osmosis
TRANSCRIPT

OsmosisOsmosis is the net movement of solvent molecules through a partially permeable membrane into a region of higher solute concentration, in order to equalize the solute concentrations on the two sides.[1][2][3] It may also be used to describe a physical process in which any solvent moves, without input of energy,[4] across a semipermeable membrane (permeable to the solvent, but not the solute) separating two solutions of different concentrations.[5] Although osmosis does not require input of energy, it does use kinetic energy [6] and can be made to do work.[7]
One frame of a computer simulation of osmosis
Net movement of solvent is from the less concentrated (hypotonic) to the more concentrated (hypertonic) solution, which tends to reduce the difference in concentrations. This effect can be countered by increasing the pressure of the hypertonic solution, with respect to the hypotonic. The osmotic pressure is defined to be the pressure required to maintain an equilibrium, with no net movement of solvent. Osmotic pressure is a colligative property, meaning that the osmotic pressure depends on the molar concentration of the solute but not on its identity.
Osmosis is essential in biological systems, as biological membranes are semipermeable. In general, these membranes are impermeable to large and polar molecules, such as ions, proteins, and polysaccharides, while being permeable to non-polar and/or hydrophobic molecules like lipids as well as to small molecules like oxygen, carbon dioxide, nitrogen, nitric oxide, etc. Permeability depends on solubility, charge, or chemistry, as well as solute size. Water molecules travel through the plasma membrane, tonoplast membrane (vacuole) or protoplast by diffusing
across the phospholipid bilayer via aquaporins (small transmembrane proteins similar to those in facilitated diffusion and in creating ion channels). Osmosis provides the primary means by which water is transported into and out of cells. The turgor pressure of a cell is largely maintained by osmosis, across the cell membrane, between the cell interior and its relatively hypotonic environment.[8]
Jean-Antoine Nollet first documented observation of osmosis in 1748.[9] The word "osmosis" descends from the words "endosmose" and "exosmose", which were coined by French physician René Joachim Henri Dutrochet (1776–1847) from the Greek words ένδον (endon : within), έξο (exo : outside), and ωσμος (osmos : push, impulsion).[10][11][12][13][14]
Basic explanations
Osmosis may occur when there is a partially permeable membrane, such
as a cell membrane. When a cell is submerged in water, the water
molecules pass through the cell membrane from an area of low solute
concentration (outside the cell) to one of high solute concentration (inside
the cell); this is called osmosis. The cell membrane is selectively
permeable, so only necessary materials are let into the cell and wastes are
left out.[8]

Water passing through a semi-permeable membrane to create an isotonic environment
When the membrane has a volume of pure water on both sides, water
molecules pass in and out in each direction at exactly the same rate; there
is no net flow of water through the membrane.
Osmosis can be explained using the concept of thermodynamic free
energy: the less concentrated solution contains more free energy, so its
solvent molecules will tend to diffuse to a place of lower free energy in
order to equalize free energy. Since the semipermeable membrane only
allows solvent molecules to pass through it, the result is a net flow of water
to the side with the more concentrated solution. Assuming the membrane
does not break, this net flow will slow and finally stop as the pressure on
the more concentrated side lessens and the movement in each direction
becomes equal: this state is called dynamic equilibrium.
Osmosis can also be explained using the notion of entropy, from statistical
mechanics. A system that has two solutions of different concentrations
separated by a semipermeable membrane has less entropy than a similar
system having two solutions of equal concentration. The system with the
differing concentrations is said to be more ordered, and thus has less
entropy. The second law of thermodynamics requires the presence of an
osmotic flow that will take the system from an ordered state of low entropy
to a disordered state of higher entropy. Thermodynamic equilibrium is
achieved when the entropy gradient between the two solutions becomes
zero.
Particle size has no bearing on osmotic pressure; this is the fundamental
postulate of colligative properties.[15]
Examples of osmosis
Effect of different solutions on blood cells
Plant cell under different environments

Osmotic pressure is the main cause of support in many plants. The
osmotic entry of water raises the turgor pressure exerted against the cell
wall, until it equals the osmotic pressure, creating a steady state.
When a plant cell is placed in a hypertonic solution, the water in the cells
moves to an area higher in solute concentration and the cell shrinks, and
in doing so, becomes flaccid. This means the cell has
become plasmolyzed – the cell membrane has completely left the cell wall
due to lack of water pressure on it; the opposite of turgid.
Also, osmosis is responsible for the ability of plant roots to draw water from
the soil. Since there are many fine roots, they have a large surface area,
and water enters the roots by osmosis.
Osmosis can also be seen when potato slices are added to a high
concentration of salt solution. The water from inside the potato moves to
the salt solution, causing the potato to shrink and to lose its 'turgor
pressure'. The more concentrated the salt solution, the bigger the
difference in size and weight of the potato slice.
In unusual environments, osmosis can be very harmful to organisms. For
example, freshwater and saltwater aquarium fish placed in water of a
different salinity than that to which they are adapted to will die quickly, and
in the case of saltwater fish, dramatically. Another example of a harmful
osmotic effect is the use of table salt to kill leeches and slugs.
Suppose an animal or a plant cell is placed in a solution of sugar or salt in
water.
1. If the medium is hypotonic — a dilute solution, with a higher water
concentration than the cell — the cell will gain water through
osmosis.
2. If the medium is isotonic — a solution with exactly the same water
concentration as the cell — there will be no net movement of
water across the cell membrane.
3. If the medium is hypertonic — a concentrated solution, with a
lower water concentration than the cell — the cell will lose water
by osmosis.
Essentially, this means that if a cell is put in a solution which has a solute
concentration higher than its own, then it will shrivel up, and if it is put in a
solution with a lower solute concentration than its own, the cell will expand
and burst. Electronucleal exchange is the
passive diffusion of cations and anions across a semi-permeable
membrane according to electrical charge.
Chemical gardens demonstrate the effect of osmosis in inorganic
chemistry.
Factors
Osmotic pressure
Main article: Osmotic pressure
As mentioned before, osmosis may be opposed by increasing the pressure
in the region of high solute concentration with respect to that in the low
solute concentration region. The force per unit area, or pressure, required
to prevent the passage of water through a selectively permeable
membrane and into a solution of greater concentration is equivalent to the
osmotic pressure of the solution, or turgor.Osmotic pressure is a colligative
property, meaning that the property depends on the concentration of the
solute, but not on its identity.

Osmotic gradient
The osmotic gradient is the difference in concentration between
two solutions on either side of a semipermeable membrane, and is used to
tell the difference in percentages of the concentration of a specific particle
dissolved in a solution.
Usually the osmotic gradient is used while comparing solutions that have a
semipermeable membrane between them allowing water to diffuse
between the two solutions, toward the hypertonic solution (the solution with
the higher concentration). Eventually, the force of the column of water on
the hypertonic side of the semipermeable membrane will equal the force of
diffusion on the hypotonic (the side with a lesser concentration) side,
creating equilibrium. When equilibrium is reached, water continues to flow,
but it flows both ways in equal amounts as well as force, therefore
stabilizing the solution.
Variation
Reverse osmosis
Main article: Reverse osmosis
Reverse osmosis is a separation process that uses pressure to force a
solvent through a semi-permeable membrane that retains the solute on
one side and allows the pure solvent to pass to the other side. More
formally, it is the process of forcing a solvent from a region of high solute
concentration through a membrane to a region of low solute concentration
by applying a pressure in excess of the osmotic pressure.
Forward osmosis
Main article: Forward osmosis
Osmosis may be used directly to achieve separation of water from a "feed"
solution containing unwanted solutes. A "draw" solution of higher osmotic
pressure than the feed solution is used to induce a net flow of water
through a semi-permeable membrane, such that the feed solution
becomes concentrated as the draw solution becomes dilute. The diluted
draw solution may then be used directly (as with an ingestible solute like
glucose), or sent to a secondary separation process for the removal of the
draw solute. This secondary separation can be more efficient than a
reverse osmosis process would be alone, depending on the draw solute
used and the feedwater treated. Forward osmosis is an area of ongoing
research, focusing on applications in desalination, water purification, water
treatment, food processing, etc.

Enzyme"Biocatalyst" redirects here. For the use of natural catalysts in organic
chemistry, see Biocatalysis.
Human glyoxalase I. Two zinc ions that are needed for the enzyme to catalyze its
reaction are shown as purple spheres, and an enzyme inhibitor called S-
hexylglutathione is shown as a space-filling model, filling the two active sites.
Enzymes are biological molecules that catalyze (i.e., increase the
rates of) chemical reactions.[1][2] In enzymatic reactions, themolecules at
the beginning of the process, called substrates, are converted into different
molecules, called products. Almost all chemical reactions in abiological
cell need enzymes in order to occur at rates sufficient for life. Since
enzymes are selective for their substrates and speed up only a few
reactions from among many possibilities, the set of enzymes made in a
cell determines which metabolic pathways occur in that cell.
Like all catalysts, enzymes work by lowering the activation energy (Ea‡) for
a reaction, thus dramatically increasing the rate of the reaction. As a result,
products are formed faster and reactions reach their equilibrium state more
rapidly. Most enzyme reaction rates are millions of times faster than those
of comparable un-catalyzed reactions. As with all catalysts, enzymes are
not consumed by the reactions they catalyze, nor do they alter
theequilibrium of these reactions. However, enzymes do differ from most
other catalysts in that they are highly specific for their substrates. Enzymes
are known to catalyze about 4,000 biochemical reactions.[3] A
few RNA molecules called ribozymes also catalyze reactions, with an
important example being some parts of the ribosome.[4][5] Synthetic
molecules called artificial enzymes also display enzyme-like catalysis.[6]
Enzyme activity can be affected by other molecules. Inhibitors are
molecules that decrease enzyme activity; activators are molecules that
increase activity. Many drugs and poisons are enzyme inhibitors. Activity is
also affected by temperature, pressure, chemical environment (e.g., pH),
and theconcentration of substrate. Some enzymes are used commercially,
for example, in the synthesis of antibiotics. In addition, some household
products use enzymes to speed up biochemical reactions (e.g., enzymes
in biological washing powders break down protein or fat stains on clothes;
enzymes inmeat tenderizers break down proteins into smaller molecules,
making the meat easier to chew).
Structures and mechanisms
Ribbon diagram showing human carbonic anhydrase II. The grey sphere is
the zinc cofactor in the active site. Diagram drawn from PDB 1MOO.
Enzymes are in general globular proteins and range from just 62 amino
acid residues in size, for the monomer of 4-oxalocrotonate tautomerase,[17] to over 2,500 residues in the animal fatty acid synthase.[18] A small
number of RNA-based biological catalysts exist, with the most common
being theribosome; these are referred to as either RNA-enzymes
or ribozymes. The activities of enzymes are determined by their three-
dimensional structure.[19]However, although structure does determine
function, predicting a novel enzyme's activity just from its structure is a
very difficult problem that has not yet been solved.[20]
Most enzymes are much larger than the substrates they act on, and only a
small portion of the enzyme (around 2–4 amino acids) is directly involved
in catalysis.[21] The region that contains these catalytic residues, binds the
substrate, and then carries out the reaction is known as the active site.
Enzymes can also contain sites that bind cofactors, which are needed for
catalysis. Some enzymes also have binding sites for small molecules,
which are often direct or indirect products or substrates of the reaction

catalyzed. This binding can serve to increase or decrease the enzyme's
activity, providing a means for feedback regulation.
Like all proteins, enzymes are long, linear chains of amino acids
that fold to produce a three-dimensional product. Each unique amino acid
sequence produces a specific structure, which has unique properties.
Individual protein chains may sometimes group together to form a protein
complex. Most enzymes can be denatured—that is, unfolded and
inactivated—by heating or chemical denaturants, which disrupt the three-
dimensional structure of the protein. Depending on the enzyme,
denaturation may be reversible or irreversible.
Structures of enzymes with substrates or substrate analogs during a
reaction may be obtained using Time resolved crystallography methods.
Specificity
Enzymes are usually very specific as to which reactions they catalyze and
the substrates that are involved in these reactions. Complementary shape,
charge and hydrophilic/hydrophobic characteristics of enzymes and
substrates are responsible for this specificity. Enzymes can also show
impressive levels of stereospecificity, regioselectivity andchemoselectivity.[22]
Some of the enzymes showing the highest specificity and accuracy are
involved in the copying and expression of the genome. These enzymes
have "proof-reading" mechanisms. Here, an enzyme such asDNA
polymerase catalyzes a reaction in a first step and then checks that the
product is correct in a second step.[23] This two-step process results in
average error rates of less than 1 error in 100 million reactions in high-
fidelity mammalian polymerases.[24] Similar proofreading mechanisms are
also found in RNA polymerase,[25] aminoacyl tRNA
synthetases [26] and ribosomes.[27]
Some enzymes that produce secondary metabolites are described as
promiscuous, as they can act on a relatively broad range of different
substrates. It has been suggested that this broad substrate specificity is
important for the evolution of new biosynthetic pathways.
"Lock and key" model
Enzymes are very specific, and it was suggested by the Nobel
laureate organic chemist Emil Fischer in 1894 that this was because both
the enzyme and the substrate possess specific complementary geometric
shapes that fit exactly into one another.[29] This is often referred to as "the
lock and key" model. However, while this model explains enzyme
specificity, it fails to explain the stabilization of the transition state that
enzymes achieve.
Diagrams to show the induced fit hypothesis of enzyme action
In 1958, Daniel Koshland suggested a modification to the lock and key
model: since enzymes are rather flexible structures, the active site is
continuously reshaped by interactions with the substrate as the substrate
interacts with the enzyme.[30] As a result, the substrate does not simply
bind to a rigid active site; the amino acid side-chains that make up the

active site are molded into the precise positions that enable the enzyme to
perform its catalytic function. In some cases, such as glycosidases, the
substrate molecule also changes shape slightly as it enters the active site.[31] The active site continues to change until the substrate is completely
bound, at which point the final shape and charge is determined.[32] Induced
fit may enhance the fidelity of molecular recognition in the presence of
competition and noise via the conformational proofreadingmechanism.[33]
Mechanisms
Enzymes can act in several ways, all of which lower ΔG‡ (Gibbs energy):[34]
Lowering the activation energy by creating an environment in which
the transition state is stabilized (e.g. straining the shape of a substrate
—by binding the transition-state conformation of the substrate/product
molecules, the enzyme distorts the bound substrate(s) into their
transition state form, thereby reducing the amount of energy required
to complete the transition).
Lowering the energy of the transition state, but without distorting the
substrate, by creating an environment with the opposite charge
distribution to that of the transition state.
Providing an alternative pathway. For example, temporarily reacting
with the substrate to form an intermediate ES complex, which would
be impossible in the absence of the enzyme.
Reducing the reaction entropy change by bringing substrates together
in the correct orientation to react. Considering ΔH‡ alone overlooks
this effect.
Increases in temperatures speed up reactions. Thus, temperature
increases help the enzyme function and develop the end product even
faster. However, if heated too much, the enzyme’s shape deteriorates
and the enzyme becomes denatured. Some enzymes like thermolabile
enzymes work best at low temperatures.
It is interesting that this entropic effect involves destabilization of the
ground state,[35] and its contribution to catalysis is relatively small.[36]
Transition state stabilization
The understanding of the origin of the reduction of ΔG‡ requires one to find
out how the enzymes can stabilize its transition state more than the
transition state of the uncatalyzed reaction. It seems that the most effective
way for reaching large stabilization is the use of electrostatic effects, in
particular, when having a relatively fixed polar environment that is oriented
toward the charge distribution of the transition state.[37] Such an
environment does not exist in the uncatalyzed reaction in water.
Dynamics and function
See also: Protein dynamics
The internal dynamics of enzymes has been suggested to be linked with
their mechanism of catalysis.[38][39][40] Internal dynamics are the movement
of parts of the enzyme's structure, such as individual amino acid residues,
a group of amino acids, or even an entire protein domain. These
movements occur at various time-scales ranging from femtoseconds to
seconds. Networks of protein residues throughout an enzyme's structure
can contribute to catalysis through dynamic motions.[41][42][43][44] This is
simply seen in the kinetic scheme of the combined process, enzymatic
activity and dynamics; this scheme can have several
independent Michaelis-Menten-like reaction pathways that are connected
through fluctuation rates.[45][46][47]
Protein motions are vital to many enzymes, but whether small and fast
vibrations, or larger and slower conformational movements are more
important depends on the type of reaction involved. However, although

these movements are important in binding and releasing substrates and
products, it is not clear if protein movements help to accelerate the
chemical steps in enzymatic reactions.[48] These new insights also have
implications in understanding allosteric effects and developing new
medicines.
Allosteric modulation
Allosteric transition of an enzyme between R and T states, stabilized by an agonist, an
inhibitor and a substrate (the MWC model)
Main article: Allosteric regulation
Allosteric sites are sites on the enzyme that bind to molecules in the
cellular environment. The sites form weak, noncovalent bonds with these
molecules, causing a change in the conformation of the enzyme. This
change in conformation translates to the active site, which then affects the
reaction rate of the enzyme.[49] Allosteric interactions can both inhibit and
activate enzymes and are a common way that enzymes are controlled in
the body.[50]
Cofactors and coenzymes
Main articles: Cofactor (biochemistry) and Coenzyme
Cofactors
Some enzymes do not need any additional components to show full
activity. However, others require non-protein molecules called cofactors to
be bound for activity.[51] Cofactors can be either inorganic (e.g., metal
ions and iron-sulfur clusters) or organic
compounds (e.g., flavin and heme). Organic cofactors can be
either prosthetic groups, which are tightly bound to an enzyme,
or coenzymes, which are released from the enzyme's active site during the
reaction. Coenzymes include NADH, NADPH and adenosine triphosphate.
These molecules transfer chemical groups between enzymes.[52]
An example of an enzyme that contains a cofactor is carbonic anhydrase,
and is shown in the ribbon diagram above with a zinc cofactor bound as
part of its active site.[53] These tightly bound molecules are usually found in
the active site and are involved in catalysis. For example, flavin and heme
cofactors are often involved in redox reactions.
Enzymes that require a cofactor but do not have one bound are
called apoenzymes or apoproteins. An apoenzyme together with its
cofactor(s) is called a holoenzyme (this is the active form). Most cofactors
are not covalently attached to an enzyme, but are very tightly bound.
However, organic prosthetic groups can be covalently bound (e.g., biotin in
the enzyme pyruvate carboxylase). The term "holoenzyme" can also be
applied to enzymes that contain multiple protein subunits, such as
the DNA polymerases; here the holoenzyme is the complete complex
containing all the subunits needed for activity.

Coenzymes
Space-filling model of the coenzyme NADH
Coenzymes are small organic molecules that can be loosely or tightly
bound to an enzyme. Tightly bound coenzymes can be called allosteric
groups. Coenzymes transport chemical groups from one enzyme to
another.[54] Some of these chemicals such as riboflavin, thiamine and folic
acid are vitamins (compounds that cannot be synthesized by the body and
must be acquired from the diet). The chemical groups carried include
the hydride ion (H-) carried by NAD or NADP + , the phosphate group carried
by adenosine triphosphate, the acetyl group carried by coenzyme A,
formyl, methenyl or methyl groups carried by folic acid and the methyl
group carried by S-adenosylmethionine.
Since coenzymes are chemically changed as a consequence of enzyme
action, it is useful to consider coenzymes to be a special class of
substrates, or second substrates, which are common to many different
enzymes. For example, about 700 enzymes are known to use the
coenzyme NADH.[55]
Coenzymes are usually continuously regenerated and their concentrations
maintained at a steady level inside the cell: for example, NADPH is
regenerated through the pentose phosphate pathway and S-
adenosylmethionine by methionine adenosyltransferase. This continuous
regeneration means that even small amounts of coenzymes are used very
intensively. For example, the human body turns over its own weight in ATP
each day.[56]
Thermodynamics
The energies of the stages of a chemical reaction. Substrates need a lot of potential
energy to reach a transition state, which then decays into products. The enzyme
stabilizes the transition state, reducing the energy needed to form products.
Main articles: Activation energy, Thermodynamic equilibrium,
and Chemical equilibrium
As all catalysts, enzymes do not alter the position of the chemical
equilibrium of the reaction. Usually, in the presence of an enzyme, the
reaction runs in the same direction as it would without the enzyme, just
more quickly. However, in the absence of the enzyme, other possible
uncatalyzed, "spontaneous" reactions might lead to different products,
because in those conditions this different product is formed faster.
Furthermore, enzymes can couple two or more reactions, so that a
thermodynamically favorable reaction can be used to "drive" a
thermodynamically unfavorable one. For example, the hydrolysis of ATP is
often used to drive other chemical reactions.[57]

Enzymes catalyze the forward and backward reactions equally. They do
not alter the equilibrium itself, but only the speed at which it is reached. For
example, carbonic anhydrase catalyzes its reaction in either direction
depending on the concentration of its reactants.
(in tissues;
high CO2 concentration)
(in lungs; low
CO2 concentration)
Nevertheless, if the equilibrium is greatly displaced in one
direction, that is, in a very exergonic reaction, the reaction is in
effect irreversible. Under these conditions, the enzyme will, in fact,
catalyze the reaction only in the thermodynamically allowed
direction.
Kinetics
Main article: Enzyme kinetics
Mechanism for a single substrate enzyme catalyzed reaction. The enzyme (E)
binds a substrate (S) and produces a product (P).
Enzyme kinetics is the investigation of how enzymes bind
substrates and turn them into products. The rate data used in
kinetic analyses are commonly obtained from enzyme assays,
where since the 90s, the dynamics of many enzymes are studied
on the level of individual molecules.
In 1902 Victor Henri proposed a quantitative theory of enzyme
kinetics,[58] but his experimental data were not useful because the
significance of the hydrogen ion concentration was not yet
appreciated. After Peter Lauritz Sørensen had defined the
logarithmic pH-scale and introduced the concept of buffering in
1909[59] the German chemist Leonor Michaelis and his Canadian
postdoc Maud Leonora Menten repeated Henri's experiments and
confirmed his equation, which is referred to as Henri-Michaelis-
Menten kinetics (termed also Michaelis-Menten kinetics).[60] Their
work was further developed by G. E. Briggs and J. B. S. Haldane,
who derived kinetic equations that are still widely considered
today a starting point in solving enzymatic activity.[61]
The major contribution of Henri was to think of enzyme reactions
in two stages. In the first, the substrate binds reversibly to the
enzyme, forming the enzyme-substrate complex. This is
sometimes called the Michaelis complex. The enzyme then
catalyzes the chemical step in the reaction and releases the
product. Note that the simple Michaelis Menten mechanism for
the enzymatic activity is considered today a basic idea, where
many examples show that the enzymatic activity involves
structural dynamics. This is incorporated in the enzymatic
mechanism while introducing several Michaelis Menten pathways
that are connected with fluctuating rates.[45][46][47] Nevertheless,
there is a mathematical relation connecting the behavior obtained
from the basic Michaelis Menten mechanism (that was indeed

proved correct in many experiments) with the generalized
Michaelis Menten mechanisms involving dynamics and
activity; [62] this means that the measured activity of enzymes on
the level of many enzymes may be explained with the simple
Michaelis-Menten equation, yet, the actual activity of enzymes is
richer and involves structural dynamics.
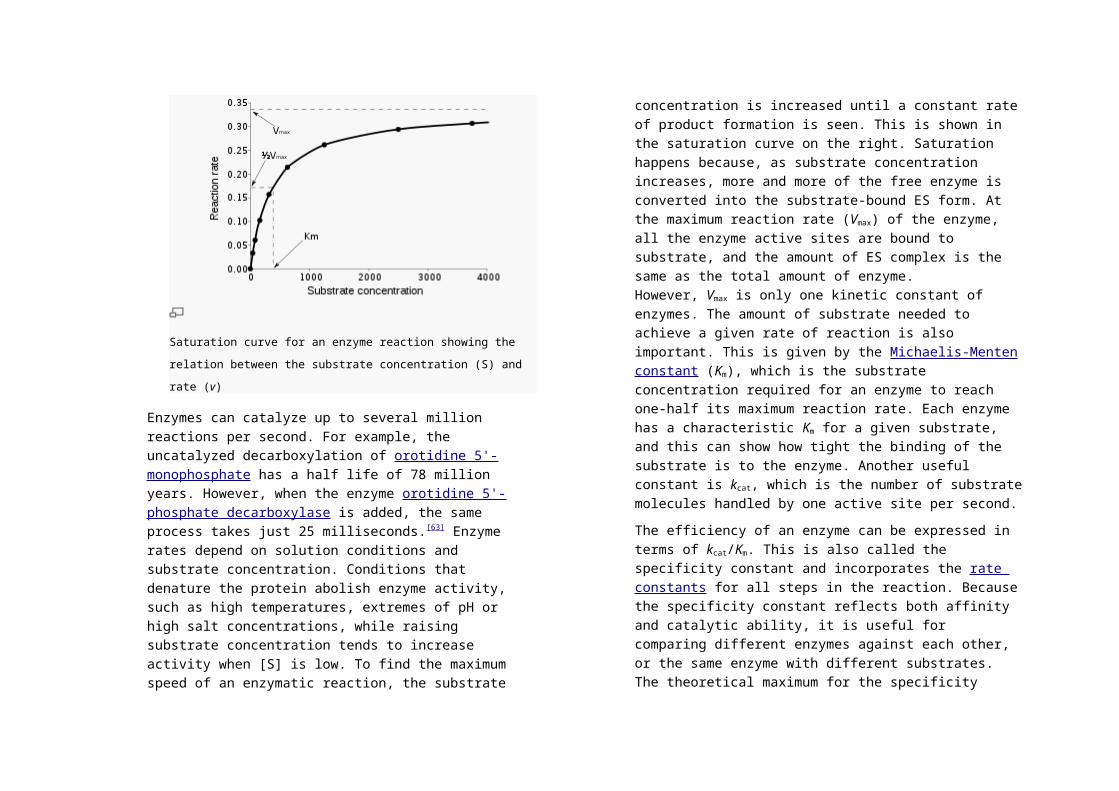
Saturation curve for an enzyme reaction showing the relation between
the substrate concentration (S) and rate (v)
Enzymes can catalyze up to several million reactions per second.
For example, the uncatalyzed decarboxylation of orotidine 5'-
monophosphate has a half life of 78 million years. However, when
the enzyme orotidine 5'-phosphate decarboxylase is added, the
same process takes just 25 milliseconds.[63] Enzyme rates depend
on solution conditions and substrate concentration. Conditions
that denature the protein abolish enzyme activity, such as high
temperatures, extremes of pH or high salt concentrations, while
raising substrate concentration tends to increase activity when [S]
is low. To find the maximum speed of an enzymatic reaction, the
substrate concentration is increased until a constant rate of
product formation is seen. This is shown in the saturation curve
on the right. Saturation happens because, as substrate
concentration increases, more and more of the free enzyme is
converted into the substrate-bound ES form. At the maximum
reaction rate (Vmax) of the enzyme, all the enzyme active sites are
bound to substrate, and the amount of ES complex is the same as
the total amount of enzyme. However, Vmax is only one kinetic
constant of enzymes. The amount of substrate needed to achieve
a given rate of reaction is also important. This is given by
the Michaelis-Menten constant (Km), which is the substrate
concentration required for an enzyme to reach one-half its
maximum reaction rate. Each enzyme has a characteristic Km for
a given substrate, and this can show how tight the binding of the
substrate is to the enzyme. Another useful constant is kcat, which
is the number of substrate molecules handled by one active site
per second.
The efficiency of an enzyme can be expressed in terms of kcat/Km.
This is also called the specificity constant and incorporates
the rate constants for all steps in the reaction. Because the
specificity constant reflects both affinity and catalytic ability, it is
useful for comparing different enzymes against each other, or the
same enzyme with different substrates. The theoretical maximum
for the specificity constant is called the diffusion limit and is about
108 to 109 (M−1 s−1). At this point every collision of the enzyme with
its substrate will result in catalysis, and the rate of product
formation is not limited by the reaction rate but by the diffusion
rate. Enzymes with this property are called catalytically
perfect or kinetically perfect. Example of such enzymes are triose-
phosphate isomerase, carbonic

anhydrase, acetylcholinesterase, catalase, fumarase, β-
lactamase, and superoxide dismutase.
Michaelis-Menten kinetics relies on the law of mass action, which
is derived from the assumptions of free diffusion and
thermodynamically driven random collision. However, many
biochemical or cellular processes deviate significantly from these
conditions, because of macromolecular crowding, phase-
separation of the enzyme/substrate/product, or one or two-
dimensional molecular movement.[64] In these situations,
a fractal Michaelis-Menten kinetics may be applied.[65][66][67][68]
Some enzymes operate with kinetics, which are faster than
diffusion rates, which would seem to be impossible. Several
mechanisms have been invoked to explain this phenomenon.
Some proteins are believed to accelerate catalysis by drawing
their substrate in and pre-orienting them by using dipolar electric
fields. Other models invoke a quantum-
mechanical tunneling explanation, whereby a proton or an
electron can tunnel through activation barriers, although for proton
tunneling this model remains somewhat controversial.[69]
[70] Quantum tunneling for protons has been observed
in tryptamine.[71] This suggests that enzyme catalysis may be
more accurately characterized as "through the barrier" rather than
the traditional model, which requires substrates to go "over" a
lowered energy barrier.
Inhibition
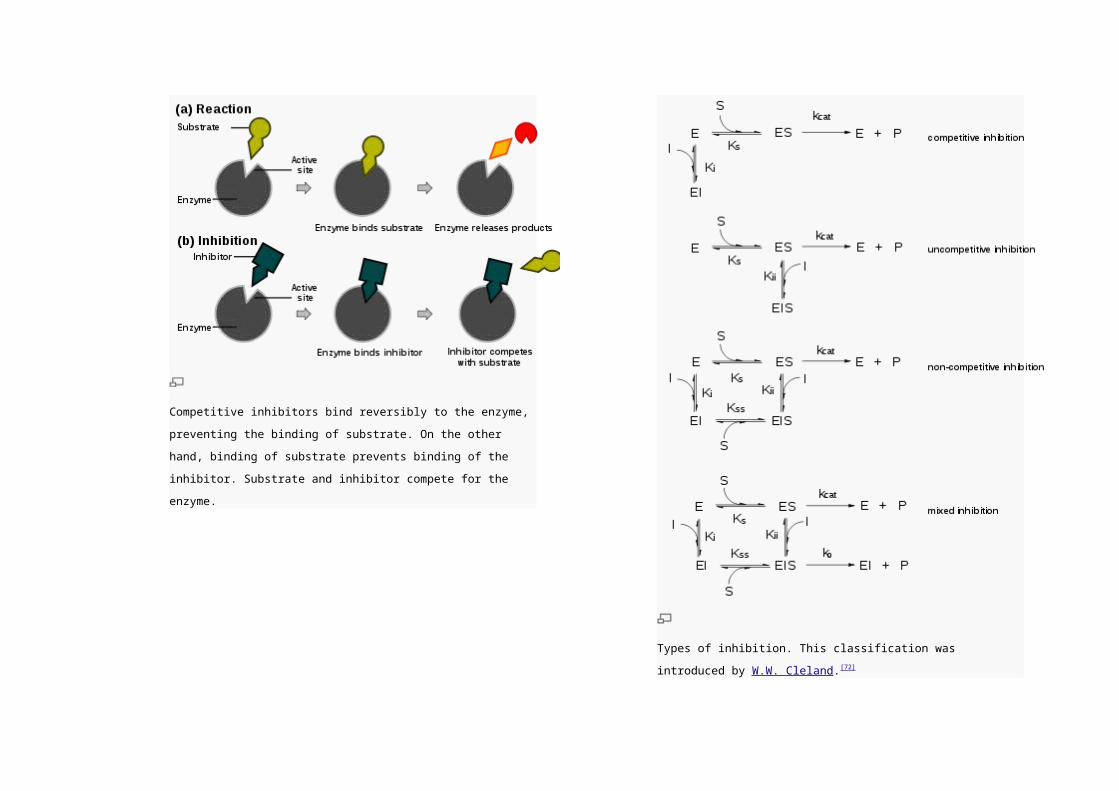
Competitive inhibitors bind reversibly to the enzyme, preventing the
binding of substrate. On the other hand, binding of substrate prevents
binding of the inhibitor. Substrate and inhibitor compete for the enzyme.

Types of inhibition. This classification was introduced by W.W. Cleland.[72]
Main article: Enzyme inhibitor
Enzyme reaction rates can be decreased by various types
of enzyme inhibitors.
Competitive inhibition
In competitive inhibition, the inhibitor and substrate compete for
the enzyme (i.e., they can not bind at the same time).[73] Often
competitive inhibitors strongly resemble the real substrate of the
enzyme. For example, methotrexate is a competitive inhibitor of
the enzyme dihydrofolate reductase, which catalyzes the
reduction of dihydrofolate to tetrahydrofolate. The similarity
between the structures of folic acid and this drug are shown in the
figure to the right bottom. In some cases, the inhibitor can bind to
a site other than the binding-site of the usual substrate and exert
an allosteric effect to change the shape of the usual binding-site.
For example,strychnine acts as an allosteric inhibitor of the
glycine receptor in the mammalian spinal cord and brain stem.
Glycine is a major post-synaptic inhibitory neurotransmitter with a
specific receptor site. Strychnine binds to an alternate site that
reduces the affinity of the glycine receptor for glycine, resulting in
convulsions due to lessened inhibition by the glycine.[74] In
competitive inhibition the maximal rate of the reaction is not
changed, but higher substrate concentrations are required to
reach a given maximum rate, increasing the apparent Km.
Uncompetitive inhibition
In uncompetitive inhibition, the inhibitor cannot bind to the free
enzyme, only to the ES-complex. The EIS-complex thus formed is
enzymatically inactive. This type of inhibition is rare, but may
occur in multimeric enzymes.
Non-competitive inhibition
Non-competitive inhibitors can bind to the enzyme at the binding
site at the same time as the substrate,but not to the active site.
Both the EI and EIS complexes are enzymatically inactive.
Because the inhibitor can not be driven from the enzyme by
higher substrate concentration (in contrast to competitive
inhibition), the apparent Vmax changes. But because the substrate
can still bind to the enzyme, the Km stays the same.
Mixed inhibition
This type of inhibition resembles the non-competitive, except that
the EIS-complex has residual enzymatic activity.This type of
inhibitor does not follow Michaelis-Menten equation.
In many organisms, inhibitors may act as part of
a feedback mechanism. If an enzyme produces too much of one
substance in the organism, that substance may act as an inhibitor
for the enzyme at the beginning of the pathway that produces it,
causing production of the substance to slow down or stop when
there is sufficient amount. This is a form of negative feedback.
Enzymes that are subject to this form of regulation are often
multimeric and have allosteric binding sites for regulatory
substances. Their substrate/velocity plots are not hyperbolar, but
sigmoidal (S-shaped).
Irreversible inhibitors react with the enzyme and form a covalent adduct
with the protein. The inactivation is irreversible. These compounds
include eflornithine a drug used to treat the parasitic disease sleeping
sickness.[75] Penicillin and Aspirin also act in this manner. With these
drugs, the compound is bound in the active site and the enzyme then
converts the inhibitor into an activated form that reacts irreversibly with one
or more amino acid residues.
Uses of inhibitors
Since inhibitors modulate the function of enzymes they are often
used as drugs. A common example of an inhibitor that is used as
a drug is aspirin, which inhibits the COX-1 and COX-2 enzymes
that produce the inflammation messenger prostaglandin, thus
suppressing pain and inflammation. However, other enzyme
inhibitors are poisons. For example, the poison cyanide is an
irreversible enzyme inhibitor that combines with the copper and
iron in the active site of the enzyme cytochrome c oxidase and
blocks cellular respiration.[76]
Biological function
Enzymes serve a wide variety of functions inside living organisms.
They are indispensable for signal transduction and cell regulation,
often via kinases and phosphatases.[77] They also generate
movement, with myosin hydrolyzing ATP to generate muscle
contractionand also moving cargo around the cell as part of
the cytoskeleton.[78] Other ATPases in the cell membrane are ion
pumps involved inactive transport. Enzymes are also involved in
more exotic functions, such as luciferase generating light
in fireflies.[79] Viruses can also contain enzymes for infecting cells,
such as the HIV integrase and reverse transcriptase, or for viral
release from cells, like theinfluenza virus neuraminidase.
An important function of enzymes is in the digestive systems of
animals. Enzymes such as amylases and proteases break down
large molecules (starch or proteins, respectively) into smaller
ones, so they can be absorbed by the intestines. Starch
molecules, for example, are too large to be absorbed from the
intestine, but enzymes hydrolyze the starch chains into smaller
molecules such asmaltose and eventually glucose, which can

then be absorbed. Different enzymes digest different food
substances. In ruminants, which have herbivorous diets,
microorganisms in the gut produce another enzyme, cellulase, to
break down the cellulose cell walls of plant fiber.[80]
Several enzymes can work together in a specific order, creating metabolic
pathways. In a metabolic pathway, one enzyme takes the product of
another enzyme as a substrate. After the catalytic reaction, the product is
then passed on to another enzyme. Sometimes more than one enzyme
can catalyze the same reaction in parallel; this can allow more complex
regulation: with, for example, a low constant activity provided by one
enzyme but an inducible high activity from a second enzyme.
Enzymes determine what steps occur in these pathways. Without
enzymes, metabolism would neither progress through the same
steps nor be fast enough to serve the needs of the cell. Indeed, a
metabolic pathway such asglycolysis could not exist
independently of enzymes. Glucose, for example, can react
directly with ATP to becomephosphorylated at one or more of its
carbons. In the absence of enzymes, this occurs so slowly as to
be insignificant. However, if hexokinase is added, these slow
reactions continue to take place except that phosphorylation at
carbon 6 occurs so rapidly that, if the mixture is tested a short
time later, glucose-6-phosphate is found to be the only significant
product. As a consequence, the network of metabolic pathways
within each cell depends on the set of functional enzymes that are
present.
Control of activity
There are five main ways that enzyme activity is controlled in the
cell.
1. Enzyme production (transcription and translation of
enzyme genes) can be enhanced or diminished by a cell
in response to changes in the cell's environment. This
form of gene regulation is called enzyme induction and
inhibition (see enzyme induction). For example, bacteria
may become resistant to antibiotics such
as penicillin because enzymes called beta-
lactamases are induced that hydrolyze the crucial beta-
lactam ring within the penicillin molecule. Another
example are enzymes in the liver called cytochrome
P450 oxidases, which are important in drug metabolism.
Induction or inhibition of these enzymes can cause drug
interactions.
2. Enzymes can be compartmentalized, with different
metabolic pathways occurring in different cellular
compartments. For example, fatty acids are synthesized
by one set of enzymes in the cytosol,endoplasmic
reticulum and the Golgi apparatus and used by a
different set of enzymes as a source of energy in
the mitochondrion, through β-oxidation.[81]
3. Enzymes can be regulated by inhibitors and activators.
For example, the end product(s) of a metabolic pathway
are often inhibitors for one of the first enzymes of the
pathway (usually the first irreversible step,
called committed step), thus regulating the amount of
end product made by the pathways. Such a regulatory
mechanism is called a negative feedback mechanism,
because the amount of the end product produced is
regulated by its own concentration. Negative feedback
mechanism can effectively adjust the rate of synthesis of
intermediate metabolites according to the demands of
the cells. This helps allocate materials and energy
economically, and prevents the manufacture of excess
end products. The control of enzymatic action helps to
maintain a stable internal environment in living
organisms.
4. Enzymes can be regulated through post-translational
modification. This can
include phosphorylation, myristoylation and glycosylation.
For example, in the response to insulin,
the phosphorylation of multiple enzymes,
including glycogen synthase, helps control the synthesis
or degradation of glycogen and allows the cell to respond
to changes in blood sugar.[82] Another example of post-
translational modification is the cleavage of the
polypeptide chain. Chymotrypsin, a digestive protease, is
produced in inactive form as chymotrypsinogen in
the pancreas and transported in this form to
the stomach where it is activated. This stops the enzyme
from digesting the pancreas or other tissues before it
enters the gut. This type of inactive precursor to an
enzyme is known as a zymogen.
5. Some enzymes may become activated when localized
to a different environment (e.g., from a reducing
(cytoplasm) to an oxidizing (periplasm) environment, high
pH to low pH, etc.). For example,hemagglutinin in
the influenza virus is activated by a conformational
change caused by the acidic conditions, these occur
when it is taken up inside its host cell and enters
the lysosome.[83]
Involvement in disease
Since the tight control of enzyme activity is essential for homeostasis, any
malfunction (mutation, overproduction, underproduction or deletion) of a
single critical enzyme can lead to a genetic disease. The importance of
enzymes is shown by the fact that a lethal illness can be caused by the
malfunction of just one type of enzyme out of the thousands of types
present in our bodies.
One example is the most common type of phenylketonuria. A
mutation of a single amino acid in the enzyme phenylalanine
hydroxylase, which catalyzes the first step in the degradation
of phenylalanine, results in build-up of phenylalanine and related
products. This can lead to mental retardation if the disease is
untreated.[84]
Another example of enzyme deficiency is pseudocholinesterase,
in which there is slow metabolic degradation of exogenous
choline.[citation needed]
Another example is when germline mutations in genes coding
for DNA repair enzymes cause hereditary cancer syndromes such
as xeroderma pigmentosum. Defects in these enzymes cause
cancer since the body is less able to repair mutations in the
genome. This causes a slow accumulation of mutations and
results in the development of many types of cancer in the sufferer.
Oral administration of enzymes can be used to treat several
diseases (e.g. pancreatic insufficiency and lactose intolerance).
Since enzymes are proteins themselves they are potentially
subject to inactivation and digestion in the gastrointestinal

environment. Therefore a non-invasive imaging assay was
developed to monitor gastrointestinal activity of
exogenous enzymes (prolyl endopeptidase as potential adjuvant
therapy for celiac disease) in vivo.[85]

Acid–base titrationAn acid-base titration is the determination of the concentration of an acid or
base by exactly neutralizing the acid/base with an acid or base of known
concentration. This allows for quantitative analysis of the concentration of an
unknown acid or base solution. It makes use of the neutralization reaction that
occurs between acids and bases and the knowledge of how acids and bases
will react if their formulas are known.
Acid–base titrations can also be used to find percent purity of chemicals.
Alkalimetry and acidimetry
Alkalimetry, sometimes spelled alkimetry, is the specialized analytic use of
acid-base titration to determine the concentration of a basic (synonymous
to alkaline) substance. Acidimetry, sometimes spelled acidometry, is the
same concept of specialized analytic acid-base titration, but for an acidic
substance.[1]
[edit]Equipment
The key equipment used in a titration are:
Burette
White tile – used to see a colour change in the solution
Pipette
pH indicator (the one used varies depending on the reactants)
Erlenmeyer flask/ Conical flask
Titrant or titrator (a standard solution of known concentration, a
common one is aqueous sodium carbonate)
Analyte or titrand (solution of unknown concentration)
[edit]Method
Before starting the titration a suitable pH indicator must be chosen. The
equivalence point of the reaction, the point at which equivalent amounts of
the reactants have reacted, will have a pH dependent on the relative
strengths of the acid and base used. The pH of the equivalence point can
be estimated using the following rules:
A strong acid will react with a strong base to form a neutral (pH=7)
solution.
A strong acid will react with a weak base to form an acidic (pH<7)
solution.
A weak acid will react with a strong base to form a basic (pH>7)
solution.
When a weak acid reacts with a weak base, the equivalence point solution
will be basic if the base is stronger and acidic if the acid is stronger. If both
are of equal strength, then the equivalence pH will be neutral. However,
weak acids are not often titrated against weak bases because the colour
change shown with the indicator is often quick, and therefore very difficult
for the observer to see the change of colour.
The point at which the indicator changes colour is called the end point. A
suitable indicator should be chosen, preferably one that will experience a
change in colour (an end point) close to the equivalence point of the
reaction.
First, the burette should be rinsed with the standard solution, the pipette
with the unknown solution, and the conical flask with distilled water.
Secondly, a known volume of the unknown concentration solution should
be taken with the pipette and placed into the conical flask, along with a
small amount of the indicator chosen.

The known solution should then be allowed out of the burette, into the
conical flask. At this stage we want a rough estimate of the amount of this
solution it took to neutralize the unknown solution. The solution should be
let out of the burette until the indicator changes colour and the value on the
burette should be recorded. This is the first (or rough) titre and should be
discluded from any calculations.
At least three more titrations should be performed, this time more
accurately, taking into account roughly where the end point will occur. The
initial and final readings on the burette (prior to starting the titration and at
the end point, respectively) should be recorded. Subtracting the initial
volume from the final volume will yield the amount of titrant used to reach
the endpoint. The end point is reached when the indicator just changes
color permanently. This is best achieved by washing a hanging drop from
the tip of the burette into the flask right at the end of the titration to achieve
a drop that is smaller in volume than what can usually be achieved by just
dripping solution off the burette.
Acid–base titration is performed with a phenolphthalein indicator, when it is
a strong acid – strong base titration, a bromthymol blue indicator in weak
acid – weak base reactions, and a methyl orange indicator for strong acid
– weak base reactions. If the base is off the scale, i.e. a pH of >13.5, and
the acid has a pH >5.5, then an Alizarine yellow indicator may be used. On
the other hand, if the acid is off the scale, i.e. a pH of <0.5, and the base
has a pH <8.5, then a Thymol Blue indicator may be used.
[edit]Titration of weak acid
The pH of a weak acid solution being titrated with a strong base solution
can be found at different points along the way. These points fall into one of
four categories [2]:
1. initial pH
2. pH before the equivalence point
3. pH at the equivalence point
4. pH after the equivalence point
1. The initial pH is approximated for a weak acid solution in water using
the equation
where Ka is the dissociation constant and F is the concentration of the
acid.

2. The pH before the equivalence point depends on the amount of
weak acid remaining and the amount of conjugate base formed. The
pH can be calculated by the following formula (which is a variation of
the Henderson-Hasselbalch equation):
where:
pKa is the negative log of the acid dissociation constant of the
weak acid.
nOH- added is the number of moles of added strong base in the
solution.
nHA initial is the number of moles the weak acid initially present.
When the numerator of the log term equals the denominator (), then the ratio
goes to 1 and the log term goes to zero. Thus the pH will equal
the pKa which occurs half-way to the equivalence point.
3. At the equivalence point, the weak acid is consumed and
converted to its weak conjugate base. The pH will be greater than
7 and can be calculated from an equation derived from the
following relationships:
1. pH + pOH = 14
2. KaKb = 10−14
3. at equivalence CaVa = CbVb
The previous 3 relationships are used to generate the equivalence
point pH formula below:
Ca = concentration of acid and Cb = concentration of base
Kw = dissociation constant for water and Ka = for the acid
Note that when an acid neutralizes a base, the pH may or
may not be neutral (pH = 7). The pH depends on the
strengths of the acid and base.
4. After the equivalence point, the solution will contain two
bases: the conjugate base of the acid and the strong base of
the titrant. However, the base of the titrant is stronger than
the conjugate base of the acid. Therefore, the pH in this
region is controlled by the strong base. As such the pH can
be found using the following:
Single formula. More accurately, a single formula[3] that
describes the titration of a weak acid with a strong base
from start to finish is given below:
φ = fraction of completion of the titration (φ
< 1 is before the equivalence point, φ = 1 is

the equivalence point, and φ > 1 is after the
equivalence point)
Ca, Cb = the concentrations of the acid and
base respectively
Va, Vb = the volumes of the acid and base
respectively
αA- = the fraction of the weak acid that is
ionized
Ka = the dissociation constant for the acid
[H+], [OH-] = concentrations of the H+ and
OH- ions respectively
This formula is somewhat cumbersome, but
does describe the titration curve as a single
equation.