pedro beltran-alvarez - uni-hannover.de · during my ph.d, and all these people have contributed to...
TRANSCRIPT

Kinetic studies on the actinorhodin
polyketide synthase
Pedro Beltran-Alvarez
A dissertation submitted to the University of Bristol
in accordance with the requirements of the degree of
Doctor of Philosophy
in the School of Chemistry, Faculty of Science.
April 2008
Word count: 41,919

ii
ABSTRACT
The actinorhodin (act) minimal polyketide synthase (PKS) from Streptomyces coelicolor
consists of three proteins: an acyl carrier protein (ACP), and two β-ketoacyl ACP synthase
components known as KSα and KSβ. The act minimal PKS catalyzes at least 18 separate
reactions, which can be divided into loading, initiation, extension, and cyclization and
release phases.
Quantitative kinetic assays were developed and used to measure individual catalytic
(kcat) and Michaelis (KM) constants for loading, initiation and extension steps. In vitro, the
reaction between malonyl CoA and ACP to form malonyl ACP (loading) is the rate-
limiting step (kcat = 0.49 min-1, KM = 207 µM). This reaction increases 5-fold in rate in the
presence of KSα/KSβ (kcat = 2.3 min-1) with unchanged KM. In the presence of S. coelicolor
malonyl CoA: ACP transacylase (MCAT), formation of malonyl-ACP is even faster: under
these conditions, it appears that decarboxylation of malonyl-ACP to form acetyl-ACP
(initiation) is the rate-limiting step (kcat = 20.6 min-1, KM = 2.4 µM). When an excess of
acetyl ACP is supplied, chain extension becomes rate limiting (kcat ~ 60 min-1). No ACP-
bound intermediates could be observed, suggesting that fully extended chains do not
accumulate, so cyclization and release are likely still faster than chain extension.
Protein-protein interactions among the components of the act minimal PKS were
investigated. It appears that KSα and KSβ have two different binding sites for ACP,
because inactive apo-ACP acts as a mixed inhibitor of malonyl-ACP binding to KSα/KSβ.
E47 and E53 in ACP are important residues for ACP: KSβ interactions. At least E47
appears to be important for ACP: KSα binding as well.
The quaternary structure of the active KSα/KSβ is most likely tetrameric.
Decarboxylation of malonyl-ACP by KSβ may be accompanied by a conformational
change in the structure of the tetramer that renders KSα ready for extension.
Addition of act KR to the minimal PKS does not affect the rates of loading, initiation or
extension, suggesting that ketoreduction is faster than chain extension. KR does not seem to
interact with KSα/KSβ, and probably acts before the first cyclisation of the polyketide
chain.

iii
To Carlos, Laura, Pedro, Pilar and Sonsoles

iv
ACKNOWLEDGEMENTS
Four years ago, Russell Cox invited me to join his group and do a Ph.D in organic and
biological chemistry, a field that is, at least at first sight, strange for a chemical engineer.
Having today arrived at the time of writing these acknowledgements, it is Russell the first
person I want to mention, because without his initial support, this moment would have
never come. Thanks Russell for the confidence you had on me from day one, for your
friendship, and for your constant support and supervision during my PhD.
My arrival at Bristol was also made possible by my family and my friends in Santander,
who understood my decision of leaving home and have made distances shorten by
travelling to and fro on every big occasion. Thanks guys for making me feel as I had never
gone whenever I was back home.
I spent three years in Bristol (2004-2007). I was welcomed by a group of people completely
different than the one I left: Kirstin, Ana, Dougal, Ludo and Jenny left soon; Pakorn, Deaw,
Chris and Chris, Kate, Elaine, Joel, Simon, David and particularly Bei, who took care of me
in the lab during my first months, and the very special Deirdre, Ursula and Song, made
Bristol a great place to live during the first years. Huge thanks also to the people that started
at the same time as me, that is you, Laura, Ellen, Tom, Marc, Carlo, Alexa, Eliza, and
specially my girlfriend Laura, because it is you with whom I shared most of the time during
these three years. You all, together with James (Dr. Goldfish), Mateusz (Prof. Scorpio), Liz
(Dr. Pepper), Andy, Ahmed, Rozida, Zafar, Persefoni and Anna deserve my gratitude for
having made these three years the best of my life. I will never forgive you.
I would also like to thank John Crosby for useful discussions, Paul Gates for help with
ESMS, and Chris, Pakorn, Eliza, Chris, Marc, Joel, Simon, Bei, Song, Laura, as well as
Gus Cameron, Tony Clarke and Annela Seddon in Biochemistry, for either materials,
training or help with sample analysis (mass spec, haddock, stopped-flow, fplc, hplc, plate
reader, pcr…).

v
Also, I am grateful to my examiners Matt Crump and Peter Leadlay for reading my thesis
and housing my viva, and to all the BRISENZ staff, i.e. Russell Cox, John Crosby, Matt
Crump, Andrea Hadfield, Adrian Mulholland, Tom Simpson and Chris Willis for
organizing the Marie Curie Programme of the European Commission, which is
acknowledged for funding (Marie Curie EST Centre BRISENZ, Contract No.: MEST-CT-
2004-504051).
Last, I thank you, the reader, for taking the time to read these acknowledgements, which are
full of names you may or may not recognize -those were the names I pronounced most
during my Ph.D, and all these people have contributed to what follows. Hope you will find
the read worthwhile.
Heidelberg, August 16th, 2008.

vi
AUTHOR’S DECLARATION
I declare that the work in this dissertation was carried out in accordance with the
Regulations of the University of Bristol. The work is original except where indicated by
special reference in the text and no part of the dissertation has been submitted for any other
academic award. Any views expressed in the dissertation are those of the author and in no
way represent those of the University of Bristol. The dissertation has not been presented to
any other University for examination either in the United Kingdom or overseas.
Pedro Beltran-Alvarez, August 16th, 2008.

vii
TABLE OF CONTENTS
1. Introduction ................................................................................................................1
1.1. Natural products ......................................................................................................................1
1.2. Polyketide biosynthesis............................................................................................................1 1.2.1. Polyketide biosynthesis is similar to fatty acid biosynthesis .......................................................... 2 1.2.2. Type II polyketide synthases ............................................................................................................ 5
1.2.2.1. The actinorhodin polyketide synthase...................................................................................... 6 1.2.3. The actinorhodin minimal PKS ........................................................................................................ 9
1.2.3.1. The actinorhodin Acyl Carrier Protein (ACP).......................................................................10 1.2.3.2. S. coelicolor malonyl-CoA: ACP transacylase (MCAT)......................................................12 1.2.3.3. The actinorhodin ketosynthase complex (KSα/KSβ). ..........................................................14 1.2.3.4. The Stanford model for the act minimal polyketide synthase in vitro .................................17 1.2.3.5. The Bristol model for the actinorhodin minimal polyketide synthase in vitro ....................19
1.2.4. Type III polyketide synthases .........................................................................................................21 1.2.5. Type I modular polyketide synthases.............................................................................................22
1.2.5.1. The 6-deoxyerythronolide B (6-dEB) polyketide synthase. .................................................23 1.2.6. Type I iterative polyketide synthases .............................................................................................24
1.3. Structure of the mammalian fatty acid synthase as a model for polyketide synthases.
26 1.3.1.1. Initial experiments: the linear model (1970s-1990s).............................................................26 1.3.1.2. Building of a 2D model (1990s) .............................................................................................26 1.3.1.3. 3D models for FAS and PKS megasynthases (1990s-2000s)...............................................28
1.4. Fundamentals of enzyme kinetics ........................................................................................30 1.4.1. Rate equations in enzyme kinetics..................................................................................................30 1.4.2. Kinetics of the act minimal PKS ....................................................................................................33
1.5. Aim of the project...................................................................................................................35
2. Studies on Acyl Carrier Proteins...............................................................................36
2.1. Protein-protein interactions in Type II FAS and PKS.....................................................37
2.2. Purification of acyl carrier proteins ....................................................................................40
2.3. Reduction of ACP dimers .....................................................................................................40
2.4. Studies on the self-malonylation activity of act holo-ACP. .............................................42

viii
2.4.1. Development of the α-ketoglutarate dehydrogenase assay to measure the rate of self-
malonylation ...................................................................................................................................................43 2.4.1.1. Development of the method to study the self-malonylation of act holo-ACP ....................45 2.4.1.2. Kinetics of the self-malonylation of act holo-ACP ...............................................................45
2.4.2. Actinorhodin minimal PKS to measure self-malonylation rate....................................................47 2.4.2.1. Development of the method....................................................................................................47 2.4.2.2. Kinetics of the self-malonylation of act holo-ACP ...............................................................50
2.5. Studies on the mechanism of self-malonylation of ACP ..................................................51 2.5.1. Mechanism of acceleration of self-malonylation rate by KSα/KSβ. ...........................................53
2.6. Discussion ................................................................................................................................56 2.6.1. Studies on the quality of ACP prior to reaction.............................................................................56 2.6.2. Both malonate and CoA are important for binding onto ACP .....................................................57 2.6.3. KSα/KSβ accelerates malonylation of ACP..................................................................................59
2.6.3.1. KSα/KSβ as a potential acyl transferase................................................................................60 2.6.3.2. KSα/KSβ activates holo-ACP towards self-malonylation ...................................................61 2.6.3.3. Insights into ACP-KSα/KSβ interactions leading to enhanced self-malonylation activity.
65
3. Initiation of polyketide synthesis. ..............................................................................67
3.1. Kinetics of KSβ can be studied in the presence of MCAT ..............................................74 3.1.1. MCAT accelerates the rate of malonylation of holo-ACP............................................................74 3.1.2. Initiation of polyketide synthesis is slower than chain elongation...............................................77
3.2. Interaction of ACP and KSβ ................................................................................................78
3.3. Discussion ................................................................................................................................79
4. Chain elongation and cyclisation and release from the PKS ....................................82 4.1.1. Kinetics of FAS systems .................................................................................................................83 4.1.2. Kinetics of Type III PKS.................................................................................................................84 4.1.3. Kinetics of Type I modular PKS (DEBS) ......................................................................................86
4.2. Kinetics of the rate-limiting reaction catalyzed by KSα ..................................................87 4.2.1. Attempts to detect polyketide intermediates..................................................................................88 4.2.2. Kinetics of the elongation step catalyzed by KSα.........................................................................89
4.3. Discussion ................................................................................................................................91
5. Stoichiometric analysis of the act minimal PKS .......................................................95
5.1. Stoichiometry of the KSα/KSβ complex –assays with mutant KSα/KSβ ....................96

ix
5.1.1. Kinetic study of a mutant CA KSα/KSβ complex ........................................................................99
5.2. Stoichiometry of the ACP: KSα/KSβ complex – inhibition of KSα/KSβ by apo-ACP.
101
5.3. Discussion ..............................................................................................................................104 5.3.1. Stoichiometry of ACP: KSα/KSβ complexes .............................................................................107
6. Kinetics of an extended act minimal PKS ...............................................................110
6.1. Structure and chemical mechanism of KR ......................................................................117
6.2. Kinetics of extended Type II minimal PKS......................................................................118
6.3. Kinetic analysis of production of mutactin by an extended act minimal PKS ...........119 6.3.1. Self-malonylation of ACP in the presence of KR .......................................................................123 6.3.2. Addition of MCAT to extended minimal PKS assays ................................................................125 6.3.3. Addition of acetyl-ACP to MCAT-supplemented, extended minimal PKS assays ..................126
6.4. Discussion ..............................................................................................................................126
7. Conclusions.............................................................................................................129
7.1. Self-malonylation of holo-ACP is aided by KSα/KSβ ....................................................130
7.2. Quaternary structure of KSα/KSβ and ACP: KSα/KSβ complexes ..........................133
7.3. Model for an extended act minimal PKS..........................................................................135
8. Experimental procedures ........................................................................................138
8.1. Bacterial strains and plasmids ...........................................................................................138 8.1.1. Escherichia coli..............................................................................................................................138 8.1.2. Streptomyces coelicolor ................................................................................................................138
8.2. DNA techniques ....................................................................................................................139 8.2.1. Site directed mutagenesis ..............................................................................................................139 8.2.2. Plasmid DNA isolation and characterization ...............................................................................140
8.3. Bacterial culture tecniques .................................................................................................140 8.3.1. Liquid media ..................................................................................................................................140
8.3.1.1. SOC medium..........................................................................................................................140 8.3.1.2. Luria-Bertani (LB) medium..................................................................................................141 8.3.1.3. Super-YEME (SY) medium..................................................................................................141
8.3.2. Solid media ....................................................................................................................................141 8.3.2.1. Luria-Bertani agar (LB agar) growth medium.....................................................................142 8.3.2.2. Mannitol Soya Flour Medium (SFM) ..................................................................................142

x
8.3.2.3. R5 Medium ............................................................................................................................142
8.4. Extraction and purification of SEK4 and SEK4b...........................................................143 8.4.1. Growth of bacteria and extraction of polyketides .......................................................................143 8.4.2. Liquid Chromatography/ Mass Spectrophotometry (LC/MS) analysis .....................................143 8.4.3. Preparative HPLC for purification of SEK4 and SEK4b............................................................144
8.5. Growth of bacteria and purification of proteins .............................................................144 8.5.1. General methods and equipment ..................................................................................................144
8.5.1.1. Centrifugation ........................................................................................................................144 8.5.1.2. Shakers ...................................................................................................................................144 8.5.1.3. UV Spectrophotometer..........................................................................................................144 8.5.1.4. Plate reader.............................................................................................................................145 8.5.1.5. Sonication...............................................................................................................................145 Cell-free extracts were obtained after lysing the cells by sonication (MSE Soniprep 150). Sonication
of E.coli was executed in 5 short bursts of 30 seconds with 1.5 min ice-cooling in between. S.
coelicolor was sonicated 10 times for 30 seconds. ...............................................................................145 8.5.1.6. Buffers ....................................................................................................................................145 8.5.1.7. NTA-His-Bind nickel affinity chromatography ..................................................................145 8.5.1.8. Fast protein liquid chromatography (FPLC)........................................................................146 8.5.1.9. Freeze-Drying ........................................................................................................................146 8.5.1.10. Sodium dodecyl sulphate Polyacrylamide gel electrophoresis (SDS-PAGE).................146
8.5.2. Growth of E. coli, and expression and purification of ACP, ACPS and MCAT ......................147 8.5.2.1. Purification of ACP ...............................................................................................................148 8.5.2.2. Purification of S. coelicolor his6-ACPS and act his6-KR....................................................148 8.5.2.3. Purification of S. coelicolor his6-MCAT .............................................................................149
8.5.3. Growth of S. coelicolor, expression and purification of his6-act-KSα/KSβ. ............................149 8.5.3.1. Growth of bacteria and expression of KSα/KSβ.................................................................149 8.5.3.2. Purification of KSα/KSβ ......................................................................................................149
8.6. Protein characterization methods......................................................................................150 8.6.1. Protein Quantification ...................................................................................................................150
8.6.1.1. Bradford Assay ......................................................................................................................150 8.6.1.2. Bicinchonic Acid Assay (BCA) ...........................................................................................150 8.6.1.3. Extinction coefficient of proteins. ........................................................................................151 8.6.1.4. Electro-Spray Mass Spectrometry (ESMS) .........................................................................151
8.7. Protein assays........................................................................................................................151 8.7.1. Phosphopantetheninylation of E47V apo-ACP to produce E47V holo-ACP............................152 8.7.2. Reduction of ACP dimers. ............................................................................................................152 8.7.3. Acylation of holo-ACP..................................................................................................................152

xi
8.7.4. Study of the self-malonylation of ACP using α-ketoglutarate dehydrogenase (KGDH) .........152 8.7.5. Minimal actinorhodin polyketide synthase assays and HPLC analysis of SEK4 and SEK4b .153 8.7.6. Kinetic studies on the actinorhodin minimal polyketide synthase .............................................154
8.7.6.1. Calibration of the method .....................................................................................................154 8.7.6.2. Minimal polyketide synthase assays to measure self-malonylation of holo-ACP ............154 8.7.6.3. Minimal polyketide synthase assays to measure inhibition of self-malonylation of holo-
ACP by CoA-SH .....................................................................................................................................155 8.7.6.4. Minimal polyketide synthase assays to measure the kinetics of KSβ ................................155 8.7.6.5. Minimal polyketide synthase assays to measure the kinetics of KSα ...............................155 8.7.6.6. Minimal polyketide synthase assays to measure inhibition of KSα/KSβ by apo-ACP....155 8.7.6.7. Minimal polyketide synthase assays to measure the kinetics of CA KSα/KSβ ................156 8.7.6.8. Data analysis ..........................................................................................................................156
References………………………………...…………………………………………..157
Appendices……………………………………………………...…………………….178

xii
LIST OF FIGURES Figure 1.1 Polyketides exhibit wide structural variety and pharmacological activities....................................... 2 Figure 1.2 Malonyl CoA is an important acyl carrier molecule. ........................................................................... 5 Figure 1.3 Natural products produced by Streptomyces species, derived from Type II PKS. The starter unit is
shown in red ............................................................................................................................................................... 6 Figure 1.4 The core of the actinorhodin (act) PKS genes.22 ................................................................................... 7 Figure 1.5 Structure of actinorhodin apo-ACP (PDB file: 2AF8), coloured from N-terminus (blue) to C-
terminus (red). ..........................................................................................................................................................11 Figure 1.6 Crystal structure of S. coelicolor MCAT, highlighting the two subdomains (blue and red) and the
catalytic active site S97 (green). PDB code: 1NM2. .............................................................................................13 Figure 1.7 Structure of KSα, showing from top to bottom the typical five layered α-β-α-β-α bundle fold of
thiolase enzymes. The active site cysteine is highlighted. PDB file: 1TQY..........................................................16 Figure 1.8 Primary sequence alignment of the active site (green star) region of a range of FAS ketosynthases
as well as KSα and KSβ from various PKS. For a complete sequence alignment see Appendix II. ..................17 Figure 1.9 Gen sequence of a DEBS PKS module. KS = ketosynthase, AT = acyl transferase, DH =
dehydratase, ER = enoyl reductase, KR = ketoreductase, ACP = acyl carrier protein. DH and ER are only
present in module 4. .................................................................................................................................................24 Figure 1.10 Head-to-tail model and linear arrangement of a dimeric FAS. Single headed arrows indicate the
order of domains in the primary structure (N-terminus to C-terminus). Blue, double headed arrows indicate
the interaction between ACP and KS domains. .....................................................................................................26 Figure 1.11 Complementation studies with mutant FAS. The inactivated domain is shown by X. A. Active
condensation intersubunit; B. Active condensation intrasubunit; C. Active acyl transfer intersubunit; D.
Active acyl transfer intrasubunit; E. No dehydration intersubunit; F. Active dehydration intrasubunit.83.......27 Figure 1.12 2-D model for the topology of Type I mammalian FAS. Single headed arrows indicate the order
of domains in primary sequence from N-terminus to C-terminus. Blue, double headed arrows indicate
interaction between ACP and KS, and between ACP and MAT.86 .......................................................................28 Figure 1.13 Structure of Type I FAS megasynthase (PDB file: 2CF2). Yellow, AT; red & green, the two
monomers of KS; blue, DH; magenta, homodimeric ER; orange, KR. The proposed reaction chambers are
marked with a star. ..................................................................................................................................................29 Figure 2.1 Primary sequence alignment of FAS and PKS ACP (green star, the PP attachment serine). .........39 Figure 2.2 Mass spectra of A. act ACP (expected, 9441 Da); B. dps ACP (expected 9603 Da); C. gris ACP
(expected, 9884 (-Me) and 10016 (+Me) Da); D. E47A-ACP (expected, 9383 Da); E. E47V-ACP (expected,
9412 Da); F. E53A-ACP (expected, 9383 Da) and G. R72A-ACP (expected, 9357 Da)....................................40 Figure 2.3 Mass spectrum of the reference act holo-ACP dimer after incubation with TCEP. Expected masses,
9441 Da (monomer), 18882 Da (dimer).................................................................................................................41 Figure 2.4 A. Incubation of malonyl-ACP with DTT. B. Incubation of malonyl-ACP with TCEP. Expected
masses: 9441 Da (holo-ACP) and 9527 Da (malonyl-ACP). ...............................................................................42 Figure 2.5. Raw data for the KGDH assay at initial holo-ACP concentrations of 15 µM (squares), 30 µM
(triangles) and 50 µM (circles). Malonyl CoA concentration was fixed at 1mM................................................46

xiii
Figure 2.6 Determination of Michaelis-Menten kinetic parameters of the self-malonylation of holo-ACP by
the KGDH coupling system. A. Plot of reaction rate vs. substrate concentration and hyperbolic fit. B. Hanes
linear plot of the data. .............................................................................................................................................47 Figure 2.7. Increase in the absorbance at 293 nm in act minimal PKS assays. Holo-ACP and malonyl CoA
concentrations were fixed at 80 and 1000 µM respectively. KSα/KSβ was 0.3 µM (diamonds), 0.5 µM
(triangles) and 1 µM (circles). ................................................................................................................................49 Figure 2.8 Dependence of initial rates of octaketide production on the concentration of KSα/KSβ at varying
ACP concentrations: squares, 10 µM; crosses, 25 µM; triangles, 50 µM; circles, 80 µM ACP.......................50 Figure 2.9 Determination of Michaelis-Menten kinetic parameters for the self-malonylation of holo-ACP by
the KSα/KSβ coupling system. A. Plot of reaction rate vs. substrate concentration and hyperbolic fit. B. Hanes
linear plot of the data. .............................................................................................................................................51 Figure 2.10 Interaction between arginine residues and malonate units.57 ..........................................................52 Figure 2.11 Determination of Kic and Kiu for the inhibition self-malonylation of ACP by coenzyme A (open
triangles, 800 µM; filled triangles, 400 µM; open circles, 200 µM; filled circles, 100 µM). ............................53 Figure 2.13 Self-malonylation of the reference act ACP (circles) and E47A ACP (open circles) as measured
by ESMS....................................................................................................................................................................54 Figure 2.14 HPLC analysis of SEK4 and SEK4b produced in 2 h by KSα/KSβ (0.3 µM) and a series of ACP
(50 µM). From bottom to top, the reference act ACP, dps ACP, E53A ACP, R72A ACP, E47A ACP, E47V
ACP, gris ACP. Yields after 2 h were 5.5, 2.3, 2.0, 2.3, 0.6, 0.1 and 0.04 pmol SEK4 and SEK4b, respectively.
...................................................................................................................................................................................55 Figure 2.15 A. Affected residues upon binding of malonyl CoA onto ACP (yellow). B. All affected residues are
solvent-exposed (blue).134 ........................................................................................................................................58 Figure 2.16 Ribbon representation of the major (A. PDB file 2FQ0) and minor (B. PDB file 2FQ2)
conformers of P. falciparum FAS ACP. Residues D57 to A60 are highlighted in magenta. ..............................61 Figure 2.17 ACP and PCP activate their substrates (for instance, malonate and alanine) as thiolesters........63 Figure 2.18 Ribbon structures of TyrC3-PCP in its A state (A. PDB file, 2GDY), A/H state (B. PDB file
2GDW) and H state (C. PDB file 2GDX). Helices are numbered in each structure...........................................64 Figure 3.1 Mass spectrum of MCAT (expected, 34113 Da); also detected as the potassium adduct (at 34152
KDa)..........................................................................................................................................................................74 Figure 3.2 A. Comparison of act PKS ‘minimal’ assays (open circles) and MCAT-supplemented assays (filled
circles). B. Hanes linear plot of the MCAT-supplemented rate data. ..................................................................76 Figure 3.3 Upon addition of MCAT, overall reaction rates become linearly dependent on KSα/KSβ
concentrations. .........................................................................................................................................................76 Figure 3.4 ESMS of acetyl-ACP (expected 9483 Da), also detected as the potassium adduct (expected 9522
Da). ...........................................................................................................................................................................77 Figure 3.5 Change in reaction rate upon titration of acetyl-ACP in MCAT-supplemented act minimal PKS.
KSα/KSβ and malonyl-ACP concentration were 0.25 µM and 20 µM respectively. ..........................................78 Figure 4.1 ESMS analysis of the ACP species in minimal PKS assays done in the presence of MCAT and
external acetyl-ACP. Expected masses: 9527 Da (malonyl-ACP), 9483 Da (acetyl-ACP)................................89

xiv
Figure 4.2. Evolution of the production of polyketides with time at saturating acetyl-ACP concentration and
2.5 µM malonyl-ACP (triangles), 5 µM malonyl-ACP (circles) and 10 µM malonyl-ACP (crosses). ..............90 Figure 5.1 Rate of polyketide production by the act PKS (supplemented with MCAT) using CQ KSα/KSβ
(0.25 µM) at a range of AQ KSα/KSβ concentrations. .........................................................................................98 Figure 5.2 Kinetics of the decarboxylation of malonyl-ACP by CA KSα/KSβ. A. Plot of reaction rate vs.
substrate concentration and hyperbolic fit. B. Hanes linear plot of the rate data. .............................................99 Figure 5.3 Rate of polyketide production by the act PKS (supplemented with MCAT) using CA KSα/KSβ (1
µM) at a range of AQ KSα/KSβ concentrations. .................................................................................................100 Figure 5.4 Mass spectrum of act C17S apo-ACP (expected, 9101 Da, also observed as the sodium and
potassium adduct at 9124 and 9140 Da, respectively)........................................................................................103 Figure 5.5 Inhibition of KSα/KSβ by apo-ACP at holo-ACP concentrations of 2.5 µM (full squares), 5 µM
(circles), 7.5 µM (squares), 15 µM (full circles) and 25 µM (triangles). ..........................................................103 Figure 5.6 Re-plot of data in Figures 5.1 and 5.3.Titration of AQ in assays performed with WT KSα/KSβ
(blue, 0.25 µM concentration) and CA (red, 1 µM concentration). ...................................................................107 Figure 6.1 Structure of a monomer of act KR, showing the β-sheet core (blue) and the catalytic residues
N114, S144, Y157 and K161 (yellow). PDB file: 1W4Z......................................................................................117 Figure 6.2 Mass Spectrum of his6-KR (expected: 29291 Da, observed: 29299 Da) and α-N-6-
phosphogluconoylation of the His-tag (expected: 29467 Da, observed: 29477 Da). .......................................120 Figure 6.3 LC/MS analysis of mutactin. A. Chromatogram showing the mutactin peak at 26.40 min. B.
Individual mass chromatogram at 303 Da (expected mass of [M+H]+). ..........................................................121 Figure 6.4 HPLC chromatograms at 280 nm showing the biosynthesis of SEK4 19, SEK4b 27 and mutactin 20
at fixed concentrations of malonyl CoA (1 mM), holo-ACP (50 µM) and KSα/KSβ (0.6 µM) and a range of
KR concentrations..................................................................................................................................................121 Figure 6.5 Effect of increasing concentrations of KR on the ratio mutactin: (SEK4 + SEK4b). .....................122 Figure 6.6. HPLC chromatograms at 280 nm of the time course study of polyketide production (SEK4 19,
SEK4b 27 and mutactin 20) in the absence (A) and presence (B) of KR (10 µM). ...........................................123 Figure 6.7 HPLC chromatograms at 280 nm showing polyketide production (SEK4 19, SEK4b 27 and
mutactin 20) upon titration of KSα/KSβ in extended minimal PKS assays at fixed KR concentration (10 µM).
.................................................................................................................................................................................124 Figure 6.8 Determination of kinetic parameters for the self-malonylation of holo-ACP in the presence of KR
by the KSα/KSβ coupling system. .........................................................................................................................125 Figure 6.9 Determination of kinetic parameters in MCAT-supplemented minimal PKS assays in the presence
of KR. ......................................................................................................................................................................126 Figure 7.1 Comparison of the behavior of MCAT-catalyzed malonylation of holo-ACP vs self-acylation of act
PKS holo-ACP. Linear plots show how the rate of self-malonylation of holo-ACP in the presence of KSα/KSβ
varies with ACP concentration at two concentrations of malonyl CoA (rate data from Figure 2.6). Curves
show the effect of saturation catalysis by MCAT at differing MCAT concentrations (for simulated MCAT with
KM of 60 µM for ACP and kcat of 450 s-1 at saturating malonyl CoA). Grey horizontal line shows demand for
malonyl ACP by KSα/KSβ at 0.75 µM. ................................................................................................................132

xv
Figure 7.2 Model for ACP: KSα/KSβ that contemplates four molecules of ACP per tetramer KSα/KSβ. .....134

xvi
LIST OF SCHEMES Scheme 1.1 Archetypical two-steps mechanism for the generation of a new carbon-carbon bond by FAS and
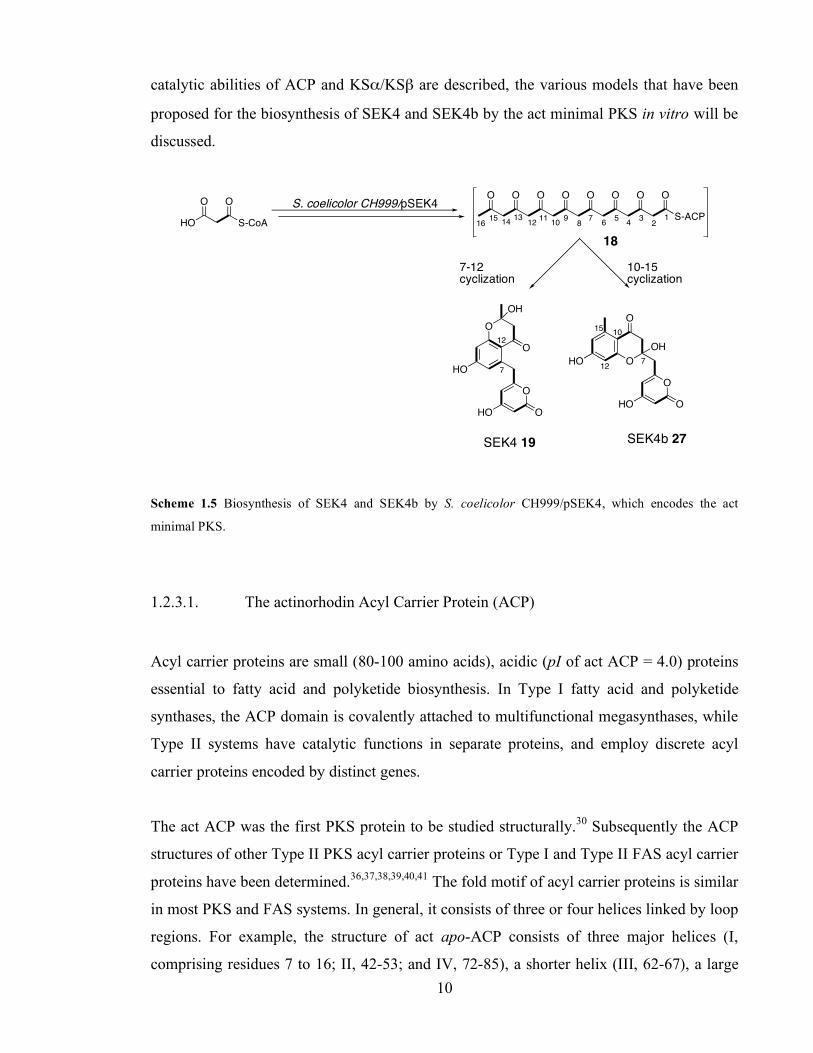
PKS: 1. Decarboxylation of activated carboxylic acid; 2. Claisen condensation. ................................................ 3 Scheme 1.2 Processing of the β-carbon after Claisen condensation in FAS. ........................................................ 4 Scheme 1.3 Proposed early steps in the biosynthesis of actinorhodin ................................................................... 7 Scheme 1.4 Proposed later steps in actinorhodin biosynthesis.29........................................................................... 9 Scheme 1.5 Biosynthesis of SEK4 and SEK4b by S. coelicolor CH999/pSEK4, which encodes the act minimal
PKS. ..........................................................................................................................................................................10 Scheme 1.6 Act holo-ACP consists of apo-ACP plus a PP cofactor (shown in red) transferred by ACPS from
CoA to Ser42 in ACP.................................................................................................................................................12 Scheme 1.7 MCAT catalyzes the malonylation of ACP in FAS systems...............................................................12 Scheme 1.8 Two-step mechanism for MCAT-catalyzed transfer of malonate to FAS-ACP. A. Loading of
malonate onto MCAT; B. Transfer of malonate to holo-ACP and regeneration of MCAT. ...............................13 Scheme 1.9 Reaction catalyzed by a general thiolase from a thiolester substrate.50 ..........................................14 Scheme 1.10 Mechanism of two-step decarboxylative Claisen condensation in thiolases (act KSα numbering),
proposed.51................................................................................................................................................................15 Scheme 1.11 The Stanford model for production of SEK4 and SEK4b by the act minimal PKS.32 A. Loading of
ACP is catalyzed by MCAT. B. Decarboxylation of malonyl-ACP is catalyzed by KSα, which subsequently
takes the acetyl starter unit. C. Claisen condensation and elongation of the polyketide chain. D. The
polyketide chain extrudes in a tunnel formed in the interface between KSα and KSβ, and finally cyclizes and it
is released from the PKS. ........................................................................................................................................18 Scheme 1.12 Self-malonylation of holo-ACP .........................................................................................................19 Scheme 1.13 Model for Type II PLS. Progressing clockwise, self-malonylation and malonyl transfer provide
the malonate substrate for initiation of polyketide synthesis by KSβ and chain elongation by KSα .................20 Scheme 1.14 Biosynthesis of chalcone and resveratrol (a stilbene) by chalcone (CHS) and stilbene (STS)
synthases.67 ...............................................................................................................................................................22 Scheme 1.15 Biosynthesis of 6-dEB by the Type I modular DEBS. Domains within each module follow the
order KS-AT-(DH)-(ER)-KR-ACP ..........................................................................................................................24 Scheme 1.16 Mechanism for enzyme-catalyzed reaction proposed by Michaelis and Menten. S = substrate, E
= enzyme, P = product. ...........................................................................................................................................31 Scheme 1.17 Briggs-Haldane mechanism. The concentration of the different species is shown underneath. ..31 Scheme 1.18 Biosynthesis of octaketides by the act minimal PKS. A. Loading of holo-ACP with malonate. B.
Initiation of polyketide synthesis by formation of acetyl-ACP. C. Elongation of the polyketide chain. D.
Formation of SEK4 and SEK4b in vitro. E. Further tailoring reactions lead to actinorhodin in vivo.1............34 Scheme 2.1 Disulphide formation between the thiols of holo-ACP......................................................................36 Scheme 2.2 Reaction scheme of KAS III. A. Condensation of acetyl CoA and malonyl-ACP to form
acetoacetyl-ACP. B. The same reaction is possible using malonyl CoA as a substrate instead of malonyl-ACP.
...................................................................................................................................................................................38 Scheme 2.3 Reaction scheme of the KGDH assay. ................................................................................................44

xvii
Scheme 2.4 Quantitative KGDH assay for CoA-SH. NADH production is followed spectrophotometrically. .45 Scheme 2.6 Two-step mechanism for the actinorhodin minimal PKS. 1. Self-malonylation of holo-ACP. 2.
Polyketide synthesis from malonyl-ACP.................................................................................................................50 Scheme 2.8 DTT reduces malonyl-ACP concentration by transthiolesterification .............................................57 Scheme 2.9 Mechanism that produces mixed inhibition of self-malonylation by CoA-SH. ................................58 Scheme 3.1 Initiation of fatty acid synthesis in E. coli, catalyzed by KASIII.144..................................................67 Scheme 3.2 Proposed mechanism for starter unit specificity in dps PKS. A. Loading of DpsC with propionate.
B. Initiation of polyketide synthesis. C. Elongation of the polyketide chain.149...................................................69 Scheme 3.3 Priming of the enc PKS with the benzoate starter unit.126.................................................................70 Scheme 3.4 Two possible models for transfer of acetyl-ACP from the KSβ to KSα active sites. .......................72 Scheme 4.1 Reactions catalyzed by KSα: A. Priming; B. Chain extension. C. Acyl transfer between the PP
thiol of ACP and the cysteine active site in KSα....................................................................................................83 Scheme 4.2 Reaction catalyzed by the pentaketide resorcylic synthase from Neurospora crassa .....................84 Scheme 4.3 Reactions catalyzed by A. Module 3 + TE; B. Modules 2, 5 and 6 + TE. .......................................86 Scheme 5.1 Dimer-tetramer KSα/KSβ equilibrium. ..............................................................................................96 Scheme 5.2 Hypothetical use of AQ KSα/KSβ to increase the rate of KSβ-mediated decarboxylation of
malonyl-ACP. KSα (red) and KSβ (green) are depicted by their corresponding active sites (see text). ...........97 Scheme 5.3 General mixed inhibition mechanism for apo-ACP as an inhibitor of KSα/KSβ ..........................102 Scheme 5.4. The addition of AQ affects the composition of A. CQ KSα/KSβ tetramers; B. CA KSα/KSβ
tetramers. ................................................................................................................................................................105 Scheme 6.1 Complementation of S. galilaeus with act KR resulted in production of a reduced polyketide (the
propionate starter unit is shown in red).198 ..........................................................................................................110 Scheme 6.2 Effect of addition of KR to act minimal PKS in in vivo experiments. .............................................111 Scheme 6.3 Production of 72 and 71 by S. galilaeus HO61 and S. galilaeus HO61 complemented with KR,
respectively. The propionate starter unit is shown in red. ..................................................................................112 Scheme 6.4 Model proposed by the group of Tsai, in which the substrate for act KR is the bicyclic
intermediate 82.206 .................................................................................................................................................114 Scheme 6.5 Proposed biosynthesis of BSM1 and BSM3.207 ................................................................................115 Scheme 6.6 Biosynthesis of wailupemycin D-G by the enc minimal PKS. Wailupemycin D: R1 = OH, R2 = Ph;
Wailupemycin E: R1 = Ph, R2 = OH.203,208 ...........................................................................................................116 Scheme 6.7 Proton relay mechanism in the act KR active site (blue arrows) showing NADPH (green), the
polyketide substrate (red) and the active site residues (black).205......................................................................118 Scheme 6.8 Biosynthesis of decaketides by tcm PKS...........................................................................................119

xviii
LIST OF TABLES Table 2.1 kcat values for the self-malonylation of ACP as measured by KGDH and the KSα/KSβ assays.........56 Table 3.1 Kinetic parameters for the decarboxylation of malonyl-ACP by KSβ (see Appendix 1 for Michaelis-
Menten analysis). .....................................................................................................................................................79 Table 4.1 Kinetic parameters for CHS.170 ..............................................................................................................85 Table 4.2 Kinetic parameters for the reaction catalyzed by AT-KS didomains from modules 3 and 6 using
ACP from every module as a substrate. .................................................................................................................87 Table 4.3 Catalytic constants in for a variety of ACP in the presence of excess acetyl-ACP. ...........................91 Table 5.1 Summary of kcat values for WT and CA KSα/KSβunder a range of conditions. ................................101 Table 6.1 Kinetic parameters for the reduction of unnatural substrates by KR.203...........................................113 Table 8.1 Plasmids transformed into E. coli ........................................................................................................138 Table 8.2 S. coelicolor A3(2) CH999 strains used in this work..........................................................................139 Table 8.3 Primers for site-directed mutagenesis of R72 ACP. Codons containing the mutation R72A are
underlined...............................................................................................................................................................139 Table 8.4 Buffers used for Nickel affinity chromatography ................................................................................146 Table 8.5 Stacking and Separating gels for SDS-PAGE. ....................................................................................146

xix
ABBREVIATIONS
Arg Arginine Asn Asp
Asparagine Aspartate
ACP ACPS
Acyl carrier protein Acyl carrier protein synthase
act ARO
actinorhodin Aromatase
AT Acyltransferase CLF Chain Length Factor CHS CYC Cys
Chalcone synthase Cyclase Cysteine
Da Daltons DH Dehydratase DNA Deoxyribonucleic acid dNTP Dau DTT
Deoxynucleotide triphosphate Daunorubicin-doxorubicin polyketide synthase Dithiothreitol
E. coli Escherichia coli EDTA Ethylenediamine tetraacetate ER ESMS
Enoyl reductase Electrospray mass spectrometry
FAS FPLC
Fatty acid synthase Fast protein liquid chromatography
fren Frenolicin Gln Glutamine Glu Glutamate Gly Glycine gra granaticin His HPLC
Histidine High performance liquid chromatography
IPTG Ile Jad KASI, II and III
Isopropylthio-β-D-galactoside Isoleucine Jadomycin β-ketoacyl:ACP synthases I, II and III
KR Ketoreductase KS Ketosynthase LB Luria-Bertani Medium – media for growth of E. coli Leu Leucine Lys Lysine MCAT Met
Malonyl CoA:ACP transacylase Methionine
NMR Nuclear magnetic resonance OD600 Optical density at 600 nm ORF Open reading frame otc/oxy Oxytetracycline PCR Polymerase chain reaction Phe Phenylalanine PKS Polyketide synthase Pro Proline rpm Revolutions per minute SDS-PAGE Ser
Sodium dodecyl sulphate – polyacrylamide gel electrophoresis Serine
STS Stilbene synthase TCA TCEP
Trichloroacetic acid Tris(2-carboxyethyl)phosphine
tcm Tetracenomycin TE Thioesterase
xx
TFA Thr Tris
Trifluoro acetic acid Threonine Tris(hydroxymethyl)aminoethane
Trp Tryptophan Tyr Val
Tyrosine Valine

Chapter 1. Introduction
1
1. Introduction
1.1. Natural products
The term ‘natural product’ is usually reserved for organic compounds of natural origin that
are unique to an organism or common to closely related organisms.1 In general, the
metabolism of natural products is carried out by specific enzymes, encoded by specific
genes, and the biosynthetic pathways involved are often distinct from the biosynthesis of
essential cell components such as fatty acids, nucleic acids and proteins, which are the
products of the primary metabolism. Thus natural products are biosynthesized by secondary
metabolic pathways that draw upon resources from primary metabolism (i.e. nutrients,
energy). Although natural products may meet the needs of specific individual producers,
they are more often seen as beneficial, or problem-solving, good for the community, be that
of micro-organisms, plants or marine or terrestrial animals (for example, pheromones and
signalling molecules, antifeedants and defense mechanisms, etc.).2
There are four main classes of natural products, according to their biosynthetic origin:
polyketides, isoprenoids, alkaloids and natural products derived from the shikimate
pathway. The latter three types of natural products are beyond the scope of this thesis.
Polyketide biosynthesis is discussed thoroughly in Section 1.2.
1.2. Polyketide biosynthesis
Polyketide natural products are secondary metabolites that are derived from the
condensation of short-chain carboxylic acids. They are produced by a wide variety of
organisms ranging from bacteria to plants, and from fungi to marine organisms. Polyketides
are also important from a pharmaceutical point of view, as many of them exhibit biological
properties such as antibiotic, antifungal, anticancer, cholesterol lowering agent or
immunosuppressant activities (Figure 1.1).

Chapter 1. Introduction
2
O
O
OOH
O OHOH
2
OH
OH
O Cl
O
O
OOO
O
OO
O
OH
O
O
O
O
HO
OOH
O
OO
O
HOH
O
ON
O O OH
O
O
OH O
OH
O
OH NH2
O
O
O OH
OHO
OHOH
OH
O
O
H
Figure 1.1 Polyketides exhibit wide structural variety and pharmacological activities.
1.2.1. Polyketide biosynthesis is similar to fatty acid biosynthesis
Despite the structural variety exhibited by polyketides (Figure 1.1), their biosynthetic
pathways are closely related to one another. Furthermore, the biosynthesis of polyketides is
very similar to that of fatty acids such as palmitate 1, which are products from primary
metabolism.
6-methylsalicylic acid antibiotic
Penicillium patulum
Daunorubicin anticancer
Streptomyces peucetius
Tetracenomycin C antibiotic
Streptomyces glaucescens
Griseofulvin antifungal
Penicillium griseofulvin
Actinorhodin antibacterial
Streptomyces coelicolor (A3)2
Rapamycin inmunosuppresant
Streptomyces hygroscopicus
Lovastatin Cholesterol-lowering agent
Aspergillus terreus

Chapter 1. Introduction
3
HO
O
1
Polyketides and fatty acids are biosynthesized by one or more enzymes that form a
polyketide or fatty acid synthase. These synthases use carboxylic acids as substrates to
make polyketide or fatty acid products, respectively. The key enzyme in polyketide and
fatty acid synthases is a β-ketoacyl synthase (KS). This enzyme catalyzes a carbon-carbon
bond-forming reaction between an acyl thiolester substrate and an acyl group attached to
KS.3,4 This reaction proceeds in two chemical steps (Scheme 1.1): first, the acyl thiolester
substrate 2, which is covalently bound to the active sulphydryl of a protein called acyl
carrier protein (ACP), is decarboxylated. This generates the carbanion 3. The second step is
a Claisen-type condensation between the carbanion 3 and a KS-bound thiolester
intermediate 4 to produce a β-ketothiolester 5. This reaction is repeated several times to
generate an ACP-bound polymer which is extended by two carbon units every
condensation.
An important difference between fatty acid synthases (FAS) and polyketide synthases
(PKS) is the nature of the starter and extender units; i.e. the initial KS-bound thiolester 4
and the acyl thiolester substrate 2. While FAS almost exclusively use acetate and malonate
for chain initiation and extension, respectively, PKS can accept a variety of starter and
extender units. In part, this accounts for the diversity of polyketide structures compared to
fatty acids.
ACP-S O-
O O
ACP-S
O
CO2
ACP-S
O
S-KSR
O
ACP-S
O
KS-S-
O
R!"
Scheme 1.1 Archetypical two-steps mechanism for the generation of a new carbon-carbon bond by FAS and
PKS: 1. Decarboxylation of activated carboxylic acid; 2. Claisen condensation.
3
4 5
2
1.
2.

Chapter 1. Introduction
4
Another important difference arises from the processing of the nascent β-ketothiolester 5.
In fatty acid biosynthesis each condensation is almost always followed by a set of three
sequential reactions at the β-carbon: a ketoreductase (KR) domain reduces the β-keto group
to form a secondary alcohol 6, a dehydratase (DH) promotes dehydration to leave an α,β
double bond 7, and an enoyl reductase (ER) catalyzes reduction of the double bond to
afford a fully saturated thiolester 8 (Scheme 1.2). On the other hand, a PKS may or may not
omit some or all of these β-carbon processing reactions after each condensation, which
results in different reduction levels at each β-carbon. This is the key difference between
FAS and PKS, and is usually known as PKS ‘programming’.5 The length of polyketide
chains (and therefore the number of condensation reactions) and the presence of additional
catalytic domains, such as C-methyltransferases (CMeT) which transfer a methyl unit to the
α-carbon in some bacterial and many fungal polyketide synthases, are further source of
diversity.
ACP-S
O O
R ACP-S
O OH
R ACP-S
O
R ACP-S
O
R!"
KR DH ER
5 6 7 8 Scheme 1.2 Processing of the β-carbon after Claisen condensation in FAS.
The two major protein architectures of FAS and PKS are named Type I and Type II. Type I
enzymes are large multifunctional polypeptides, while type II systems catalyze the
individual reactions using separate, distinct proteins. Type I FAS are found in animals and
fungi, whereas Type II FAS are typical of bacteria and plants. Correspondingly, Type I
PKS are typical of fungi, while bacteria employ Type I and Type II PKS. There is a third
type of PKS, namely Type III PKS, which is typical of plants (and also found in some
bacteria and fungi) and does not have a counterpart in fatty acid biosynthesis.
In the following sections, the enzymology of the different types of PKS is discussed
starting with the most relevant to this thesis, i.e. Type II PKS. Later, Type I PKS in bacteria
and fungi, as well as Type III PKS, will be overviewed by comparison to Type II PKS. As
fatty acid synthases can provide useful information for the characterization of polyketide
synthases, fatty acid biosynthesis will be discussed in parallel with polyketide biosynthesis
when appropriate.

Chapter 1. Introduction
5
1.2.2. Type II polyketide synthases
Actinomycetes are a rich source of bioactive polyketides. Streptomyces species in particular
produce more than one third of all known antibiotics to date.6 Streptomyces employ three
types of PKS organisations; i.e Type I, Type II and Type III PKS.
Bacterial Type II PKS can be defined as complexes of several monofunctional proteins
that convert specific carboxylic acids into polycyclic aromatic polyketides.7 Type II
polyketide synthases are then organised as an assembly of enzymes, brought together by
non-covalent forces, that acts iteratively. This feature as well as the availability of genetic
and molecular tools amenable to actinomycetes prompted the study of aromatic polyketides
as a model for more complicated bacterial or fungal polyketide synthases.
In most cases, the carboxylic acids used as a substrate by Type II PKS are activated as
coenzyme A (CoA) thiolesters. The terminal sulphydryl group in the phosphopantetheine
(PP) arm of CoA is the point to which acyl groups are attached, and then form an acyl-
CoA, such as malonyl-CoA (Figure 1.2).
OH
P
O
O O
N
NN
N
NH2
O
O OH
OP
O
OH
PO
OH
OH
NH
OH
O
NH
S
O
HO
O O
Figure 1.2 Malonyl CoA is an important acyl carrier molecule.
There is some variety in the choice of starter unit for Type II PKS-derived polyketides. For
example, actinorhodin 9,8 jadomycin 10,9 granaticin 1110 and griseusin 1211 are synthesized
from an acetate starter unit. Other Type II PKS employ alternative starter units, such as
propionate (daunorubicin 13,9 doxorubicin 14,9 aclacinomycins12), (iso)butyrate (R1128
1513), malonamate (oxytetracycline 1614) and benzoate (enterocin 1715), (Figure 1.3). On
the other hand, chain elongation is carried out exclusively from malonyl CoA.
Malonate
Phosphopantetheine arm
3’Phosphoadenosine monophosphate

Chapter 1. Introduction
6
These and other natural products formed by Type II PKS comprise an important and
structurally diverse group of secondary metabolites, and many of them (or their
biosynthetic derivatives) have clinically useful anticancer (e.g. daunorubicin, doxorubicin)
and antibiotic (e.g. oxytetracycline) activities. The first Type II PKS-derived polyketide
studied was actinorhodin, whose biosynthesis is reviewed in the next subsection.
Figure 1.3
1.2.2.1. The actinorhodin polyketide synthase
The genome of Streptomyces coelicolor A3(2) was sequenced in 2002. More than 20 gene
clusters were found which corresponded to known or predicted secondary metabolites,
including polyketides, non-ribosomal peptides and terpenes.16 One of them, the polyketide
antibiotic actinorhodin 9 had been known since much earlier (1970s), when the cloning of
bacterial polyketide synthase genes began.
In early work by Rudd and Hopwood, an actinorhodin-producing strain of S. coelicolor
was subjected to random UV mutation. Selection of the mutants that did not produce
actinorhodin was possible due to the absence of the characteristic blue colour of
actinorhodin. A series of 76 S. coelicolor A3(2) mutants were selected in this way.17 These
actinorhodin-blocked mutants were classified into seven classes according to their
phenotype; i.e. the compounds that they produced. For instance, the so-called actIII mutants
failed to secrete any metabolite that could be converted by other mutants to actinorhodin, so
they were deduced to be interrupted in an early step of the actinorhodin biosynthesis.
Whereas the actIII mutants produced a red pigment, the actI mutants were non-pigmented.
Thus, the actI mutants were thought to be incapable of assembling a polyketide, whereas
the actIII mutants would produce a polyketide but would not carry out an essential
modification in the polyketide chain. Moreover, actIII mutants could not convert
compounds secreted by other classes of mutants to actinorhodin. These further mutants
(actIV, VA, Vb, VI, VII) were deduced to be blocked at later stages in actinorhodin
biosynthesis.17
Malpartida and Hopwood then isolated a large (30 Kbp), continuous segment of DNA
from actinorhodin-producing S. coelicolor and showed that this DNA was able to
complement the seven classes of act mutants (actI-actVII) and restore production of
actinorhodin.18 Furthermore, introduction of this DNA on an actinorhodin-sensitive strain

Chapter 1. Introduction
7
of S. parvulus not only reconstituted actinorhodin production but also conferred immunity
to the host.18 The entire actinorhodin gene cluster had been cloned.
The notation of the actinorhodin genes and proteins had been devised according to the
phenotype of the mutants.17 Later, when the actIII region was sequenced, an open reading
frame (ORF) encoding a ketoreductase was found.19 The actI region revealed three open
reading frames encoding three different proteins: ORF1 (ketosynthase, KSα), ORF2 (β
component of the ketosynthase, KSβ) and ORF3 (acyl carrier protein, ACP). ActVII
encoded an aromatase (ARO) and actIV a cyclase (CYC) (Figure 1.4).20
actI
actIII actVII actIV
1 2 3
Figure 1.4 The core of the actinorhodin (act) PKS genes.21
These studies were carried out by means of mutation or deletion in regions of the
actinorhodin PKS cluster, which consists of 23 genes (including structural, regulatory,
export and self-resistance conferring genes). Further understanding of the PKS required a
more convenient strategy. The generation of a derivative strain of S. coelicolor A3(2) that
lacked the entire set of genes of the actinorhodin cluster, namely S. coelicolor A3(2)
CH999, provided an actinorhodin non-producing host for subsequent in vivo studies.22 The
introduction of the desired sets of PKS subunits into this deficient strain of S. coelicolor
allowed the identification of the genes and proteins responsible for the early steps in
actinorhodin biosynthesis (Scheme 1.3).
Three proteins, namely ACP, KSα and KSβ were found to be essential to assemble an
hypothetical octaketide intermediate 18 from malonyl CoA. This set of proteins was termed
the act ‘minimal’ PKS.23 This minimal system was also presumed to catalyze the formation
of the first ring. In the absence of any other enzyme, spontaneous cyclisation and
dehydration of the resulting polyketide intermediate resulted in the isolable SEK4 19.24
Scheme 1.3

Chapter 1. Introduction
8
A ketoreductase (KR, actIII) was found to reduce 18 at C-9 to give an alcohol, and then
mutactin 20 was produced if no other enzymes were present.24 An aromatase (ARO, actVII)
was needed to aromatase the first ring of the polyketide, which spontaneously cyclised to
SEK34 21 in the absence of a cyclase (CYC, actIV).25 ActIV cyclased the second ring to
generate the hypothetic bicyclic intermediate 22.25 In the absence of tailoring enzymes, 3,8-
dihydroxy-1-methylanthraquinone-2-carboxylic acid (DMAC) 23 was produced.25 Further
modifications of the intermediate 22 lead to actinorhodin 9.
Studies have since moved to the area of enzymology and biochemical characterization of
the actinorhodin PKS. Efforts have been made to shed light onto the biochemistry of further
steps in actinorhodin biosynthesis.10,26,27,28,29 In particular, the group of Ichinose has
recently proposed a complete biosynthetic pathway for actinorhodin (Scheme 1.4).28
Following production of the bicyclic intermediate 22, ketoreduction at carbon 3 produces
the hydroxyacid 24, which cyclises and is dehydrated to give the intermediate 4-dihydro-9-
hydroxy-1-methyl-10-oxo-3-H-naphto-[2,3-c]-pyran-3(S)-acetic acid (DNPA) 25.
Oxidation of 25 at carbon 6 affords dehydrokalafungin 26, which is two steps from
actinorhodin: hydroxylation of C-8 of 26, and the dimerization reaction. Valton, Fontecave
and co-workers demonstrated that actVA-ORF5 and actVB catalyze the
monohydroxylation at C-8 of 27 in vitro.26 To date, the enzyme responsible for the
dimerization reaction has not been identified.
During the last decade, much effort has been dedicated to solve the X-ray crystal structure
or NMR structure of the proteins that constitute the act PKS. To date, the NMR solution
structure of the act ACP,30 and crystal structures of ketosynthase KSα/KSβ complex,31 the
KR32 and the monooxygenase enzyme ActVA ORF633 have been published.
This has facilitated the design and interpretation of experiments, as well as the
generation of mutant proteins in order to prove the chemical mechanism of the enzymes.
This is especially true in the case of the act minimal PKS. In vitro studies of the act
minimal PKS have been performed for more than ten years, and the following section
reviews the ‘state of the art’.

Chapter 1. Introduction
9
OOH
OH
O
OO
OOH
OH
O
OHO
OOH
OH
O
OH
O
OOH
OH
O O
OOH
OH
O O
OOH
OH
O O
O
OH
H
O
O
OH
OH
OH
H
O
O
OH
OH
OOHO HO
Scheme 1.4 Proposed later steps in actinorhodin biosynthesis.28
1.2.3. The actinorhodin minimal PKS
As described in Section 1.2.2, early in vivo experiments showed that the act minimal PKS
consisted on an acyl carrier protein (ACP), and two β-ketoacylthiolester synthase
(KSα/KSβ) components.17,20,23 These experiments led to the generation of S. coelicolor
CH999/pSEK4, the strain carrying the genes that encode the act minimal PKS. This strain
was capable of assembling the octaketide SEK4 19.23 SEK4 was assumed to be the product
of a C7-C12 intramolecular aldol condensation of a hypothetical octaketide intermediate 18
(Scheme 1.5). Fu, Khosla and co-workers then described the isolation of a second product
of S. coelicolor CH999/pSEK4, and named it SEK4b.34 SEK4b 27 arises from a C10-C15
cyclisation of 18. In vivo, the ratio 19:27 was approximately 1:1.24
In vitro studies were facilitated by the cloning, expression and purification of ACP and
KSα/KSβ during late 1990s. This greatly contributed to the understanding of the catalytic
properties of each enzyme, which are described in the following subsections. Once the
22 24
25
9
26
ActVI-ORF1
ActVI-ORF1 ?
ActVI-ORF1 + ActVI-ORFA?
ActVI-ORF2
ActVA-ORF6
ActVA-ActVB?
Chapter 1. Introduction
10
catalytic abilities of ACP and KSα/KSβ are described, the various models that have been
proposed for the biosynthesis of SEK4 and SEK4b by the act minimal PKS in vitro will be
discussed.
HO
O O
S-CoA
HO
HO
O
O
OH
O
O
O
OHO
O
OH
HO
O
S-ACP
O O O O O O O O
12
34
56
78
910
1112
1314
1516
7
12
127
1015
Scheme 1.5 Biosynthesis of SEK4 and SEK4b by S. coelicolor CH999/pSEK4, which encodes the act
minimal PKS.
1.2.3.1. The actinorhodin Acyl Carrier Protein (ACP)
Acyl carrier proteins are small (80-100 amino acids), acidic (pI of act ACP = 4.0) proteins
essential to fatty acid and polyketide biosynthesis. In Type I fatty acid and polyketide
synthases, the ACP domain is covalently attached to multifunctional megasynthases, while
Type II systems have catalytic functions in separate proteins, and employ discrete acyl
carrier proteins encoded by distinct genes.
The act ACP was the first PKS protein to be studied structurally.30 Subsequently the ACP
structures of other Type II PKS acyl carrier proteins or Type I and Type II FAS acyl carrier
proteins have been determined.36,37,38,39,40,41 The fold motif of acyl carrier proteins is similar
in most PKS and FAS systems. In general, it consists of three or four helices linked by loop
regions. For example, the structure of act apo-ACP consists of three major helices (I,
comprising residues 7 to 16; II, 42-53; and IV, 72-85), a shorter helix (III, 62-67), a large
18
SEK4 19 SEK4b 27
7-12 cyclization
10-15 cyclization
S. coelicolor CH999/pSEK4

Chapter 1. Introduction
11
first loop separating helices I and II, and a shorter second loop connecting helices II and III
(Figure 1.5). Helices I and IV run antiparallel and are stabilized by numerous contacts
between each other. Helix II runs almost parallel to helix IV. Helices I, II, and IV pack
around a central hydrophobic cleft.30
Figure 1.5 Structure of actinorhodin apo-ACP (PDB file: 2AF8), coloured from N-terminus (blue) to C-
terminus (red).
Inactive, apo-ACP species need be post-translationally modified to afford active, holo acyl
carrier proteins. In vivo, the apo form of ACP is post-translationally modified to holo-ACP
by transfer of 4’-phosphopantetheine (PP) from CoA to a strictly conserved serine residue
located at the bottom of helix II (S42 in act ACP). This reaction is catalysed by a holo-acyl
carrier protein synthase (ACPS), (Scheme 1.6). The PP arm houses an end sulphydryl that
is the attachment point of the acyl groups to holo-ACP. For this reason, holo-ACP is
represented as ACP-SH in biosynthetic schemes throught this thesis.
Heterologous expression and purification of S. coelicolor ACPS by the Bristol group
allowed the study of its catalytic properties in vitro.42 S. coelicolor ACPS is a versatile
phosphopantetheinyl transferase, capable of recognizing a wide range of bacterial FAS and
PKS acyl carrier proteins, including act ACP.
Helix I Helix II
Helix III
Helix IV
Loop I
Loop II

Chapter 1. Introduction
12
OH
P
O
O O
NH
OH
O
N
NN
N
NH2
O
O OH
PO
OH
OH
OP
O
OH
SHNH
O
Scheme 1.6 Act holo-ACP consists of apo-ACP plus a PP cofactor (shown in red) transferred by ACPS from
CoA to Ser42 in ACP.
Acylation of an ACP occurs when a starter or extender unit is attached to the sulphydryl
nucleophilic group at the end of the PP arm. In fatty acid biosynthesis, this reaction is
catalyzed by an enzyme known as malonyl CoA: ACP transacylase (MCAT), (Scheme 1.7).
By analogy to FAS systems, Type II polyketide synthases were expected to utilise discrete
MCAT activity to load the ACP with malonyl units. Since genes encoding MCAT are
absent in most type II PKS genes, it was proposed that FAS MCAT enzymes in
Streptomyces were able to activate both FAS and PKS acyl carrier proteins, in a process
often referred to as ‘cross-talk’ between primary and secondary metabolism.43,44 In this
model, type II PKS systems would recruit MCAT from fatty acid biosynthesis, and the
actinorhodin minimal PKS would involve a fourth component, i.e. S. coelicolor MCAT.
S OH
OO
CoA
S OH
OO
ACP
FASHS+
ACP
FAS
Scheme 1.7 MCAT catalyzes the malonylation of ACP in FAS systems.
1.2.3.2. S. coelicolor malonyl-CoA: ACP transacylase (MCAT)
In S. coelicolor fatty acid metabolism, the corresponding acyl carrier protein (FAS ACP) is
loaded with a malonyl unit in an MCAT-catalyzed reaction (Scheme 1.7). S. coelicolor
ACPS
Apo-ACP Holo-ACP
MCAT

Chapter 1. Introduction
13
MCAT is a 32 KDa monomeric protein. Its crystal structure at 2.0 Å resolution was solved
in 2003 by Stroud and co-workers.45 MCAT is composed of two subdomains (which are
coloured red and blue in Figure 1.6), with the catalytic S97 on a nucleophilic elbow at the
core of a conserved GHSXG motif within the large subdomain.
Figure 1.6 Crystal structure of S. coelicolor MCAT, highlighting the two subdomains (blue and red) and the
catalytic active site S97 (green). PDB code: 1NM2.
The Bristol group studied the malonyl transferase activity of S. coelicolor MCAT in
detail.46 The mechanism and kinetics of the MCAT-catalysed malonylation of S. coelicolor
FAS ACP were investigated. The transfer of malonate from CoA to ACP was found to
operate via a two-step mechanism where MCAT is first acylated by malonate at S97, and
then malonate is transferred to the sulphydryl of the PP arm in holo-ACP (Scheme 1.8).
MCAT
ACP
S OH
OO
CoA+
MCAT
SH
S OH
OO
MCATS OH
OO
+SH
S OH
OO
ACP
CoA
MCAT
Scheme 1.8 Two-step mechanism for MCAT-catalyzed transfer of malonate to FAS-ACP. A. Loading of
malonate onto MCAT; B. Transfer of malonate to holo-ACP and regeneration of MCAT.
A.
B.

Chapter 1. Introduction
14
1.2.3.3. The actinorhodin ketosynthase complex (KSα/KSβ).
In the actinorhodin PKS, polyketide chains are synthesized by a two-component β-
ketoacylthiolester synthase. The two subunits are separate proteins each of ca 45 KDa.
They are usually known as KSα/KSβ or KS-CLF. The former nomenclature will be used
here.
By sequence comparison with other known members of the thiolase superfamily, it
soon became clear that KSα catalyzed the formation of carbon-carbon bonds via Claisen
condensation reactions (Scheme 1.9).20
S
O
thiolase
S
O
R
Substrate carrier
Scheme 1.9 Reaction catalyzed by a general thiolase from a thiolester substrate.48
The most important groups of thiolase enzymes are fatty acid and polyketide β-
ketoacylsynthases (KAS or KS). The active site architecture of thiolases includes a
conserved Cys-His-His (or Cys-His-Asn in Type III PKS) catalytic triad to perform
decarboxylative Claisen condensations.
Rock, White and collaborators have recently proposed a mechanism for the Claisen
condensation catalyzed by KASII of Streptococcus pneumoniae, based on a combination of
structural and biochemical information.49 They generated a series of mutant enzymes
(including mutation of the residues in the catalytic triad, i.e. C169A, H309A and H346A,
act KSα numbering). The catalytic properties of these enzymes, as well as the effect of
mutations in protein structure and ACP binding, were assessed.
For instance, while C169 and H346 were essential for condensation, the H309A mutant
retained some activity. Comparison of the crystal structures of the wild type (WT) and
H309A S. pneumoniae KASII led to the observation that there was a water molecule in the
active site of the WT protein, which the H309A mutant lacked. This water molecule was
hydrogen-bonded to the Nε of H309.

Chapter 1. Introduction
15
In their model, H309 is the anchor that positions a catalytic water molecule W1 in exactly
the right place to perform a nucleophilic attack on the carboxylate 28 of the substrate and
release bicarbonate (Scheme 1.10). There was evidence that bicarbonate was the product of
the decarboxylation reaction, rather than CO2, because bicarbonate inhibited malonyl-ACP
decarboxylation. His346 promotes the formation of the carbanion 29 at C2 of the malonate
by stabilizing the enolate intermediate. The carbanion reacts with the acyl-Cys169
intermediate, and the tetrahedral transition state resolves to form the β-ketoacyl-ACP
product.
NH
O
ACP-S
O
OH
N
HN
His309His346
O
H
H
N
NH
O
ACP-S
O
OH
N
HN
His309His346
H
HO
O
R S
Cys169
HN
N
Scheme 1.10 Mechanism of two-step decarboxylative Claisen condensation in thiolases (act KSα
numbering), proposed.49
The structure of act KSα resembles that of the thiolase superfamily fold. KSα has two
halves, the N-terminal half and the C-terminal half, which have a βαβαβαββ secondary
structure. The two halves are assembled in a five layered α-β-α-β-α bundle structure
(Figure 1.7). The essential and fully conserved cysteine (C169) active site is in the Nβ3-
Nα3 loop, in a position in which the electrostatics of helix Nα3 lower the pKa of the
reactive cysteine, making it more nucleophilic.50,51
1.
2.
W1
28
29

Chapter 1. Introduction
16
Figure 1.7 Structure of KSα, showing from top to bottom the typical five layered α-β-α-β-α bundle fold of
thiolase enzymes. The active site cysteine is highlighted. PDB file: 1TQY
Most enzymes of the thiolase superfamily are dimers, tetramers or hexamers, and
monomers have not been observed.48 Thus before direct experimental evidence had been
obtained, it was accepted that KSα and KSβ formed at least a heterodimer. While the role
of ACP and KSα became apparent ever since their sequence were known, the function of
KSβ was unclear.20 KSα and KSβ are very similar to each other (40% similarity, 26%
identity) nevertheless a fundamental difference between the proteins is that the KSα active
site has an essential cysteine (Cys, C) active site, whereas in the KSβ the corresponding
residue is a highly conserved glutamine (Gln, Q). This glutamine is conserved along KSβ
from many Type II PKS as well as the loading domain (KQ) on Type I modular PKS such
as niddamycin (nid) and tylosin (tyl) (Figure 1.8).
Early in vivo experiments by McDaniel, Khosla et al provided the first insights into the
properties of KSβ. The KSα, KSβ and ACP from the octaketides actinorhodin 9 and
granaticin 10 PKS, and the decaketide tetracenomycin (tcm) 30 polyketide synthases were
combined. The chain length of the polyketides produced was then analyzed. It appeared
that KSβ was the main determinant of chain length because, when tcm KSβ was coupled
with act KSα, a decaketide was produced. Conversely, when tcm KSα was paired with gra
KSβ, an octaketide was detected.22 The Stanford group hence baptized KSβ ‘chain length
factor’ (CLF).
α bundle
β bundle
α bundle β bundle
α bundle

Chapter 1. Introduction
17
Figure 1.8 Primary sequence alignment of the active site (green star) region of a range of FAS ketosynthases
as well as KSα and KSβ from various PKS. For a complete sequence alignment see Appendix II.
OH
CH3OHO
O
O
O
OHOH
OO
O
30
1.2.3.4. The Stanford model for the act minimal polyketide synthase in vitro
Within the Stanford group, Carreras and Khosla first reconstituted PKS activity using act
KSα/KSβ, holo-acyl carrier proteins from a variety of Type II polyketide synthases (but not
act ACP), and MCAT.53 In the absence of MCAT, no activity was detected. This result
apparently supported the idea of a ‘cross-talk’ between fatty acid and polyketide
biosynthesis in S. coelicolor,43,44 because an enzyme from the primary metabolism was
necessary to enable production of polyketides in vitro.
Therefore, in the Stanford model, the formation of malonyl-ACP from holo-ACP and
malonyl CoA is catalyzed by MCAT (Scheme 1.11.A). Initiation of polyketide synthesis
proceeds via decarboxylation of malonyl-ACP to acetyl-ACP, catalyzed by KSα. The
acetyl unit generated is transferred to the active site cysteine in KSα (Scheme 1.11.B).
Iterative loading and Claisen condensation steps lead to elongation of the chain (Scheme
1.11.C). The elongating chain extrudes into a ‘polyketide tunnel’ created between KSα and

Chapter 1. Introduction
18
KSβ (Scheme 1.11.D). When the end of the tunnel is reached, the chain buckles, and
cyclization and release occur.
ACPS OH
OO
CoA+SH
S OH
OO
ACP
CoA
MCAT
S
HS
KS! KS"
HO
O O
CO2
S
HS
O
ACP ACP
KS! KS"
S
S
HO
O O
CO2
S
HS
O
ACP ACP
O
R
O
R
KS! KS" KS! KS"
S
OO
ACP
O O O O O O
Scheme 1.11 The Stanford model for production of SEK4 and SEK4b by the act minimal PKS.31 A. Loading
of ACP is catalyzed by MCAT. B. Decarboxylation of malonyl-ACP is catalyzed by KSα, which
subsequently takes the acetyl starter unit. C. Claisen condensation and elongation of the polyketide chain. D.
The polyketide chain extrudes in a tunnel formed in the interface between KSα and KSβ, and finally cyclizes
and it is released from the PKS.
In remarkable work, the Stanford group identified two key residues in KSβ that help form
the polyketide tunnel.54 Two phenylalanines (F109 and F116) in KSβ were mutated to
alanines, and the corresponding single and double mutant KSα/KSβ complexes were
purified. These mutations introduced a less bulky aminoacid in the position of two aromatic
residues; thus they aimed to increase the chain length of the polyketides produced. In vitro
assays with purified ACP, MCAT and KSα/KSβ complexes showed that the mutant F116A
KSβ
KSα
SEK4 + SEK4b Cyclization and release
A.
B.
C.
D.

Chapter 1. Introduction
19
produced a ratio 64:36 of decaketides to octaketides (as determined by HPLC analysis),
while the F109A/F116A double mutant synthesized decaketides as >95% of total
polyketides.54
1.2.3.5. The Bristol model for the actinorhodin minimal polyketide synthase in vitro
The Bristol group was able to reconstitute PKS activity in vitro using only holo-ACP and
KSα/KSβ, thus in the absence of MCAT.8 Indeed, the loading of holo-ACP with malonate
from malonyl CoA to form malonyl-ACP was shown to be catalyzed by holo-ACP itself
(Scheme 1.12).55 This ‘self-acylation’ activity of holo-ACP from the actinorhodin 9,
griseusin 12 and oxytetracycline 16 PKS was proven with a range of substrates, CoA and
SNAC derivatives.55
CoA-SHS OH
OO
ACPHS
CoA
S OH
OO
ACP
Scheme 1.12 Self-malonylation of holo-ACP
Awkwardly, the debate about the capability of Type II acyl carrier proteins to catalyse their
own acylation was continuous in the literature during the following years. The Stanford
group could not reconstitute PKS activity using only KSα/KSβ and ACP, and argued that
the reported self-malonylation of ACP was attributed to contamination of ACP preparations
with E. coli MCAT due to the use of E. coli-based over-expression systems.53,56
Reynolds and collaborators observed the self-malonylation ability of the
tetracenomycin and frenolicin acyl carrier proteins.57 Moreover, they observed that these
PKS acyl carrier proteins were able to catalyze the malonylation of FAS ACP, which
suggested that PKS ACP and MCAT could catalyze the same chemical reactions. However,
these reports were later retracted as an artefact of heterologous expression and purification
of ACP from E. coli.58 This was based on two facts: first, that consecutive purification steps
of ACP preparations apparently reduced the self-malonylation activity of the

Chapter 1. Introduction
20
tetracenomycin ACP. Second, that an ACP purified from insect cells (which lack discrete
MCAT activity) was unable to self-malonylate.58
Conclusive evidence in support of the ability of, at least, act ACP to self-malonylate was
gathered when the Bristol group used a chemically synthesized ACP. The self-malonylation
activity of this synthetic ACP unequivocally proved the inherent self-malonylation activity
of act ACP, as such protein could not be contaminated with E. coli MCAT.59
Furthermore, the initial observation by the group of Reynolds that PKS acyl carrier
proteins can catalyze the malonylation of FAS ACP, was also proved true by the Bristol
group. Mass spectrometric studies showed that act and otc malonyl-ACP could transfer
their malonyl groups to a recipient PKS or FAS holo-ACP in vitro.60
Recently, the self-malonylation activity of other Type II ACP (i.e. FAS acyl carrier proteins
from Plasmodium falciparum and Brassica napus) has been reported.61 This is striking
because these FAS systems also employ distinct MCAT activities to catalyze malonylation
of ACP. It is possible that MCAT could ensure the specificity of ACP malonylation
permitting only malonyl CoA to be utilized as substrate.
In the model proposed by the Bristol group for the act minimal PKS,60 the minimal PKS
consists of the KSα/KSβ heterodimer and two molecules of holo-ACP, one of which is
complexed with KSα/KSβ (ACP1) and the other is free in solution (ACP2). In a first step,
the ACP in solution undergoes self-malonylation and loads itself with malonate from
malonyl CoA (Scheme 1.13).60 ACP2 then transfers its malonate to the active thiol of the
complexed ACP1. Malonyl-ACP1 is decarboxylated by KSβ (see Chapter 3) to generate
acetyl-ACP,62 which in turn primes KSα with an acetyl starter unit. Meanwhile, ACP2 has
undergone self-malonylation again, and transferred malonate to ACP1. Then a Claisen
condensation catalysed by KSα generates the first polyketide intermediate. This is
transferred to KSα, and ACP1 is ready to receive an extender unit once more. Six further
condensations need be carried out to produce the first isolable polyketides, SEK4 and
SEK4b. This model describes an associative, self-sufficient minimal Type II PKS. Scheme 1.13

Chapter 1. Introduction
21
1.2.4. Type III polyketide synthases
Polyketides produced by Type III PKS recently received much attention due to their
biological activities, which include antioxidant, anti-inflammatory and anti-cancer
properties.3,63 Type III polyketide synthases are typical of plants, but have also been found
in bacteria and fungi.
The most remarkable feature of Type III PKS is that all the catalytic activities needed
for making the final product are contained within a single enzyme. Indeed, selection of
starter and extender units, decarboxylation of substrates, carbon-carbon bond formation and
processing of polyketides (cyclisation) occur in the same active site of the enzyme. Thus,
Type III PKS possess β-ketoacyl synthase (KS) activity, as well as the ability to cyclise and
release the polyketide, but there is no requirement for any further processing step (e.g.
reduction, elimination). Type III PKS also lack an ACP domain, and starter and extender
units are activated as CoA thiolesters, instead of ACP thiolesters. There are only a few
examples of Type III PKS that accept ACP-bound starter units (e.g. Type III PKS
SCO7221 and SCO7671 from S. coelicolor).64
The architecture of the active site in Type III PKS is very similar to that in KSα
components of Type II PKS.65 The universal cysteine is maintained, as well as the
corresponding histidine to H309 (act KSα numbering). However, in Type III PKS an
asparagine residue substitutes for the second histidine (H346).
The chalcone and stilbene plant polyketide synthases are the best studied Type III
PKS.4 The X-ray structures of chalcone synthase from alfalfa67 and stilbene synthase from
pine68 and peanut69 have been published. These show that type III PKS are small (40-45
KDa), homodimeric enzymes which usually share a high degree of similarity in primary
sequence (50-90% sequence identity within plant PKS, and 25% within bacterial PKS).
The stilbene and chalcone PKS both use p-coumaryl CoA 31 as starter unit and extend it
with three repetitive, decarboxylative condensations with acetate extender units derived
from malonyl CoA (Scheme 1.14). A most remarkable difference between the two enzymes
is the cyclisation of the final polyketide intermediate, which results in the production of two
different polyketides, chalcone 32 and resveratrol 33. The mechanism of this cyclisation is
not fully understood;3,68,69 in fact, despite the high degree of similarity between the two

Chapter 1. Introduction
22
enzymes, the group of Noel needed to mutate eighteen residues in order to convert a
chalcone synthase into a stilbene synthase.68
HO
S-CoA
O
CoA-S
O O
OH
HO
O O
S-Enz
HO
O O O
O S-Enz
HO
OH
HO
OH
O
OH
HO
OH
O
HO
O
O
O
Enz-S
HO
O
O
OO
Enz-S
CO2
Scheme 1.14 Biosynthesis of chalcone and resveratrol (a stilbene) by chalcone (CHS) and stilbene (STS)
synthases.65
1.2.5. Type I modular polyketide synthases
Type I modular PKS, usually found in bacteria such as actinomycetes, are responsible for
the biosynthesis of macrolide compounds such as niddamycin 34 from Streptomyces
caelestis and tylosin 35 from Streptomyces fradiae and polyethers such as monensin 36
from Streptomyces cinnamonensis. In this type of PKS, several large proteins, each
containing more than one catalytic active site, combine in a processive fashion to produce a
polyketide. Each protein is subdivided in subunits called ‘modules’, which are covalently
bound to each other. Each module possesses the required catalytic domains for a round of
chain extension. A classic example of this modular Type I PKS is that of 6-
deoxyerythronolide B (6-dEB) synthase (DEBS) from Saccharopolyspora
erythraea.4,70,71,72,73
33
32
31
Type III PKS
2 x malonyl CoA Type III PKS
CHS
STS

Chapter 1. Introduction
23
O
O
O
O
CH3
H3C
O
O
CH3
OH
OO
HON(CH2)2
OH
O
OH
O
O
34
O
O
O
O
O
CH3
OH
OO
HON(CH2)2
OH
O
OH
OH
OO
OMeOMe
HO
35
OO
HO
MeO O O
O
HO
OHO
HO 36
1.2.5.1. The 6-deoxyerythronolide B (6-dEB) polyketide synthase.
6-dEB 37 is the polyketide part of the commercially used antibiotic erythromycin A 38.
The DEBS consists of three proteins (denoted DEBS1, -2 and -3), each of which is
organised in two PKS modules. Each module contains the necessary domains for a round of
chain extension and modification. In every module, at least an acyltransferase (AT), acyl
carrier protein (ACP) and ketosynthase (KS) domains are present, as well as the necessary
active sites for the modification of the β-keto groups in each round (KR, DH and ER). In
addition, DEBS1 and DEBS3 contain the catalytic domains capable of selecting an
appropriate starter unit (loading) and releasing the final product (thiolesterase, TE),
respectively.
The primary sequence of each module is organised in the same fashion (Figure 1.9).
Linker regions between domains and between modules within the same enzyme are
covalent. Interactions between subunits (between DEBS1 and DEBS2, and between
DEBS2 and DEBS3) occur between the ACP of the previous enzyme and the KS of the
following enzyme. This means that biosynthetic intermediates probably undergo direct

Chapter 1. Introduction
24
transthiolesterification between ACP and KS and are not released into solution. Thus KS
both accepts the growing polyketide chain from the previous module and catalyzes the
subsequent decarboxylative condensation between this substrate and an ACP-bound
methylmalonyl extender unit, introduced by AT.
Figure 1.9 Gen sequence of a DEBS PKS module. KS = ketosynthase, AT = acyl transferase, DH =
dehydratase, ER = enoyl reductase, KR = ketoreductase, ACP = acyl carrier protein. DH and ER are only
present in module 4.
The first of the DEBS proteins contains the loading and two PKS modules. A propionyl
starter unit is loaded onto the ACP by the dedicated AT domain, Scheme 1.15. Module 1
then catalyses the decarboxylation of a methylmalonyl CoA and condensation with the
starter propionyl unit in the KS active site. Reduction of the β-carbonyl by KR yields the
first ACP-bound polyketide intermediate, 3-hydroxy-2-methylpentanoate 39. The second
module is analogous to the first module and contains the same catalytic domains.
DEBS2 contains modules 3 and 4. The third module elongates the polyketide chain
with a new methylmalonyl CoA. Although it carries a ketoreductase domain (KRi, Scheme
1.15), this appears to be inactive. In the fourth module, reduction of the β-carbonyl by KR
is followed by dehydration (DH) and reduction of the double bond (ER) to afford a fully
saturated thiolester.
DEBS3 houses module 5, which elongates the chain with a further methylmalonyl unit
and reduces the corresponding ketone. Module 6 is analogous to module 5. Finally, the
extra TE domain cleaves the thiolester between the polyketide and the ACP, leading to
deoxyerythronolide B 37.
Scheme 1.15
1.2.6. Type I iterative polyketide synthases
Type I iterative polyketide synthases are only found in fungi. Although fungi may use for
example Type III polyketide synthases, most polyketide synthases found in fungi are
KS AT (DH) (ER) KR ACP

Chapter 1. Introduction
25
multifunctional enzymes. These megasynthases (> 200 KDa) house the several catalytic
activities needed for the assembly of polyketides.
As described in Section 1.2.5, each module of a Type I modular PKS acts only once
during polyketide synthesis. Conversely, PKS activity in fungi is iterative, much more like
Type II PKS. Thus, the same multifunctional enzyme acts once and again in successive
rounds of chain extension. In Type II polyketide synthases, ACP and KSα probably build
up the full-length polyketide prior to any tailoring step such as reduction and cyclization
(Scheme 1.11). On the other hand, a fungal PKS may or may not process the nascent β-
carbon after each condensation, leading to different levels of reduction after each round
(see Scheme 1.2 in Section 1.2.1).
As a result, fungal polyketides show huge structural variety. Orsellinic acid 40 from
Aspergillus and Penicillium genera, tetrahydroxynaphthalene 41 from Colletotrichum
lagenarium and norsolorinic acid 42 produced by many Aspergilli are examples of the
simple, non-reduced fungal polyketides; whereas fusarin C 43 from Fusarium species and
tenellin 44 from Beauveria bassiana represent more complex polyketide structures fused to
amino acid derivatives.
OHO
HO
OH
OH OH
HO OH 40 41
OH
HO
O
O
OH O
OH
O
N
OH
O
OH
HO
42 43
OMeO
O
NH
O
O
OH
HO 44

Chapter 1. Introduction
26
1.3. Structure of the mammalian fatty acid synthase as a model for polyketide
synthases.
The similarity between fatty acid and polyketide biosynthesis has been emphasized above.
FAS and PKS share common biochemical and structural features, and insights into Type I
FAS structures have important implications for understanding polyketide synthases.74 Ever
since the study of the biochemistry of fatty acids started 50 years ago, scientists have
speculated about FAS structure and mechanism.
1.3.1.1. Initial experiments: the linear model (1970s-1990s)
Type I FAS and PKS are multifunctional proteins with individual functional domains.
Initially, fatty acid synthases were thought to be large, linear structures75 but it soon (i.e.
1975) became clear that only the dimer form of the synthase was functional.76 During the
decades of 1980 and 1990, first inhibition studies77 and then low resolution electron
microscopy78 suggested that the two linear polypeptides were arranged in a head-to-tail
fashion. In this model, the ACP of one subunit would interact with the KS of the other
subunit (Figure 1.10).
ACP TEERMATKS DH KR
ACPTE ER MAT KSDHKR
Figure 1.10 Head-to-tail model and linear arrangement of a dimeric FAS. Single headed arrows indicate the
order of domains in the primary structure (N-terminus to C-terminus). Blue, double headed arrows indicate
the interaction between ACP and KS domains.
1.3.1.2. Building of a 2D model (1990s)
New experimental data obtained by Joshi, Smith, Witkowski and collaborators during the
late 1990s urged a revision of the linear model. They found that mutant FAS lacking

Chapter 1. Introduction
27
functional KS, DH, ACP or TE domains in both subunits of the dimer (i.e. homodimers), as
well as heterodimers formed between mutant ACP and DH, were unable to synthesize fatty
acids. However, heterodimers formed between mutant KS and DH or ACP regained partial
FAS activity (Figure 1.11).79,80 Thus, the linear model for the animal FAS had to be revised
to reflect the finding that the two constituent polypeptides were not simply positioned side-
by-side in a fully extended conformation, but were coiled in a manner that allowed, for
example, the KS and the DH domains of one polypeptide to access the ACP domain located
thousands of residues away on the same polypeptide. This implied the necessity to account
for head-to-tail contacts both inter and intrasubunits.
Direct experimental evidence in support of the new model was obtained by
dibromopropanone cross-linking experiments. This showed that the PP arm in ACP could
interact with the cysteine in KS of both subunits.82 In the subsequent new proposed model,
the two polypeptide chains are intertwined head-to-tail and head-to-head thus facilitating
the interaction of the ACP of both monomers with the catalytic active sites situated at both
sides of the dimer, especially the MAT and the KS domains (Figure 1.12).79,83,84
ACP TEERMATKS DH KR
ACPTE ER MAT KSDHKR X
X ACP TEERMATKS DH KR
ACPTE ER MAT KSDHKR XX
ACP TEERMATKS DH KR
ACPTE ER MAT KSDHKR X
X ACP TEERMATKS DH KR
ACPTE ER MAT KSDHKRX X
X
ACP TEERMATKS DH KR
ACPTE ER MAT KSDHKR X
X ACP TEERMATKS DH KR
ACPTE ER MAT KSDHKR X
X
Figure 1.11 Complementation studies with mutant FAS. The inactivated domain is shown by X. A. Active
condensation intersubunit; B. Active condensation intrasubunit; C. Active acyl transfer intersubunit; D.
Active acyl transfer intrasubunit; E. No dehydration intersubunit; F. Active dehydration intrasubunit.81
A.
C.
B.
D.
E. F.

Chapter 1. Introduction
28
ACP
TE
ER
ACP
TE
ER
KS
MATDH
KR
KS
MAT
DH
KR
Figure 1.12 2-D model for the topology of Type I mammalian FAS. Single headed arrows indicate the order
of domains in primary sequence from N-terminus to C-terminus. Blue, double headed arrows indicate
interaction between ACP and KS, and between ACP and MAT.84
1.3.1.3. 3D models for FAS and PKS megasynthases (1990s-2000s)
The publication of the high-resolution (4.5 Å) X-ray structure of the porcine FAS by Maier,
Jenni and Ban was a breakthrough.85 The enzyme was revealed as an iterative
nanomachine, with overall dimensions of 210 Å x 180 Å x 90 Å that are in good agreement
with previous low-resolution electron microscopy observations (Figure 1.13).86 The
positioning of domains revealed the complex architecture of the multienzyme, which forms
an intertwined dimer with two lateral semicircular reaction chambers. Each monomer
contains a full set of catalytic domains required for fatty acid elongation. The complex
adopts an X-shape with a central body extended at the upper and lower ends by so-called
‘arms’ and ‘legs’ (Figure 1.13). The lower part is comprised by the homodimeric KS
domains, and AT forms the legs. This lower body is connected at the waist with an upper
part formed by the DH, ER (upper body), and KR domains (arms). Distances between the
active sites of catalytic domains are in the nm order which means that the ACP possesses an
extraordinary inherent flexibility to perform its function of acyl (i.e. starter and extender
units and intermediates) carrier protein. In fact, neither the ACP or the TE domains could
be assigned, presumably due to their high mobility.
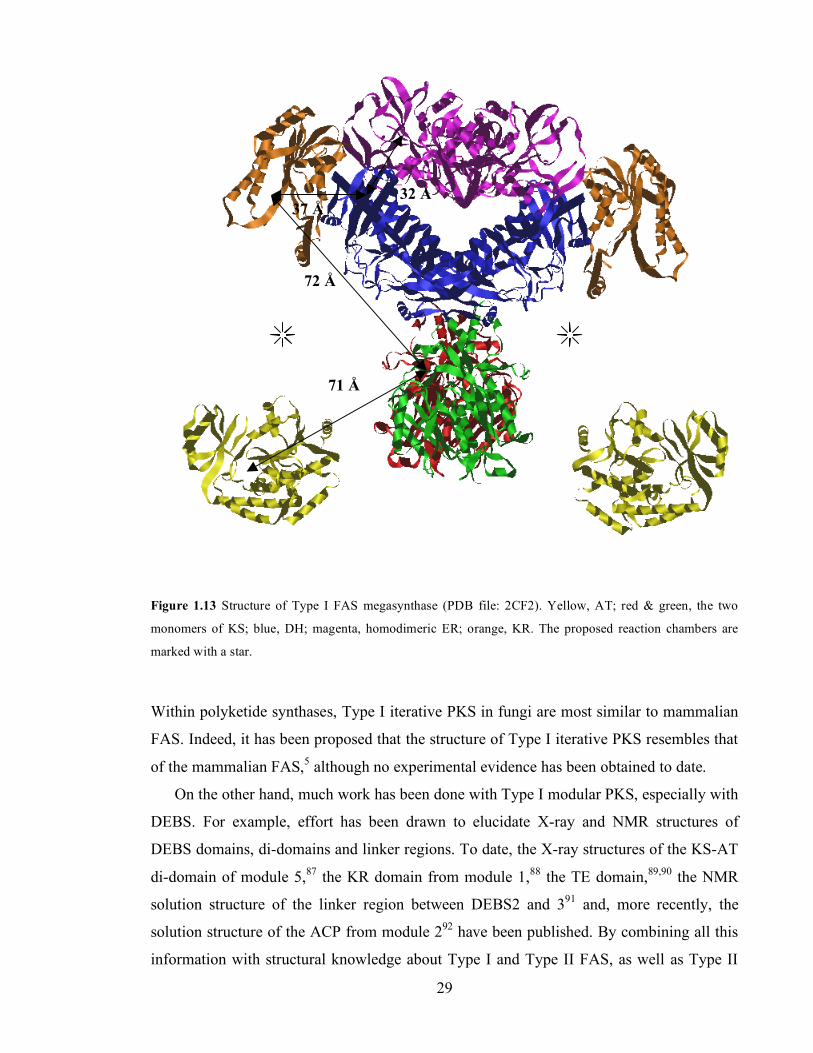
Chapter 1. Introduction
29
Figure 1.13 Structure of Type I FAS megasynthase (PDB file: 2CF2). Yellow, AT; red & green, the two
monomers of KS; blue, DH; magenta, homodimeric ER; orange, KR. The proposed reaction chambers are
marked with a star.
Within polyketide synthases, Type I iterative PKS in fungi are most similar to mammalian
FAS. Indeed, it has been proposed that the structure of Type I iterative PKS resembles that
of the mammalian FAS,5 although no experimental evidence has been obtained to date.
On the other hand, much work has been done with Type I modular PKS, especially with
DEBS. For example, effort has been drawn to elucidate X-ray and NMR structures of
DEBS domains, di-domains and linker regions. To date, the X-ray structures of the KS-AT
di-domain of module 5,87 the KR domain from module 1,88 the TE domain,89,90 the NMR
solution structure of the linker region between DEBS2 and 391 and, more recently, the
solution structure of the ACP from module 292 have been published. By combining all this
information with structural knowledge about Type I and Type II FAS, as well as Type II
71 Å
72 Å
37 Å 32 Å

Chapter 1. Introduction
30
PKS, it has recently been proposed that each module adopts a homodimeric X-shape much
as that of the animal fatty acid synthase.73 This is especially useful for module 4, which
shares with FAS all the reductive domains.
1.4. Fundamentals of enzyme kinetics
All enzymatic kinetic research is based ultimately on the experimental determination of the
catalytic activity of the enzymes. A kinetic test is reliable if (a) the signal is directly
proportional to the variation of the compound measured, (b) the monitoring of reaction
signal is in convenient time range for determining the initial rate unambiguously, and (c)
the reaction rate is linear with respect to enzyme concentration.93 A wide range of
conditions (e.g. enzyme and substrate concentrations, temperature, pH, etc.) must be
assayed in order to characterise the kinetic behaviour of the enzymes.94
A discontinuous assay requires the removal of samples from the reaction mixture at time
intervals and subsequent analysis to determine the extent of reaction, for instance, by
HPLC. Conversely, a continuous assay monitors the progress of the reaction continuously
with an automatic recording device. For example, a well known spectrophotometric effect
in biochemistry is the change in the absorbance at 340 nm when the reduced form of
NAD(P) is oxidised. Changes in fluorescence is another physical property the experimenter
can take advantage of. A change in the pH of the reaction can also be followed and related
to enzyme activity. Even if no spectroscopic or pH change takes place, a coupled enzymatic
assay can often be devised. Here, the reaction of interest is coupled with another reaction,
faster than the target reaction, which does cause a measurable change in some of the
properties outlined above.94
1.4.1. Rate equations in enzyme kinetics
The theory of enzyme kinetics was developed at the beginning of the last century. First
Henri, and then Michaelis and Menten proposed a mechanism where the substrate bound to

Chapter 1. Introduction
31
the enzyme to generate an enzyme-substrate complex, which then released the product and
regenerated the enzyme (Scheme 1.16).94
E + S ES E + P
Kd k2
Scheme 1.16 Mechanism for enzyme-catalyzed reaction proposed by Michaelis and Menten. S = substrate, E
= enzyme, P = product.
Michaelis and Menten then carried out a mass balance assuming that the reversible step was
a true equilibrium, and the concentration of the intermediate ES was simply calculated
through the dissociation constant, Kd. The rate equation derived by Michaelis and Menten
was:
!
rate =k
2E
0[ ] S[ ]Kd + S[ ]
(Eq. 1)
where E0 is the initial enzyme concentration.
Later, Briggs and Haldane examined a more general mechanism in which the reversible
step was not treated as an equilibrium (Scheme 1.17).94
E + S ES E + P
k-1
k2k1
E0-x xS P
Scheme 1.17 Briggs-Haldane mechanism. The concentration of the different species is shown underneath.
The concentration of the intermediate, x, was assumed to reach a constant, steady state
value (Eq. 2).
!
dx
dt= k1( E0 " x )S " k"1x " k2x = 0 (Eq. 2)

Chapter 1. Introduction
32
Collecting terms in x and calculating the rate as k2x gives Eq. 3.
!
rate =k
2E
0[ ] S[ ]k
-1 +k2
k1
+ S[ ] (Eq. 3)
Eq. 3 was derived for a particular mechanism (Scheme 1.17), but it applies to much more
complex systems than the simplest two-steps Michaelis-Menten or Briggs-Haldane
mechanism (Scheme 1.16). It usually takes the general form:
!
rate =kcat
appE0[ ] S[ ]
KM
app+ S[ ]
(Eq. 4)
where kcatapp and KM
app are not true first order rate constant and equilibrium
constants, but the apparent catalytic constant and Michaelis constant, respectively.
If the concentration of substrate is much higher than KMapp, the equation of the rate is as
follows:
!
rate " kcat
appE[ ] (Eq. 5)
Under these circumstances, the rate is the maximum reaction rate achievable at a certain
enzyme concentration and is generally referred to as Vmax. In turn, KMapp can be defined as
the substrate concentration that results in a reaction rate which is half the value of Vmax.
Similarly, at low substrate concentrations (much lower than KM) the quantity kcat / KM is
the second-order reaction rate constant:
!
rate " kcat
/KM
[E][S] (Eq. 6)
kcat / KM is usually referred to as specificity constant because it determines the ratio of rates
for two competing substrates A and B when they are mixed together:

Chapter 1. Introduction
33
!
rateA
rateB
"(V/K
M)S
A
(V"/K"M
)SB
=(k
cat/K
M)S
A
(kcat
"/K"M
)SB
(Eq. 7)
where SA and SB are the concentrations of A and B respectively.
The value of kcat / KM is limited by the diffusion-controlled encounter of the enzyme-
substrate, which is between 108 and 109 M-1 s-1.95 Some of the fastest enzymes known, for
example carbonic anhydrase, triose phosphate isomerase and acetylcholinesterase have kcat /
KM values in that range.
A final, important consideration is that in deriving the rate equations described in this
section, the concentration of the substrate, S, was not corrected to account for the formation
of the intermediate ES or product. This is an essential condition of the Michaelis-Menten
and Briggs-Haldane approaches: the substrate concentration was treated as constant and
equal to the initial value. Consequently, the rate that appears in Eq. 4 is the initial rate, i.e.
the rate of the enzymatic reaction when the substrate concentration either a) is much higher
than KM (>5-10 fold) or b) does not vary considerably (ideally less than 10%) during the
length of the experiment.
1.4.2. Kinetics of the act minimal PKS
The actinorhodin Type II PKS from Streptomyces coelicolor A3(2) has been the model for
Type II PKS in the literature. The act minimal PKS has been extensively studied by the
Bristol and Stanford groups during the last decade.
The act minimal PKS catalyses a number of individual reactions. Firstly malonate must
be transferred from CoA to holo-ACP (Scheme 1.18.A). In the Bristol model for the act
minimal PKS, this loading reaction is catalyzed by ACP itself (Section 1.2.3.5). The
kinetics of self-malonylation of holo-ACP were studied by means of a radioactive,
discontinuous method to measure the transfer of 14C labelled malonate units.55 A catalytic
constant, kcat = 0.34 min-1 and Michaelis constant KM = 219 µM were measured. Malonyl

Chapter 1. Introduction
34
transfer between acyl carrier proteins has also been studied in the Bristol group by
discontinuous mass spectrometry methods, though no kinetic parameters were calculated.
Initiation of polyketide synthesis proceeds via decarboxylation of malonyl-ACP to form
acetyl-ACP, the starter unit for polyketide biosynthesis (Scheme 1.18.B).62 The polyketide
chain is then extended to 16 carbon atoms by seven extension reactions using malonyl-ACP
(Scheme 1.18.C). These reactions are catalyzed by iterative cycles of KSα.96 It has been
traditionally assumed that the polyketide chain is fully formed before the chain cyclizes and
it is released from the synthase to form SEK4 19 and SEK4b 27 (Scheme 1.18.D).
HO
O O
S-CoA
holo-ACP
CoA-SH
HO
O O
S-ACP
CO2
O
S-ACP
x 7 x 1
O O O O O O O
S-ACP
O
OH
H
COOHO
O
OH
OH
OH
H
COOHO
O
OH
OH
HO
HO
O
O
OH
O
O
O
O
HO O
OH
HO
O
Scheme 1.18 Biosynthesis of octaketides by the act minimal PKS. A. Loading of holo-ACP with malonate. B.
Initiation of polyketide synthesis by formation of acetyl-ACP. C. Elongation of the polyketide chain. D.
Formation of SEK4 and SEK4b in vitro. E. Further tailoring reactions lead to actinorhodin in vivo.97
Remarkably, little is known of the absolute or relative rates of the component reactions of
the act minimal PKS, although the overall transformation from malonyl CoA to SEK4 and
SEK4b has been studied. The Stanford group measured SEK4 and SEK4b production in
MCAT-supplemented act minimal PKS.56 A discontinuous thin layer chromatography
based method for detection of 14C labelled SEK4/4b was used. Under their experimental
conditions (and using an ACP from the frenolicin PKS), a KM of 5 µM for holo-ACP was
given as well as a catalytic constant kcat of 0.31 ± 0.11 min-1. Subsequent publications from
19
27 9
A. Loading B. Initiation
C. Extension
D. In vitro: cyclisation and release
E. In vivo: act PKS

Chapter 1. Introduction
35
the same group have reported similar kinetic parameters, in the range kcat = 0.11-0.27 min-1
and KM = 2.7-6.4 µM with ACP from a variety of Type II PKS, including act ACP.98,99
However, the ACP used in these studies were not able to load themselves with malonate
units and questions have been arisen towards the quality of these proteins due to the
absence of ACP characterization (e.g. levels of folded, active holo-ACP).60 In any event,
the reaction rates of the initiation and extension steps could not be disclosed.
Within the Bristol group, Matharu performed actinorhodin minimal PKS assays and
measured the initial rate of SEK4/4b production. She varied the concentration of ACP from
2.5 to 200 µM, with constant malonyl-CoA (1 mM) and KSα/KSβ (1.5 µM) concentrations.8
A discontinuous assay was used to follow the reaction: at time intervals, a sample was
taken from the reaction mixture and analysed by HPLC. It appeared that production of
SEK4 and SEK4b occurred at the rate predicted by Hitchman55 for the self-malonylation of
act holo-ACP.8 Therefore, it was hypothesized that the self-malonylation of ACP was the
rate limiting step in the overall production of octaketides by the minimal PKS, even at the
highest ACP concentrations used in the study (200 µM holo-ACP).
1.5. Aim of the project
The aim of this work was to further understand the process of polyketide biosynthesis using
the S. coelicolor Type II actinorhodin PKS as a model system. The first objective of the
project was to develop a method amenable for kinetic purposes to measure the rate of the
reactions catalyzed by each component of the actinorhodin minimal PKS. In this thesis,
Chapters 2, 3 and 4 are devoted to the study of the catalysis of ACP (loading step), KSβ
(initiation step) and KSα (extension step), (Scheme 1.18). Subsequently, the second
objective was to generate a series of ACP, KSα and KSβ mutant proteins and subject them
to these assays in order to identify specific residues responsible for the catalysis of each
protein.
In parallel, we aimed to use a kinetic approach to investigate the stoichiometry of the
act minimal PKS (Chapter 5), and the effect of the addition of further act proteins to the
minimal system (Chapter 6).

Chapter 3. Initiation of polyketide synthesis
36
2. Studies on Acyl Carrier Proteins
Acyl carrier proteins are essential components of Type I and Type II polyketide and fatty
acid synthases. In vivo, acyl carrier proteins are expressed as inactive apo forms, and need
to be postranslationally modified to the active holo forms by addition of a
4’phosphopantetheine (PP) cofactor with its terminal sulphydryl (Section 1.2.3.1).
Previous studies within the Bristol group had shown the rapid disulphide formation
between the thiols of the PP of holo-ACP to form ACP dimers in solutions lacking a
sulphydryl reducing agent (Scheme 2.1).8,52 The preservation of the reactive sulphydryls of
ACP is critical to the maintenance of their function, as the malonate units required for
initiation and extension of the polyketide chain are attached to the sulphydryl at the end of
the PP arm.
Another reaction that leads to inactive ACP forms, at least in vitro, is an intramolecular
disulfide formation between C17 in act ACP and the terminal thiol of the PP arm. For this
reason, a mutant C17S act ACP was engineered within the Bristol group.52 In this thesis,
this C17S ACP will be referred to as the ‘reference’ act ACP, because it was used
throughout our work instead of the wild type act ACP.
ACP
SHACP
SHACP
S S
ACP
Scheme 2.1 Disulphide formation between the thiols of holo-ACP
The classical model of fatty acid and polyketide biosynthesis pictures a semi-static ACP
with a highly mobile 18-20 Å ‘swinging’ PP arm to deliver intermediates as well as starter
and extender units between catalytic domains. However, recent studies with DEBS and the
mammalian fatty acid synthase have provided compelling evidence that ACP has to interact
with catalytic active sites which are separated by distances as long as 80 Å.85 In fact, in a
round of chain extension in which the nascent β-carbon is fully reduced to saturation (e.g.
module 4 in DEBS and Type I FAS), the ACP-bound substrate must be shuttled
successively from AT to KS, then to KR, DH and ER. Finally, the extended polyketide is
ACP monomers ACP dimer

Chapter 3. Initiation of polyketide synthesis
37
either passed to the KS of the following module (e.g. in DEBS) or cleaved from its anchor
ACP by TE (e.g. in mammalian FAS). Thus as many as six partner enzymes have to be
recognized by ACP. Therefore ACP-partner enzymes interactions are of utter importance to
understand the mechanism of polyketide and fatty acid synthesis.
2.1. Protein-protein interactions in Type II FAS and PKS
Type II fatty acid and polyketide synthases consist of several monofunctional enzymes
which are presumed to form a protein complex stabilized by non-covalent forces (Section
1.2.2). In order to form a Type II PKS-derived polyketide such as actinorhodin, a series of
10-15 proteins need be at the same time in the same place of the cell and interact with each
other efficiently. Thus it holds that efficient protein-protein recognition is vital in Type II
polyketide synthases.
The FAS counterpart of Type II polyketide synthases has been studied thoroughly.
Type II fatty acid synthases also consist of a collection of individual enzymes encoded by
separate genes. The E. coli FAS is the model for Type II fatty acid synthases.101 Very
important structural information has been gathered: to date, the NMR structure of ACP102
and the crystal structures of MCAT,104,105 the elongation β-ketoacyl synthases KASI and
KASII,103,106,107 the initiation ketoacylsynthase KASIII,108 as well as those of KR,109 DH,111
and ER110 have been reported.
Despite this wealth of structural information, little is known about protein-protein
interactions between the Type II FAS components. Seminal work by Zhang, Rock and co-
workers was carried out by in silico modelling of the docking of E. coli ACP on one of the
ketosynthases used by E. coli in the synthesis of fatty acids, i.e. KASIII.111 KASIII
catalyzes the formation of acetoacetyl-ACP from acetyl CoA and malonyl-ACP (Scheme
2.1.A). They identified a positively charged pocket in KASIII in close interactions with
negatively charged residues in helix II of ACP. Significantly, R249KASIII was observed to
interact with E41ACP (which is situated within helix II of ACP and corresponds to E47 in
act ACP). They proposed that, this ion pair had a critical role in orientating the ACP-bound
substrate towards the catalytic active site in KASIII.
Biochemical assays were performed by the same group to validate this model. Zhang,
Rock and co-workers harnessed the fact that KASIII can also accept malonyl CoA instead

Chapter 3. Initiation of polyketide synthesis
38
of malonyl-ACP as a substrate (although much less efficiently), (Scheme 2.1.B). The
hypothesis that holo-ACP could be an inhibitor of this second reaction was addressed.111 In
this model, holo-ACP could dock onto KASIII and inhibit KASIII activity towards malonyl
CoA. In other words, if holo-ACP docked onto KASIII, its PP arm would enter the active
site of KASIII, and the ketosynthase would not be able to process malonyl CoA as a
substrate. When the wild type (WT) KASIII was subjected to this assay, strong inhibition
by ACP was observed (20% activity at 150 µM ACP and 5 mM malonyl CoA). A mutant
R249A KASIII showed 80% activity under the same conditions.111 This lack of inhibition
suggested inefficient binding of ACP to KASIII, and therefore supported the role of R249
in KASIII as binding site for ACP.
ACP--S OH
OO
CoA-S
O ACP--S
OO
CoA
CoA-S OH
OO
CoA-S
O CoA-S
OO
CoA
Scheme 2.2 Reaction scheme of E. coli KAS III. A. Condensation of acetyl CoA and malonyl-ACP to form
acetoacetyl-ACP. B. The same reaction is possible using malonyl CoA instead of malonyl-ACP.
Models for the binding of act ACP onto S. coelicolor MCAT and for the docking of
Helicobacter pylori ACP to MCAT have also been done in silico.113,114 The importance of
charged residues along helix II of ACP, as well as hydrophobic forces, were postulated to
determine these protein-protein interactions.113,115 A further model of B. subtilis ACP
bound to ACPS also appointed helix II of ACP as the docking region upon ACPS.116
Similarly, in silico analysis of the structure of ACP bound to ER in E. coli FAS showed
several acidic residues in and close to the ACP helix II (D41, D44 and E47, act ACP
numbering throughout) interacting with basic amino acids in E. coli ER (L201, R204,
L205).117 Experimental evidence for the role of helix II of E. coli ACP in the binding to E.
coli KR was gathered by the group of Rock.118 Using a combination of inhibition studies,
A.
B.
KAS III
KAS III

Chapter 3. Initiation of polyketide synthesis
39
surface plasmon resonance, FRET assays and NMR titrations, a pair of aspartate residues
on helix II of ACP (D37 and D41) and the conserved serine S42 as well as I60 in the loop
between helix II and helix III were identified as the docking residues of ACP.118 The group
of Rock then hypothesized that highly conserved negatively charged residues in helix II are
a universal recognition motif that could be responsible for electrostatic interactions between
ACP and its partner enzymes. In a recent study by Cronan, the importance of negatively
charged and hydrophobic residues in helix II of E. coli FAS ACP has also been shown in
vivo.119 Scarcer information is available in Type I fatty acid and polyketide synthases,
although it has been proposed that, electrostatic (via recognition helix II) and hydrophobic
forces are the key players in protein-protein interactions in DEBS73,92 and yeast FAS.121
In fact, the majority of acyl carrier proteins from Type I and Type II FAS and PKS
contain a pair of negatively charged residues six or seven amino acids apart (Figure 2.1).
Within the Bristol group, Arthur studied the interaction of act ACP with act KSα/KSβ, act
KR and S. coelicolor ACPS by tryptophan fluorescence assays. He measured the
dissociation constants of act ACP and a series of mutants, and identified E47 and E53 as
essential residues that support ACP-KSα/KSβ and ACP-KR interactions. E47 was also
involved in ACP-ACPS binding.120
Figure 2.1 Primary sequence alignment of FAS and PKS ACP (green star, the PP attachment serine).

Chapter 3. Initiation of polyketide synthesis
40
2.2. Purification of acyl carrier proteins
All act acyl carrier proteins are mutant proteins in which the cysteine 17 has been mutated
to serine (see introduction to Chapter 2). The acyl carrier proteins used in this work are the
act mutants C17S ACP (referred to as ‘reference act ACP’), E47A/C17S ACP, E47V/C17S
ACP, E53A/C17S ACP and R72A/C17S ACP as well as the acyl carrier proteins from the
griseusin (gris) and daunorubicin/doxorubicin (dps) polyketide synthases.
All acyl carrier proteins come from dual expression plasmids that harbour the
corresponding acyl carrier protein gene as well as the gene encoding E. coli ACPS.47 All
acyl carrier proteins were over-expressed in E. coli BL21 (DE3) and purified to
homogeneity as described previously in the literature for act ACP52 and in the Experimental
Chapter (Section 8.5.2.1). Most of the acyl carrier proteins were obtained in their holo form
as judged by ESMS analysis (Figure 2.2), except E47V ACP, where the ratio holo:apo was
1:2. Phosphopantetheinylation of this ACP was then performed in vitro with purified S.
coelicolor ACPS and CoA (in a ratio of ACPS:ACP:CoA concentrations of 1:10:100) in the
presence of 10 mM Mg2+.42 Final holo:apo relative abundance was 1:1. Separation of the
apo and holo forms of act E47V ACP was achieved by anion exchange chromatography
(Q-Sepharose) and the ACP was eluted as described for ACP purification.
Figure 2.2
2.3. Reduction of ACP dimers
Dithiothreitol (DTT) 45 has been traditionally used to carry out the reduction of the
intermolecular disulphide bond that can occur between the terminal thiols of the PP arms of
holo-ACP in vitro (see introduction to Chapter 2). Incubation of holo-ACP dimer with DTT
(1 mM, 30 ˚C, overnight) yields the active holo-ACP monomer. Within the Bristol group,
act minimal PKS assays where ACP was treated with DTT, and then DTT was removed by
gel-filtration prior to assay, were previously performed.8,62,96,122 Because this is a tedious
and time-consuming procedure, the Bristol55 and other groups have also used DTT in situ at
concentrations between 1 and 2 mM.56,57,58,98,99
However, thiol reducing agents such as DTT can potentially be incompatible with
thiolesters, as they can act as nucleophiles and decrease thiolester intermediate
concentrations by transthiolesterification. On the other hand, tris(2-carboxyethyl)phosphine

Chapter 3. Initiation of polyketide synthesis
41
(TCEP) 46 has been described as an alternative reductant in the literature,123 and recently
Moore and co-workers have used TCEP as reducing agent in PKS assays.124
HS
OH
OH
SH
HO
O
P
OH
O
OHO
45 46
When act holo-ACP is lyophilized in air, it rapidly forms a disulfide dimer via the linking
of the PP thiols.8 Dimer prepared in this way was then treated with TCEP (1 mM) for 1 h at
30 ˚C to test for the ability of TCEP to reduce the disulfide bonds. Subsequent analysis by
ESMS confirmed that >95% of the holo-ACP was in its monomeric form (Figure 2.3).
Figure 2.3 Mass spectrum of the reference act holo-ACP dimer after incubation with TCEP. Expected
masses, 9441 Da (monomer), 18882 Da (dimer).
We then investigated the effect of DTT and TCEP on acylated ACP by ESMS. Malonyl-
ACP was made using the method described by Hitchman for the self-malonylation of
ACP:55 purified act holo-ACP monomer (200 µM) was incubated with malonyl CoA (1
mM) for 2 h at 30 ˚C. Then, the protein solution was desalted by gel filtration to remove the
excess malonyl CoA. About 90% of the total ACP was converted to malonyl-ACP, as
judged by ESMS analysis.
monomer
dimer

Chapter 3. Initiation of polyketide synthesis
42
The desalted malonyl-ACP (50 µM) was treated with TCEP or DTT (1 mM) for 1 h at 30
˚C, and the ACP species then analysed by ESMS. Incubation of malonyl-ACP (9527 Da)
with DTT mostly led to transthiolesterification of the acyl group and production of holo-
ACP (9441 Da) as observed by ESMS (Figure 2.4.A). We could only detect traces of
malonyl-ACP after 60 min indicating almost complete removal of the malonate.
This contrasted to the use of TCEP as reductant -TCEP did not react with malonyl-ACP
(Figure 2.4.B). Thus, stock solutions of holo-ACP were routinely treated with TCEP to
ensure production of ACP monomers, and PKS assays were performed in the presence of
TCEP.
Figure 2.4 A. Incubation of malonyl-ACP with DTT. B. Incubation of malonyl-ACP with TCEP. Expected
masses: 9441 Da (holo-ACP) and 9527 Da (malonyl-ACP).
2.4. Studies on the self-malonylation activity of act holo-ACP.
Act ACP has the ability to catalyze its own malonylation from malonyl CoA, to form
malonyl-ACP (see Section 1.2.3.5 in Chapter 1). Within the Bristol group, a radioactive
assay was implemented to study the kinetics of self-malonylation of act holo-ACP with 14C
labelled malonyl CoA.55 The assay involved the reaction of 14C-labeled malonyl CoA with
holo-ACP over time. At set time points the reaction was sampled, protein precipitated,
excess radio-label washed away, protein resolubilised and 14C incorporation estimated by
scintillation counting. This enabled the kinetic parameters of kcat = 0.34 min-1 and KM = 219
µM to be measured.55
A. B.

Chapter 3. Initiation of polyketide synthesis
43
This assay required discontinuous, individual measurements and used significant amounts
of pure protein. The assay was quantitative, but the very large number of measurements
required, and the use of large amounts of protein made this method impractical for further
routine use, for instance to study mutant acyl carrier proteins.
A second, mass spectrometric assay to measure the extent of self-malonylation was
developed by the Bristol group.60,120 Following the incubation of malonyl CoA with holo-
ACP, the reaction was quenched at time intervals and the protein fraction analyzed by mass
spectrometry. Although this assay did not involve radioactive materials, it was also
discontinuous and not suitable for the determination of kinetic parameters due to the
qualitative nature of protein mass spectrometry. The aim of this section is to develop an
assay amenable for the kinetic study of the self-malonylation of ACP.
2.4.1. Development of the α-ketoglutarate dehydrogenase assay to measure the rate of
self-malonylation
α-Ketoglutarate (or 2-oxoglutarate) dehydrogenase (KGDH) is an enzyme involved in the
citric acid cycle.125 The KGDH complex is composed of multiple copies of three
component proteins: α-ketoglutarate decarboxylase (E1), lipoate succinyltransferase (E2)
and dihydrolipoamide dehydrogenase (E3).125,126 Each enzyme needs a cofactor for catalytic
activity, namely thiamine pyrophosphate (TPP) 47, lipoic acid (lip) 48 and flavin adenine
dinucleotide (FAD) 49 respectively.
Oxidation of α-ketoglutarate 50 is catalysed by the consecutive action of E1, E2 and
E3. First, 50 is decarboxylated by E1, which uses 47 as a cofactor (Scheme 2.3-A). Second,
the succinyl unit is transferred to E2 with partial reduction of 48 and regeneration of the
active E1 complex (Scheme 2.3-B). The succinyl unit is then transferred to coenzyme A
(CoA-SH) 51 by E2 (Scheme 2.3-C).
Regeneration of E2 activity is catalysed by E3, which uses 49 as a cofactor (Scheme
2.3-D). E3 itself is reactivated by reduction of nicotinamide adenine dinucleotide (NAD+)
to form NADH 52, Scheme 2.3-E. Overall, α-ketoglutarate and CoA-SH are converted to
succinyl CoA using and equivalent of NAD+ - it is the production of NADH (from NAD+)
which is then followed spectrophotometrically.

Chapter 3. Initiation of polyketide synthesis
44
A number of groups have used the α-ketoglutarate dehydrogenase complex as a coupling
enzyme for assaying the generation of CoA-SH. For example, Khandekar et al studied the
kinetics of the β-ketoacyl-acyl carrier protein synthase III (FabH) from Streptococcus
pneumoniae.127 Molnos et al coupled the reaction of the E. coli malonyl CoA: acyl carrier
protein transacylase (MCAT, FabD) to α-ketoglutarate dehydrogenase activity.128 More
recently, a Type III PKS from Pseudomonas fluorescens and MCAT from Helicobacter
pylori have been characterised by the same method.129,130 The respective FAS or PKS
reaction generates coenzyme A, which is converted to succinyl CoA by the KGDH
complex (Scheme 2.4).
O
HO
O
O-O
E1(TPP) TPP
O
HO
O
E1CO2
E2-lip(S-S)TPP
O
HO
O
E1 E1(TPP) S
O
HO
O
lip(S-H)-E2
S
O
HO
O
lip(S-H)-E2CoA-SH S
O
HO
O
CoAE2-lip(SH2)2
E2-lip(SH2)2 E3(S-S)-FAD E2-lip(S-S) E3(SH2)2-FAD
E3(S-S)-FADE3(SH2)2-FAD NAD+ NADH H+
Scheme 2.3 Reaction scheme of the KGDH assay.
A.
B.
C.
D.
E.
50

Chapter 3. Initiation of polyketide synthesis
45
O
OH
O
O
HO
O
HO
O
SCoA
NAD+ NADH
CoA-SH
Acetyl or malonyl CoA
Scheme 2.4 Quantitative KGDH assay for CoA-SH. NADH production is followed spectrophotometrically.
2.4.1.1. Development of the method to study the self-malonylation of act holo-ACP
The rate of NADH production can be monitored either by measuring absorbance at 340 nm
(ε = 6230 M-1 cm-1) or fluorescence (λexcitation = 340 nm, λemission = 465 nm). Though
fluorescence measurements have increased sensitivity, the ready availability of an
ultraviolet absorbance plate reader in our laboratory led us to choose the latter method.
The concentrations of all assay components were as described in the Experimental
Chapter (Section 8.7.4). To ensure the coupling enzyme was in its holo form, KGDH was
incubated with KGA, TPP and NAD+ for 5 minutes before the assays were started. Previous
studies had reported an initial burst of activity when assays were started by addition of
malonyl CoA, presumably due to small amounts of CoA-SH in malonyl CoA stocks.128,130
In order to avoid this, all assays were started by addition of the enzyme, holo-ACP.
2.4.1.2. Kinetics of the self-malonylation of act holo-ACP
We aimed to adapt the KGDH assay to the kinetic study of the self-malonylation of ACP.
First, we incubated all assay components with KGDH and started the experiment with act
holo-ACP in several concentrations. We observed an increase in the absorbance at 340 nm
due to reduction of NAD+. The initial change in absorbance was dependent on the initial
ACP concentration, and all reactions were complete in ten minutes (Figure 2.5).
FAS or PKS component
KGDH

Chapter 3. Initiation of polyketide synthesis
46
The self-malonylation reaction produces 1 mol of malonyl-ACP and CoA-SH per mol of
holo-ACP and malonyl CoA (see Scheme 1.12 in Chapter 1). If the concentration of
malonyl CoA is in excess, then the equilibrium will lie on the right side and the final
concentration of malonyl-ACP will be essentially identical to the initial holo-ACP
concentration. In turn, the KGDH assay produces 1 mol of NADH per mol of CoA-SH
(Scheme 2.3). Thus it holds that the molar concentration of NADH generated must be equal
to the concentration of malonyl-ACP, and the change in absorbance at 340 nm due to
production of NADH can be directly related to the concentration of malonyl-ACP.
Therefore this method provided a very accurate measurement of the final concentration of
malonyl-ACP and also of the initial concentration of holo-ACP.
Figure 2.5. Raw data for the KGDH assay at initial holo-ACP concentrations of 15 µM (squares), 30 µM
(triangles) and 50 µM (circles). Malonyl CoA concentration was fixed at 1mM.
A kinetic study of the self-malonylation of act holo-ACP was carried out by measuring the
initial reaction rate at a range of substrate concentrations. Malonyl CoA concentration was
varied from 10 to 1000 µM at a fixed holo-ACP concentration of 60 µM and kinetic
parameters calculated according to the Michaelis-Menten model. A turnover number kcat =
50 µM holo-ACP
30 µM holo-ACP
15 µM holo-ACP

Chapter 3. Initiation of polyketide synthesis
47
0.49 ± 0.01 min-1 and Michaelis constant KM = 207 ± 29 µM were determined by direct fit
of the experimental data points to a hyperbolic function (Figure 2.6).
Figure 2.6 Determination of Michaelis-Menten kinetic parameters of the self-malonylation of holo-ACP by
the KGDH coupling system. A. Plot of reaction rate vs. substrate concentration and hyperbolic fit. B. Hanes
linear plot of the data.
2.4.2. Actinorhodin minimal PKS to measure self-malonylation rate
Kinetic analysis of Type II minimal PKS have been published by the Bristol and other
groups. In Bristol, an HPLC method to detect SEK4 and SEK4b was used.8 A radioactive
thin layer chromatography method has been used by other groups to measure polyketide
production by a variety of Type II PKS.56,98,99,131 However, these assays are unsuitable for
kinetic assays because they are discontinuous, thus only allow to measure the extent of
reaction at discrete time points.
Instead, we aimed to monitor the production of the octaketides directly and
continuously. We reasoned that formation of aromatic octaketides could be directly
observable using ultraviolet spectrophotometry.
2.4.2.1. Development of the method
All spectrophotometric measurements were carried out in an absorbance plate reader, as
described in Section 8.7.6. SEK4 and SEK4b were isolated from Streptomyces coelicolor
CH999/pSEK4 following published procedures8,24,34 and used as standards for calibration.
A. B.

Chapter 3. Initiation of polyketide synthesis
48
The absorbance spectra of SEK4 and SEK4b show maxima in the aromatic region at 280
nm. However, the adenine group of CoA also absorbs strongly at this wavelength at pH =
7.3 and A280 was not appropriate for polyketide detection in minimal PKS assays. On the
other hand, our assays rely on the absorbance of SEK4 and SEK4b at 293 nm. The
detection limit of polyketides was estimated as 100 pmol (corresponding to a change of 6
mAU under our experimental conditions), and the calibration was linear up to 30 nmol
SEK4/4b (corresponding to 150 µM polyketides in 200 µl experiments, and a change in
absorbance of 1.89 AU).
The extinction coefficients of SEK4 and SEK4b at 293 nm were determined
experimentally as 13,200 and 12,100 M-1 cm-1 (measured in 100 mM phosphate buffer, pH
= 7.3) respectively. The Bristol and Stanford groups have shown by HPLC analysis of the
products of actinorhodin minimal PKS assays that the ratio SEK4:SEK4b is approximately
1:1 in actinorhodin minimal PKS assays in vitro;8,34 thus a mean extinction coefficient of
12,600 M-1 cm-1 was assumed for kinetic measurements.
The KSα/KSβ heterodimer was purified as described in the Experimental section. Typical
yields were 75-100 mg L-1. When we incubated the act minimal PKS with malonyl CoA, an
increase in the absorbance at 293 nm was observed, due to the assembly of the aromatic
SEK4 and SEK4b (Figure 2.7). A change in A293 was only observed in the presence of
malonyl CoA, holo-ACP and KSα/KSβ; and controls in which individual assay
components were absent did not show any activity.
Having established the detection conditions, we first investigated the dependence of
polyketide production rate on the concentration of KSα/KSβ. Thus, the concentration of
KSα/KSβ was varied while maintaining holo-ACP and malonyl CoA concentrations fixed
at 80 µM and 1 mM respectively; and we observed the reaction at 293 nm. The initial
change in absorbance (i.e. the initial reaction rate) depended on the concentration of
KSα/KSβ (Figure 2.7).

Chapter 3. Initiation of polyketide synthesis
49
Figure 2.7. Increase in the absorbance at 293 nm in act minimal PKS assays. Holo-ACP and malonyl CoA
concentrations were fixed at 80 and 1000 µM respectively. KSα/KSβ was 0.3 µM (diamonds), 0.5 µM
(triangles) and 1 µM (circles).
This experiment was repeated for a variety of holo-ACP and KSα/KSβ concentrations, and
initial reaction rates were measured (Figure 2.8). For KSα/KSβ concentrations less than 0.5
µM, initial reaction rates increased linearly with the concentration of the ketosynthase
components. In these conditions, initial rates depended on the concentrations of both
KSα/KSβ and holo-ACP. At higher KSα/KSβ concentrations the reaction rate depended
solely on the concentration of holo-ACP, indicating that a holo-ACP dependent process
(i.e. self-malonylation) was rate-limiting at concentrations of KSα/KSβ higher than 2 µM.
These results indicated that the actinorhodin minimal PKS followed a two-step mechanism.
In this model, first holo-ACP self-malonylated (from malonyl CoA) to form malonyl-ACP.
In a second step, KSα/KSβ catalyzed the synthesis of SEK4 and SEK4b from malonyl-
ACP (Scheme 2.5). Therefore, KSα/KSβ (at concentrations higher than 3 µM) could
effectively be used as a coupling enzyme for the production of malonyl-ACP in order to
study the kinetics of self-malonylation. In these circumstances, production rates of aromatic
polyketides could be related to the self-malonylation rate by a stoichiometric factor of 8.
1 µM KSα/KSβ
0.5 µM KSα/KSβ
0.3 µM KSα/KSβ
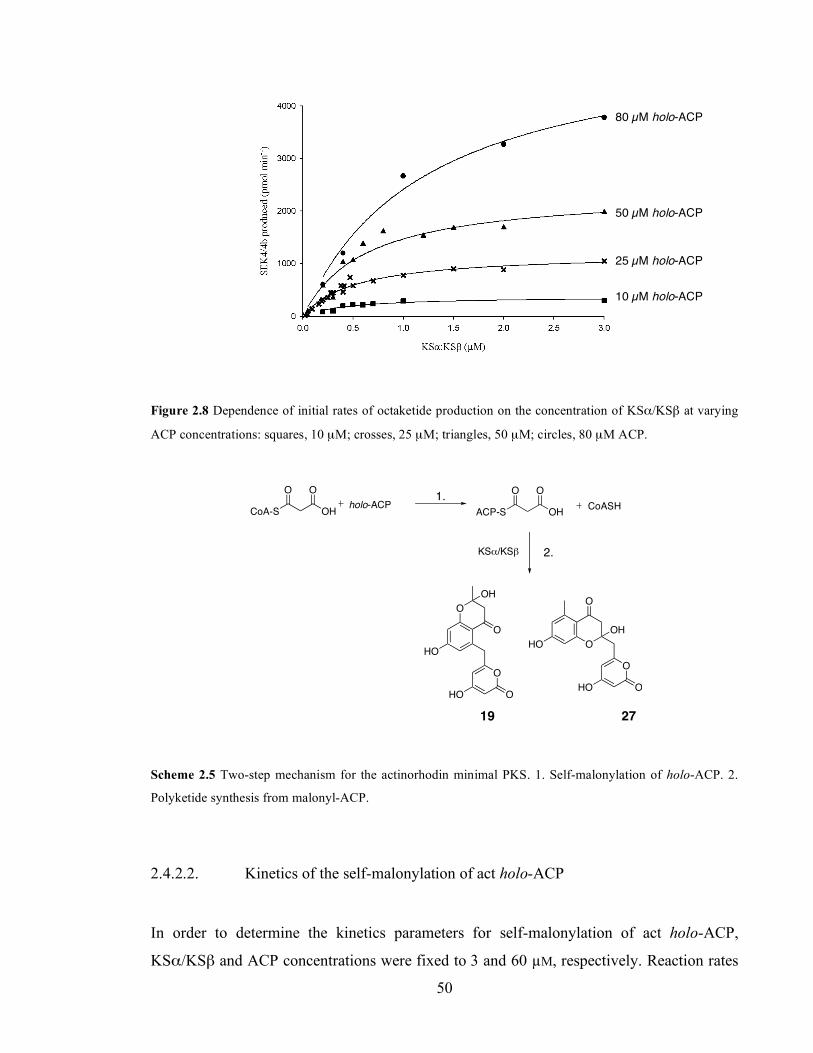
Chapter 3. Initiation of polyketide synthesis
50
Figure 2.8 Dependence of initial rates of octaketide production on the concentration of KSα/KSβ at varying
ACP concentrations: squares, 10 µM; crosses, 25 µM; triangles, 50 µM; circles, 80 µM ACP.
ACP-S OH
O O
holo-ACP
KS!/KS"
HO
O
O
OH
O
HO
O
OHO
O
OH
HO
O
O
CoA-S OH
O O
CoASH
19 27
Scheme 2.5 Two-step mechanism for the actinorhodin minimal PKS. 1. Self-malonylation of holo-ACP. 2.
Polyketide synthesis from malonyl-ACP.
2.4.2.2. Kinetics of the self-malonylation of act holo-ACP
In order to determine the kinetics parameters for self-malonylation of act holo-ACP,
KSα/KSβ and ACP concentrations were fixed to 3 and 60 µM, respectively. Reaction rates
80 µM holo-ACP
50 µM holo-ACP
25 µM holo-ACP
10 µM holo-ACP
2.
1.

Chapter 3. Initiation of polyketide synthesis
51
were then studied for a range of malonyl CoA concentrations (Figure 2.9). Kinetic
parameters of kcat = 2.30 ± 0.27 min-1 and KM = 215 ± 66 µM malonyl CoA were measured
by direct fit to a hyperbolic function.
Comparison of self-malonylation rates determined by the KGDH (Figure 2.6) and the
KSα/KSβ (Figure 2.9) coupling systems indicated that the rate of ACP self-malonylation
increased by a factor of five when KSα/KSβ was present in saturating concentrations, due
to a change in the kcat component of the rate equation. As a control, we used the KGDH
assay to measure the rate of self-malonylation of holo-ACP in the presence of a mutant
KSα/KSβ (AQ mutant) which is incapable of producing polyketides. Under these
conditions, the catalytic number was calculated as kcat = 2.4 min-1. Again, this is ca. 5-fold
higher than the rate of self-malonylation in the absence of the ketosynthase components.
Figure 2.9 Determination of Michaelis-Menten kinetic parameters for the self-malonylation of holo-ACP by
the KSα/KSβ coupling system. A. Plot of reaction rate vs. substrate concentration and hyperbolic fit. B. Hanes
linear plot of the data.
2.5. Studies on the mechanism of self-malonylation of ACP
The mechanism of the self-malonylation reaction of holo-ACP has not yet been fully
elucidated. Early studies speculated that conserved arginine residues within the ACP were
able to bind malonate moieties via salt bridges between the guanidine and carboxylate
A. B.

Chapter 3. Initiation of polyketide synthesis
52
functionalities (Figure 2.10).55 This could immobilize the malonyl group and allow the PP
thiol of ACP to intercept it.
O
CoA-S
O
O
HN
H
NH
HN
ACP
Figure 2.10 Interaction between arginine residues and malonate units.55
A primary sequence alignment of FAS and PKS acyl carrier proteins shows a high degree
of similarity (Figure 2.1). Notably, there is a conserved arginine residue in helix IV of PKS
acyl carrier proteins that is not present in FAS acyl carrier proteins (G74 in S. coelicolor
FAS ACP and R71, R73, R72 and R71 in fren, gris, act and otc acyl carrier proteins
respectively).
Within the Bristol group, ESMS was used to estimate the rates of self-malonylation of
act ACP by following the conversion of holo-ACP to malonyl-ACP.120 Although ESMS
cannot be used to determine precise kinetic parameters, it is useful for qualitative
comparisons between different mutant acyl carrier proteins. Using this method, a R72A
mutant ACP was shown to self-malonylate at a lower pace than the reference act ACP.
Using the continuous method which utilizes KGDH as a coupling enzyme to self-
malonylation, we determined the kinetic parameters for the self-malonylation of act R72A
ACP as kcat = 0.22 ± 0.04 min-1 and KM = 302 ± 63 µM; i.e. kcat was reduced by half with
increased KM when compared with act ACP.
We then investigated if the CoA moiety of malonyl CoA was also important in the self-
malonylation reaction. If the CoA moiety was involved then CoA-SH itself should inhibit
self-malonylation. Of the two spectrophotometric methods we have developed to measure
the kinetics of self-malonylation, only the coupling of SEK4/4b production to generation of
malonyl-ACP could be used in this investigation, as the KGDH assay uses CoA-SH as a
substrate. Thus, KSα/KSβ in high concentrations was used to measure the self-
malonylation activity of holo-ACP in the presence of several CoA-SH concentrations. We
observed a decrease in rate when increasing CoA-SH concentrations were added to our

Chapter 3. Initiation of polyketide synthesis
53
assays. CoA-SH showed a mixed inhibition pattern of self-malonylation, and inhibition
constants of Kic = 72 ± 5 µM and Kiu = 650 ± 150 µM were determined (Figure 2.11).
To rule out the possibility of KSα/KSβ being inhibited by CoA-SH, four control assays
were executed in the presence of malonyl CoA: ACP transacylase (MCAT), so that the rate
of KSα/KSβ could be measured (see Chapter 3). No inhibition of KSα/KSβ was
measurable at a CoA-SH concentration of 2 mM.
Figure 2.11 Determination of Kic and Kiu for the inhibition self-malonylation of ACP by coenzyme A (open
triangles, 800 µM; filled triangles, 400 µM; open circles, 200 µM; filled circles, 100 µM).
2.5.1. Mechanism of acceleration of self-malonylation rate by KSα /KSβ.
In general, the tertiary structure of ACP is composed of three major helices (plus a shorter
helix between the second and the fourth helices in some models) arranged around a central
hydrophobic core. In the majority of acyl carrier proteins, helix II contains a highly
conserved pair of negatively charged residues six amino acids apart so placing them on the
same face of the helix (E47 and E53 in act ACP). These residues have been proposed to
have a role in protein-protein interactions (see introduction to Chapter 2).
We studied three mutant act acyl carrier proteins available within the group, i.e. E47A,
E47V and E53A ACP in terms of their self-malonylation ability. The three mutations
introduced a hydrophobic residue instead of a negatively charged glutamate. Previous
studies within the Bristol group had shown that these mutant acyl carrier proteins self-
malonylated essentially as the reference act ACP.60,120 Moreover, E47 and E53 are located
A. B.
Kic Kiu

Chapter 3. Initiation of polyketide synthesis
54
on helix II of act ACP, within the proposed recognition motif of ACP. Therefore these
mutants presented a good chance to study ACP: KSα/KSβ interactions during the self-
malonylation step.
First, we evaluated the rate of self-malonylation of these acyl carrier proteins in
isolation using the KGDH assay. Surprisingly, the self-malonylation activity of E47-
mutants and E53A holo-ACP was reduced by ten and two-fold respectively when compared
to act holo-ACP, using the same concentration of malonyl CoA. KM for E53A remained
unchanged. Due to the low activity of E47A and E47V ACP, it would have been necessary
to use very high ACP concentrations (>300 µM) to determine KM.
The reason for the disagreement between the results presented here and previous
observations using a mass spectrometry assay is not clear at this stage. In order to obtain
direct physical evidence, we also did the mass spectrometry assay to allow for qualitative
comparison between act ACP and an E47A mutant ACP. Each protein (at 50 µM) was
incubated with malonyl CoA (1 mM) and then malonyl-ACP analyzed by ESMS at time
intervals. In agreement with our spectrophotometric assay (Figure 2.5) the reaction
catalyzed by act ACP reached equilibrium in 5-10 minutes, whereas E47A was much less
efficient (Figure 2.13), (see Appendix III for raw ESMS data).
Figure 2.12 Self-malonylation of the reference act ACP (circles) and E47A ACP (open circles) as measured
by ESMS.
Act ACP
Act E47A ACP
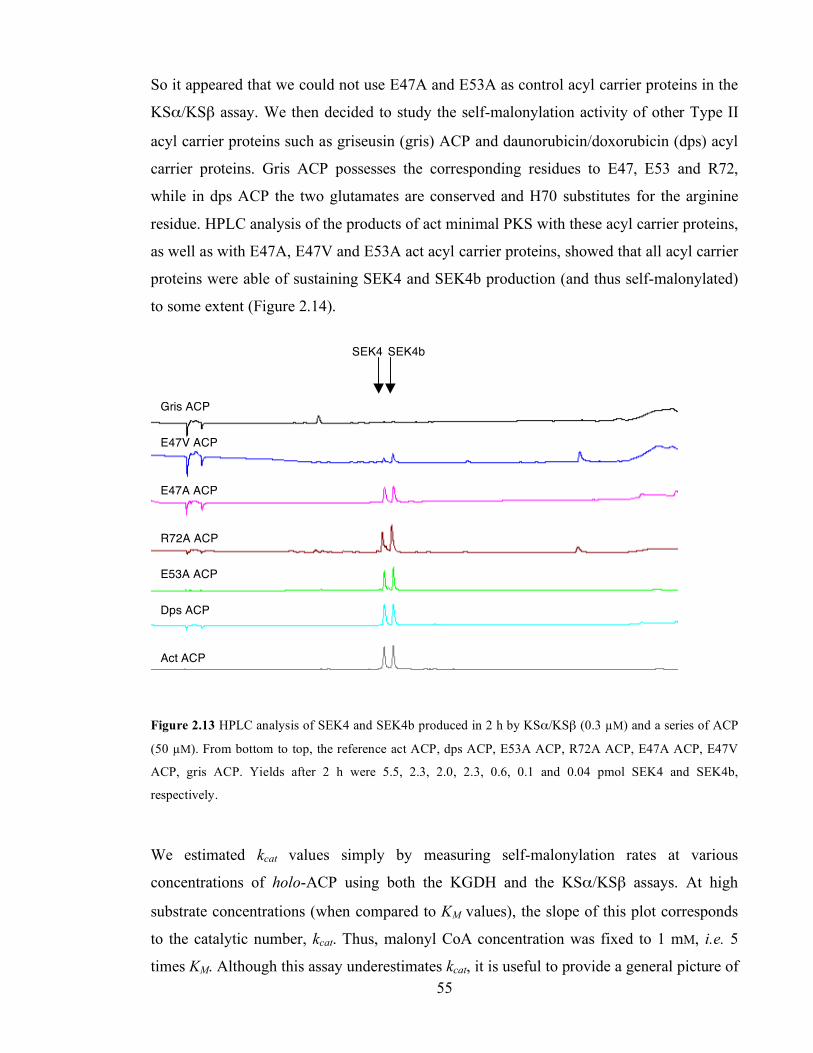
Chapter 3. Initiation of polyketide synthesis
55
So it appeared that we could not use E47A and E53A as control acyl carrier proteins in the
KSα/KSβ assay. We then decided to study the self-malonylation activity of other Type II
acyl carrier proteins such as griseusin (gris) ACP and daunorubicin/doxorubicin (dps) acyl
carrier proteins. Gris ACP possesses the corresponding residues to E47, E53 and R72,
while in dps ACP the two glutamates are conserved and H70 substitutes for the arginine
residue. HPLC analysis of the products of act minimal PKS with these acyl carrier proteins,
as well as with E47A, E47V and E53A act acyl carrier proteins, showed that all acyl carrier
proteins were able of sustaining SEK4 and SEK4b production (and thus self-malonylated)
to some extent (Figure 2.14).
Figure 2.13 HPLC analysis of SEK4 and SEK4b produced in 2 h by KSα/KSβ (0.3 µM) and a series of ACP
(50 µM). From bottom to top, the reference act ACP, dps ACP, E53A ACP, R72A ACP, E47A ACP, E47V
ACP, gris ACP. Yields after 2 h were 5.5, 2.3, 2.0, 2.3, 0.6, 0.1 and 0.04 pmol SEK4 and SEK4b,
respectively.
We estimated kcat values simply by measuring self-malonylation rates at various
concentrations of holo-ACP using both the KGDH and the KSα/KSβ assays. At high
substrate concentrations (when compared to KM values), the slope of this plot corresponds
to the catalytic number, kcat. Thus, malonyl CoA concentration was fixed to 1 mM, i.e. 5
times KM. Although this assay underestimates kcat, it is useful to provide a general picture of
SEK4 SEK4b
Gris ACP
E47V ACP
E47A ACP
R72A ACP
E53A ACP
Dps ACP
Act ACP

Chapter 3. Initiation of polyketide synthesis
56
self-malonylation rates, as long as the affinity of the mutant ACP for malonyl CoA is
similar to the reference act ACP. As a control, we studied the self-malonylation of act ACP
by this method. kcat was underestimated by 8% (KGDH assay) and 12% (KSα/KSβ assay).
With this restriction in mind, we determined kcat for WT gris ACP, WT dau ACP and act
mutants E47A, E47V, E53A and R72A ACP (Table 2.1).
ACP Act E47A E47V E53A R72A Gris Dps
kcat (min-1) KGDH 0.45 0.05 0.02 0.27 0.22 0.04 0.23
kcat (min-1) KSα /KSβ 2.03 0.09 <0.06 0.12 0.16 <0.06 0.23
Table 2.1 kcat values for the self-malonylation of ACP as measured by KGDH and the KSα/KSβ assays.
Two conclusions could be drawn from the data in Table 2.1. First, that the self-
malonylation rate did not increase in the presence of KSα/KSβ with either the act mutants
or gris or dps ACP. Second, that the self-malonylation activity of act E53A ACP and dps
ACP in isolation (as measured by the KGDH assay) was around half that of the reference
act ACP, whereas E47A, E47V and gris ACP self-malonylated more slowly, at between 5
and 10% of the rate of act ACP.
2.6. Discussion
2.6.1. Studies on the quality of ACP prior to reaction
In order to undertake a kinetic study of a Type II PKS, it is essential to assess the levels of
holo-ACP vs. apo-ACP, the levels of dimeric and monomeric holo-ACP species present in
act minimal PKS assays and the extent of acylation of the monomeric holo-ACP
component prior to reaction. This may be achieved by ESMS.
Here we have presented evidence that the traditional disulfide reductant DTT is
incompatible with malonyl-ACP as incubation of DTT with malonyl-ACP resulted in
production of holo-ACP due to transthiolesterification (Scheme 2.6). Thus, DTT reduces
the concentration of thiolester intermediates such as malonyl-ACP in polyketide

Chapter 3. Initiation of polyketide synthesis
57
biosynthesis and should be avoided in kinetic studies. On the other hand, TCEP is useful as
an alternative reductant. TCEP efficiently reduces ACP dimers and does not react with
malonyl-ACP. Therefore TCEP can be included in PKS assays, whereas DTT must be
avoided especially if the kinetics of the PKS are going to be assessed.
OH
OH
SHHS
ACP
S
O
HO
O
Scheme 2.6 DTT reduces malonyl-ACP concentration by transthiolesterification
2.6.2. Both malonate and CoA are important for binding onto ACP
The mechanism of self-malonylation has been proposed to rely on electrostatic interactions
between the guanidine group of positively charged arginine residues in the surface of ACP
and the carboxylate functionality of the malonate substrate (Figure 2.10).55 This hypothesis
was tested within the Bristol group by the generation of the act ACP mutants R11A, R34A
and R72A. The self-malonylation activity of these proteins was assessed by ESMS. It was
found that, while the self-malonylation activity of R11A and R34A ACP was unaffected,
R72A ACP self-malonylated at around half the rate of the wild type ACP.60 Herein, we
have validated this qualitative estimation by determining kcat for R72A as 0.22 min-1, in
comparison to 0.49 min-1 for act ACP. KM for R72A ACP was 302 µM, in comparison with
207 µM for the reference act ACP. In short, mutation of R72 decreased the specificity
constant kcat / KM from 2.4 mM-1 min-1 to 0.7 mM-1 min-1.
We also studied the importance of the CoA moiety for malonyl CoA: ACP binding. Within
the Bristol group, the binding of malonyl CoA to ACP was studied by NMR. Anna
Szafranska studied the effect of adding malonyl CoA to apo-ACP, and identified a series of
residues whose chemical shift changed with respect to apo-ACP alone.132 No residues on
helix I or the loop between helix I and II were affected; on the other hand, 13 residues
situated on helix II, the loop between helix II and helix III, helix III itself and the loop
between helix III and helix IV were identified as a putative malonyl CoA binding site

Chapter 3. Initiation of polyketide synthesis
58
(Figure 2.15). These residues included hydrophobic (L43, M46, A50, Y56, V58, S59, V64),
positively charged (R67 and R72) and negatively charged (E47, E53, D62, D63, D69)
amino acids.
Figure 2.14 A. Affected residues upon binding of malonyl CoA onto ACP (yellow). B. All affected residues
are solvent-exposed (blue).132
We showed here that CoA-SH competes with malonyl CoA for binding sites within ACP,
thus inhibiting self-malonylation. This is important as CoA-SH itself is produced in the
self-malonylation reaction. The general mechanism for enzyme inhibition is one in which
the inhibitor can bind both to the free enzyme and also to the enzyme-substrate complex
(Scheme 2.9). This general scheme is known as ‘mixed inhibition mechanism’.94
ACP + malonyl CoA ACP.malonyl CoA malonyl-ACP + CoA-SH
CoA-SH
+
ACP.CoA-SH
CoA-SH
+
CoA-SH.ACP.malonyl CoA
KicKiu
+ malonyl CoA
Scheme 2.7 Mechanism that produces mixed inhibition of self-malonylation by CoA-SH.
I
II
III
IV A. B.

Chapter 3. Initiation of polyketide synthesis
59
Kic and Kiu are known as the competitive and uncompetitive inhibition constants,
respectively. We expected CoA-binding to the free enzyme, ACP, to form a complex
ACP.CoA-SH. Our data reported a Kic = 72 µM, which is lower than the Michaelis constant
for self-malonylation of ACP (ca. 214 µM malonyl CoA). This suggests that, the small,
acidic ACP has three times lower affinity for coenzyme A when a negatively charged unit
such as malonate is bound to its phosphopantetheine arm, even taking into account the salt
bridges between R72 and the malonate unit. Unexpectedly, there was an uncompetitive
component in the inhibition, which probably means that ACP can bind two CoA moieties at
any time. This effect, though measurable, was ten times less important than its competitive
counterpart.
2.6.3. KSα /KSβ accelerates malonylation of ACP
We measured the kinetics of self-malonylation spectrophotometrically by two methods.
The α-ketoglutarate dehydrogenase (KGDH) assay measures the production of CoA-SH
from malonyl CoA catalyzed by holo-ACP. Our kinetic data (kcat = 0.49 min-1, KM = 207
µM) are in good agreement with those previously reported by the Bristol group using a
radioactive discontinuous method (kcat = 0.34 min-1, KM = 219 µM).55 The difference in kcat
could be attributed to the use of DTT in previous studies, which is likely to reduce the
concentration of malonyl ACP (Section 2.3).
The second assay to measure self-malonylation of holo-ACP employed KSα/KSβ as a
coupling enzyme for the production of malonyl-ACP. The minimal PKS produces the
aromatic polyketides SEK4 and SEK4b which can be quantitated by their absorbance at
293 nm. Unexpectedly, we observed a change in kcat when self-malonylation was measured
in the presence of KSα/KSβ (from 0.49 to 2.30 min-1) with unchanged KM leading to a five
times higher specificity constant kcat/KM in the presence of KSα/KSβ. This may be due to
either of two possible reasons, i.e. that KSα/KSβ has some acyltransferase activity, or that
KSα/KSβ activates holo-ACP towards self-malonylation.

Chapter 3. Initiation of polyketide synthesis
60
2.6.3.1. KSα/KSβ as a potential acyl transferase
In type II FAS, MCAT catalyzes the malonylation of FAS ACP. In general, type II PKS
lack a dedicated MCAT. When the gene encoding act KSα was sequenced, a putative
acyltransferase motif GHSLG was found in KSα (italics, the putative AT serine active site).
It was then speculated that KSα might be a bifunctional enzyme, exhibiting malonyl CoA:
ACP transacylase as well as β-ketoacylthiolester synthase activities.20 Interestingly,
mutation of the putative serine active site (S347) to leucine reduced the production of
polyketides in vivo, although it did not completely abrogate it.133 At the same time, Meurer
and Hutchinson showed that a mutation S347A (act KSα numbering) in the tetracenomycin
KSα did not have any effect in polyketide production and it was consequently assumed that
AT activity was not an essential feature of KSα.134 Years later, when the self-malonylation
ability of act, gris and otc ACP was discovered by the Bristol group,55 it became clear that
production of polyketides could be supported in the absence of any acyltransferase activity.
The determination of the crystal structure of act KSα/KSβ by the Stanford group
showed that, although S347 was close to the active site cysteine, its potential involvement
in loading was hampered by its buried position within the hydrophobic core of the
enzyme.31
It was conceivable that the increase in observed rate of malonylation of ACP in the
presence of KSα/KSβ could be accounted for if KSα/KSβ were contaminated with S.
coelicolor MCAT. If KSα/KSβ were contaminated with MCAT, then, under conditions
where malonylation was rate limiting, one would expect to observe a decrease in the
measured KM value towards that for the MCAT catalysed reaction (KM of malonyl CoA as a
substrate for MCAT is estimated as ca 60 µM).46 However, the same KM value for malonyl
CoA was measured in the presence and in the absence of KSα/KSβ. Thus we concluded
that the increase in measured kcat value represented a genuine increase in the catalytic
efficiency of the ACP malonylation reaction when the ACP was in the presence of
KSα/KSβ.

Chapter 3. Initiation of polyketide synthesis
61
2.6.3.2. KSα/KSβ activates holo-ACP towards self-malonylation
The alternative to a KSα-catalyzed malonylation of ACP is a KSα/KSβ-mediated
conformational change in ACP structure, which renders ACP more active towards self-
malonylation. NMR and crystal structures of some FAS and PKS acyl carrier proteins (but
not act ACP)30 have shown that some holo-ACP exists in at least two different
conformational states in solution in ratios from 90:10 to 65:35 major to minor
isomers.36,40,41 For instance, it has been shown that Plasmodium falciparum FAS holo-ACP
exists in two different conformational states in solution in a ratio 65:35. This is due to
conformational changes, in particular, of residues V41, A60 and L61.41 These changes have
two important effects on the conformation of ACP: first, whereas in the major
conformation there are three residues that interact with the phosphopantetheine arm (S37 –
the attachment point-, L38 and D39), V41 poses an additional restraint to the movement of
the PP arm only in the minor isomer. Second, helix III (D57-A60) unwinds due to the
changes in A60 and L61, rendering a more flexible loop (Figure 2.16).
Figure 2.15 Ribbon representation of the major (A. PDB file 2FQ0) and minor (B. PDB file 2FQ2)
conformers of P. falciparum FAS ACP. Residues D57 to A60 are highlighted in magenta.
It has been suggested that these two ACP conformations correspond to ‘open’ and ‘closed’
forms of ACP.41,135 In the closed conformation (the major P. falciparum isomer), the loop
A. B.

Chapter 3. Initiation of polyketide synthesis
62
between helices II and III is packed against the three helix bundle and prevents the insertion
of the hydrophobic acyl group into the protein core. In this state, the PP arm is thought to
be flexible and in an extended conformation. In the open conformation, a more flexible
secondary structure due to the unwinding of helix III moves away from the three-helix
bundle, and allows the insertion of acyl chains into the hydrophobic pocket. This is
important because FAS ACP has to shuttle the growing fatty acid chain to the active sites
of enzymes (KR, DH, ER). Consequently, it was assumed that ACP must undergo this
conformational change in each successive cycle of chain elongation.41
The first structure of a PKS ACP was obtained by the Bristol group in 1997. The
actinorhodin apo-ACP was structurally similar to FAS ACP, and was composed of three
major helices (plus a shorter one, comprising residues D62-R67) packed into a bundle, and
a flexible loop between helices I and II. It was reported that act ACP only existed in one
conformation, and it was speculated that conformational variability may only be a feature
of FAS ACP.30 However, the solution structure of frenolicin (fren) PKS ACP also
evidenced two conformational states.39 In this case A56, T58, D59 and G63 were found to
exchange slowly (50-100 ms) between two states. This rendered the loop between helices II
and III as well as helix III itself intrinsic flexibility to determine the conformation of PKS
ACP.
The general fold motif of FAS and PKS acyl carrier proteins is also conserved in a third
group of proteins, the peptidyl carrier proteins (PCP) from non-ribosomal peptide synthases
(NRPS). Peptidyl carrier proteins are also small proteins of ca. 8-10 KDa and, like acyl
carrier proteins, they house a universal serine nucleophilic residue upon which a PP arm is
attached. The role of the carrier protein in polyketide and non-ribosomal peptide
biosynthesis is to shuttle substrates and intermediates between catalytic domains. Whereas
in PKS the substrates are carboxylic acids such as malonate, in NRPS the substrates are
amino acids, for example alanine. In both carrier proteins, substrates are activated as
thiolesters (Figure 2.17).

Chapter 3. Initiation of polyketide synthesis
63
S OH
O OACP
SNH2
OPCP
Figure 2.16 ACP and PCP activate their substrates (for instance, malonate and alanine) as thiolesters.
The tyrocidine 53 NRPS is an assembly line very much like typical modular PKS, in which
several catalytic domains are enclosed in each module, and every module carries out the
elongation of the non-ribosomal peptide via the generation of a new peptide bond between
the lengthening oligopeptide and an extender peptidyl unit. Weber, Marahiel, Holak and co-
workers studied the PCP from module 3 of the tyrocidine NRPS (TyrC3-PCP). Initially, the
structure was released as a four-helix bundle analogous to FAS and PKS ACP.136
Upon recent examination, however, the NMR spectrum of apo-PCP indicated the existence
of two different and slowly exchanging conformations. The changing residues were at the
N terminus of helix II and adjacent loop, including the active site residue S45; and between
helices II and IV. When the holo form was studied, it was found to exist in two
conformations as well. Interestingly, while the apo and the holo forms shared one
conformational state (termed A/H), the two remaining states (A for apo and H for holo)
were distinct from each other and from the structurally identical common state.137
The three states of TyrC3-PCP differ mainly in the orientation and length of the helices.
The purely apo (A) state was achieved when the active site serine was mutated to alanine.
While helices I and II are shortened with respect to the A/H conformation, helix III does
not exist at all. In the A/H state, the helices are more stable and longer, and the loop
between helices II and IV now forms helix III. This is the most compact structure. On the
other hand, in the H state helix III unravels itself and prompts a reorganization of the
globular protein core (Figure 2.18).
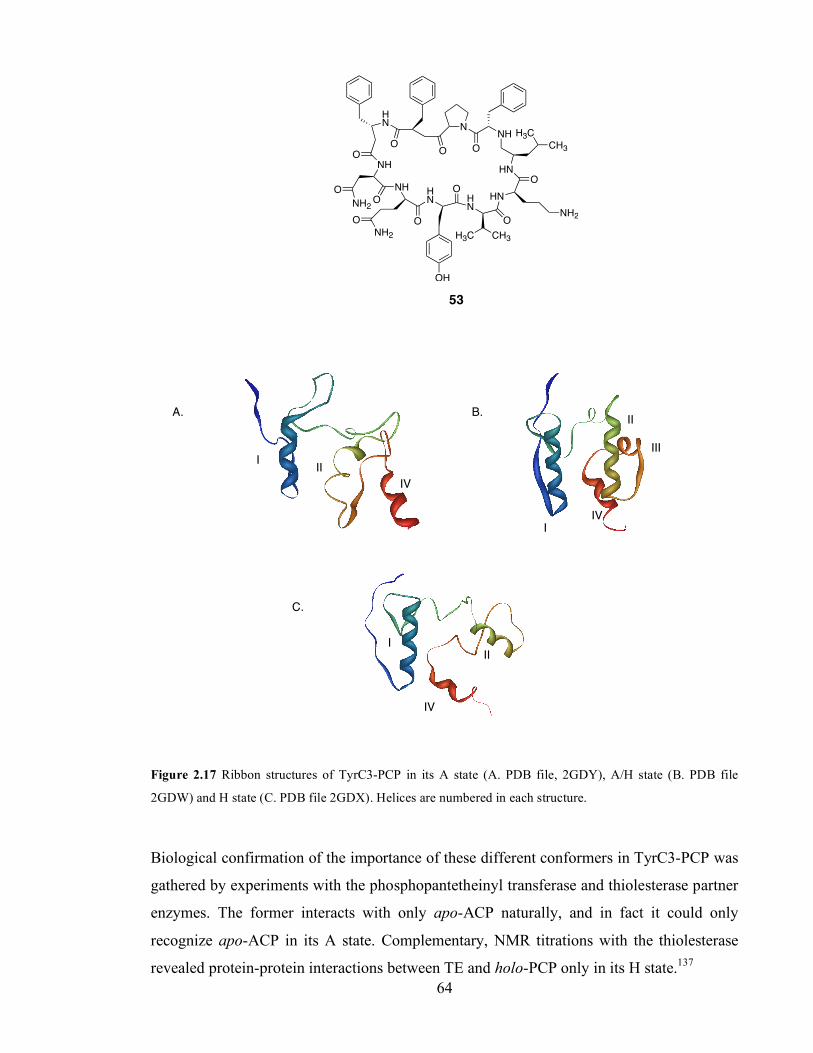
Chapter 3. Initiation of polyketide synthesis
64
O
N
O
NH
CH3
H3C
HN
O
HN
NH2O
HN
O
CH3H3C
HN
OH
O
NH
O
NH2
O
NH
HN
O
O
O
NH2
53
Figure 2.17 Ribbon structures of TyrC3-PCP in its A state (A. PDB file, 2GDY), A/H state (B. PDB file
2GDW) and H state (C. PDB file 2GDX). Helices are numbered in each structure.
Biological confirmation of the importance of these different conformers in TyrC3-PCP was
gathered by experiments with the phosphopantetheinyl transferase and thiolesterase partner
enzymes. The former interacts with only apo-ACP naturally, and in fact it could only
recognize apo-ACP in its A state. Complementary, NMR titrations with the thiolesterase
revealed protein-protein interactions between TE and holo-PCP only in its H state.137
I
I
I
II
II
III
II
IV
IV
IV
A. B.
C.

Chapter 3. Initiation of polyketide synthesis
65
Altogether, this information points towards the conformational flexibility of ACP and PCP
as an important mode of controlling the interactions between the carrier protein and partner
enzymes in complex biosynthetic pathways,115 and it could account for our observations
that catalytic properties of ACP are enhanced by the presence of its known counterpart,
KSα/KSβ. In any event, our kinetic studies provide evidence for a close association of ACP
and KSα/KSβ during the self-malonylation step.
2.6.3.3. Insights into ACP-KSα/KSβ interactions leading to enhanced self-
malonylation activity.
In order to gain understanding of the association between ACP and KSα/KSβ during the
self-malonylation step, we proceeded to study act ACP mutants such as E47A, E47V and
E53A in terms of their self-malonylation activity in the presence and absence of KSα/KSβ.
Unexpectedly, neither of the act mutant acyl carrier proteins studied performed as well
as the reference act ACP in isolation, when the self-malonylation was studied by the
KGDH assay. kcat values decreased two-fold (E53A) and 10-fold (E47A and E47V ACP).
This was striking because previous evidence within the Bristol group using an ESMS
assays had shown that act E47A and E53A ACP had similar activities to the reference act
ACP. When we repeated the ESMS assay with our E47A ACP, we observed a clear
reduction in self-malonylation activity, in agreement with our spectrophotometric assay.
It is not clear why E47A and E53A acyl carrier proteins performed as well as the
reference act ACP in the past. In our hands, these acyl carrier proteins were clearly
deficient in self-malonylation. We have shown that the CoA moiety is important for
appropriate ACP- malonyl CoA interactions (Section 2.5), so the possibility that E47 and
E53 are involved in CoA binding, as proposed by Szafranska,132 might be valid. Although it
seems unlikely that negatively charged residues are directly involved in binding the CoA
moiety, they may play an important role in creating a net of hydrogen bonds leading to
efficient interaction between ACP and CoA.
We then included two acyl carrier proteins from other type II PKS, i.e. gris ACP and dps
ACP, in our studies. Gris ACP contains the corresponding residues to E47, E53 and R72
(act ACP numbering). Dps ACP possesses the corresponding E47 and E53 but a histidine

Chapter 3. Initiation of polyketide synthesis
66
residue substitutes for R72. Gris ACP and dps ACP in isolation self-malonylated more
slowly than act ACP. We did not observe any increase in rate in the presence of KSα/KSβ.
In conclusion, E47, E53 and R72 appear to be essential residues to support self-
malonylation of act ACP, both in the absence and in the presence of KSα/KSβ.
Unfortunately, we could not confirm that E47 and E53 are involved in protein-protein
interactions between act ACP and KSα/KSβ because the self-malonylation activity of
E47A, E47V and E53A was impaired when studied in isolation. The study of the self-
malonylation of gris and dps ACP, which possess the equivalent to E47 and E53, suggested
that there are still more features that prompt an efficient ACP: KSα/KSβ association which
have not been identified.
Future work should adapt the KGDH assay for the measurement of NADH production by
fluorescence spectrophotometry. When excited at 340 nm, NADH emits energy at 465 nm
that can be monitored continuously in a fluorescence spectrophotometer. Fluorescence
measurements are intrinsically more sensitive than absorbance measurements, and therefore
it should be possible to use the KGDH assay for the study of the self-malonylation of ACP
mutant proteins by detection of NADH by fluorescence. This is most important for a full
kinetic characterization of acyl carrier proteins with low activity such as act E47A and
E47V ACP, and gris ACP.
The characterization of the activation of self-malonylation by KSα/KSβ should be
continued, first, by the study of more ACP mutants which self-malonylate as the reference
act ACP in isolation. Eventually, residues responsible for ACP: KSα/KSβ interactions in
KSα/KSβ could also be identified.

Chapter 3. Initiation of polyketide synthesis
67
3. Initiation of polyketide synthesis.
Type II FAS systems found in most bacteria consist of a series of discrete proteins, each of
which catalyzes an individual reaction of the fatty acid biosynthetic pathway. In organisms
using a type II pathway, for instance E. coli and S. coelicolor, there are at least two β-
ketoacyl ACP synthase (KAS) enzymes, known as KASII and KASIII.138,139 KASII is the
generic enzyme responsible for the elongations required for synthesis of long-chain fatty
acids,140 analogous to act KSα. KASIII catalyzes the first condensation from acetyl CoA
and malonyl-ACP to produce butyryl-ACP 54 (Scheme 3.1), the starter unit for FAS.141
Butyryl-ACP is then the substrate for the KASII-catalyzed elongations that result in long-
chain fatty acids.142
S
OH
O
O
ACP
S
O
CoA
S
O
O
ACPCO2, CoA-SH
+
54
Scheme 3.1 Initiation of fatty acid synthesis in E. coli, catalyzed by KASIII.142
We have seen in previous chapters that the actinorhodin PKS does not require KASIII
activity, and the only ketosynthase capable of catalyzing carbon-carbon bond forming
reactions is KSα. However, an analogous mechanism to bacterial FAS, in which additional
KASIII (also known as priming ketosynthase), and also AT and sometimes ACP proteins
are responsible for the biosynthesis and attachment of short-chain fatty acids, has been
proposed for other Type II PKS. Particularly, this mechanism has been proposed to operate
in polyketide synthases that employ a starter unit other than acetate.
For example, the daunorubicin (dps) PKS employs a propionyl starter unit (see Figure 1.3
in Chapter 1). How the dps PKS chooses propionate is currently the subject of intense
research.142 Initial in vivo complementation studies were carried out by Meurer and
Hutchinson. They used the KSα and KSβ components from the daunorubicin PKS with
KASIII

Chapter 3. Initiation of polyketide synthesis
68
ACP and tailoring enzymes from the tetracenomycin PKS.144 Only acetyl-derived
polyketides were produced such as 55 (starter unit shown in red), indicating that dps KSα
and KSβ did not have any requirement for propionyl-ACP as a starter unit.
HO
OH OH OH
O
O
OH
55
The same group used cell-free extracts of cultures expressing dps KSα/KSβ, ACP as well
as KR and CYC to feed labelled propionyl-CoA and analyze the metabolites produced.
Incorporation of radioactivity was not observed. When a cell-free extract of a culture
expressing DpsC and DpsD was added, the propionate-derived polyketide UWM5 56
(starter unit shown in red) was produced. This result suggested that DpsC and DpsD were
essential for substrate specificity in cell-free systems.145 Hutchinson are co-workers then
hypothesized that DpsC and DpsD (homologues to bacterial KASIII -although substituting
the active site cysteine for serine- and AT, respectively), were the primary determinants of
starter unit specificity.
O
OH
O
O OH
OH
HO
56
In vitro studies with purified DpsC showed that DpsC could use propionyl CoA as substrate
and was acylated by propionate at Ser118 (Scheme 3.2.A).146 DpsC also catalyzed the
condensation of propionyl CoA and malonyl-ACP to form β-ketovaleryl-ACP 57 (Scheme
3.2.B).146 Subsequently, the valerate unit would be loaded onto KSα for further elongation
with malonate units (Scheme 3.2.C).

Chapter 3. Initiation of polyketide synthesis
69
S
O
CoA
HS
DpsC
S
O
DpsC
S
OH
O
O
ACP
S
O
DpsC
S
O
O
ACP
S
O
O
ACP
S
O
KS!
O
S
OH
O
O
ACP
OMe
O
O
OH
OH
O
O
OH
O
NH2HO
HS
KS!
Scheme 3.2 Proposed mechanism for starter unit specificity in dps PKS. A. Loading of DpsC with propionate.
B. Initiation of polyketide synthesis. C. Elongation of the polyketide chain.147
Other examples of related PKS priming mechanisms include the biosynthesis of frenolicin
(frn), benastatin 58 (ben) and R1128.148,149,150 In all cases, genes encoding KASIII
homologues were identified in the respective gene clusters (FrnI, BenQ and ZhuH
respectively).
In the case of the R1128 PKS, an additional enzyme, i.e. ZhuC (which corresponds to
DpsD in the daunorubicin PKS) was initially appointed a malonyl CoA: ACP transacylase
activity.151 Later, however, the role of ZhuC was revised: when it was added to tcm
minimal PKS assays in vitro, hexanoyl-ACP was preferred to acetyl-ACP as the starter
unit, unequivocally pointing towards a role for ZhuC in initiation of polyketide synthesis.
Tang, Khosla and co-workers proposed that ZhuC (and by analogy DpsD) are thiolesterases
that target acetyl-ACP selectively thus eliminating competitive acetate substrates. Acetyl
and propionyl-ACP were indeed 5, 100 and 200 times better substrates for ZhuC than
butyryl, hexanoyl and malonyl-ACP, respectively.152
Furthermore, an additional ACP (ZhuG, termed priming ACP) has been shown to be
essential for the incorporation of non-acetate starter units in the R1128 PKS. This does not
A.
B.
C.
57
13

Chapter 3. Initiation of polyketide synthesis
70
result in the other ACP being redundant, as the priming ketosynthase ZhuH and the
elongating ketosynthase ZhuA have orthogonal ACP specificity showing preference for the
priming and elongating ACP partners, respectively.98 This is striking because, whereas
there are two acyl carrier proteins in the frenolicin PKS gene cluster, only one ACP is
present in dps and ben PKS. OHO
HO
OHOOH
HO
58
Neither the frenolicin, benastatin and R1128 gene clusters encode the necessary KR, DH
and ER catalysts for the reduction of the β-ketoester generated by KASIII to the butyrate
(frn), hexanoate (ben) or (iso)butyrate, valerate or 4-methylvalerate (R1128 complexes)
starter units (see Figure 1.3 in Chapter 1), which has led to the assumption that these
activities are recruited from fatty acid metabolism.149,151 In remarkable work, Khosla and
co-workers harnessed the fact that, when the R1128 gene cluster is heterogously expressed
in S. coelicolor, it produces the same polyketides as in the original producer (S. sp. R1128),
to identify and purify an enzyme (SCO1815) that reduces the ACP-tethered β-ketoacyl
substrates produced by the initiation module of R1128 PKS. SCO 1815 was highly
homologous to FAS KR.153
A second, alternative priming pathway in Type II PKS that do not use KASIII-like activity,
is the direct loading of the starter unit onto the minimal PKS. For instance, in the enterocin
(enc) PKS from Streptomyces maritimus, it has been proposed that an enzyme with ACP
ligase activity, namely EncN, is capable of loading ACP with the benzoate starter unit
directly from benzoic acid 58 to form benzoyl-ACP 59. The benzoate is then transferred to
KSα.124
OHACP+
SH
O
S
O
ACPS
O
KS!
Scheme 3.3 Priming of the enc PKS with the benzoate starter unit.124
EncN KSα/KSβ 58 59

Chapter 3. Initiation of polyketide synthesis
71
The priming of Type II PKS with acetate units proceeds in a different manner, also
different to fatty acid biosynthesis. First, ACP is loaded with malonate from the
corresponding CoA thiolester either by self-malonylation or via MCAT (see Sections
1.2.3.2 and 1.2.3.5). Malonyl-ACP is then decarboxylated to acetyl-ACP and transferred to
KSα to yield acetyl-S-KSα and initiate polyketide building. This mechanism is unique in
fatty acid and polyketide synthases in that it generates acetyl-ACP. This differs from FAS
where the first ACP-tethered intermediate is acetoacetyl-ACP (see Scheme 1.1 in Chapter
1).
The mechanism for decarboxylation of malonyl-ACP to generate acetyl-ACP has been a
matter of debate in the literature. This originated from the discovery of the ‘mystery’ gene
encoding KSβ,20 which was then thought to occur only in Type II PKS systems (later,
similar KSQ proteins were described in some modular Type I PKS). Initial in vivo
experiments by Hopwood showed that a mutation in KSβ abolished polyketide production
in actinorhodin-producing S. coelicolor.154 Once established that functional KSβ were
essential for PKS activity, the Stanford group speculated that KSβ provided the template
for the growing polyketide chain and was the principal determinant of chain length (see
Section 1.2.3.3 in Chapter 1).
Thus, in the Stanford model for the act minimal PKS (see Section 1.2.3.4 in Chapter 1)
KSβ exerts a structural (rather than catalytic) role. Based on the crystal structure of the
KSα/KSβ complex, the Stanford group proposed that the corresponding residue to the
cysteine active site of KSα in KSβ (Q161) was buried and had a structural role, i.e. the
formation of a salt bridge between D297 and R332 (KSβ residues). By default, they argued
that decarboxylation of malonyl-ACP must be catalyzed by KSα.31 This would proceed
with the aid of two histidines (H309 and H346, KSα residues), in analogy to the
mechanism proposed for the yeast FAS,155 KASII from E. coli156 and similar to that in the
chalcone synthase.157
However, biochemical studies with a series of mutant KSα/KSβ within the Bristol and
Cambridge groups in vitro suggested that Q161 in KSβ was essential for efficient malonyl-
ACP decarboxylation activity of the ketosynthase complex. KSα/KSβ mutant complexes
were generated in which either active site (C169 in KSα or Q161 in KSβ) were replaced

Chapter 3. Initiation of polyketide synthesis
72
with an alanine residue.62,122 The following notation of mutant proteins was devised: each
protein complex was termed according to the corresponding residue in the active site of
KSα and KSβ. For example, the wild type KSα/KSβ was referred to as CQ, as there is a
cysteine in the active site of KSα and a glutamine in the active site of KSβ. The following
mutant proteins were generated: AQ, CA and AA KSα/KSβ in which one or the two active
sites of KSα and/or KSβ were replaced by an alanine residue.
First, these mutants, as well as the WT complex, were tested for their ability to produce
polyketides. Thus, each mutant was incubated in vitro with malonyl CoA and holo-ACP
and the products analyzed by HPLC. As expected, only the CA KSα/KSβ mutant was able
to produce polyketides; however, there was a lag of about 5 minutes without activity. This
lag was not observed in the control experiment using the wild type CQ KSα/KSβ. When
external acetyl-ACP was added to the assay using CA KSα/KSβ, the lag was
abolished.62,122 This observation leads to important conclusions. First, that KSα can be
primed with acetate directly by adding acetyl-ACP into solution. This may suggest that the
acetyl-ACP generated by decarboxylation of malonyl-ACP is probably released into
solution, rather than transferred directly to the KSα active site (Scheme 3.4). Second, that
the concentration of acetyl-ACP appears to build up to a certain value before polyketide
synthesis could occur, which suggests that the decarboxylation activity of the CA KSα/KSβ
is not high enough to provide this critical acetyl-ACP concentration at the beginning of the
assay.
S
O
ACP
KS! KS"
S
OH
O
O
ACPS
O
ACP
Scheme 3.4 Two possible models for transfer of acetyl-ACP from the KSβ to KSα active sites.
The fact that acetyl-ACP was released into solution after decarboxylation of malonyl-ACP
was harnessed by the Cambridge and Bristol groups to estimate the malonyl decarboxylase
Bulk solution
Acetyl-ACP released into bulk solution
‘Internal’ transfer of acetyl-ACP

Chapter 3. Initiation of polyketide synthesis
73
activity of the AQ and AA KSα/KSβ, which are incapable of polyketide synthesis.62,122
These mutants were incubated with malonyl-ACP, and the relative abundance of acetyl-
ACP after a certain period of time was determined by mass spectrometry by calculating the
ratio malonyl-ACP: acetyl-ACP. Whereas AQ KSα/KSβ could decarboxylate malonyl-
ACP at 20 µM h-1, the AA mutant did not show any activity towards malonyl-ACP. This
latest result, however, was not reproducible in the Cambridge group: an AA mutant was
later shown to decarboxylate malonyl-ACP essentially at the same rate as the AQ
complex.158
Alternative starter units, such as butyryl-ACP and hexanoyl-ACP, have also been fed to
the act minimal PKS. These acyl-acyl carrier proteins were utilized as substrate by KSα
only when the CA KSα/KSβ was used. When the WT (i.e. CQ) KSα/KSβ was used, the
‘fake’ starter units were out of compete and only acetate-derived polyketides were
detected.96
Taken together, these results showed that KSβ (via its active site Q161, and maybe
D297 and R332, the corresponding residues to the KSα catalytic triad Cys169-His309-His346)
initiates the synthesis of polyketides catalysing the first decarboxylation of malonyl-ACP to
generate the starter unit, acetyl-ACP. This mechanism was also proposed to operate in the
loading module (KSQ) of Type I modular PKS, such as those of monensin 19, tylosin 18
and niddamycin 17.62,122 Once KSα has been primed with an acetyl unit, it catalyses the
condensation of seven further acetates derived from malonyl-ACP.
The ultimate evidence towards the elucidation of the role of KSβ should rely on the
purification of this protein on its own, and subsequent structural studies and biochemical
assays in vitro. However, KSα and KSβ form intimate contacts with each other (more than
20% of their respective surface areas are buried)31 and attempts to purify KSα and KSβ
individually have been unsuccessful.142
The hypothesis, introduced by the Bristol and Cambridge groups, that KSβ is responsible
for the initiation step, was taking here as the starting point. The aim of Chapter 3 is to adapt
the continuous method we developed in Section 2.4.2 to measure the production of
polyketides by the act minimal PKS, to the kinetic study of the reactions catalyzed by KSα
and KSβ.

Chapter 3. Initiation of polyketide synthesis
74
3.1. Kinetics of KSβ can be studied in the presence of MCAT
In Section 2.4.2 we developed a continuous, spectrophotometric method to study the
kinetics of self-malonylation of ACP by coupling the formation of malonyl ACP to the
production of SEK4 and SEK4b by KSα/KSβ. However, this experiment did not provide
useful information about the kinetics of KSα/KSβ itself. In order to be able to measure the
rate of the reaction catalyzed by KSα/KSβ, we needed to find the appropriate conditions in
which the rate limiting step in the overall reaction executed by the act minimal PKS was
not self-malonylation of ACP, but turnover of malonyl-ACP by the ketosynthase complex.
3.1.1. MCAT accelerates the rate of malonylation of holo-ACP
The Bristol group reported that MCAT accelerates the transfer of a malonate group from
malonyl CoA to the PP arm of act holo-ACP, when compared to self-malonylation at low
concentrations of ACP (see Sections 1.2.3.2 and 1.2.3.5 in Chapter 1).46 Matharu studied
the production of polyketides by the act minimal PKS and found that self-malonylation was
the likely rate limiting step in the overall reaction; and that the presence of MCAT (5 nM)
increased the rate of polyketide production only when ACP concentration was lower than
20 µM.8 In the light of this information, we aimed to compare SEK4/4b production by the
act minimal PKS, in the presence and absence of MCAT. MCAT was thus expressed in E.
coli and purified following literature procedures,46 and characterized by ESMS (Figure 3.1).
Figure 3.1 Mass spectrum of MCAT (expected, 34113 Da); also detected as the potassium adduct.

Chapter 3. Initiation of polyketide synthesis
75
As described previously (Section 2.4.2), our assays rely on the direct spectrophotometric
observation of the polyketides produced by the act minimal PKS. The rate of polyketide
synthesis was studied by determining initial changes in the absorbance. We thus generated
reaction rate data for several ratios of ACP: KSα/KSβ concentrations (see Figure 2.8 in
Chapter 2).
In order to compare polyketide production rate in the presence and absence of MCAT, a
concentration of 0.3 µM KSα/KSβ was chosen which lay within the linear range of the
dependence of the rate with enzyme concentration (see Figure 2.8 in Chapter 2). This meant
that the polyketide synthesis step was rate limiting. Initial rates of reaction were then
measured for a range of holo-ACP initial concentrations. In the absence of MCAT, a linear
dependence of the rate on the concentration of holo-ACP was observed for ACP
concentrations lower than 50 µM (Figure 3.2). This indicated that, under these conditions,
an ACP dependent process was the rate limiting step, which agreed with the hypothesis that
self-malonylation of ACP was the slowest step in the actinorhodin minimal PKS in vitro.
In order to measure the kinetic parameters for malonyl-ACP as a substrate for KSα/KSβ,
the rate of ACP malonylation was increased so that this was no longer the rate limiting step.
This was achieved by using a high concentration of MCAT. We therefore conducted assays
at 0.3 µM KSα/KSβ with various concentrations of holo-ACP, in the presence of MCAT
(0.5 µM). Under these reaction conditions, the concentration of malonyl-ACP in the assays
was constant and equal to the initial, known concentration of holo-ACP. The same
maximum velocity Vmax was eventually achieved both in the absence and presence of
MCAT (Figure 3.2), as the catalysis by KSα/KSβ was not affected by the malonylation of
ACP.
However, at low ACP concentrations the formation of octaketides was much faster in
the presence of MCAT. This confirmed earlier observations by the Bristol group using an
HPLC assay.8 Kinetic parameters for the reaction catalyzed by KSα/KSβ were determined
as kcat = 20.6 ± 0.9 min-1 and KM = 2.39 ± 0.38 µM malonyl-ACP by direct fit to a
hyperbolic Michaelis-Menten equation (Figure 3.2).
To independently determine the turnover number (kcat) of the ketosynthase, we measured
the dependence of the reaction rate on the concentration of KSα/KSβ when MCAT (0.5

Chapter 3. Initiation of polyketide synthesis
76
µM) was added to the assays. At saturating ACP and MCAT concentrations, initial rates
were linear with increasing concentration of KSα/KSβ even at the highest concentration (3
µM), (Figure 3.3). From the slope of this plot, the catalytic constant kcat was independently
calculated as 16.9 ± 0.9 min-1.
In this section we have shown that the kinetics of polyketide production by KSα/KSβ can
be studied in MCAT-supplemented act minimal PKS. Nevertheless, in this assay we were
unable to de-couple the reactions catalysed by KSβ (decarboxylation of malonyl-ACP) and
KSα (elongation of the chain), so we decided to study this further.
Figure 3.2 A. Plot of reaction rate vs. holo-ACP concentration for act PKS ‘minimal’ assays (open circles)
and MCAT-supplemented assays (filled circles). B. Hanes linear plot of the MCAT-supplemented rate data.
Figure 3.3 Upon addition of MCAT, reaction rates become linearly dependent on KSα/KSβ concentrations.
A. B.

Chapter 3. Initiation of polyketide synthesis
77
3.1.2. Initiation of polyketide synthesis is slower than chain elongation
Initiation of polyketide synthesis proceeds via decarboxylation of malonyl-ACP to acetyl-
ACP. If this were the rate-limiting step in the production of SEK4 and SEK4b from
malonyl-ACP, then addition of exogenous acetyl-ACP to MCAT-supplemented act
minimal PKS assays would increase the overall rate of production of polyketides. In other
words, if the concentration of acetyl-ACP were limiting, then addition of external acetyl-
ACP would enhance the overall reaction rate.
To test for this hypothesis, acetyl-ACP was made by the method of Bisang simply by
incubating TCEP-treated holo-ACP (500 µM) with 1-acetylimidazole (5 mM).62 This
reaction was monitored by ESMS. Acetylation of holo-ACP was complete after 2 h (Figure
3.4).
Figure 3.4 ESMS of acetyl-ACP (expected 9483 Da), also detected as the potassium adduct (expected 9522
Da).
Acetyl-ACP was then purified by gel filtration to remove excess 1-acetylimidazole, and
added to PKS assays in increasing concentrations (up to 40 µM). Initial reaction rates were
then measured. In the absence of extra acetyl-ACP octaketides were produced at 4 µM min-
1. When acetyl-ACP was added, polyketide production rates increased (Figure 3.5). This
indicated that acetyl-ACP concentration was limiting under these conditions. When acetyl-
ACP was added in concentrations higher than 20 µM, polyketides were produced ca. 3
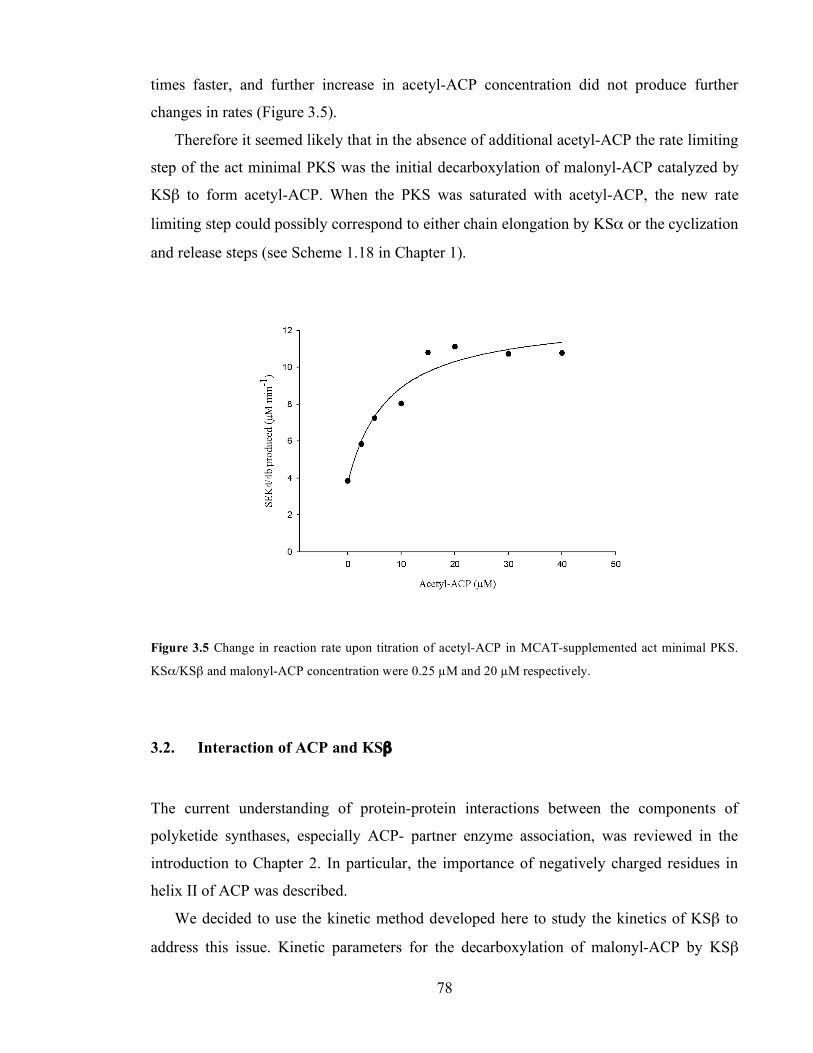
Chapter 3. Initiation of polyketide synthesis
78
times faster, and further increase in acetyl-ACP concentration did not produce further
changes in rates (Figure 3.5).
Therefore it seemed likely that in the absence of additional acetyl-ACP the rate limiting
step of the act minimal PKS was the initial decarboxylation of malonyl-ACP catalyzed by
KSβ to form acetyl-ACP. When the PKS was saturated with acetyl-ACP, the new rate
limiting step could possibly correspond to either chain elongation by KSα or the cyclization
and release steps (see Scheme 1.18 in Chapter 1).
Figure 3.5 Change in reaction rate upon titration of acetyl-ACP in MCAT-supplemented act minimal PKS.
KSα/KSβ and malonyl-ACP concentration were 0.25 µM and 20 µM respectively.
3.2. Interaction of ACP and KSβ
The current understanding of protein-protein interactions between the components of
polyketide synthases, especially ACP- partner enzyme association, was reviewed in the
introduction to Chapter 2. In particular, the importance of negatively charged residues in
helix II of ACP was described.
We decided to use the kinetic method developed here to study the kinetics of KSβ to
address this issue. Kinetic parameters for the decarboxylation of malonyl-ACP by KSβ

Chapter 3. Initiation of polyketide synthesis
79
were estimated for a range of act mutant acyl carrier proteins and other Type II acyl carrier
proteins. Mutant acyl carrier proteins such as act E47A, E53A and R72A ACP gave the
same turnover number kcat as the reference act ACP, although the Michaelis constant for
E47A and E53A (and E47V) was 3 times higher (Table 3.1). E47V showed a decreased
catalytic constant. Interestingly, the three WT acyl carrier proteins studied gave the same
specificity constant, kcat / KM, suggesting that act KSβ does not show any specificity for act
ACP in relation to gris and dps ACP.
ACP kcat (min-1) KM (µM) kcat / KM (min-1 µM-1) Act 20.6±0.9 2.39±0.38 8.62±2.37
E47A 20.0±1.9 8.03±2.00 2.49±0.95 E47V 3.5±0.4 6.81±1.05 0.51±0.38 E53A 17.5±1.4 8.07±1.39 2.17±1.00 R72A 20.3±0.8 1.95±0.32 10.41±2.5 Gris ACP 9.5±0.3 1.25±0.17 7.62±1.76 Dps ACP 16.4±1.3 1.86±0.49 8.82±2.65
Table 3.1 Kinetic parameters for the decarboxylation of malonyl-ACP by KSβ (see Appendix 1 for
Michaelis-Menten analysis).
3.3. Discussion
Chain initiation in the act PKS is known to proceed via decarboxylation of malonyl-ACP to
acetyl-ACP. This reaction was previously shown to be catalyzed by KSβ.62 In Chapter 3,
we have used a continuous method to measure the kinetics of this initiation step. This
methodology has been applied to study the interaction of ACP with KSβ.
In the actinorhodin minimal PKS, self-malonylation of ACP is the rate limiting step (See
Chapter 2). Thus, in order to measure the kinetics of the other components of the act
minimal PKS this loading reaction must be accelerated. To this end, we used an enzyme
from the primary metabolism of S. coelicolor, i.e. MCAT, known to malonylate S.
coelicolor FAS and PKS ACP.46 We showed that, upon addition of MCAT (0.5 µM),
loading of the PKS with malonate was no longer rate limiting, and the kinetics of the
ketosynthase complex could be characterized. Furthermore, we were able to de-couple the

Chapter 3. Initiation of polyketide synthesis
80
reactions of KSα and KSβ: we added exogenous acetyl-ACP to MCAT-supplemented
minimal PKS assays and observed a clear increase in rate, meaning that acetyl-ACP
concentration was limiting under these conditions. Therefore it appeared that
decarboxylation of malonyl-ACP catalyzed by KSβ was a slower process than the chain
elongation catalyzed by KSα, and cyclisation and release steps. Thus the kinetic values
determined here (kcat = 20.6 ± 0.9 min-1 and KM = 2.39 ± 0.38 µM) most likely correspond to
the initiation of polyketide synthesis by KSβ.
Previous attempts have been made to measure the kinetics of Type II minimal PKS. For
example, the group of Khosla studied heterologous PKS complexes consisting of the
actinorhodin, tetracenomycin, S. halstedii spore pigment, and pradimycin KSα/KSβ paired
with the frenolicin, R1128, granaticin, S. coelicolor spore pigment and pradimycin
ACP.98,99 In all these cases their assays have relied on the addition of MCAT and are thus
comparable with our assays described above. However, the kcat values reported in their
studies ranged between 0.11 and 0.95 min-1, i.e. at least 20 times lower than those reported
here. Although the reason for this difference is not clear, the fact that they used high
concentrations of DTT in these assays likely reduced the concentration of acyl-ACP species
(see Section 2.3 in Chapter 2). Also, only discontinuous assays (e.g. HPLC, TLC) were
used, and this may have hampered the gathering of genuine initial rates -therefore their
numbers are likely to be underestimations. As expected, there is no difference in the
reported KM values (ranging 1.4-6.4 µM malonyl-ACP), as the determination of affinity
constants only depends on the relative rates at several substrate concentrations.
Protein-protein interactions between PKS components were discussed in the introductory
material to Chapter 2. In short, electrostatic and hydrophobic forces have been proposed to
determine ACP: partner protein interactions; in particular, negatively charged residues in
helix II of ACP have been appointed as the key players in ACP recognition by other
components of Type I and Type II PKS.
In this work, we have studied the interaction of ACP with KSβ by measuring the kinetic
parameters of decarboxylation of mutant malonyl-ACP by KSβ. All act mutants (except
E47V) achieved the same maximum velocity (kcat ~ 20 min-1), meaning that E47, E53 and
R72 are not involved in catalysis during initiation of polyketide synthesis. Notably, the

Chapter 3. Initiation of polyketide synthesis
81
introduction of a bulky group in the E47V mutant severely impaired its suitability as a
substrate, suggesting a less-favoured docking position of E47V ACP onto KSβ. R72A
behaved essentially as the reference ACP. Remarkably, the same specificity constant kcat /
KM was measured for the reference act ACP and WT gris and dps ACP, in agreement with
previous reports showing successful complementation of Type II KSα/KSβ with
heterologous ACP.98,99
Also, we have provided further evidence to support the role of helix II as a recognition
motif in acyl carrier proteins: Michaelis constants of E47A, E47V and E53A ACP
increased three to four-fold when compared with act ACP (KM ~ 7-8 µM vs. 2-2.5 µM),
indicating less binding affinity of KSβ towards these mutants.
Our kinetic studies have also allowed us to achieve a better understanding of polyketide
biosynthesis by Type II PKS. In order to prime KSα with the acetate starter unit, two
possible mechanisms could be conceivable. First, an ‘internal transfer’ of acetyl-ACP
occurs directly from the KSβ active site to the KSα active site. The second scenario depicts
the release of acetyl-ACP into solution, and its subsequent encountering with KSα (Scheme
3.4). Whereas perhaps the first option would appear more effective, our results are
consistent with previous evidence that support the ‘external transfer’ of starter units,62,96 as
KSα is clearly able to load itself with acetate delivered by ACP from solution.
Nevertheless, our results cannot rule out the coexistance of the ‘internal’ and ‘external’
acetyl-ACP transfer pathways.

Chapter 3. Initiation of polyketide synthesis
82
4. Chain elongation and cyclisation and release from the PKS
The biosynthesis of SEK4 and SEK4b by the actinorhodin minimal PKS in vitro composes
at least 20 different chemical reactions, including loading, initiation of polyketide
synthesis, chain elongation and cyclisation and release steps (see Scheme 1.18 in Chapter
1). Chapter 4 deals with the reactions catalyzed by KSα.
The first reaction catalyzed by KSα is its own priming with an acetate starter unit from
acetyl-ACP onto its active site cysteine, to form acetyl-S-KSα (Scheme 4.1.A). This occurs
via an acyl transfer step between the thiols of the PP arm in ACP and the active C169 in
KSα. Elongation of the polyketide chain occurs via decarboxylative Claisen condensations
between the acyl-S-KSα intermediate and malonyl-ACP (Scheme 4.1.B). After each
condensation, the growing polyketide chain must be transferred back to the active site of
KSα, again through an acyl transfer between the PP in ACP and KSα (Scheme 4.1.C).
It has been proposed by the Stanford group that, as the polyketide chain is elongated, it
extrudes into a ‘polyketide tunnel’ formed between KSα and KSβ (Section 1.2.3.4).31 This
may keep the reactive ketide groups far from each other, thus preventing spontaneous,
aberrant cyclizations. The polyketide tunnel can also provide an actual basis for the acyl
transfer from the terminal thiol of ACP to the active thiol in KSα, which must occur after
every condensation.
The acyl transfer between ACP and KSα has not been investigated in this work. We
have assumed that it quickly reaches equilibrium. This acyl transfer step as well as the
seven Claisen condensations have been considered here as the ‘elongation’ or ‘extension’
steps.
Finally, production of SEK4 and SEK4b must involve cyclization of the polyketide
chain and release of polyketides from the PKS into solution. It is unknown if and when the
cyclisation and release reactions are catalyzed by the ketosynthase complex (see Chapter 6
for further discussion).
KSα belongs to the thiolase superfamily of enzymes (see Section 1.2.3.3 in Chapter 1). The
kinetics of reactions catalyzed by other thiolases, such as the ketosynthases from Type I

Chapter 3. Initiation of polyketide synthesis
83
and Type II FAS and other PKS, have been measured; and some examples are provided in
the following sections.
ACP
S
O
+ KS! KS"KS! KS"
Cys169S
O
KS! KS"
Cys169S
O
ACP
S
O+
O
KS! KS"
Cys169HS
ACP
S
O+
OHO
KS! KS"
Cys169HS
ACP
S
O+
O
KS! KS"
Cys169S
OOACP
SH
+
Scheme 4.1 Reactions catalyzed by KSα: A. Priming; B. Chain extension. C. Acyl transfer between the PP
thiol of ACP and the cysteine active site in KSα
4.1.1. Kinetics of FAS systems
Type I fatty acid synthases from mammals were the first fatty acid synthases studied
kinetically. In most cases, this was done by direct monitoring of the oxidation of NADPH
by spectrophotometry. For example, during the decades of the 70s and 80s, the Michaelis
constants for pigeon liver FAS (1.3 and 30 µM for acetyl and malonyl CoA,
respectively),159 bovine FAS (16.5 and 20 µM for acetyl-CoA and malonyl-CoA,
respectively),160 and chicken liver FAS (KM = 2.5 µM malonyl CoA)161 had been estimated.
Human FAS was studied later (KM = 6 µM malonyl CoA).162 Type I fatty acud synthases
from mycobacteria such as Mycobacterium smegmatis (KM =30 µM for acetyl CoA)163 and
Euglena gracillis (KM =1.3 and 5.1 µM for acetyl and malonyl CoA, respectively)164 were
also characterized. In summary, it appeared that the Michaelis constants ranged from 1 to
A.
B.
Acyl transfer: priming
extension
C.
Acyl transfer

Chapter 3. Initiation of polyketide synthesis
84
30 µM for most of the fatty acid synthases studied. kcat values varied from 160 to 1200 min-1
in human and chicken liver FAS, respectively.161,162
Radioactive substrates have traditionally been used for the kinetic study of Type II fatty
acid synthases. The first Type II FAS studied was that of E. coli. KASIII exhibited a KM of
40-150 µM for acetyl-CoA, and 5-25 µM for malonyl-ACP.165,166 KASI and KASII showed
affinities of 30-80 µM for a variety of substrates, saturated and unsaturated C-12 to C-14
acyl-ACP.167
Other Type II systems have produced different kinetic parameters; for instance, KASIII
from Staphylococcus aureus (kcat = 16 min-1 and KM = 6.18 µM for acetyl CoA, and 1.76 µM
for malonyl-ACP)168 and KASI and KASII from Mycobacterium tuberculosis (kcat = 4.8
and 1.4 min-1 respectively, and KM = 13.5 µM for malonyl-ACP).169 The study of the plant
Type II FAS from Cuphea wrightii gave similar Michaelis constants (1.4 and 4.1 µM for
acetyl and malonyl CoA, respectively).170
4.1.2. Kinetics of Type III PKS
Type III polyketide synthases are very simple enzymes that catalyze repetitive
decarboxylative condensations between a starter unit and one or more molecules of malonyl
CoA (see Section 1.2.4 in Chapter 1). Therefore the kinetic study of these enzymes
provides a very useful model for other thiolases.
Type III polyketide synthases from plants and bacteria have been biochemically
characterized in vitro. Additionally, there is one Type III PKS from fungi that has been
kinetically studied.171 Although it accepted a wide range of starter units (and consequently
produced a variety of products), the most favourable substrate was stearoyl CoA 59 to
produce a pentaketide resorcylic acid 60 (Scheme 4.2). This proceeded with kcat = 3.1 min-1
and KM = 20.0 µM.171
S-CoA
O
15
OHO
O
OH
OH15
4 x malonyl CoA
Scheme 4.2 Reaction catalyzed by the pentaketide resorcylic synthase from Neurospora crassa
59 60
Type III PKS

Chapter 3. Initiation of polyketide synthesis
85
Similar kinetic values had been reported for plant polyketide synthases. For instance, Jez,
Noel and co-workers had thoroughly studied the biosynthesis of chalcone by the Medicago
sativa (alfalfa) chalcone synthase (CHS). Using radioactive methods, they independently
measured the rate of decarboxylation of malonyl CoA by monitoring the conversion to
acetyl CoA, and the rate of product formation by counting radioactivity in thin layer
chromatography spots (Table 4.1).172
Decarboxylation of
malonyl CoA CHS assay (p-
coumaroyl CoA) CHS assay (malonyl
CoA) kcat (min-1 ) 4.1 2.2 3.7
KM (µM) 19.1 4.5 4.1
Table 4.1 Kinetic parameters for CHS.172
Abe, Noguchi and co-workers have reported similar kinetic values for the chalcone
synthases from Huperzia serrata (kcat = 2.0 min-1 and KM = 19.7 µM)173 and Scutellaria
baicalensis (kcat = 1.3 min-1 and KM = 36.1 µM).174 The same group has described the
activity of two Type III polyketide synthases from aloe (Aloe arborescens), a pentaketide
chromone synthase (PCS) and an octaketide synthase (OKS). While PCS uses 5 molecules
of malonyl CoA to afford 61, OKS produces the octaketides SEK4 19 and SEK4b 27 from
eight acetate units derived from malonyl CoA. These were less efficient enzymes working
at kcat = 0.4 and 0.1 min-1 and KM = 71 and 95 µM (PCS and OKS respectively). 175,176
OHO
OH O 61
The first bacterial Type III PKS to be characterized was RppA from Streptomyces
griseus.177 It catalyzes the formation of 1,3,6,8-tetrahydroxynaphtalene 62 from 5
molecules of malonyl CoA. Further characterization yielded similar kinetic parameters as
its plant relatives, with kcat = 0.77 min-1 and KM = 0.93 µM.178 Recently, a similar enzyme
has been found in Streptomyces coelicolor (kcat = 1.3 min-1, KM = 3.9 µM).179 The fastest
Type III enzyme has been found in the bacterium Pseudomonas fluorescens. PhID produces

Chapter 3. Initiation of polyketide synthesis
86
phloroglucinol 63 from 3 molecules of malonyl CoA. A kcat value of 24 min-1 was
determined.180
OHOH
HO OH
OH
HO OH 62 63
4.1.3. Kinetics of Type I modular PKS (DEBS)
In vitro kinetic analysis of modular PKS was prompted by the discovery that individual
DEBS modules could be expressed and purified to homogeneity.181 Subsequent steady-state
kinetic analyses of DEBS modules were undertaken with engineered systems consisting of
a DEBS module supplemented with the TE domain.181,182 Using this strategy, the
conversion of the natural diketide substrate analogue, (2S, 3R)-2-methyl-3-
hydroxypentanoyl SNAC 64, to the corresponding triketide lactone 65 or 66 in the presence
of methylmalonyl CoA and NADPH was assessed by HPLC for modules 2 to 6 (Scheme
4.3).182 The catalytic constant kcat varied from 1.5 (module 3) to 9.5 (module 6) min-1, with
KM in the millimolar range (ca 4 mM).
O
O
O
O
OH
O
SNAC
OOH
HO S-CoA
OO
Scheme 4.3 Reactions catalyzed by A. Module 3 + TE; B. Modules 2, 5 and 6 + TE.
Reconstitution of the ‘natural’ ACP-bound substrates was performed by two methods. First,
by co-expression of the loading domain and module 1 with module 2, 5 or 6 + TE. Second,
by addition of the purified ACP domain and linker from module 4, which was
phosphopantetheinylated in vitro from the CoA analogue of 64.183 Both methods resulted in
64
65
66 + NADPH

Chapter 3. Initiation of polyketide synthesis
87
a 100-fold decrease in the KM value with respect to the SNAC substrates, showing the
importance of the protic body of ACP in shuttling substrates to the PKS.
The Stanford group has also studied the performance of AT-KS didomains of modules
3 and 6 with the ACP from all six modules (termed ACP1 to ACP6 in Table 4.2).184 The
best ACP substrate for each didomain was that of the same module. For instance, ACP
from modules 1 and 6 were much worse substrates than ACP3 for the AT-KS of module 3;
and, in turn, ACP3 itself was not recognized efficiently by AT-KS from module 6. As
discussed in Chapter 2, the same group has recently described some of the structural basis
for this difference.92
AT-KS module 3 ACP1 ACP2 ACP3 ACP4 ACP5 ACP6 kcat (min-1) n.d. 0.03 0.18 0.21 0.16 n.d. KM (µM) n.d. 9 16 179 56 n.d. kcat / KM (min-1 mM-1) n.d. 3.2 11.0 1.2 2.8 n.d. AT-KS module 6 ACP1 ACP2 ACP3 ACP4 ACP5 ACP6 kcat (min-1) 0.005 0.012 n.d. 0.008 0.021 0.050 KM (µM) 16 31 n.d. 119 37 27 kcat / KM (min-1 mM-1) 0.28 0.39 n.d. 0.07 0.56 1.85
Table 4.2 Kinetic parameters for the reaction catalyzed by AT-KS didomains from modules 3 and 6 using
ACP from every module as a substrate.
4.2. Kinetics of the rate-limiting reaction catalyzed by KSα
We have used the methodology described in Chapter 3 to study the kinetics of the rate
limiting step within the reactions catalyzed by KSα. In Chapter 3 we showed that, if act
minimal PKS assays are supplemented with MCAT, then the kinetics of KSβ could be
studied. Addition of exogenous acetyl-ACP to MCAT-supplemented assays further
increased the rate of production of aromatic polyketides; in fact, saturation with acetyl-ACP
produced a 3-fold increase in rate (Section 3.1.2). Under these circumstances, reaction rates
depended linearly on the concentration of KSα/KSβ. In principle, this new rate limiting
step could be any of the intervening reactions, i.e. elongation of the chain catalyzed by
KSα, cyclisation or release. We first investigated whether under fast malonylation
conditions, and in the presence of excess acetyl ACP, chain extension or chain termination
steps (i.e. cyclization and release) were rate limiting.

Chapter 3. Initiation of polyketide synthesis
88
4.2.1. Attempts to detect polyketide intermediates
In an attempt to establish whether chain extension or chain termination steps were rate
limiting, we searched for any polyketide intermediate prior to cyclization and release. If
only early intermediates could be observed, then that would suggest that elongation of the
chain was rate limiting. On the other hand, if a full octaketide prior to SEK4/4b formation
could be detected, then it would be more likely that cyclisation and release were slower
processes than chain elongation.
After each condensation, the resulting polyketide intermediate is transferred from the PP
arm in ACP to the active site cysteine in KSα (Scheme 4.1-C). In principle, polyketide
intermediates could be attached to either ACP or KSα, and the number of polyketide chains
under construction at any given time point was unlikely to significantly exceed the number
of active synthases. We thus conducted assays at high KSα/KSβ concentrations (3 µM),
hoping to observe part-completed acylated acyl carrier proteins or KSα by ESMS.
However, increasing KSα/KSβ concentrations also increased the overall rate of octaketide
production and malonyl CoA consumption, so aliquots of reaction mixtures containing
KSα/KSβ, holo-ACP, MCAT, acetyl-ACP and malonyl CoA were rapidly quenched by
addition of C4 resin,185 desalted and examined by ESMS. Only acetyl and malonyl ACP
species were observed (Figure 4.1). The detection of KSα by ESMS failed. This was
unfortunate, because, assuming that the acyl transfer step between the thiols of ACP and
KSα is in equilibrium (see introduction to Chapter 4), early polyketide intermediates most
likely are attached to KSα.
The same experiment was repeated and polyketides extracted and analyzed by HPLC to
search for triketides to heptaketides released into solution. We could only detect SEK4 and
SEK4b.
These experiments suggested no accumulation of polyketide intermediates either
attached to ACP (as judged by ESMS) or released into solution (as observed by HPLC).
This led us to speculate that the cyclisation and release steps are likely to be faster than the
elongation steps.

Chapter 3. Initiation of polyketide synthesis
89
Figure 4.1 ESMS analysis of the ACP species in minimal PKS assays done in the presence of MCAT and
external acetyl-ACP. Expected masses: 9527 Da (malonyl-ACP), 9483 Da (acetyl-ACP).
4.2.2. Kinetics of the elongation step catalyzed by KSα .
In order to study the kinetics of KSα, we first tried to measure the kinetic constants for
malonyl-ACP as a substrate for KSα (i.e. the elongation step). The determination of kinetic
parameters in assays where excess acetyl-ACP was externally added was difficult due to a
range of circumstances. First, the rapid consumption of acetyl-ACP released holo-ACP into
solution. At 0.25 µM KSα/KSβ consumption rates of acetyl-ACP are ca. 12 µM min-1 under
saturating concentrations of malonyl-ACP (Section 3.1.2). In the presence of MCAT, the
generated holo-ACP was readily converted to malonyl-ACP. This introduced a degree of
uncertainty in the concentration of malonyl-ACP. Second, we assumed that the binding site
for acetyl-ACP onto KSα was the same as that for malonyl-ACP. Thus we anticipated that,
at high concentrations of acetyl-ACP and low concentrations of malonyl-ACP, we might
observe inhibition of the elongation reaction, due to the non-productive binding of acetyl-
ACP onto malonyl-ACP docking sites within KSα.
In an attempt to estimate the Michaelis constant for malonyl-ACP, we studied reaction
rates at fixed acetyl-ACP concentration (30 µM) and a range of malonyl-ACP
concentrations between 0.5 and 60 µM. When low malonyl-ACP concentrations were used,
we observed a lag in the production of polyketides which lasted 20-30 s. This agreed with
our predictions that acetyl-ACP was probably inhibiting polyketide synthesis when it was
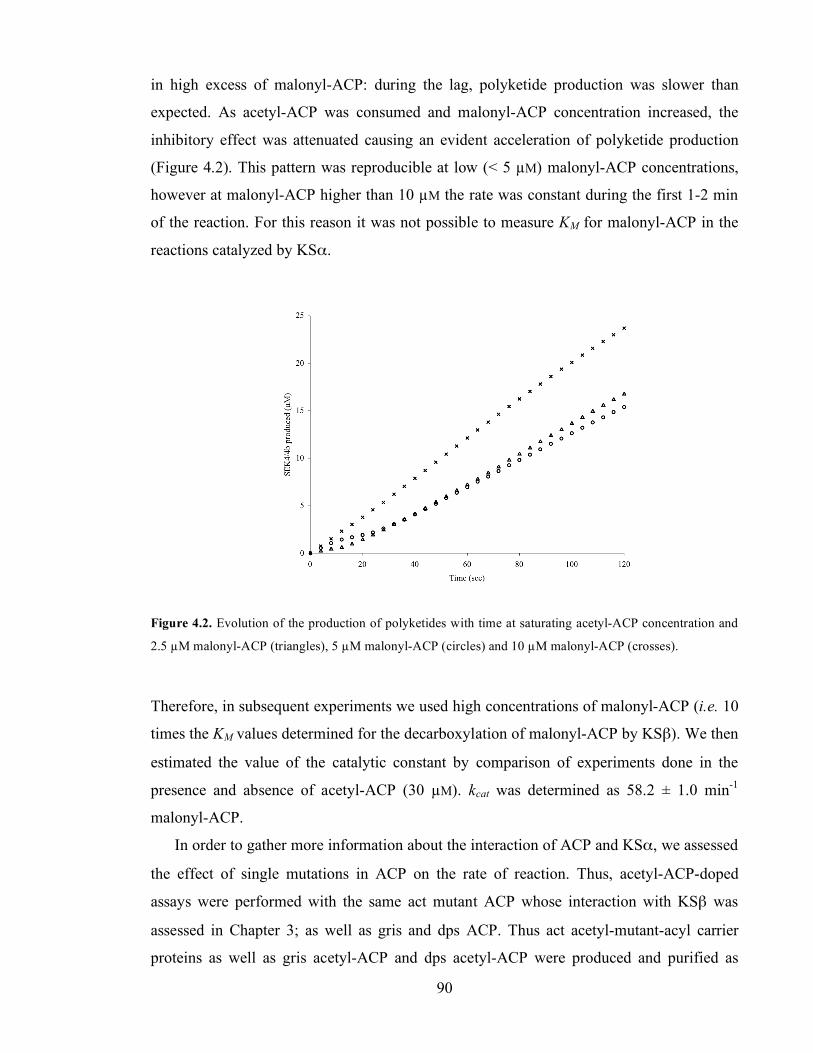
Chapter 3. Initiation of polyketide synthesis
90
in high excess of malonyl-ACP: during the lag, polyketide production was slower than
expected. As acetyl-ACP was consumed and malonyl-ACP concentration increased, the
inhibitory effect was attenuated causing an evident acceleration of polyketide production
(Figure 4.2). This pattern was reproducible at low (< 5 µM) malonyl-ACP concentrations,
however at malonyl-ACP higher than 10 µM the rate was constant during the first 1-2 min
of the reaction. For this reason it was not possible to measure KM for malonyl-ACP in the
reactions catalyzed by KSα.
Figure 4.2. Evolution of the production of polyketides with time at saturating acetyl-ACP concentration and
2.5 µM malonyl-ACP (triangles), 5 µM malonyl-ACP (circles) and 10 µM malonyl-ACP (crosses).
Therefore, in subsequent experiments we used high concentrations of malonyl-ACP (i.e. 10
times the KM values determined for the decarboxylation of malonyl-ACP by KSβ). We then
estimated the value of the catalytic constant by comparison of experiments done in the
presence and absence of acetyl-ACP (30 µM). kcat was determined as 58.2 ± 1.0 min-1
malonyl-ACP.
In order to gather more information about the interaction of ACP and KSα, we assessed
the effect of single mutations in ACP on the rate of reaction. Thus, acetyl-ACP-doped
assays were performed with the same act mutant ACP whose interaction with KSβ was
assessed in Chapter 3; as well as gris and dps ACP. Thus act acetyl-mutant-acyl carrier
proteins as well as gris acetyl-ACP and dps acetyl-ACP were produced and purified as

Chapter 3. Initiation of polyketide synthesis
91
described for the reference act ACP. Polyketide production rates in the presence of excess
malonyl-ACP and the corresponding acetyl-ACP were then measured and compared with
control experiments lacking acetyl-ACP.
Thus holo-(mutant)-ACP concentration was set to 20 µM (reference act ACP and R72A,
gris and dps ACP) or 50 µM (E47A, E47V, E53A ACP). All assays (with and without
acetyl-ACP) were done at least in triplicate. kcat was around three times higher for each of
the act mutants (Table 4.3). Gris ACP and dps ACP behaved somewhat differently, and kcat
increased by a factor of 4.7 and 2.3 respectively. Higher concentrations of gris and dps
acetyl-ACP (up to 60 µM) did not produce further increase in rate.
ACP kcat (KSα) min-1
Increase with respect to KSβ
Act 58.2±1.0 2.8 E47A 56.3±4.0 2.8 E47V 10.6±0.5 3.0 E53A 62.3±2.0 3.6 R72A 71.3±2.1 3.5 Gris 45.1±2.3 4.7 Dps 38.4±4.2 2.3
Table 4.3 Catalytic constants in for a variety of ACP in the presence of excess acetyl-ACP.
4.3. Discussion
In Chapter 4 we have exploited the methodology developed in preceding chapters to
measure the kinetics of KSα. Thus we have used acetyl-ACP in high concentrations to
overcome the limitation in polyketide production rate imposed by KSβ (Chapter 3). Under
these conditions, the new rate limiting step could in principle correspond to either the
slowest of the reactions catalyzed by KSα, or the cyclization and release steps (see Scheme
1.18 in Chapter 1).
Here we hypothesizee that the elongation step catalyzed by KSα is the rate limiting step
in act minimal PKS in the presence of MCAT and exogenous acetyl-ACP, because SEK4
and SEK4b were the first isolable octaketides produced by the act minimal PKS. Attempts
to trap late polyketide intermediates either bound to ACP or released into solution failed.

Chapter 3. Initiation of polyketide synthesis
92
On the other hand, there is evidence in the literature of early polyketide intermediates
attached to ACP189 or KSα.31 Taken together, this results suggested that the cyclization and
release steps proceeded at a higher rate than the chain elongation catalyzed by KSα.
First, we approached the study of the new rate limiting step by the determination of kinetic
parameters for malonyl-ACP as a substrate for KSα. When we assessed the rate of reaction
for a range of malonyl-ACP concentrations, we observed complex evolution of the rate as
the reaction proceeded, particularly during the first 20-30 sec. This we attributed to
inhibition of polyketide synthesis due to non-productive binding of acetyl-ACP to KSα,
when acetyl-ACP was in high excess of malonyl-ACP. This result evidences that the
docking site for acetyl and malonyl-ACP in KSα is the same.
Although these experiments could have been useful for estimating KM for acetyl and
malonyl-ACP in KSα-catalyzed reactions, the effect could not be studied further with the
methods developed in this work. In our steady state measurements, the first 5-10 seconds of
reaction could not be monitored, because the instrument required that time to start the
measurements. Future work should address the need for stopped-flow kinetic measurements
in order to study the reaction rate at low (< 5 µM) malonyl-ACP concentrations and a range
of acetyl-ACP concentrations. Under stopped-flow conditions, initial reaction rates can be
studied during the first milliseconds of reaction, when the pool of acetyl-ACP is still intact
and the concentration of malonyl-ACP corresponds to the concentration of holo-ACP
added.
Furthermore, radioactive acetyl-ACP could be used to learn the extent of acetyl starter
unit that is incorporated into products from exogenous acetyl-ACP, rather than from
decarboxylation of malonyl-ACP. This would be definitive evidence to assess if there is
any pathway for an ‘internal’ transfer of acetyl-ACP from KSβ to the active site of KSα
(Scheme 3.4). For that matter, these studies should be carried out with a KSα/KSβ complex
which is inefficient in catalyzing the initiation reaction (e.g. CA mutant – see Chapter 5).
We then decided to do assays under saturating concentrations of both acetyl-ACP and
malonyl-ACP. Reaction rates upon addition of acetyl-ACP depended on the nature of the
ACP. For example, act ACP supported a kcat value of ca. 60 min-1, whereas gris and dps
ACP were less effective (ca. 45 and 38 min-1 respectively). Interestingly, the reaction rate

Chapter 3. Initiation of polyketide synthesis
93
in the presence of all act mutant-ACP experienced a 3-fold increase with respect to assays
in the absence of acetyl-ACP, i.e. in which the reaction catalyzed by KSβ is rate limiting.
For instance, act E47V ACP was 6 times worse a substrate than the reference act ACP in
KSβ, and also in KSα. This probably means that the binding of ACP onto KSβ and KSα is
very similar, and the introduction of a bulky valine residue has the same disadvantageous
effect on the catalysis of both enzymes.
The priming reaction of KSα with acetyl-ACP is similar to the priming of FAS with an
acetyl starter unit. Witkowski, Smith and co-workers studied the reaction rate of the
mammalian FAS in the absence and presence of added acetyl CoA.190 Interestingly, a 5-
fold increase in the reaction rate was also observed when exogenous acetyl-CoA was added
to their assays. Smith and co-workers suggested that, in the absence of added acetyl-CoA,
FAS exhibited a weak affinity for malonyl-ACP and decarboxylation of malonyl-ACP
occurred inefficiently. In contrast, in the presence of added acetyl-CoA, a stronger malonyl-
ACP binding site was created so that decarboxylation, coupled with condensation, took
place efficiently. They speculated that the β-ketoacyl synthase domain would exist in two
distinct conformations, one favouring the acylation of the active site cysteine and the other
favouring the decarboxylation of malonyl-ACP.
Likewise, act KSα/KSβ could exist in two different conformations, one favouring
decarboxylation of malonyl-ACP in the KSβ active site, and the other favouring the
acylation of the active site cysteine in KSα. This model would explain why the active site
of KSβ seemed to be buried in the crystal structure of KSα/KSβ published by the Stanford
group, which led them to dismiss the catalytic role of KSβ in the initiation of polyketide
synthesis: the published KSα/KSβ structure may represent a complex ready for extension,
rather than initiation. Indeed, electron density of intermediates attached to KSα was
observed in the Stanford crystal structure.
In this dynamic model, decarboxylation of malonyl-ACP by KSβ would trigger a
conformational change in the whole dimer to render a KSα that would then be in an ‘open’
conformation and ready to load itself with the acetyl starter unit. This model suggests that
the rate limiting step in the presence of exogenous acetyl-ACP is probably the priming of
KSα with an acetyl starter unit, because the rate of the KSα-catalyzed reaction seemed to

Chapter 3. Initiation of polyketide synthesis
94
depend on the rate of malonyl-ACP decarboxylation by KSβ. This makes sense, because
KSα has to encounter acetyl-ACP in the bulk solution (see discussion to Chapter 3) in order
to catalyze the acetyl transfer from ACP onto its active site (Scheme 4.1.A).
The priming of KSα is analogous to the priming of Type III polyketide synthases with
the corresponding starter unit; also to that of DEBS modules primed with synthetic starter
units such as SNAC derivatives; and to that of priming of ZhuH KASIII-type initiation
ketosynthase in the Type II PKS of R1128 (see introduction to Chapters 3 and 4). Typical
kcat values for plant and bacterial Type III PKS range from 0.7 to 3.1 min-1. To date, the
fastest Type III enzyme characterized (PhID from the bacterium Pseudomonas fluorescens)
has a kcat value of 24 min-1. The DEBS Type I modular PKS catalyzes polyketide synthesis
at a similar rate with reported kcat values between 1.5 (module 3) and 9.5 (module 6) min-1.
The priming ketosynthase ZhuH is a faster enzyme, and catalyzes the condensation of
butyryl, propionyl or acetyl CoA with malonyl-ACP at 113, 74 and 58 min-1
respectively.131 These values are comparable to the catalytic constant determined here (kcat
~ 60 min-1).

Chapter 3. Initiation of polyketide synthesis
95
5. Stoichiometric analysis of the act minimal PKS
The organization of a protein complex is defined at four levels. The amino acid sequence of
the protein is called its primary structure. In a folded protein, different regions form local
secondary structures, such as α-helices and β-sheets. The tertiary structure is formed by
packing such secondary structural elements into domains with defined topology, which, for
instance, can bring together amino acids far apart in the primary sequence. Finally, many
protein complexes exist as an arrangement of several polypeptide chains, same or different
from each other, which is known as the quaternary structure.95 In this case, the
stoichiometry of the complex is the relative abundance of each component polypeptide.
The quaternary structure is therefore the ultimate molecular organization of an enzyme
or complex of enzymes. In the latter case, it provides essential information about the
interaction and biomolecular communication among its different components.
Most enzymes of the thiolase superfamily exhibit a dimeric quaternary structure. For
example, in bacterial fatty acid synthases the ketosynthase typically exists in a dimeric form
(such as E. coli KASI, KASII and KASIII enzymes).48 Type III polyketide synthases are
also homodimeric, as are the ketosynthase domains of animal FAS85 and Type I modular
PKS.92 Other members of the thiolase superfamily, such as the biosynthetic thiolase from
Zoogloea ramigera, exist as tetramers.192
Initial studies by the Stanford group on the quaternary structure of the act KSα/KSβ
suggested that it had a tetrameric conformation in solution. Carreras and Khosla subjected
purified KSα/KSβ preparations to gel filtration chromatography, and found that the
complex eluted at an apparent molecular weight of ~168 kDa.53 Because KSα and KSβ
were co-purified as an equimolar mixture, this indicated that the major KSα/KSβ species
was probably the tetramer (which has an expected molecular weight of ca. 180 KDa).
Within the Cambridge and Bristol groups, a number of Ph.D students have also studied
the quaternary structure of KSα/KSβ. For example, gel filtration studies in the Cambridge
group also indicated that the predominant KSα/KSβ species eluted at the expected
molecular weight for the α2β2 tetramer.158 In the Bristol group, analytical
ultracentrifugation, gel filtration and electron microscopy studies also suggest that

Chapter 3. Initiation of polyketide synthesis
96
KSα/KSβ exists mainly as a tetramer in solution (Dr. John Crosby, personnal
communication and ref. 120).
In preceeding chapters, KSα/KSβ complexes have been represented simply as a dimer. In
order to study the stoichiometry of the act minimal PKS, we may now consider that the
dimeric and tetrameric forms of KSα/KSβ are in equilibrium (Scheme 5.1), and that the
major KSα/KSβ species in solution seems to be the tetramer.
KS! KS"
KS! KS"
KS! KS" KS! KS"+
Scheme 5.1 Dimer-tetramer KSα/KSβ equilibrium.
There is little evidence about the stoichiometry of ACP: KSα/KSβ association. In the
Bristol group, tryptophan fluorescence experiments were performed to address the question
of how many acyl carrier proteins bound each KSα/KSβ complex. ACP titrations in
KSα/KSβ solutions suggested a 1:1 stoichiometry relation between ACP and KSα/KSβ.120
Although these analytical methods are of course very useful, they only provide information
about the state of proteins in solution, not about the catalytic activity of the enzyme
complexes. It seemed necessary to study and characterize these protein complexes under
conditions upon which they were active, and polyketides were produced in an effective
manner. Thus we set ourselves to investigate the relationship between the catalysis and the
quaternary structure of KSα/KSβ and ACP: KSα/KSβ stoichiometry, using the quantitative
methods described in previous chapters of this thesis.
5.1. Stoichiometry of the KSα /KSβ complex –assays with mutant KSα /KSβ
The study of the stoichiometry of KSα/KSβ in solution is intrinsically difficult due to the
protocol we use to purify them. KSα contains an N-terminal hexa-histidine tag which

Chapter 3. Initiation of polyketide synthesis
97
allows its purification by nickel-affinity purification.8 Because KSα and KSβ share
extensive surface area,31 KSβ is not washed away during the affinity chromatography
procedure; in other words, KSβ is co-purified in KSα preparations. Therefore it is not
feasible to vary the relative concentrations of the ketosynthase components in order to
study their respective kinetic properties. Previous attempts within the Bristol group to
express and purify soluble and active KSα and KSβ as individual proteins, have been
unsuccessful to date.142
The Bristol and Cambridge groups had previously studied the activity of a series of
KSα and KSβ mutant proteins, namely CA, AQ and AA mutant KSα/KSβ complexes (see
introduction to Chapter 3).62 A combination of HPLC analysis of polyketides as well as
ESMS analysis of acyl-ACP species, suggested that a CA KSα/KSβ complex, in which the
glutamine active site of KSβ had been mutated to alanine, was deficient in decarboxylation
of malonyl-ACP to acetyl-ACP.62 Conversely, an AQ KSα/KSβ complex was an efficient
decarboxylase of malonyl-ACP, as demonstrated by detection of acetyl-ACP by ESMS.62
Using this qualitative method, it was estimated that AQ KSα/KSβ decarboxylated malonyl-
ACP at approximately 40 µM h-1. Acetyl-ACP most likely was released into solution, rather
than transferred ‘internally’ to KSα (see introduction to Chapter 3 and Section 3.3).
Therefore we speculated that addition of AQ KSα/KSβ to act minimal PKS assays
using the wild type KSα/KSβ (i.e. CQ KSα/KSβ), would effectively increase the relative
concentration of KSβ (Scheme 5.2). If this hypothesis held, then we would be able to fix
the concentration of KSα (by fixing CQ KSα/KSβ concentration), and increase the
concentration of KSβ simply by increasing AQ KSα/KSβ concentration in PKS assays.
QCQA
ACP
S
HO
O
O
ACP
S
O
Polyketide synthesis
Scheme 5.2 Hypothetical use of AQ KSα/KSβ to increase the rate of KSβ-mediated decarboxylation of
malonyl-ACP. KSα (red) and KSβ (green) are depicted by their corresponding active sites (see text).
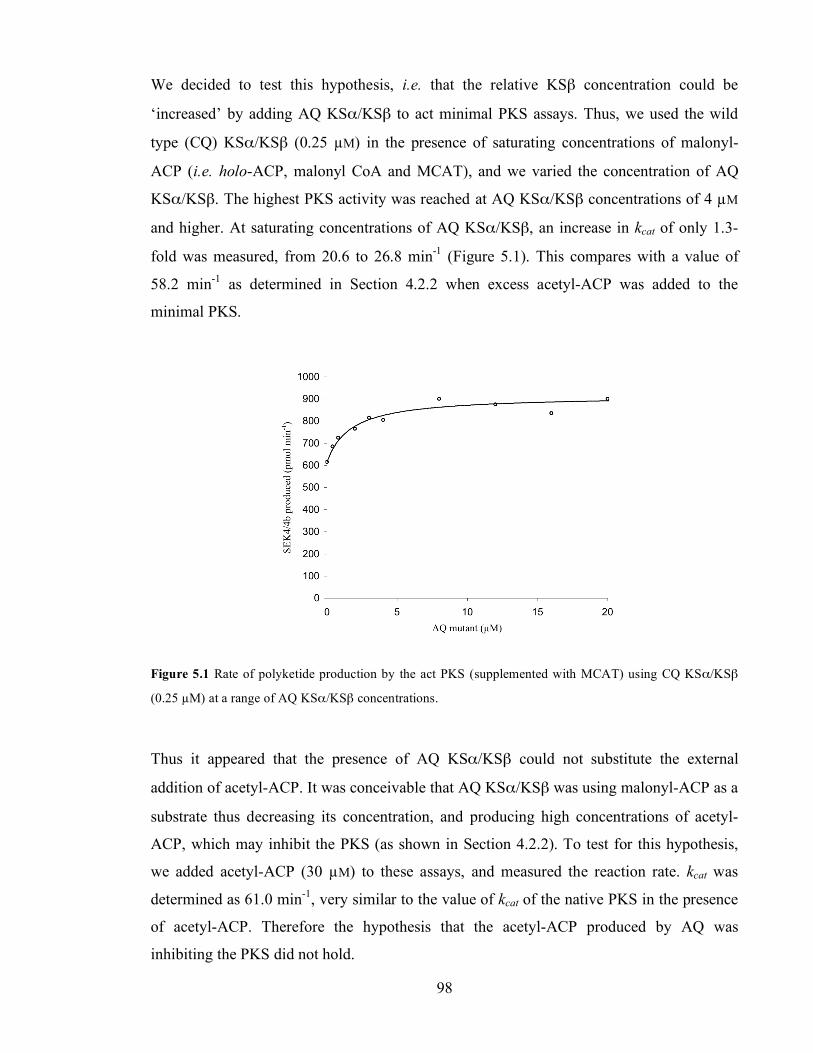
Chapter 3. Initiation of polyketide synthesis
98
We decided to test this hypothesis, i.e. that the relative KSβ concentration could be
‘increased’ by adding AQ KSα/KSβ to act minimal PKS assays. Thus, we used the wild
type (CQ) KSα/KSβ (0.25 µM) in the presence of saturating concentrations of malonyl-
ACP (i.e. holo-ACP, malonyl CoA and MCAT), and we varied the concentration of AQ
KSα/KSβ. The highest PKS activity was reached at AQ KSα/KSβ concentrations of 4 µM
and higher. At saturating concentrations of AQ KSα/KSβ, an increase in kcat of only 1.3-
fold was measured, from 20.6 to 26.8 min-1 (Figure 5.1). This compares with a value of
58.2 min-1 as determined in Section 4.2.2 when excess acetyl-ACP was added to the
minimal PKS.
Figure 5.1 Rate of polyketide production by the act PKS (supplemented with MCAT) using CQ KSα/KSβ
(0.25 µM) at a range of AQ KSα/KSβ concentrations.
Thus it appeared that the presence of AQ KSα/KSβ could not substitute the external
addition of acetyl-ACP. It was conceivable that AQ KSα/KSβ was using malonyl-ACP as a
substrate thus decreasing its concentration, and producing high concentrations of acetyl-
ACP, which may inhibit the PKS (as shown in Section 4.2.2). To test for this hypothesis,
we added acetyl-ACP (30 µM) to these assays, and measured the reaction rate. kcat was
determined as 61.0 min-1, very similar to the value of kcat of the native PKS in the presence
of acetyl-ACP. Therefore the hypothesis that the acetyl-ACP produced by AQ was
inhibiting the PKS did not hold.

Chapter 3. Initiation of polyketide synthesis
99
Another possibility was that our AQ KSα/KSβ preparation was not active. We decided to
test for this hypothesis using another system; and we purified the CA KSα/KSβ mutant
complex, i.e. a KSα/KSβ complex which is defective in decarboxylation, because the
change in rate should be even more dramatic.
5.1.1. Kinetic study of a mutant CA KSα /KSβ complex
In the light of the continuous method we developed in Chapter 3 to measure the kinetics of
KSβ, we decided to study the activity of the CA KSα/KSβ complex in depth. Thus
production of polyketides by CA KSα/KSβ was assessed in the presence of malonyl CoA,
MCAT and a variety of holo-ACP concentrations. Initial reaction rates were then measured
(Figure 5.2). Kinetic parameters of kcat = 2.2 ± 0.3 min-1 and KM = 2.00 ± 0.66 µM malonyl-
ACP were determined, i.e. the CA KSα/KSβ exhibited the same affinity as the CQ
KSα/KSβ for malonyl-ACP, but could only function ca. 10 times slower than the wild type
protein.
Figure 5.2 Kinetics of the decarboxylation of malonyl-ACP by CA KSα/KSβ. A. Plot of reaction rate vs.
substrate concentration and hyperbolic fit. B. Hanes linear plot of the rate data.
Following the methodology developed in Chapter 3, we then turned our efforts to
complement this decarboxylase-deficient CA KSα/KSβ by addition of extra starter unit,
acetyl-ACP. We anticipated that addition of external acetyl-ACP would surpass the lack of
decarboxylation activity of the CA KSα/KSβ. Therefore we expected to observe similar
reaction rates as measured with the wild type CQ KSα/KSβ (kcat ~ 60 min-1). Surprisingly,
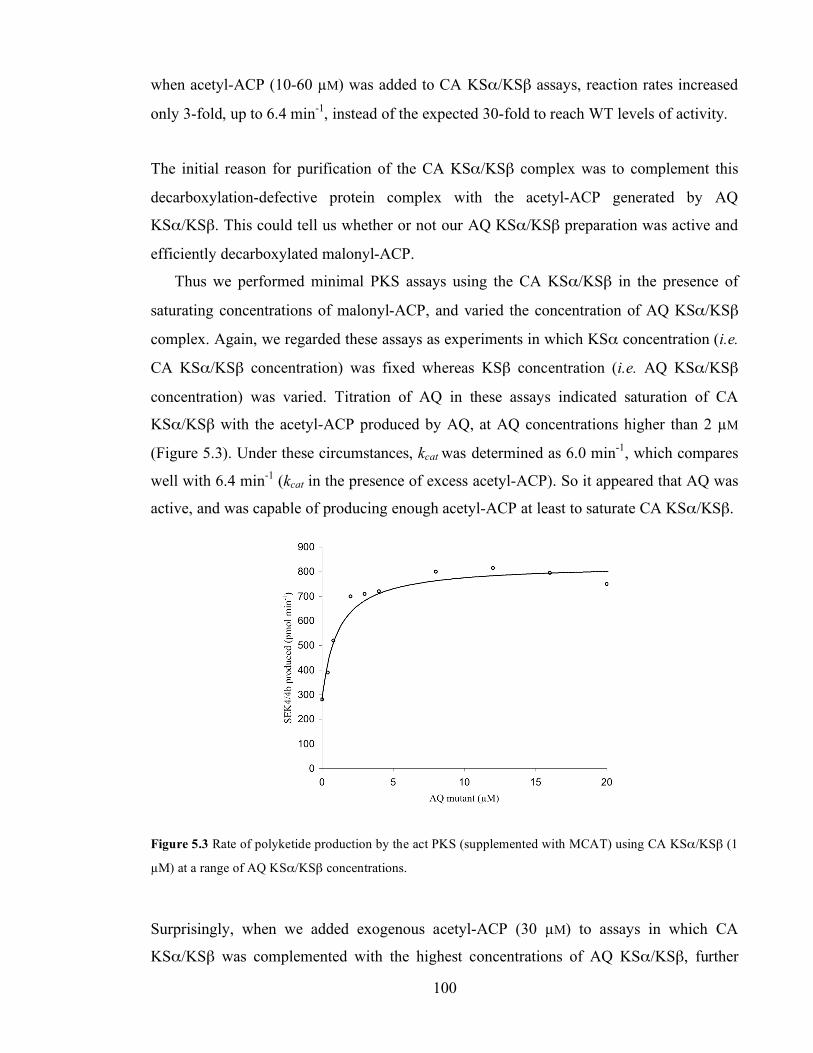
Chapter 3. Initiation of polyketide synthesis
100
when acetyl-ACP (10-60 µM) was added to CA KSα/KSβ assays, reaction rates increased
only 3-fold, up to 6.4 min-1, instead of the expected 30-fold to reach WT levels of activity.
The initial reason for purification of the CA KSα/KSβ complex was to complement this
decarboxylation-defective protein complex with the acetyl-ACP generated by AQ
KSα/KSβ. This could tell us whether or not our AQ KSα/KSβ preparation was active and
efficiently decarboxylated malonyl-ACP.
Thus we performed minimal PKS assays using the CA KSα/KSβ in the presence of
saturating concentrations of malonyl-ACP, and varied the concentration of AQ KSα/KSβ
complex. Again, we regarded these assays as experiments in which KSα concentration (i.e.
CA KSα/KSβ concentration) was fixed whereas KSβ concentration (i.e. AQ KSα/KSβ
concentration) was varied. Titration of AQ in these assays indicated saturation of CA
KSα/KSβ with the acetyl-ACP produced by AQ, at AQ concentrations higher than 2 µM
(Figure 5.3). Under these circumstances, kcat was determined as 6.0 min-1, which compares
well with 6.4 min-1 (kcat in the presence of excess acetyl-ACP). So it appeared that AQ was
active, and was capable of producing enough acetyl-ACP at least to saturate CA KSα/KSβ.
Figure 5.3 Rate of polyketide production by the act PKS (supplemented with MCAT) using CA KSα/KSβ (1
µM) at a range of AQ KSα/KSβ concentrations.
Surprisingly, when we added exogenous acetyl-ACP (30 µM) to assays in which CA
KSα/KSβ was complemented with the highest concentrations of AQ KSα/KSβ, further

Chapter 3. Initiation of polyketide synthesis
101
increase in rate was observed. In these assays kcat reached 9.6 min-1. This result was
unexpected, because it meant that the presence of AQ KSα/KSβ somehow increased the
activity of CA KSα/KSβ (but not the wild type CQ KSα/KSβ), (Table 5.1) in the presence
of saturating concentrations of acetyl-ACP.
Addition (saturating concentrations)
kcat WT KSα /KSβ (min-1)
kcat CA KSα/KSβ (min-1)
none 20.6 ± 0.9 2.2 ± 0.3
AQ 26.8 ± 4.0 6.0 ± 1.0
Acetyl-ACP 58.2 ± 1.0 6.4 ± 0.7 AQ and acetyl-ACP
61.0 ± 4.8
9.6 ± 1.0
Table 5.1 Summary of kcat values for WT and CA KSα/KSβ under a range of conditions.
5.2. Stoichiometry of the ACP: KSα /KSβ complex – inhibition of KSα/KSβ by apo-
ACP.
Inhibition studies can provide important information about the stoichiometry of protein
complexes. The general mechanism for enzyme inhibition describes two possible types of
behaviour. An inhibitor which is analogous to the substrate may only bind to the free
enzyme, and this would suggest that there is only one binding site for the inhibitor (as well
as for the substrate to which the inhibitor is analogous). In this case, the inhibition is termed
‘competitive’ (as substrate and inhibitor compete for the same binding site on the enzyme)
and it is characterized by a competitive inhibition constant, Kic (Scheme 5.2).94
On the other hand, an uncompetitive inhibitor only binds to the enzyme-substrate
complex, and it does so with an affinity described by an uncompetitive inhibition constant,
Kiu (Scheme 5.2).94 In this scenario, the inhibitor does not compete with the substrate,
which suggests that inhibitor and substrate bind to different sites on the protein.
Finally, if the inhibitor binds to both the free enzyme and the complex formed between the
enzyme and the substrate, then this may be evidence for multiple substrate binding sites. In
this situation, the enzyme would be able to bind (at least) two substrates at the same time.

Chapter 3. Initiation of polyketide synthesis
102
Apo-ACP is the inactive form of ACP that lacks the PP arm (see Section 1.2.3.1). NMR
studies of act apo-ACP and malonyl-ACP suggest that both species have very similar
structures in solution.193 Therefore apo-ACP was a good entry point in order to perform
inhibition studies with the act PKS. This could be detected by a decrease in polyketide
production rates.
We expected that apo-ACP would at least compete with malonyl-ACP for binding sites
on KSα/KSβ (Scheme 5.3). For instance, apo-ACP could compete with malonyl-ACP for
binding sites only on KSβ; or with acetyl and/or malonyl-ACP for binding sites only on
KSα. Competition could also occur in both subunits, KSα and KSβ. In these three
situations, only competitive inhibition would be observed. The presence of an
uncompetitive component in the inhibition pattern would be evidence for an allosteric
relation between the catalysis of KSα and KSβ.
KS!/KS" + malonyl-ACP KS!/KS". malonyl-ACP polyketide production
apo-ACP
+
KS!/KS". apo-ACP
+
KS!/KS". apo-ACP. malonyl-ACP
KicKiu
+ malonyl-ACP polyketide production??(see discussion)
apo-ACP
Scheme 5.3 General mixed inhibition mechanism for apo-ACP as an inhibitor of KSα/KSβ
Act apo-ACP was produced by expression in E. coli and purified essentially as holo-ACP.
The yield was 30-40 mg L-1. When apo-ACP was incubated with malonyl CoA and
KSα/KSβ, a background rate of polyketide production was observed, which must be
attributed to the presence of minor amounts of holo-ACP formed by
phosphopantetheinylation of apo-ACP by E. coli ACPS, although the presence of holo-
ACP was not observed by ESMS (Figure 5.1). By comparing the reaction rate in assays
with apo-ACP, with our previous kinetic results using holo-ACP (Chapter 3), it was
estimated that 0.5-1% of the total ACP concentration was holo-ACP.
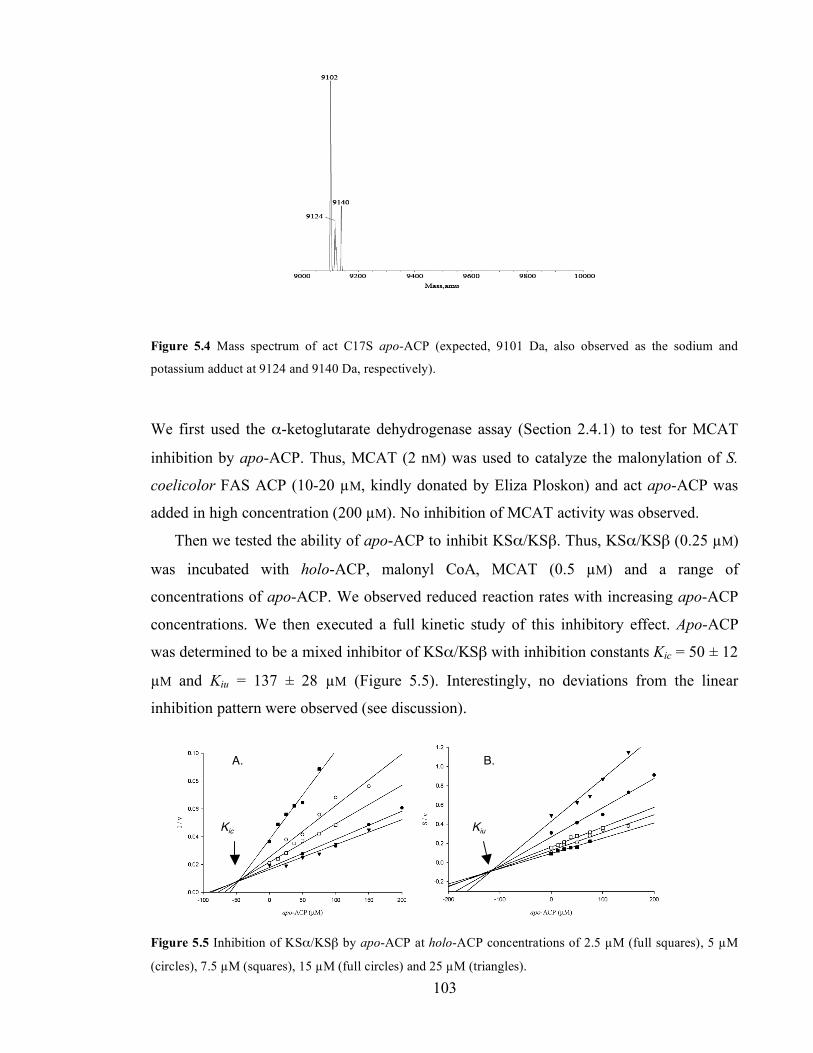
Chapter 3. Initiation of polyketide synthesis
103
Figure 5.4 Mass spectrum of act C17S apo-ACP (expected, 9101 Da, also observed as the sodium and
potassium adduct at 9124 and 9140 Da, respectively).
We first used the α-ketoglutarate dehydrogenase assay (Section 2.4.1) to test for MCAT
inhibition by apo-ACP. Thus, MCAT (2 nM) was used to catalyze the malonylation of S.
coelicolor FAS ACP (10-20 µM, kindly donated by Eliza Ploskon) and act apo-ACP was
added in high concentration (200 µM). No inhibition of MCAT activity was observed.
Then we tested the ability of apo-ACP to inhibit KSα/KSβ. Thus, KSα/KSβ (0.25 µM)
was incubated with holo-ACP, malonyl CoA, MCAT (0.5 µM) and a range of
concentrations of apo-ACP. We observed reduced reaction rates with increasing apo-ACP
concentrations. We then executed a full kinetic study of this inhibitory effect. Apo-ACP
was determined to be a mixed inhibitor of KSα/KSβ with inhibition constants Kic = 50 ± 12
µM and Kiu = 137 ± 28 µM (Figure 5.5). Interestingly, no deviations from the linear
inhibition pattern were observed (see discussion).
Figure 5.5 Inhibition of KSα/KSβ by apo-ACP at holo-ACP concentrations of 2.5 µM (full squares), 5 µM
(circles), 7.5 µM (squares), 15 µM (full circles) and 25 µM (triangles).
A. B.
Kic Kiu

Chapter 3. Initiation of polyketide synthesis
104
5.3. Discussion
The decarboxylation of malonyl-ACP by KSα/KSβ had previously been assessed by
qualitative ESMS analysis of the acetyl-ACP generated by mutant KSα and KSβ proteins
in which the corresponding cysteine (C169) and glutamine (Q161) active sites had been
mutated to alanine.62 The underlying assumption was that mutation of the respective active
sites completely abrogated the activity of KSα or KSβ towards decarboxylation of
malonyl-ACP, while leaving the activity of the other component unaffected. Here we
decided to test this hypothesis.
First, we studied the effect of ‘increasing’ KSβ concentration in the system by adding
AQ KSα/KSβ to PKS assays in which either CQ or CA KSα/KSβ was used. In Chapter 3
we showed that external addition of acetyl-ACP to act minimal PKS assays caused a 3-fold
increase in rate (kcat varied from 20.6 to 58.2 min-1), thus meaning that acetyl-ACP was the
limiting species in the presence of excess malonyl-ACP (Section 3.1.2). By adding AQ CQ
KSα/KSβ to the assays, we expected the same increase in rate due to AQ-catalyzed
production of acetyl-ACP, however, we only observed a 1.3-fold increase.
We then supplemented CA KSα/KSβ with AQ KSα/KSβ. The reaction rate under these
conditions corresponded to that observed under saturation of KSα/KSβ with external
acetyl-ACP. We expected that the addition of external acetyl-ACP (or AQ KSα/KSβ for
that matter) to assays using CA KSα/KSβ would surpass the lack of a functional KSβ and
thus an increase of 30-fold in the reaction rate would be observed, i.e. back to WT levels of
activity. Surprisingly, when extra acetyl-ACP (or AQ KSα/KSβ) was added to CA
KSα/KSβ assays, the increase in rate was only three-fold. This meant either that CA
KSα/KSβ was not active, or that the lack of a functional KSβ in the ketosynthase complex
somehow impaired the catalytic activity of CA KSα/KSβ.
Unexpectedly, addition of external acetyl-ACP to this assay (i.e. to minimal PKS assays
using CA KSα/KSβ and in the presence of saturating concentrations of AQ KSα/KSβ)
produced further increase in rate and kcat raised to 9.6 min-1. This was an important result,
as it suggested that CA KSα/KSβ was damaged at yet another stage than just the initial
decarboxylation of malonyl-ACP. This also showed that KSβ affected the rate of the
reactions catalyzed by KSα, which is in agreement with our results using the wild type
KSα/KSβ and mutant acyl carrier proteins (see discussion to Chapter 4), where we

Chapter 3. Initiation of polyketide synthesis
105
observed a dependence of the rate of KSα-catalyzed reactions on the rate of the initiation
step catalyzed by KSβ.
The interpretation of these results requires consideration of the quaternary structure of
KSα/KSβ. Let us suppose that the active KSα/KSβ species is the dimer of heterodimers,
(i.e. α2β2 tetramer), as suggested by the early work of Khosla53 and ongoing research in the
Bristol group (Scheme 5.1).120
In this case, the addition of AQ KSα/KSβ to PKS assays would affect the composition
of KSα/KSβ tetramers. Assuming the equilibrium constant K = 1, and for concentrations of
CQ and CA KSα/KSβ of 0.25 and 1 µM respectively, > 95% of the total enzyme capable of
sustaining polyketide production would be in complex with AQ at AQ KSα/KSβ
concentrations higher than 5 µM (Scheme 5.4).
Q
C
C
Q A
A
Q A
C
Q
+K = 1Q Q
C
C
A A
A
Q A
C
Q
+K = 1Q AA
2 x
2 x
Scheme 5.4. The addition of AQ affects the composition of A. CQ KSα/KSβ tetramers; B. CA KSα/KSβ
tetramers.
Our results could be explained if the active KSα/KSβ species is indeed tetrameric and, as
speculated in Chapter 4, the catalysis of KSα and KSβ is accompanied by a conformational
change. For example, in assays using CA KSα/KSβ, the species in solution would be
{CA|CA}. In the presence of AQ KSα/KSβ, the major species with PKS activity would be
{CA|AQ}, (Scheme 5.4). The species {CA|AQ} was found to be more active than
{CA|CA}: the presence of an active KSβ favoured the catalysis of the tetramer at
saturating concentrations of acetyl-ACP (i.e. when acetyl-ACP was externally supplied).
This implicated the presence of Q in the active site of KSβ in one dimer in improving the
catalytic behaviour of KSα in the other dimer.
A.
B.

Chapter 3. Initiation of polyketide synthesis
106
Thus, the generation of an active KSβ in the complex {CA|AQ} probably enhances the
catalysis of the tetramer by a different means than production of acetyl-ACP as a substrate
for KSα (because the system is saturated with acetyl-ACP). The active KSβ may prompt a
more favoured conformational state in its KSα counterpart. Thus a conformational change
in an active KSβ subsequently would affect the whole structure of the tetramer.
Furthermore, this is consistent with our observations on the supplementation of CA
KSα/KSβ complex with external acetyl-ACP, when we found that the reaction rate
increased only 3-fold, and not 30-fold to reach WT levels. If the role of KSβ were only the
decarboxylation of malonyl-ACP to acetyl-ACP, then CA KSα/KSβ should perform at ~ 60
min-1 in the presence of excess acetyl-ACP. However, the 10-fold decrease in
decarboxylation activity (kcat changes from 20.6 to 2.2 min-1) of KSα/KSβ due to the
substitution of the KSβ active site glutamine for alanine probably renders a ‘static’ KSβ,
which lacks the conformational flexibility that facilitates the catalysis of KSα. As a result,
saturating concentrations of acetyl-ACP produced an increase in kcat from 2.2 only to 6.4
min-1 (far from WT levels at 60 min-1).
In the presence of excess malonyl-ACP, production of acetyl-ACP by AQ KSα/KSβ should
depend linearly on the concentration of AQ KSα/KSβ. A re-plot of the data in Figures 5.1
and 5.3 shows a linear increase in polyketide production reaction rate with AQ KSα/KSβ
concentration at AQ KSα/KSβ concentrations lower than 2 µM (Figure 5.6). This is most
likely due to increasing acetyl-ACP concentrations. However, at AQ KSα/KSβ
concentrations higher than 2 µM, the linear relationship is not maintained.
We think this is due to lack of decarboxylation activity of the AQ KSα/KSβ complex
when compared to the wild type. As KSα in AQ KSα/KSβ complexes is not active, it also
lacks the conformational flexibility which is necessary for an efficient catalytic activity of
the tetramer. It may be possible, for instance, that the absence of a conformational change
in KSβ leads to product inhibition. In this case, acetyl-ACP (the product of the reaction
catalyzed by KSβ) may decrease the activity of KSβ, when acetyl-ACP concentrations
reach certain threshold. This would render inactive AQ KSα/KSβ complexes. This may
explain why, for example, in assays using the wild type CQ KSα/KSβ, the increase in rate
due to addition of AQ KSα/KSβ was only 1.3-fold, instead of 3-fold to reach the level of
activity when CQ KSα/KSβ is saturated with acetyl-ACP.

Chapter 3. Initiation of polyketide synthesis
107
Figure 5.6 Re-plot of data in Figures 5.1 and 5.3.Titration of AQ in assays performed with WT KSα/KSβ
(blue, 0.25 µM concentration) and CA (red, 1 µM concentration).
5.3.1. Stoichiometry of ACP: KSα /KSβ complexes
Inhibition of FAS and PKS by inactive forms of ACP has previously been reported. For
example, the group of Cronan reported a decrease in cell growth when FAS apo-ACP was
overexpressed in E. coli. They further showed that apo-ACP inhibited the enzyme sn-
glycerol-3-phosphate acyltransferase in vitro, resulting in the inability to transfer the mature
fatty acid to sn-glycerol-3-phosphate.194 Another example of ACP inhibition of a
ketosynthase was observed in the R1128 PKS system. The apo form of ACP was reported
to inhibit the priming ketosynthase, ZhuH.13 Also, the Stanford group reported that act apo-
ACP was a competitive inhibitor of KSα/KSβ with a Kic = 13 µM.53 However, this study
was done using holo-ACP from the frenolicin PKS. Furthermore, it was not ruled out that
the inhibitory effect observed was in fact due to apo-ACP inhibition of MCAT rather than
of KSα/KSβ.
The study of the inhibition of MCAT and KSα/KSβ by apo-ACP (Section 5.2) showed that
apo-ACP did not inhibit MCAT, but was a mixed inhibitor of KSα/KSβ. This suggested
that there are two different binding sites for ACP in the KSα/KSβ complex. In effect, the
general scheme for mixed inhibition (Scheme 5.3) envisages an uncompetitive inhibition

Chapter 3. Initiation of polyketide synthesis
108
component due to the binding of the inhibitor to the enzyme-substrate complex. As
KSα/KSβ binds malonyl ACP for two different reactions (initiation and extension) it
seemed likely that the observed pattern of inhibition represented apo-ACP binding at the
extension (i.e. KSα) and initiation (i.e. KSβ) sites. Kic for inhibition by apo-ACP was 50
µM, a factor of 20-fold higher than KM for malonyl ACP as a substrate (2.39 µM). This
difference suggested that KSα/KSβ recognizes malonyl-ACP preferentially, meaning either
a specific binding interaction to malonyl-phosphopantetheine, or that malonyl-ACP takes
up a different conformation to apo-ACP. The latter idea is not supported by structural
studies but it may still be valid because CoA, which possesses the same
phosphopantetheine moiety as holo-ACP, does not appear to inhibit this step (see Section
2.5 in Chapter 2).
The inhibition behaviour shown in Figure 5.5 corresponds to the classical pattern of a
mixed inhibitor: linear relationships between the inverse of the reaction rate and the
inhibitor concentration. Within the range of concentrations studied, no deviations from this
linear behaviour were observed. This can also be useful for discussion and interpretation of
results.
Cooperativity is a property of multimeric enzymes that results from ‘cooperation’
between different active sites. It can either be ‘positive’, when substrate binding on one
active site increases the activity of neighbouring sites (i.e. it increases the affinity of the
enzyme towards the substrate); or ‘negative’, when the activity of neighbouring sites is
decreased upon substrate binding to one site. Cooperative effects can be detected from
inhibitions studies. They usually appear as deviations from the linear behaviour in the
inhibition plots described above.
Many enzymes that show allosteric regulation are also cooperative. We have
hypothesized in Chapters 4 and 5 that a conformational change in KSβ produces an
allosteric modification in KSα. If this was essential for efficient PKS activity of KSα/KSβ,
then cooperative effects between the active sites of KSα and KSβ could also occur. For
example, if binding of apo-ACP onto KSβ produced a change in KSα conformation, then a
change in the affinity of KSα towards ACP could be expected. However, the study of
inhibition of KSα/KSβ by apo-ACP did not support this idea because only linear
relationships between the inverse of the rate and the inhibitor concentration were observed.

Chapter 3. Initiation of polyketide synthesis
109
This suggests that the conformational change that renders active KSα occurs as a result of
malonyl-ACP decarboxylation, rather than upon substrate binding onto KSβ.
Deviations from the linear behaviour in inhibition patterns are not only a sign of
cooperative effects. They also point towards hyperbolic inhibition.94 In Scheme 5.3, the
intermediate {KSα/KSβ. apo-ACP. malonyl-ACP} may or may not be a dead-end species.
If this species were still capable of producing polyketides, then apo-ACP would be a
hyperbolic inhibitor of KSα/KSβ and the linear relationships between the inverse rate and
inhibitor concentration would not be maintained.94
Assuming that KSα and KSβ have indeed two different binding sites for ACP, then
there are two possibilities to yield such {KSα/KSβ. apo-ACP. malonyl-ACP} species: first,
that apo-ACP binds to KSα; second, that apo-ACP binds to KSβ. Interestingly, we only
observed linear relationships between rate and inhibitor concentration. These results
suggest that both possibilities render inactive KSα/KSβ complexes, incapable of polyketide
synthesis. It is clear that if apo-ACP binds KSα, then PKS activity is inhibited. It appears
that polyketide synthesis is also inhibited if apo-ACP binds KSβ. Again, this could support
our hypothesis of the conformational change: if apo-ACP binds KSβ, it may prompt a
conformational flip in KSβ that probably renders a KSα/KSβ complex ready for
decarboxylation of malonyl-ACP, rather than for polyketide synthesis.
Future work on the study of ACP: KSα/KSβ complexes should investigate whether or not
KSα and KSβ have indeed two different binding sites for ACP. A simple experiment could
illustrate whether apo-ACP inhibits KSα, KSβ or both: inhibition assays could be
performed in the presence of excess acetyl-ACP. If no decrease in rate could be detected,
then it would be clear that apo-ACP does not inhibit the reaction catalyzed by KSα. The
opposite would hold if polyketide production rates decreased with increasing apo-ACP
concentration. Comparison of these experiments with the assays performed in this work
could identify which ketosynthase subunit is inhibited by apo-ACP.

Chapter 3. Initiation of polyketide synthesis
110
6. Kinetics of an extended act minimal PKS
In preceding chapters the production of the octaketides SEK4 19 and SEK4b 27 in vitro by
the actinorhodin minimal polyketide synthase from malonyl CoA was studied. Although
WT S. coelicolor A3 (2) does indeed produce some SEK4 and SEK4b (Dr. Greg Challis,
personal communication), it is generally assumed that, in vivo, the hypothetical octaketide
intermediate 18 is mostly processed to the final product actinorhodin 9 by the remaining
components of the act polyketide synthase. Early modifications of 18 include
ketoreduction, aromatization and cyclization (see Scheme 1.3 in Chapter 1).70
Identification of the gene product responsible for ketoreductase activity in the act cluster
was initially approached by in vivo experiments. Bartel, Keller and collaborators used S.
galilaeus 31671, which produces the polyketide 9-hydroxy-aklavinone 67 from a
propionate starter unit and 10 malonate extender units, as a host for genetic
manipulation.196 Transformation of S. galilaeus 31671 with a plasmid that contained the
actIII gene resulted in production of 68 (Scheme 6.1). This suggested that actIII encoded a
ketoreductase that catalyzed the reduction of C-9 of the hypothetical intermediate 69,
resulting in the formation of a secondary alcohol (the stereochemistry of the act KR-
catalyzed reduction has not been determined to date).32
O O O
O
O O O O
COOH
O
O O O
HO
O O O O
COOH
O99
OCOCH3
OH
OHOH
O
OOH
HO
OCOCH3
OH
OHOH
O
OOH
Scheme 6.1 Complementation of S. galilaeus with act KR resulted in production of a reduced polyketide (the
propionate starter unit is shown in red).196
act KR
S. galilaeus S. galilaeus
67 68
69

Chapter 3. Initiation of polyketide synthesis
111
In vivo experiments with S. coelicolor A3(2) mutants also suggested that the necessary KR
activity was provided by actIII. Using an actVII S. coelicolor mutant which was able to
produce polyketides (but not actinorhodin), Zhang, He and co-workers isolated and
characterized mutactin 20.197 Mutactin was hypothesized to arise from reduction of C-9 of
the intermediate 18. The corresponding reduced octaketide 70 then cyclised to form
mutactin 20, which exhibited the same cyclisation pattern as 19 and indeed actinorhodin 9
in the formation of the first ring between the carbons C-7 and C-12 (Scheme 6.2).
S-ACP
OOOO
O
O OO
HO
O
O
O
OH
O
OHO
O
OH
OH
HO
O
O
OHO
O
OH
HO
O
S-ACP
OOOO
HO
O OO
99
7
7
12
12 12
12
7
10
10
15
15
7
Scheme 6.2 Effect of addition of KR to act minimal PKS in in vivo experiments.
Further in vivo studies using S. coelicolor CH999 as the host for transformation with the
pRM5 vector (see Section 1.2.2.1) showed that, in the absence of KR, a second product of
the act minimal PKS was SEK4b 27, which originated by an alternative cyclisation of the
octaketide backbone 18, i.e. between carbons C-10 and C-15.198 As mutactin 20 was the
major product when KR was co-expressed with the act minimal PKS,199 a second role for
KR was assumed to be the control of the regiospecificity of the first cyclisation.200
All Type II PKS-derived polyketides that undergo ketoreduction are only reduced at the
ninth carbon from the carboxy terminus of the assembled polyketide. This strict
regiochemistry is independent from polyketide chain length (octaketide to decaketide) and
the nature of the starter unit.200 In vivo experiments could not explain this regio-
especificity, nor could they identify the substrate for act KR: was it the full-length
18 70
19 27 20
KR
Cyclisation Cyclisation

Chapter 3. Initiation of polyketide synthesis
112
octaketide 18 (as assumed above)196 or an earlier intermediate? Furthermore, if KR used an
octaketide as a substrate, which was the timing of the first cyclization (i.e. before or after
reduction)?
The role of KR in directing the first cyclization was evidenced by work by Kunnari,
Hilionko et al, using S. galilaeus 31365 mutants for metabolite analysis studies.201 S.
galilaeus 31365 produces aklavinone 71 (which shows reduction at C-9, and C-7 to C-12
cyclization pattern analogous to mutactin) from a propionate starter unit and 9 malonate
extender units. The mutant S. galilaeus 31365 HO61 produces 72, which results from a
C10-C15 first cyclization (Scheme 6.3), analogous to that of SEK4b. When this mutant was
transformed with a plasmid carrying a gene capable of providing KR activity, formation of
72 was abrogated, and production of aklavinone 71 was recovered (Scheme 6.3).
Since the ketoreduction changes the hybridization of the substrate carbonyl (from sp2 to
sp3), it introduces a defined bend of the poly-β-keto intermediate chain. Thus, Kunnari,
Hilionko et al speculated that reduction occurred when the polyketide chain was complete,
but before the first cyclization. In this model, the chain, once reduced at C-9, takes up a
favoured orientation for the subsequent aldol condensation between C-7 and C-12.201
Alternatively, one could argue that KR may prompt a conformational change in the PKS
complex, rendering a more rigid struture that prevents the C-10 to C-15 cyclization.
O O O
O
O O O O
COOHO
O O O
O
O O O O
COOH
O9
10
OH
O
OOH
HO
OCOCH3
OH
OHOH
O
OOH
7
12
12
7
O
OH
O10
15
15
72 71
Scheme 6.3 Production of 72 and 71 by S. galilaeus HO61 and S. galilaeus HO61 complemented with KR,
respectively. The propionate starter unit is shown in red.
S. galilaeus HO61
S. galilaeus HO61 + KR

Chapter 3. Initiation of polyketide synthesis
113
An alternative model, in which the first cyclization occurs prior to reduction was proposed
by Korman, Tsai and co-workers, who constructed a series of in silico models of substrate
binding to act KR, and speculated that the first cyclisation must occur prior to interaction of
the polyketide with KR.202 This model has been recently supported by experimental
evidence. The group of Tsai studied the activity of act KR in vitro with a series of possible
substrates, 74 to 81.203 They could not detect activity of KR towards the products 77 to 81,
or indeed towards acetyl CoA or acetoacetyl CoA. On the other hand, the bicyclic
compounds 74, 75 and 76 were used by KR as a substrate with unequal specificity (Table
6.1).
HO
H
H
H
OO
O
O
O
OO
O
O
O
O
O
O
O
O
79 80 81
Substrate kcat (s-1) KM (mM) kcat / KM (s-1 mM-1)
74 2.6 0.79 3.2 75 0.16 3.9 0.04 76 0.073 5.1 0.013
Table 6.1 Kinetic parameters for the reduction of unnatural substrates by KR.203
Overall, it was clear that, within the range of compounds studied, act KR showed a
preference for bicyclic substrates. This lead to Korman, Tsai and co-workers to speculate
that the most likely substrate for KR was a hypothetical intermediate 82, where the first
cyclization (C7 to C-12 to form 83) and the second cyclization (C-6 to C-15 to form 82)
have already occurred (Scheme 6.4). The product of KR, a hypothetical intermediate 84,
would spontaneously cyclize to afford mutactin 20.
74 75 76 77 78

Chapter 3. Initiation of polyketide synthesis
114
S-ACP
OOOO
O
O OO
O
O
OH
OH
HO
O
S-ACP
OOO
O
O OO
99
7
12 12
12
7
7
9
O O
O
O
O
12
79
OHOH
OS-ACP
O O
O
HO
O
12
79
OHOH
OS-ACP
6 6
66
6
15 15
151515
Scheme 6.4 Model proposed by the group of Tsai, in which the substrate for act KR is the bicyclic
intermediate 82.203
The model proposed by the group of Tsai has been challenged by Kalaitzis and Moore, who
heterologously expressed the actinorhodin minimal PKS genes, together with actIII (KR),
actVII (ARO) and actIV (CYC) (see Figure 1.6 in Chapter 1), in S. lividans.204 Two new
hexaketides BSM1 85 and BSM3 86 were characterized (Scheme 6.5) which arose from
partially completed polyketide structures, in which the ketoreduction had already taken
place.
Kalaitzis and Moore then proposed an alternative pathway for the biosynthesis of
polyketides, where ketoreduction occurs during the elongation of the polyketide chain
rather than after the assembly of the full length octaketide (Scheme 6.5).204 In this model,
the substrate for KR would be the pentaketide 87. After reduction at the C-3, the reduced
pentaketide 88 would be extended with a further malonate unit to the hexaketide 89. And
aldol condensation between C-3 and C-8 then would lead to 85 and 86.
The detection of BSM1 and BSM3 is unique in all Type II PKS studies in that it
describes the isolation of partially complete polyketides. This has never been reported in in
vitro assays. In fact, Moore left open the possibility that these hexaketides may be
degradation products resulting from the catabolism of S. lividans.
C-7 to C-12 cyclization
C-6 to C-15 cyclization
KR spontaneous
83
82 84 20
18

Chapter 3. Initiation of polyketide synthesis
115
O
COOH
O
OH
O
COOH
O
S-ACP
O
O
O OO
3
1
65
CoA-S
O
OH
O
5 x
S-ACP
O
HO
O OO
3
1
65
O
HO
O OO
5
3
86
O
S-ACP
malonyl CoA
CoA
3
8
H2O
Scheme 6.5 Proposed biosynthesis of BSM1 and BSM3.204
Another example where ketoreduction was proposed to operate before the completion of
the polyketide chain was that of the enterocin (enc) 17 PKS. Hertweck, Moore and
collaborators expressed the enc ACP, KSα and KSβ in S. lividans in the presence of
benzoyl CoA. No polyketides could be detected.205 On the other hand, when KR was co-
expressed with ACP, KSα and KSβ, wailupemycins D-G 90-93 were produced (Scheme
6.6). It thus appeared that the corresponding enc minimal PKS would need the inclusion of
a fourth component, i.e. KR.
Indeed, polyketide production by enc ACP, KSα and KSβ was not recovered either by
complementation with act KR or with a mutant enc KR in which the catalytic serine active
site had been mutated to alanine.205 Thus it was clear, first, that KR was an essential
component of the minimal enc PKS and exerted a catalytic, rather than structural, role.
Second, that ketoreduction must occur during polyketide elongation. Although Hertweck,
Moore and co-workers proposed that the likely substrate for KR could be an hypothetical
pentaketide (Scheme 6.6), no evidence for this could be gathered –indeed, absolutely no
truncated polyketides could be observed. Therefore, if KR was not present, either the
85 86
89 88
87 KR
Act Min PKS
Act Min PKS
Chapter 3. Initiation of polyketide synthesis
116
hypothetic polyketides resulting from 94 (or other intermediate) were not released from the
PKS, or they were simply catabolyzed by the host.
S-ACP
O
HO
Ph
O OO
CoA-S
O
OH
O
4 x
O
HO
Ph
O OO
O
S-CoA
O
S-ACP
OOO
O
OH
O
OH
HO
O
OO
OH
O
O
OH
R2
OH
HO
OH
O
O
OH
Ph
OH
HO
O
O
O
OH
Ph
OH
HO
OH
R1
Scheme 6.6 Biosynthesis of wailupemycin D-G by the enc minimal PKS. Wailupemycin D: R1 = OH, R2 =
Ph; Wailupemycin E: R1 = Ph, R2 = OH.205
In summary, no conclusive evidence has been found to date that unequivocally identifies
the substrate of act KR, the timing of the first and second cyclizations of the polyketide
chain, or how KR interacts with the rest of the components of the act PKS. This is
especially true in in vitro systems. Given the quantitative assay to measure reaction rates
which was developed in this work, we decided to adapt the method to measure mutactin
production by an extended act minimal PKS consisting of ACP, KSα/KSβ and KR. The
aim of this Chapter is therefore to gain insight into the catalytic properties of KR.
In the following sections, the structure and chemical mechanism of KR, as well as
seminal work by the Stanford group on the kinetics of the ketoreduction, are described.
Then, our results will be presented.
17
Enc ACP, KSα/KSβ and KR
Min enc PKS + 3 x malonyl CoA
Min enc PKS
Spontaneous
94
90, 91 D, E 92
F 93 G

Chapter 3. Initiation of polyketide synthesis
117
6.1. Structure and chemical mechanism of KR
The first three-dimensional structure of a Type II PKS reductase, the actinorhodin KR, was
determined at 2.5 Å resolution by the Bristol group.32 The quaternary structure of act KR
was investigated by native PAGE. In the absence of NADPH, the enzyme appeared to exist
in different oligomeric forms (i.e. tetramer, dimer and monomer).206 Addition of NADPH
(or indeed NADP+) resulted in only one protein band corresponding to the expected
tetrameric molecular weigh.206 Crystallization of KR was therefore done in the presence of
NADP+ to ensure an active conformation of the enzyme.
KR belongs to the short-chain dehydrogenases/ reductases (SDR) protein family. These
are relatively small (250-350 residues) enzymes that show medium (15-30%) sequence
identity and display highly similar folding patterns, with a central β-sheet core flanked by α
helices.207 The Asn-Ser-Tyr-Lys catalytic tetrad of the SDR family is conserved in act KR
(Figure 6.1).
Figure 6.1 Structure of a monomer of act KR, showing the β-sheet core (blue) and the catalytic residues
N114, S144, Y157 and K161 (yellow). PDB file: 1W4Z.
Reduction of the ketone substrate is accompanied by a proton relay mechanism, which is
similar to that in other enzymes of the SDR family.202 Following hydride transfer from
NADPH to the substrate carbonyl, the alkoxide is stabilized by the oxyanion hole formed
by S144 and Y157 (Scheme 6.7), and the proton from the hydroxyl group of Y157 is

Chapter 3. Initiation of polyketide synthesis
118
transferred to the carbonyl oxygen. A proton relay then occurs to account for the proton
extracted from Y157, sequentially involving the hydroxyl group of the NADPH ribose, the
amino group of K161 and four water molecules.
Y157
OH
O
OO
R NH
H
H
K161
NH H
R'
R''O
OH
S144
H
O
HOH
H
O
H
H
O
H
H
O
H
N114
Scheme 6.7 Proton relay mechanism in the act KR active site (blue arrows) showing NADPH (green), the
polyketide substrate (red) and the active site residues (black).202
6.2. Kinetics of extended Type II minimal PKS.
The Stanford group studied the production of polyketides when the act 9 and tcm 30
minimal PKS were complemented with auxiliary PKS enzymes such as act KR, gris ARO
and tcm ARO.11 The tcm minimal PKS assembles a decaketide from malonyl CoA, which
in the absence of further tailoring enzymes spontaneously cyclises to form mostly SEK15
95 and SEK15b 96, as well as RM80 97. Kinetic studies were performed using a
discontinuous, radioactive thin layer chromatography assay. The kcat value for total
decaketide production by the tcm minimal PKS was determined as 1.40 ± 0.25 min-1,11
substantially higher than kcat for the act minimal PKS (kcat = 0.31 ± 0.11 min-1).56
Addition of tcm ARO resulted in a relative increase in production of 97 with respect to
95 and 96 (Scheme 6.8). Furthermore, production of polyketides was enhanced by the
presence of ARO, and an increase in kcat to 2.55 ± 0.31 min-1 was measured by HPLC
analysis of products. This agreed with early work by Shen and Hutchinson which indicated
that the presence of further PKS enzymes also enhanced the rate of polyketide
production.188

Chapter 3. Initiation of polyketide synthesis
119
O
OH
HO
O OH
OH
O OH
O
OH
HO
O OH
O
O OH
OH
HO
O
O
O OH
OH
O
10 x malonyl CoA
OH
CH3OHO
O
H3CO
O
OHOH
OO
OCH3
CH3
Scheme 6.8 Biosynthesis of decaketides by tcm PKS
In the actinorhodin PKS, addition of KR to minimal assays results in production of
mutactin 20 (Scheme 1.13). Further supplementation with actVII (an aromatase) affords
SEK34 21. When the corresponding aromatase from the griseusin PKS substituted actVII,
SEK34 21 was still produced.208
The Stanford group then assessed the production of mutactin and SEK34 by HPLC.
They found that inclusion of act KR in act minimal assays resulted in a decrease in the rate
of polyketide production (kcat = 0.11 ± 0.02 min-1),208 when compared with previous kinetic
data for the act minimal PKS (kcat = 0.31 ± 0.11 min-1)56
When gris ARO was added to KR-extended act minimal PKS assays, an increase in rate
was measured (kcat = 0.44 ± 0.04 min-1). Obviously this meant that the rate of ketoreduction
had been increased at least by a factor of four due to the presence of gris ARO, which they
attributed to KR-ARO channelling of polyketide intermediates.
6.3. Kinetic analysis of production of mutactin by an extended act minimal PKS
The actinorhodin his6-KR was expressed in E. coli and purified by nickel affinity
chromatography as described previously in the literature.32 6 mg pure KR per litre of
SEK15 95 SEK15b 96
RM80 97 30
Tcm min PKS
Tcm min PKS + ARO
Tcm PKS

Chapter 3. Initiation of polyketide synthesis
120
culture were obtained. ESMS analysis of the enzyme showed the peak corresponding to KR
and a peak probably corresponding to the α-N-6-phosphogluconoylation of the His-tag
(Figure 6.2).209
Figure 6.2 Mass Spectrum of his6-KR (expected: 29291 Da, observed: 29299 Da) and α-N-6-
phosphogluconoylation of the His-tag (expected: 29467 Da, observed: 29477 Da).
Mutactin was purified from in vitro assays. 0.6 mg of mutactin were obtained by HPLC
separation of polyketides produced in the assays. This sample was further analyzed by
LC/MS (Figure 6.3). We used this standard for calibration of the HPLC system and for the
calculation of the extinction coefficient of mutactin at 293 nm (ε = 6,700 M-1 cm-1) for
kinetic purposes.
Production of mutactin by the act minimal PKS in the presence of KR was reconstituted
following the procedure of Zawada and Khosla.208 First, we studied the dependence of the
ratio mutactin vs. total polyketide production at a range of KR concentrations. Thus, holo-
ACP, KSα/KSβ and malonyl CoA were incubated with increasing concentrations of KR
and production of polyketides after 2 h assay was assessed by HPLC analysis, by
measuring the absorbance of the samples at 280 nm. In agreement with previous reports,208
we needed excess KR to favour the production of mutactin over SEK4/4b (Figure 6.4).
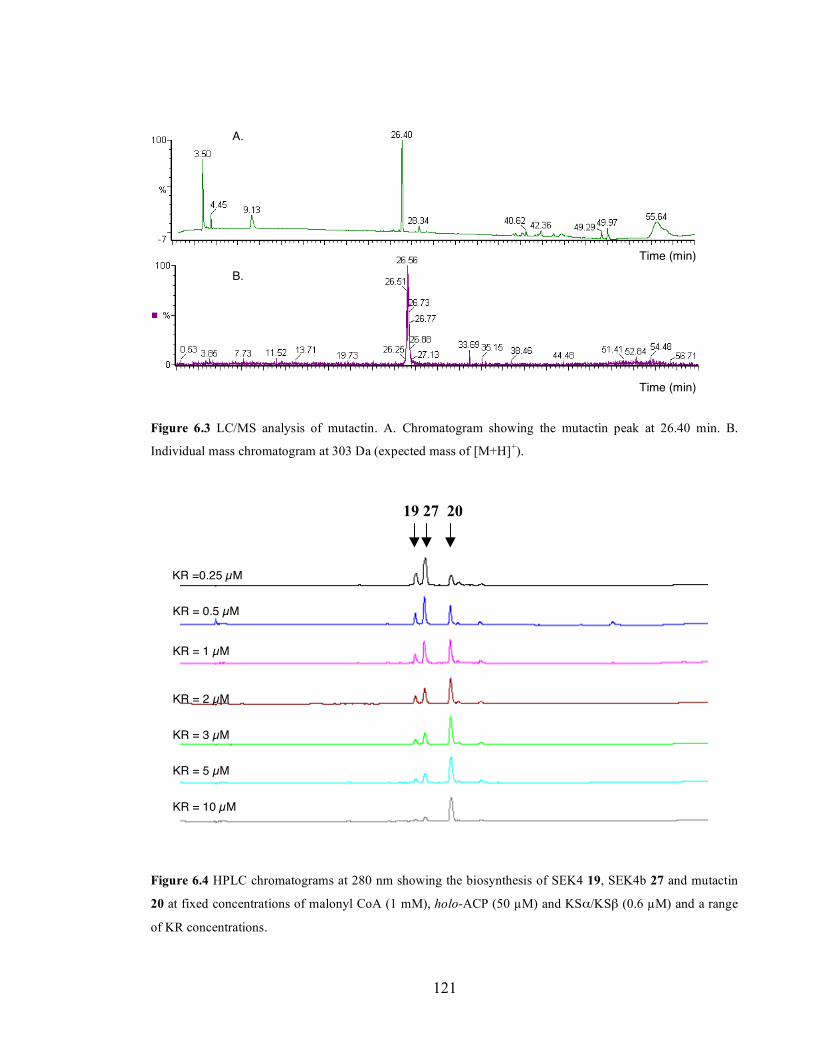
Chapter 3. Initiation of polyketide synthesis
121
Figure 6.3 LC/MS analysis of mutactin. A. Chromatogram showing the mutactin peak at 26.40 min. B.
Individual mass chromatogram at 303 Da (expected mass of [M+H]+).
19 27 20
Figure 6.4 HPLC chromatograms at 280 nm showing the biosynthesis of SEK4 19, SEK4b 27 and mutactin
20 at fixed concentrations of malonyl CoA (1 mM), holo-ACP (50 µM) and KSα/KSβ (0.6 µM) and a range
of KR concentrations.
A.
B.
KR =0.25 µM
KR = 0.5 µM
KR = 1 µM
KR = 2 µM
KR = 3 µM
KR = 5 µM
KR = 10 µM
Time (min)
Time (min)
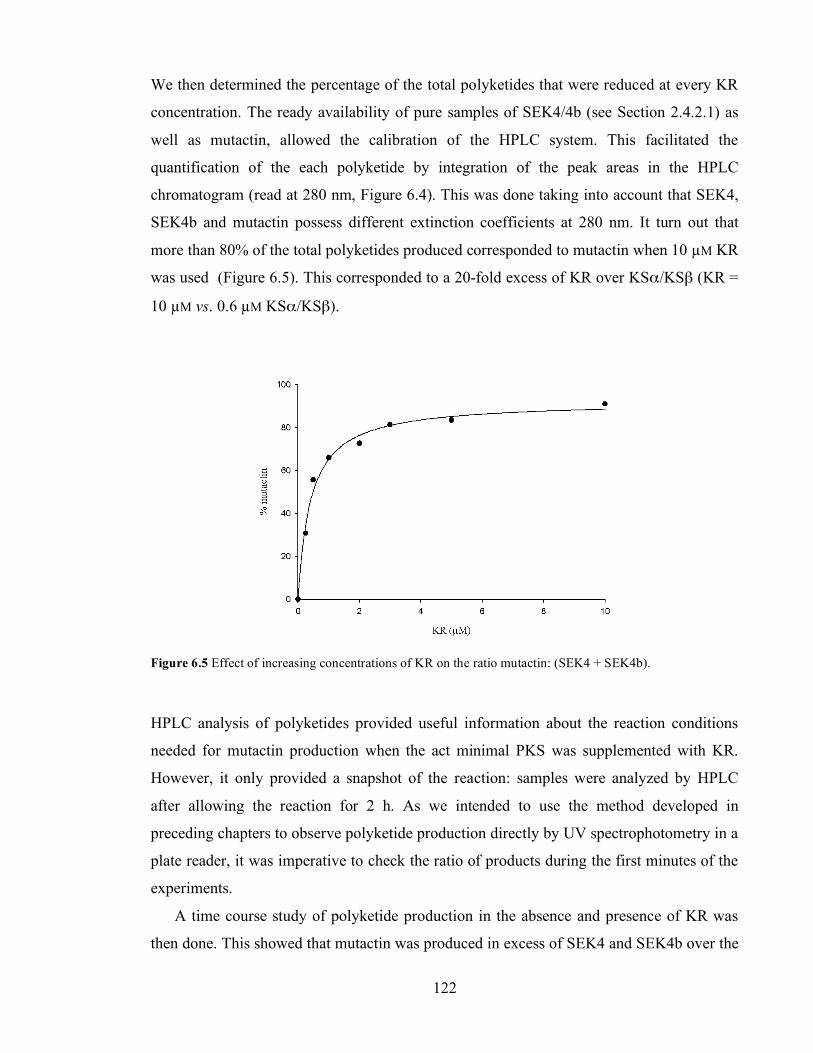
Chapter 3. Initiation of polyketide synthesis
122
We then determined the percentage of the total polyketides that were reduced at every KR
concentration. The ready availability of pure samples of SEK4/4b (see Section 2.4.2.1) as
well as mutactin, allowed the calibration of the HPLC system. This facilitated the
quantification of the each polyketide by integration of the peak areas in the HPLC
chromatogram (read at 280 nm, Figure 6.4). This was done taking into account that SEK4,
SEK4b and mutactin possess different extinction coefficients at 280 nm. It turn out that
more than 80% of the total polyketides produced corresponded to mutactin when 10 µM KR
was used (Figure 6.5). This corresponded to a 20-fold excess of KR over KSα/KSβ (KR =
10 µM vs. 0.6 µM KSα/KSβ).
Figure 6.5 Effect of increasing concentrations of KR on the ratio mutactin: (SEK4 + SEK4b).
HPLC analysis of polyketides provided useful information about the reaction conditions
needed for mutactin production when the act minimal PKS was supplemented with KR.
However, it only provided a snapshot of the reaction: samples were analyzed by HPLC
after allowing the reaction for 2 h. As we intended to use the method developed in
preceding chapters to observe polyketide production directly by UV spectrophotometry in a
plate reader, it was imperative to check the ratio of products during the first minutes of the
experiments.
A time course study of polyketide production in the absence and presence of KR was
then done. This showed that mutactin was produced in excess of SEK4 and SEK4b over the

Chapter 3. Initiation of polyketide synthesis
123
whole period of study (Figure 6.6). Interestingly, the ratio SEK4/SEK4b changed over
time; but the pattern was the same in both experiments.
19 27 19 27 20
Figure 6.6. HPLC chromatograms at 280 nm of the time course study of polyketide production (SEK4 19,
SEK4b 27 and mutactin 20) in the absence (A) and presence (B) of KR (10 µM).
6.3.1. Self-malonylation of ACP in the presence of KR
Two continuous assays are described in Chapter 2 to measure the rate of self-malonylation
of ACP. In principle, we could use the α-ketoglutarate dehydrogenase assay in the presence
of a mutant KSα/KSβ which is incapable of polyketide synthesis (Section 2.4.2.2) and
measure the rate of self-malonylation in the presence of both KSα/KSβ and KR. However,
KR needs NADPH (or at least NADP+), to be in the active, tetrameric conformation206 and
this complicated the use of α-ketoglutarate dehydrogenase, which itself uses NAD+ as a
substrate, as a coupling enzyme.
We then decided to use KSα/KSβ at high concentrations (3 µM) to couple the
production of polyketides to the formation of malonyl-ACP from holo-ACP and malonyl
CoA. First, we tested whether an increase in the concentration of KSα/KSβ affected the
ratio mutactin: SEK4/4b determined in Figure 6.7. Therefore the dependence of the ratio
mutactin vs. (SEK4+SEK4b) on the ratio KSα/KSβ: KR at a fixed concentration of KR was
B.
15 min
30 min
60 min
120 min
15 min
30 min
60 min
120 min
A.

Chapter 3. Initiation of polyketide synthesis
124
assessed. Thus KR was fixed to 10 µM, and nine different concentrations of KSα/KSβ
between 0.3 and 3 µM were studied. After allowing reaction for 2 h, polyketides were
analyzed by HPLC. Integration of the peak areas and comparison with standards showed
that mutactin accounted for 81 ± 1 % of the total polyketides produced at all KSα/KSβ
concentrations (Figure 6.7).
This experiment suggested that the ratio KSα/KSβ: KR was unimportant to determine
the production of mutactin, when compared to SEK4 and SEK4b. However, the absolute
KR concentration was an essential variable to achieve an excess of mutactin, as shown in
Figure 6.4. Therefore the hypothesis that KR interacts with ACP, as proposed by the Bristol
and Tsai groups,32,202 might be valid.
Figure 6.7 HPLC chromatograms at 280 nm showing polyketide production (SEK4 19, SEK4b 27 and
mutactin 20) upon titration of KSα/KSβ in extended minimal PKS assays at fixed KR concentration (10 µM).
The study of the self-malonylation of act holo-ACP by the KSα/KSβ coupling system in
the presence of KR rendered kinetic parameters kcat = 2.41 ± 0.22 min-1 and KM = 226 ± 51
µM (Figure 6.8), which are in excellent agreement with those determined in the absence of
KR (Section 2.4.2.2).
19 27 20
0.3 µM
0.5 µM
0.75 µM
1 µM
1.25 µM
1.5 µM
2 µM
2.5 µM
3 µM
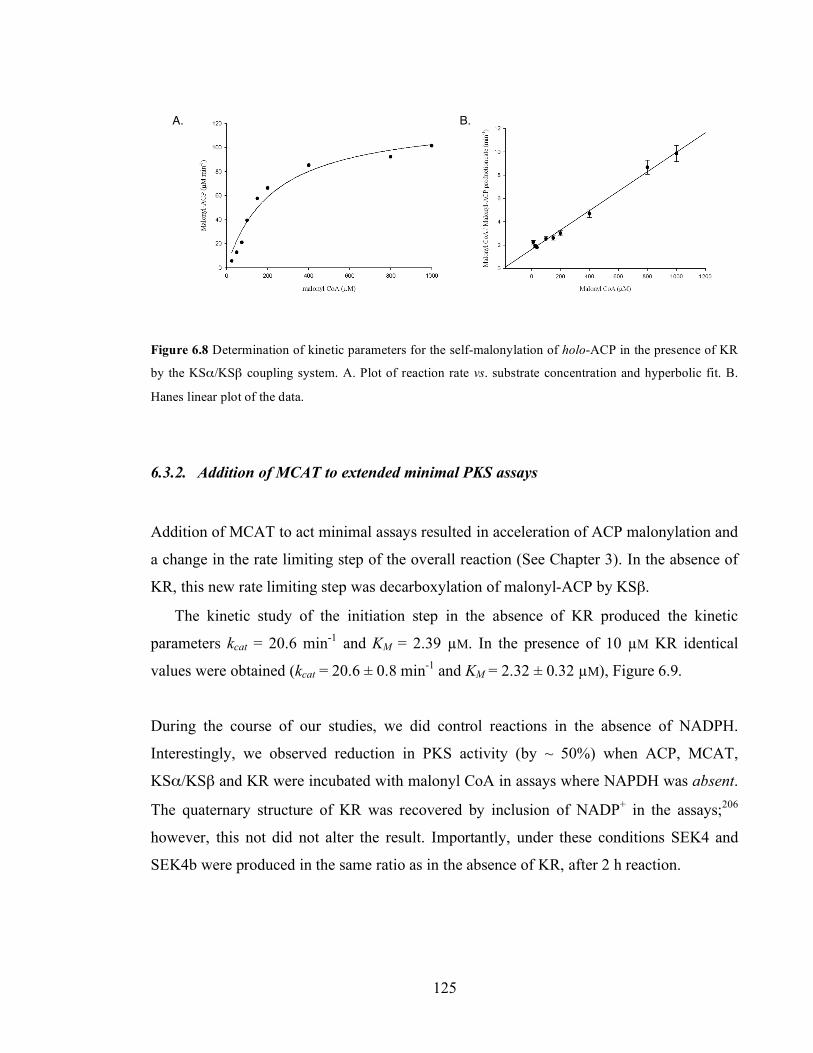
Chapter 3. Initiation of polyketide synthesis
125
Figure 6.8 Determination of kinetic parameters for the self-malonylation of holo-ACP in the presence of KR
by the KSα/KSβ coupling system. A. Plot of reaction rate vs. substrate concentration and hyperbolic fit. B.
Hanes linear plot of the data.
6.3.2. Addition of MCAT to extended minimal PKS assays
Addition of MCAT to act minimal assays resulted in acceleration of ACP malonylation and
a change in the rate limiting step of the overall reaction (See Chapter 3). In the absence of
KR, this new rate limiting step was decarboxylation of malonyl-ACP by KSβ.
The kinetic study of the initiation step in the absence of KR produced the kinetic
parameters kcat = 20.6 min-1 and KM = 2.39 µM. In the presence of 10 µM KR identical
values were obtained (kcat = 20.6 ± 0.8 min-1 and KM = 2.32 ± 0.32 µM), Figure 6.9.
During the course of our studies, we did control reactions in the absence of NADPH.
Interestingly, we observed reduction in PKS activity (by ~ 50%) when ACP, MCAT,
KSα/KSβ and KR were incubated with malonyl CoA in assays where NAPDH was absent.
The quaternary structure of KR was recovered by inclusion of NADP+ in the assays;206
however, this not did not alter the result. Importantly, under these conditions SEK4 and
SEK4b were produced in the same ratio as in the absence of KR, after 2 h reaction.
A. B.

Chapter 3. Initiation of polyketide synthesis
126
Figure 6.9 Determination of kinetic parameters in MCAT-supplemented minimal PKS assays in the presence
of KR. A. Plot of reaction rate vs. substrate concentration and hyperbolic fit. B. Hanes linear plot of the data.
6.3.3. Addition of acetyl-ACP to MCAT-supplemented, extended minimal PKS assays
Addition of acetyl-ACP (30 µM) to MCAT-supplemented act minimal assays in the
presence of 10 µM KR led to a 2.6-fold increase in rate, in agreement with our findings in
the absence of KR. Under these conditions, reactions rates depended linearly on the
concentration of KSα/KSβ.
6.4. Discussion
Numerous questions remain about the mechanism and specificity of PKS KR. For instance,
does KR interact with ACP, KSα/KSβ or both? How is the regiospecificity of the
ketoreduction at C-9 controlled? Does KR use as a substrate a full length octaketide? If this
is the case, is it the previously cyclised octaketide or the uncyclised intermediate? And
consequently, how does KR exert its influence on the regioespecificity of the first
cyclisation?
In Chapter 6 we have applied the methodology developed in preceding sections to study
the effect of KR on the kinetics of the act minimal PKS in vitro. The kinetic
characterization of the act minimal PKS in the presence of KR done here aimed to
contribute to the understanding of the reaction catalyzed by KR. The Stanford group
A. B.

Chapter 3. Initiation of polyketide synthesis
127
observed a decrease in rate when act KR was added to act minimal PKS supplemented with
MCAT.11 In our hands, the rates of self-malonylation of holo-ACP, decarboxylation by
KSβ and chain elongation by KSα appeared to be unchanged upon addition of KR. Thus
the reduction of the polyketide catalyzed by KR must be faster than any of the preceding
steps and a full kinetic analysis of the ketoreduction catalyzed by KR was not possible by
the methods we have developed.
In the absence of KR, the act minimal PKS produces the unreduced octaketides SEK4 and
SEK4b. KR must interact with the minimal PKS, because the C10-C15 cyclisation route of
SEK4b is lost when KR is added to minimal PKS in vitro assays. In agreement with
previous reports by the Stanford group,11 we needed high concentrations of KR (10 µM) in
the assays in order to observe predominant production of the reduced polyketide mutactin.
The interaction between KR and KSα/KSβ cannot be limiting in this respect, because
the ratio KR: KSα/KSβ did not seem to alter the relative production of mutactin with
respect to SEK4 and SEK4b: the same 80% excess mutactin (over SEK4/4b) was observed
for a range of KR: KSα/KSβ ratios varying between 30 and 3.
Therefore the idea that KR interacts with ACP might be valid.32 KR could induce a
more rigid ACP-PP structure, thus restraining the degrees of freedom of the polyketide
chain in the tunnel between KSα and KSβ. This could favour the C7-C12 cyclisation
pattern of mutactin and prevent the ‘unnatural’ C10-C15 cyclisation of SEK4b.
The idea that KR interacts with ACP and not with KSα/KSβ is supported by the
structure of the mammalian FAS (see Section 1.3). In this type I FAS structure, the KS
domains form the body of the megasynthase whereas KR forms the ‘arms’, and their
corresponding active sites are 72 Å apart.85 ACP, on the other hand, is supposed to lie
between KS and KR, in the middle of each reaction chamber (see Figure 1.13 in Chapter 1).
Therefore, in the mammalian FAS it is reasonable to think that ACP is the link between KS
and KR domains, and direct interaction between KR and KS seems unlikely. In the act
PKS, ACP interacts with KSα/KSβ (Chapters 2, 3 and 4) and it also probably acts as the
link between KSα/KSβ and KR.
An interesting observation was the reduction in PKS activity when ACP, MCAT, KSα/KSβ
and KR were incubated with malonyl CoA in the absence of NADPH (but in the presence
of NADP+). Under these conditions, SEK4 and SEK4b were produced in the same ratio as

Chapter 3. Initiation of polyketide synthesis
128
in the absence of KR, but at a lower rate. Assuming that the quaternary structure of KR is
conserved in the presence of NADP+, as suggested by the Bristol group,206 this result would
mean that an ‘inactive’ KR partially impairs polyketide production by the act PKS.
This finding is reminiscent of the Hertweck, Moore et al work when they could not
reconstitute the enc minimal PKS in vivo, either in the absence of enc KR, or indeed in the
presence of an inactive enc KR (see introduction to Chapter 6).205 Therefore it appeared
that ketoreduction in the enc PKS must occur in order for enterocin-wailupemycins
polyketides to be produced.
Similarly, we could argue that ketoreduction must occur in order for the C-7 to C-12
cyclisation to be predominant in the act PKS. This would imply that cyclisation of the
polyketide chain occurs after the KR step. Evidence for this comes from our finding that, if
a structurally competent but catalytically inactive act KR (i.e. KR without NADPH) is
present in act in vitro assays, SEK4b is still produced. Therefore KR must perform a
catalytic role in the cyclisation of the polyketide chain, and this is irrespective of any
structural constrain KR may impose on the ACP-PP-polyketide chain structure, as
speculated above.
This hypothesis is in agreement with recent findings from Ma, Zhang and collaborators,
who found that act KR was able to reduce a polyketide produced by a fungal PKS.210 PSK4
from Gibberella fujikoroi synthesizes the nonaketide 97 from malonyl CoA. When act KR
was added to the assay, mutactin 20 was also produced. This is striking because 97 does not
show a C-7 to C-12 cyclisation. However, the action of act KR induced production of
mutactin, a C-7 to C-12 cyclised polyketide. This result strongly suggests that
ketoreduction by act KR occurs prior to cyclisation of the polyketide chain.
O
O OH OH
OHHO
2
79
1012
13
17
97

Chapter 3. Initiation of polyketide synthesis
129
7. Conclusions
The act PKS has proven to be a very useful model for understanding polyketide
biosynthesis. For example, the act PKS is a good model for the iterative Type I PKS found
in fungi, because the act PKS is also iterative; that is, the single set of proteins acts several
times to produce a single final product (i.e. actinorhodin). Also, the individual reactions
catalyzed by the act PKS are the same as those catalyzed by the chain extension domains of
single modules of bacterial Type I modular polyketide synthases. Finally, the act PKS is
also a model for other Type II polyketide synthases in bacteria. Thus, understanding of the
act PKS serves to underpin advances in understanding the main classes of polyketide
synthase proteins in bacteria and fungi.
The act PKS has been studied for more than 20 years, from the identification and
cloning of the corresponding gene cluster to the study of the enzymology of purified
proteins. Within the act PKS, there are three proteins essential for production of
polyketides. ACP, KSα and KSβ thus form the act minimal PKS. During the last decade,
production of polyketides by the act minimal PKS has been studied mainly by two groups,
i.e. the Stanford and the Bristol groups. In 2006, the Bristol group published a model for
the production of SEK4 and SEK4b by the act minimal PKS in vitro (Scheme 1.13).60 We
accepted this model as the starting point.
In this work, we have developed continuous methods to measure the rate of succeeding
steps in the act minimal PKS. The chemical reactions catalyzed by the act minimal PKS
were grouped into loading, initiation, chain extension, and cyclisation and release steps
(Scheme 1.18). The loading step (i.e. self-malonylation of holo-ACP) was the rate limiting
step in act minimal PKS assays, as suggested by early work in the Bristol group.8
Experimentally, loading rates could be studied under conditions where KSα/KSβ was
added in high concentrations (Chapter 2).
In order to accelerate the rate of the loading step, MCAT was added to our assays.
MCAT provided rapid formation of malonyl-ACP, so that loading was no longer rate
limiting. Under these conditions, it appeared that initiation of polyketide synthesis by
decarboxylation of malonyl-ACP by KSβ was slower than chain elongation catalyzed by
KSα (Chapter 3). Addition of excess starter unit in the form of acetyl-ACP allowed the

Chapter 3. Initiation of polyketide synthesis
130
measurement of the slowest reaction catalyzed by KSα. It appeared that cyclisation and
release of the polyketide chain to form SEK4 and SEK4b occurred at an even higher rate
(Chapter 4).
Therefore a methodology was developed to study the kinetics of each of the enzymes
that constitute the act minimal PKS, as well as KR (Chapter 6). During the course of our
studies, we have obtained evidence for protein-protein interactions among the components
of the act minimal PKS, which have contributed to understanding the molecular
architecture of the complex.
7.1. Self-malonylation of holo-ACP is aided by KSα/KSβ
The study of protein-protein interactions between ACP and KSα/KSβ was prompted by the
discovery that ACP and KSα/KSβ were closely associated during the loading step. The rate
of self-malonylation increased five-fold when holo-ACP was in the presence of KSα/KSβ
(Section 2.4.2). We attempted to gain insights into the ACP: KSα/KSβ association by the
study of the self-malonylation of act mutant acyl carrier proteins. Thus we chose two act
mutants (E47A and E53A) previously shown to self-malonylate with the same rate as the
reference act ACP by a mass spectrometry assay. E47 and E53 had been proposed to have a
role in ACP: KSα/KSβ interactions.120
In our hands, however, these mutant acyl carrier proteins were deficient in self-
malonylation when studied in isolation, and thus we lacked of an appropriate control to
study ACP: KSα/KSβ interactions during the loading step (Section 2.5.1). Because of this,
the nature of this association remains unclear. It is tempting to speculate that holo-ACP
adopts a different, perhaps more rigid, conformation in the presence of KSα/KSβ, leading
to enhanced catalytic activity. But structural studies performed within the Bristol group
have shown that holo-ACP in isolation is itself a rigid protein, and the inherent flexibility of
other FAS and PKS acyl carrier proteins has not been observed with act ACP.193
The ACP: KSα/KSβ interaction during the loading step was highly specific: the rates of
self-malonylation of gris ACP and dps ACP did not undergo any change in the presence of
act KSα/KSβ. Future work should investigate this further by the generation of more mutant
acyl carrier proteins and kinetic characterization of their self-malonylation rates. NMR

Chapter 3. Initiation of polyketide synthesis
131
titrations of KSα/KSβ to WT and mutant holo-ACP solutions could help proving the
hypothesis of the conformational change. Eventually, residues on the surface of KSα/KSβ
could also be mutated.
A key question raised by our in vitro results is their relevance to the situation in vivo. It is
clear that the minimal act PKS consisting of ACP and KSα/KSβ is competent to produce
octaketides over a wide range of protein and malonyl CoA concentrations (Chapter 2), but
does it do this efficiently? In the absence of MCAT the rate limiting step is the self-
malonylation of ACP. The rate of this reaction depends on the concentrations of malonyl
CoA and holo-ACP and increases linearly with holo-ACP concentration (Figure 2.12). We
have also studied how the overall rate of octaketide production depends on holo-ACP
concentration (Figure 3.2). At concentrations above about 80 µM holo-ACP and saturating
concentrations of malonyl CoA, the rate of self-malonylation is sufficient to match or
outpace the requirement for malonyl-ACP by KSα/KSβ. At lower holo-ACP
concentrations, MCAT accelerates malonyl transfer, and for concentrations of holo-ACP
below 80 µM, the presence of MCAT speeds up the overall reaction.
The determination of kinetic parameters in the absence and presence of MCAT allows
the prediction on how the behaviour of the MCAT-catalyzed malonylation of holo-ACP
and the self-malonylation of holo-ACP in the presence of KSα/KSβ vary with the
concentration of ACP, MCAT, and malonyl CoA (Figure 7.1). The kinetic parameters for
act holo-ACP as a substrate for MCAT have not been measured because of the self-
malonylation reaction, but by comparison with S. coelicolor FAS ACP it is reasonable to
estimate a KM of 60 µM and kcat of 450 s-1.46 It is clear that under conditions such as high
MCAT concentration (>10 nM), low ACP concentration (<80 µM), and low malonyl CoA
concentration (<100 µM), the MCAT catalyzed reaction would be expected to dominate
(Figure 7.1). However it is also possible for the self-malonylation reaction to dominate, for
example at high ACP (>80 µM) and malonyl CoA concentrations (>400 µM) and low
MCAT concentrations (<5 nM).
The in vivo concentrations of malonyl CoA and act holo-ACP have never been
measured in S. coelicolor, but Rock and co-workers have estimated the concentration of E.
coli FAS ACP as between 17 and 130 µM in vivo, and malonyl CoA concentrations have
been estimated between 110 and 1000 µM.166 Few in vivo experiments have been performed
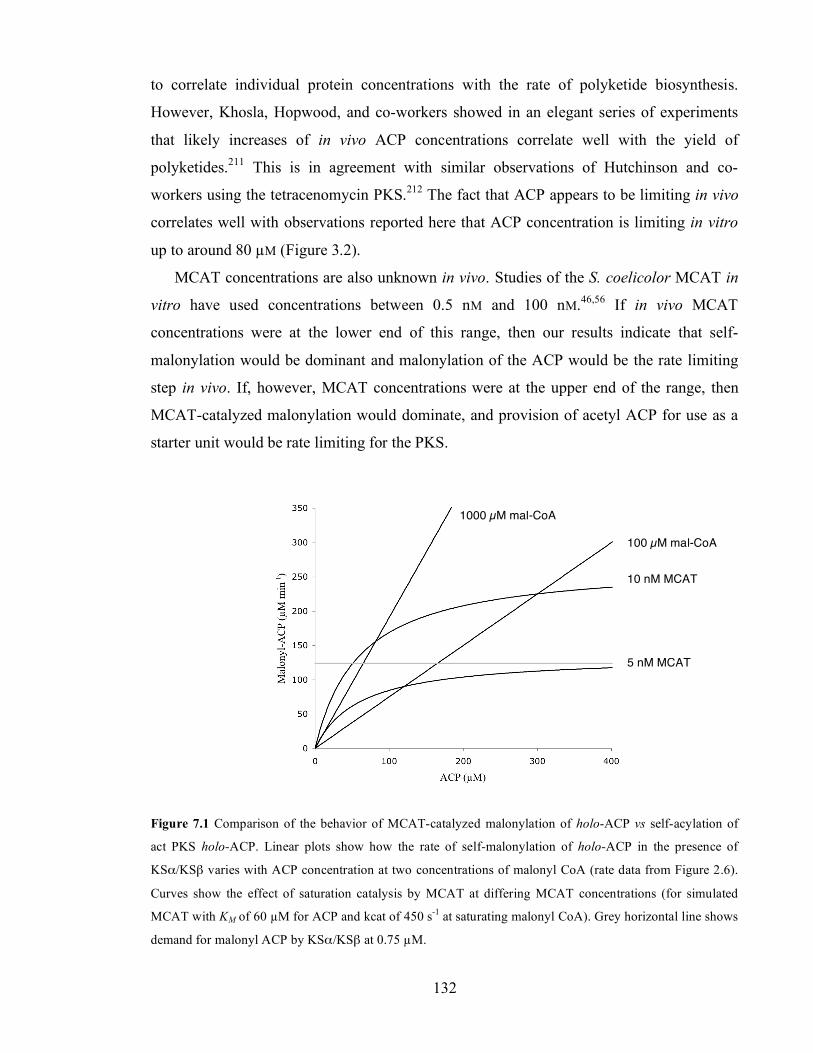
Chapter 3. Initiation of polyketide synthesis
132
to correlate individual protein concentrations with the rate of polyketide biosynthesis.
However, Khosla, Hopwood, and co-workers showed in an elegant series of experiments
that likely increases of in vivo ACP concentrations correlate well with the yield of
polyketides.211 This is in agreement with similar observations of Hutchinson and co-
workers using the tetracenomycin PKS.212 The fact that ACP appears to be limiting in vivo
correlates well with observations reported here that ACP concentration is limiting in vitro
up to around 80 µM (Figure 3.2).
MCAT concentrations are also unknown in vivo. Studies of the S. coelicolor MCAT in
vitro have used concentrations between 0.5 nM and 100 nM.46,56 If in vivo MCAT
concentrations were at the lower end of this range, then our results indicate that self-
malonylation would be dominant and malonylation of the ACP would be the rate limiting
step in vivo. If, however, MCAT concentrations were at the upper end of the range, then
MCAT-catalyzed malonylation would dominate, and provision of acetyl ACP for use as a
starter unit would be rate limiting for the PKS.
Figure 7.1 Comparison of the behavior of MCAT-catalyzed malonylation of holo-ACP vs self-acylation of
act PKS holo-ACP. Linear plots show how the rate of self-malonylation of holo-ACP in the presence of
KSα/KSβ varies with ACP concentration at two concentrations of malonyl CoA (rate data from Figure 2.6).
Curves show the effect of saturation catalysis by MCAT at differing MCAT concentrations (for simulated
MCAT with KM of 60 µM for ACP and kcat of 450 s-1 at saturating malonyl CoA). Grey horizontal line shows
demand for malonyl ACP by KSα/KSβ at 0.75 µM.
1000 µM mal-CoA
100 µM mal-CoA
10 nM MCAT
5 nM MCAT

Chapter 3. Initiation of polyketide synthesis
133
The demand for malonyl ACP must also be considered as well as the supply. Our results
show that in the absence of excess acetyl ACP, octaketides are produced by KSα/KSβ at
20.6 min-1 which corresponds to the use of malonyl ACP at 165 min-1. For a KSα/KSβ
concentration of 0.75 µM (typical for in vitro studies), this corresponds to a malonylation
rate of 123 µM·min-1; this is shown as the grey line in Figure 7.1. It is clear that this could
be supplied by ACP at approximately 70 µM and malonyl CoA at 1000 µM in the absence
of MCAT or by 55 µM ACP and saturating malonyl CoA in the presence of 10 nM MCAT.
Thus, the concentrations of all catalytic and substrate participants are required in order to
determine which pathway dominates in vivo. While the results reported here show which
factors are important and give limits for each process, further experiments will be required
to determine these concentrations in vivo during actinorhodin biosynthesis.
7.2. Quaternary structure of KSα /KSβ and ACP: KSα /KSβ complexes
In order to study the quaternary structure of KSα/KSβ, we assayed mutant KSα/KSβ using
the methods developed to study the kinetics of KSα and KSβ separately. For example, the
decarboxylation activity of a CA KSα/KSβ was assessed (Section 5.1.1). As expected, this
mutant was deficient in decarboxylation of malonyl-ACP, due to the mutation of the active
site in KSβ. Addition of a mutant KSα/KSβ incapable of polyketide synthesis (i.e. AQ
KSα/KSβ) to CA KSα/KSβ assays resulted in an increase of the rate of SEK4/4b
production under saturating concentrations of acetyl-ACP (Section 5.3) This meant that the
presence of AQ enhanced the production of polyketides by CA KSα/KSβ by a different
means than production of acetyl-ACP. This result could be explained if the catalytic
KSα/KSβ species were the dimer of heterodimers, and then addition of {AQ|AQ} to
{CA|CA} resulted in some {CA|CQ} complex being formed. This {CA|CQ} complex
would then produce polyketides more efficiently than {CA|CA}.
We speculate that decarboxylation of malonyl-ACP by KSβ is accompanied by a
conformational change of KSα/KSβ (see discussion to Chapter 5). This could account for
the observed increase in rate caused by introduction of an active KSβ in the tetramer. This
conformational change is not prompted by substrate binding onto KSβ (see discussion to
Chapter 5), and may happen as a result of malonyl-ACP decarboxylation. This makes sense

Chapter 3. Initiation of polyketide synthesis
134
because the following reactions are catalyzed by KSα, which is now in an active
conformation. Further evidence to support the hypothesis of the conformational change was
provided by the experiments in which we added excess acetyl-ACP to CA KSα/KSβ
assays. In these assays, an increase in rate of only 3-fold (kcat changed from 2.2 to 6.4 min-
1) was observed, instead of the expected 30-fold to reach WT levels. This suggested that
CA KSα/KSβ was deficient in more than just the initial decarboxylation of malonyl-ACP
(see discussion to Chapter 4).
We propose here that the crystal structure of KSα/KSβ published by the Stanford group
in 2004,31 corresponds to the ‘closed’ (inactive) form of KSβ and ‘open’ (active) form of
KSα; i.e. it represents a complex ready for extension, rather than for initiation. This could
explain why the active site of KSβ appeared to be buried. Indeed, electron density of
intermediates bound to KSα was identified.31
The stoichiometry of ACP: KSα/KSβ interactions was addressed by the study of the
inhibition of polyketide production by apo-ACP (Section 5.2). Whereas apo-ACP did not
appear to inhibit holo-ACP self-malonylation or indeed MCAT, it was a mixed inhibitor of
KSα/KSβ. Thus it seemed that KSα/KSβ possessed two different binding sites for ACP. In
principle, this could mean either that ACP interacted with each KSα/KSβ dimer (i.e. a 1:1
ratio between ACP and KSα/KSβ); or that KSα and KSβ within each dimer had different
binding sites for ACP (i.e. a 2:1 ratio between ACP and KSα/KSβ). In the first scenario, we
would expect very similar values for competitive and uncompetitive inhibition constants.
However, Kic was 50 µM whereas Kiu was 137 µM. These data are consistent with a model
in which each ketosynthase subunit in the KSα/KSβ tetramer has the ability of binding a
molecule of ACP (Figure 7.2).
KS! KS"
KS! KS"
ACP
ACP
ACP
ACP
Figure 7.2 Model for ACP: KSα/KSβ that contemplates four molecules of ACP per tetramer KSα/KSβ.

Chapter 3. Initiation of polyketide synthesis
135
The interaction of ACP with KSβ was then studied. In agreement with previous models,120
we identified a couple of negatively charged residues within helix II of ACP as key players
in ACP: KSβ interations. Indeed, mutation of E47 or E53 to alanine residues resulted in a
3-4 fold increase in KM with unchanged kcat (Section 3.2) Moreover, the assays revealed
that KSβ binds malonyl-ACP (KM = 2.4 µM) selectively over apo-ACP, despite the little
structural difference between both species.
The interaction of ACP and KSα could not be fully studied due to the uncertainty in the
determination of KM for malonyl-ACP (Section 4.2). However, the fact that the introduction
of a bulky valine residue in position 47 of ACP had the same disadvantageous kinetic effect
in KSα and KSβ suggested that the binding site for ACP in both proteins is very similar.
Finally, the interaction of KR with ACP: KSα/KSβ complexes was studied (Section 6.3).
The ratio mutactin: SEK4/4b depended strongly on the absolute concentration of KR, but
not on the ratio of KR: KSα/KSβ, meaning that KR may not interact with KSα/KSβ.
Consisting with this, the self-malonylation rate of ACP in the presence of KSα/KSβ did not
change when KR was added: KR did not have an effect on the ACP: KSα/KSβ interactions
that lead to enhanced self-malonylation activity of holo-ACP. These results are consistent
with previous models described in the literature,32,202 which suggest that KR interacts with
ACP rather than with KSα/KSβ. This also makes sense when the structure of the
mammalian FAS (best current model for the act PKS) is taken into account (see discussion
to Chapter 6): ACP might be the link between the ketosynthase and the ketoreductase
activities in both models.
7.3. Model for an extended act minimal PKS
Based on the currently accepted model for the act minimal PKS,60 we propose here simple
refinements that can account for our observations (Scheme 7.1). Self-malonylation of holo-
ACP seems to be more efficient in association with KSα/KSβ (kcat ~ 2.3 min-1), rather than
in solution (kcat ~ 0.5 min-1). Therefore the loading step is likely to occur when ACP is
bound to the KSα/KSβ complex. Consequently, the ACP-ACP transfer activity which
occurs between acyl carrier proteins,60 might not be of biological importance.

Chapter 3. Initiation of polyketide synthesis
136
The initiation step is catalyzed by KSβ, which generates acetyl-ACP. This acetyl-ACP most
likely is released into solution. Simultaneously, there is a conformational change in the
KSα/KSβ tetramer which renders a KSα that is now in an ‘open’ conformation. KSα then
binds acetyl-ACP and is primed with the acetyl starter unit. Extension of the polyketide
chain then occurs.
The reduction by KR probably occurs before the first cyclisation, because when KR
was added to minimal PKS assays in the absence of NADPH, SEK4b was still produced.
This means that the ‘correct’ C7-C12 first cyclisation is probably only favoured after
reduction of C-9 by KR (Section 6.3).
HS
ACP
KS! KS"
CO2
KS! KS"
ACP
S
O O
OH
ACP
S
O
ACP
S
O
ACP
S
O
mal CoA
CoA
SHO
OO
ACP
KR
CoA
Mal CoA
ACP
HO
O
O
O
OH
O O O
S
S
O
O
O
O
O
n
KS! KS"
KS! KS"
KS! KS"
KS! KS"
KS! KS"
KS! KS"
KS! KS"
KS! KS"
KS! KS"
KS! KS"
KS! KS"
KS! KS"
Scheme 7.1. Model for an extended act minimal PKS. Progressing clockwise from bottom left, self-
malonylation of ACP in association with KSα/KSβ; initiation reaction and dissociation of acetyl-ACP into
solution; priming of KSα with acetyl; extension of the polyketide chain by Claisen-type condensations;
reduction of polyketide by KR; and cyclisation and release. For clarity, only one reaction chamber is shown.
The timing of the reduction has not been elucidated in this work and n can vary from 1 to 4.
Loading
Initiation
KSα encounters acetyl-ACP in solution
Priming
Extension
Mutactin 20

Chapter 3. Initiation of polyketide synthesis
137
Our kinetic data demonstrate that the loading step is the rate limiting step in the act minimal
PKS in vitro (kcat ~ 2.3 min-1). In the presence of excess malonyl-ACP, the initiation step
catalyzed by KSβ becomes rate-limiting (kcat ~ 20 min-1). If acetyl-ACP is present in
excess, the rate increases 3-fold more (kcat ~ 60 min-1), which most likely represents the rate
of the extension steps catalyzed KSα. We were unable to measure rates for the cyclization
and release steps, but these are likely higher still, because no accumulation of intermediates
attached to ACP or released into solution could be detected. The observed reaction rate in
the presence of excess malonyl and acetyl-ACP remained unchanged upon addition of KR,
which also indicates that cyclisation and release are not rate limiting, because the addition
of KR necessarily affects the cyclisation of the polyketide chain.
The results presented here show that individual catalytic steps of the act PKS can be
dissected. The act minimal PKS appears to be optimized for the conversion of malonyl
CoA to octaketides, because individual rate constants measured for the individual catalytic
steps show an increase in value for each succeeding step.
The biosynthesis of malonyl CoA from acetyl CoA (which is obtained from pyruvate in
the citric acid cycle) is catalyzed by an enzyme known as acetyl CoA carboxylase. This
reaction requires energy in the form of ATP.101 Therefore it makes sense that the
malonylation of ACP is the rate limiting step in the act PKS, ensuring that malonyl-ACP is
produced at the rate it is consumed by the PKS. This guarantees no accumulation of
malonyl-ACP in the cell, and no waste of malonyl CoA or ATP.
In vitro, the loading step can be accelerated by addition of MCAT. In these conditions,
the initiation step (i.e. decarboxylation of malonyl to acetyl-ACP) becomes rate limiting.
Because the cell needs energy to make malonyl-ACP, it also makes metabolic sense that
acetyl-ACP is not accumulated, and rapidly used by KSα to form polyketides. In vitro, the
elongation step catalyzed by KSα is ca. three times as fast as the initiation step catalyzed
by KSβ. Successive reactions catalyzed by the act PKS might be even faster. For instance,
the ketoreduction catalyzed by KR appeared to be faster than the elongation step (Section
6.3.3). Other methods than those developed in this work need be introduced to enable the
characterization of the kinetics of KR and further tailoring enzymes in the act PKS.

Chapter 3. Initiation of polyketide synthesis
138
8. Experimental procedures
8.1. Bacterial strains and plasmids
8.1.1. Escherichia coli
RapidTrans™ TAM1 competent cells (Active Motif) were used for high-throughput
bacterial transformation and isolation of plasmids. BL21(DE3)pLysS competent cells
(Promega) were used for high efficiency protein expression of the desired genes (Table
8.1).
Plasmid Product, Resistance Reference / Supplier
pRJC001 Act C17S holo-ACP, Amr Dr. Russell J. Cox
pIJ2367 Act C17S apo-ACP, Amr Dr. Russell. J. Cox
pCJAII136/1 Act E47A C17S holo-ACP, Amr Dr. Christopher J. Arthur pCJAII124/1 Act E47V C17S holo-ACP, Amr Dr. Christopher J. Arthur pCJAII81/1 Act E53A C17S holo-ACP, Amr Dr. Christopher J. Arthur pRJC002 Gris WT holo-ACP, Amr Dr. Russell J. Cox
pPBIV165 Act R72A C17S holo-ACP Pedro Beltran
pPWDpsG WT holo-DpsG, Spmr Dr. Pakorn Wattana-Amon
pLHSIII67A S. coelicolor ACPS, Amr Dr. Leah Smith
pIJ2371 S. coelicolor MCAT, Amr Dr. Russell J. Cox
Table 8.1 Plasmids transformed into E. coli
8.1.2. Streptomyces coelicolor
S. coelicolor A3(2) CH999 lacks the entire set of genes of the actinorhodin cluster. The
group of Cambridge carried out the transformation of this strain with the plasmid carrying
wild type of mutant KSα/KSβ and provided mycelium for further growth and purification
of protein in our laboratory.

Chapter 3. Initiation of polyketide synthesis
139
Plasmid Product, Resistance Reference / Supplier pCB84 his6-act KSα:KSβ, Kmr Dr. Christian Bisang
pCB91 his6-act KSα:[Q161A]KSβ, Kmr Dr. Christian Bisang
pCB56 his6-act [C169A]KSα:KSβ, Kmr Dr. Christian Bisang
pSEK4 act KSα:KSβ, act ACP, act
CYC, actIV, Ampr
24
Table 8.2 S. coelicolor A3(2) CH999 strains used in this work.
8.2. DNA techniques
8.2.1. Site directed mutagenesis
Site-directed mutagenesis of ACP was performed using the Stratagene QuikChange™
Mutagenesis Kit. Mutagenesis reactions were performed according to the manufacturer’s
instructions and contained 0.2 mM dNTP mixture, 50 ng dsDNA template (pRJC001), 4%
v/v dimethylsulphoxyde (DMSO), 125 ng forward primer, 125 ng reverse primer (Table
8.3), 1 unit KOD Hot Start polymerase enzyme, 1 mM MgSO4 and 10% v/v polymerase
reaction buffer in 50 µl total volume (made up with dH2O). The reaction was temperature
cycled using a MJ Research PTC-100 programmable temperature controller. Samples were
heated at 95°C for 30 seconds, followed by 18 cycles of 95 ˚C for 30 seconds, 60°C for 1
min and 72 °C for 2.5 min. 9 units of DpnI restriction enzyme were added once the PCR
reaction had completed and the template digested for 4 hours. The PCR product was
transformed into E. coli TAM1 competent cells for plasmid isolation.
Forward primer GTC GAC ACG CCG GCT GAG CTG CCT GAC
Reverse primer GTC GAG CAG CTC AGC CGG CGT GTC GAC
Table 8.3 Primers for site-directed mutagenesis of R72 ACP. Codons containing the mutation R72A are
underlined.

Chapter 3. Initiation of polyketide synthesis
140
8.2.2. Plasmid DNA isolation and characterization
Plasmid DNA (1-10µg) was isolated from overnight cultures of E. coli TAM1 competent
cells (LB medium, 4 ml) using GeneElute™ Plasmid Miniprep Kit (Sigma), according to
the manufacturer’s instructions.
DNA agarose electrophoresis was performed using a Sigma horizontal gel
electrophoresis tank at an applied voltage of 200V (100A) connected to a Consort
microcomputer power supply unit. Staining of DNA fragments by soaking the agarose gel
in ethidium bromide (1 mg/l) allowed for the visualization of DNA bands under ultraviolet
light. Size and concentration of DNA plasmids were estimated by comparison with
standards (Hyperladder I, Sigma). pET15b plasmid based DNA samples of concentration
50-100 ng/µl were sequenced by Lark Technologies using T7 promotor.
8.3. Bacterial culture tecniques
Media ingredients were purchased from Sigma, Difco or Fisher, unless otherwise stated.
Distilled water was used for all media and solutions. All solid and liquid media was
sterilised prior to use (Astell Cientific ASA270, 121 ˚C, 15 psi, 15 min). All pH
determinations of media and buffers were undertaken using a Gelplas double junction
combination electrode (BDH) attached to a model 292 pH-meter (Pye Unicam).
8.3.1. Liquid media
8.3.1.1. SOC medium
SOC medium was used for the post-transformation recovery of E. coli. Tryptone (2 g),
yeast extract (1.5 g) and NaCl (0.5 g) were dissolved in water, then KCl (1 ml from 250
mM stock) was added and the volume made up to 90 ml. The pH was adjusted to 7.0 with 6
M NaOH and the total volume made up to 100 ml. After sterilisation, 2 ml of sterile 1 M
glucose and 1 ml sterile 1 mg/ml MgCl2 were added.

Chapter 3. Initiation of polyketide synthesis
141
8.3.1.2. Luria-Bertani (LB) medium
LB medium was used for growth of E. coli either for plasmid isolation (3-5 ml) or protein
expression (1-4 L). Tryptone (10 g), yeast extract (5g) and NaCl (10 g) were dissolved in
900 ml water, and the pH adjusted to 7.5 with 6 M NaOH. The total volume was then made
up to 1 L. 200 ml were dispensed into 500 ml flasks which were covered with sponge
bungs and aluminium foil, and autoclaved.
8.3.1.3. Super-YEME (SY) medium
SY medium was used for growth of S. coelicolor. Tryptone (5 g), yeast extract (3 g), malt
extract (3 g), MgCl2 (1.1 g), glycine (5 g), L-proline (75 mg), L-arginine (75 mg), L-
cysteine (75 mg), L-histidine (100 mg) and uracil (15 mg) were dissolved in 800 ml water
(Solution A). Separately, glucose (10 g) and sucrose (340 g) were dissolved in 200 ml
water (Solution B). All solutions were autoclaved. Before use, 20 ml of Solution B were
dispensed in 10 x 500 ml sterilised flasks containing coiled springs and 80 ml of Solution
A.
8.3.2. Solid media
Solid media have been used to allow for selection of a single bacterial colony under
antibiotic selection markers. Antibiotic (100 µg/ml carbenicillin and kanamycin or 50
µg/ml spectinomycin and thiosthrepton according to the resistance gene in the
corresponding plasmid) was added after first melting and then cooling the medium to
approximately 60°C. The media was then poured (20 ml) into Petri dishes and allowed to
solidify.

Chapter 3. Initiation of polyketide synthesis
142
8.3.2.1. Luria-Bertani agar (LB agar) growth medium
LB agar was the solid media used to plate out E. coli after transformation. 1.5% agar was
added to LB liquid medium and autoclaved. Plates were kept at 37 ˚C for 30 min prior to
plating out bacteria.
8.3.2.2. Mannitol Soya Flour Medium (SFM)
SFM was used to select a single colony of S. coelicolor for protein expression purposes. 2 g
agar, 2 g mannitol and 2 g soya flour were dissolved in 100 ml tap water and autoclaved
twice.
8.3.2.3. R5 Medium
R5 Medium was used to grow S. coelicolor A3(2) CH999/pSEK4 for isolation of SEK4 and
SEK4b. A solution was made up with 103 g/l sucrose, 0.25 g/l potassium sulphate, 10.12
g/l magnesium chloride hexahydrate, 10 g/l glucose, 0.1 g/l Difco casaminoacids, 3 ml
trace element solution (40 mg ZnCl2, 200 mg FeCl3.6H2O, 10 mg CuCl2.2H2O, 10 mg
MnCl2.4H2O, 10 mg Na2B4O7.10H2O and 10 mg (NH4)6Mo7O24.4H2O in 1000 ml distilled
water), 5 g/l yeast extract and 5.73 g/l TES buffer. A total volume of 1.5 L was made. 100
ml of this solution was poured into 250 ml flasks each containing 2.2 g agar and
autoclaved. At time of use, the medium was re-melted and the following ingredients were
added: 1 ml KH2PO4 (0.5%), 0.4 ml CaCl2.2H2O (5 M), 1.5 ml L-proline (20%), 0.7 ml
NaOH (1 M) and 100 µl thiosthrepton.

Chapter 3. Initiation of polyketide synthesis
143
8.4. Extraction and purification of SEK4 and SEK4b
8.4.1. Growth of bacteria and extraction of polyketides
Growth of bacteria and extraction and purification of aromatic polyketides was executed
following published methods.8,24 Frozen S. coelicolor A3(2) CH999/pSEK4 mycelium was
grown up in 100 ml SY medium for 4 days at 30 ˚C, 220 rpm under thiostrepton selection
and then transferred to 60 plates (1.5 L R5 medium with 50 µg/ml thiostrepton). Plates
were incubated at 28 ˚C for 9 days and then diced and extracted in 3 litres of ethyl acetate
and methanol (4:1 EtOAc:MeOH) with 1% acetic acid. The solvent was dried (MgSO4) and
evaporated. The solid, dark brown residue was re-suspended in EtOAc, the solution then
filtered and evaporated. The weight of the remaining products was 3 g. This was re-
suspended in EtOAc and loaded into a Silica 60 column, which was flushed with 200 ml of
a EtOAc:Hexane:Acetic acid (80:17:3) mixture and then with 750 ml of acetonitrile. Thin
layer chromatography (70% EtOAc, 30% petroleum ether, 1% acetic acid) was used to
follow the aromatic rich fractions (by UV absorbance). The MeCN fractions showing
aromatic absorbance with an Rf of 0.35 were gathered together, the solvent evaporated and
the products re-suspended in EtOAc and applied to a second Silica column, run under
EtOAc:Hexane:Acetic acid (80:17:3). The same solvent was used to perform the TLC
analysis, and the aromatic compounds detected with an Rf of 0.61.
8.4.2. Liquid Chromatography/ Mass Spectrophotometry (LC/MS) analysis
LC/MS was used throughout the purification protocol to chase SEK4 and SEK4b. LC was
run in a Luna 5u C8(2) 150 x 2 mm column (Phenomenex) with water and acetonitrile
(both + 0.05% TFA) as mobile phase at a rate of 0.5 ml/min. A gradient from 5 to 95 %
acetonitrile was executed over 20 min (Waters 600 HPLC Pump). The UV absorbance of
the sample was monitored in a Waters 996 Photodiode Array Detector. Mass spectrometry
data was subsequently acquired by a Platform Micromass Electrospray Mass
Spectrophotometry facility (positive ion mode).

Chapter 3. Initiation of polyketide synthesis
144
8.4.3. Preparative HPLC for purification of SEK4 and SEK4b
The separation of SEK4 and SEK4b was achieved by HPLC (Beckman). An Anachem C18
column was used with flow rate of 4 ml min-1. The mobile phase was water/acetonitrile (30
to 50% MeCN over 10 min) controlled by a Programmable Solvent Module 126. Detection
of polyketides by UV absorbance (Diode Array Detector Module 168) allowed the
collection of SEK4 and SEK4b (Autosampler System Gold 157).
8.5. Growth of bacteria and purification of proteins
8.5.1. General methods and equipment
8.5.1.1. Centrifugation
Large volumes were centrifuged either in the SLA-1500, SLA-3000 or GS-3 rotors in a
RC5C centrifuge (Sorvall instruments) at maximum speeds of 14,500, 11,000 and 9,000
rpm respectively. Medium volumes were centrifuged in a SS-34 rotor at 18,000 rpm. Small
volumes were centrifuge in eppendorf-centrifuges at 13,000 rpm (MSE microcentaur
centrifuges).
8.5.1.2. Shakers
Shakers were from New Brunswick Scientific, and allowed the control of temperature and
loads of 20 x 500 ml flasks.
8.5.1.3. UV Spectrophotometer
The UV Spectrophotometer was an Ultrospec III from Pharmacia. 1.5 ml plastic cuvettes
were purchased from Sarsdted.

Chapter 3. Initiation of polyketide synthesis
145
8.5.1.4. Plate reader
Supplied by Molecular Devices, a SpectraMax 190 Plate Reader was used to measure the
absorbance of small volumes (up to 200 µl) in Corning UV-Transparent 96 wells
microplates.
8.5.1.5. Sonication
Cell-free extracts were obtained after lysing the cells by sonication (MSE Soniprep 150).
Sonication of E.coli was executed in 5 short bursts of 30 seconds with 1.5 min ice-cooling
in between. S. coelicolor was sonicated 10 times for 30 seconds.
8.5.1.6. Buffers
All buffers were made up with distilled buffer. Buffers for FPLC were filtered (0.45 µm) at
reduced pressure and degassed. Buffers for HPLC were filtered (0.2 µm) and degassed.
Buffers for proteins assays were filtered-sterilised (0.2 µm).
8.5.1.7. NTA-His-Bind nickel affinity chromatography
Nickel chromatography chromatography (NTA His-Bind resin -Novagen) was executed
following the manufacturer’s instructions. The column, which was stored in 50% ethanol,
was charged with 50 mM NiSO4 and equilibrated in binding buffer. The sample (typically
100 ml) was then loaded and the column subsequently washed with binding and washing
buffers. The protein of interest (ACPS, MCAT or KSα/KSβ) was collected in elution
buffer.

Chapter 3. Initiation of polyketide synthesis
146
Addition Binding Buffer Washing Buffer Elution Buffer Stripping Buffer Phosphate Buffer 100 mM 100 mM 100 mM 20 mM Tris-Cl NaCl 0.5 M 0.5 M 0.5 M 0.5 M Imidazole 5 mM 60 mM 1 M 100 mM EDTA Glycerol 10% 10% 10% - pH 7.5 7.5 7.5 7.9
Table 8.4 Buffers used for Nickel affinity chromatography
8.5.1.8. Fast protein liquid chromatography (FPLC)
All FPLC equipment was purchased from Pharmacia. The AKTA FPLC working station
was controlled by UNICORN v. 4.0 software packet. Columns for anion exchange
chromatography (Q-Sepharose 26/10 or 16/10) and gel filtration (Superdex 75 130/10 and
HiPrep Desalt 26/10) were used.
8.5.1.9. Freeze-Drying
Lypholysation of protein was done in an Edwards freeze dryer Modulo (-40°C, 6 mbar).
8.5.1.10. Sodium dodecyl sulphate Polyacrylamide gel electrophoresis (SDS-PAGE)
The Acrylamide solutions were prepared according to Table 8.5. The separating gel was
pre-set between the plates for 30-45 min and then the stacking gel was pipetted in and
assembled within the 15 wells comb.
Ingredient 10 % Separating Gel (ml) 4 % Stacking Gel (ml) 40 % Acrylamide:Bis 37:1 2.5 1 Gel Buffer 3.33 2.5 50% glycerol in water 4.1 --- Distilled water --- 6.5 10 % AMPS 50 µl 50 µl TEMED 20 µl 10 µl TOTAL 10 ml 10 ml
Table 8.5 Stacking and Separating gels for SDS-PAGE.

Chapter 3. Initiation of polyketide synthesis
147
Concentrated protein samples were directly added (2:1 v/v) to loading buffer. Diluted
samples (100 µl) were treated with trichloroacetic acid (10 µl, 72%, TCA) and incubated at
0 ˚C, 15 min. The resultant protein precipitate was pelleted by centrifugation (13,000 rpm,
5 min) and the supernatant discarded. The protein precipitate was resuspended in loading
buffer (5 µl) and TRIS base (2 M, 5 µl). The protein solution was then heated for 15 min at
100 ˚C in a Grant BT3 heat block to denature the protein and allow adhesion of SDS.
SDS-PAGE was run at constant current (80 mA). Buffers were prepared as follows:
Loading buffer: 9.456 g Trizma base, 1 ml mercaptoethanol, 1 g SDS, 10 ml glycerol,
0.01 g bromophenol blue, and made up to 100 ml with water, pH = 6.8.
Cathode buffer: 6.055 g Trizma base, 8.96 g Tricine, 0.5 g SDS made up to 500 ml with
distilled water, pH = 8.25.
Anode buffer: 12.11 g Trizma base, 1.5 g SDS to 500 ml with distilled water (pH 8.9).
Gel buffer: 181.7 g Trizma base and 1.5 g SDS to 500 ml with distilled water, pH = 8.4.
Staining solution: 2.5g/l Coomassie ble in 45% MeOH/45% glacial AcOH.
Destaining solution: 45% MeOH / 10% glacial AcOH in water.
8.5.2. Growth of E. coli, and expression and purification of ACP, ACPS and MCAT
E. coli BL21(DE3) competent cells were transformed with the appropriate plasmid for
expression of the corresponding ACP, ACPS or MCAT according to the supplier protocol
(Promega). They were then selected in carbenicillin-supplemented agar plates overnight. A
single colony was picked up using sterile toothpicks. This seed culture was amplified for 2-
4 h into at 37 ˚C, 220 rpm and used to inoculate the starter culture (LB medium, 200 ml).
The starter culture was grown until the optical density at 595 nm (A595) was 0.6, and then
centrifuged (10 min, 10,000 rpm).
Cells were re-suspended and then grown in 2 litres of LB medium (plus 100 µg/l
carbenicillin) at 37 ˚C, 250 rpm. IPTG (1 mM) was added when A595 was 0.6-0.8, and the
cultures incubated for further 4h at 32 ˚C, then harvested by centrifugation. Cells were
lysed by sonication and cell-free extracts subjected to the corresponding purification
protocol.

Chapter 3. Initiation of polyketide synthesis
148
8.5.2.1. Purification of ACP
The purification of heterologous C17S and C17S/other mutant ACP has been carried out
following the procedure described by Crosby and Cox.52 Cell-free extracts were treated
with streptomycin sulphate (20 g/l) to precipitate DNA (9,000 rpm, 15 min). Ammonium
sulphate precipitation was then undertaken. First, 60% cut was performed and the pellet
(9,000 rpm, 15 min) discarded. The supernatant was subjected to a 100% cut and the pH
adjusted to 3.4. This solution was then stored at 4˚C for 48 hr to allow for complete
precipitation of ACP. Subsequent centrifugation (18,000 rpm, 20 min) caused a pellet
which was resuspended and dyalised (Spectra/Pore 3,000 Da cut molecular porous
membrane tubing, Spectrum Laboratories) against TRIS buffer (50 mM, pH = 8.0),
overnight.
This sample was then purified by anionic exchange fast protein liquid chromatography
(Q-Sepharose HiLoad 26/10 or 16/10). A NaCl gradient from 0 to 1 M was performed, the
apo-ACP peak observed at around 0.5 M salt, whereas holo-ACP (dimer) was eluted at
around 0.6 M NaCl. If the sample required further purification (as judged by SDS-PAGE),
it was injected into a Superdex 75 130/10 to separate proteins by size. The ACP-containing
fractions (SDS-PAGE) were desalted (HiPrep Desalt 26/10), flash-frozen in liquid nitrogen
and freeze-dried. The freeze-dried protein was weighed and dissolved in 100 mM phosphate
buffer, pH = 7.3. Yields of purification of holo-ACP were in the range 5-10 mg/l. Apo-ACP
was recovered in higher yields (30 mg/l).
8.5.2.2. Purification of S. coelicolor his6-ACPS and act his6-KR
Purification of S. coelicolor ACPS and act KR was carried out following the procedure of
Cox47 and Hadfield,32 respectively. Cell-free extracts were subjected to fractional
precipitation by streptomycin sulphate (20g/l) to precipitate DNA (9,000 rpm, 15 min). The
supernatant was then filtered (0.45 µm) and applied to a Nickel chromatography column
(NTA His-Bind resin -Novagen), which was run as described in Section 8.5.1.7. ACPS or
KR-containing fractions were immediately desalted by FPLC (HiPrep Desalt 26/60). The
desalted protein solution was flash-frozen in liquid nitrogen and stored at -80 ˚C.

Chapter 3. Initiation of polyketide synthesis
149
8.5.2.3. Purification of S. coelicolor his6-MCAT
Purification of S. coelicolor MCAT was carried out following the procedure of
Szafranska.46 Cell-free extracts were subjected to fractional precipitation by streptomycin
sulphate (20g/l) to precipitate DNA (9,000 rpm, 15 min). The supernatant was then filtered
(0.45 µm) and applied to a Nickel chromatography column (NTA His-Bind resin -
Novagen), which was run as described in Section 8.5.1.7. MCAT-containing fractions were
subjected to further purification by anion exchange chromatography (Q-Sepharose HiLoad
26/10) and finally desalted by FPLC (HiPrep Desalt). The desalted protein solution was
flash-frozen in liquid nitrogen and stored at -20 ˚C.
8.5.3. Growth of S. coelicolor, expression and purification of his6-act-KSα /KSβ .
8.5.3.1. Growth of bacteria and expression of KSα/KSβ
The liquid medium for the growth of S. coelicolor A3(2) CH999 carrying the genes for
expression of wild type or mutant KSα/KSβ was Super-Yeme (SY) medium. 2 ml of frozen
mycelium in glycerol were amplified for 2-5 days at 30 ˚C, 220 rpm in SY medium
supplemented with kanamycin (100 µg/ml). This was used as starter culture for 2 litres of
SY medium (plus 100 µg/l kanamycin), which were subsequently grown at 30 ˚C, 220 rpm
for 2-3 days and then induced (thiostrepton, 10 µg/ml) and the culture incubated for a
further couple of days. Then the cultures were harvested (20x2 min, 9,000 rpm, diluted to
50% with water) and the cells lysed by sonication.
8.5.3.2. Purification of KSα/KSβ
Purification of KSα/KSβ was done essentially as described previously by the Bristol
group.8 Fractional precipitation by streptomycin sulphate was followed by
ultracentrifugation (43,000 rpm, 30 min). His6-(mutant)-KSα/KSβ was then purified by
nickel-affinity chromatography as described in 8.5.1.7. KSα/KSβ-containing fractions were

Chapter 3. Initiation of polyketide synthesis
150
immediately desalted by FPLC (HiPrep Desalt). The desalted protein solution was flash-
frozen in liquid nitrogen and stored at -80 ˚C.
8.6. Protein characterization methods
8.6.1. Protein Quantification
8.6.1.1. Bradford Assay
The Bradford reagent was made up as follows: 10 mg Brilliant Blue G, 10 ml 85%
phosphoric acid, 5 ml 95% ethanol, up to 100 ml with distilled water, then filtered. A blank
sample was routinely tested (100 µl distilled water in 1 ml Bradford reagent), as well as
Bovine Serum Albumin (Sigma) standard samples for calibration (20 µl protein solution
and 80 µl distilled water in 1 ml Bradford reagent). Protein samples (KSα/KSβ and ACP)
were prepared in the same manner. All the samples were incubated for 5 min at room
temperature and then the absorbance at 595 nm was measured.
8.6.1.2. Bicinchonic Acid Assay (BCA)
The BCA Protein Assay Kit was purchased from Pierce. The BCA reagent was made up as
follows: 50 parts of reagent A (1%BCA-Na2, 2% Na2CO3, 0.16% Na2 tartrate, 0.4% NaOH
and 0.95 NaHCO3) were mixed with 1 part of reagent B (4% CuSO4.5 H2O). A blank
sample was routinely tested (20 µl distilled water in 1 ml BCA reagent), as well as Bovine
Serum Albumin (Sigma) standard samples for calibration. Protein samples (KSα/KSβ and
ACP) were prepared in the same manner. All the samples were incubated for 30 min at 60˚
C and the absorbance at 562 nm read.

Chapter 3. Initiation of polyketide synthesis
151
8.6.1.3. Extinction coefficient of proteins.
The extinction coefficients at 280 nm for ACP (2400 M-1cm-1) and KSα/KSβ (84200 M-1
cm-1) have been previously determined within the group (Arthur, 2004). Protein
concentrations were calculated from Beer’s law:
A280 = ε c L
where A280 is the absorbance at a given wavelength, ε is the extinction coefficient, c
is the concentration of the protein and L is the pathlength of the UV beam through the
solution.
8.6.1.4. Electro-Spray Mass Spectrometry (ESMS)
In order to desalt and concentrate protein samples for analysis by ESMS, protein solutions
were prepared by hydrophobic interaction with a C4 resin as described by Winston and
Fitzgerald.185 C4 resin (3-5 mg, Jupiter 15u 300 Å, Phenomenex) was equilibrated in HPLC
grade methanol (100 µl) and the protein solution added. The sample was vortexed (5 min)
and centrifuged (13,000 rpm, 5 min). The supernatant was discarded, the pellet washed
twice with 0.1% aqueous trifluoroacetic acid (TFA, 1 ml), then vortexed and spun down
(13,000 rpm, 5 min). The final pellet was eluted in acetonitrile (25 µl).
ESMS analysis was performed on a QSTAR® XL system (Applied Biosystems, Foster
City, California) equipped with a chip based NanoESI source, Nanomate® 100 (Advion
biosciences, Ithaca NY) using Chipsoft software (version 5.1). The Nanomate® 100 source
was operated using a 400 nozzle chip, with a delivery pressure of 0.3 psi and spray voltage
of 1.40 kV. The QSTAR® XL was controlled using AnalystTM QS software (version 1.1) in
positive TOF MS mode.
8.7. Protein assays.
All assays were performed in phosphate buffer (pH = 7.3) at 30 ˚C, unless otherwise
described.

Chapter 3. Initiation of polyketide synthesis
152
8.7.1. Phosphopantetheninylation of E47V apo-ACP to produce E47V holo-ACP.
ACPS was incubated with E47V apo-ACP and CoA in a 1:10:100 ratio in TRIS buffer, pH
= 8.8 in the presence of MgCl2 (10 mM). The reaction was monitored by ESMS and
allowed for 12 h. Purification of holo-ACP from ACPS and unreacted apo-ACP was
achieved by anion-exchange chromatography using a gradient of NaCl 0-1 M as described
in Section 8.5.2.1.
8.7.2. Reduction of ACP dimers.
Holo-ACP monomer was produced after reduction of the disulfide bonds between PP arms
of two ACP by a sulphydryl reducing agent. ACP (50-500 µM) was treated for 1 h, 30 ˚C
with either tris(2-carboxyethyl)phosphine (TCEP) or dithiothreitol (DTT), at a final
concentration of 1-2 mM. ESMS of ACP species was then performed as described in
Section 8.6.1.4.
8.7.3. Acylation of holo-ACP
Following the procedure of Hitchmann, acetyl and malonyl-ACP were produced by
incubation of ACP (500 µM) with either 1-acetylimidazole (5 mM) or malonyl CoA (2 mM)
for 2-4 h, and the reactions monitored by ESMS.
8.7.4. Study of the self-malonylation of ACP using α-ketoglutarate dehydrogenase
(KGDH)
The kinetic study of self-malonylation of ACP by the α-ketoglutarate dehydrogenase
(KGDH) coupling system was carried out by measuring the change in A340 due to the
absorbance of NADH. All enzymatic reactions were in a final volume of 200 µl and
performed in 96-well microplates in the plate reader. Assays were performed essentially as
described in the literature. The final concentrations of each ingredient in the assay were as

Chapter 3. Initiation of polyketide synthesis
153
follows: 100 mM phosphate buffer pH = 7.3, 1 mM EDTA, 1 mM TCEP, 2 mM α-
ketoglutaric acid (KGA), 0.5 mM NAD+, 0.4 mM thiamine pyrophosphate (TPP), 1 mM
MgCl2 and 80 mU/ 100 µl KGDH. A working stock of 10 times their final concentrations
of EDTA, TCEP, KGA, TPP and NAD+ was made up. Assays were started by addition of
monomer holo-ACP and initial rates approximated from the linear regressions of the
absorbance traces during the first 0.5 to 2 min (6 to 24 time points).
8.7.5. Minimal actinorhodin polyketide synthase assays and HPLC analysis of SEK4
and SEK4b
All minimal PKS assays were performed in 100 mM phosphate buffer, pH = 7.3 in the
presence of 2 mM EDTA and 1 mM TCEP at 30 ˚C. The protocol for production of
polyketides by the act minimal PKS was adapted from Matharu.8 In general, malonyl CoA
was added to the reaction buffer prior to addition of KSα/KSβ. The enzyme was
equilibrated for 1 min under reaction conditions and assays were then started by addition of
monomer holo-ACP.
At designated time points, aliquots (50-100 µl) were taken from the general reaction
volume (up to 1 ml), acidified (NaH2PO4, 500 mg/ml) and extracted with 2 x 500 µl ethyl
acetate, as described by Matharu.8 A gentle stream of nitrogen helped evaporating the
organic solvent. The concentrated products were re-suspended in methanol (50 µl) and
injected into the HPLC system.
Analysis of polyketides was performed in a Dionex HPLC equipped with a pump
system P680, a Thermosthated Column Compartment TCC-100 and a PDA-100 Photodiode
Array Detector. The column used for detection and quantitative analysis of polyketides was
a Luna 5u C18 250 x 4.60 mm (Phenomenex), thermosthated at 25 ˚C. The mobile phase
for RP-HPLC was water/acetonitrile. A gradient from 5 to 75% acetonitrile was carried out
over 35 min at 1 ml/min, followed by washing (95% acetonitrile) and re-equilibration (95%
H2O) steps. SEK4 was detected after 19.6 min and SEK4b after 20.3 min. A slight
difference in retention times was observed when a Luna 5u C18(2) by Phenomenex was
used. The elution volume of SEK4 was 19.1 min; SEK4b was detected after 19.8 min.

Chapter 3. Initiation of polyketide synthesis
154
8.7.6. Kinetic studies on the actinorhodin minimal polyketide synthase
Kinetics of the actinorhodin minimal polyketide synthase were studied in a Spectramax 190
plate reader (Molecular devices), by monitoring the absorbance of the polyketides
produced.
8.7.6.1. Calibration of the method
Solutions of purified SEK4 and SEK4b were used as standards for calibration. Working
solutions of 0.1 mg/ml (314 µM) SEK4 and 1 mg/ml (3.14 mM) SEK4b in 1:1 water:
acetonitrile were made up, and diluted in 100 mM phosphate buffer, pH = 7.3 to the desired
concentrations in a total volume of 200 µl. The absorbance at 293 nm was read and the
calibration curve (1-100 µM) constructed. The experimental error was estimated as less
than 7% by repeating the calibration procedure with three independent series.
8.7.6.2. Minimal polyketide synthase assays to measure self-malonylation of holo-
ACP
All enzymatic reactions were in a final volume of 200 µl and performed in an absorbance
plate reader. The final concentrations of all components were 100 mM phosphate buffer, 2
mM EDTA, 1 mM TCEP, 5 to 1000 µM malonyl CoA, 3 µM KSα/KSβ and 0.2 to 160 µM
holo-ACP. KSα/KSβ was incubated for 2 minutes in the reaction mixture containing
malonyl CoA, EDTA and TCEP, which was pre-equilibrated at 30 ˚C in the plate reader.
Assays were started by addition of monomer holo-ACP. The absorbance of the samples
was immediately measured for 3-20 minutes, every 4 seconds. Initial reaction rates were
calculated from the linear regressions of the absorbance traces during the linear period,
usually 1 to 5 minutes (15 to 60 time points).

Chapter 3. Initiation of polyketide synthesis
155
8.7.6.3. Minimal polyketide synthase assays to measure inhibition of self-
malonylation of holo-ACP by CoA-SH
CoA-SH was purchased from Sigma, aliquoted in 100 mM fractions and kept at -20 ˚C. The
concentration of CoA-SH in inhibition assays ranged from 500 to 2000 µM. Concentrations
of holo-ACP and KSα/KSβ were 50 µM and 3 µM respectively.
8.7.6.4. Minimal polyketide synthase assays to measure the kinetics of KSβ
To study the kinetics of KSβ, S. coelicolor MCAT (0.5-2.5 µM) was added to the reaction
mixture to accelerate the malonylation of ACP. The concentration of KSα/KSβ in these
assays was 0.2-0.5 µM. ACP concentrations were varied as needed for the determination of
kinetic parameters. MCAT-supplemented act minimal assays were then carried out as
described in Section 8.7.6.2.
8.7.6.5. Minimal polyketide synthase assays to measure the kinetics of KSα
In order to study the kinetics of KSα, acetyl-ACP was added to a concentration of 30 µM,
and assays then started by addition of malonyl CoA (1 mM). MCAT was added in
concentrations of 0.5 µM. Holo-ACP concentration in the assays was about 5 to 10-fold the
KM value for the corresponding ACP species.For the rest, assays were executed as
described in Section 8.7.6.2.
8.7.6.6. Minimal polyketide synthase assays to measure inhibition of KSα/KSβ by
apo-ACP
Assays were carried out as described in Section 8.7.6.2. MCAT concentration was 0.5 µM.
KSα/KSβ concentration was 0.25 µM. Holo-ACP concentration was 20 µM. Apo-ACP was
added in concentrations of 25-200 µM.

Chapter 3. Initiation of polyketide synthesis
156
8.7.6.7. Minimal polyketide synthase assays to measure the kinetics of CA KSα/KSβ
To study the kinetics of CA KSα/KSβ, S. coelicolor MCAT (0.5-2.5 µM) was added to
assays in which the concentration of mutant KSα/KSβ was 1-2 µM. and assays were then
carried out as described in Section 8.7.6.2.
8.7.6.8. Data analysis
Initial reaction rates were deduced from absorbance data by fitting to a straight line using
the software provided by the manufacturer of the plate reader (Molecular Devices). In
general, assays were done at least in triplicate, and the average value is shown throughout
this thesis as a mean plus or minus an error.
For the determination of Michaelis-Menten parameters, these data were used to produce
plots showing the hyperbolic dependence of reaction rates on substrate concentration.
Kinetic parameters were then calculated by fitting this plots to a hyperbolic function using
Sigmaplot 8TM.
Hanes linear plots were also produced to assess the quality of the kinetic data. Hanes
plots result from Eq. 4 (see Section 1.4.1), with some re-arrangement:
!
[ S ] / rate =KM
kcat [ Eo]+
1
kcat [ Eo][ S ] (Eq. 8)
This means that a plot of [S] / rate vs. [S] should be a straight line. This plot was used as it
is recommended by modern enzyme kinetics textbooks as the less biased linear treatment of
kinetic data.94

Chapter 3. Initiation of polyketide synthesis
157
1 Mann, J. (1987) Secondary Metabolism, 2nd edition, Clarendon Press, Oxford.
2 Luckner, M. (1990) Secondary metabolism in microorganism, plants and animals,
3rd edition, Springer-Verlag, Berlin.
3 Staunton, J., and Weissman, K. J. (2001) Polyketide Biosynthesis: a millennium
review, Nat Prod Rep. 18, 380-416.
4 Cox, R. J. (2002) Biosynthesis, Annu Rep Prog Chem, Sect. B, 98, 223-251.
5 Cox, R. J. (2007) Polyketides, proteins and genes in fungi: programmed nano-
machines begin to reveal their secrets, Org Biomol Chem. 5, 2010-2026.
6 Kieser, T., Bibb, M.J., Buttner, M.J., Chater, K.F. and Hopwood, D.A. (2000)
Practical Streptomyces genetics, The John Innes Foundation, Chichester.
7 Richardson, M. and Khosla, C. (1999) Comprehensible Natural Products Chemistry
v.1. Elsevier.
8 Matharu, A.L., Cox, R.J., Crosby, J., Byrom, K.J. and Simpson, T.J. (1998) MCAT
is not required for in vitro polyketide synthesis in a minimal actinorhodin
polyketide synthase from Streptomyces coelicolor, Chem Biol 5, 699-711.
9 Wohlert, S., Wendt-Pienkowski, E., Bao, W. and Hutchinson, C.R. (2001)
Production of Aromatic Minimal Polyketides by the Daunorubicin Polyketide
Synthase Genes Reveals the Incompatibility of the Heterologous DpsY and JadI
Cyclases, J Nat Prod. 64, 1077-1080.
10 Taguchi, T., Ebizuka, Y., Hopwood, D.A. and Ichinose, K. (2001) A new mode of
stereochemical control revealed by analysis of the biosynthesis of dihydrogranaticin
in Streptomyces violaceoruber Tu22, J Am Chem Soc. 123, 11376-11380.
11 Yu, T-W., Bibb, M. J., Revill, W. P. and Hopwood, D. A. (1994) Cloning,
Sequencing, and Analysis of the Griseusin Polyketide Synthase Gene Cluster from
Streptomyces griseus, J Bacteriol 176, 2627-2634.
12 Räty, K., Kantola, J., Hautala J., Hakala, J., Ylihonko, K. and Mäntsälä, P. (2002)
Cloning and characterization of Streptomyces galilaeus aclacinomycins polyketide
synthase (PKS) cluster, Gene 293, 115-122.
13 Meadows E.S. and Khosla,C. (2001) In Vitro Reconstitution and Analysis of the
Chain Initiating Enzymes of the R1128 Polyketide Synthase, Biochemistry 40,
14855-14861.
14 Petkovi, H., Thamchaipenet, A., Zhou, L., Hranueli� , D., Raspor, P., Waterman,
P.G., and Hunter, I.S. (1999) Disruption of an aromatase/cyclase from the

Chapter 3. Initiation of polyketide synthesis
158
oxytetracycline gene cluster of Streptomyces rimosus results in production of novel
polyketides with shorter chain lengths, J Biol Chem. 274, 32829-32834.
15 Piel, J., Hertweck, C., Shipley, P. R., Hunt, D. M., Newman, M. S., and Moore, B.
S. (2000) Cloning, sequencing and analysis of the enterocin biosynthesis gene
cluster from the marine isolate Streptomyces maritimus: evidence for the derailment
of an aromatic polyketide synthase, Chem Biol. 7, 943–955.
16 Bentley, S.D., Chater, K.F., Cerdeno-Tarraga, A.M., Challis, G.L., Thomson, N.R.,
James, K.D., Harris, D.E., Quail, M.A., Kieser, H., Harper, D., Bateman, A.,
Brown, S., Chandra, G., Chen, C.W., Collins, M., Cronin, A., Fraser, A., Goble, A.,
Hidalgo, J., Hornsby, T., Howarth, S., Huang, C.H., Kieser, T., Larke, L., Murphy,
L., Oliver, K., O'Neil, S., Rabbinowitsch, E., Rajandream, M.A., Rutherford, K.,
Rutter, S., Seeger, K., Saunders, D., Sharp, S., Squares, R., Squares, S., Taylor, K.,
Warren, T., Wietzorrek, A., Woodward, J., Barrell, B.G., Parkhill, J. and Hopwood
D.A. (2002) Complete genome sequence of the model actinomycete Streptomyces
coelicolor A3(2), Nature 417, 141-147.
17 Rudd, B.A and Hopwood, D. A. (1979) Genetics of actinorhodin biosynthesis by
Streptomyces coelicolor A3(2), J Gen Microbiol. 114, 35-43.
18 Malpartida, F. and Hopwood, D. A. (1984) Molecular-Cloning of the whole
Biosynthetic-Pathway of a Streptomyces Antibiotic and its Expression in a
Heterologous Host, Nature 309, 462-464
19 Hallam, S. E., Malpartida, F. and Hopwood, D. A. (1988) Nucleotide sequence,
transcription and deduced function of a gene involved in polyketide antibiotic
synthesis in Streptomyces coelicolor, Gene 74, 305-320.
20 Fernandez-Moreno, M. A., Martinez, E., Boto, L., Hopwood, D. A. and Malpartida,
F. (1992) Nucleotide-Sequence and Deduced Functions of a Set of Co-transcribed
Genes of Streptomyces coelicolor A3(2), including the Polyketide Synthase for the
Antibiotic Actinorhodin, J Biol Chem. 267, 19278-19290.
21 Hopwood, D.A. (1997) Genetic contributions to understanding polyketide
synthases, Chem Rev. 97, 2465-2497.
22 McDaniel, R., Ebert-Khosla, S., Hopwood, D.A. and Khosla, C. (1993) Engineered
biosynthesis of novel polyketides, Science 262, 1546-1550.
23 McDaniel, R., Ebert-Khosla, S., Hopwood, D. A. and Khosla, C. (1994) Engineered
biosynthesis of novel polyketides: influence of a downstream enzyme on the

Chapter 3. Initiation of polyketide synthesis
159
catalytic specificity of a minimal aromatic polyketide synthase, P Natl Acad Sci
U.S.A. 91, 11542-11546.
24 Fu, H., Ebert-Khosla, S., Hopwood, D.A. and Khosla, C. (1994) Engineered
Biosynthesis of Novel Polyketides: Dissection of the Catalytic Specificity of the act
Ketoreductase, J Am Chem Soc. 116, 4166-4170.
25 McDaniel, R., Evert-Khosla, S., Hopwood, D. A. and Khosla, C. (1994) Engineered
Biosynthesis of Novel Polyketides: actVII and actIV Genes Encode Aromatase and
Cyclase Enzymes, Respectively, J Am Chem Soc. 116, 10855-10859.
26 Valton, J., Fontecave, M., Douki, T., Kendrew, S.G and Niviere, V. (2006) An
aromatic hydroxylation reaction catalyzed by a two-component FMN-dependent
monooxygenase, J Biol Chem. 281, 27-35.
27 Taguchi, T., Kunieda, K., Takeda-Shitaka, M., Takaya, D., Kawano, N., Kimberley,
M.R., Booker-Milburn, K.I., Stephenson, G.R., Umeyama, H., Ebizuka, Y. and
Ichinose, K. (2004) Remarkably different structures and reaction mechanisms of
ketoreductases for the opposite stereochemical control in the biosynthesis of BIQ
antibiotics, Bioorg Med Chem. 12, 5917-5927.
28 Taguchi, T., Okamoto, S., Lezhava, A., Li, A., Ochi, Y. Ebizuka, Y. and Ichinose,
K. (2007) Possible involvement of ActVI-ORFA in transcriptional regulation of
actVI tailoring-step genes for actinorhodin biosynthesis, FEMS Microbio Lett. 269,
234-239.
29 Booker-Milburn, K.I., Gillan, R., Kimberley, M., Taguchi, T., Ichinose, K.,
Stephenson, G.R., Ebizuka, Y. and Hopwood, D.A. (2005) Enantioselective
reduction of β-keto acids with engineered Streptomyces coelicolor, Angew Chem Int
Edit. 44, 1121-1125.
30 Crump, M.P., Dempsey, C.E., Parkinson, J.A., Murray, M., Hopwood, D.A. and
Simpson, T.J. (1997) Solution structure of the actinorhodin polyketide synthase acyl
carrier protein from Streptomyces coelicolor A3(2), Biochemistry 36, 6000-60088.
31 Keating-Clay, A.T., Maltby, D.A., Medzihradszky K.F., Khosla, C. and Stroud,
R.M. (2004) An antibiotic factory caught in action, Nat Struc Mol Biol. 11, 888-
893.
32 Hadfield, A.T., Limpkin, C., Teartasin, W., Simpson, T.J., Crosby, J. and Crump,
M. (2004) The crystal structure of the actIII actinorhodin polyketide reductase:
proposed mechanism for ACP and polyketide binding, Structure 12, 1865-1875.

Chapter 3. Initiation of polyketide synthesis
160
33 Sciara, G., Kendrew, S.G., Miele, A.E., Marsh, N.G., Federici, L., Malatesta, F.,
Schimperna, G., Savino, C. and Vallone, B. (2003) The structure of ActVA-Orf6, a
novel type of monooxygenase involved in actinorhodin biosynthesis, EMBO J. 22,
205.
34 Fu, H., Hopwood, D.A. and Khosla, C. (1994) Engineered biosynthesis of novel
polyketides: evidence for temporal, but not regiospecific, control of cyclization of
an aromatic polyketide precursor, Chem Biol. 1, 205-210.
35 McDaniel, R., Ebert-Khosla, S., Hopwood, D.A. and Khosla, C. (1993) Engineered
biosynthesis of novel polyketides, Science 262, 1546-1550.
36 Holak, T.A., Kearsley, S.K., Kim, Y. and Prestegard, J.H (1988) Three-dimensional
structure of acyl carrier protein determined by NMR pseudoenergy and distance
geometry calculations, Biochemistry 27, 6135-6142.
37 Xu, G-Y., Tam, A., Lin, L., Hixon, J., Fritz, C.C. and Powers, R (2001) Solution
structure of B. subtilis acyl carrier protein, Structure 9, 277-287.
38 Wong, H.C., Liu, G., Zhang, Y., Rock, C.O. and Zheng, J. (2002) The solution
structure of acyl carrier protein from Mycobacterium tuberculosis, J Biol Chem.
277, 15874-15880.
39 Li, Q., Khosla, C., Puglisi, J.D and Liu, C.W. (2003) Solution structure and
backbone dynamics of the holo form of the frenolicin acyl carrier protein,
Biochemistry 42, 4648-4657.
40 Kim, Y. and Prestegard, J.H. (1990) Demostration of a conformational equilibrium
in acyl carrier protein from spinach using rotating frame nuclear magnetic
resonance spectrometry, J Am Chem Soc. 112, 3707-3709.
41 Sharma, A.K., Sharma, S.K., Surolia, A., Surolia, N. and Sarma, S.P. (2006)
Solution structures of conformationally equilibrium forms of holo-acyl carrier
protein (PfACP) from Plasmodium falciparum provides insight into the mechanism
of activation of ACPs, Biochemistry 45, 6904-6916.
42 Cox, R. J., Crosby, J., Daltrop, O., Glod, F., Jarzabek, M., Nicholson, T. P,
Simpson, T. J., Soulas, F. and Westcott, J. (2002) Cloning, expression and substrate
activity of Streptomyces coelicolor holo-Acyl Carrier Protein synthase, J. Chem.
Soc., Perkin Trans 1, 2002.

Chapter 3. Initiation of polyketide synthesis
161
43 Revill, W. P., Bibb, M. J. and Hopwood, D. A. (1995) Purification of a
malonyltransferase from Streptomyces coelicolor A3(2) and analysis of its genetic
determinant, J Bacteriol. 177, 3946-3952.
44 Summers, R. G., Ali, A., Shen, B., Wessel, W. A. and Hutchinson, C. R. (1995)
Malonyl-coenzyme A:acyl carrier protein acyltransferase of Streptomyces
glaucescens: a possible link between fatty acid and polyketide biosynthesis,
Biochemistry 34, 9389-9402.
45 Keatinge-Clay, A. T., Shelat, A. A., Savage, D. F., Tsai, S. C., Miercke, L. J.,
O'Connell, J. D. 3rd, Khosla, C. and Stroud, R. M. (2003) Catalysis, specificity, and
ACP docking site of Streptomyces coelicolor malonyl-CoA:ACP transacylase,
Structure 11, 147-154.
46 Szafranska, A.E., Hitchman T.S., Cox, R.J., Crosby, J. and Simpson, T.J. (2002)
Kinetic and mechanistic analysis of the malonyl CoA: ACP transacylase from
Streptomyces coelicolor indicates a single catalytically competent serine
nucleophile at the active site, Biochemistry 41, 1421-1427.
47 Cox, R.J., Hitchman, T.S., Byrom, K.J., Findlow, S.C., Tanner, J.A., Crosby, J. and
Simpson, T.J. (1997) Post-translational modification of heterologously expressed
Streptomyces Type II polyketide synthase acyl carrier proteins, FEBS Lett. 405,
267-272.
48 Haapalainen, A.M., Merilainen, G. and Wierenga, R.K. (2006) The thiolase
superfamily: condensing enzymes with diverse reaction specificities, Trends
Biochem Sci. 31, 64-71.
49 Zhang, Y-M., Hurlbert, J., White, S. W., and Rock, C. O. (2006) Roles of the
Active Site Water, Histidine 303, and Phenylalanine 396 in the Catalytic
Mechanism of the Elongation Condensing Enzyme of Streptococcus pneumoniae, J
Biol Chem. 281, 17390-17399.
50 Price, A.C., Choi, K-H., Heath, R.J., Li, Z., White, S.W. and Rock, C.O. (2001)
Inhibition of β-ketoacyl-acyl carrier protein synthases by thiolactomycin and
cerulenin. Structure and mechanism, J Biol Chem. 276, 6551-6559.
51 Olsen, J.G., Kadziola, A., von Wettstein-Knowles, P., Siggaard-Andersen, M. and
Larsen, S. (2001) Structures of β-ketoacyl-acyl carrier protein synthase I complexed
with fatty acids elucidate its catalytic machinery, Structure 9, 233-243.

Chapter 3. Initiation of polyketide synthesis
162
52 Crosby, J., Byrom, K., Hitchman, T.S., Cox, R.J., Crump, M.P., Findlow, S.C.,
Bibb, M.J. and Simpson, T.S. (1998) Acylation of Streptomyces Type II polyketide
synthase acyl carrier proteins, FEBS Lett. 433, 132-138.
53 Carreras, C.W. and Khosla, C. (1998) Purification and in Vitro Reconstitution of the
Essential Protein Components of an Aromatic Polyketide Synthase, Biochemistry
37, 2084.
54 Tang, Y., Tsai, S.C. and Khosla, C. (2003) Polyketide chain length control by chain
length factor, J Am Chem Soc. 125, 12708-12709.
55 Hitchman, T.S., Crosby, J., Byrom, K.J., Cox, R.J. and Simpson, T.J. (1998)
Catalytic self-acylation of type II polyketide synthase acyl carrier proteins, Chem
Biol. 5, 35-47.
56 Dreier, J., Shah, A.N. and Khosla, C. (1999) Kinetic analysis of the actinorhodin
aromatic polyketide synthase, J Biol Chem. 274, 25108-25112.
57 Zhou, P., Florova, G., and Reynolds, K.A. (1999) Polyketide synthase acyl carrier
protein (ACP) as a substrate and a catalyst for malonyl ACP biosynthesis, Chem
Biol. 6, 577-584.
58 Florova, G., Kazanina, G. and Reynolds, K.A. (2002) Enzymes involved in fatty
acid and polyketide biosynthesis in Streptomyces glaucescens: role of FabH and
FabD and their acyl carrier protein specificity Biochemistry 41, 10462-10471.
59 Arthur, C.J., Szafranska, A., Evans, S.E., Findlow, S.C., Burston, S.G., Owen, P.,
Clark-Lewis, I., Simpson, J., Crosby, J. and Crump, M.P. (2005) Self-malonylation
is an intrinsic property of a chemically synthesized type II polyketide synthase acyl
carrier protein, Biochemistry 44, 15414-15421.
60 Arthur, C.J., Szafranska, A., Long, J., Mills, J., Cox, R.J., Findlow, S.C., Simpson,
T.J., Crump, M.P. and Crosby, J. (2006) The malonyl transfer activity of Type II
PKS acyl carrier proteins, Chem Biol. 13, 587-596.
61 Misra, A., Sharma, S. K., Surolia, N. and Surolia, A. (2007) Self-Acylation
Properties of Type II Fatty Acid Biosynthesis Acyl Carrier Protein, Chem Biol. 14,
775-783.
62 Bisang, C., Long, P.F., Cortes, J., Westcott, J., Crosby, J., Matharu, A.L., Cox, R.J.,
Simpson, T.J., Staunton, J. and Leadlay, P.F. (1999) A chain initiation factor
common to both modular and aromatic polyketide synthases, Nature 401, 502-505.

Chapter 3. Initiation of polyketide synthesis
163
63 Tsai, S-C. (2004) A Fine Balancing Act of Type III Polyketide Synthases, Chem
Biol. 11, 1174-1175.
64 Gryschow, S., Buchholz, T. J., Seufert, W., Dordick, J. S. and Sherman, D. H.
(2007) Substrate Profile Analysis and ACP-Mediated Acyl Transfer in Streptomyces
coelicolor Type III Polyketide Synthases, ChemBioChem 8, 863-868.
65 Austin, M. B. and Noel, J.P. (2003) The chalcone synthase superfamily of type III
polyketide synthases, Nat Prod Rep. 20, 79–110.
66 Katsuyama, Y. Matsuzawa, M. Funa, N. and Horinouchi, S. (2007) In Vitro
Synthesis Of Curcuminoids By Type III Polyketide Synthase From Oryza Sativa, J
Biol Chem. 282, 37702-37709.
67 Jez, J. M., Ferrer, J. L., Bowman, M. E., Dixon, R.A and Noel, J.P. (2000) Structure
of chalcone synthase and the molecular basis of plant polyketide biosynthesis, Nat
Struc Biol. 6, 775-785.
68 Austin, M. B., Bowman, M.E., Ferrer, J. L., Schröder, J. and Noel, J.P. (2004) An
aldol switch discovered in stilbene synthases mediates cyclization specificity of type
III polyketide synthases, Chem Biol. 11, 1179-1194. 69 Shomura, Y., Torayama, I., Suh, D. Y., Xiang, T., Kita, A., Sankawa, U., Miki, K.
(2005) Crystal structure of stilbene synthase from Arachis hypogaea, Proteins 60,
803-806.
70 Hopwood, D.A. (1997) Genetic contributions to understanding polyketides
synthases, Chem Rev. 97, 2465-2497.
71 Hill, A.M. (2006) The biosynthesis, molecular genetics and enzymology of the
polyketide-derived metabolites, Nat Prod Rep. 23, 256-320.
72 McDaniel, R., Welch, M. and Hutchinson, C.R. (2005) Genetic approaches to
polyketide antibiotics 1, Chem Rev. 105, 543-558.
73 Khosla, C., Tang, Y., Chen, A. Y., Schnarr, N. and Cane, D. (2007) Structure and
mechanism of the 6-Deoxyerythronolide B Synthase, Annu Rev Biochem 76, 11.1-
11.27
74 Townsend, C. A., Crawford, J. M and Bililign, T. (2006) New images evoke
FAScinating questions, Chem Biol. 13, 349-351.
75 Wakil, S. J., Pugh, E. L., and Sauer, F. (1964) The Mechanism of Fatty Acid
Synthesis, P Natl Acad Sci U. S. A. 52, 106-114.

Chapter 3. Initiation of polyketide synthesis
164
76 Stoops, J.K., Arslanian, A.W., Oh, Y.H., Aune, K.C., Vanaman, T.C., and Wakil,
S.J. (1975) Presence of Two Polypeptide Chains comprising Fatty Acid Synthetase,
P Natl Acad Sci U. S. A. 75, 1940-1944.
77 Stoops, J. K and Wakil, S. J. (1981) Animal Fatty Acid Synthetase: A Novel
Arrangement of the β-Ketoacyl Synthetase Sites Comprising Domains of the Two
Subunits, J Biol Chem. 256, 5128-5133.
78 Stoops, J. K., Wakil, S. J., Uberbacherl, E. C., and Bunick, G. J. (1987) Small-angle
Neutron-scattering and Electron Microscope Studies of the Chicken Liver Fatty
Acid Synthase, J Biol Chem. 262, 10246-10251.
79 Joshi, A. K., Witkowsky, A. and Smith, S. (1997) Mapping of functional
interactions between domains of the animal fatty acid synthase by mutant
complementation in vitro, Biochemistry 36, 2316-2322.
80 Joshi, A. K., Witkowsky, A. and Smith, S. (1998) The Malonyl/Acetyltransferase
and β-Ketoacyl Synthase Domains of the Animal Fatty Acid Synthase Can
Cooperate with the Acyl Carrier Protein Domain of Either Subunit, Biochemistry
37, 2515-2523.
81 Smith, S. and Tsai, S-C (2007) The type I fatty acid and polyketide synthases: a tale
of two megasynthases, Nat Prod Rep. 24, 1041-1072.
82 Witkowski, A., Joshi, A. K., Rangan, V. S., Falick, A. M., Witkowska, H. E. and
Smith, S. (1998) Dibromopropanone Cross-linking of the Phosphopantetheine and
Active-site Cysteine Thiols of the Animal Fatty Acid Synthase Can Occur Both
Inter- and Intrasubunit, J Biol Chem 274, 11557-11563.
83 Chirala S. S. and Wakil S. J. (2004) Structure and Function of Animal Fatty Acid
Synthase, Lipids 39, 1045-1053.
84 Smith, S., Witkowski, A. and Joshi, A. K. (2003) Structural and functional
organization of the animal fatty acid synthase, Prog Lipid Res. 42, 289-317.
85 Maier, T., Jenni, S and Ban, N. (2006) Architecture of the mammalian fatty acid
synthase at 4.5 Å resolution, Science 311, 1258-1262.
86 Asturias, F. J., Chadick, J. Z., Cheung, I. K., Stark, H., Witkowski, A., Joshi, A. K.
and Smith, S. (2005) Structural and functional organization of the animal fatty acid
synthase, Nat Struc Mol Biol. 12, 225–232.

Chapter 3. Initiation of polyketide synthesis
165
87 Tang, Y., Kim, C.Y., Mathews, I.I., Cane, D.E. and Khosla, C. (2006) The 2.7-Å
crystal structure of a 194-kDa homodimeric fragment of the 6-deoxyerythronolide B
synthase. P Natl Acad Sci U. S. A. 103, 11124-11129.
88 Keatinge-Clay, A.T. and Stroud R.M. (2006) The Structure of a Ketoreductase
Determines the Organization of the β-Carbon Processing Enzymes of Modular
Polyketide Synthases, Structure 14, 737-748.
89 Tsai, S.-C., Lu, H., Cane, D.E., Khosla, C. and Stroud, R.M. (2003) Insights into
channel architecture and substrate specificity from crystal structures of two
macrocycle-forming thioesterases of modular polyketide synthases, Biochemistry
41,12598-12606.
90 Tsai, S.C., Miercke, L.J., Krucinski, J., Gokhale, R., Chen, J.C., Foster, P.G., Cane,
D.E., Khosla, C. and Stroud, R.M. (2001) Crystal structure of the macrocycle-
forming thioesterase domain of the erythromycin polyketide synthase: versatility
from a unique substrate channel, P Natl Acad Sci U. S. A. 98,14808-14813.
91 Broadhurst, R.W., Nietlispach, D., Wheatcroft, M.P., Leadlay, P.F. and Weissman,
K.J. (2003) The structure of docking domains in modular polyketide synthases,
Chem Biol. 10, 723-731.
92 Alekseyev, V.V., Liu, C. W., Cane, D. E., Puglisi, J. D. and Khosla, C. (2007)
Solution structure and proposed domain–domain recognition interface of an acyl
carrier protein domain from a modular polyketide synthase, Protein Sci. 16, 2093-
2107.
93 Buc, J. and Giordani, R. (1998) A spectrophotometric method for kinetic studies
with quinone-dependent oxidoreductases. Application to detection in membranes of
nitrate reductase activity with menadione and duroquinone as electron donors
Enzyme Microb Tech. 22, 165-169.
94 Cornish-Bowden, A. (2004) Fundamentals of Enzyme Kinetics, 3rd ed. Portland
Press, London.
95 Branden, C. and Tooze, J. (1999) Introduction to Protein Structure, 2nd ed, Garland
Publishing Inc., New York.
96 Nicholson, T.P., Winfield, C., Westcott, J., Crosby, J., Simpson, T.J. and Cox, R.J.
(2004) First in vitro directed biosynthesis of new compounds by a minimal Type II
polyketide synthase: evidence for the mechanism of chain determination, Chem.
Comm. 6, 686-687.

Chapter 3. Initiation of polyketide synthesis
166
97 Beltran-Alvarez, P., Cox, R. J., Crosby, J. and Simpson, T. J. (2007) Dissecting the
component reactions catalyzed by the actinorhodin minimal polyketide synthase,
Biochemistry 46, 14672-14681.
98 Tang, Y., Lee, T-S., Kobayashi, S. and Khosla, C. (2003) Ketosynthases in the
initiation and elongation modules of aromatic polyketide synthases have orthogonal
acyl carrier protein specificity, Biochemistry 42, 6588-6595.
99 Lee, T-S., Khosla, C. and Tang, Y. (2005) Orthogonal protein interactions in spore
pigment producing and antibiotic producing polyketide synthases, J Antibiot. 58,
663-666.
100 Wu, N., Cane, D. E. and Khosla, C. (2002) Quantitative analysis of the relative
contributions of donor acyl carrier proteins, acceptor ketosynthases, and linker
regions to intermodular transfer of intermediates in hybrid polyketide synthases,
Biochemistry 41, 5056-5066.
101 Mathews, D.K. and van Holde, K.E. (2000) Biochemistry, 2nd ed. McGraw-Hill,
Boston.
102 Holak, T. A., Kearsley, S.K., Kim, Y. and Prestegard, J. H. (1998) Three-
dimensional structure of acyl carrier protein determined by NMR pseudoenergy and
distance geometry calculations, Biochemistry 27, 6135-6142.
103 Olsen, J. G., Kadziola, A., Wettstein-Knowles, P., Siggaard-Andersen, M.,
Lindquist, Y., and Larsen, S. (1999) The X-ray crystal structure of β-ketoacyl [acyl
carrier protein] synthase I, FEBS Lett. 460, 46–52
104 Serre, L., Swenson, L., Green, R., Wei, Y., Verwoert, I.I.G.S., Verbree, E.C.,
Stuitje, A.R. and Derewenda, Z.S. (1994) Crystallization of the malonyl coenzyme
A-acyl carrier protein transacylase from Escherichia coli. J Mol Biol. 242, 99-102.
105 Serre, L., Verbree, E.C., Dauter, Z., Stuitje, A.R. and Derewenda, Z.S. (1995) The
Escherichia coli malonyl-CoA:acyl carrier protein transacylase at 1.5-Å resolution.
Crystal structure of a fatty acid synthase component, J Biol Chem. 270, 12961-
12964.
106 Moche, M., Schneider, G., Edwards, P., Dehesh, K. and Lindqvist, Y. (1999)
Structure of the complex between the antibiotic cerulenin and its target, β-ketoacyl-
acyl carrier protein synthase, J Biol Chem. 274, 6031-6034.

Chapter 3. Initiation of polyketide synthesis
167
107 Huang W., Jia J., Edwards P., Dehesh K., Schneider G. and Lindqvist Y. (1998)
Crystal structure of β-ketoacyl-acyl carrier protein synthase II from E.coli reveals
the molecular architecture of condensing enzymes, EMBO J. 17, 1183-1191.
108 Davies, C., Health, R.J., White, S.W. and Rock, C.O (2000) The 1.8 Å crystal
structure and active-site architecture of β-ketoacyl-acyl carrier protein synthase III
(FabH) from Escherichia coli, Structure 8, 185-195.
109 Price, A. C., Zhang, Y. M., Rock, C. O. and White, S. W. (2001) Structure of β-
ketoacyl-acyl carrier protein reductase from Escherichia coli: negative cooperativity
and its structural basis, Biochemistry 40, 417-428.
110 Baldock C., Rafferty J. B., Stuitje A. R., Slabas A. R. and Rice D. W. (1998) The
X-ray structure of Escherichia coli enoyl reductase with bound NAD+ at 2.1 Å
resolution, J Mol Biol. 284, 1529-46.
111 Leesong, M., Henderson, B. S., Gillig, J. R., Schwab, J. M. and Smith, J. L. (1996)
Structure of a dehydratase-isomerase from the bacterial pathway for biosynthesis of
unsaturated fatty acids: two catalytic activities in one active site, Structure 4, 253-
264.
112 Zhang, Y-M., Rao, M.S., Heath, R.J., Price, A.C., Olson, A.J., Rock, C.O. and
White, S.W. (2001) Identification and analysis of the acyl carrier protein (ACP)
docking site on β-ketoacyl-ACP synthase III, J Biol Chem. 276, 8231-8238.
113 Keatinge-Clay, A.T., Shelat, A. A., Savage, D. F., Tsai, S-C., Miercke, L. J.,
O'Connell, J. D., Khosla, C. and Stroud, R. M. (2003) Catalysis, Specificity, and
ACP Docking Site of Streptomyces coelicolor Malonyl-CoA:ACP Transacylase,
Structure 11, 147-154.
114 Zhang, L., Liu, W., Xiao, J., Hu, T., Chen, J., Chen, K., Jiang, H., and Shen, X.
(2007) Malonyl-CoA: acyl carrier protein transacylase from Helicobacter pylori:
Crystal structure and its interaction with acyl carrier protein, Protein Sci. 16, 1184-
1192.
115 Lai, J. R., Koglin, A and Walsh, C. T. (2006) Carrier Protein Structure and
Recognition in Polyketide and Nonribosomal Peptide Biosynthesis, Biochemistry
45, 14869-14879.
116 Parris, K. D., Lin, L., Tam, A., Mathew, R., Hixon, J., Stahl, M., Fritz, C. C.,
Seehra, J., and Somers, W. S. (2000) Crystal structures of substrate binding to

Chapter 3. Initiation of polyketide synthesis
168
Bacillus subtilis holo-(acyl carrier protein) synthase reveal a novel trimeric
arrangement of molecules resulting in three active sites, Structure 8, 883–895.
117 Rafi, S., Novichenok, P., Kolappan, S., Zhang, X., Stratton, C. F., Rawat, R.,
Kisker, C., Simmerling, C. and Tonge, P. J. (2006) Structure of acyl carrier protein
bound to FabI, the FASII Enoyl Reductase from Escherichia coli, J Biol Chem. 281,
39285-39293.
118 Zhang, Y. M., Wu, B. N., Zheng, J. and Rock, C.O. (2003) Key residues responsible
for acyl carrier protein and β-ketoacyl-acyl carrier protein reductase (FabG)
interaction, J Biol Chem. 278, 52935-52943.
119 De Lay, N. R. and Cronan, J. E. (2007) In Vivo Functional Analyses of the Type II
Acyl Carrier Proteins of Fatty Acid Biosynthesis, J Biol Chem. 282, 20319-20328.
120 Arthur, C. J. (2004) PhD thesis, University of Bristol, UK.
121 Leibundgut, M., Jenni, S., Frick, C., Ban, N. (2007) Structural Basis for Substrate
Delivery by Acyl Carrier Protein in the Yeast Fatty Acid Synthase, Science 316,
288-290.
122 Westcott, J. (2001) PhD thesis, University of Bristol.
123 Burmeister Getz E., Xiao, M., Chakrabarty, T., Cooke, R. and Selvin, P. (1999) A
Comparison between the Sulfhydryl Reductants Tris(2-carboxyethyl)phosphine and
Dithiothreitol for Use in Protein Biochemistry, Anal Biochem. 273, 73-80.
124 Izumikawa, M., Cheng, Q. and Moore, B. S. (2006) Priming Type II Polyketide
Synthase via a Type II Nonribosomal Peptide Synthetase Mechanism, J Am Chem
Soc. 128, 1428-1429.
125 Hamada, M., Koike, K., Nakaula, Y., Hiraoka, T., Koike, M. and Hashimoto, T.
(1975) A kinetic study of the α-keto acid dehydrogenase complexes from pig heart
mitochondria, J Biochem 77, 1047-1056.
126 Bunik, V., Westpahl, A.H and de Kok, A. (2000) Kinetic properties of the 2-
oxoglutarate dehydrogenase complex from Azotobacter vinelandii evidence for the
formation of a precatalytic complex with 2-oxoglutarate, Eur J Biochemi. 267,
3583-3591.
127 Khandekar, S.S., Gentry, D.R., van Aller, G.S., Warren, P., Xiang, H., Silverman,
C., Doyle, M.L., Chambers, P.A., Konstantinidis, A.K., Brandt, M., Daines, R.A
and Lonsdale, J.T. (2001) Identification, substrate specificity, and inhibition of the

Chapter 3. Initiation of polyketide synthesis
169
Streptococcus pneumoniae β-ketoacyl-acyl carrier protein synthase III (FabH), J
Biol Chem. 276, 30024-30030.
128 Molnos, J., Gardiner, R., Dale, G.E. and Lange, R. (2003) A continuous coupled
enzyme assay for bacterial malonyl-CoA:acyl carrier protein transacylase (FabD),
Anal Biochem. 319, 171-176.
129 Zha, W., Rubin-Pitel, S.B. and Zhao, H. (2006) Characterization of the substrate
specificity of PhlD, a type III polyketide synthase from Pseudomonas fluorescens, J
Biol Chem. 281, 32036-32047.
130 Liu, W., Han, C., Hu, L., Chen, K., Shen, X. and Jiang, H. (2006) Characterization
and inhibitor discovery of one novel malonyl-CoA: acyl carrier protein transacylase
(MCAT) from Helicobacter pylori, FEBS Lett. 580, 697-702 (2006).
131 Meadows, E.S. and Khosla, C. (2001) In vitro reconstitution and analysis of the
chain initiating enzymes of the R1128 polyketide synthase, Biochemistry 40, 14855-
14861.
132 Szafranska, A. E. PhD thesis, University of Bristol, 1999.
133 Kim, E-U., Cramer, K. D., Shreve, A. L. and Sherman, D. H. (1995) Heterologous
Expression of an Engineered Biosynthetic Pathway: Functional Dissection of Type
II Polyketide Synthase Components in Streptomyces Species, J Bacteriol. 177,
1202-1207.
134 Meurer, G. and Hutchinson C. R. (1995) Functional Analysis of Putative β-
Ketoacyl:Acyl Carrier Protein Synthase and Acyltransferase Active Site Motifs in a
Type II Polyketide Synthase of Streptomyces glaucescens, J Bacteriol. 177, 477-
481.
135 Roujeinikova, A., Baldock, C., Simon, W. J., Gilroy, J., Baker, P. J., Stuitje, A. R.,
Rice, D. W., Slabas, A. R., and Rafferty, J. B. (2002) X-ray crystallographic studies
on butyryl-ACP reveal flexibility of the structure around a putative acyl chain
binding site, Structure 10, 825-835.
136 Weber, T., Baumgartner, R., Renner, C., Marahiel, M. A. and Holak, T. A. (2000)
Solution structure of PCP, a Prototype for the Peptidyl Carrier Domains of Modular
Peptide Synthethases, Structure 8, 401-418.
137 Koglin, A., Mofid, M. R., Löhr, F., Schäfer, B., Rogov, V. V., Blum, M-M., Mittag,
T., Marahiel, M .A., Bernhard, A. and Dötsch, V. (2006) Conformational Switches

Chapter 3. Initiation of polyketide synthesis
170
Modulate Protein Interactions in Peptide Antibiotic Synthetases, Science 312, 273-
276.
138 D’Agnolo, G., Rosenfeld, I. S. and Vagelos, P. R. (1975) Multiple forms of β-
ketoacyl-acyl carrier protein synthetase in Escherichia coli, J Biol Chem. 250, 5289-
5294.
139 Rosenfeld, I. S., D’Agnolo, G. and Vagelos, P. R. (1973) Synthesis of unsaturated
fatty acids and the lesion in fabB mutants, J Biol Chem. 248, 2452-2460.
140 Garwin J. L., Klages, A. L. and Cronan, J. E. Jr. (1980) Structural, Enzymatic, and
Genetic Studies of β-Ketoacyl-Acyl Carrier Protein Synthases I and II of
Escherichia coli, J Biol Chem. 255, 11949.
141 Jackowski, S. and Rock, C. O. (1987) Acetoacetyl-Acyl Carrier Protein Synthase, a
Potential Regulator of Fatty Acid Biosynthesis in Bacteria, J Biol Chem. 262, 7927-
7931.
142 Jackowski, S., Murphy, C. M., Cronan, J. E. Jr. and Rock, C. O. (1989)
Acetoacetyl-acyl Carrier Protein Synthase: a target for the antibiotic thiolactomycin,
J Biol Chem 264, 7624-7629..
143 Wattana-Amorn, P. (2007) PhD thesis, University of Bristol, UK.
144 Meurer, G., and Hutchinson, C. R. (1995) Daunorubicin type II polyketide synthase
enzymes DpsA and DpsB determine neither the choice of starter unit nor the
cyclization pattern of aromatic polyketides, J Am Chem Soc. 117, 5899–5900.
145 Bao, W., Sheldon, T. J., Wendt-Pienkowski, E. and Hutchinson, C. R. (1999) The
Streptomyces peucetius dpsC Gene Determines the Choice of Starter Unit in
Biosynthesis of the Daunorubicin Polyketide, J Bacteriol. 181, 4690-4695.
146 Bao, W., Sheldon, T. J. and Hutchinson, C. R. (1999) Purification and Properties of
the Streptomyces peucetius DpsC β-Ketoacyl:Acyl Carrier Protein Synthase III that
Specifies the Propionate-Starter Unit for Type II Polyketide Biosynthesis,
Biochemistry 38, 9752-9757.
147 Hertweck, C., Luzhetskyy, A., Reberts, Y and Bechthold, A. (2007) Type II
polyketide synthases: gaining a deeper insight into enzymatic teamwork, Nat Prod
Rep. 24, 162-190.

Chapter 3. Initiation of polyketide synthesis
171
148 Bibb, M. J., Sherman, D. H., Omura, S. and Hopwood, D. A. (1994) Cloning,
sequencing and deduced functions of a cluster of Streptomyces genes probably
encoding biosynthesis of the polyketide antibiotic frenolicin, Gene 142, 31-39.
149 Xu, Z., Schenk, A and Hertweck, C. (2007) Molecular Analysis for the Benastatin
Biosynthetic Pathway and Genetic Engineering of Altered Fatty Acid-Polyketide
Hybrids, J Am Chem Soc. 129, 6022-6030.
150 Marti, T., Hu, Z., Pohl, N. L., Shah, A. N., and Khosla, C. (2000) Cloning,
nucleotide sequence, and heterologous expression of the biosynthetic gene cluster
for R1128, a non-steroidal estrogen receptor antagonist. Insights into an unusual
priming mechanism, J Biol Chem. 275, 33443-33448.
151 Meadows, E. S and Khosla, C. (2001) In Vitro Reconstitution and Analysis of the
Chain Initiating Enzymes of the R1128 Polyketide Synthase, Biochemistry 40,
14855-14861.
152 Tang, Y., Koppisch, A. C and Khosla, C. (2004) The acyltransferase homologue
from the initiation module of the R1128 polyketide synthase is an acyl-ACP
thioesterase that edits acetyl primer units, Biochemistry 43, 9546-9555.
153 Tang, Y., Lee, H. Y., Tang, Y., Kim, C-Y., Mathews, I. and Khosla, C. (2006)
Structural and Functional Studies on SCO1815: A β-Ketoacyl-Acyl Carrier Protein
Reductase from Streptomyces coelicolor A3(2), Biochemistry 45, 14085-14093.
154 Sherman, D. H., Kim, E-S., Bibb, M. J. and Hopwood, D. A. (1992) Functional
Replacement of Genes for Individual Polyketide Synthase Components in
Streptomyces coelicolor A3(2) by Heterologous Genes from a Different Polyketide
Pathway, J Bacteriol. 174, 6184-6190.
155 Kresze, G-B., Steber, L., Oesterhelt, D. and Lynen, F. (1997) Reaction of yeast fatty
acid synthetase with iodoacetamide. 1. Kinetics of inactivation and extent of
carboxamidomethylation, Eur J Biochem. 79, 173-180.
156 Huang W., Jia J., Edwards P., Dehesh K., Schneider G. and Lindqvist Y. (1998)
Crystal structure of β-ketoacyl-acyl carrier protein synthase II from E. coli reveals
the molecular architecture of condensing enzymes. EMBO J. 17, 1183-1191.
157 Jez, J.M., Ferrer, J.L., Bowman, M.E., Dixon, R.A. and Noel, J.P. (2000) Dissection
of malonyl-coenzyme A decarboxylation from polyketide formation in the reaction
mechanism of a plant polyketide synthase, Biochemistry 39, 890-902.
158 Thwaite, J. S. (2003) PhD thesis, University of Cambridge, UK.

Chapter 3. Initiation of polyketide synthesis
172
159 Katnar, S. S., Cleland, W. W. and Porter, J. W. (1975) Fatty Acid Synthetase: A
steady state kinetic analysis of the reaction catalyzed by the enzyme from
pigeon liver, J Biol Chem. 250, 2709-2717.
160 Ghayourmanesh, S. and Kumar, S. (1981) Synthesis of acetoacetyl-CoA by bovine
mammary fatty acid synthase, FEBS Lett. 132, 231-4.
161 Cox, B. G. and Hammes, G. G. (1983) Steady-state kinetic study of fatty acid
synthase from chicken liver, P Natl Acad Sci U. S. A 80, 4233-7.
162 Carlisle-Moore, L., Gordon, C. R., Machutta, C. A., Miller, W. T. and Tonge, P. T.
(2005) Substrate recognition by the human fatty acid synthase, J Biol Chem. 280,
42612-42618.
163 Wood, W. I., Peterson, D. O. and Bloch, K. (1977) Mycobacterium smegmatis
Fatty Acid Synthetase: A Mechanism Based On Steady State Rates And
Product Distributions, J Biol Chem. 252, 5745-5749.
164 Walker, T. A., Jonak, Z. L., Worsham, L. M. and Ernst-Fonberg M. L. (1981)
Kinetic studies of the fatty acid synthetase multienzyme complex from Euglena
gracilis variety bacillaris, Biochem J. 199, 383-392.
165 Jackowski, S., Murphys, C. M., Cronan, J. E. and Rock, C. O. (1989) Acetoacetyl-
acyl Carrier Protein Synthase: a target for the antibiotic thiolactomycin, J Biol
Chem. 264, 7624-7629.
166 Heath, R. J. and Rock, C. O. (1996) Inhibition of β-ketoacyl-acyl carrier protein
synthase III (FabH) by acyl-acyl carrier protein in Escherichia coli, J Biol Chem.
271, 10996-1000.
167 Garwin J. L., Klages, A. L. and Cronan, J. E. Jr. (1980) Structural, enzymatic, and
genetic studies of β-ketoacyl-acyl carrier protein synthases I and II of Escherichia
coli, J Biol Chem. 255, 11949-56.
168 He, X. and Reynolds, K. A. (2002) Purification, Characterization, and Identification
of Novel Inhibitors of the β-Ketoacyl-Acyl Carrier Protein Synthase III (FabH)
from Staphylococcus aureus, Antimicrob Agents Ch. 46, 1310-1318.
169 Schaeffer M. L., Agnihotri G., Volker C., Kallender H., Brennan P. J. and Lonsdale
J. T. (2001) Purification and biochemical characterization of the Mycobacterium
tuberculosis β-ketoacyl-acyl carrier protein synthases KasA and KasB, J Biol Chem.
270, 47029-47037.

Chapter 3. Initiation of polyketide synthesis
173
170 Abbadi, A., Brummel, M., Schutt, B. S., Slabaugh, M. B., Schuch, R. and Spener, F.
(2000) Reaction mechanism of recombinant 3-oxoacyl-(acyl-carrier-protein)
synthase III from Cuphea wrightii embryo, a fatty acid synthase type II condensing
enzyme, Biochem J. 345, 153-160.
171 Funa, N., Awakawa, T. and Horinouchi, S. (2007) Pentaketide resorcylic acid
synthesis by type III polyketide synthase from Neurospora crassa, J Biol Chem.
online.
172 Jez, J. M., Ferrer, J.L., Bowman, M. E., Dixon, R. A. and Noel, J. P. (2000)
Dissection of malonyl-coenzyme A decarboxylation from polyketide formation in
the reaction mechanism of a plant polyketide synthase, Biochemistry 39, 890-902.
173 Wanibuchi, K., Zhang, P., Abe, T., Morita, H., Kohno, T., Chen, G., Noguchi, H.
and Abe, I. (2007) An acridone-producing novel multifunctional type III polyketide
synthase from Huperzia serrata. FEBS J. 274, 1073-1082.
174 Abe, I., Morita, H., Nomura, A. and Noguchi, H. (2000) Substrate specificity of
chalcone synthase: Enzymatic formation of unnatural aromatic polyketides from
synthetic cinnamoyl-CoA analogues, J Am Chem Soc. 122, 11242-11243.
175 Abe, I., Utsumi, Y., Oguro, S., Morita, H., Sano, Y. and Noguchi, H. (2005) A plant
type III polyketide synthase that produces pentaketide chromone, J Am Chem Soc.
127, 1362-1363.
176 Abe, I., Utsumi, Y., H., Sano, Y. and Noguchi, H. (2005) Engineered biosynthesis
of plant polyketides: chain length control in an octaketide-producing plant type III
polyketide synthase, J Am Chem Soc. 127, 12709-12716.
177 Funa, N., Ohnishi, Y., Fujii, I., Shibuya, M., Ebizuka, Y. and Horinouchi, S. (1999)
A new pathway for polyketide biosynthesis in microorganisms, Nature 400, 897-
899.
178 Funa, N., Ohnishi, Y., Ebizuka, Y. and Horinouchi, S. (2002) Properties and
substrate specificity of RppA, a chalcone synthase-related polyketide synthase in
Streptomyces griseus, J Biol Chem. 277, 4628-4635.
179 Li, S., Gruschow, S., Dordick, J. S. and Sherman, D. H. (2007) Molecular Analysis
of the Role of Tyrosine 224 in the active site of Streptomyces coelicolor RppA, a
Bacterial Type III Polyketide Synthase, J Biol Chem. 282, 12765-12772.

Chapter 3. Initiation of polyketide synthesis
174
180 Zha, W., Rubin-Pitel, S B. and Zhao, H. (2006) Characterization of the Substrate
Specificity of PhID, a Type III Polyketide Synthase from Pseudomonas fluorescens,
J Biol Chem. 281, 32036-32047.
181 Gokhale, R. S., Tsuji, S. Y., Cane, D. E. and Khosla, C. (1999) Dissecting and
Exploiting Intermodular Communication in Polyketide Synthases, Science 284,
482-485.
182 Wu, N., Kudo, F., Cane, D. E and Khosla, C. (2000) Analysis of the Molecular
Recognition Features of Individual Modules Derived from the Erythromycin
Polyketide Synthase, J Am Chem Soc. 122, 4847-4852
183 Wu, N., Tsuji, S. Y., Cane, D. E and Khosla, C. (2001) Assessing the Balance
between Protein-Protein Interactions and Enzyme-Substrate Interactions in the
Chanelling of Intermediates between Polyketide Synthase Modules, J Am Chem
Soc. 123, 6465-6474.
184 Chen, A. Y., Schnarr, N. A., Kim, C-Y. Cane, D. C. and Khosla, C. (2006) Extender
Unit and Acyl Carrier Protein Specificity of Ketosynthase Domains of the 6-
Deoxyerythronolide B Synthase, J Am Chem Soc. 128, 3167-3074.
185 Winston, R. L. and Fitzgerald, M. C. (2000) Concentration and desalting of protein
samples for mass spectrometry analysis, Anal Biochem. 262, 83-85.
186 Lau, J., Cane, D. E. and Khosla, C. (2000) Dissecting the Role of Acyltransferase
Domains of Modular Polyketide Synthases in the Choice and Stereochemical Fate
of Extender Units, Biochemistry 39, 10514-10520.
187 Liou, G. F., Lau, J., Cane, D. E., Khosla, C. (2003) Quantitative analysis of loading
and extender acyltransferases of modular polyketide synthases, Biochemistry 42,
200-207.
188 Shen, B. and Hutchinson, C. R. (1996) Deciphering the mechanism for the assembly
of aromatic polyketides by a bacterial polyketide synthase, P Natl Acad Sci. U. S. A.
93, 6600-6604.
189 Dreier, J. and Khosla, C. (2000) Mechanistic Analysis of a Type II Polyketide
Synthase. Role of Conserved Residues in the β-Ketoacyl Synthase-Chain Length
Factor Heterodimer, Biochemistry 39, 2088-2095.
190 Witkowski, A, Joshi, A .K., Lindqvist, Y and Smith, S. (1999) Conversion of a β-
Ketoacyl Synthase to a Malonyl Decarboxylase by Replacement of the Active-Site
Cysteine with Glutamine, Biochemistry 38, 11643-11650.

Chapter 3. Initiation of polyketide synthesis
175
191 Austin, M. B. and Noel, J. P. (2003) The chalcone synthase superfamily of Type III
polyketide synthases, Nat Prod Rep. 20, 79-110.
192 Modis, Y. and Wierenga, R. K. (1999) A biosynthetic thiolase in complex with a
reaction intermediate: the crystal structure provides new insights into the catalytic
mechanism, Structure 7, 1279-1290.
193 Evans, S. E. (2007) PhD thesis, University of Bristol, UK.
194 Keating, D. H., Carey, M. R. and Cronan, J. E. (1995) The Unmodified (Apo) Form
of E. coli Acyl Carrier Protein is a Potent Inhibitor of Cell Growth, J Bacteriol. 270,
22229-22235.
195 McGuire, K. A., Siggard-Andersen, M., Bangera, M. G., Olsem J. G. and von
Wettstein-Knowles, P. (2001) β-Ketoacyl-[Acyl Carrier Protein] Synthase I of
Escherichia coli: Aspects of the Condensation Mechanism Revealed by Analyses of
Mutations in the Active Site Pocket, Biochemistry 40, 9836-9845.
196 Bartel, P. L., Zhu, C-B., Lampel, J. S., Dosch, D. C., Connors, N. C., Strohl, W. R.,
Beale, J. M. and Floss, H. G. (1990) Biosynthesis of Anthraquinones by
Interspecies Cloning of Actinorhodin Biosynthesis Genes in Streptomycetes:
Clarification of Actinorhodin Gene Functions, J Bacteriol. 172, 4816-4826.
197 Zhang, X., He, A., Adefarati, J., Gallucci, S. P., Cole, J. M., Beale, P. J., Keller, H.
G., and Floss, H. (1990) Mutactin, a Novel Polyketide from Streptomyces
coelicolor. Structure and Biosynthetic Relationship to Actinorhodin, J Org Chem.
55, 1682-1684.
198 Fu, H., Hopwood, D. A. and Khosla, C. (1994) Engineered biosynthesis of novel
polyketides: evidence for temporal, but not regiospecific, control of cyclization of
an aromatic polyketide precursor, Chem Biol. 1, 205-210.
199 McDaniel, R., Ebert-Khosla, S., Fu, H., Hopwood, D. A. and Khosla, C. (1994)
Engineered biosynthesis of novel polyketides: Influence of a downstream enzyme
on the catalytic specificity of a minimal aromatic polyketide synthase, P Natl Acad
Sci U.S.A 91, 11542-11546.
200 McDaniel, R., Ebert-Khosla, S., Fu, H., Hopwood, D. A. and Khosla, C. (1993)
Engineered biosynthesis of novel polyketides, Science 262, 1546-1550.
201 Kunnari, T., Ylihonko, C., Hautala, A., Klika, K. D., Matntsatla P. and Hakala, J.
(1999) Incorrectly folded aromatic polyketides from polyketide reductase deficient
mutants, Bioorg Med Chem Lett. 9, 2639-2642.

Chapter 3. Initiation of polyketide synthesis
176
202 Korman, T. P., Hill, J. A., Vu, T. N. and Tsai, S-C. (2004) Structural Analysis of
Actinorhodin Polyketide Ketoreductase: Cofactor Binding and Substrate
Specificity, Biochemistry 43, 14529-14538.
203 Korman, T. P., Tan, J. H., Wong, J., Luo, R. and Tsai, S-C. (2008) Inhibition
Kinetics and Emodin Cocrystal Structure of a Type II Polyketide Ketoreductase,
Biochemistry 47, 1837-1847.
204 Kalaitzis, J. A. and Moore, B. S. (2004) Heterologous Biosynthesis of Truncated
Hexaketides Derived from the Actinorhodin Polyketide Synthase, J Nat Prod. 67,
1419-1422
205 Hertweck, C., Xiang, L., Kalaitzis, J. A., Cheng, Q., Palzer, M. and Moore, B. S.
(2004) Context-Dependent Behavior of the Enterocin Iterative Polyketide Synthase:
A New Model for Ketoreduction, Chem Biol. 11, 461–468.
206 Teartasin, W., Limpkin, C., Glod, F., Cox, R. J., Simpson, T. J., Crosby, J., Crump,
M. P. and Hadfield, A. T. (2004) Expression, purification, and preliminary X-ray
diffraction analysis of a ketoreductase from a Type II polyketide synthase, Acta
Crystallogr D60, 1137-1138.
207 Oppermann, U., Filling, C., Hult, M., Shafqat, N., Wu, X., Lindh, M., Shafqat, J.,
Nordling, E., Kallberg, Y., Persson, B, and Jorrnvall, H. (2003) Short-chain
dehydrogenases / reductases: the 2002 update, Chem-Biol Interact. 143, 247-253.
208 Zawada, R. J. and Khosla, C. (1999) Heterologous expression, purification,
reconstitution and kinetic analysis of an extended type II polyketide synthase, Chem
Biol. 6, 607-615.
209 Geoghegan, K. F., Dixon, H. B., Rosner, P. J., Hoth, L. R., Lanzetti, A. J.,
Borzilleri, K. A., Marr, E. S., Pezzullo, L. H., Martin, L. B., LeMotte, P. K.,
McColl, A. S., Kamath, A. V. and Stroh, J. G. (1999) Spontaneous α-N-6-
phosphogluconoylation of a "His tag" in Escherichia coli: the cause of extra mass of
258 or 178 Da in fusion proteins, Anal Biochem 267, 169-184.
210 Ma, S. M., Zhan, J., Xie, X., Watanabe, K., Tang, Y. and Zhang, W. (2008)
Redirecting the Cyclization Steps of Fungal Polyketide Synthase, J Am Chem Soc.
130, 38-39.
211 Khosla, C., McDaniel, R., Ebert-Khosla, S., Torres, R., Sherman, D. H., Bibb, M. J.,
and Hopwood, D. A (1993) Genetic construction and functional analysis of hybrid

Chapter 3. Initiation of polyketide synthesis
177
polyketide synthases containing heterologous acyl carrier proteins, J Bacteriol. 175,
2197-2204.
212 Decker, H., Summers, R. G., and Hutchinson, C. R. (1994) Overproduction of the
acyl carrier protein component of a Type II polyketide synthase stimulates
production of tetracenomycin biosynthetic intermediates in Streptomyces
glaucescens, J Antibiot. 47, 54-63.
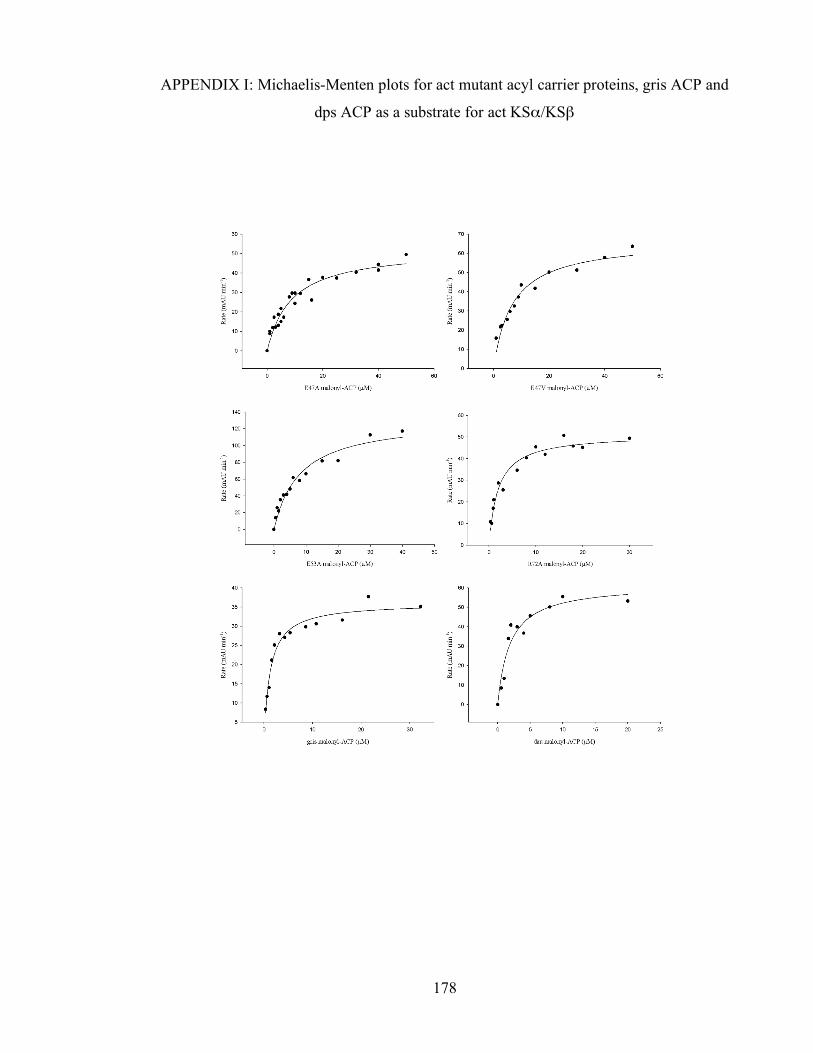
Chapter 3. Initiation of polyketide synthesis
178
APPENDIX I: Michaelis-Menten plots for act mutant acyl carrier proteins, gris ACP and
dps ACP as a substrate for act KSα/KSβ

Chapter 3. Initiation of polyketide synthesis
179
APPENDIX II: Sequence alignment of FAS and PKS ketosynthases
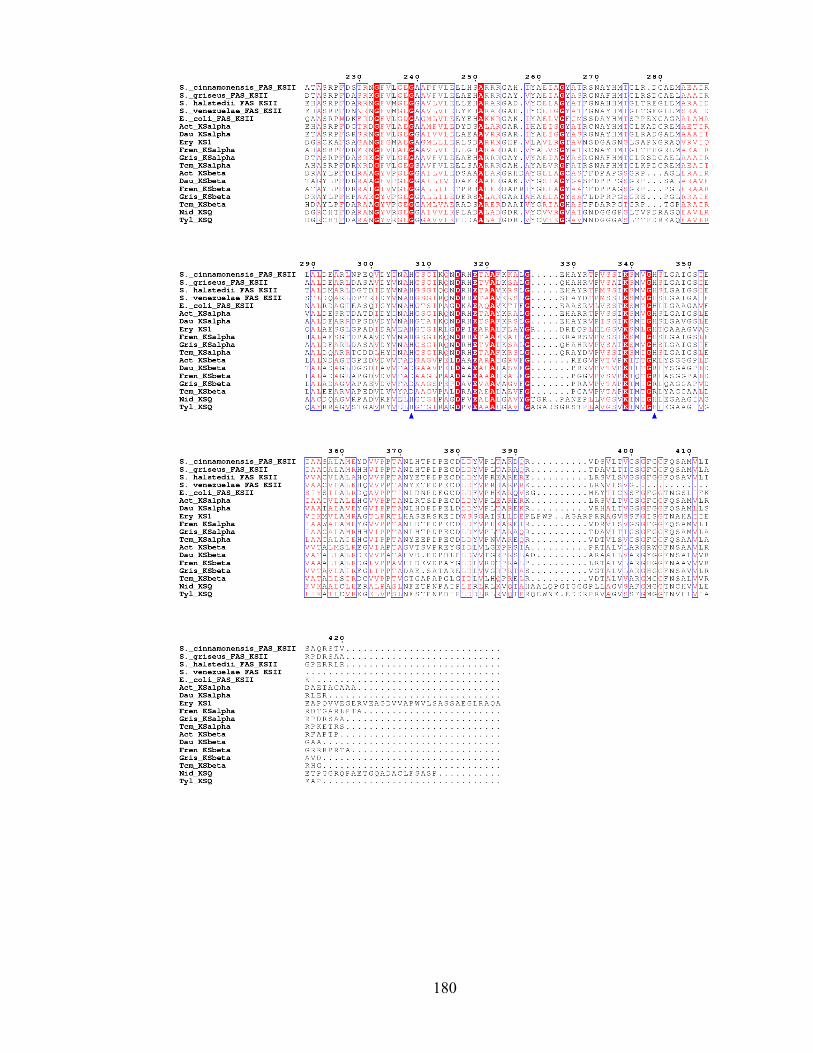
Chapter 3. Initiation of polyketide synthesis
180

Chapter 3. Initiation of polyketide synthesis
181
APPENDIX III. ESMS time-course study of self-malonylation of the reference act ACP
and an E47A ACP mutant. A: initial sample; B: 5 min; C: 15 min; D: 30 min; E: 120 min
A B
C D
E
Figure III.1. 50 µM ACP C17S ACP. Expected masses: 9441 (holo-ACP) and 9527
(malonyl-ACP).
holo
holo
holo holo
holo
malonyl
malonyl
malonyl
malonyl

Chapter 3. Initiation of polyketide synthesis
182
A B
C D
Figure III.2. 25 µM C17S ACP. Expected masses: 9441 (holo-ACP) and 9527 (malonyl-
ACP).
malonyl
malonyl malonyl
holo
holo
holo
holo
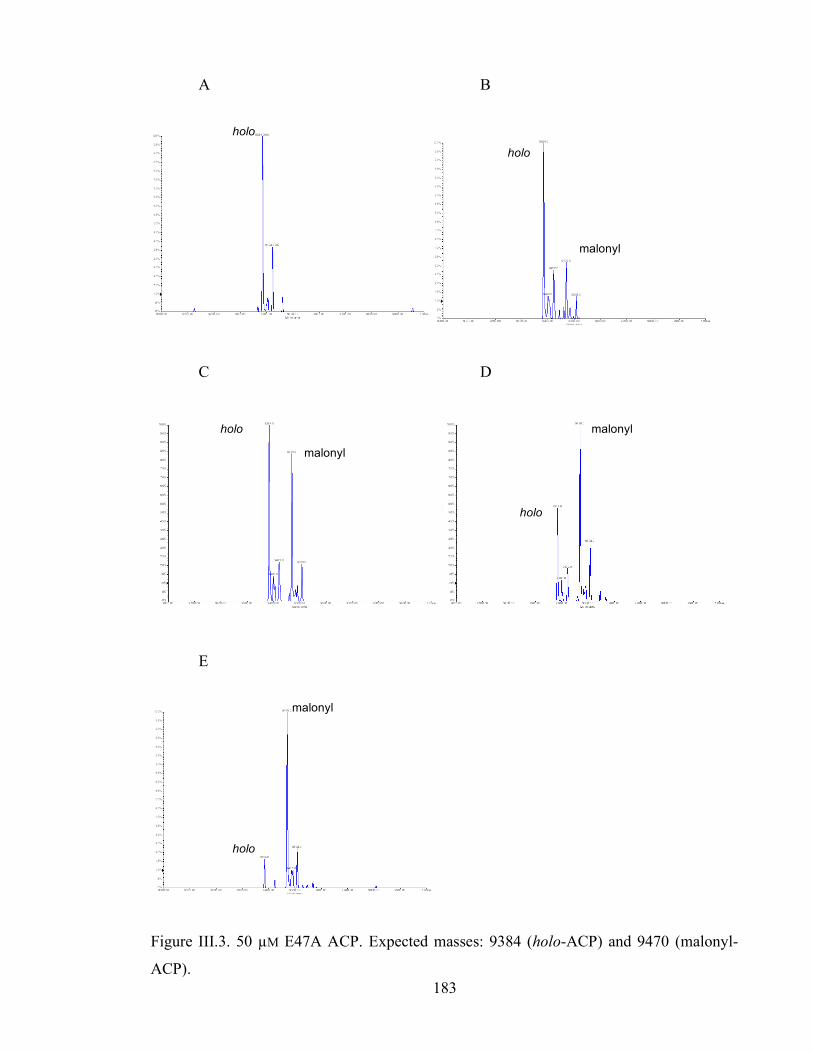
Chapter 3. Initiation of polyketide synthesis
183
A B
C D
E
Figure III.3. 50 µM E47A ACP. Expected masses: 9384 (holo-ACP) and 9470 (malonyl-
ACP).
holo
holo
holo
holo
holo
malonyl
malonyl
malonyl
malonyl

Chapter 3. Initiation of polyketide synthesis
184
A B
C D
Figure III.4. 25 µM E47A ACP. Expected masses: 9384 (holo-ACP) and 9470 (malonyl-
ACP).
malonyl
malonyl
malonyl
holo
holo
holo
holo