perbandingan metode dan verifikasi analisis total ... · karbohidrat yang akurat, cepat dan dapat...
TRANSCRIPT

PERBANDINGAN METODE DAN VERIFIKASI ANALISIS TOTAL KARBOHIDRAT DENGAN METODE LUFF-SCHOORL DAN ANTHRONE
SULFAT
SKRIPSI
MANIKHARDA
F24061217
FAKULTAS TEKNOLOGI PERTANIAN INSTITUT PERTANIAN BOGOR
BOGOR2011

METHOD COMPARATION AND VERIFICATION OF TOTAL CARBOHYDRATE ANALYSIS WITH LUFF-SCHOORL AND ANTHRONE SULFURIC ACID
Manikharda, Hanifah Nuryani Lioe and Dian HerawatiDepartment of Food Science and Technology, Faculty of Agricultural Technology,
Bogor Agricultural University, IPB Darmaga Campus, PO Box 16680, Bogor, West Java, Indonesia.
Phone +62852 13 374 396, E-mail : [email protected]
ABSTRACT
Carbohydrate plays crucial role in food industry. Therefore an accurate, direct and reliable carbohydrate analysis is needed. Among many colorimetric methods for carbohydrate determination, the Anthrone-sulfuric acid is the most commonly used. The Anthrone-sulfuric method for carbohydrate analysis is simple and sensitive. However, the SNI official method for carbohydrate analysis employing the Luff-Schoorl method which is time consuming, difficult for untrained staff and the reduction reactions are seldom stoichiometric. Therefore a new candidate method employing Anthrone sulfuric acid was proposed to replace the SNI 01-2891-1992 total carbohydrate analysis.
In this research both methods were compared using three matrices which represent general food matrices in liquid form based on AOAC proposed triangle scheme. Samples from the low, medium and high content of carbohydrate from the triangle scheme were selected. The selected samples were coconut milk, soy sauce and sweet soy sauce. Based on the comparation result, Anthrone method as a new proposed method proved ineligible to replace the SNI 01-2891-1992. Thus the next step taken was to verify the SNI 01-2891-1992 method through its repeatability and accuracy. Accuracy was accessed using reference material and standard addition. The repeatability showed acceptable precision. But the standard addition exhibited poor recovery value in SNI 01-2891-1992 method of total carbohydrate.
Keywords: total carbohydrate, carbohydrate analysis, Anthrone method, Luff-Schoorl method, methodvalidation

MANIKHARDA. F24061217. Perbandingan Metode dan Verifikasi Analisis Total Karbohidrat
dengan Metode Luff-Schoorl dan Anthrone Sulfat. Di bawah bimbingan Hanifah Nuryani Lioe dan
Dian Herawati. 2011
RINGKASAN
Karbohidrat memegang peranan penting dalam bidang pangan. Oleh karena itu analisis
karbohidrat yang akurat, cepat dan dapat dipercaya diperlukan untuk mengetahui kandungan total
karbohidrat dalam produk. Diantara banyak metode kolorimetri yang ada untuk menganalisis
karbohidrat, yang paling banyak digunakan adalah Anthrone sulfat. Analisis total karbohidrat dengan
Anthrone sulfat cukup sederhana dan sensitif. Tetapi metode analisis untuk total karbohidrat dalam SNI
01-2891-1992 menggunakan metode Luff Schoorl yang menggunakan prinsip titrimetri, banyak
memakan waktu, sulit dikerjakan bagi analis yang tidak terlatih dan reaksi reduksinya tidak
stoikiometris. Metode kandidat yang menggunakan Metode Anthrone sulfat diajukan untuk dapat
menggantikan metode total karbohidrat Luff Schoorl dalam SNI 01-2891-1992.
Penelitian dilakukan dengan memilih sampel yang dapat mewakili matriks sampel pangan
secara umum yang bentuknya cair. Pemilihan sampel berdasarkan komposisi kimia pangan cair
mengandung karbohidrat rendah, sedang dan tinggi dari studi literatur. Selanjutnya dari sampel yang
terpilih, komposisinya dikonfirmasi melalui analisis proksimat. Kecap manis, kecap kedelai asin dan
santan menjadi sampel yang terpilih dan dikonfirmasi komposisinya, masing-masing merupakan
matriks yang tinggi, sedang dan rendah kadar karbohidratnya. Perbandingan analisis total karbohidrat
menggunakan kedua metode yaitu Anthrone sulfat dan Luff-Schoorl pada ketiga sampel yang terpilih
dilakukan. Berdasarkan hasil uji statistik dengan menggunakan uji F pada tingkat kepercayaan 95%
terlihat bahwa kedua metode tidak memiliki perbedaan yang nyata dalam hal presisi untuk sampel
kecap manis dan kecap asin, tetapi pada sampel santan terdapat perbedaan presisi pada kedua metode.
Berdasarkan hasil uji t menunjukkan bahwa hasil analisis dari kedua metode berbeda nyata pada tingkat
kepercayaan 95%. Uji korelasi menggunakan regresi linear dilakukan dengan menggunakan tambahan
data sekunder dari matriks sampel pangan yang berwujud padat. Hasil regresi liniear menunjukkan
bahwa adanya estimasi error diantara kedua metode. Karena Metode Anthrone sulfat dan Metode
Luff-Schoorl tidak memiliki kesesuaian hasil yang dapat diterima, Metode Anthrone sulfat tidak dapat

menggantikan metode Luff Schoorl dalam SNI 01-2891-1992 untuk total karbohidrat.
Verifikasi metode baku untuk analisis karbohidrat total menurut SNI 01-2891-1992 (dengan
Metode Luff-Schoorl) dilakukan menggunakan bahan acuan. Bahan acuan yang digunakan adalah susu
bubuk dengan rentang kadar karbohidrat 59,61-59,67g/100g (hasil analisis dari satu lab); tepung kacang
hijau dengan rentang kadar karbohidrat 14,02-19,26 g/100g (hasil analisis dari 8 lab) dan tepung kacang
kedelai rentang kadar karbohidrat 49,26-57,96g/100g (hasil analisis dari 6 lab). Dilihat dari
ripitabilitasnya Metode Luff-Schoorl yang diterapkan pada ketiga bahan acuan memiliki presisi yang
dapat diterima yaitu yaitu RSD 0,51-2,58% yang lebih kecil dari RSD hitung menurut Horwitz. Uji
reprodusibilitas yang dilakukan dengan selang waktu lebih dari dua bulan menunjukkan bahwa dengan
independent student t- test hasil analisis bahan acuan kedelai dan susu bubuk menghasilkan nilai yang
berbeda nyata, sedangkan untuk hasil analisis bahan acuan kacang hijau nilainya tidak berbeda nyata
dengan hasil analisis dua bulan sebelumnya. Hal ini kemungkinan disebabkan karena kandungan
protein dari susu bubuk dan kedelai menurunkan keakuratan analisis.
Hasil analisis matriks bahan pangan cair yang diuji dengan independent student t test
menunjukkan hasil analisis sampel kecap manis berbeda nyata dengan hasil analisis yang dilakukan dua
bulan sebelumnya, yaitu hasil analisis menunjukkan nilai lebih rendah. Hal ini dapat disebabkan oleh
kandungan gula yang tinggi dan kemungkinan kadar gula kecap manis mengalami perubahan selama
dua bulan penyimpanan. Hasil analisis sampel kecap asin dan santan nilainya tidak berbeda nyata
dengan hasil analisis dua bulan sebelumnya.
Uji akurasi menggunakan rentang bahan acuan dan uji rekoveri. Hasil analisis bahan acuan
tepung kacang hijau dan tepung kacang kedelai masih berada dalam rentang tersebut, tetapi nilai
rekoveri yang diperoleh yaitu 62-97%. Nilai rekoveri matriks sampel pangan cair yaitu kecap manis,
kecap asin dan santan memiliki rentang -57-122%. Sedangkan rentang rekoveri yang dapat diterima
menurut AOAC (2002) yaitu 95-102%. Hal ini menunjukkan bahwa besarnya rekoveri juga sangat
dipengaruhi oleh matriks.

PERBANDINGAN METODE DAN VERIFIKASI ANALISIS TOTAL
KARBOHIDRAT DENGAN METODE LUFF-SCHOORL DAN ANTHRONE
SULFAT
SKRIPSI
Sebagai salah satu syarat untuk memperoleh gelar
SARJANA TEKNOLOGI PERTANIAN
Pada Departemen Teknologi Industri Pertanian,
Fakultas Teknologi Pertanian,
Institut Pertanian Bogor
Oleh:
MANIKHARDA
F24061217
FAKULTAS TEKNOLOGI PERTANIAN
INSTITUT PERTANIAN BOGOR
BOGOR
2011

Judul Skripsi : Perbandingan Metode dan Verifikasi Analisis Total Karbohidrat
dengan Metode Luff-Schoorl dan Anthrone Sulfat
Nama : Manikharda
NIM : F24061217
Menyetujui,
Pembimbing I
(Dr.Ir Hanifah Nuryani Lioe, M.Si.)
NIP 19680809.199702.2.001
Pembimbing II
(Dian Herawati, S.TP, M.Si.)
NIP 19750111.020070.2.001
Mengetahui :
Ketua Departemen Ilmu dan Teknologi Pangan,
(Dr.Ir. Feri Kusnandar. M.Sc.)
NIP 19680526.199303.1.004
Tanggal lulus: 23 September 2011

PERNYATAAN MENGENAI SKRIPSI DAN SUMBER INFORMASI
Saya menyatakan dengan sebenar-benarnya bahwa skripsi dengan judul Perbandingan
Metode dan Verifikasi Analisis Total Karbohidrat Dengan Metode Luff-Schoorl dan Anthrone
Sulfat adalah hasil karya saya sendiri dan belum diajukan dalam bentuk apapun pada perguruan tinggi
manapun. Skripsi ini merupakan hasil arahan dari Dosen Pembimbing Akademik dari akademisi IPB.
Sumber informasi yang berasal atau dikutip dari karya yang diterbitkan maupun tidak diterbitkan dari
penulis lain telah disebutkan dalam teks dan dicantumkan dalam Daftar Pustaka di bagian akhir
skripsi ini.
Bogor, Desember 2011
Yang membuat pernyataan
Manikharda
F24061217

© Hak cipta milik Manikharda, tahun 2011
Hak cipta dilindungi
Dilarang mengutip dan memperbanyak tanpa seizin tertulis dari
Institut Pertanian Bogor, sebagian atau seluruhnya dalam bentuk apapun, baik cetak,
fotokopi, mikrofilm dan sebagainya.

iii
BIODATA PENULIS
Manikharda dilahirkan di Bogor, 17 Januari 1989, dari ayah
Sumardjo dan ibu Tri Sawarni, sebagai anak kedua dari dua
bersaudara. Penulis menamatkan pendidikan dasar di SDN Polisi 5,
Bogor pada tahun 2000. Sekolah lanjutan pertama di SLTPN 1 Bogor
pada tahun 2003 dan SMAN 1 Bogor pada tahun 2006. Penulis
diterima masuk Institut Pertanian Bogor (IPB) melalui jalur Undangan
Seleksi Masuk IPB (USMI) tahun 2006. Setelah melewati tahun
pertama di Tingkat Persiapan Bersama, penulis memilih mayor Ilmu dan Teknologi Pangan.
Selama mengikuti perkuliahan, penulis mengikuti aktivitas sebagai anggota Badan
Eksekutif Mahasiswa Fakultas Teknologi Pertanian IPB dan berbagai kegiatan kepanitiaan yang
diselenggarakan oleh Himpunan Mahasiswa Teknologi Pangan (HIMITEPA) IPB seperti HACCP
dan LCTIP. Pada tahun 2010, penulis mengikuti University of Ryukyus Short Term Exchange
Program selama 10 bulan. Penulis juga pernah menjadi asisten praktikum untuk mata kuliah
Teknologi Pengolahan Pangan dan Evaluasi Nilai Biologis Komponen Pangan. Sebagai tugas akhir
penulis melakukan penelitian mengenai validasi metode analisis total karbohidrat di bawah
bimbingan Dr. Ir. Hanifah Nuryani Lioe, M.Si. dan Dian Herawati, S.TP, MSi.

iv
KATA PENGANTAR
Puji syukur dipanjatkan atas kehadirat Allah SWT atas karunia-Nya sehingga skripsi ini
berhasil diselesaikan. Penelitian dengan judul “Validasi Metode Analisis Total Karbohidrat dengan
Metode Anthrone Sulfat” ini dilaksanakan di Laboratorium Departemen Ilmu dan Teknologi
Pangan IPB.
Dengan telah selesainya penelitian hingga tersusunnya skripsi ini, penulis ingin
menyampaikan penghargaan dan terima kasih yang sebesar-besarnya kepada:
1. Ibu, Ayah dan kakak tercinta, Leonard Dharmawan yang selalu memberikan dukungan
dan bantuannya kepada penulis baik berupa moril maupun materil serta kesabarannya
selama ini.
2. Dr.Ir. Hanifah Nuryani Lioe, M.Si. selaku dosen pembimbing utama atas arahan,
bimbingan dan bantuannya selama penulis menyelesaikan kuliah di Departemen Ilmu dan
Teknologi Pangan IPB dan menyelesaikan tugas akhir. Petuah, teladan dan masukan beliau
sangat berharga buat penulis baik untuk bidang akademik maupun dalam kehidupan
pribadi.
3. Dian Herawati, S.TP, M.Si. selaku dosen pembimbing kedua yang atas semua bantuan
yang diberikan dan kesabaran beliau dalam membimbing penulis terutama dalam tugas
akhir.
4. Dr. Ir. Feri Kusnandar, M.Sc. atas kesediaan dan waktunya sebagai dosen penguji pada
ujian akhir.
5. Teman-teman yang telah banyak membantu dan berbagi susah dan senang bersama
penulis di ITP 43: Rachmat Widyanto, Sarah Fathia, Zatil Afrah, Stella Kristanti, Siti Sri
Utami, Dhimas Satrio, Ipan Permadi, Siti Kholifah, Awaliyatus Sholihah dan teman-teman
lain yang tidak penulis sebutkan satu persatu. Terima kasih atas dukungan dan moment
susah dan senang yang kita jalani bersama. Teman-teman satu penelitian dan satu lab:
Dhina, Tiara, Ricky Sinaga, Desir, Khafid, Marissa, Mbak Ilul, Alya, Ronald, Cipi, Bu
Elmi, dan Nida atas semangat, dukungan dan bantuannya selama ini di saat penulis sangat
membutuhkannya.
6. Laboran yang telah banyak membantu dalam penelitian ini: Pak Wahid, Mbak Vera, Pak
Gatot, Bu Rubiyah, Mas Aldi, Pak Sobirin, dan Pak Rozak.
7. Teman-teman yang penulis kenal selama di Okinawa Kak Nina, Kak Tiyu, Kak Gebol, Mas
Fadry, Pak Armid, Pak Basyuni, Bu Santi, Mas Idham, Bu Dyah, Mbak Dudu, Pak Ricky,
Pak Agus, Bu Kusumiyati dan Takara sensei, Wada sensei, dan teman-teman satu lab di

v
Okinawa yang telah memberikan dukungan, download jurnal dan banyak pelajaran hidup
bagi penulis.
8. Seluruh dosen dan staf ITP yang telah memberikan ilmu dan masukan kepada penulis
selama penulis berkuliah di ITP. Serta seluruh pihak yang tidak dapat penulis sebutkan satu
persatu. Terima kasih banyak atas bantuan, yang telah diberikan
Akhirnya penulis berharap semoga tulisan ini bermanfaat dan dapat memberikan kontribusi
yang nyata terhadap perkembangan ilmu pengetahuan.
Bogor, Desember 2011
Penulis

vi
DAFTAR ISI
KATA PENGANTAR..................................................................................................................... iv
I. PENDAHULUAN ....................................................................................................................1
1.1. Latar Belakang ...........................................................................................................................1
1.2. Tujuan.........................................................................................................................................3
1.2.1. Tujuan Umum..........................................................................................................................3
1.2.2. Tujuan Khusus.........................................................................................................................3
1.3. Manfaat Penelitian......................................................................................................................3
1.4. Hipotesis.....................................................................................................................................3
II. TINJAUAN PUSTAKA ...........................................................................................................4
2.1. Karbohidrat.................................................................................................................................4
2.1.1 Struktur karbohidrat..................................................................................................................4
2.1.2. Monosakarida ..........................................................................................................................5
2.1.3. Oligosakarida...........................................................................................................................5
2.1.4. Polisakarida .............................................................................................................................5
2.2. Pentingnya Analisis Total Karbohidrat ......................................................................................5
2.3. Total Karbohidrat dalam Bahan Pangan dan Metode Analisisnya .............................................6
2.3.1. Definisi total karbohidrat.........................................................................................................6
2.3.2. Metode analisis total karbohidrat.............................................................................................6
2.3.2.1. Analisis karbohidrat langsung ..............................................................................................6
2.3.2.1.1. Analisis total karbohidrat dalam SNI 01-2891-1992 .........................................................7
2.3.2.1.2. Analisis total karbohidrat dengan Metode Anthrone sulfat ...............................................8
2.4. Validasi dan Verifikasi Metode................................................................................................10
2.4.1. Akurasi ..................................................................................................................................11
2.4.2. Presisi ....................................................................................................................................12
2.4.3. Spesifisitas.............................................................................................................................14
2.4.4. Limit Deteksi dan Limit Kuantitasi .......................................................................................14
2.4.5. Linieritas................................................................................................................................14
2.5. Matriks Sampel.........................................................................................................................15
2.5.1. Kecap manis ..........................................................................................................................17

vii
2.5.2. Kecap kedelai asin.................................................................................................................18
2.5.3. Santan ....................................................................................................................................18
2.5.4. Bahan Acuan .........................................................................................................................19
III. METODOLOGI PENELITIAN .........................................................................................21
3.1. Bahan dan Alat .........................................................................................................................21
3.1.1 Bahan......................................................................................................................................21
3.1.2. Alat ........................................................................................................................................21
3.2. Metode Penelitian.....................................................................................................................21
3.2.1. Penentuan matriks sampel .....................................................................................................22
3.2.1.1. Pemilihan sampel untuk uji perbandingan metode berdasarkan studi literatur...................22
3.2.1.2. Analisis proksimat ..............................................................................................................22
3.2.2. Perbandingan metode ............................................................................................................22
3.2.3. Validasi Metode Anthrone sulfat...........................................................................................23
3.2.3.1. Presisi .................................................................................................................................23
3.2.3.2. Akurasi ...............................................................................................................................23
3.2.3.3. Linieritas.............................................................................................................................24
3.2.4. Verifikasi metode SNI 01-2891-1992 ...................................................................................24
3.2.4.1. Presisi .................................................................................................................................24
3.2.4.2. Akurasi ...............................................................................................................................25
IV. HASIL DAN PEMBAHASAN ..........................................................................................26
4.1. Pemilihan Matriks Sampel .......................................................................................................26
4.2. Perbandingan metode ...............................................................................................................27
4.3. Verifikasi metode SNI 01-2891-1992 ......................................................................................33
4.3.1. Aspek presisi .........................................................................................................................34
4.3.1.1. Ripitabilitas bahan acuan....................................................................................................34
4.3.1.2. Reprodusibilitas bahan acuan dan matriks sampel .............................................................36
4.3.2. Aspek akurasi ........................................................................................................................39
4.3.2.1. Akurasi berdasarkan bahan acuan ......................................................................................39
4.3.2.2. Akurasi berdasarkan uji rekoveri........................................................................................40
4.3.2.2.1. Rekoveri dengan bahan acuan .........................................................................................41
4.3.2.2.2. Rekoveri dengan sampel matriks uji................................................................................41
4.4. Faktor-Faktor Kesalahan Pada Analisis Total Karbohidrat SNI 01-2891-1992 .......................43
4.5. Kelemahan Analisis Total Karbohidrat SNI 01-2891-1992 .....................................................45

viii
V. KESIMPULAN DAN SARAN ..............................................................................................48
5.1. Kesimpulan...............................................................................................................................48
DAFTAR PUSTAKA......................................................................................................................50
LAMPIRAN ....................................................................................................................................56

ix
DAFTAR TABEL
Tabel 1 Persentase rekoveri yang dapat diterima sesuai dengan konsentrasi analat .....................12
Tabel 2 Nilai presisi (RSD) sesuai dengan konsentrasi analat ...................................................13
Tabel 3 Komposisi kimia kecap manis, kecap asin dan santan ..................................................17
Tabel 4. Kandungan asam amino kecap asin dan kecap manis (g/100g) ....................................18
Tabel 5. Komposisi proksimat matriks sampel cair yang terpilih untuk uji perbandingan metode
analisis total karbohidrat (N=2) ................................................................................27
Tabel 6. Perbandingan Metode Anthrone sulfat dan Luff-Schoorl untuk analisis karbohidrat total
pada 3 matriks sampel pangan cair (N=3) ..................................................................28
Tabel 7. Karbohidrat total dari tiga sampel matriks pangan cair dengan beberapa metode...........30
Tabel 8 Komposisi proksimat bahan acuan yang digunakan dalam verifikasi metode karbohidrat
total SNI 01-2891-1992 ...........................................................................................34
Tabel 9. Ripitabilitas metode karbohidrat SNI 01-2891-1992 pada berbagai bahan acuan (N=7) .35
Tabel 10. Ripitabilitas metode karbohidrat SNI 01-2891-1992 pada berbagai bahan acuan dengan
penambahan kadar glukosa (N=7).............................................................................35
Tabel 11. Reprodusibilitas metode karbohidrat SNI 01-2891-1992 pada berbagai bahan acuan ...37
Tabel 12 Reprodusibilitas metode karbohidrat SNI 01-2891-1992 pada berbagai sampel pangan
cair (N=3)...............................................................................................................37
Tabel 13. Akurasi metode karbohidrat total SNI 01-2891-1992 pada berbagai bahan acuan (N=7)
..............................................................................................................................39
Tabel 14. Hasil uji rekoveri pada berbagai bahan acuan dengan spike glukosa (N=7) .................40
Tabel 15. Hasil uji rekoveri pada berbagai sampel pangan cair dengan spike glukosa (N=7) .......40

x
DAFTAR GAMBAR
Gambar 1. Matriks pangan berdasarkan kadar protein, lemak dan karbohidrat (Nielsen 2010). .16
Gambar 2. Tahapan penelitian validasi metode analisis karbohidrat..........................................22
Gambar 3. Hasil penempatan sampel matriks berdasarkan studi literatur...................................26
Gambar 4. Perbandingan hasil analisis karbohidrat total pada tiga matriks sampel pangan cair
ditambah dengan tiga matriks sampel pangan padat (N=18) dengan metode SNI
(Luff-Schoorl) dan Metode Anthrone sulfat..........................................................28
Gambar 5. Diagram kesalahan analisis metode karbohidrat total SNI 01-2891-1992 ..................45

xi
DAFTAR LAMPIRAN
Lampiran 1. Data hasil analisis perbandingan metode…………………………………………. 56
Lampiran 2. Uji statistik perbandingan metode dengan SPSS 17.0…………………………… 58
Lampiran 3. Prosedur analisis………………………………………………………………… 60
Lampiran 4. Metode yang divalidasi…………………………………………………………… 62
Lampiran 5 Verifikasi metode karbohidrat total SNI 01-2891-1992……………………….... 65
Lampiran 6. Uji statistik reprodusibilitas intralab…………………………………………….. 70

1
I. PENDAHULUAN
1.1. Latar Belakang
Karbohidrat merupakan komponen yang sering kita jumpai dalam bahan pangan.
Karbohidrat dalam pangan ada dalam berbagai macam bentuk dari glukosa sederhana hingga
bentuk polisakarida yang kompleks. Contoh bahan pangan yang banyak mengandung karbohidrat
diantaranya serealia dan umbi-umbian. Karbohidrat berkontribusi besar dalam menyusun produk
pangan pada umumnya (Fennema 1996) dan merupakan salah satu makronutrien yang dibutuhkan
oleh tubuh. Lebih dari 70% kebutuhan energi manusia dipenuhi dengan karbohidrat (BeMiller
2010). Sifat fungsional karbohidrat yang penting dalam proses pengolahan pangan, menyebabkan
keberadaan karbohidrat menjadi komponen yang perlu diperhatikan dan dianalisis.
Analisis total karbohidrat telah lama dilakukan pada berbagai sampel seperti ekstrak
tanaman (Yemm dan Willis 1954), tanah (Safarik dan Satruckova 1992), feses (Ameen and Powell
1985), produk farmasi (Leyva et al 2007) dan produk pangan (BeMiller 2009). Jumlah karbohidrat
dalam produk pangan perlu diketahui, antara lain untuk: standardisasi identitas pangan, label
nutrisi, deteksi adanya adulterasi dan untuk pengembangan suatu produk pangan. Peran
karbohidrat yang signifikan terutama dalam produk pangan menjadikan analisis total karbohidrat
penting.
Pengukuran karbohidrat sejak dahulu hingga sekarang masih dilakukan adalah
menggunakan metode by difference dalam sistem analisis proksimat Weende yaitu dengan
mengurangi kadar air, kadar protein, kadar lemak dan kadar abu dari total bahan pangan yang
diujikan (Southgate 1976). Akan tetapi pada metode by difference terdapat kelemahan yaitu dapat
menyebabkan hasil yang kurang akurat. Hasil yang kurang akurat diakibatkan oleh akumulasi dari
kesalahan pada metode yang digunakan untuk menganalisis komponen lain, seperti protein dan
lemak, sehingga nilai yang didapat semakin jauh dari nilai sebenarnya. Selain itu juga ada
kemungkinan komponen nonkarbohidrat seperti asam organik, lignin dan tanin ikut terhitung
sebagai karbohidrat.
Berbagai bidang yang spesifik seperti industri pemurnian gula dan penghasil minuman
anggur, muncul kebutuhan untuk mengembangkan pengukuran gula secara langsung. Hal ini
memicu berkembangnya kajian metodologis mengenai karbohidrat terlarut, diantaranya dengan

2
metode refraktometri, gravimetri, polarimetri, titrimetri dan kolorimetri kondensasi (Southgate
1976). Banyaknya metode analisis yang dikembangkan tentu dapat menimbulkan kebingungan
karena setiap metode dapat menghasilkan nilai yang berbeda. Dengan demikian, perlu ditetapkan
persetujuan untuk menggunakan satu metode.
Metode yang digunakan untuk analisis total karbohidrat langsung yang ditetapkan oleh
BSN (Badan Standardisasi Nasional) melalui SNI 01-2891-1992, yaitu tentang cara uji makanan
dan minuman, adalah Metode Luff-Schoorl. Namun terdapat kelemahan pada Metode
Luff-Schoorl karena dapat menimbulkan hasil yang kurang konsisten (Faulks dan Timms 1985)
sehingga tingkat kepercayaan terhadap hasil kurang. Selain itu Metode Luff-Schoorl juga
membutuhkan pekerjaan yang tidak sederhana dan lebih banyak memakan waktu dibanding
metode analisis kolorimetri.
Beberapa metode yang digunakan untuk menganalisis total karbohidrat secara langsung
selain Luff–Schoorl, yaitu Metode Anthrone sulfat, fenol sulfat, orsinol dan resorsinol. Metode
Anthrone sulfat adalah yang paling umum digunakan (Leyva et al 2008) dengan menggunakan
instrument spektofotometer UV-Visible. Metode Anthrone ini memiliki banyak keunggulan
antara lain kesederhanaan ujinya, spektrumnya yang luas dan sensitifitasnya yang cukup baik
(Koehler 1952).
Analis pangan sampai sekarang masih terikat dengan prosedur analisis yang telah
ditetapkan oleh peraturan yaitu SNI (Standard Nasional Indonesia) 01-2891-1992. Penggunaan
metode yang baku merupakan hal yang penting untuk menjamin bahwa hasil yang diperoleh sesuai
dengan persyaratan yang ditetapkan oleh pemerintah (Nielsen, 2010). Beberapa metode analisis
pangan bersifat empiris yaitu metode itu masih digunakan hingga saat ini karena memang metode
itu yang sudah digunakan sejak dulu dan hasil yang didapat cukup konsisten (Sawyer 1984).
Begitu pula halnya dengan Metode Luff-Schoorl yang dijadikan metode standard dalam SNI
01-2891-1992 karena sifatnya yang empiris.
Metode analisis total karbohidrat dengan menggunakan Metode Anthrone sulfat bukan
merupakan metode standard, maka perlu divalidasi sebelum digunakan. Selain itu, validasi metode
terutama untuk matriks pangan yang spesifik penting untuk menjamin ketepatan dari metode yang
digunakan (Nielsen, 2010). Dengan adanya validasi, kita dapat mengetahui bahwa hasil dari
analisis itu dapat dipercaya pada matriks pangan yang dianalisis.
Sampai sejauh ini belum pernah dilakukan perbandingan metode antara Metode
Luff-Schoorl dengan Metode Anthrone sulfat untuk menganalisis total karbohidrat pada bahan

3
pangan cair dan belum diketahui validitas Metode Anthrone sulfat dengan hidrolisis asam untuk
menganalisis karbohidrat total secara langsung terutama pada matriks pangan cair untuk dapat
menggantikan Metode Luff-Schoorl. Oleh karena itu tujuan dari penelitian ini adalah
membandingkan kedua metode pada matriks pangan cair dengan tingkat karbohidrat rendah,
sedang dan tinggi dan menentukan metode mana yang lebih baik untuk digunakan dalam analisis
rutin dan melakukan validasi Metode Anthrone atau verifikasi metode yang sudah baku yaitu Luff
Schoorl berdasarkan hasil perbandingan metode.
1.2. Tujuan
1.2.1. Tujuan Umum
Tujuan umum dari penelitian ini adalah menentukan metode yang lebih baik untuk
analisis total karbohidrat antara metode SNI 01-2891-1992 secara titrimetri dan metode kandidat
dengan Anthrone sulfat secara spektrofotometri.
1.2.2. Tujuan Khusus
Tujuan khusus dari penelitian ini adalah:
1. Melakukan perbandingan hasil analisis total karbohidrat dengan menggunakan dua
metode berbeda yaitu metode SNI 01-2891-1992 secara titrimetri dengan metode
kandidat yang menggunakan Anthrone sulfat secara spektrofotometri.
2. Melakukan validasi Metode Anthrone sulfat atau verifikasi metode SNI berdasarkan hasil
yang diperoleh dari perbandingan metode pada berbagai matriks.
1.3. Manfaat Penelitian
Manfaat dari penelitian ini adalah:
1. Mendapatkan informasi mengenai metode analisis mana yang lebih baik untuk
digunakan pada analisis total karbohidrat secara rutin.
2. Mendapatkan informasi mengenai tingkat validitas metode yang digunakan
1.4. Hipotesis
Hasil pengukuran dengan Metode Anthrone tidak berbeda nyata dengan dengan hasil
pengukuran dengan Metode Luff-Schoorl, sehingga Metode Anthrone dapat diadopsi sebagai
metode alternatif. Selanjutnya diperlukan Metode Anthrone diuji validitasnya untuk analisis total
karbohidrat.

4
II. TINJAUAN PUSTAKA
2.1. Karbohidrat
Kebanyakan ahli kimia kesulitan dalam mengelompokkan bahan apa saja yang termasuk ke
dalam karbohidrat. Definisi klasik karbohidrat berdasarkan asal katanya yaitu carbo dari bahasa
Latin dan hydros dari bahasa Yunani adalah ‘hidrat dari karbon’ yang mengandung hidrogen dan
oksigen dengan perbandingan 2:1 (Southgate 1978) atau elemen yang terdiri dari air dan karbon
dengan perbandingan 1:1 (Kennedy dan White 1988). Karbohidrat adalah senyawa organik yang
mengandung karbon, hidrogen dan oksigen baik dalam bentuk molekul sederhana maupun
kompleks (Christian dan Vaclavik 2003).
Karbohidrat telah menjadi sumber energi utama untuk metabolisme pada manusia dan sarana
untuk memelihara kesehatan saluran pencernaaan manusia. Karbohidrat adalah penyumbang
utama dari komponen yang membentuk produk pangan baik sebagai komponen alami maupun
bahan yang ditambahkan. Karbohidrat meliputi lebih dari 90% dari berat kering tanaman.
Karbohidrat banyak tersedia dan murah. Penggunaannya sangat luas dan jumlah penggunaannya
cukup besar (Fennema 1996) baik untuk pemanis, pengental, penstabil, gelling agents dan fat
replacer (Christian dan Vaclavik 2003). Karbohidrat dapat dimodifikasi baik secara kimia dan
biokimia dan modifikasi itu digunakan untuk memperbaiki sifat dan memperluas penggunaannya.
2.1.1 Struktur karbohidrat
Karbohidrat digunakan dalam kimia untuk senyawa dengan formula Cm(H2O)n, tetapi kini
rumus molekul itu tidak secara kaku digunakan untuk mendefinisikan karbohidrat (Kennedy dan
White 1988). Sebelumnya beberapa ahli kimia memasukkan formaldehid dan glikoaldehid sebagai
karbohidrat, namun sekarang istilah karbohidrat dalam biokimia, tidak mengikutsertakan senyawa
yang kurang dari tiga atom karbon. Southgate (1978) menggunakan definisi karbohidrat sebagai
senyawa yang tersusun oleh polihidroksi aldehid, keton, alkohol, asam dan turunan sederhananya
serta polimernya yang memiliki ikatan polimer tipe asetal.
Menurut strukturnya karbohidrat dapat dibagi menjadi kelompok sakarida: monosakarida,
oligosakarida dan polisakarida. Monosakarida adalah gula sederhana yang tidak dapat dipecah lagi
menjadi molekul yang lebih kecil dan monosakarida inilah yang menjadi unit penyusun dari
oligosakarida dan polisakarida. Oligosakarida dan polisakarida tersusun dari monosakarida yang
dihubungkan dengan ikatan glikosidik.

5
2.1.2. Monosakarida
Monosakarida terdiri dari tiga sampai delapan karbon atom, tetapi umumnya hanya lima atau
enam yang biasa ditemukan. Biasanya monosakarida digolongkan berdasarkan jumlah atom
karbonnya, misalnya triosa (C3H6O3), tetrosa (C4H8O3), pentosa (C5H10O5) dan heksosa (C6H12O6).
Dari golongan tersebut dapat dibagi lagi berdasarkan gugus fungsional yang ada, misalnya dari
golongan heksosa ada aminoheksosa (C6H13O5N), deoksiheksosa (C6H12O5) dan asam heksuronat
(C6H10O7). Contoh monosakarida adalah glukosa dan fruktosa.
2.1.3. Oligosakarida
Oligosakarida terdiri dari beberapa monosakarida (2-10) yang saling terikat oleh ikatan
glikosidik. Tetapi ada juga yang mengklasifikasikan sendiri karbohidrat dengan dua gugus gula
sebagai disakarida. Menurut Christian dan Vaclavik (2003) disakarida terdiri dari dua molekul
monosakarida yang bergabung dengan ikatan glikosidik. Contoh disakarida di pangan adalah
maltosa, selubiosa, dan sukrosa. Oligosakarida yang memiliki lebih dari tiga gugus gula contohnya
adalah rafinosa dan stakiosa.
2.1.4. Polisakarida
Polisakarida merupakan polimer dari gula sederhana yang tersusun atas lebih dari sepuluh
monomer gula sederhana. Contoh polisakarida di makanan adalah pati, pektin dan gum. Ketiganya
adalah polimer karbohidrat kompleks dengan sifat yang berbeda, tergantung unit gula
penyusunnya, tipe ikatan glikosidik dan derajat percabangan molekul.
2.2. Pentingnya Analisis Total Karbohidrat
Total karbohidrat yang ada dalam bahan pangan perlu diketahui dengan alasan: standards of
identity (pangan harus memiliki komposisi yang sesuai dengan regulasi pemerintah); nutritional
labelling (menginformasi konsumen mengenai kadar nutrisi dalam bahan pangan); detection of
adulteration (tiap tipe pangan memiliki 'fingerprint' karbohidrat); food quality (sifat fisikokimia
dari pangan seperti kemanisan, penampakan, stabilitas dan tekstur tergantung tipe dan stabilitas
karbohidrat yang ada); ekonomi (agar lebih dapat menghemat biaya produksi bahan yang
digunakan pada industri) dan food processing (efisiensi dari proses pangan banyak tergantung
pada jenis dan kadar karbohidrat). Dalam berbagai studi mengenai bahan makanan penting untuk
mengetahui persentasi kadar karbohidrat pada pangan yang diujikan sehingga nilai karbohidrat
pada bahan lain dapat dikonversi menjadi nilai total pangan.

6
2.3. Total Karbohidrat dalam Bahan Pangan dan Metode Analisisnya
2.3.1. Definisi total karbohidrat
Total karbohidrat atau total karbohidrat menurut Badan Pengawasan Obat dan Makanan
(2005) meliputi gula, pati, serat pangan dan komponen karbohidrat lain. Pernyataan jumlah total
karbohidrat dalam gram penyajian yang dinyatakan dengan nilai gram terdekat, jika penyajian
kurang dari 0,5 gram, jumlah kadarnya dapat dinyatakan sebagai nol dan jika penyajian lebih dari
0,5 gram dibulatkan ke kelipatan 1 gram terdekat. Total karbohidrat dapat dinyatakan dengan total
karbohidrat by difference.
Total karbohidrat dalam pengukuran karbohidrat dengan metode langsung dinyatakan dalam
bentuk persen yang setara dengan glukosa. Satuan glukosa (glucose equivalent) juga dapat diganti
dengan larutan gula lain yang dijadikan sebagai larutan standar.
2.3.2. Metode analisis total karbohidrat
Sejumlah teknik analisis telah dikembangkan untuk mengukur jumlah dan tipe karbohidrat
yang ada di bahan pangan. Kadar karbohidrat di bahan pangan dapat diketahui dengan menghitung
persentase yang tersisa setelah semua komponen lain telah diukur (total carbohydrate by
difference), yaitu dengan persamaan (1.1) (SNI 01-2891-1992):
(1.1)
Metode by difference ini masih digunakan oleh FDA, tetapi metode ini dapat menghasilkan
nilai yang salah karena ada kemungkinan terjadi akumulasi kesalahan dari metode-metode yang
digunakan untuk mengukur komponen lain, dan kemungkinan adanya komponen non karbohidrat
yang terukur sebagai karbohidrat menyebabkan penyimpangan yang lebih besar. Pengukuran kadar
karbohidrat secara langsung lebih baik karena didapat hasil lebih yang akurat.
2.3.2.1. Analisis karbohidrat langsung
Metode yang telah dikembangkan untuk analisis karbohidrat sangat banyak, dan tergantung
juga oleh jenis analisis (kuantitatif atau kualitatif) dan tipe karbohidrat yang dianalisis. Sehingga
metode pengukuran karbohidrat sangat beragam mulai dari metode kromatografi dan elektroforesis
(Kromatografi Lapis Tipis, Kromatografi Likuid Kinerja Tinggi dan Kromatografi Gas); metode
kimia (metode titrasi Lane Eynon, metode gravimetri Munson Walker, metode Luff Schoorl,
metode kolorimetri seperti anthrone sulfat dan fenol sulfat); metode enzimatis; metode fisik
(polarimetri, indeks refraktif, densitas dan infra merah) serta metode immunoassay.

7
Uji karbohidrat yang resmi ditetapkan oleh BSN dalam SNI 01-2891-1992 yaitu analisis total
karbohidrat dengan menggunakan metode Luff Schoorl. Pada tahun 1936 International
Commission for Uniform Methods of Sugar Analysis mempertimbangkan Metode Luff-Schoorl
sebagai salah satu metode yang digunakan untuk menstandarkan analisis gula pereduksi karena
metode Luff Schoorl saat itu menjadi metode yang resmi dipakai di pulau Jawa, di samping
nominator lainnya yaitu metode Lane-Eynon. Tetapi pada saat itu metode kolorimetri belum
banyak berkembang dan dalam catatan komisi itu terdapat agenda untuk melakukan penyeragaman
analisis gula dengan metode kolorimetri.
Berikut ini adalah beberapa jenis analisis total karbohidrat langsung:
2.3.2.1.1. Analisis total karbohidrat dalam SNI 01-2891-1992
Seluruh senyawa karbohidrat yang ada dipecah menjadi gula-gula sederhana (monosakarida)
dengan bantuan asam yaitu HCl dan panas. Monosakarida yang terbentuk kemudian dianalisis
dengan Metode Luff-Schoorl. Prinsip analisis dengan Metode Luff-Schoorl yaitu reduksi Cu2+
menjadi Cu 1+ oleh monosakarida. Monosakarida bebas akan mereduksi larutan basa dari garam
logam menjadi bentuk oksida atau bentuk bebasnya. Kelebihan Cu2+ yang tidak tereduksi
kemudian dikuantifikasi dengan titrasi iodometri (SNI 01-2891-1992).
Reaksi yang terjadi (1.2):
Karbohidrat kompleks → gula sederhana (gula pereduksi)
Gula pereduksi+ 2 Cu2+→ Cu2O(s)
2 Cu2+ (kelebihan) + 4 I-→ 2 CuI2 → 2 CuI- + I2
I2 + 2S2O32-→ 2 I- + S4O6
2-
(1.2)
Osborne dan Voogt (1978) mengatakan bahwa Metode Luff-Schoorl dapat diaplikasikan untuk
produk pangan yang mengandung gula dengan bobot molekuler yang rendah dan pati alami atau
modifikasi.
Kemampuan mereduksi dari gugus aldehid dan keton digunakan sebagai landasan dalam
mengkuantitasi gula sederhana yang terbentuk. Tetapi reaksi reduksi antara gula dan tembaga
sulfat sepertinya tidak stoikiometris dan sangat tergantung pada kondisi reaksi. Faktor utama yang
mempengaruhi reaksi adalah waktu pemanasan dan kekuatan reagen. Penggunaan luas dari metode
ini dalam analisis gula adalah berkat kesabaran para ahli kimia yang memeriksa sifat empiris dari
reaksi dan oleh karena itu dapat menghasilkan reaksi yang reprodusibel dan akurat (Southgate
1976).

8
2.3.2.1.2. Analisis total karbohidrat dengan Metode Anthrone sulfat
Penggunaan Metode Anthrone untuk analisis total karbohidrat mulai berkembang sejak
penggunaan pertama kali oleh Dreywood pada tahun 1946 untuk uji kualitatif. Dasar dari reaksi ini
adalah kemampuan karbohidrat untuk membentuk turunan furfural dengan keberadaan asam dan
panas, yang kemudian diikuti dengan reaksi dengan anthrone yang menghasilkan warna biru
kehijauan (Sattler dan Zerban 1948) dalam Brooks et al (1986).
Anthrone, C6H4COC6H4CH2, adalah turunan dari anthraquinone. Senyawa ini diproduksi
oleh reduksi katalitik dari anthraquinone oleh asam hidroklorat dengan keberadaan logam timah.
Senyawa ini mungkin ada dalam bentuk keto atau enol, yang masing-masing dikenal dengan nama
anthrone and anthranol. Reaksinya dapat dilihat pada persamaan (1.3):
(1.3)
Mekanisme pembentukan warna anthrone dengan gula telah diteliti. Hurd dan Isenhour
(1932) dan Wolfrom et al (1948) mempostulasikan bahwa karbohidrat dan turunannya mengalami
pembentukan cincin dalam keberadaan asam kuat dari mineral, seperti yang ditunjukkan untuk
glukosa (1.4):
(1.4)
Tiap tahap adalah pemecahan dari glukosa(I) menjadi 5-(hydroxymethyl)-2-furaldehyde(IV)
menunjukkan dehidrasi baik pada double bond atau pembentukan cincin. Wolfrom et al. (1948)
menunjukkan bukti spektroskopik untuk senyawa intermediate (II) dan (III) pada reaksi ini Sattler

9
and Zerban (1948) menyarankan bahwa pembentukan warna hijau pada reaksi anthrone tergantung
oleh keberadaan 5-(hidroksimetil)-2-furaldehid, atau senyawa furfural yang mirip, yang dibentuk
oleh reaksi asam sulfat pada karbohidrat.
Momose et al. (1957) melakukan kromatografi pada ekstrak benzene dari pewarna terhadap
alumina dan menunjukkan bahwa bagian yang dapat larut dari benzene-terdiri dari beberapa
pewarna yang memberikan pewarnaan yang berbeda dengan asam sulfat. Mereka menentukan
berat molekul dari salah satu pewarna utama yaitu kurang lebih 530, dan mempostulasikan
formula dari pewarna itu (C47H30O3). Mereka menyimpulkan bahwa 3 mol anthrone bereaksi
dengan 1 mol glukosa, yang digambarkan dalam persamaan (1.5):
3C14H10O + C6H12O6 C47H3O30 + 5H2O + CH2O (1.5)
Dari data analisis dan spektrum inframerah dari pewarna, dan mekanisme reaksinya
dipertimbangkan, mereka menduga struktur yang mungkin adalah 1,2,5,- atau
1,3,5,-trianthronylidenepentane.
Ludwig dan Goldberg (1956) melaporkan adaptasi dari Metode Anthrone kolorimetri untuk
analisis total karbohidrat secara kuantitatif pada pangan. Metode yang digunakan relatif cepat dan
akurat serta lebih baik daripada metodologi analisis karbohidrat sebelumnya, yaitu metode
Somogyi-Shaffer-Hartmann yang menggunakan teknik teknik iodometri dan prinsip gula
pereduksi. Mereka menunjukkan bahwa persiapan hidrolisis dan deproteinisasi tidak perlu
dilakukan ketika teknik anthrone digunakan.
Uji Anthrone ini memiliki kelebihan dalam hal sensitifitas dan kesederhanaan ujinya
(Koehler 1952).Sejumlah kecil karbohidrat dapat memberikan warna yang terdeteksi dengan
menggunakan spektrofotometer. Dreywood (1946) melakukan uji spesifisitas dari reaksi dan
membuat daftar 18 jenis karbohidrat, termasuk beberapa turunan selulosa, yang memberikan hasil
positif. Dia juga melaporkan hasil negatif terhadap kelompok besar nonkarbohidrat, termasuk
sejumlah resin sintetik nonselulosa, asam organik, aldehid, fenol, lemak, terpena, alkaloid, dan
protein. Nonkarbohidrat yang menunjukkan hasil positif hanya furfural, tetapi hasil positif ini
cepat menghilang karena warna hijau dikaburkan oleh presipitat coklat. Morris (1948) juga
menunjukkan spesifisitas anthrone untuk karbohidrat sangat tinggi, dan dia melaporkan reaksi
positif untuk semua mono-, di-, dan polisakarida murni yang diujikan, juga sampel of dekstrin,
dekstran, pati, polisakarida tumbuhan dan gum, polisakarida tipe II dan II dari pneumococcus,
glukosida, dan senyawa asetat dari mono-, di-, dan polisakarida.

10
Kekurangan dari Metode Anthrone adalah ketidakstabilan dari reagen (anthrone yang
dilarutkan dalam asam sulfat), sehingga perlu dilakukan persiapan reagen yang baru setiap hari.
Dreywood (1946) memperhatikan bahwa panas yang dihasilkan oleh pelarutan asam sulfat
merupakan bagian yang penting dalam uji. Morris (1948) melihat signifikansi dari panas pada
reaksi anthrone dan menunjukkan bahwa pada sejumlah karbohidrat yang diberikan, intensitas
warna bervariasi dengan jumlah panas yang dihasilkan. Oleh karena itu kurva standar juga perlu
dibuat setiap hari.
Nilai total karbohidrat tidak dapat dinyatakan dalam persen karbohidrat, tetapi lebih baik
dinyatakan dengan istilah glucose equivalents per cent, karena kepekatan warna yang dihasilkan
dari reaksi anthrone bervariasi dengan tipe gula yang ada. Kepekatan warna yang sama contohnya,
ditunjukkan oleh 100 µg. glukosa, 105 µg. maltosa, dan 111 µg glikogen. Gula murni lain selain
glukosa dapat dikalkulasi dengan faktor konversi. Tetapi jika terdapat campuran karbohidrat yang
tidak diketahui pada bahan pangan faktor konversi itu tidak dapat digunakan, dan hasilnya bukan
persentase karbohidrat absolut, melainkan ekuivalen glukosa, yang dapat bervariasi dari nilai
persentasi karbohidrat yang sebenarnya dengan jumlah yang tidak dapat ditentukan. Keganjilan ini
tidak signifikan ketika nilai glucose equivalents per cent digunakan hanya sebagai basis untuk
mengkonversi nilai total karbohidrat menjadi nilai total pangan (Beck dan Bibby 1961). Untuk
tujuan ini glucose equivalents per cent hanya sebagai indeks dari persentasi absolute dari
masing-masing karbohidrat dalam pangan.
2.4. Validasi dan Verifikasi Metode
Metode analisis memiliki beberapa atribut, seperti ketepatan, ketelitian, spesifisitas,
sensitivitas, kemandirian, dan kepraktisan, yang harus dipertimbangkan ketika akan digunakan
(Garfield et al. 2000). Informasi yang digunakan untuk mengambil keputusan harus seimbang
dengan pertimbangan praktis seperti biaya, waktu, risiko, kesalahan, dan tingkat keahlian yang
diperlukan. Selain itu suatu laboratorium yang akan menerapkan suatu metode perlu
mempertimbangkan apakah data validasi yang ada mengenai metode tersebut cukup memadai atau
apakah masih membutuhkan tindakan validasi ulang sebelum metode itu digunakan. Selanjutnya
jika data validasi telah cukup memadai, laboratorium perlu mengetahui apakah level performa
yang ditunjukkan oleh data validasi tersebut mampu dilaksanakan. Untuk mencapai level performa
itu dibutuhkan analis yang kompeten serta peralatan dan fasilitas yang memadai (Jelita 2011).
Data validasi yang kurang memadai biasanya ada pada metode yang baru dikembangkan baik
oleh laboratorium itu sendiri atau yang dikembangkan oleh pihak lain; metode yang digunakan

11
oleh laboratorium lain atau metode yang telah dipublikasi tetapi belum menjadi metode baku.
Ketika data validasi yang ada telah memadai, yaitu seperti pada metode yang telah divalidasi oleh
organisasi terstandarisasi seperti AOAC (Association of Official Analytical Chemists)
Internasional, laboratorium umumnya hanya menjaga performa data dengan cara melakukan
verifikasi metode.
Validasi metode analisis adalah suatu tindakan penilaian terhadap parameter tertentu,
berdasarkan percobaan laboratorium, untuk membuktikan bahwa parameter tersebut memenuhi
persyaratan untuk penggunaannya (Harmita, 2004). Berdasarkan Harvey (2000), validasi
merupakan suatu proses evaluasi kecermatan dan keseksamaan yang dihasilkan oleh suatu
prosedur dengan nilai yang dapat diterima. Sebagai tambahan, validasi memastikan bahwa suatu
prosedur tertulis memiliki detail yang cukup jelas sehingga dapat dilaksanakan oleh analis atau
laboratorium yang berbeda dengan hasil yang sebanding. Menurut AOAC (2002) validasi metode
menunjukkan apakah suatu metode sesuai dengan tujuan yang diinginkan. Dalam praktiknya,
memungkinkan untuk merancang percobaan yang akan dilakukan sehingga karakteristik validasi
yang sesuai dapat diterapkan untuk mendapatkan hasil yang cukup dan menyeluruh mengenai
kemampuan suatu prosedur analisis, seperti: spesifisitas, linearitas, rentang, akurasi (kecermatan),
dan presisi (keseksamaan) (EMA, 1995).
Verifikasi metode adalah suatu tindakan validasi metode tetapi hanya pada beberapa beberapa
karakteristik performa saja. Laboratorium harus menentukan karakteristik performa yang
dibutuhkan. Spesifikasi analisis dapat menjadi acuan untuk merancang proses verifikasi.
Rancangan yang baik akan menghasilkan informasi yang dibutuhkan serta meminimalisir tenaga,
waktu, serta biaya. Pemilihan parameter validasi atau verifikasi tergantung pada beberapa faktor
seperti aplikasi, sampel uji, tujuan metode, dan peraturan lokal atau internasional.
Adapun beberapa parameter analisis yang harus dipertimbangkan dalam validasi metode
analisis :
2.4.1. Akurasi
Akurasi atau kecermatan adalah seberapa dekat suatu hasil pengukuran kepada nilai
sebenarnya. Terkadang masalah dalam menentukan akurasi adalah ketidaktahuan terhadap nilai
yang sebenarnya. Dalam beberapa tipe sampel kita dapat menggunakan sampel yang telah
diketahui nilainya dan mengecek metode pengukuran yang kita gunakan untuk menganalisis
sampel itu sehingga kita mengetahui akurasi dari prosedur yang diujikan, metode ini disebut
dengan CRM (Certified Reference Method). Pendekatan lain adalah dengan membandingkan

12
hasilnya dengan hasil yang dilakukan oleh lab lain (Smith, 2010) atau dengan menggunakan
metode referen (Walton 2001). Akurasi juga dapat diketahui dengan melakukan uji rekoveri
(Walton 2001). Hasil uji ini akurasi dapat dinyatakan sebagai persen perolehan kembali (recovery)
analat yang ditambahkan pada sampel. Sampel ditambahkan (spiking) dengan standar yang telah
diketahui jumlah dan kadarnya (EMA, 1995). Rentang nilai penerimaan kecermatan suatu metode
akan bervariasi sesuai kebutuhannya (FAO, 1998). Adapun AOAC menetapkannya seperti dalam
Tabel 1.
Tabel 1 Persentase rekoveri yang dapat diterima sesuai dengan konsentrasi analat
(%) analat Unit Rata-rata rekoveri (%)
100 100% 98-102
10 10% 95-102
1 1% 97-103
0.1 0.10% 95-105
0.01 100 ppm 90-107
0.001 10 ppm 80-110
0.0001 1 ppm 80-110
0.00001 100 ppb 80-110
0.000001 10 ppb 60-115
0.0000001 1 ppb 40-120
(sumber: AOAC 2002)
2.4.2. Presisi
Presisi adalah ukuran yang menunjukkan derajat kesesuaian antara hasil uji individual, diukur
melalui penyebaran hasil individual dari rata-rata jika prosedur diterapkan secara berulang pada
sampel-sampel yang diambil dari campuran yang homogen (Harmita, 2004). Presisi dapat dibagi
dalam dua kategori: keterulangan atau ripitabilitas (repeatability) dan ketertiruan (reproducibility).
Ripitabilitas adalah nilai presisi yang diperoleh jika seluruh pengukuran dihasilkan oleh satu orang
analis dalam satu periode tertentu, menggunakan pereaksi dan peralatan yang sama dalam
laboratorium yang sama. Ketertiruan adalah nilai presisi yang dihasilkan pada kondisi yang
berbeda, termasuk analis yang berbeda, atau periode dan laboratorium yang berbeda dengan analis
yang sama. Karena ketertiruan dapat memperbanyak sumber variasi, ketertiruan dari analisis tidak
akan lebih baik hasilnya dari nilai keterulangan (Harvey, 2000).

13
Presisi dalam hal ripitabilitas diukur dengan menghitung relative standard deviation atau
simpangan baku relatif (RSD) dari beberapa ulangan dengan menggunakan rumus (1.6):
(1.6)
Standar deviasi ripitabilitas bervariasi tergantung pada konsentrasi (AOAC 2002). Oleh karena itu
hasil yang didapat dari perhitungan dibandingkan hasilnya dengan nilai yang ada di Tabel 2.
Tabel 2 Nilai presisi (RSD) sesuai dengan konsentrasi analat
(%) analat Konsentrasi RSD (%)
100 100% 1
10 10% 1.5
1 1% 2
0.1 0.10% 3
0.01 100 ppm 4
0.001 10 ppm 6
0.0001 1 ppm 8
0.00001 10 ppb 15
(sumber: AOAC 2002)
Nilai yang didapat juga dapat dibandingkan atau dengan menggunakan rumus (1.7):
(1.7)
dengan C adalah konsentrasi yang didapat dari rataan.
Nilai yang dapat diterima untuk ripitabilitas adalah antara 1/2 dan 2 kali dari nilai yang dijadikan
sebagai pembanding. Ada juga yang menggunakan RSD Horwitz sebagai nilai pembanding, RSD
Horwitz dihitung dengan rumus (1.8):
(1.8)
Dengan menggunakan pembanding RSD Horwitz nilai yang dapat diterima untuk ripitabilitas
adalah RSD yang terhitung dari ulangan yang ada harus kurang dari 2/3 dari nilai RSD Horwitz
(Garfield 2000).

14
2.4.3. Spesifisitas
Spesifisitas dari metode analitik tertentu berarti metode itu hanya mendeteksi komponen yang
diinginkan. Metode analitis dapat bersifat sangat spesifik untuk komponen tertentu atau pada
beberapa kasus dapat menganalisis spektrum komponen yang luas (Smith, 2010).
Spesifisitas suatu metode diuji dengan membandingkan hasil dari sampel yang mengandung
pengotor dengan hasil sampel yang tidak mengandung pengotor. Pada dasarnya, spesifisitas dapat
diuji secara langsung atau tidak langsung. Pendekatan secara tidak langsung ditinjau dari
penerimaan parameter akurasi. Pendekatan secara langsung ditinjau dari keberadaan komponen
pengganggu (Ermer dalam Ermer dan Miller, 2005). Cara yang terakhir dilakukan dengan
menambahkan sejumlah tertentu komponen pengganggu pada larutan standar murni. Jika
diperkirakan tidak adanya komponen pengganggu pada sampel, spesifisitas dapat ditunjukkan
dengan membandingkan hasil uji sampel dengan standar (EMA, 1995).
2.4.4. Limit Deteksi dan Limit Kuantitasi
Limit deteksi atau Limit of Detection (LOD) suatu metode analisis adalah jumlah terkecil dari
analat yang dapat dideteksi namun jumlah ini belum tentu dapat dikuantisasi dengan presisi yang
baik oleh metode tersebut. Limit kuantitasi atau Limit of Quantitation (LOQ) yang disebut juga
limit determinasi adalah konsentrasi terendah dari analat yang dapat ditentukan secara kuantitatif
dengan presisi dan akurasi yang dapat diterima (Ermer dalam Ermer dan Miller, 2005).
Giese (2004) menyatakan bahwa terdapat dua cara untuk menentukan LOD dan LOQ, yaitu
dengan menentukan kurva kalibrasi menggunakan sepuluh level konsentrasi, atau melakukan
analisis blanko berulang. Tetapi ada masalah dalam pendekatan menggunakan blanko karena
seringkali sulit diukur dan variasinya sangat tinggi. Lebih lanjut, nilai yang didapat dengan
pendekatan seperti ini tidak bergantung dari analat (AOAC 2002).
Limit deteksi hanya berguna untuk mengontrol ketidakmurnian yang tidak diinginkan yang
konsentrasinya harus tidak lebih dari level tertentu dan mengontrol kontaminan dengan konsentrasi
rendah, sedangkan materi yang bermanfaat harus ada pada konsentrasi yang cukup tinggi agar
dapat menjadi fungsional. Limit deteksi dan determinasi seringkali bergantung pada kemampuan
instrumen (AOAC 2002).
2.4.5. Linieritas
Linearitas metode analisis menunjukkan kemampuan suatu metode untuk memperoleh hasil
uji, yang baik langsung maupun dengan definisi transformasi matematis yang baik, proporsional

15
dengan konsentrasi analat dalam sampel pada range tertentu (Leyva et al 2008). Linieritas dapat
diuji secara informal dengan membuat plot residual yang dihasilkan oleh regresi linier pada respon
konsentrasi dalam satu seri kalibrasi (Thompson et al. 2002).
Linieritas harus dievaluasi dengan pemeriksaan visual terhadap plot absorbansi yang
merupakan fungsi dari konsentrasi analat. Jika hubungannya linier, hasil uji dievaluasi lebih lanjut
secara statistik dengan perhitungan garis regresi. Dalam penentuan linieritas, sebaiknya
menggunakan minimum lima konsentrasi (EMA, 1995). Rentang penerimaan linieritas tergantung
dari tujuan pengujian. Pada kondisi yang umum, nilai koefisien regresi (r2) ≥ 0,99.
2.5. Matriks Sampel
Suatu metode harus dapat menunjukkan rekoveri dan ripitabilitas yang dapat diterima
pada konsentrasi dan matriks yang mewakili kelompok sampel dimana metode itu hendak
diterapkan (AOAC 2002). Suatu metode yang hendak diterapkan pada “pangan” secara umum,
metode tersebut perlu diujikan pada jenis pangan yang dianggap mewakili kelompok pangan
secara umum. Sampel yang yang dianggap mewakili dapat dipilih berdasarkan skema segitiga atau
triangle scheme yang disarankan AOAC Internasional (Gambar 1) (Sullivan dan Carpenter 1993).
Skema segitiga ini berdasarkan kadar karbohidrat, protein dan lemaknya yang mana dianggap
memiliki pengaruh terbesar terhadap kemampuan metode analisis. Suatu kelompok pangan, yang
diwakili oleh segitiga kecil, dikatakan memiliki kadar yang “tinggi”, “sedang” dan “rendah”
berdasarkan kadar karbohidrat, protein dan lemaknya. Pangan kompleks diposisikan pada salah
satu segitiga kecil—menurut kadar karbohidrat, lemak dan proteinnya (dengan persentase yang
telah dinormalisasi menurut perbandingan dari ketiga komponen). Pemetaan ini dilakukan dengan
meniadakan persentase kadar air dan kadar abu. Tiap sudut segitiga merupakan kelompok pangan
yang terdiri dari 100% lemak, 100%protein, dan 100% karbohidrat.

16
Gambar 1. Matriks pangan berdasarkan kadar protein, lemak dan karbohidrat (Nielsen 2010).
Nielsen (2010) mengatakan bahwa kemampuan suatu metode analisis dipengaruhi oleh
matriks pangan (misalnya komponen dari pangan tersebut terutama lemak, protein dan
karbohidrat). Matriks pangan merupakan tantangan terbesar bagi para analis pangan. Makanan
dengan kadar lemak tinggi dan kadar gula tinggi dapat menghasilkan interferensi yang berbeda
dengan makanan dengan kadar lemak rendah dan kadar gula rendah. Prosedur digesti dan tahap
ekstraksi sangat penting bagi hasil analisis yang akurat. Hal ini tergantung pada matriks pangan.
Kompleksitas dari berbagai sistem pangan seringkali membutuhkan lebih dari satu teknik dan
prosedur untuk komponen spesifik tertentu, termasuk pengetahuan mengenai teknik mana yang
sesuai untuk matriks pangan yang spesifik.
Metode analitik yang umum harus dapat menganalisis kesembilan kombinasi yang ada,
menggantikan metode yang spesifik pada matriks tertentu (matrix dependent method). Misalnya
dengan menggunakan metode yang dipengaruhi oleh matriks, kita mungkin dapat
menggunakannya untuk menganalisis bahan yang rendah protein, dengan karbohidrat dan lemak
sedang seperti coklat dan keripik kentang. Tetapi untuk bahan dengan protein tinggi, lemak rendah
dan karbohidrat tinggi seperti susu rendah lemak, harus digunakan metode analisis yang lain. Hal
ini cukup merepotkan dan kemungkinan nilai yang didapat dari hasil analisis kedua metode perlu
dievaluasi (Nielsen 2010).
Validasi metode memerlukan pengetahuan mengenai identitas dari sampel yang akan
dianalisis, karena jika tidak, meski banyak informasi berguna yang didapat, tetapi informasi itu
akan terombang-ambing bagaikan kapal di lautan yang luas, tidak mengetahui dimana
keberadaannya, tanpa penanda yang menunjukkan posisinya (AOAC 2002). Oleh karena itu selain
melakukan studi literatur dilakukan uji proksimat terhadap sampel yang akan dianalisis untuk
17
mengonfirmasi komposisi dari sampel. Berikut data mengenai sampel yang akan digunakan dalam
perbandingan metode:
2.5.1. Kecap manis
Kecap manis merupakan produk olahan kedelai, yang teksturnya kental dan berwarna
coklat kehitaman (Suprapti 2005). Komposisi kimia kecap manis dapat dilihat pada Tabel 3.
Tabel 3 Komposisi kimia kecap manis, kecap asin dan santan
KomponenKadar (%)
Kecap manis Kecap asin Santan
Air 29,61a 63, 84a 54,9c
Protein kasar 1,46a 6,55a 4,20b
Lemak 0,14a 0,35a 34,30b
Abu 7,64a 18,48a 1-1,3c
Karbohidrat 61,15a 10,78a 5,60b
Garam (NaCl) 6,27a 18,43a (tidak ada informasi)
Sumber: aJudoamidjojo (1987) , bDirektorat Gizi (1967), c Woodroof (1979)
Kandungan gula dan viskositas yang tinggi dari produk ini disebabkan karena penambahan gula
dalam proses pembuatannya. Komponen terbesar kecap manis adalah karbohidrat, terutama
sukrosa, glukosa dan fruktosa (Kusumadewi, 2011). Kandungan gula kecap manis, yaitu 26-61%,
lebih banyak dari kecap asin yang hanya 4-19% (Judoamidjojo 1987). Kandungan asam amino
yang cukup tinggi dari kecap manis karena salah satu bahan yang digunakan untuk membuatnya
adalah kedelai yang memiliki kandungan protein yang tinggi (Santoso 1994). Rincian jenis asam
amino kecap manis dapat dilihat pada Tabel 4.
Dalam kecap manis, selain dari kedelai senyawa organik yang ada juga berasal dari gula
merah. Senyawa organik dalam kecap manis adalah asam sitrat, tartarat, suksinat, laktat, format,
piroglutamat, propionate dan butirat (Judoamidjojo et al 1985). Kecap yang bermutu tinggi
berkadar garam 18%, gula minimal 40% dan pHnya berkisar antara 4,7-4,8 (Buckle et al 1988).
Adapun persyaratan BSN untuk kecap manis (SNI 01-2543-1999) kadar garam minimal 3% dan
total gula (dihitung sebagai sakarosa) minimal 40%.
18
Tabel 4. Kandungan asam amino kecap asin dan kecap manis (g/100g)
Asam amino Kecap Asin Kecap Manis
Asam aspartat 0,42 0,03
Treonin 0,21 0,01
Serin 0,29 0,01
Glutamat 0,63 0,10
Prolin 0,16 0,01
Glisin 0,15 0,00
Alanin 0,30 0,02
Valin 0,30 0,02
Metionin 0,08 0,00
Isoleusin 0,29 0,02
Leusin 0,41 0,02
Tirosin 0,15 0,02
Fenilalanin 0,24 0,02
Lisin 0,27 0,01
Histidin 0,09 0,00
Arginin 0,27 0,00
Triptofan 0,00 0,00
Sistein 0,00 0,00
Sumber: Judoamidjojo et al (1985)
2.5.2. Kecap kedelai asin
Kecap kedelai asin atau yang biasa dikenal dengan nama kecap asin merupakan hasil
fermentasi dari kedelai. Menurut definisi SNI 01-3543-994 kecap kedelai adalah produk cair yang
diperoleh dari hasil fermentasi dan atau cara kimia (hidrolisis) kacang kedelai (Glycine max. L)
dengan atau tanpa penambahan bahan makanan lain dan bahan tambahan makanan yang diizinkan.
Warna dari kecap asin adalah coklat gelap. Tetapi warna ini bergantung pada proses penuaan atau
agingnya. Kecap asin mirip dengan kecap manis, hanya tanpa penambahan gula. Komposisi kimia
dari kecap kedelai dapat dilihat dari Tabel 3 dan kandungan asam aminonya dapat dilihat pada
Tabel 4.
2.5.3. Santan
Berdasarkan SNI 01-3816-1995, santan adalah produk cair yang diperoleh dengan
menyaring daging buah kelapa (Cocos nucifera) dengan atau tanpa penambahan bahan tambahan
makanan yang diizinkan. Santan merupakan emulsi lemak dalam air (Kirk dan Otmer 1950) yang

19
distabilisasi secara alamiah oleh protein (globulin dan albumin) dan fosfolipida (Tangsuphoom dan
Coupland, 2008). Senyawa δ-C8-laktone, δ-C10-laktone, dan n-oktanol merupakan komponen
volatil utama dan memberikan karakteristik aroma pada santan kelapa (Lin dan Wilkens 2006),
Adapun komposisi kimia santan dapat dilihat di Tabel 3. Tetapi komposisi kimianya
masih bervariasi tergantung pada varietas lokasi tumbuh, cara budi daya, kematangan buah, dan
metode ekstraksi seperti jumlah penambahan air dan suhu ekstraksi. Menurut Seow dan Gwee
(1997), komposisi kimia santan kelapa yang diekstraksi dengan tanpa penambahan air terdiri atas
protein 2.6-4.4%; lemak 32-40%; air 50-54%; dan abu 1-1.5%.
2.5.4. Bahan Acuan
Semua metode instrumental membutuhkan bahan acuan, sekalipun untuk metode yang
mengukur analat yang empiris. Analat yang empiris adalah analat yang nilainya tidak seperti
senyawa kimia yang stoikiometris yang bersifat tetap. Analat empiris merupakan hasil dari
penerapan prosedur yang biasa digunakan untuk mengukurnya, contohnya untuk kadar air, kadar
abu, kadar lemak, kadar karbohidrat (by difference) dan kadar serat (AOAC 2002).
Bahan acuan memainkan peranan penting untuk mengetahui akurasi dalam melakukan
validasi. Bahan acuan disini dapat diartikan sebagai bahan atau zat yang memiliki sifat-sifat
tertentu yang cukup homogen dan stabil, yang telah ditetapkan untuk dapat digunakan dalam
pengukuran atau dalam pengujian suatu contoh. Bahan acuan dapat digunakan untuk mengontrol
presisi pengukuran walaupun bahan acuan tersebut tidak memiliki nilai acuan (assigned value),
sedangkan untuk kalibrasi atau untuk mengontrol kebenaran pengukuran hanya bahan acuan yang
memiliki nilai acuan yang dapat digunakan (Dara 2010). Kalibrasi dan pengontrolan analisis
sangat penting, karena menyangkut kehandalan hasil pengujian. Untuk pengambilan keputusan
yang krusial diperlukan hasil pengujian yang dapat dipercaya (Nuryatini 2010). Bahan acuan ini
dapat diperoleh dari berbagai produsen bahan acuan seperti Puslit Kimia LIPI yang telah
mengembangkan beberapa bahan acuan (in-house reference materials) khususnya untuk pengujian
dalam bidang lingkungan dan pangan (Dara 2010).
Bahan acuan dapat dibagi menjadi dua yaitu Certified Reference Material (CRM) dan
Standard Reference Material (SRM). CRM dapat ditelusur hingga standard internasional dengan
ketidakpastian yang telah diketahui dan oleh karena itu dapat digunakan untuk mengukur semua
aspek bias (bias metode, bias antarlab, and intralab) secara bersamaan, dengan asumsi bahwa tidak
ada ketidaksesuaian matriks. Perlu dipastikan bahwa nilai ketidakpastian yang dimiliki cukup kecil
sehingga dapat mendeteksi bias pada kisaran tertentu. Tetapi jika nilainya tidak cukup kecil,

20
penggunaan CRM masih dianjurkan, tetapi dengan disertai dengan pengujian tambahan. Jika
diperlukan dan dapat dilakukan, sejumlah CRM yang sesuai dengan matriks dan konsentrasi analit
sebaiknya diujikan (Thompson et al 2002).
SRM dapat digunakan jika tidak ada CRM. SRM adalah material yang telah
dikarakterisasi dengan baik untuk tujuan validasi. Hal yang perlu diperhatikan adalah jika nilai
bias tidak signifikan, hal ini bukan berarti merupakan bukti bahwa tidak adanya bias sama sekali.
Akan tetap jika terdapat bias yang signifikan, hal ini menandakan perlunya investigasi lebih lanjut.
SRM dapat berupa material yang telah dikarakterisasi oleh produsen CRM tetapi tidak dilengkapi
dengan dokumen mengenai nilai ketidakpastiannya atau material yang telah terkualifikasi oleh
sebuah manufakturer; materials yang dikarakterisasi dalam lab sebagai reference material; dan
material yang didistribusikan dalam proficiency test. Meskipun ketertelusuran dari material
tersebut dipertanyakan, jauh lebih baik untuk menggunakan material tersebut dibandingkan tidak
melakukan pengukuran terhadap bias sama sekali. Material dapat digunakan dengan cara yang
sama seperti CRM, sekalipun tidak ada nilai ketidakpastian yang tercantum, seluruh pengujian
yang signifikan bergantung seluruhnya pada presisi yang dapat diamati dari hasil (Thompson et al
2002).

21
III. METODOLOGI PENELITIAN
3.1. Bahan dan Alat
3.1.1 Bahan
Seluruh bahan kimia yang digunakan memiliki grade analitik. Asam sulfat terkonsentrasi
(H2SO4 98%), reagen anthrone, KI, HCl 37%, Na2CO3, asam sitrat, standar glukosa, CH3COOH
100%, Na2S2O3.5H2O, heksana, HgO dan indikator pati berasal dari Merck, Jerman. Kalium
dikromat (K2CrO7), Cu2SO4.5H2O, H3BO3, K2SO4 dan NaOH berasal dari CICA, Jepang. Standar
amilosa (potato amylose) berasal dari Sigma-Aldrich. Es, indikator fenolftalein, kapas bebas lemak
dan air distilasi. Sampel matriks pangan cair yang digunakan untuk penelitian perbandingan
metode analisis yaitu kecap asin, kecap manis dan santan. Selain itu juga untuk verifikasi
digunakan sampel berupa bahan acuan tepung kedelai dan tepung kacang hijau yang diperoleh dari
LIPI Kimia Bandung dan bahan acuan susu bubuk dari BBIA Bogor.
3.1.2. Alat
Alat yang digunakan pada penelitian ini adalah hot plate (Cimarec 3 Thermolyne USA),
oven vakum (V0-7-3 Ogawa Seiki Japan), tanur (4800 Furnace Barnstead Thermolyne USA),
waterbath (Type 1008, GFL Gesselschaft fur Labortechnik mbH D-30938 Burgwedel Germany),
kertas saring, alat ekstraksi soxhlet (kondensor dan pemanas listrik), labu lemak, desikator berisi
bahan pengering, batang pengaduk, tabung reaksi, tabung reaksi bertutup, gelas piala, labu takar,
baskom plastik, sudip, batang pengaduk, pipet tetes, pipet ukur, pH meter (Orion model 210 A,
Thermo Electron Corp. USA), erlenmeyer, neraca analitik (Precisa XT 220A, Swiss), bulb, vortex,
spektrofotometer (UV Mini 1240, UV-Vis Spectrophotometer, Shimadzu Japan), stopwatch, buret
(volume 25 mL), cawan porselen, cawan alumunium dan labu Kjeldahl.
3.2. Metode Penelitian
Penelitian ini memiliki tiga tahapan yaitu tahap penentuan matriks sampel, tahap
perbandingan metode dan tahap validasi atau verifikasi metode. Bagan alir dari tahapan penelitian
yang dilakukan dapat dilihat pada Gambar 2

22
3.2.1. Penentuan matriks sampel
Penentuan matriks sampel dilakukan untuk mendapatkan sampel yang mewakili segitiga
pangan. Selain itu juga digunakan untuk mendapatkan informasi mengenai komponen lain yang
terdapat pada sampel yang akan digunakan.
3.2.1.1. Pemilihan sampel untuk uji perbandingan metode berdasarkan studi
literatur
Studi literatur dilakukan untuk memetakan beberapa sampel berdasarkan ke dalam
skema segitiga matriks pangan. Dari hasil pemetaan akan dipilih sampel yang dapat mewakili
matriks dengan kadar karbohidrat rendah, sedang dan tinggi.
3.2.1.2. Analisis proksimat
Hasil pemilihan sampel berdasarkan literatur dikonfirmasi komposisinya dengan
analisis proksimat. Selain untuk konfirmasi, analisis proksimat juga berfungsi untuk identifikasi
komponen yang ada dalam sampel. Analisis proksimat yang dilakukan meliputi kadar air, kadar
lemak, kadar abu, kadar protein dan kadar karbohidrat menggunakan metode dari SNI
01-2891-1992 (Cara Uji Makanan dan Minuman).
3.2.2. Perbandingan metode
Perbandingan metode dilakukan untuk mengevaluasi sejauh mana kedua metode yang
diperbandingkan menghasilkan kesesuaian nilai. Hasil dari perbandingan metode dapat digunakan
untuk melihat apakah metode yang baru (metode kandidat) dapat menggantikan metode yang
digunakan sebelumnya.
Sebanyak tiga kali ulangan dilakukan menggunakan metode kandidat dan metode SNI
01-2891-1992 pada tiga matriks yang telah ditentukan. Setelah itu hasil dari metode kandidat dan
metode SNI 01-2891-1992 dibandingkan dan disesuaikan dengan data analisis proksimat.
Gambar 2. Tahapan penelitian validasi metode analisis karbohidrat
Validasi Metode Anthrone sulfat Verifikasi metode SNI 01-2891-1992
Tidak berbeda nyata Berbeda nyata
Penentuan matriks sampel
Perbandingan metode

23
Perbandingannya meliputi uji varian (uji F), independent student t-test dan korelasi kedua metode
dengan regresi linear. Jika hasil analisis metode kandidat tidak berbeda nyata dengan hasil analisis
metode SNI 01-2891-1992 serta sesuai dengan hasil uji proksimat, maka akan dilakukan validasi
metode kandidat. Jika hasil yang didapatkan berbeda jauh, maka akan dilakukan verifikasi pada
metode SNI 01-2891-1992. .
3.2.3. Validasi Metode Anthrone sulfat
Validasi dilakukan pada matriks sampel yang terpilih yaitu sampel yang mewakili kadar
karbohidrat rendah, kadar karbohidrat sedang dan kadar karbohidrat tinggi dan bahan acuan.
Sampel dari matriks karbohidrat rendah, sedang dan tinggi diukur kadar karbohidratnya untuk
mengetahui tingkat validitas dari Metode Anthrone sulfat. Penentuan tingkat validasi ini meliputi:
3.2.3.1. Presisi
Ripitabilitas merupakan salah satu aspek presisi yang menggambarkan keseragaman
nilai yang diperoleh dari rangkaian pengukuran berulang terhadap analat dengan menggunakan
prosedur analisis yang sama (Leyva et al 2008). Sebanyak 7 kali ulangan dengan prosedur yang
sama, hari yang sama dan analis yang sama dilakukan pada sampel kemudian dihitung RSDnya.
Besarnya RSD dalam satuan % menunjukkan ripitabilitas. Keberterimaan RSD analisis ditentukan
sebesar 2/3 RSD Horwitz (Garfield 2000) atau 1/2 sampai 2 kali RSD AOAC (AOAC 2002).
Reprodusibilitas diukur dengan melakukan analisis yang sama setelah dua bulan sejak dilakukan
analisis pertama. Hasil analisis dibandingkan lalu diuji secara statistik untuk melihat apakah hasil
berbeda signifikan atau tidak.
3.2.3.2. Akurasi
Akurasi dilaksanakan dengan mengggunakan bahan acuan tepung kedelai dan tepung
kacang hijau dari LIPI Kimia Bandung dan bahan acuan susu bubuk dari BBIA Bogor. Selain itu
uji rekoveri juga dilakukan.
Tujuan uji rekoveri adalah memeriksa adanya interferensi kompetitif dan efek dari
matriks sampel (Koch dan Peter 1999; Cembrowski dan Sullivan 1992). Uji rekoveri dilakukan
dengan menggunakan sampel yang dispike (ditambahkan) standard glukosa. Percobaan spiking
dilakukan sebanyak tujuh ulangan pada sampel bahan acuan. Sebelumnya juga dilakukan uji
terhadap sampel yang tidak dispiking. Akurasi dilihat dari nilai rekoveri yang diperoleh. Recovery
dihitung dengan rumus (2.1):

24
(2.1)
3.2.3.3. Linieritas
Linieritas dari metode analitis yang menggambarkan kemampuan suatu metode untuk
hasil analisis yang proporsional dengan konsentrasi analat pada sampel dalam range tertentu baik
secara langsung maupun melalui transformasi matematik (Leyva et al 2008). Untuk mengetahui
linieritas metode, sebanyak tujuh kali ulangan dilakukan pada standar glukosa dengan 6-8
konsentrasi. Kemudian tiap kali ulangan dihitung rataan, SD1 dan RSD1. Selain itu tiap ulangan
diplotkan persamaan garis dari kurva kalibrasi dan dihitung koefisien korelasinya (r2). Selanjutnya
ditabulasikan nilai y yang baru berdasarkan persamaan garis yang ada. Dari nilai y yang baru
dihitung rataan, standar deviasinya (yang kemudian disebut SD2) dan RSDnya. Uji F digunakan
untuk mengetahui apakah ada perbedaan signifikan pada variansi kurva pada tiap kelompok
konsentrasi.
3.2.4. Verifikasi metode SNI 01-2891-1992
Verifikasi dilakukan dengan mengukur kadar karbohidrat matriks sampel yang terpilih
yaitu sampel yang mewakili kadar karbohidrat rendah, kadar karbohidrat sedang dan kadar
karbohidrat tinggi dan beberapa sampel yang telah diketahui nilainya yaitu bahan acuan (reference
material). Verifikasi ini meliputi atribut presisi (ripitabilitas) dan akurasi (dengan bahan acuan uji
rekoveri).
3.2.4.1. Presisi
Ripitabilitas merupakan salah satu aspek presisi yang menggambarkan keseragaman
nilai yang diperoleh dari rangkaian pengukuran berulang terhadap analat dengan menggunakan
prosedur analisis yang sama (Leyva et al 2008). Sebanyak 7 kali ulangan dengan prosedur yang
sama, hari yang sama dan analis yang sama dilakukan pada sampel kemudian dihitung RSDnya.
Besarnya RSD dalam satuan % menunjukkan ripitabilitas. Keberterimaan RSD analisis ditentukan
sebesar 2/3 RSD Horwitz (Garfield 2000) atau 1/2 sampai 2 kali RSD AOAC (AOAC 2002).
Reprodusibilitas diukur dengan melakukan analisis yang sama setelah dua bulan sejak dilakukan
analisis pertama. Hasil analisis dibandingkan lalu diuji secara statistik untuk melihat apakah hasil
berbeda signifikan atau tidak.

25
3.2.4.2. Akurasi
Akurasi dilaksanakan dengan mengggunakan bahan acuan tepung kedelai dan tepung
kacang hijau dari LIPI Kimia Bandung dan bahan acuan susu bubuk dari BBIA Bogor. Selain itu
uji rekoveri juga dilakukan. Tujuan uji rekoveri adalah memeriksa adanya interferensi kompetitif
dan efek dari matriks sampel (Koch dan Peter 1999; Cembrowski dan Sullivan 1992). Uji rekoveri
dilakukan dengan menggunakan sampel yang dispike standard glukosa. Spiking dilakukan
sebanyak tujuh ulangan pada sampel bahan acuan. Sebelumnya juga dilakukan uji terhadap sampel
yang tidak dispiking. Akurasi dilihat dari nilai rekoveri yang diperoleh. Recovery dihitung dengan
rumus (2.2):
(2.2)

26
IV. HASIL DAN PEMBAHASAN
4.1. Pemilihan Matriks Sampel
Matriks pangan sangat mempengaruhi performa suatu metode, terutama komponen
mayor seperti protein, karbohidrat, dan lemak, oleh karena itu beberapa sampel pangan cair dari
hasil studi literatur dipilih berdasarkan tiga kriteria karbohidratnya yaitu mewakili matriks sampel
dengan kadar karbohidrat rendah, sedang dan tinggi menurut skema segitiga yang disusun oleh
AOAC International seperti pada Gambar 1. Penempatan sampel menurut studi literatur dapat
dilihat pada Gambar 3. Sampel kecap manis dimasukkan pada kelompok pangan dengan
karbohidrat tinggi, sampel kecap asin dimasukkan pada kelompok pangan dengan karbohidrat
sedang, lemak rendah dan protein sedang serta santan dimasukkan pada kelompok pangan dengan
karbohidrat rendah, protein rendah dan lemak tinggi. Kemudian dilakukan analisis proksimat
dengan menggunakan metode SNI 01-2891-1992 untuk melakukan konfirmasi terhadap komposisi
dan identitasnya. Hasil analisis proksimat dapat dilihat pada Tabel 5. Hasil analisis proksimat
sesuai dengan penempatan yang dilakukan berdasarkan studi literatur.
Gambar 3 Hasil penempatan sampel matriks berdasarkan studi literatur

27
Tabel 5. Komposisi proksimat matriks sampel cair yang terpilih untuk uji perbandingan metode
analisis total karbohidrat (N=2)
No SampelKadar Air
(g/100g)
Kadar Abu
(g/100g)
Kadar Protein
(g/100g)
Kadar Lemak
(g/100g)
Kadar Karbohidrat
by difference (g/100g)
1 Kecap Manis 27.92 5.37 1,45 0,30 64,96
2 Kecap Asin 72.50 19.01 4.78 0,06 3,65
3 Santan 53.15 0.52 3,55 41,78 1,00
4.2. Perbandingan metode
Hasil analisis total karbohidrat dengan menggunakan Metode Luff-Schoorl dan Metode
Anthrone sulfat pada tiga matriks sampel pangan cair (kecap manis, kecap asin dan santan), yang
mewakili skema segitiga matriks pangan, diuji statistik dengan SPSS 17.0 dengan menggunakan
uji F menunjukkan bahwa varian kedua metode tidak berbeda nyata pada tingkat kepercayaan 95%
untuk sampel kecap asin, kecap manis, dan santan. Hasil uji F dapat dilihat pada Lampiran 2. Hal
ini menunjukkan tidak ada perbedaan signifikan dalam segi presisi dari Metode Luff-Schoorl
dengan Metode Anthrone sulfat untuk sampel kecap manis dan kecap asin dan santan.
Uji lanjut dilakukan dengan menggunakan independent student t-test, seperti yang
terlihat pada Tabel 6. Perbedaan signifikan pada hasil analisis sampel kecap manis, kecap asin dan
santan dengan Metode Luff-Schoorl dan Metode Anthrone sulfat terlihat pada tingkat kepercayaan
95%. Secara spesifik, hasil ini menunjukkan bahwa hasil analisis total karbohidrat dengan metode
Luff-Schoorl berbeda nyata dengan hasil analisis total karbohidrat dengan Metode Anthrone sulfat
pada ketiga matriks sampel yang digunakan.
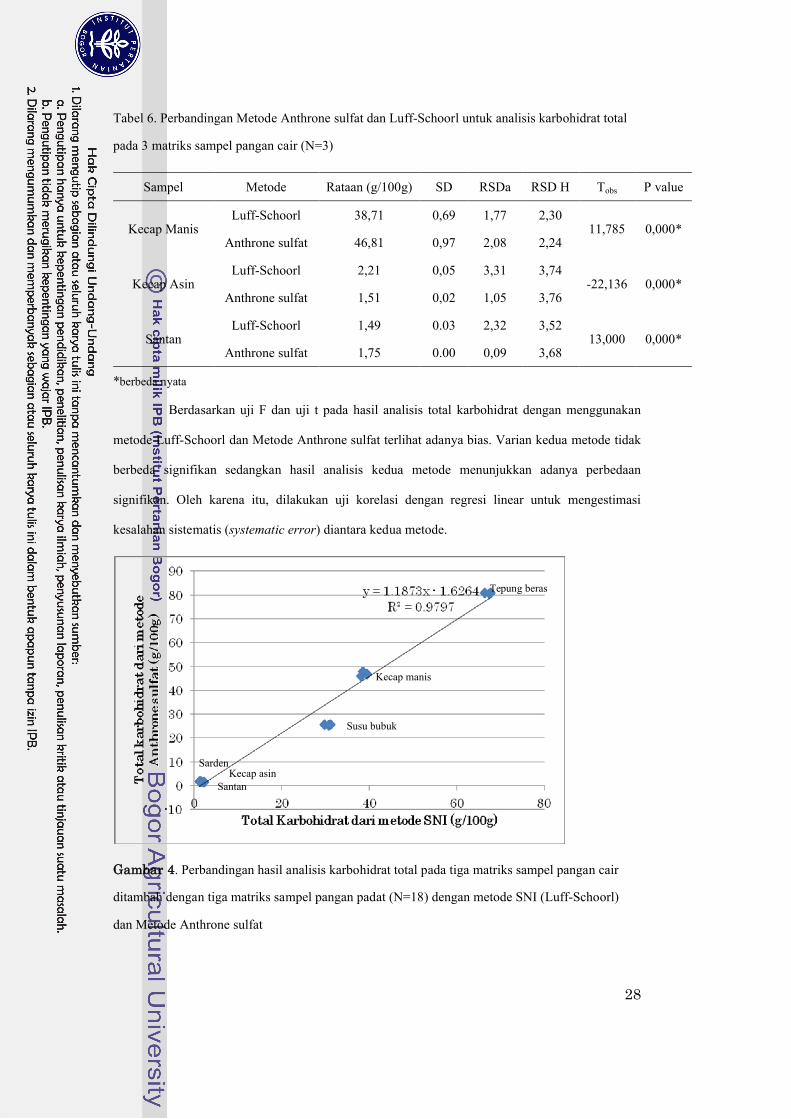
28
Tabel 6. Perbandingan Metode Anthrone sulfat dan Luff-Schoorl untuk analisis karbohidrat total
pada 3 matriks sampel pangan cair (N=3)
Sampel Metode Rataan (g/100g) SD RSDa RSD H Tobs P value
Kecap ManisLuff-Schoorl 38,71 0,69 1,77 2,30
11,785 0,000*Anthrone sulfat 46,81 0,97 2,08 2,24
Kecap AsinLuff-Schoorl 2,21 0,05 3,31 3,74
-22,136 0,000*Anthrone sulfat 1,51 0,02 1,05 3,76
SantanLuff-Schoorl 1,49 0.03 2,32 3,52
13,000 0,000*Anthrone sulfat 1,75 0.00 0,09 3,68
*berbeda nyata
Berdasarkan uji F dan uji t pada hasil analisis total karbohidrat dengan menggunakan
metode Luff-Schoorl dan Metode Anthrone sulfat terlihat adanya bias. Varian kedua metode tidak
berbeda signifikan sedangkan hasil analisis kedua metode menunjukkan adanya perbedaan
signifikan. Oleh karena itu, dilakukan uji korelasi dengan regresi linear untuk mengestimasi
kesalahan sistematis (systematic error) diantara kedua metode.
Gambar 4. Perbandingan hasil analisis karbohidrat total pada tiga matriks sampel pangan cair
ditambah dengan tiga matriks sampel pangan padat (N=18) dengan metode SNI (Luff-Schoorl)
dan Metode Anthrone sulfat
Tepung beras
Kecap manis
SardenKecap asin
Santan
Susu bubuk

29
Perbandingan antara kedua metode dilakukan dengan menggunakan suplemen data dari
penelitian Novitri (2011). Hasil regresi linier dapat dilihat pada Gambar 4; dengan koefisien
korelasi (r2) dari kurva regresi (y=1.1873x-1.6264) menunjukkan nilai yang memuaskan yaitu
0.9797 (n=18). Nilai ini menunjukkan bahwa range konsentrasi yang digunakan memadai untuk
analisis regresi sederhana, tetapi nilai ini tidak digunakan untuk menentukan apakah suatu metode
akurat, relatif terhadap metode baku (Walton 2001; Westgard 1998), yang dalam hal ini adalah
Luff Schoorl. Slope kurva regresi (1.1873) memperlihatkan bahwa kurva sedikit lebih curam
dibandingkan kurva regresi yang ideal yaitu 1:1. Hal ini menunjukkan adanya proportional
systematic error diantara metode yang digunakan (Walton 2001) dan terlihat bahwa Metode
Anthrone sulfat sedikit lebih sensitif dibandingkan metode Luff-Schoorl. Dari intercept kurva
regresi (-1.6264) kita dapat melihat bahwa Metode Anthrone menghasilkan nilai analisis 1.63%
lebih rendah dibanding metode Luff-Schoorl pada sampel dengan nilai karbohidrat terendah
(intercept 1.6264 pada nilai total karbohidrat Metode Anthrone= 0). Nilai ini juga menunjukkan
estimated constant error diantara kedua metode (Walton 2001). Dari penjelasan ini menunjukkan
bahwa, meskipun korelasi cukup baik, terdapat mutual bias diantara kedua metode. Tetapi karena
konsentrasi dari populasi sampel kurang mewakili seluruh populasi matriks pangan secara umum,
kesimpulan regresi linear pada perbandingan metode ini belum dapat dijadikan landasan yang
kokoh. Regresi ini hanya memberikan gambaran sepintas dari populasi yang diujikan yaitu kecap
manis, kecap asin, santan, sarden, susu bubuk dan tepung beras.
Hasil analisis menggunakan uji F, independent student t-test dan regresi linear
sederhana tehadap perbandingan hasil analisis menggunakan metode Luff-Schoorl dan Metode
Anthrone sulfat pada tiga sampel matriks pangan cair, menunjukkan bahwa dengan presisi yang
tidak berbeda nyata, nilai hasil yang didapat oleh kedua metode berbeda nyata. Oleh karena itu
penyebab bias dari kedua metode dianalisis.
Bias dapat juga karena pengaruh interferensi dari komponen yang ada pada matriks dari
sampel yang dianalisis. Bisa jadi suatu komponen dapat menginterferensi analisis pada suatu
metode tapi tidak menganggu metode yang lain. Adanya interferensi dapat menyebabkan nilai
yang terukur berbeda dari nilai sebenarnya. Tabel 7 menunjukkan nilai kadar karbohidrat dengan
menggunakan metode by difference, SNI 01-2891-1992 dan metode kandidat. Perlu ditegaskan

30
lagi bahwa nilai analisis metode by difference dapat mengandung akumulasi kesalahan, oleh
karena itu nilai yang ada hanya dijadikan perbandingan.
Tabel 7. Karbohidrat total dari tiga sampel matriks pangan cair dengan beberapa metode
Sampel Kadar karbohidrat (g/100g)
by difference Luff-Schoorl Anthrone sulfat
Kecap manis 64,96 38,71 46,81
Kecap asin 3,65 1,57 1,51
Santan 1,00 1,49 1,75
Dilihat dari Tabel 7 pada sampel kecap manis dan kecap asin, hasil metode pengukuran
karbohidrat secara langsung yaitu baik Luff-Schoorl maupun Metode Anthrone sulfat, nilainya
lebih kecil dibandingkan metode by difference. Metode by difference dapat memiliki kesalahan
positif karena metode ini tidak dapat membedakan komponen non karbohidrat seperti asam
organik, tanin dan lignin. Baik kecap asin dan kecap manis merupakan produk hasil fermentasi
oleh kapang, oleh karena itu produk samping hasil metabolit, seperti asam organik, dapat
terkandung dalam kecap manis dan kecap asin.
Hal lain yang dapat menyebabkan lebih rendahnya nilai pengukuran karbohidrat secara
langsung dibandingkan dengan metode by difference adalah tahap hidrolisis karbohidrat yang
digunakan pada metode pengukuran karbohidrat secara langsung. Hidrolisis yang digunakan
menggunakan asam kuat encer yaitu HCl 3% dan pemanasan pada ±99oC selama 3 jam untuk
menghidrolisis sampel keseluruhan. Hidrolisis asam sampel seperti ini memiliki kelemahan dan
dapat menjadi tidak akurat bahkan dapat menghasilkan nilai yang keliru karena pada kondisi yang
dibutuhkan untuk dapat memecah pati dan dekstrin dapat menyebabkan destruksi dari fruktosa
(Loomys dan Shull 1937); atau gula-gula lain (Shriner 1932). Glukosa juga terdegradasi perlahan
jika dipanaskan dengan asam, laju destruksi ini dipercepat oleh asam sulfat dan jauh lebih cepat
dengan HCl (Whelan dan Pirt 2006) sedangkan HCl digunakan pada tahap hidrolisis sampel. Jadi
hal ini juga dapat menyebabkan nilai analisis dengan metode by difference nilainya lebih tinggi
dibandingkan dengan Metode Anthrone sulfat maupun metode Luff-Schoorl pada sampel kecap
asin.

31
Adapun nilai analisis sampel santan baik metode by difference dan Luff-Schoorl
menunjukkan nilai yang hamper sama, yaitu jika dibulatkan nilainya 1%. Adapun Metode
Anthrone nilainya sedikit lebih besar dibandingkan metode by difference maupun Luff-Schoorl.
Hal ini dapat disebabkan karena kandungan gula sederhana (terutama dalam bentuk glukosa dan
fruktosa) yang ada pada santan tidak sebanyak pada kecap manis maupun kecap asin, sehingga
pengaruh degradasi gula sederhana pada tahap hidrolisis asam tidak terlalu terlihat. Selain itu
komponen non karbohidrat yang dapat terhitung sebagai karbohidrat oleh metode by difference,
seperti asam organik, tidak terlalu banyak terdapat pada sampel santan yang digunakan.
Metode by difference tidak dapat dijadikan sebagai acuan karena metode ini tidak lepas
dari banyak bias. Perbedaan nilai antara metode by difference dengan metode lainnya
menunjukkan bahwa ada kemungkinan nilai yang didapat baik oleh Metode Anthrone sulfat
maupun Metode Luff-Schoorl, terutama untuk sampel kecap asin dan kecap manis, bukanlah nilai
kadar total karbohidrat karena serat kasar seperti selulosa juga tidak dapat dihidrolisis dengan
asam kuat encer saja (Southgate 1976) dan juga tidak dapat dikatakan sebagai nilai total available
karbohidrat juga karena sulit untuk memisahkan fraksi pati dari karbohidrat struktural (Loomys
dan Shull 1937). Nilai yang didapat lebih cocok jika disebut sebagai nilai total karbohidrat yang
dapat terhidrolisis oleh asam (Weinmann 1946).
Pengaruh faktor konversi yang digunakan juga dapat berdampak pada perbedaan nilai
yang didapat antara metode kandidat, Luff-Schoorl dan metode by difference. Tanpa melihat jenis
karbohidrat yang banyak terkandung pada matriks, faktor konversi 0.9 diterapkan untuk semua
matriks. Adapun dalam perbandingan metode ini pengaruh komponen lain seperti lemak dan
protein belum dapat diketahui melalui penelitian ini.
Perbedaan nilai yang terlihat pada Metode Luff-Schoorl dengan Metode Anthrone
seperti yang terlihat pada Tabel 7 dapat disebabkan karena Metode Luff-Schoorl hanya
mengidentifikasi gula pereduksi saja, kompleks karbohidrat yang ada belum tentu dihidrolisis
sempurna seluruhnya menjadi gula pereduksi. Hal ini menyebabkan hasil analisis dari Metode
Anthrone sulfat menunjukkan nilai yang lebih besar pada sampel kecap manis dan santan. Selain
itu juga, nilai yang lebih besar dari Metode Anthrone sulfat dapat juga terkait dengan penguatan
warna oleh ion Cl (Fales et al 1961, Jermyn 1975). Sedangkan untuk kecap asin, Metode

32
Luff-Schoorl menunjukkan nilai yang sedikit lebih besar dibandingkan Metode Anthrone sulfat
(selisih rataan 0.06%). Ada juga kemungkinan interferensi komponen pereduksi yang bukan gula
yang menyebabkan kesalahan positif pada metode Luff Schoorl.
Tiap metode memang memiliki keterbatasan. Metode Anthrone sulfat rentan terhadap
interferensi non spesifik (Faulks dan Timms 1985) salah satunya keberadaan ion halida (Fales et al
1961) terutama ion Cl yang berasal dari tahap hidrolisis dengan HCl. Intensitas warna yang
dihasilkan oleh reaksi Anthrone juga berbeda-beda untuk gula yang berbeda (Yemm dan Willis
1954). Selain itu reaksi senyawa Anthrone cenderung lebih baik untuk senyawa heksosa dan reaksi
dengan pentose kurang menghasilkan warna yang stabil (Koehler 1952; Southgate 1976).
Penggantian suatu metode dengan metode lain dapat dilakukan jika kedua metode
memiliki kesesuaian hasil yang dapat diterima. Meski presisi kedua metode tidak berbeda nyata
berdasarkan uji F, uji T yang dilakukan menunjukkan Metode Anthrone sulfat dan Metode
Luff-Schoorl menghasilkan nilai yang berbeda nyata pada aplikasinya untuk sampel kecap manis,
kecap asin dan santan yang mewakili matriks pangan cair. Karena kedua metode berbeda nyata
dan tidak ada acuan bahwa Metode Anthrone sulfat memiliki nilai yang lebih akurat dibanding
metode yang telah baku (Luff-Schoorl dalam SNI 01-2891-1992), maka Metode Anthrone sulfat
dianggap tidak dapat menggantikan Metode Luff-Schoorl, sehingga tahap selanjutnya yang
dilakukan adalah verifikasi Metode Luff-Schoorl yang telah baku. Selain karena Metode Anthrone
pada tahap yang telah dilakukan dianggap tidak dapat menggantikan Metode Luff-Schoorl,
keputusan untuk melakukan verifikasi ini diambil karena Metode Luff-Schoorl merupakan metode
yang telah baku (ditetapkan dalam SNI 01-2891-1992).
Penggunaan metode yang baku yang telah disepakati berdasarkan konsensus merupakan
hal yang penting untuk menjamin bahwa hasil yang diperoleh sesuai dengan persyaratan yang
ditetapkan oleh pemerintah (Nielsen, 2010) dan dapat diterima sehingga dapat memenuhi
permintaan dalam label pangan. Hasil perbandingan metode yang menunjukkan bahwa nilai yang
didapat antara metode baku (Luff-Schoorl) dan metode kandidat (Anthrone) tidak menunjukkan
kesesuaian (nilai berbeda nyata menurut uji statistik). Jika lab tetap memutuskan untuk
menggunakan Metode Anthrone, maka hasil yang diperoleh dapat bertentangan dengan hasil yang
diperoleh lab lain untuk sampel yang sama sehingga kemungkinan hasil analisis tidak diakui atau

33
diterima. Sampai saat ini uji profisiensi lab untuk pemenuhan persyaratan SNI 19-17025-2000
masih menggunakan nilai konsensus dari peserta lab uji, maka penggunaan metode baku manual
SNI masih menjadi alternatif yang lebih baik untuk mendapatkan hasil analisis dengan performa
yang memenuhi standard. Oleh karena itu, tahap validasi Metode Anthrone tidak dilakukan dan
dan hanya dilakukan verifikasi terhadap metode baku yaitu Luff-Schoorl.
4.3. Verifikasi metode SNI 01-2891-1992
Tingkat validasi tergantung status dari suatu metode pada struktur analitik (AOAC
2002), yang dimaksud disini adalah validasi seperti apakah yang harus diterapkan pada suatu
metode tergantung status metode itu sendiri. Metode yang telah baku hanya memerlukan verifikasi
dari kemampuan suatu laboratorium untuk mencapai karakteristik performa yang ditetapkan,
sedangkan di sisi lain untuk metode yang masih baru atau aplikasi suatu metode pada matriks yang
baru memerlukan validasi (AOAC 2002). Karena Metode Luff-Schoorl dalam SNI 01-2891-1992
sudah baku maka hanya dilakukan verifikasi. Karakteristik yang akan dinilai dalam verifikasi
adalah aspek presisi dan akurasi
Verifikasi dilakukan dengan matriks sampel uji dan bahan acuan pengendalian mutu
hasil analisis (quality control reference material). Karena adanya kesulitan dalam mendapatkan
bahan acuan, maka bahan acuan dipilih berdasarkan bahan acuan yang tersedia dan dapat
diperoleh yaitu tepung kacang hijau dan tepung kedelai dari Lembaga Ilmu Pengetahuan Indonesia
(LIPI) Kimia Bandung serta susu bubuk dari Balai Besar Industri Agro (BBIA) di Bogor. Kadar
karbohidrat yang ada pada bahan acuan kacang hijau berdasarkan nilai konsensus dari 8 lab dan
pada bahan acuan kedelai berdasarkan konsensus dari 6 lab dengan menggunakan uji Luff-Schoorl.
Untuk sampel susu bubuk, karena masih dalam tahap percobaan, maka nilai yang dicantumkan
pada sampel susu bubuk bukanlah nilai konsensus dari beberapa lab seperti pada sampel tepung
kacang hijau dan tepung kedelai, melainkan nilai yang didapat oleh satu lab saja (Lab Jasa Analisis
Pangan (LDITP) IPB) sehingga rentang nilainya sempit. Informasi lengkap dapat dilihat pada
Tabel 8.

34
Tabel 8 Komposisi proksimat bahan acuan yang digunakan dalam verifikasi metode karbohidrat
total SNI 01-2891-1992
Parameter
Kedelaia Kacang hijaua Susu bubukb
Nilai g/100g
rata-rata Rentang rata-rata Rentang rata-rata Rentang
Air 7.24 6.60-7.87 9.49 8.66-10.31 3.14 3.14-3.15
Abu 4.73 4.53-4.93 3.07 2.89-3.25 4.48 4.47-4.50
Protein 33.26 31.24-35.28 23.49 21.69-25.28 14.48 14.46-14.50
Karbohidrat 16.64 14.02-19.26 53.61 49.26-57.96 59.64 59.61-59.67
Lemak 21.07 20.22-21.91 NA NA 18.25 18.24-18.26
aberdasarkan nilai yang tercantum pada bahan acuan LIPI Kimia
bberdasarkan hasil analisis proksimat Lab Kimia LD-ITP
Bahan acuan yang dipakai jika dimasukkan ke dalam matriks segitiga pangan akan
terbagi menjadi dua kelas matriks dalam segitiga pangan. Kedelai masuk ke dalam kelas dengan
kadar karbohidrat rendah, lemak rendah dan protein sedang (yang ditandai dengan nomor 8 pada
matriks segitiga pangan di Gambar 1). Sedangkan kacang hijau dan susu bubuk akan masuk ke
dalam kelas protein rendah, lemak rendah dan karbohidrat sedang (yang ditandai dengan nomor 6
pada matriks segitiga pangan di Gambar 1). Sebelumnya pada perbandingan metode digunakan
sampel yang mewakili tiga kelas matriks dalam segitiga pangan (Gambar 3). Sehingga kalau
dijumlah sampel dan bahan acuan yang digunakan telah mewakili 5 dari 9 matriks segitiga pangan
yang ada.
4.3.1. Aspek presisi
Walton (2001) merekomendasikan evaluasi terhadap presisi sebagai langkah pertama
dalam validasi metode. Jika presisi metode sudah tidak baik, maka sulit untuk mendapatkan hasil
yang dapat dipercaya. Salah satu aspek yang umum digunakan dalam verifikasi adalah ripitabilitas
(Mullins 2003). Tetapi dalam pengujian presisi metode untuk validasi satu lab (single laboratory
validation) dapat berupa ripitabilitas dan reprodusibilitas intralab.
4.3.1.1. Ripitabilitas bahan acuan
Ripitabilitas memungkinkan variasi terkecil dapat ditemukan pada sebuah analisis
(Jelita 2011). Ripitabilitas dapat dilihat dari nilai RSD. Nilai RSD dan RSDR(Horwitz) analisis
total karbohidrat dengan menggunakan metode SNI 01-2891-1992 ditunjukkan pada Tabel 9
untuk analisis beberapa bahan acuan, Tabel 10 untuk uji ripitabilitas dengan spike glukosa.

35
Tabel 9. Ripitabilitas metode karbohidrat SNI 01-2891-1992 pada berbagai bahan acuan (N=7)
Bahan acuan
Hasil analisis (g/100g) RSD
analisis
(%)
2/3 RSDH
(%)
2xRSD
AOAC
(%)Rataan Range SD
Susu Bubuk 45.72 45.11-46.08 0.43 0.93 1.50 2.25
Kacang kedelai 15.90 15.19-16.50 0.41 2.58 1.76 2.64
Kacang hijau 55.66 55.45-56.16 0.28 0.51 1.45 2.18
Tabel 10. Ripitabilitas metode karbohidrat SNI 01-2891-1992 pada berbagai bahan acuan dengan
penambahan kadar glukosa (N=7)
Bahan acuan
Hasil analisis yang terbaca
(g/100g)
RSD
analisis
(%)
2/3
RSDH
(%)a
2x RSD
AOAC
(%)b
Rataan Range SD
Susu Bubuk 47,65 47,37-48,55 0,43 0,91 1,49 2,24
Kedelai 23,44 22,98-24,05 0,42 1,80 1,66 2,49
Kacang hijau 58,50 58,37-58,66 0,12 0,22 1,47 2,17
aGarfield (2000)
bAOAC (2002)
Koefisien variasi atau relatif standard deviasi yang diperoleh berkisar antara 0,51-2,58%
untuk sampel bahan acuan (n=7) dan 0,22-1,80% untuk sampel bahan acuan yang mengalami
penambahan kadar glukosa (n=7). Nilai ini menunjukkan variasi yang kecil dalam ulangan yang
dilakukan pada tiap bahan acuan. Garfield (2000) mengatakan bahwa ripitabilitas dikatakan baik
jika memiliki nilai RSD yang lebih kecil dari 2/3 RSDR yang dihitung dari rumus Horwitz. Tetapi
AOAC (2002) mengatakan bahwa nilai yang dapat diterima untuk ripitabilitas adalah antara 0,5
sampai 2 kali dari nilai yang terhitung berdasarkan rumus atau di Tabel 2. Bahkan nilai RSD di
bawah 5% dapat diterima, meskipun terkadang batas itu tergantung tipe dari analisis (Smith 2010).
Hasil analisis yang didapat pada bahan acuan susu bubuk dan kacang hijau
menunjukkan nilai yang didapat kurang dari 2/3 RSDR yang dihitung dari rumus Horwitz, kecuali
pada analisis bahan acuan tepung kedelai. Nilainya masih lebih kecil dari RSDR Horwitz tetapi
lebih besar dari 2/3 RSDR Horwitz. Tetapi jika kita mengikuti acuan AOAC (2002) nilai ini masih

36
dalam range yang dapat diterima. Begitupula jika mengikuti acuan Smith (2010), yaitu RSD masih
di bawah 5%.
Nilai RSD kedelai cenderung lebih besar dibanding kacang hijau dan susu bubuk baik
pada bahan acuan dengan penambahan glukosa maupun bahan acuan tanpa penambahan glukosa.
Hal ini dapat disebabkan oleh konsentrasi karbohidrat pada kedelai yang lebih kecil dibandingkan
susu bubuk dan kacang hijau. Akan tetapi jika dilihat dari nilai standard deviasi(SD)nya sendiri,
kedelai memiliki SD yang hampir sama bahkan cenderung lebih kecil dibandingkan susu bubuk.
Hal ini mengindikasikan bahwa konsentrasi karbohidrat yang lebih kecil (hingga pada range lebih
dari ±15,90 gram karbohidrat setara glukosa/100 gram sampel) bukan berarti menyebabkan
keterulangan yang lebih buruk dibandingkan konsentrasi karbohidrat yang lebih tinggi. Adanya
kecenderungan bahwa nilai SD susu bubuk lebih besar dari kedelai lebih besar dari kacang hijau
perlu diteliti lebih lanjut untuk mengetahui komponen apa dari tiap bahan acuan yang mungkin
dapat menyebabkan variasi yang ada. Dalam penelitian ini, range konsentrasi ±15,90-58.50 gram
karbohidrat setara glukosa/100 gram sampel pada sampel kacang hijau, kedelai dan susu bubuk
masih memiliki kerterulangan (ripitabilitas) yang dapat diterima terutama pada lab tempat
penelitian dilaksanakan telah dikonfirmasi.
4.3.1.2. Reprodusibilitas bahan acuan dan matriks sampel
Reprodusibilitas dapat digunakan untuk memperkirakan bias yang terjadi jika analisis
dilakukan pada hari yang berbeda. Reprodusibilitas yang diukur adalah reprodusibilitas intralab,
yaitu dengan lab yang sama hanya selang waku yang berbeda. Selang waktu yang digunakan untuk
mengukur reprodusibilitas intralab yang dilakukan dalam penelitian ini adalah lebih dari 2 bulan.
Reprodusibilitas intralab diukur pada bahan acuan yang dapat dilihat pada Tabel 11 dan sampel
matriks pangan cair pada Tabel 12.
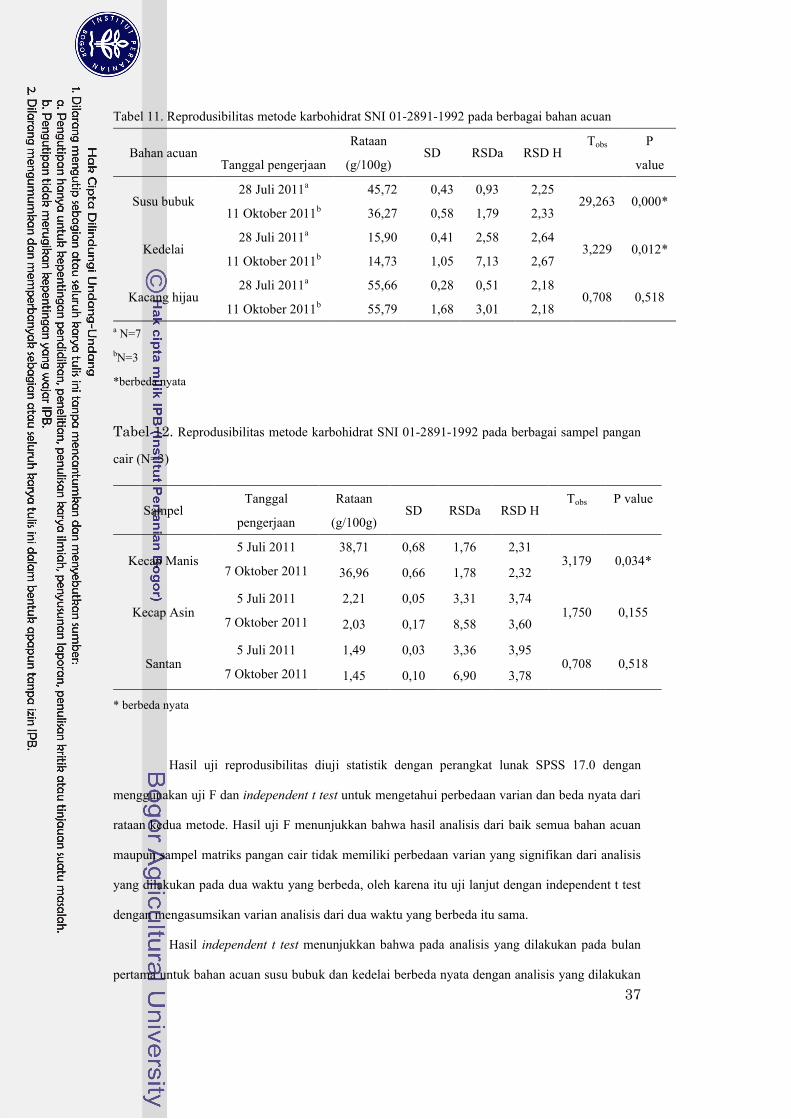
37
Tabel 11. Reprodusibilitas metode karbohidrat SNI 01-2891-1992 pada berbagai bahan acuan
Bahan acuanTanggal pengerjaan
Rataan
(g/100g)SD RSDa RSD H
Tobs P
value
Susu bubuk28 Juli 2011a 45,72 0,43 0,93 2,25
29,263 0,000*11 Oktober 2011b 36,27 0,58 1,79 2,33
Kedelai28 Juli 2011a 15,90 0,41 2,58 2,64
3,229 0,012*11 Oktober 2011b 14,73 1,05 7,13 2,67
Kacang hijau28 Juli 2011a 55,66 0,28 0,51 2,18
0,708 0,51811 Oktober 2011b 55,79 1,68 3,01 2,18
a N=7
bN=3
*berbeda nyata
Tabel 12. Reprodusibilitas metode karbohidrat SNI 01-2891-1992 pada berbagai sampel pangan
cair (N=3)
SampelTanggal
pengerjaan
Rataan
(g/100g)SD RSDa RSD H
Tobs P value
Kecap Manis5 Juli 2011 38,71 0,68 1,76 2,31
3,179 0,034*7 Oktober 2011 36,96 0,66 1,78 2,32
Kecap Asin5 Juli 2011 2,21 0,05 3,31 3,74
1,750 0,1557 Oktober 2011 2,03 0,17 8,58 3,60
Santan5 Juli 2011 1,49 0,03 3,36 3,95
0,708 0,5187 Oktober 2011 1,45 0,10 6,90 3,78
* berbeda nyata
Hasil uji reprodusibilitas diuji statistik dengan perangkat lunak SPSS 17.0 dengan
menggunakan uji F dan independent t test untuk mengetahui perbedaan varian dan beda nyata dari
rataan kedua metode. Hasil uji F menunjukkan bahwa hasil analisis dari baik semua bahan acuan
maupun sampel matriks pangan cair tidak memiliki perbedaan varian yang signifikan dari analisis
yang dilakukan pada dua waktu yang berbeda, oleh karena itu uji lanjut dengan independent t test
dengan mengasumsikan varian analisis dari dua waktu yang berbeda itu sama.
Hasil independent t test menunjukkan bahwa pada analisis yang dilakukan pada bulan
pertama untuk bahan acuan susu bubuk dan kedelai berbeda nyata dengan analisis yang dilakukan

38
pada bulan kedua yang berselang lebih dari dua bulan sejak analisis pertama, sedangkan untuk
bahan acuan kacang hijau tidak berbeda nyata. Adapun hasil independent t test pada analisis yang
dilakukan pada bulan pertama untuk sampel matriks pangan cair yaitu kecap asin dan santan tidak
berbeda nyata dengan analisis yang dilakukan pada bulan kedua, sedangkan untuk sampel kecap
manis berbeda nyata. Nilai yang berbeda nyata ini mengindikasikan reprodusibilitas yang buruk.
Jumlah total karbohidrat yang ada pada bahan acuan seharusnya tidak akan banyak
berubah karena lingkungan. Jika diasumsikan bahwa bahan acuan cenderung bersifat stabil, maka
perubahan atau ketidakkonsistenan dapat berasal dari analis, reagen, atau lingkungan yang
mempengaruhi performa metode itu sendiri. Meskipun reagen seperti natrium tiosulfat dan reagen
lain disiapkan segar, reagen Luff yang digunakan untuk analisis pada bulan kedua sama dengan
yang digunakan pada bulan pertama karena diasumsikan reagen ini bersifat stabil. Tetapi ternyata
hasil analisis menunjukkan adanya ketidakkonsistenan dalam ripitabilitas dan reprodusibilitas,
sehingga ada kemungkinan jika reagen kurang stabil dalam penyimpanan lebih dari 2 bulan. Hal
ini juga dapat menyebabkan bias. Adapun ketidakkonsistenan dari analis dan perubahan kondisi
pada lingkungan juga dapat mempengaruhi performa metode.
Koefisien variasi atau relatif standard deviasi yang diperoleh untuk analisis yang
dilakukan pada bulan pertama cenderung lebih baik dibandingkan hasil analisis yang dilakukan
pada bulan kedua. Hal ini juga yang dapat menunjukkan bahwa adanya ketidakkonsistenan pada
analisis yang dilakukan pada bulan kedua. Hal ini kemungkinan besar dapat disebabkan karena
adanya perubahan pada reagen, matriks, analis dan lingkungan. Reagen dapat mengalami
perubahan seperti yang disebutkan sebelumnya. Dari segi analis, metode yang memiliki tahapan
yang panjang dan melelahkan dapat menyebabkan performa metode kurang konsisten. Selain itu
perubahan dari matriks sampel (dalam hal ini terutama matriks sampel pangan cair) baik secara
biologis atau kimia dapat menyebabkan hasil kurang konsisten baik untuk ripitabilitas maupun
reprodusibilitas. Dari sini dapat dilihat juga bahwa reprodusibilitas metode dipengaruhi oleh
matriks sampel yang dianalisis.
Faulks dan Timms (1985) mengatakan bahwa metode dengan prinsip gula pereduksi
memiliki reprodusibilitas yang buruk. Hal ini juga telah dikonfirmasi dalam percobaan ini, yaitu
dimana pada matriks kecap manis serta bahan acuan susu bubuk dan kedelai, nilai

39
reprodusibilitasnya buruk (analisis yang dilakukan dalam selang waktu dua bulan hasilnya berbeda
nyata).
4.3.2. Aspek akurasi
Akurasi dari metode SNI 01-2891-1992 dilakukan dengan menggunakan bahan acuan
dan uji rekoveri. Hasil analisis terhadap bahan acuan dapat dilihat pada Tabel 13, dan uji rekoveri
dengan menggunakan standard glukosa dapat dilihat pada Tabel 14.
Tabel 13. Akurasi metode karbohidrat total SNI 01-2891-1992 pada berbagai bahan acuan (N=7)
Bahan acuanRentang bahan
acuan(g/100g)
Hasil analisis (g/100g)
Rataan Range SD
Susu Bubuk 59,61-59,67a 45,72 45,11-46,08 0,43
Kedelai 14,02-19,26b 15,90 15,19-16,50 0,41
Kacang hijau 49,26-57,96b 55,66 55,45-56,16 0,28
a berdasarkan hasil analisis by difference Lab Kimia LD-ITP
b berdasarkan nilai yang tercantum pada bahan acuan LIPI Kimia (analisis Luff-Schoorl)
4.3.2.1. Akurasi berdasarkan bahan acuan
Bahan acuan yang digunakan bukanlah Certified Reference Material (CRM), melainkan
hanya bahan acuan yang nilai (assigned value) komposisinya berdasarkan konsensus beberapa lab
dan digunakan untuk uji profisiensi. Sekalipun demikian, bahan acuan seperti ini masih dapat
digunakan untuk mengetahui adanya bias (Thompson et al 2002). Hasil analisis terhadap bahan
acuan menunjukkan nilai yang masih dalam rentang yang tercantum pada bahan acuan, kecuali
untuk bahan acuan susu bubuk. Khusus untuk susu bubuk rentangnya masih sempit karena nilai
yang ditampilkan merupakan hasil uji dari satu lab saja dan itupun masih menggunakan metode by
difference.
Hasil analisis menunjukkan bahwa analisis total karbohidrat dengan Metode
Luff-Schoorl untuk kedelai dan kacang hijau masih dalam rentang pengukuran. Hal ini juga
mengonfirmasi bahwa pada rentang konsentrasi karbohidrat 15,90-55,66 gram karbohidrat setara
glukosa/100 gram sampel untuk bahan acuan kacang hijau, kedelai dan susu bubuk masih
dimungkinkan untuk dianalisis dengan Metode Luff-Schoorl dengan menghasilkan nilai akurasi
yang masih dapat diterima sesuai dengan rentang bahan acuan empiris. Bahan acuan empiris yang
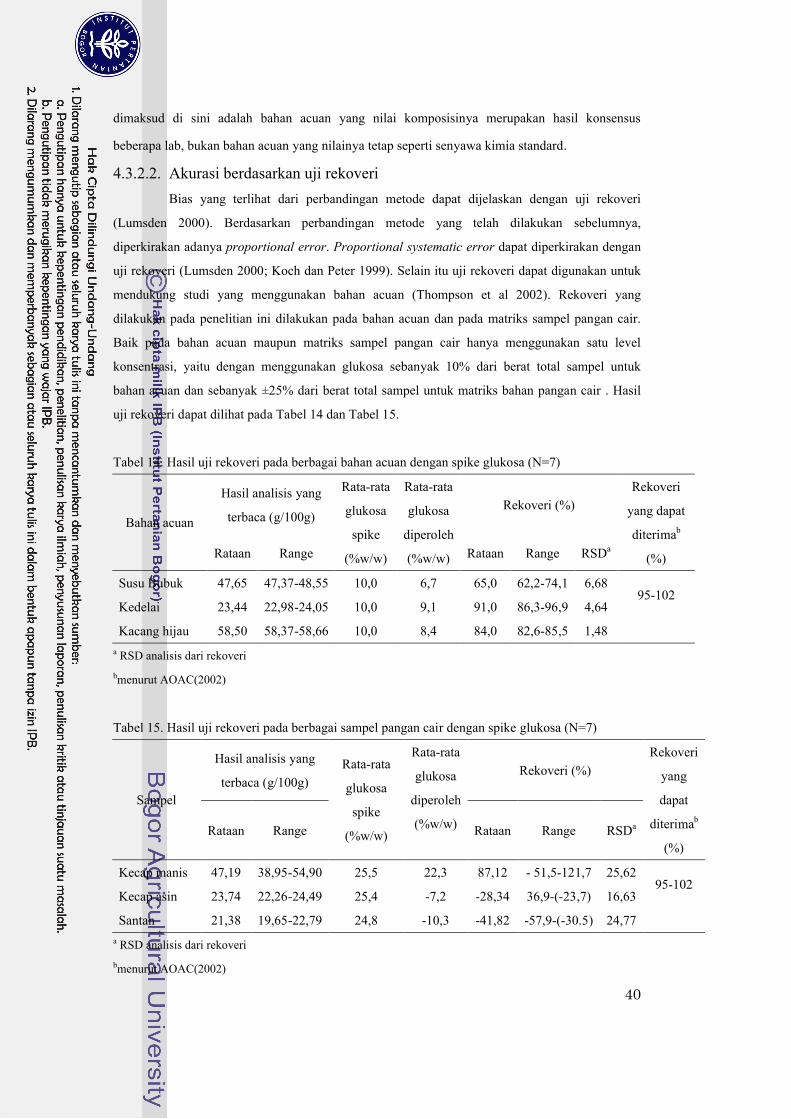
40
dimaksud di sini adalah bahan acuan yang nilai komposisinya merupakan hasil konsensus
beberapa lab, bukan bahan acuan yang nilainya tetap seperti senyawa kimia standard.
4.3.2.2. Akurasi berdasarkan uji rekoveri
Bias yang terlihat dari perbandingan metode dapat dijelaskan dengan uji rekoveri
(Lumsden 2000). Berdasarkan perbandingan metode yang telah dilakukan sebelumnya,
diperkirakan adanya proportional error. Proportional systematic error dapat diperkirakan dengan
uji rekoveri (Lumsden 2000; Koch dan Peter 1999). Selain itu uji rekoveri dapat digunakan untuk
mendukung studi yang menggunakan bahan acuan (Thompson et al 2002). Rekoveri yang
dilakukan pada penelitian ini dilakukan pada bahan acuan dan pada matriks sampel pangan cair.
Baik pada bahan acuan maupun matriks sampel pangan cair hanya menggunakan satu level
konsentrasi, yaitu dengan menggunakan glukosa sebanyak 10% dari berat total sampel untuk
bahan acuan dan sebanyak ±25% dari berat total sampel untuk matriks bahan pangan cair . Hasil
uji rekoveri dapat dilihat pada Tabel 14 dan Tabel 15.
Tabel 14. Hasil uji rekoveri pada berbagai bahan acuan dengan spike glukosa (N=7)
Bahan acuan
Hasil analisis yang
terbaca (g/100g)
Rata-rata
glukosa
spike
(%w/w)
Rata-rata
glukosa
diperoleh
(%w/w)
Rekoveri (%)
Rekoveri
yang dapat
diterimab
(%)Rataan Range Rataan Range RSDa
Susu Bubuk 47,65 47,37-48,55 10,0 6,7 65,0 62,2-74,1 6,6895-102
Kedelai 23,44 22,98-24,05 10,0 9,1 91,0 86,3-96,9 4,64
Kacang hijau 58,50 58,37-58,66 10,0 8,4 84,0 82,6-85,5 1,48
a RSD analisis dari rekoveri
bmenurut AOAC(2002)
Tabel 15. Hasil uji rekoveri pada berbagai sampel pangan cair dengan spike glukosa (N=7)
Sampel
Hasil analisis yang
terbaca (g/100g)
Rata-rata
glukosa
spike
(%w/w)
Rata-rata
glukosa
diperoleh
(%w/w)
Rekoveri (%)
Rekoveri
yang
dapat
diterimab
(%)Rataan Range Rataan Range RSDa
Kecap manis 47,19 38,95-54,90 25,5 22,3 87,12 - 51,5-121,7 25,6295-102
Kecap asin 23,74 22,26-24,49 25,4 -7,2 -28,34 36,9-(-23,7) 16,63
Santan 21,38 19,65-22,79 24,8 -10,3 -41,82 -57,9-(-30.5) 24,77
a RSD analisis dari rekoveri
bmenurut AOAC(2002)

41
4.3.2.2.1. Rekoveri dengan bahan acuan
Uji rekoveri dengan spiking glukosa untuk susu bubuk pada Tabel 13 menunjukkan
rata-rata rekoveri 65,0%, untuk kedelai didapatkan rata-rata rekoveri 91,0% dan untuk kacang
hijau didapatkan rata-rata rekoveri 84,0%. Uji rekoveri dengan spiking glukosa untuk sampel
kecap manis pada Tabel 14 menunjukkan rata-rata rekoveri 87,12%; untuk sampel kecap asin
didapatkan rata-rata rekoveri -28,34%, dan untuk sampel santan didapatkan rata-rata rekoveri
-41,82%. Berdasarkan uji rekoveri tidak ada hasil yang menunjukkan nilai rekoveri yang dapat
diterima berdasarkan batas yang ditetapkan oleh AOAC (2002). Meski nilai rekoveri yang baik
belum tentu menandakan bahwa nilai analisis merupakan nilai yang sebenarnya karena efek dari
analat yang ditambahkan dengan analat dalam bentuk alaminya mungkin berbeda, tetapi nilai
rekoveri yang buruk jelas menunjukkan adanya bias dari nilai yang sebenarnya (Thompson et al
2002).
Nilai rekoveri sampel kedelai (91,03%) lebih besar daripada kacang hijau (83,95%) dan
lebih besar daripada susu bubuk (65,0%). Hal ini menunjukkan bahwa efek matriks yang dapat
mengganggu analisis paling besar terlihat pada bahan acuan susu bubuk. Selain itu nilai rekoveri
yang kurang dari 60-70% perlu pemeriksaan yang mengarah pada perbaikan (AOAC 2002) karena
kemungkinan nilai rekoveri ini menunjukkan bahwa ada kesalahan sistematis akibat adanya
komponen matriks lain yang menganggu dalam analisis seperti maltodekstrin yang digunakan
sebagai bahan pengisi pada susu bubuk. Courtin et al (2000) mengatakan bahwa nilai yang
dihasilkan oleh analisis maltodekstrin dengan metode gula pereduksi cenderung lebih kecil
dibandingkan dengan metode kolorimetri dan adanya komponen lain yang memiliki kemampuan
mereduksi dapat mempengaruhi gula pereduksi yang ada. Nilai rekoveri rata-rata untuk bahan
acuan susu bubuk adalah 65%, sehingga jika Metode Luff-Schoorl seperti dalam prosedur SNI
01-2891-1992 diaplikasikan sampel yang komposisinya mirip seperti pada bahan acuan susu
bubuk diperkirakan ada kemungkinan kesalahan sistematis dapat terjadi.
4.3.2.2.2. Rekoveri dengan sampel matriks uji
Nilai rekoveri kecap asin dan santan lebih buruk dibandingkan pada bahan acuan. Nilai
rekoveri yang negatif kemungkinan disebabkan adanya substansi yang dapat menginterferensi
pada sampel. Adapun kandungan lemak yang tinggi (±42%, Tabel 5) pada santan diduga dapat
menganggu analisis karena Shaffer dan Hartman (1920) mengatakan bahwa analisis dengan

42
metode gula pereduksi dianjurkan untuk melakukan presipitasi protein dan lemak dengan asam
tungstat seperti pada analisis sampel susu. Sama halnya dengan nilai rekoveri bahan acuan, untuk
matriks sampel pangan cair tidak ada hasil yang menunjukkan nilai rekoveri yang dapat diterima
berdasarkan batas yang ditetapkan oleh AOAC (2002).
Kemungkinan efek perbedaan matriks sampel terhadap perbedaan besarnya nilai
rekoveri telihat dalam penelitian ini. Karena uji rekoveri dapat memeriksa adanya interferensi
kompetitif dan efek dari matriks sampel (Koch dan Peter 1999; Cembrowski dan Sullivan 1992),
sehingga kemungkinan diperkirakan pada susu bubuk ada substansi yang dapat menginterferensi.
Hal ini juga diperkuat oleh koefisien variasi (RSD) yang ditunjukkan pada nilai perolehan rekoveri
yaitu 6.68%, yang merupakan nilai yang paling besar dibandingkan nilai RSD yang didapat pada
bahan kedelai (4,64%) dan kacang hijau (1,48%). Selain pada susu bubuk, kecap asin dan santan
juga memiliki rata-rata nilai rekoveri yang buruk, yaitu masing-masing -28,34%dan -41,82%.
Keduanya juga memiliki nilai RSD yang besar yaitu masing-masing 6,68% dan 24,77%.
Substansi yang dapat menginterferensi pada susu bubuk, kecap asin atau santan, dapat
menganggu baik pada saat proses hidrolisis polisakarida menjadi gula-gula pereduksi atau pada
saat kuantifikasi dari gula pereduksi. Karena nilai rekoveri yang rendah dapat mengindikasikan
adanya kesalahan negatif. Kesalahan negatif dari tahap hidrolisis asam dapat disebabkan oleh
destruksi glukosa atau gula lain oleh adanya asam dan panas (Whelan dan Pirt 2006; Loomys dan
Shull 1937; Shriner 1932) atau terbentuk produk dari reaksi antara asam amino dan karbohidrat
(Southgate 1976). Karena pada metode karbohidrat total SNI 01-2891-1992 tidak ada tahap
deproteinisasi atau upaya lain untuk menghilangkan substansi yang dapat menginterferensi.
Shaffer dan Hartman (1920) menyarankan untuk melakukan presipitasi protein dan lemak dengan
asam tungstat untuk analisis sampel susu menggunakan metode gula pereduksi, tetapi hal ini tidak
dilakukan pada analisis karbohidrat total metode SNI 01-2891-1992. Kemungkinan karena tidak
adanya deproteinisasi dan rusaknya gula sederhana pada saat hidrolisis juga yang dapat menjadi
penyebab nilai rekoveri pada bahan acuan lain yaitu kacang hijau dan kacang kedelai serta matriks
sampel pangan cair (kecap manis, kecap asin dan santan) tidak mencapai range rekoveri yang
dapat diterima. Sampel kecap manis yang banyak mengandung gula yang ditambahkan dalam
proses pembuatannya menyebabkan adanya kemungkinan destruksi gula saat pemanasan sehingga
nilai rekoveri yang didapat kecil bahkan negatif.

43
4.4. Faktor-Faktor Kesalahan Pada Analisis Total Karbohidrat SNI
01-2891-1992
Diagram Ishikawa adalah diagram sebab-akibat yang merupakan salah satu dari tujuh
pengendali mutu. Faktor-faktor kesalahan yang digambarkan dalam diagram Ishikawa diperoleh
melalui pengamatan selama penelitian dilakukan dan studi literatur. Faktor-faktor kesalahan yang
dapat terjadi selama analisis total karbohidrat metode SNI 01-2891-1992 digambarkan melalui
diagram Ishikawa (Gambar 5). Faktor-faktor kesalahan digolongkan ke dalam lima kategori utama
yaitu reagen, metode, alat, matriks, lingkungan dan analis.
Masing-masing kategori terbagi menjadi beberapa faktor. Pada faktor reagen dibagi
menjadi reagen yang rentan seperti reagen Luff-Schoorl (reagen tembaga sulfat dalam asam sitrat
dan natrium karbonat), natrium tiosulfat yang digunakan sebagai titer serta reagen lain seperti
larutan KI, H2SO4 dan larutan yang digunakan untuk standardisasi. Kontaminasi atau kemurnian,
umur simpan, serta stabilitas reagen merupakan kemungkinan penyebab terjadinya kesalahan
selama analisis. Reagen yang digunakan ada beberapa yang tidak stabil seperti natrium tiosulfat,
oleh karena itu perlu pengecekan konsentrasi (standardisasi) minimal dua minggu sekali. Selain itu
reagen sitrat yang digunakan sebagai salah satu komponen campuran reagen Luff memiliki
kekurangan. Reagen dianjurkan menggunakan tartarat untuk menstabilkan ion tembaga (Southgate
1976). Penggunaan sitrat dibanding tartarat menyebabkan berkurangnya jumlah tembaga yang
tereduksi dan sensitifitas menjadi lebih buruk. Selain itu reagen Luff yang digunakan tidak
mengandung iodida menunjukkan adanya pemisahan sejumlah kecil tembaga oksida dan kenaikan
tingkat autoreduksi selama pemanasan yang meningkat seiring dengan usia reagen (Shaffer dan
Somogyi 1932). Oleh karena itu diperlukan pemeriksaan blanko secara berkala. Fluktuasi jumlah
titer yang digunakan untuk mentitrasi blanko juga dikonfirmasi dalam penelitian ini. Adapun
reagen yang paling tidak stabil adalah natrium tiosulfat.
Pada faktor metode terbagi menjadi sesuai tahapan analisis. Mulai dari persiapan
sampel, hidrolisis, penetralan, penepatan volume, penyaringan, pemipetan, homogenisasi,
pengisian buret, suhu dan waktu pemanasan reaksi Luff, waktu tunggu sebelum titrasi,
pendinginan sebelum ditambahkan KI, penambahan reagen, pembacaan buret, penentuan titik
akhir, dan kalkulasi gula pereduksi merupakan bagian dari faktor kesalahan metode. Persiapan

44
sampel yang tidak tepat dapat menyebabkan sampel tidak homogen sehingga hasil analisis
memiliki keragaman yang tinggi. Hidrolisis asam memerlukan kestabilan suhu dari waterbath,
homogenitas panas dan ketepatan waktu hidrolisis. Proses pemanasan untuk reaksi reduksi
tembaga harus dilakukan dengan sangat hati-hati karena sangat laju reaksi reduksi sangat
dipengaruhi oleh suhu dan waktu pemanasan. Pemanasan yang terlalu lama akan menyebabkan
gula terdestruksi dan pemanasan yang terlalu sebentar akan menyebabkan hasil kurang
reprodusibel dan proporsionalitas antara gula yang teroksidasi dan tembaga yang tereduksi kurang
konstan (Shaffer dan Hartmann 1920). Selain itu laju kinetika reaksi juga berbeda-beda untuk
kadar gula yang berbeda (Faulks dan Timms 1985). Kondisi saat pemanasan itu juga harus
dikontrol agar tidak terjadi reoksidasi tembaga yang telah tereduksi, oleh karena itu kontaminasi
dengan O2 harus dihindari (Shaffer dan Somogyi 1932). Titrasi harus dilakukan dengan cepat
tetapi dengan hati-hati dan waktu tunggu antar sampel tidak boleh terlalu lama. Pembacaan titik
akhir juga harus tepat, titik akhir titrasi kadang tidak terlalu jelas dan warna biru dapat muncul
kembali (Shaffer dan Hartmann 1920) sehingga menyulitkan titrasi. Penambahan reagen KI dan
H2SO4 harus sesuai urutan agar reaksi berjalan dengan benar (Shaffer dan Somogyi 1932).
Pembacaan buret dan penambahan indikator pati harus dilakukan dengan tepat. Selain itu blanko
harus dibuat berkala karena adanya kemungkinan autoreduksi yang meningkat perlahan seiring
dengan usia reagen (Shaffer dan Somogyi 1932). Pembuatan dan penyimpanan reagen perlu
diperhatikan agar menghindari kontaminasi. Standardisasi untuk reagen yang rentan seperti
natrium tiosulfat perlu dilakukan secara berkala dan akurat.
Untuk faktor alat dapat dibagi menjadi alat gelas, neraca analitik, buret, pHmeter,
waterbath, dan hotplate. Pencegahan alat-alat gelas dari kontaminasi baik debu maupun reagen
lain dan penjagaan kebersihannya perlu diperhatikan karena akan mengganggu analisis (Shaffer
dan Somogyi 1932). Neraca analitik, pH-meter dan waterbath adalah alat yang mungkin dapat
menjadi penyebab kesalahan analisis. Neraca analitik dan pH-meter harus dikalibrasi terlebih
dahulu karena dapat menyebabkan keragaman pada data yang dihasilkan. Waterbath harus
memiliki suhu yang stabil dan homogenitas panas yang baik agar hidrolisis terkontrol. Buret juga
harus dijaga agar tidak terkonaminasi serta mencegah tip buret dari kebocoran, adanya udara di
dalam tube dan stopcock yang longgar.

45
Gambar 5. Diagram kesalahan analisis metode karbohidrat total SNI 01-2891-1992
Faktor analis yaitu ketrampilan, sikap atau perilaku dan faktor kelelahan menjadi
penentu hasil analisis. Prosedur yang panjang dan memakan waktu mengharuskan analis mengatur
waktu dengan baik agar hasil analisis tidak terpengaruh oleh ketrampilan yang tidak konstan akibat
kelelahan.
Faktor lingkungan dapat berupa fluktuasi suhu, yang berpengaruh pada sampel dan
titran. Terdapat juga faktor kesalahan dari sampel berupa efek interferensi dari matriks.
4.5. Kelemahan Analisis Total Karbohidrat SNI 01-2891-1992
Analisis total karbohidrat SNI 01-2891-1992 memiliki beberapa kelemahan, selain
banyaknya faktor kesalahan yang mungkin terjadi dalam analisisnya. Salah satu kelemahannya ada
pada tahap hidrolisis. Selain ada kemungkinan bahwa seluruh karbohidrat tidak terhidrolisis
sempurna, hidrolisis asam yang dilakukan dapat menyebabkan destruksi dari fruktosa (Loomys
dan Shull 1937); atau gula-gula lain (Shriner 1932). Glukosa juga terdegradasi perlahan jika
dipanaskan dengan asam, laju destruksi ini dipercepat oleh asam sulfat dan jauh lebih cepat dengan
Kesalahan
analisis
Analis
Keterampilan
Kelelahan
Sikap/perilaku
Lingkungan
Fluktuasi suhu
Matriks sampel
Reagen
Umur simpan
Kemurnian/kontaminasi
Sifat kimia reagen
Standardisasi
Pembuatan reagen
Buret
Alat
Alat gelas
waterbath
pHmeter
Hotplate
Neraca analitik
Metode
Persiapan sampel
Hidrolisis asam
PenetralanPemipetan
Penepatan volume
Homogenisasi
Pembacaan buret
TitrasiSuhu &waktu pemanasan
Pembuatan & penambahan reagenPendinginan

46
HCl (Whelan dan Pirt 2006) terutama jika terdapat protein atau asam amino (Southgate 1976).
Dekstruksi gula pada tahap hidrolisis dapat menyebabkan kesalahan negatif, nilai yang didapat
menjadi tidak akurat bahkan dapat menghasilkan nilai yang keliru.
Nilai yang didapat dari analisis kadar karbohidrat dengan menggunakan hidrolisis asam
tidak dapat dikatakan sebagai nilai kadar total karbohidrat maupun nilai total available karbohidrat
juga karena sulit untuk memisahkan fraksi pati dari karbohidrat struktural (Loomys dan Shull
1937) dan kemungkinan keberadaan serat kasar juga tidak dapat dihidrolisis dengan asam kuat
encer saja. Serat contohnya, selulosa cenderung tahan terhadap hidrolisis asam kuat encer
(Southgate 1976). Dengan demikian, nilai yang didapat lebih cocok jika disebut sebagai nilai total
karbohidrat yang dapat terhidrolisis oleh asam (Weinmann 1946).
Kelemahan metode SNI 01-2891-1992 lainnya terdapat pada tahap analisis gula
pereduksi dengan Metode Luff-Schoorl. Metode Luff Schoorl yang berprinsip pada reduksi Cu2+
oleh gula pereduksi, memiliki kelemahan yaitu reaksi reduksi antara gula dan tembaga sulfat
tampaknya tidak stoikiometris (Davidson 1967; Southgate 1976), kondisi reaksi kritis (Miller
1959; Southgate 1976), dan laju reaksi tiap gula berbeda-beda (Miller et al 1961). Faktor utama
yang mempengaruhi reaksi adalah pemanasan, alkalinitas, konsentrasi gula dan kekuatan reagen
(Southgate 1976).
Faulks dan Timms (1985) mengatakan bahwa metode dengan prinsip gula pereduksi
selain menunjukkan respon yang bervariasi, reprodusibilitasnya sering sekali buruk, sekalipun
dengan menggunakan sistem yang terotomatisasi. Reagen yang diperlukan untuk analisis ini cukup
banyak, dan beberapa reagennya rentan terhadap oksidasi oleh oksigen (Faulks dan Timms 1985)
dan memerlukan standardisasi berkala. Reagen yang memerlukan standardisasi berkala salah
satunya natrium tiosulfat. Selain itu pekerjaan yang diperlukan untuk metode SNI 01-2891-1992
cukup banyak (labourous), alat gelas yang banyak, memakan waktu dan memerlukan tenaga yang
terampil.
Kesalahan dapat terjadi jika ada substansi dari sampel yang menghambat proses
hidrolisis dari karbohidrat menjadi gula-gula pereduksi atau bereaksi dengan produk akhir hasil
hidrolisis. Selain itu ada juga kemungkinan bahwa adanya substansi yang menghambat
kuantifikasi dari gula pereduksi, misalnya ada agen pengoksidasi yang mengoksidasi kembali
tembaga (Cu+) yang telah tereduksi oleh gula-gula pereduksi; gula pereduksi yang ada malah

47
mereduksi senyawa yang lain bukannya tembaga atau ada substansi yang mengganggu
kesetimbangan reaksi reversible dari residu garam tembaga. Reaksi residu garam tembaga yang
membebaskan iodin adalah sebagai berikut (3.1):
(3.1)
Iodin yang terbentuk kemudian akan dititrasi dengan tiosulfat (Shaffer dan Hartmann
1920). Jika terjadi reoksidasi pada tembaga yang telah tereduksi oleh gula pereduksi maka residu
garam tembaga ( akan semakin banyak dan iodine yang dibebaskan akan semakin besar.
Hal ini berdampak pada nilai yang didapat menjadi lebih kecil dibanding nilai yang sebenarnya.
Kelemahan lain ada pada faktor konversi yang digunakan dalam perhitungan yang
mengonversi total gula menjadi total karbohidrat, yaitu 0,9. Faktor ini seharusnya berbeda sesuai
dengan jenis karbohidrat yang banyak terkandung pada matriks sampel. Faktor konversi 0,9 yang
ditetapkan dalam analisis pati seharusnya tidak disamakan dengan analisis total karbohidrat,
karena bisa saja komposisi karbohidrat yang terdapat pada matriks sampel tertentu lebih banyak
dalam bentuk gula sederhana (monosakarida) dan bukan polisakarida, Sehingga faktor konversi
0,9 bisa jadi membuat nilai total karbohidrat lebih kecil dari yang seharusnya. Dari sini dapat
terlihat bahwa pengaruh matriks terhadap hasil analisis salah satunya dipengaruhi komposisi
(jenis) karbohidrat penyusun matriks itu sendiri. Konsentrasi dari analat (karbohidrat) suatu
sampel diduga tidak terlalu mempengaruhi selama konsentrasinya masih dalam rentang yang dapat
dianalisis oleh metode. Adapun pengaruh komponen lain seperti lemak dan protein belum dapat
disimpulkan dalam percobaan ini.

48
V. KESIMPULAN DAN SARAN
5.1. Kesimpulan
Perbandingan metode analisis karbohidrat total menggunakan dua metode yang berbeda
yaitu metode karbohidrat total SNI 01-2891-1992 yang menggunakan metode Luff Schoorl secara
titrimetri dan metode kandidat yang menggunakan Anthrone sulfat secara spektrofotometri pada
sampel pangan cair terpilih yang mewakili kadar karbohidrat rendah, sedang dan tinggi yaitu kecap
manis, kecap asin dan santan yang dilakukan menunjukkan nilai presisi yang tidak berbeda nyata
berdasarkan uji F tetapi hasil yang berbeda nyata berdasarkan uji t pada tingkat kepercayaan 95%.
Estimasi error antara kedua metode dikonfirmasi melalui uji korelasi menggunakan regresi linear
yang dilakukan dengan menggunakan tambahan data sekunder dari matriks sampel pangan yang
berwujud padat. Metode Anthrone sulfat tidak dapat menggantikan Metode Luff-Schoorl dalam
SNI 01-2891-1992 untuk total karbohidrat, karena Metode Anthrone sulfat dan metode Luff Schoorl
tidak memiliki kesesuaian hasil yang dapat diterima.
Verifikasi terhadap metode karbohidrat total SNI 01-2891-1992 menggunakan presisi
dan akurasi. Presisi dievaluasi berdasarkan ripitabilitas dan reprodusibilitas. Akurasi dievaluasi
dengan uji rekoveri pada matriks sampel pangan cair dan bahan acuan serta membandingkan nilai
hasil analisis bahan acuan dengan rentang nilai pada bahan acuan.
Ripitabilitas metode pada bahan acuan dan matriks pangan cair menunjukkan nilai
presisi yang dapat diterima untuk semua sampel dan bahan acuan yang dianalisis bulan pertama
sedangkan untuk sampel dan bahan acuan yang dianalisis pada bulan kedua dengan selang waktu
dua bulan dari bulan pertama, hanya satu dari tiga bahan acuan yang memiliki ripitabilitas yang
baik dan hanya satu dari tiga sampel matriks pangan cair yang memiliki ripitabilitas yang baik.
Hasil uji reprodusibilitas juga menunjukkan bahwa dua dari tiga bahan acuan yang dianalisis pada
bulan pertama dan kedua memiliki nilai yang berbeda nyata dan satu dari tiga sampel matriks
pangan cair yang dianalisis pada bulan pertama dan kedua memiliki nilai yang berbeda nyata.
Sehingga dapat disimpulkan bahwa reprodusibilitas metode SNI 01-2891-1992 tidak begitu baik.
Hal ini dapat disebabkan adanya perubahan atau ketidakstabilan dari reagen Luff-Schoorl, sampel
pangan cair atau ketidakkonsistenan analisis yang disebabkan oleh prosedur analisis yang panjang.

49
Uji rekoveri pada matriks pangan cair yaitu kecap manis, kecap asin dan santan
menunjukkan nilai rekoveri yang tidak masuk dalam range rekoveri yang dapat diterima.
Begitupula halnya dengan uji rekoveri pada bahan acuan yaitu susu bubuk, tepung kedelai dan
tepung kacang hijau memperlihatkan nilai rekoveri yang tidak masuk dalam range rekoveri yang
dapat diterima, meski hasil analisis bahan acuan masih masuk dalam rentang dari nilai bahan
acuan. Dengan demikian, meski memiliki presisi yang dapat diterima akurasi dari metode
karbohidrat total SNI 01-2891-1992 itu sendiri masih diragukan karena kemungkinan masih rentan
terhadap interferensi dan pengaruh matriks.
5.2. Saran
Pengembangan dan validasi metode analisis terhadap total karbohidrat yang dapat
diaplikasikan untuk pangan secara umum perlu dilakukan. Perhatian lebih perlu diberikan pada
analisis total karbohidrat dengan metode kolorimetri kondensasi tanpa melakukan hidrolisis asam
terlebih dahulu seperti pada prosedur karbohidrat total SNI 01-2891-1992. Perlakuan pendahuluan
untuk menghilangkan substansi yang menginterferensi perlu dilakukan sesuai dengan matriks
dimana metode itu hendak diterapkan. Investigasi lebih lanjut terhadap penyebab bias dari metode
dan analisis mengenai ruggedness dan selektivitas dari metode juga perlu dilakukan. Selain
Anthrone sulfat, metode analisis dengan fenol sulfat juga perlu dipertimbangkan karena
diperkirakan menganalisis jenis karbohidrat yang lebih luas dibandingkan Anthrone sulfat.

50
DAFTAR PUSTAKA
Ameen VZ dan Powell GK. 1985. A simple spectrophotometric method for quantitative fecal
carbohydrate measurement. Clinica Chimica Acta, 152: 3-9.
Badan Standardisasi Nasional. 1992 Uji Makanan dan Minuman. SNI 01-2891-1992
Badan Pengawas Obat dan Makanan. 2005. Pedoman Pencantuman Informasi Nilai Gizi Pada
Label Pangan Jakarta:Direktorat Standardisasi Produk Pangan, Badan Pengawas Obat dan
Makanan
Beck, DJ dan BG Bibby. 1961. A modified anthrone colorimetric technique for use in
investigations related to cariogenicity of foodstuffs. Journal of Dental Research 40: 161-170.
BeMiller JN dan Whistler RL. 1996. Carbohydrate. Di dalam: Fennema O.(ed). 1996. Food
Chemistry. New York: Marcel Dekker.
BeMiller, JN. 2010. Carbohydrate analysis. Di dalam: S. Nielsen (eds). 2010. Food Analysis. New
York: Springer Science.
Brooks JR, Griffin VK dan Kattan MW. 1986. A modified method for total carbohydrate analysis
of glucose syrups, maltodextrins, and other starch hydrolysis products. Cereal Chemistry. 63
(5): 465-466.
Cembrowski GS dan Sullivan AM. 1992. Quality Control and Statistics. Di dalam. Bishop ML,
Duben-Engelkirk JL dan Fody EP (eds). 1992. Clinical Chemistry Principles Procedures,
Correlation. Philadelphia: Lippincott.
Christian VA dan Vaclavik EW. 2003. Essentials of Food Science 2nd Edition. London: Kluwer
Academic.
Courtin CM, Van den Broeck H dan Delcour JA. 1999. Determination of reducing end sugar
residues in oligo- and polysaccharides by gas–liquid chromatography. Journal of
Chromatography A, 866: 97–104

51
Dara F. 2010. Bahan Acuan (Reference Material) dalam Metrologi.
http://kimia.lipi.go.id/wp-content/uploads/2010/05/certified-reference-material-fitri.pdf [21
Agustus 2011]
Davidson, E. A. (1967). Carbohydrate reactions. In: Carbohydrate Chemistry. New York: Holt,
Rinehart and Winston Inc.
Direktorat Gizi. 1967. Daftar Komposisi Bahan Makanan. Jakarta: Bharata.
Dreywood, R. 1946. Qualitative test for carbohydrate material. Industrial. and Engineering
Chemistry, Analytical Edition 18: 499.
[EMA] The European Agency for the Evaluation of Medicinal Products. 1995. ICH Topic Q 2 B.
Validation of Analytical Procedures: Methodology.
http://www.pharmacontract.ch/support/pdf_support/Q2a.pdf. [17 Maret 2010]
Ermer, J. 2005. Performance parameters, calculations and tests. Di dalam : Method Validation in
Pharmaceutical Analysis (J. Ermer dan J.H.McB.Miller, eds.). Weinheim: WILEY-VCH
Verlag GmbH & Co. KGaA.
Fales FW, Russel JA dan Fain JN. Some applications and limitations of the enzymic, reducing
(Somogyi), and Anthrone methods for estimating sugar. Clinical Chemistry 7(4): 289-303
Faulks RM dan Timms SB. 1985. A rapid method for determining the carbohydrate component of
dietary fibre. Food Chemistry 17:273-287
Fennema, OR. 1996. Food Chemistry. New York: Marcel Dekker.
Garfield FGE, Klesta dan J Hirsch. 2000. Quality Assurance Principles for Analytical
Laboratories. USA:AOAC International.
Giese, G. 2004. Method Validation. Institute of Hygiene and Environment, City of Hamburg.
http://www.havakalitesi.cevreorman.gov.tr/english/training_4-6/paper_method_validation.pd
f. [1 Juni 2010]

52
Harmita. 2004. Petunjuk pelaksanaan validasi metode dan cara perhitungannya. Majalah Ilmu
Kefarmasian 1(.3): 117 – 135..
Harvey, D. 2000. Modern Analytical Chemistry. McGraw-Hill Companies, Inc., USA.
Hurd CD dan Isenhour. 1932. Pentose reactions I furfural formation, Journal of American
Chemical Society 54:317.
Jelita K. 2011. Verifikasi Metode Analisis Serat Pangan dengan Metode AOAC dan ASP terhadap
Parameter Repeatabilitas, Selektivitas dan Ruggedness. [Skripsi]. Departemen Ilmu dan
Teknologi Pangan Institut Pertanian Bogor.
Jermyn AH. 1975. Increasing of the Anthrone method for carbohydrate. Analytical Biochemistry
68: 332-335.
Judoamidjojo RM, Itoh T, Tomatsu A dan Matsuyama A. 1985. The analytical study of
kecap—Indonesian soy sauce. Di dalam Makalah Internasional Symposium on Agriculture
Product, Processing and Technology, Bogor.
Judoamidjodjo RM. 1987. Studies on chemical and microbiological aspect of kecap as
fundamental to improve ITS quality. Di dalam Kumpulan Seminar Bioteknologi Pertanian
PAU Bioteknologi, IPB.
Kennedy JF dan White Ca. 1988. Classification and description of monosaccharides,
oligosaccharides, and polysaccharides. Di dalam: Kennedy JF (ed). 1988. Carbohydrate
chemistry. Oxford: Clarendon express.
Kirk, R. E. dan O. F. Othmer. 1950. Encyclopedia of Chemical Technology. The Interscience
Encyclopedia, inc. New York.
Koch DD dan Peters T. Selection and Evaluation of Methods. Di dalam Burtis CA dan Ashwood
ER(eds). 1999. Tietz Textbook of Clinical Chemistry. Philadelphia: Saunders Elsevier.
Koehler, LH. 1952. Differentiation of carbohydrates by anthrone reaction rate and color intensity.
Analytical Chemistry 24: 1576-1579.

53
Leyva A, Quintana A, Sanchez M, Rodriguez EN, Cremata J, Sanchez JC. 2008. Rapid and
sensitive Anthrone—sulfuric acid assay in microplate format to quantity carbohydrate in
biopharmaceutical product: method development and validation. Biologicals 36: 134-141.
Lin FM dan Wilkens WF.. 2006. Volatile Flavor Components of Coconut Meat. Journal of Food
Science 35(5): 538-539
Loomis WE dan Shull CA. 1937. Methods in Plant Physiology. New York: Mc-Graw Hill
BookCo., Inc.
Ludwig, TG dan HJ Goldberg. 1956. The Anthrone Method for Determination of Carbohydrate in
Oral Rinsing. Journal of Dental Research 35: 90
Lumsden JH. 2000. Laboratory test method validation. Revue de Medicine Veterinaire 151 (7):
623-630
Miller, G. L. 1959. Use of dinitrosalicyclic acid reagent for determination of reducing sugars.
Analytical Chemistry 31: 426-8.
Miller, G. L., Slater, R., Birzgalis, R. dan Blum, R. 1961. Application of different colorimetric
tests to cellodextrins. Analytical. Biochemistry, 2: 521-528.
Momose, T, Ueda, Y, Sawada, K, dan Sugi, A.et al. 1957. Organic analysis VIII reaction
mechanism of anthrone with sugars . Pharm Bull (Tokyo) 5: 31.
Morris, DL. 1948. Quantitative determination of Carbohydrate with Dreywood’s anthrone reagent.
Science, 107: 254.
Mullins E. 2003. Statistics for the Quality Control Chemistry. Laboratory. UK: Royal Society of
Chemistry.
Nielsen, S. 2010. Introduction to food analysis. Di dalam: S. Nielsen (eds). 2010. Food Analysis.
New York: Springer Science.
Nuryatini. 2010. Ketertelusuran Pengukuran.
http://isjd.pdii.lipi.go.id/admin/jurnal/1518101215_0251-0476.pdf [21Agustus 2011]

54
Pirt SJ dan Whelan WJ. 2006. The determination of starch by acid hydrolysis. Journal of the
Science of Food and Agriculture 2(5):224-228.
Safarik I dan Satruckova H. 1992. Direct determination of total soil carbohydrate content. Plant
and Soil 143: 109-114.
Santoso HB. 1994. Kecap dan Tauco Kedelai. Yogyakarta: Kanisius.
Sattler L dan FW. Zerban. 1948. The Dreywood anthrone reaction as affected by carbohydrate
structure, Science, 108:207.
Sawyer, R. 1984. Food composition and analytical accuracy. Di dalam: Birch GG dan K.J. Parker
(eds). Control of Food Quality and Food Analysis. New York: Elsevier.
Seow CC dan Gwee CN. 1997. Coconut milk: chemistry and technology. Journal of Food Science
32: 189-201.
Shaffer PA dan Hartmann AF. 1920. The Iodometric determination of copper and its use in sugar
analysis. Journal of Biological Chemistry 45: 365-390.
Shaffer PA dan Somogyi M. 1932. Copper-iodometric reagents for sugar determination. Journal of
Biological. Chemistry 100: 695-713
Shriner RL. 1932. The determination of starch by acid hydrolysis. Plant Physiology 7(3):541-546.
Smith, JS. 2010. Evaluation of analytical data. Di dalam: S. Nielsen (eds). 2010. Food Analysis.
New York: Springer Science.
Southgate DAT. 1976. Determination of Food Carbohydrates. London: Applied Science Publisher
Ltd.
Sullivan DM dan Carpenter DE. 1993. Methods of Analysis for Nutritional Labeling.
Gaithersburg: AOAC International.
Suprapti MS. 2005. Kecap Tradisional. Yogyakarta: Kanisius.

55
Tangsuphoom N dan Coupland JN. 2008. Effect of heating and homogenization on the stability of
coconut milk emulsions. Journal of Food Science 70(8): 466-470.
Thompson M, Ellison SLR dan Wood R. 2002. Harmonized guidelines for single laboratory
validation of methods of analysis (IUPAC Technical report). Pure Applied Chemistry 74(5):
835-855.
Walton RM. 2001. Validation of laboratory tests and methods. Seminars in Avian and Exotic Pet
Medicine 10(2):59-65.
Weinmann H. 1946. Determination of total available carbohydrate in plants. Plant Physiology 22:
279-290.
Westgard, JO. 1998. Points of care in using statistics in methods comparison studies (editorial).
Clinical Chemistry 44: 2240-2242.
Wolfrom, ML et al. 1948. Chemical interaction of amino compounds and sugars III the conversion
of D-glucose to 5-(hydroxymethyl)-2-furaldehyde. Journal of American Chemical Society 70:
514
Yemms EW dan Willis AJ. 1954. The estimation of carbohydrates in plant extracts by anthrone.
Biochemistry Journal 57: 508-514
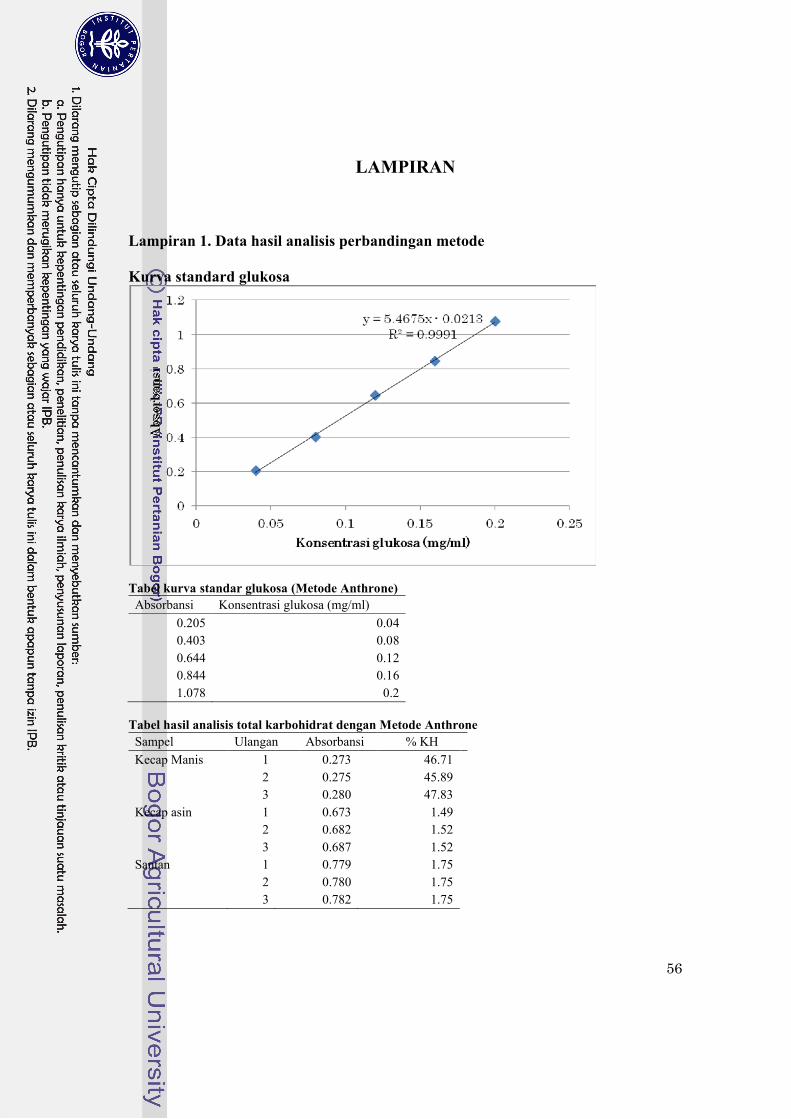
56
LAMPIRAN
Lampiran 1. Data hasil analisis perbandingan metode
Kurva standard glukosa
Tabel kurva standar glukosa (Metode Anthrone)Absorbansi Konsentrasi glukosa (mg/ml)
0.205 0.040.403 0.080.644 0.120.844 0.161.078 0.2
Tabel hasil analisis total karbohidrat dengan Metode AnthroneSampel Ulangan Absorbansi % KH
Kecap Manis 1 0.273 46.712 0.275 45.893 0.280 47.83
Kecap asin 1 0.673 1.492 0.682 1.523 0.687 1.52
Santan 1 0.779 1.752 0.780 1.753 0.782 1.75

57
Tabel hasil analisis total karbohidrat dengan metode SNI 01-2891-1992
Sampel UlanganPembacaan volume Volume
titranselisih
blanko-titran%Karbohidrat
Awal Akhir
Kecap Manis 1 0.25 7.80 7.55 17.35 39.48 2 7.9 15.50 7.60 17.30 38.17 3 15.65 23.50 7.85 17.05 38.48
Kecap asin 1 0.1 19.80 19.70 5.20 2.24 2 0.3 20.00 19.70 5.20 2.24 3 0.1 20.00 19.90 5.00 2.15
Santan 1 0 21.55 21.55 3.35 1.45 2 4.05 21.40 17.35 7.55 1.51 3 0 21.40 21.40 3.50 1.51
Keterangan: blanko 24, 90 ml
Data pendamping (matriks pangan padat)
Tabel hasil analisis total karbohidrat dengan Metode Anthrone
Sampel Ulangan Berat sampel (g) Absorbansi %KH
Tepung beras 1 0.1062 0.514 80.842 0.1076 0.515 80.69
3 0.1069 0.517 80.92Susu bubuk 1 0.5470 0.757 25.45
2 0.5455 0.760 25.52
3 0.5453 0.761 25.56Sarden 1 5.1782 0.763 1.72
2 5.1384 0.797 1.79
3 5.1337 0.797 1.79
Tabel hasil analisis total karbohidrat dengan metode SNI 01-2891-1992
Sampel Ulangan Berat sampel (g) Volume titran %KH
Tepung beras 1 0.1062 21.50 67.712 0.1076 21.50 67.46
3 0.1069 21.55 66.37Susu bubuk 1 0.5470 17.90 30.75
2 0.5455 18.10 29.82
3 0.5453 17.80 31.18Sarden 1 5.1782 22.00 1.25
2 5.1384 21.95 1.27
3 5.1337 22.00 1.25Keterangan: blanko 24, 90 ml
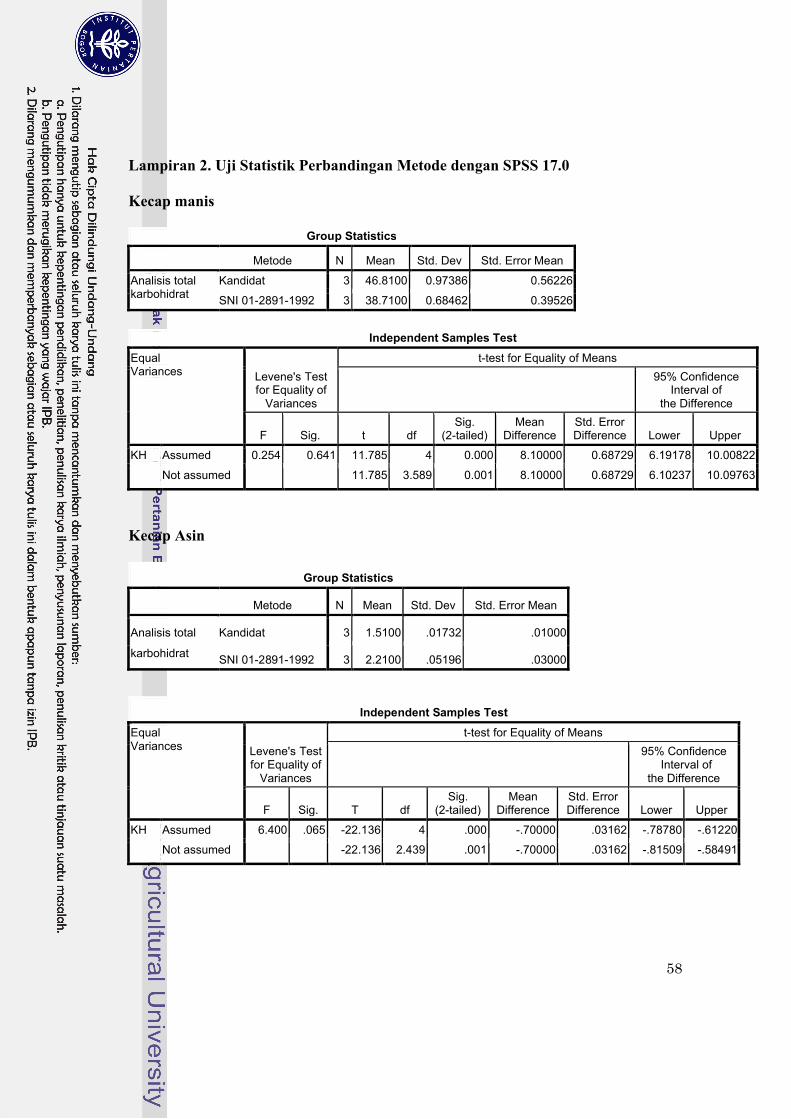
58
Lampiran 2. Uji Statistik Perbandingan Metode dengan SPSS 17.0
Kecap manis
Group Statistics
Metode N Mean Std. Dev Std. Error Mean
Analisis total karbohidrat
Kandidat 3 46.8100 0.97386 0.56226
SNI 01-2891-1992 3 38.7100 0.68462 0.39526
Independent Samples Test
Equal Variances Levene's Test
for Equality of Variances
t-test for Equality of Means
95% Confidence Interval of
the Difference
F Sig. t dfSig.
(2-tailed)Mean
DifferenceStd. Error Difference Lower Upper
KH Assumed 0.254 0.641 11.785 4 0.000 8.10000 0.68729 6.19178 10.00822
Not assumed 11.785 3.589 0.001 8.10000 0.68729 6.10237 10.09763
Kecap Asin
Group Statistics
Metode N Mean Std. Dev Std. Error Mean
Analisis total
karbohidrat
Kandidat 3 1.5100 .01732 .01000
SNI 01-2891-1992 3 2.2100 .05196 .03000
Independent Samples Test
Equal Variances Levene's Test
for Equality of Variances
t-test for Equality of Means
95% ConfidenceInterval of
the Difference
F Sig. T dfSig.
(2-tailed)Mean
DifferenceStd. Error Difference Lower Upper
KH Assumed 6.400 .065 -22.136 4 .000 -.70000 .03162 -.78780 -.61220
Not assumed -22.136 2.439 .001 -.70000 .03162 -.81509 -.58491
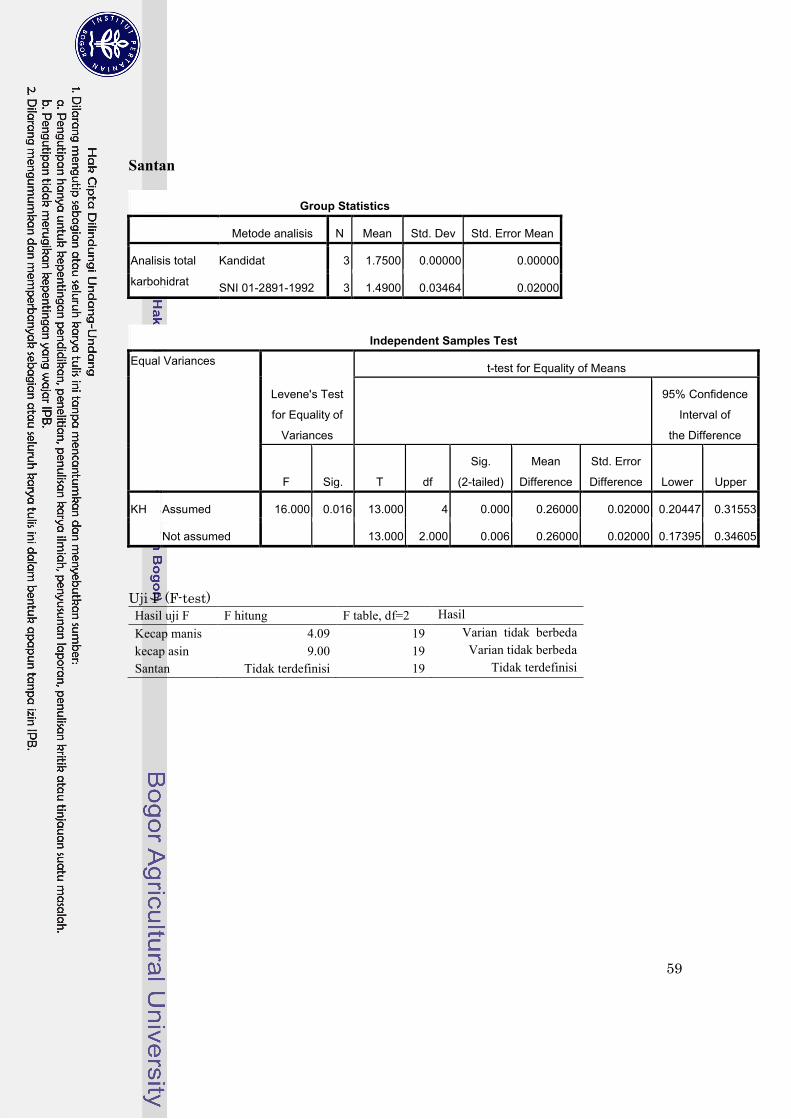
59
Santan
Group Statistics
Metode analisis N Mean Std. Dev Std. Error Mean
Analisis total
karbohidrat
Kandidat 3 1.7500 0.00000 0.00000
SNI 01-2891-1992 3 1.4900 0.03464 0.02000
Independent Samples Test
Equal Variances
Levene's Test
for Equality of
Variances
t-test for Equality of Means
95% Confidence
Interval of
the Difference
F Sig. T df
Sig.
(2-tailed)
Mean
Difference
Std. Error
Difference Lower Upper
KH Assumed 16.000 0.016 13.000 4 0.000 0.26000 0.02000 0.20447 0.31553
Not assumed 13.000 2.000 0.006 0.26000 0.02000 0.17395 0.34605
Uji F (F-test)Hasil uji F F hitung F table, df=2 Hasil
Kecap manis 4.09 19 Varian tidak berbeda
kecap asin 9.00 19 Varian tidak berbeda
Santan Tidak terdefinisi 19 Tidak terdefinisi

60
Lampiran 3. Prosedur Analisis Proksimat
A. PROSEDUR ANALISIS1. Analisis Kadar Air, Metode Oven (AOAC, 1995)
Kadar air diukur dengan metode oven biasa karena kandungan bahan volatil pada sampel rendah dan sampel tidak terdegradasi pada suhu 100 oC. Cawan aluminium kosong dikeringkan dalam oven suhu 105 oC selama 15 menit lalu didinginkan dalam desikator selama 5 menit atau sampai tidak panas lagi. Cawan ditimbang dan dicatat beratnya. Lalu ditimbang sampel sebanyak 5 g di dalam cawan tersebut. Dikeringkan sampel dalam oven sampai beratnya konstan (perubahan berat tidak lebih dari 0.003 g). Setelah itu cawan didinginkan di dalam desikator, ditimbang berat akhirnya, dan dihitung kadar airnya dengan persamaan berikut
Keterangan :x = berat cawan dan sampel sebelum dikeringkan (g)y = berat cawan dan sampel setelah dikeringkan (g)a = berat cawan kosong (g)
2. Analisis Kadar Abu, Metode Oven (AOAC, 1995)Cawan porselen dibakar dalam tanur selama 15 menit kemudian didinginkan di dalam desikator. Setelah cawan dingin, ditimbang. Kemudian sampel sebanyak 5 g ditimbang di dalam cawan lalu diabukan di dalam tanur hingga diperoleh abu berwarna putih dan beratnya tetap. Pengabuan dilakukan dalam 2 tahap yaitu tahap pertama pada suhu 400 oC lalu dilanjutkan pada suhu 550 oC, kemudian didinginkan di dalam desikator lalu ditimbang.
Perhitungan :Keterangan :W1 = berat sampel (g)W2 = berat abu (g)
3. Analisis Kadar Protein (AOAC, 1995)Sampel sebanyak 0.1 – 0.2 g dimasukkan ke dalam labu Kjedahl 100 ml, lalu ditambahkan 2 g K2SO4, 40 mg HgO, dan 2.5 ml H2SO4 pekat. Setelah itu, didestruksi selama 30 menit sampai cairan berwarna jernih dan dibiarkan sampai dingin. Selanjutnya ditambahkan air suling secukupnya dan 10 ml NaOH pekat sampai berwarna coklat kehitaman dan didestilasi. Hasil destilasi ditampung dalam erlenmeyer 125 ml yang berisi H2BO3 dan indikator, kemudian dititrasi dengan HCl 0.02 N. Larutan blanko juga dianalisis seperti sampel. Kadar nitrogen dihitung berdasarkan rumus :
Kadar
4. Analisis Kadar Lemak, Metode Soxhlet (AOAC, 1995)Labu lemak yang telah bebas lemak dikeringkan di dalam oven kemudian ditimbang setelah dingin. Sampel sebanyak 5 g dibungkus dalam kertas saring kemudian ditutup kapas yang bebas lemak. Sampel dimasukkan ke dalam alat ekstraksi soxhlet, kemudian pasang kondensor dan labu pada ujung-ujungnya. Pelarut heksana dimasukkan ke dalam alat lalu sampel direfluks selama 6 jam. Setelah itu pelarut didestilasi dan ditampung pada wadah lain. Labu lemak dikeringkan di dalam oven pada suhu 105oC sampai diperoleh berat tetap.
×100%

61
Kemudian labu lemak dipindahkan ke desikator, lalu didinginkan dan ditimbang. Perhitungan :
Keterangan : W1 = Berat sampel (g)W2 = Berat lemak (g)
5. Analisis Kadar Karbohidrat By Difference (AOAC, 1995)
Pengukuran kadar karbohidrat menggunakan metode by difference, dilakukan dengan cara :
Kadar karbohidrat (%b/b) = 100% - (kadar air + kadar protein + kadar lemak + kadar abu)

62
Lampiran 4. Metode Anthrone dan Luff Schoorl
1. Analisis total karbohidrat Metode Kandidat (dengan Anthrone Sulfat)Penyiapan reagenReagen disiapkan baru setiap hari dengan melarutkan 0.1 g anthrone dalam 100 mL asam sulfat 98% pada suhu ruang.
Persiapan contoh1. Timbang dengan seksama lebih kurang 5 g cuplikan ke dalam Erlenmeyer 500 mL.2. Tambahkan 50 mL larutan HCl 3%, didihkan selama 3 jam dengan pendingin tegak.3. Dinginkan dan netralkan dengan larutan NaOH 30% (dengan lakmus atau fenolftalein)
dan ditambahkan sedikit CH3COOH 3% agar suasana larutan sedikit asam4. Pindahkan isinya ke dalam labu 500 mL dan impitkan hingga tanda garis kemudian
saring dan encerkan seperlunya
Prosedur analisis1. Masukkan 5,0 ml contoh (dari persiapan contoh) ke dalam labu takar 100 ml dan
diencerkan sampai tanda tera dengan air destilasi2. Masukkan sebanyak 1 ml contoh tersebut ke dalam tabung reaksi bertutup3. Tambahkan 5 ml pereaksi Anthrone dan ditutup.Vortex dan kocok hingga merata4. Panaskan tabung reaksi di atas penangas air 1000C selama 12 menit5. Setelah didinginkan, pindahkan larutan ke dalam kuvet dan baca absorbansinya dengan
UV-Vis spektrofotometer pada panjang gelombang 630 nm6. Tentukan konsentrasi gula dalam contoh dengan menggunakan kurva standar hubungan
antara konsentrasi glukosa standar dengan absorbansinya dan dengan memperhitungkan pengenceran yang dilakukan, yaitu dengan menggunakan rumus sebagai berikut:
dimana:
G = konsentrasi gula dari kurva standar (g)FP = faktor pengenceranW = berat contoh (g)
2. Analisis total karbohidrat dengan metode SNI 01-2891-1992Pembuatan pereaksi Luff-Schoorl
1. Larutkan 143,8 g Na2CO3 anhidrat dalam 300 ml air suling. 2. Larutkan 50 g asam sitrat dengan 50 mL air suling. 3. Tambahkan 25 gram Cu2SO4.5H2O ke dalam 100 ml air suling4. Pindahkan larutan tersebut larutan tersebut ke dalam labu 1 liter, tepatkan sampai tanda
garis dengan air suling dan kocok.5. Biarkan semalam dan saring bila perlu. Larutan ini mempunyai kepekatan Cu2+ 0,1 N
Na2CO3
6. Larutkan kalium iodide KI 20%7. Larutkan asam sulfat H2SO4 25%8. Larutkan natrium tiosulfat Na2S2O3, 0, 1 N9. Penunjukkan larutan kanji 0,5%
Pengujian kepekatan larutan Luff-Schoorl1. Pipet 25 mL larutan Luff, tambahkan 3 g KI dan 25 mL larutan H2SO4 6N2. Titar dengan larutan natrium tiosulfat 0,1 M dengan petunjuk larutan kanji 0,5%3. Larutan natrium tiosulfat yang dipergunakan untuk titrasi 25x2 mL4. Pipet 10 mL larutan Luff, masukkan ke dalam labu ukur 100 mL, encerkan dengan air

63
suling dan kocok5. Pipet 10 mL larutan hasil pengenceran tersebut dan masukkan ke dalam erlenmeyer
berisi 25 mL HCl 0,1 N6. Masukkan erlenmeyer tersebut ke dalam penangas air mendidih dan biarkan selama 1
jam, kemudian angkat dan dinginkan.7. Encerkan dengan air suling dan titar dengan larutan NaOH 0,1 N dengan indikator
fenolftalein8. Pipet 10 mL larutan hasil pengenceran (b) masukkan ke dalam erlenmeyer dan titar
dengan HCl 0,1 M dengan indikator fenolftalein9. Larutkan HCl 0,1 M yang dipergunakan untuk titrasi harus sekitar 6,0 sampai 7,6 mL10. Larutan Luff harus mempunyai pH 9,3-9,4
Cara kerja1. Timbang dengan seksama lebih kurang cuplikan ke dalam Erlenmeyer 500 mL.2. Tambahkan 50 mL larutan HCl 3%, didihkan selama 3 jam dengan pendingin tegak.3. Dinginkan dan netralkan dengan larutan NaOH 30% (dengan lakmus atau fenolftalein)
dan ditambahkan sedikit CH3COOH 3% agar suasana larutan sedikit asam4. Pindahkan isinya ke dalam labu 500 mL dan impitkan hingga tanda garis kemudian
saring5. Pipet 10 mL saringan ke dalam Erlenmeyer 500 mL, tambahkan 25 mL larutan Luff
(dengan pipet) dan beberapa butir batu didih serta 15 mL air suling.6. Panaskan campuran tersebut dengan nyala tetap. Usahakan agar larutan dapat mendidih
dalam waktu 3 menit (gunakan stopwatch), didihkan terus selama tepat 10 menit (dihitung dari saat mulai mendidih dan gunakan stopwatch) kemudian dengan cepat dinginkan dalam bak berisi es.
7. Setelah dingin tambahkan 15 mL larutan KI 20% dan 25 mL H2SO4 25% perlahan-lahan.8. Titar secepatnya dengan larutan tiosulfat 0,1 M (gunakan penunjuk larutan kanji 0,5%)
Perhitungan:(blanko-penitar) x N tiosulfat x 10, setara dengan terusi yang tereduksi. Kemudian lihat dalam daftar Luff –Schoorl berapa mg gula yang terkandung untuk mL tio yang dipergunakan
dimana: W1 = bobot cuplikan (mg)W = glukosa yang terkandung untuk ml tio yang dipergunakan (mg)FP = faktor pengenceran
Standardisasi larutan tiosulfat1. Sebanyak 0,5 gram K2Cr2O7 ditimbang dan dilarutkan dengan akuades2. Ditepatkan hingga 100 ml dengan labu takar3. Ambil 25 ml ke dalam erlenmeyer4. Ditambahkan 10mL KI, 25mL HCl, dan akuades hingga 200 ml5. Titar dengan natrium tiosulfat hingga berwarna kuning6. Tambahkan indikator kanji7. Titar dengan natrium tiosulfat hingga berwarna hijau toska (hijau jamrud)8. Dilakukan sebanyak 3 kali ulangan

64
Tabel penetapan gula Luff-Schoorl
Na2S2O3, 0,1N (ml)
Glukosa, Fruktosa, Gula Inversi (mg)
1 2.4
2 4.83 7.24 9.75 12.26 14.77 17.28 19.89 22.410 25.011 27.612 30.313 33.014 35.715 38.516 41.317 44.218 47.119 50.020 53.021 56.022 59.123 62.2

65
Lampiran 5 Verifikasi Metode Karbohidrat Total SNI 01-2891-1992Analisis tanggal 28 Juli 2011Kacang hijauUlangan Berat sampel (g) Titer (ml) blanko (ml) mg glukosa %KH % recovery
1 0,5018 11,95 24,78 28,1840 56,1658 104,76732 0,5014 12,10 24,78 27,8322 55,5091 103,54243 0,5016 12,00 24,78 28,0667 55,9544 104,37314 0,5014 12,10 24,78 27,8322 55,5091 103,54245 0,5014 12,10 24,78 27,8322 55,5091 103,54246 0,5015 12,10 24,78 27,8322 55,4980 103,52177 0,5019 12,10 24,78 27,8322 55,4538 103,4392
%KH (Karbohidrat) Rataan : 55,66%Standard deviasi : 0,2827RSD analisis : 0,51
2x RSD AOAC : 2,18
2/3 RSD Horwitz : 1,45
Kedelai
UlanganBerat sampel
(g)Titer (ml)
blanko (ml)
mg glukosa %KH% recovery
1 0,5002 20,85 24,78 8,2522 16,4977 99,144912 0,5003 21,15 24,78 7,6008 15,1924 91,300763 0,5002 21,00 24,78 7,9265 15,8466 95,231964 0,5002 21,00 24,78 7,9265 15,8466 95,231965 0,5005 20,90 24,78 8,1436 16,2709 97,781956 0,5005 21,00 24,78 7,9265 15,8371 95,174887 0,5003 21,00 24,78 7,9265 15,8434 95,21293
%KH (Karbohidrat)
Rataan : 15,90%
Standard deviasi : 0,4099
RSD analisis : 2,58
2x RSD AOAC : 2,64
2/3 RSD Horwitz : 1,76
Susu bubuk
Ulangan Berat sampel (g) Titer (ml)Blanko
(ml)bl-titer (ml) mg glukosa %KH
1 0,5006 15,45 25,50 10,1807 22,9227 45,7905
2 0,5004 15,40 25,50 10,2313 23,0412 46,0456
3 0,5003 15,60 25,50 10,0287 22,5672 45,1073
4 0,5000 15,60 25,50 10,0287 22,5672 45,1343
5 0,5002 15,45 25,50 10,1807 22,9227 45,8271
6 0,5000 15,40 25,50 10,2313 23,0412 46,0825
7 0,5002 15,40 25,50 10,2313 23,0412 46,0641
%KH (Karbohidrat)
Rataan : 45,72%
Standard deviasi : 0,4264
RSD analisis : 0,93
2x RSD AOAC : 2,25
2/3 RSD Horwitz : 1,50

66
Analisis tanggal 11 Oktober 2011
Kacang Hijau
Ulangan Berat sampel (g) Titer (ml) Vblanko-titrant (ml) %KH
1 5,1494 7,05 7,05 53,852 5,0049 7,10 7,20 56,653 5,2058 7,10 7,50 56,86
Kedelai
Ulangan Berat sampel (g) Titer (ml) Vblanko-titrant (ml) %KH
1 5,0613 6,90 13,52 13,522 5,0143 7,08 15,38 15,383 5,1475 7,95 15,29 15,29
Susu Bubuk
Ulangan Berat sampel (g) Titer (ml) Vblanko-titrant (ml) %KH
1 5,0587 7,65 17,40 35,872 5,2166 7,10 17,95 36,003 5,0854 7,10 17,95 36,93
Analisis tanggal 7 Oktober 2011
Kecap manis
Ulangan Berat sampel (g) Titer (ml) Vblanko-titrant (ml) %KH
1 5,2178 15,30 9,75 37,332 5,1232 15,75 9,30 36,203 5,0744 15,55 9,50 37,36
Kecap asin
Ulangan Berat sampel (g) Titer (ml) Vblanko-titrant (ml) %KH
1 5,3262 15,30 9,75 1,832 5,3239 13,60 11,45 2,163 5,0430 14,55 10,50 2.09
Santan
Ulangan Berat sampel (g) Titer (ml) Vblanko-titrant (ml) %KH
1 5,2093 17,90 7,15 1,352 5,0331 17,70 7,35 1,443 5,0410 17,15 7,90 1,55

67
Hasil uji rekoveri pada berbagai bahan acuan dengan spike glukosaBahan acuan kacang hijau
UlanganGlukosa
(g)
Berat bahan
acuan(g)
W total(g)
Titer(ml)
Blanko(ml)
mg glukosa
%KH %rekoveri
1 0,0505 0,4500 0,5005 12,00 24,30 29,2334 58,41 82,902 0,0503 0,4500 0,5003 11,95 24,30 29,3501 58,66 85,553 0,0500 0,4501 0,5001 12,00 24,30 29,2334 58,46 83,624 0,0506 0,4503 0,5009 11,95 24,30 29,3501 58,59 84,715 0,0503 0,4504 0,5007 11,95 24,30 29,3501 58,62 85,116 0,0501 0,4504 0,5005 12,00 24,30 29,2334 58,41 83,127 0,0504 0,4504 0,5008 12,00 24,30 29,2334 58,37 82,62
%KH (Karbohidrat)
Rataan : 58,50%
Standard deviasi : 0,1189
RSD analisis : 0,22
2x RSD AOAC : 2,17
2/3 RSD Horwitz :1,47
%Rekoveri
Rataan : 83,95%
RSD analisis : 1,48
Bahan acuan kedelai
UlanganGlukosa
(g)
Berat bahan
acuan(g)
W total(g)
Titer(ml)
Blanko(ml)
mg glukosa
%KH %rekoveri
1 0,0500 0,4500 0,5000 18,65 24,30 11,9340 23,87 95,582 0,0504 0,4501 0,5005 18,70 24,30 11,8260 23,63 92,653 0,0505 0,4504 0,5009 18,75 24,30 11,7180 23,39 90,234 0,0502 0,4500 0,5002 18,80 24,30 11,6100 23,21 88,755 0,0500 0,4504 0,5004 18,85 24,30 11,5020 22,99 86,816 0,0503 0,4503 0,5006 18,85 24,30 11,5020 22,98 86,337 0,0504 0,4502 0,5006 18,60 24,30 12,0420 24,06 96,90
%KH (Karbohidrat)
Rataan : 23,44%
Standard deviasi : 0,4229
RSD analisis : 1,80
2x RSD AOAC : 2,49
2/3 RSD Horwitz : 1,66
%Rekoveri
Rataan : 91,03%
RSD analisis : 4,64

68
Bahan acuan susu bubuk
(9,0066 g susu bubuk+1,0002g glukosa sebagai sampel)
Ulangan Berat sampel (g)W total
(g)Titer(ml)
Blanko(ml)
mg glukosa
%KH %rekoveri
1 0,5011 0,5011 14,50 24,90 23,7524 47,40 62,532 0,5014 0,5014 14,50 24,90 23,7524 47,37 62,253 0,5013 0,5013 14,50 24,90 23,7524 47,38 62,344 0,5012 0,5012 14,45 24,90 23,8709 47,63 64,805 0,5014 0,5014 14,25 24,90 24,3450 48,55 74,076 0,5012 0,5012 14,50 24,90 23,7524 47,39 62,447 0,5015 0,5015 14,40 24,90 23,9894 47,84 66,88
%KH (Karbohidrat)
Rataan : 47,65%
Standard deviasi : 0,4341
RSD analisis : 0,91
2x RSD AOAC : 2,24
2/3 RSD Horwitz : 1,49
%Rekoveri
Rataan : 65,05%
RSD analisis : 6,68
Hasil uji rekoveri pada berbagai sampel matriks pangan cair dengan spike glukosa
Kecap manis
UlanganGlukosa
(g)
Berat bahan
acuan(g)
W total(g)
Titer(ml)
Titer-Blanko(ml)
%KH %rekoveri
1 1,0337 4,0164 5,0501 15,75 9,30 46,17 81,962 1,0639 4,0512 5,1151 15,20 9,50 48,60 92,923 1,0031 4,1541 5,1572 14,65 10,40 50,15 104,774 1,0853 4,0417 5,1270 14,00 11,05 54,90 121,715 1,0680 4,1988 5,2668 15,55 9,50 45,16 77,406 1,0046 4,0529 5,0575 16,90 8,15 39,85 51,517 1,0335 4,1212 5,1547 15,65 9,40 45,50 79,55
%KH (Karbohidrat)
Rataan : 47,19%
Standard deviasi : 4,69
RSD analisis : 9,94
RSD Horwitz : 2,24
%Rekoveri
Rataan : 87,12%
RSD analisis : 22,32

69
Kecap asin
UlanganGlukosa
(g)
Berat bahan
acuan(g)
W total(g)
Titer(ml)
Titer-Blanko(ml)
%KH %rekoveri
1 1,0403 4,0355 5,0758 15,35 9,70 24,01 -26,22522 1,0266 4,0238 5,0504 15,25 9,80 24,34 -25,12463 1,1044 4,4452 5,5496 15,15 9,90 22,26 -36,90744 1,0538 4,0372 5,0910 15,30 9,75 24,13 -25,02295 1,0088 4,0046 5,0134 15,75 9,30 23,15 -31,67116 1,0559 4,0839 5,1398 15,05 10,00 24,49 -23,74027 1,0172 4,1365 5,1537 15,20 9,50 23,80 -29,7159
%KH (Karbohidrat)
Rataan : 23,74%
Standard deviasi : 0,78
RSD analisis : 3,30
RSD Horwitz : 2,48
%Rekoveri
Rataan : -28,34%
RSD analisis : 16,63
Santan
UlanganGlukosa
(g)
Berat bahan
acuan(g)
W total(g)
Titer(ml)
Titer-Blanko(ml)
%KH %rekoveri
1 1,0122 4,0625 5,0747 16,05 9,00 22,06 -37,74172 1,0123 4,5343 5,5466 16,10 8,95 19,65 -57,88513 1,0189 4,1624 5,1813 16,40 8,65 20,65 -45,97944 1,0646 4,3420 5,4066 15,15 9,90 22,79 -35,00275 1,1356 4,0577 5,1933 16,00 9,05 22,21 -30,49446 1,0054 4,1519 5,1573 16,06 9,00 21,50 -42,34367 1,0335 4,0895 5,1230 16,50 8,55 20,77 -43,2929
%KH (Karbohidrat)
Rataan : 21,38%
Standard deviasi : 1,08
RSD analisis : 5,07
RSD Horwitz : 2,52
%Rekoveri
Rataan : 87,12%
RSD analisis : -21,14

70
Lampiran 6. Analisis Statistik Reprodusibilitas intralab
Kecap manis
Group Statistics
Tanggal pengerjaan N Mean Std. Deviation Std. Error Mean
Karbohidrat 5 Juli 2011 3 38.7100 .68462 .39526
7 Oktober 2011 3 36.9633 .66124 .38176
Independent Samples Test
Equal variances
Levene's Test for Equality of
Variances
t-test for Equality of Means
95% ConfidenceInterval of
the Difference
F Sig. t dfSig.
(2-tailed)Mean
DifferenceStd. ErrorDifference Lower Upper
KH Assumed .000 .983 3.179 4 .034 1.74667 .54953 .22094 3.27239
not assumed 3.179 3.995 .034 1.74667 .54953 .22021 3.27312
Kecap asinGroup Statistics
Tanggal pengerjaan N Mean Std. Deviation Std. Error Mean
Karbohidrat 5 Juli 2011 3 2.2100 .05196 .03000
7 Oktober 2011 3 2.0267 .17388 .10039
Independent Samples Test
Equal variances
Levene's Test for Equality of Variances
t-test for Equality of Means
95% Confidence Interval of
the Difference
F Sig. t DfSig.
(2-tailed)Mean
DifferenceStd. ErrorDifference Lower Upper
KH Assumed 5.245 .084 1.750 4 .155 .18333 .10477 -.10757 .47424
not assumed 1.750 2.354 .203 .18333 .10477 -.20830 .57497

71
SantanGroup Statistics
Tanggalpengerjaan N Mean
Std. Deviation Std. Error Mean
Karbohidrat 5 Juli 2011 3 1.4900 .03464 .02000
7 Oktober 2011 3 1.4467 .10017 .05783
Independent Samples Test
Equal variances
Levene's Test for Equality of Variances
t-test for Equality of Means
95% ConfidenceInterval of
the Difference
F Sig. t dfSig.
(2-tailed)Mean
DifferenceStd. ErrorDifference Lower Upper
KH assumed 1.755 .256 .708 4 .518 .04333 .06119 -.12656 .21323
not assumed .708 2.472 .540 .04333 .06119 -.17721 .26388
Susu bubukGroup Statistics
Tanggal pengerjaan N Mean Std. Deviation Std. Error Mean
Karbohidrat 5 Juli 2011 7 45.7200 .42521 .16071
7 Oktober 2011 3 36.2667 .57813 .33378
Independent Samples Test
Equal variances
Levene's Test for Equality of Variances
t-test for Equality of Means
95% Confidence Interval of
the Difference
F Sig. t dfSig.
(2-tailed)Mean
DifferenceStd. Error Difference Lower Upper
KH Assumed .483 .507 29.263 8 .000 9.45333 .32305 8.70838 10.19829
not assumed 25.518 2.981 .000 9.45333 .37046 8.27020 10.63647

72
KedelaiGroup Statistics
Tanggal pengerjaan N Mean Std. Deviation Std. Error Mean
Karbohidrat 5 Juli 2011 7 16.0071 .26731 .10104
7 Oktober 2011 3 14.7300 1.04886 .60556
Independent Samples Test
Equal variances
Levene's Test for Equality of Variances
t-test for Equality of Means
95% Confidence Interval of
the Difference
F Sig. t dfSig.
(2-tailed)Mean
DifferenceStd. Error Difference Lower Upper
KH Assumed 16.729 .003 3.229 8 .012 1.27714 .39558 .36493 2.18936
not assumed 2.080 2.112 .166 1.27714 .61393 -1.23420 3.78849
Kacang hijau
Group Statistics
Tanggal pengerjaan N Mean Std. Deviation Std. Error Mean
Karbohidrat 5 Juli 2011 7 55.6557 .28023 .10592
7 Oktober 2011 3 55.7867 1.68049 .97023
Independent Samples Test
Equal variances
Levene's Test for Equality of Variances
t-test for Equality of Means
95% ConfidenceInterval of
the Difference
F Sig. t dfSig.
(2-tailed)Mean
DifferenceStd. Error Difference Lower Upper
KH assumed 25.177 .001 -.217 8 .834 -.13095 .60352 -1.52268 1.26077
not assumed -.134 2.048 .905 -.13095 .97599 -4.23766 3.97575