-~ cm-l, respectively), and the dissociation constant of the enzyme-DC1 complex, determined by a spectrophotometric method, is 1.3 x low6 M.
The nature of the DC1 binding groups of the enzyme is not, however, firmly established from the competitive binding stud- ies. Of the various alternatives, one which appears possible is an enzyme-sulfhydryl group at or near the substrate-binding site. The evidence favoring such a DCI-sulfhydryl interaction is as follows.
Previous workers have shown that DC1 can interact with free sulfhydryl groups of glutathione, cysteine, and of mitochondrial proteins (7, 8, 10). Certain sulfhydryl-containing soluble pro- teins also interact with DCI, after treatment with the denaturing agent sodium dodecyl sulfate, although not in the native state (9). The DCI-sulfhydryl interaction described by these workers appears to differ from the DCI-xanthine oxidase interaction described in the present paper, however, in that the former in- volves covalent bond formation (at least in the case of glutathione and cysteine), and is not reversible on chromatography or dialy- sis, while on the other hand, as shown above, the DCI-xanthine oxidase binding appears to be noncovalent, being reversible both by dialysis and by gel filtration. The DCI-glutathione conjugate does, however, show a bathochromic shift, similar to that of the DCI-enzyme complex, of approximately 40 nm of the absorp- tion maximum of DC1 in the visible region (8), although the DCI-cysteine conjugate does not show significant absorption in this region (7).
The interaction of DC1 and xanthine oxidase is blocked by PMB. Xanthine oxidase is known to contain free sulfhydryls which can be titrated by PMB (23), and the prevention of DC1 binding by this reagent is therefore compatible with DCI-thiol interaction. Evidence against this possibility, however, is the observation that a large molar excess of PMB over enzyme was required to block DC1 binding completely, with only “partial” blocking being seen after a a-hour incubation of the enzyme with PMB at a concentration of 0.12 mM. Furthermore, PMB, in addition to reacting with xanthine oxidase sulfhydryls, is also known to be a noncompetitive inhibitor of the liver enzyme (24). Thus, the blocking of the DCI-enzyme interaction by PMB is not necessarily due to blocking of enzyme sulfhydryls by the latter reagent, but, if it is assumed that the milk enzyme re- sembles the liver enzyme in susceptibility to PMB inhibition, could also be due to competition between PMB and DC1 for a common nonsulfhydryl binding site.
In addition, PMB, if allowed to react with milk xanthine oxi- dase for a sufficient length of time, is known to be able to titrate at least 35 sulfhydryls per mole of enzyme (23). With DCI, on the other hand, after the “immediate” spectral shift seen on addition of the dye to the enzyme, no time-dependent increase in the spectrally detectable interaction is seen. It is evident, therefore, that if DCI-xanthine oxidase interaction represents
sulfhydryl binding, thiol groups titrated in this way must differ in some fundamental way from the remaining free thiols known to be present in the enzyme.
The interaction between DC1 and xanthine oxidase is blocked by arsenite. Arsenite is a commonly used sulfhydryl reagent and can form a linkage with single thiols or with closely adjacent dithiols. In the case of xanthine oxidase, however, it has not been established that the inhibition of the enzyme by arsenite is due to sulfhydryl binding. Recent spectral and EPR evi- dence indicates that such inhibition may, in fact, be due to inter- action between arsenite and the enzyme molybdenum (21). Thus, the blocking of the DCI-enzyme interaction by arsenite is susceptible to other possible explanations than competition between DC1 and arsenite for an enzyme thiol.
The stoichiometry of DCI-xanthine oxidase binding indicates 2 DC1 molecules bound per molecule of enzyme. In a recent paper, Massey et al. (6) have cited unpublished thiol titration data with 5,5’-dithiobis(2-nitrobenzoic acid) indicating the pres- ence of one reactive sulfhydryl group per molecule of enzyme- bound FAD, i.e. two reactive sulfhydryls per enzyme molecule. If it is assumed that these thiols are involved in the binding of DC1 also, the stoichiometry of DCI-xanthine oxidase interac- tion would be appropriate. However, a similar argument could also be used to indicate interaction of DC1 with the enzyme molybdenum or FAD, both of which are present at a 2: 1 molar ratio.
An alternate possibility is that the spectrophotometrically detectable DCI-xanthine oxidase interaction results from bind- ing to the enzyme molybdenum. The evidence for this possi- bility can be summarized as follows.
In the 4,6-diHPP-inactivated enzyme, molybdenum is present in a reduced form (5, 6). Furthermore, with such enzyme, the spectral shift occurs only as the molybdenum is reoxidized (Fig. 6). These experiments could be interpreted as indicating that DC1 interacts with enzyme molybdenum when the latter is in the oxidized form (Mov’), but not when in the reduced form (MoIV or MoV).
DCI, in addition to acting as an electron acceptor for xanthine oxidase, is an effective inhibitor for the enzyme (Reference 12 and Fig. 7). I f DCI-MoV1 interaction takes place, it is likely that the molybdenum participating in such complex formation would be less readily reducible, or even nonreducible, by sub- strate. As a consequence, the total concentration of catalyti- cally active, substrate-reducible enzyme would be lowered and inhibition would result.
DCI-xanthine oxidase interaction is blocked by arsenite, and, as noted above, arsenite appears to bind the enzyme molybde- num. Thus, the interference with DCI-enzyme complex forma- tion by arsenite could result from competition between arsenite and DC1 for molybdenum. It is of interest in this connection that these two possibilities for the DCI-binding site considered above (i.e. enzyme molybdenum and enzyme sulfur) are not mutually exclusive alternatives. Bray et al. (25) have suggested the presence of a molybdenum-sulfur linkage in the noncycling enzyme. If the interaction with DC1 results from complex formation between the dye and one of the members of this link- age, an influence on the other member of the pair would be anticipated, even if actual cleavage of the molybdenum-sulfur linkage as a result of the DC1 binding does not occur.
Irrespective of the nature of the group involved in DC1 bind- ing, it is likely that such binding takes place at or in close juxta-
by guest on April 13, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Issue of January 25, 1971 H. L. Gurtoo and D. G. Johns 293
position to the substrate-binding site of the enzyme. Evidence supporting DC1 binding at or near the substrate site is the ob- servation that the DC1 spectral shift is blocked by the competi- tive inhibitor ammeline, and also the earlier observation of Fridovich (12) that, by kinetic criteria, the inhibition by DC1 is competitive with the substrate salicylaldehyde.
Of considerable interest is the observation that the DCI-en- zyme interaction is not prevented by prior treatment of the en- zyme with cyanide. That cyanide binds to and inhibits xanthine oxidase has been known for many years; the stoichiometry ap- pears to be 2 molecules of cyanide bound per molecule of enzyme (26). The site of inhibition by cyanide is not fully established, however; Coughlan et al. (21) have suggested that cyanide, like arsenite, binds the enzyme molybdenum. Interaction of cyanide with an enzyme disulfide linkage, or with the enzyme flavin, is also a possibility (22, 27). From the present observation that cyanide does not prevent the DCI-induced spectral change, and that DC1 does not protect against cyanide inhibition (Table I), it would appear that cyanide and DC1 react with different groups. Since DC1 and arsenite appear to share the same binding sit,e, it would follow from these observations that arsenite and cyanide do not bind at the same locus, a conclusion somewhat at variance with that of Coughlan et al. (21).
The other major point of interest concerning the DCI-xan- thine oxidase interaction is whether the spectrally demonstrable DCI-enzyme complex represents the Michaelis complex between enzyme and DC1 which participates in xanthine oxidase-cata- lyzed oxidations when DC1 is used as acceptor. The evidence would appear to be against this possibility.
As shown by Fridovich (12), the value of the Km of DC1 when considered as an oxidizing substrate for the enzyme (approxi- mately 1.5 x lo-’ M) is of a different order of magnitude from Dhe value 0” the Ki of DC1 as a xanthine oxidase inhibitor (approximately 1 X 1OV M).
In the 4,6-diHPP-inhibited enzyme, the spectral change characteristic of DC1 complex formation is not detectable, but electron transfer from the reduced molybdenum to the dye still takes place, i.e. spectrally detectable DCI-enzyme complex formation is not obligatory for electron transfer from the enzyme to DCI.
Another point of interest arising from the experiments with 4,6-diHPP-inhibited enzyme is the requirement of approxi- mately 1 mole of DC1 to bring about reoxidation of the reduced molybdenum of the 4,6-diHPP-reduced enzyme complex. Massey et al. (6), on the basis of ferricyanide titration experi- ments, have suggested that the molybdenum of the 4,6-diHPP- inhibited enzyme is present in the MoIv form. In the latter experiments, the interpretation of the ferricyanide titration data was complicated, however, by the simultaneous reduction of this agent in a competing side reaction with enzyme sulfhydryls. Assuming that both molybdenum atoms of xanthine oxidase combine with 4,8diHPP, and that all the enzyme is in the form of the xanthine oxidase-4,6-diHPP complex, the stoichiometry of DC1 reduction in the present experiments would indicate that the 4,6-diHPP-complexed molybdenum is in the Mov rather than the MoIV oxidation state.
Whether these observations of DCI-xanthine oxidase complex formation have a wider relevance is not known. DC1 is widely used in enzymology as an electron acceptor, but other reported
instances of DC1 inhibition appear to be rare. Hadler et al. (10) have shown that DC1 will bind to mitochondrial proteins, and inhibition by DC1 of 32Pi-ATP exchange catalyzed by mito- chondria has been reported by Lijw et al. (28). Gross, Wagen- knecht, and Hahn (29) have reported inhibition by DC1 of a soluble NADH dehydrogenase from rat liver (EC 1.6.99.3) ; as with xanthine oxidase, the dye served also as an electron ac- ceptor for the enzyme. The latter authors did not carry out spectrophotometric studies of the DC1 binding; the dissociation constant of the DCI-enzyme complex was found by kinetic methods to be 3 X lo-+ M, i.e. similar in magnitude to that found for the DCI-xanthine oxidase complex in the studies of Fridovich (12) and in the present study.
Acknozuledgment-The valuable technical assistance of Miss Barbara Moroson is gratefully acknowledged.
1.
2.
3. 4.
5.
6.
7.
8.
9.
10.
11.
12. 13.
14.
15. 16. 17. 18.
19. 20. 21.
22.
23. 24.
25.
26.
27.
28.
29.
REFERENCES AVIS, P. G., BERGEL, F., AND BRAY, R. C., J. Chem. Sot.,
1219 (1956). BRAY, R. C., PALMER, G., AND BEINERT, H., J. Biol. Chem.,
239, 2667 (1964). PALMER, G., AND MASSEY, V., J. Biol. Chem., 244, 2614 (1969). KOMAI. H., MASSEY, V., AND PALMER. G.. J. Biol. Chem.. 244.
1692 ‘(1969). ’ I I , I
SPECTOR, T., AND JOHNS, D. G., Biochem. Biophys. Res. Com- mun., 38, 583 (1970).
MASSEY, V., KOMAI, H., PALMER, G., AND ELION. G. B.. J. Biol. Chem., 246,2$37 (1970).
HADLER. H. I.. ERWIN. M. J.. AND LARDY. H. A.. J. Amer. Chem.‘Soc., &, 458 (i963). ’
COFFEY, D. S., AND HELLERMAN, L., Biochemistry, 3, 394 (19G4).
COFFEY, D. S., AND HELLERMAN, L., Biochim. Biophys. Acta, 100, 98 (1965).
HADLER, H. I., ALT, S. K., AND FALCONE, A. B., J. Biol. Chem., 241, 2886 (1966).
JOHNS, D. G., Biochem. Biophys. Res. Commun., 31, 197 (1968).
FRIDOVICH, I., J. Biol. Chem., 241, 3624 (1966). GURTOO, H. L., AND JOHNS, D. G., Abstracts of the American
Chemical Society Meeting, Chicago, September 19Y0, p. 165B. HART, L. I., MCGARTOLL, M. A., CHAPMAN, H. R., AND BRAY,
B. C., Biochem. J., 116, 851 (1970). FRIDOVICH, I., J. Biol. Chem., 237, 584 (1962). GILBERT, D. A., AND BERGEL, F., Biochem. J., 90, 350 (1964). SPECTOR, T., AND JOHNS, D. G., J. Biol. Chem., 246,5079 (1970). JOHNS, D. G., SPECTOR, T., AND ROBINS, R. K., Biochem.
Phurmacol., 18, 2371 (1969). MEITES, L., AND MEITES, T., Anal. Chem., 20, 984 (1948). FRIDOVICH, I., Biochemistry, 4, 1098 (19G5). COUGHLAN, M. P., RAJAGOPALAN, K. V., AND HANDLER, P.,
J. Biol. Chem., 244, 2658 (1969). RAJAGOPALAN, K. V., AND H.QNDLER, P., J. Biol. Chem., 242,
4097 (1967). BERGEN, F.,.AND BRAY, R. C., Biochem. J., 73, 182 (1959). BRUMBY, P. E., MILLER, R. W., AND MASSEY, V., J. Biol.
Chem., 240, 2222 (1965). BRAY, R. C., KNOWLES, P. F., PICK, F. M., AND VXNNGHRD,
T., Biochem. J., 107, 601 (1968). FRIDOVICH, I., AND HANDLER, P., J. Biol. Chem., 231, 899
(1958). BRAY, R. C., in P. 0. BOYER, H. LARDY, AND K. MYRBXCK
(Editors), The enzymes, Tiol. 7, Academic Press, New York, 1963, p. 533.
Law, H., SIEKEVITZ, P., ERNSTER, L., AND LINDBERG, O., Biochim. BioDhus. Acta, 29. 392 (19581.
GROSS, J., WA~E~KNECH~, C:, AND‘HA~N, V., Acta Biol. Med. Ger., 22, 241 (1969).
by guest on April 13, 2018
http://ww
w.jbc.org/
Dow
nloaded from

H. L. Gurtoo and D. G. JohnsBovine Milk Xanthine Oxidase
On the Interaction of the Electron Acceptor 2,6-Dichlorophenolindophenol with
1971, 246:286-293.J. Biol. Chem.
http://www.jbc.org/content/246/2/286Access the most updated version of this article at
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/246/2/286.full.html#ref-list-1
This article cites 0 references, 0 of which can be accessed free at
by guest on April 13, 2018
http://ww
w.jbc.org/
Dow
nloaded from