polymer-metal complexes as catalysts - inflibnet
TRANSCRIPT

CHAPTER 1
POLYMER-METAL COMPLEXES AS CATALYSTS

Polymer-meld Complexes as Caldysts --
8
n a polvmeric- c-atalyst or polymer-supported catalyst a conventional
catal\,tic syccles IS attached to a macromolecular backbone. Natural and I synthetic polymers have been widely used as carriers of bioactive molecules
for modelling, e.g., enzyme catalysis.' Despite the' fact that they usually show
a much lower c,atalytic activity than a true enzyme,2 they proved to be
valuable in understanding some basic features of enzymic processes and in
the developing of other types of synthetic polymers. An important class of
these model compounds is that formed by transition rnptal ions or complr\
ions anchored to a polymer. When the matrix in polypeptide, the
corresponding polymer-supported complexes are supposed to mimic the
activity of metal lo enzyme^.^,^ The preparation and investigation of chemically
active species bound to polymeric supports continue to be an active
interdisciplinary field of research with applications in organic chemistry,
inorganic chemistry, chemical technology, biochemistry and biology." A
biopolymer catalyst, such as enzyme, exhibits high selectivity and activity for
several reactions. It is well known that their specificities are caused bv an
intramolecular k,ooperative catalysis between functional groups in the
enzyme. The use of polymer as supports for chelates and catalysts, as well as
synthetic reagents, has grown tremendously since Merrifield demonstrated
their use in peptide synthesis. The catalytic activities of metal complexes
bound on polymer matrices are entirely different from the corresponding
monomeric analogue due to the specificities induced by the macromolecular
ligands." a polymer-metal complex, the polymer support provide a specific
catalytic site and act as an effectivematrix for the formation of active centres9
The catalytic activity of a polymer-metal complex is generally lower
than the corresponding monomeric analogue due to steric effects. However,
the activity can sometimes be higher than the monomer complexes due to the

unsaturated coordination structure or by the strain in the formation of
inactive dimeric complex due to steric effect.10," Polymer-metal complexes
are widelv used as immobilised catalysts, in which a specific catalytic
behaviour was induced by the polymer matrix.12 In addition to this catalytic
activity, polymeric ligands have immense application in various areas like
cond~ct iv ih i ,~ '~~ ' modification of material surface by binding of metal
c~mplexes ,~"~^ separation of metal ions1820 and in the mimicking of
bio-inorgan~c s v s t e m ~ . ~ ~ ~ ~ ~
2.A. Polymer-metal Complexes: Synthesis and Classification
Recently there has been growing attention to the molecular function of
macromolecular complexes as a new frontier in material systems.
Macromolecular complexes are defined as complexes of macromolecules. A
typical example of macro molecular^ metal complexes is illustrated in
Figure 2.1. Their synthesis represents an attempt to give inorganic function to
organic polymer
Figure 2.1. Schematic illustration of macromolecular metal complex

Polymer-mefd Complexes as Ca'dysts 10
Intermacromolecular complexes and molecular complexes between
macromolecules and organic molecules are also included in the category of
macromolecular complexes. Recause the macromolecular complexes are
combination of macromolecules, small organic molecules and metal ions a t
the molecular level and because they are structurally labile, there are
unlimited possibilities for providing a wide variety of previously unknown
molecular funcbons.
In the macromolecular complexes, physicochemical properties and
chemical reactivities of the complex moieties are often strongly affected by
interactions with the surrounding polymer matrix. These interactions are
weak but significant and act multiply and dynamically. They not only
construct the rnacromolecular complexes themselves, but also control their
higher-order structure based on a dynamic conjugation between the complex
moieties and the polymer matrices. Thus a weak and soft profile in the
coordination reactions, a cooperative interactions behveen the complex
moieties and a multiplied or enhanced interactions are observed as
characteristic features of the macromolecular complexes.
Polymer-metal complex is formed by the interaction of a polymer
containing coordinating groups with metal ions. The polymeric ligand can be
obtained by the polymerisation of monomer containing coordinating sites or
by the reaction between a polymer and a low molecular weight compound
having coordinahng ability. The general methods for incorporating active
functional groups are classified into four different groups: (i) direct
polymerisation or copolymerisation of monomers containing the desired
functional groups; (ii) chemical modification of preformed polymer; (iii)
functional monomer idubition into or grafting onto the prepared polymer,
followed by polymerisation; and (iv) immobilisation of hydrophobic
chelating extractants during polymerisation or post-polymerisation.

, , Polymer-mefal Complexes as Caldysts i , . 1 1
, ' 7, 1 ,, T' , : .
2.A.I kndant complexes .- . .~~
In pendant complexes the metal ion is attached to the ligand function
which is appended on the polymer chain. Based on the chelating abilities it is
classified as monodentate and polydentate pendant complexes.
(a) Monodentate pedant contpkxes
A monodentate pendant complex is formed when a metal ion or a
stable metal complex in which central metal ion is already masked with low
molecular weight ligands except for one coordination site that remains vacant
is coordinated to a polymeric ligand. Such complexes have a simple structure
of the monodentate type as shown in Scheme 2.1. Even if the metal ion or the
metal complex has more than two labile ligands, it is often possible to form a
monodentate complex by selecting appropriate reaction conditions.
I I 1 &6pfJ Scheme 2.1. Monodentate pendant complexes
An example of the formation of a pendant monodentate complex is the
complexatton of < IS-[Co(en)zXz]X with poly(4-vinyl pyridine) (l).Li

Polymer-meld Complexes as Caldysls 12
(1) X = C1, Br, N3 en = ethylenediamim
Polydentate ligands often result in the formation of stable metal
complexes with bridged structures. Most of the chelating resins come under
this category. Pendant polymer.-metal complexes are characterised by their
relatively well-defined coordination structure. Structure of the polydentate
pendant metal complexes is given in Scheme 2.2.
Scheme 2.2. Polydentate pendnnt metal compleves
2.A.2 Inter- and intra-molecularly bridged cpmplexes
In the case of the complexation of a polymeric ligand with a metal ion,
which has four or six coordinating binding hands, the polymer-metal
complex formed may be of the intra-polymer chelate type or inter-polymer
chelate type (Scheme 2.3).
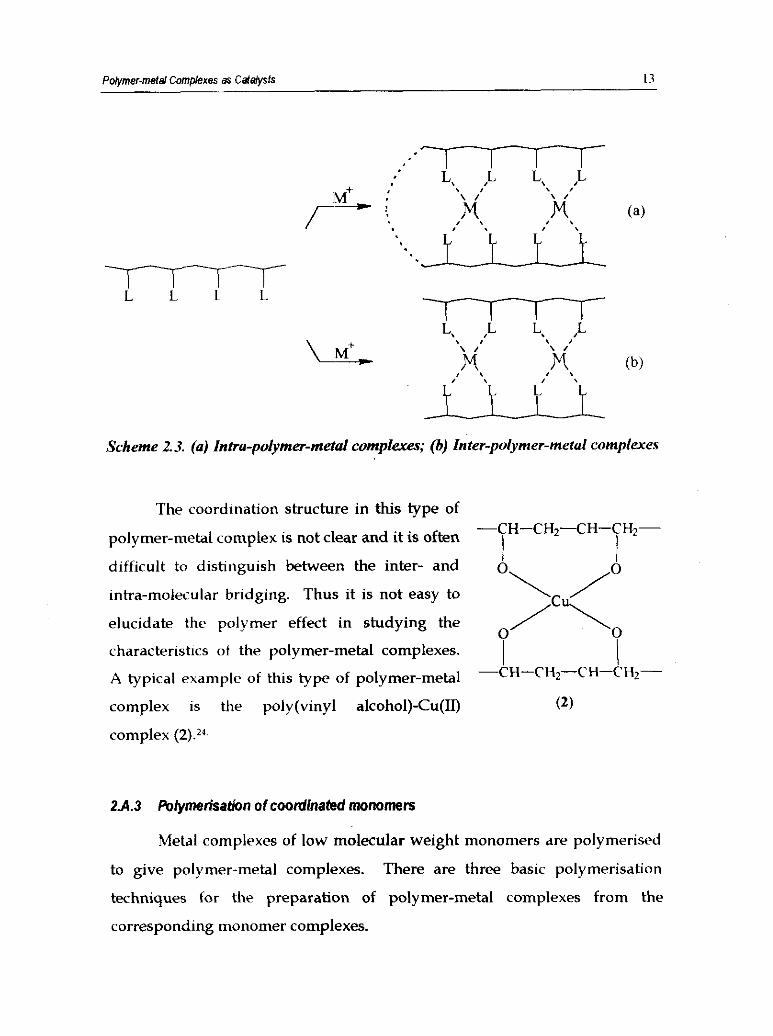
Pdymer-meld Complexes as Caldysfs 13
Scheme 2.3. (a) Intra-polymer-metal complexes; (b) Inter-polymer-metal complexes
The coordination structure in this type of -CH-CH2-CH-CH2- polymer-metal complex is not clear and it is often
difficult to distinguish between the inter- and 1 I
intra-molecular bridging. Thus it is :not easy to "\ / elucidate the polymer effect in studying the
0 characteristics ot the polymer-metal complexes.
/c\o
A typical example of this type of polymer-metal I
-CH-CH2-CH-CH2-
complex is the poly(viny1 alcohol)-Cu(I1) (2)
complex (2).24.
2.A.3 Polymerisation of coordinaled monomers
Metal complexes of low molecular weight monomers are poly merised
to give polymer-metal complexes. There are three basic polymerisation
techniques for the preparation of polymer-metal complexes from the
correspond~ng monomer complexes.

Pdyrner-metd Complexes as C&+sls 14
(a) Polyn~erisatiotr of vinyl cmirpounds cmitaitritig metal cmtrplexes
The vinyl monomers which can be polymerised are limited and the
polymerisation depends on the electrostatic interaction between the
coordinated monomers and the growing radical. This type of polymer-metal
complex is characterised by a clear coordination structure. The
polymerisation occurs by the radical of ionic initiation to form a polymer of
high molecular weight as depicted in Scheme 2.4.
Scheme 2.4. Polymerisation of coordinated vinyl monomcr.v
Some transition metal salts and model complexes act as inhibitors in
the polymerisation of vinyl monomers. Thus the polymerisation of 4-vinyl
pyridine is strongly inhibited in the presence of Cu(I1) or Fe(IIIJL5 But Zn(II),
Co(I1) or Cu(I)-4-vinyl pyridine complexes can be polymerised to form the
corresponding poly(4-vinyl pyridine)-metal complexes.26 Methacrylate
coordinated with amine-Co(I1I) c:omplexes to form methacrylato-pentamine-
Co(II1) perchlorate (3) and cis-din~ethacrylatotetramine-Co(II1) perchlorate (4).

(b) Polycortdeirsation of ~ le ta l compkxes corltairrirrg pee jurlrtiornl groups
'The prepdratlon of polymer-metal complex by condensation is
reported by Hartwell and Bailer (Scheme 2.5).27 Polyesters and polyamides
have been synthesised by interfacial polycondensation with aryl acid
chlorides.
Scheme 2.5. Polycondensation of low moleculur weight metul complexes
The polymer-metal complexes obtained by polycondensation and ring-
opening polymerisation are considered as coordination polymers since the
polymer chain is composed of coordinate bonds. This involves the
polymerisation of a metal complex containing a strained ring which is
capable of ring-opening polymerisation as shown in Scheme 2.6. An example
of this type of polymer-metal complex formation is the polymerisation of
beryllium complex of bis-P-carbonyl compound (Scheme 2.7).26
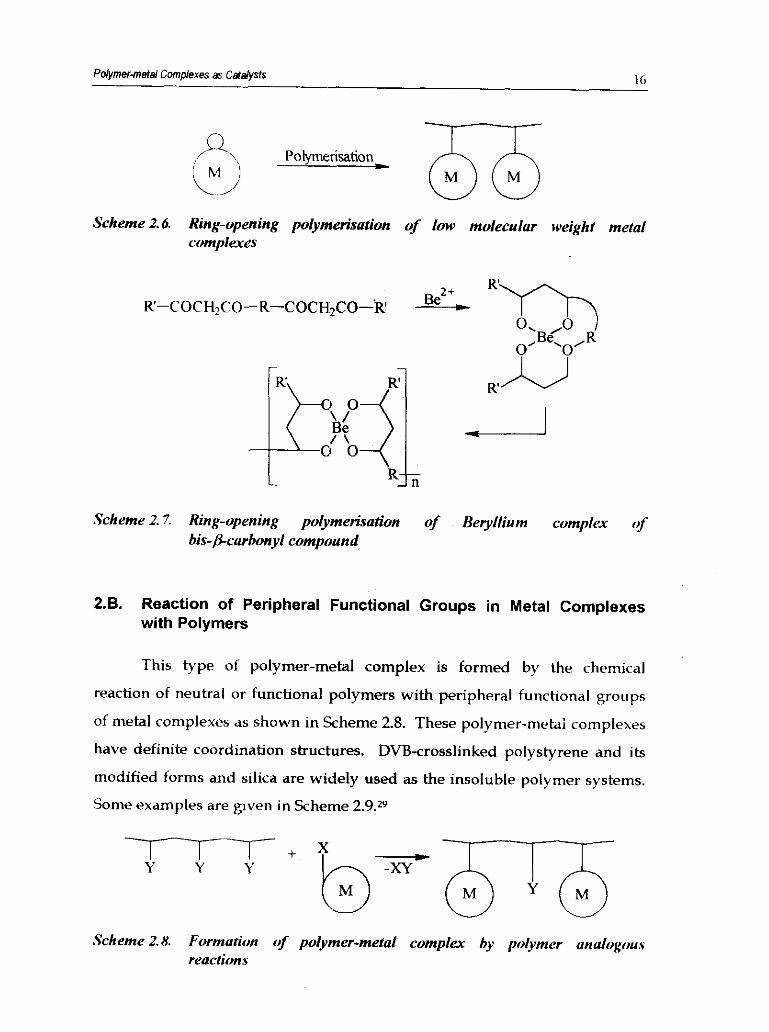
Polymer-metal Complexes as Ca(dys1s I6
Scheme2.6. Ring-opening polymerisafion of low molecular weight metal complexes
Scheme 2.7. Ring-opening polymerisation of Beryllium complex of
bis-gcarbonyl compound
2.B. Reaction of Peripheral Functional Groups in Metal Complexes with Polymers
This type of polymer-metal complex is formed by the chemical
reaction of neutral or functional polymers with peripheral functional groups
of metal complexes as shown in Scheme 2.8. These polymer-metai complexes
have definite coordination structures. DVB-crosslinked polystyrene and its
modified forms and silica are widely used as the insoluble polymer systems.
Some examples are given in Scherne 2.9.29
Scheme 2.8. Formation of polymer-metal complex by polymer analogous reacn'ons
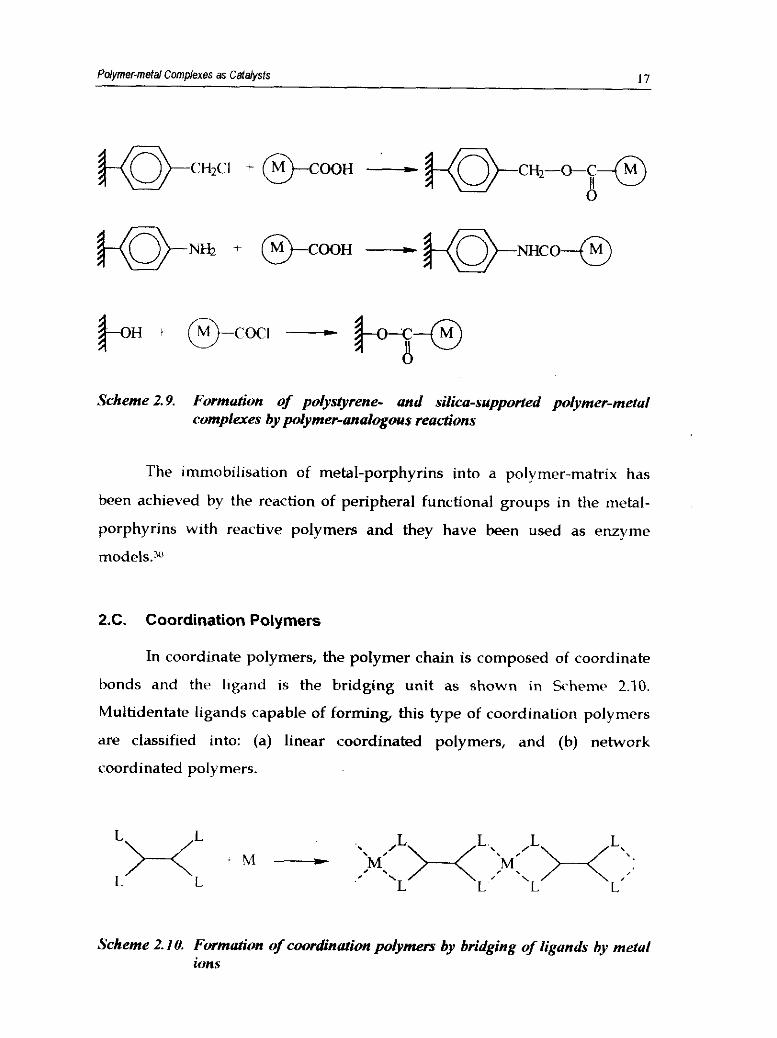
Pdymermefal Complexes as Catalysts 17
Scheme 2.9. Formation of polystyrene- and silica-supported polymer-metul compleres by polymer-analogous reactions
The ~mmobilisat~on of metal-porphyrins into a polymer-mahix has
been achieved by the reaction of peripheral functional groups in the metal-
porphyrins with reactive polymers and they have been used as enzyme
mode1s.N'
2.C. Coordination Polymers
In coordinate polymers, the polymer chain is composed of coordinate
bonds and the l~gand is the bridging unit as shown in Schemr 2.10.
Muttidentate ligands capable of forming, this type of coordination polymers
are classified into: (a) linear coordinated polymers, and (b) network
coordinated polymers.
Scheme 2.10. Formation of coordination polymers by bridging of ligunds by metal ions

Powmer-meld Complexes as Cdalysts 18
2.C. 1 Linear coordinated polymers
Linear coordinated polymers can be prepared by two methods, from
bifunctional ligands and from simple compound or ion as ligands.
IIerr the 1)olvrner chain is composed of bifunrtional ligands and metal
ions. A typical example for the coordination complex formed between a
bifunctional ligand and metal ion is the complex between dithiooxamide and
metal ions (Scheme 2.11).31
Scheme 2.11. Iormurton cf net~vork-courdinuied polymer-tneful complex (4 dithiooxamide
(b) Sitrrple cmtrpouird or ion us ligands
A simple compound or ion can function as a bridging ligand giving
rise to a polymeric structure. Cupric chloride forms an associated structure in
which the chloride occupying as bridging ligands on both the axial and
equatorial coordination sites of <3u(II) (5).32

Polymer-meld Complexes as Caldysts I9
This type of polymer-metal complex is formed by a 'template reaction'
between two functional groups of the ligands induced by their coordination
to metal ion resulting in a chelate type metal complex as depicted in
Scheme 2.12. Poly(methy1 phthalocyanine) is an example for this type of
network-coordinated polymers.. It is formed by the reaction of
tetracyanobenzene with metal halides (Scheme 2.13).
L-L
Scheme 2.12. Schematic representation. of the formation (4 nehvork-coordinated polymers
Scheme 2.13. Formation of network-cwrdinated polymer-metul complex o j tetracyanobenzene

2.D. Catalytic Activities of Polymer-metal Complexes
Catalytically active polymers can be obtained by introducing a
catalytic centre to a polymer backbone and it is reasonable to assume that the
catalyst bound to the polymer will show specific catalytic activity, reflecting
the properties of polymer chain. In the case of metalloenzyme such as
oxidase and haemoglobin where a metal complex is the active site, the
macromolecular protein is that which plays a significant role. Polymeric
catalyst reduces the possibility of catalyst poisoning since the catalytic site is
somehow protected by the polymer matrix. In a polymer-metal complex,
aggregation is phvsically prevented by the rigidity of the polymer matrix and
has the advantage of maintaining its catalytic activity over a wide range of
concentration. In polymer-metal complexes, the selectivity arises from the
steric hindrance and/or chemical environment of the polymer matrix."
Polymer-metal complexes are markedly useful as immobilised catalyst
for practical use because it is more reactive than the corresponding monomer
analogous due to the specificities of their large ligand molecules. It is mainly
used in oxidations,j5 hydroge~~ation,~~ hydro ly~is ,~~ hydr~formylation,~
decomposi&n of H202," initiation of radical polymerisation,~l' asymmetric
synthesisll and optical resolution.~2
2.D.1 Catalytic activity of polymer-metal complex
The catalytic cycle of a metal complex catalysed reaction is
ML, + !j + L,.I M-S
M'L + S* * LI M'S*
M - metal ion; L - ligand; S - substrate

In the first step substrate coordinate to a metal catalyst and form
intermediate mixed complex (LMS). The substrate is activated by metal ion
and dissociates from the catalyst, having accomplished its purpose, is
regenerated to the original complex.
The main characteristics of polymer supported catalysts are:
(1) Polymers can be used as insoluble carriers for catalyst. The domain in
the solid phase or the boundary of its surface provides a specific
catalytic stte. The isolation of the products from the catalyst is
markedly facilitated and the catalyst can be used repeatedly by a
simple procedure.
(2) Though the activities of the polymer-metal complexes are sometimes
lower than those of corresponding monomeric analogous, due to steric
lundrances, they can sometimes be higher than the monomer
complexes. For instance, unsaturated coordination structures or sterical
prevention of the formation of an inactive dimeric complex will
enhance the catalytic activity.
(3) Selectivities of the reaction by polymer complexes are often different
from the monomer complexes.
(4) Polymer can be used as an effective matrix for the formation of active
metal parbcles.
2. D.2 Application of polymer-metal complexes as catalysts
Oxidation of hydroquinone catalysed by C u ( v complex of
poly(1-vinylimidazole), vinylamine-vinyl acetarnide copolymers and
polyethylene imine were s t~died.4~ The poly(1-vinyl imidazole) complex
showed catalytic activity 20 times higher than cupric ions. The oxidation
mechanism can be represented as:

Polymermeld Complexes as Calalysls 22
Oq f u(II) complex JCH H2Q
0 2 C u(1) complex % Q
(H2Q: hydroquinone; Q: quinone)
Dehydrogenation of alcohols catalysed by the solid phase copper
complex of poly(e carbobenzoxy-L-lysine) was carried out at 150-250°C.44
The advantages of polymer complexes were:
Selectivity for the substrate due to steric effect of polymer ligand was
observed.
The metal ions are monodispersed monoatomically giving high
catalybc effic~ency.
The synthetic procedure is excellent on account of the insolubility of
the polvmer catalyst.
In the autooxidation of cumene45 H catalysed by the metal complex of ,,,,,,,
polymer-supported Schiff base, the
activity decreased in the order: Mn > co > . Ni > Cu > Zn and decreased with
increasing molecular weight of the Schiff base. In this example, the polymer
complexes showed lower activity than the complexes of the monomeric Schiff
bases, probably due to steric reasons.
Polymer complexes having more than two kinds of active sites are
found to have high catalytic activity. In this example, polymer-bonded
metalloporphyrin whch contains both an oxidation site (Co) and a proton
acceptor site (amine) are effective catalysts for the oxidation of thiol to
disulfides.46

Since the catalytic hydrogenation proceeds via a mixed complex of
catalyst and substrate, at least one coordination site of the catalyst must be
free or labile in order that the substrate be coordinated to the catalyst Such a
free or labile coordination site, so called "unsaturation" can be realised in a
polymer-metal complex due to steric effects. The Pd complex of the diphenyl
phosphinated polystyrene showed
remarkably high catalytic activity for CH2 P(Pb- -h--..PdC12
the hydrogenation of the methyl
ester of soybean fatty acid and the
monomer~r complex PdC12(PPh7)2.47 The polymer metal complexes mainly
used for hydrogenation are Rh, Ru or Ni carbonyl complexes of biphenyl
phosphonated supported on polystyrene.

The polvmer complex of titanocene A <
dichloride showed 15-120 times high
catalytic activity for the homogeneous
hydrogenation of olefins than the
monomeric titanocene dichloride.*"
Selective hydrogenation of olefins
catalysed by polymer-metal complexes PPh2-----RuClz(CO)z
was observed. The diphenyl
phosphinated polystyrene-Ru complex or
diacetyl iminomethylated polystyrene-Pd CH2N(CH2COOhPd
complex catalysed the hydrogenation
of octadiene giving selectively
cy c lo~c tene .~~
TIus is because the rate of hydrogenation of the diene to rnonoene is
much higher than that of the morloene cyclooctane.
Recent interesting works are the use of polymer as matrix for the
dispersion of metal particles. A colloid has been prepared by reduction of
Rh(III)-chloride with methyl alcohol in presence of PVA.9 The colloidal
dispersion of (Rh) in methyl alcohol-water solution was effective fur Lhe
hydrogenation of olefins at 30°C: under an atmospheric hydrogen pressure.
The colloidal dispersion contains rhodium particles of remarkably small size
of 8 and 40 A.51 The reactions were considered as like this:
RhCL A Rh(II1)-PVA complex CHJOH (reductiotl)
Rh particles (diameter: 8 A) (growk!) +
Rh particles (diameter: 40 .A)
Porphyrin containing polymers were found to be remarkably effective
as a catalyst in photo redox systems.52 In such systems, various porphyrin
groups such as protoporphyrins and chlorine as well as metalloporphyrin

Polymermet# Complexes 6s Caldysts 25
groups such as Mg(II)- and Cu(Q-chlorophyllin and Fe(IIl) and Co(1I)
protoporphyrin were incorporated into vinyl polymers.
(c) Hydrolysis
The hydrolysis of sodium pyrophosphate was effected by using
somemetal complexes of poly(methacry1acetone) as catalysts.53 The
catalytic activity of the polymer complexes declined in the following order:
Zr(IV) 0 > U(V1) 0 2 > Cr(m) 2 Ce(1II) E Cu(II). The enhancement of activity
was ascribed not only due to the electrostatic effect of the polymer complexes
but also to a possible multiplying effect by ZrOL+ and ZrOL(0H) along the
polymer-ligand chains. The catalytic
hydrolysis of oligophosphates by
poly(L-Lysine)-Cu(Q complexes has
been reported." Breslow et ~ 1 . ~ ~ \ ' ~i
attached the Ni(1I) complex to , . ,
cyclohexaamylose, which formed a
hydrophobic cavity, as shown as (6), @:"-
and studied the hydrolysis of p-nitro ( 6 )
phenyl acetate. The reaction was
accelerated by a factor of about 103 over the uncatalysed system, the increased
reactivity being the result of binding of the substrate in the hydrophobic
cavity.
Hydroformylation is the reaction of olefins, carbonmonoxide and
hydrogen producing saturated aldehyde or alcohols. Insoluble catalysts of
polymer-metal complexes are remarkably useful for this reactions, since they
are easily separated from the products and used repeatedly. They sometimes
show selectivities for the reaction due to the polymeric structure.

Polymer-metal Complexes as Catalysts 26
L-+R& I C,
CHO
The complex of [Rh(C0)2Cl]z bound to the crosslinked polystyrene
containing the -P(Ph)z, -N(CIt)z or -CHzN(CEt)z group catalysed the
hydroformylation of 1-hexane.M The polymeric phosphine-Rh complex
showed selectivity for the aldehyde synthesis and the polymeric amine-Rh
complex, selectivity for the alcohol synthesis. The complex of RhCI(C0)
bound to phosphinated silica gel coated with polystyrene was a stable
heterogeneous catalyst for the hydroformylation of ethylene.57
The cobalt resin derived from a crosslinked polv(chloromethylated
styrene) is a good hydroformylation catalyst for 1-pentene and cyclohexene
under mild condition.%
Catalyst recovery can be achieved by simple filtration and its catalytic
activity is retained after prolonged exposure of the resin to air. Such
polymer-supported insoluble catalysts are especially suitable for commercial
process, because expensive catalyst can be recovered and used repeatedly by
simple procedures.
(e) ltiitiation of radical polyiletisation
Polymer-metal complexes initiate radical polymerisation and often
show higher activity than the corresponding monomeric analogous.
Nylon-Cu(1I) systems initiate the radical polymerisation of methyl
methacrylate (MMA) in presenre of CQ and water.59 The study of

Polymer-metal Complexes as Cefdysfs 27
polymerisation by Cu(Il) complex of (E amino caproic acid) and other
oligomeric nvlon showed the increase of the catalytic activity with the
increase of the degree of polymerisation of the nylon. This in fact, the low
activity of the N-substituted oligomer, and the importance of the end amino
group for this catalysis suggested the mechanism given below."
Natural productsb' such as cellulose and soluble starch were found to
initiate the polymerisation of MMA in aqueous solution in presence of CuZ'
ions, probably through the complex formation of CuZ+ with the natural
polymer.
(f) Asymrtretric synthesis and optical resolutiorr
Steric hndrance of polymer ligand can provide T
asymmetric structure whch bring about asymmetric
reaction.
Hateno (1969, 1970) and Nozawa (1971)
succeeded in carrying out an asymmetrically selective H---.
oxidation of 3,4-dihydroxy phenyl alanine DOPA by
poly(l1ysine) (PLL)-Cu(I1) complex."@ D-DOPA was
6" &H
Ph2P PPh2 found to be oxidised by the PLI,Cu(II) complex more
rapidly than L-DOPA. Such an asymmetric oxidation
was ascribed to this asymrnelrically selective interaction of DOPA with PLL-
Cu(II) complex. The combination of two or more asymmetric cupric complex
units held on the a-helical structure of PLL was considered to cause the
asymmetric catalysis.

Rh complex of diphosphine compounds bound to polystyrene
catalysed the asymmetric hydrogenation of ketones such as acetophenone,
methyl benzyl ketone with the optical yield of 58%.62
Asymmetr~c structure provided by polymer-metal complexes makes
optical resolution of the adsorbed organic molecules. The Cu2+ complex of
the polymer containing N,N-dibenzyl leucine adsorbed D-isomer of amino
acids more than L- i~omer .~~ The skreoselectivity was opposite to the
complex of the monomeric analogue. The crowded structure composed of
isobutyl and benzyl groups would cause selective adsorption of D-isomer
(Scheme 2.14).
Scheme 2.14. Selective adsorption of D-valine by ihe crowded copper complex of N, N ' -d iben~l leucine polymer

The decomposition of Hz02 is effected using various metal complexes
as catalysts. 'The catalysis has its origin in redox action of a metal complex. A
transient intermediate is formed between the metal catalyst and the substrate,
at least in the initial step of the reaction, but the reaction also proceeds
through chain decomposition. 'The decomposition of hydrogen peroxide is
often employed as a standard reaction to determine the catalytic activity of
polymer-metal complexes because the experimental technique is simple.
Polymer metal complexes exhibit high efficiency in the catalytic
decomposition of H202 because:
(i) Some polymer-metal complex contain incomplete complexes due to
steric hindrance and increases catalytic activity.M.65
(ii) In other polymer complexes, the coordinate bond between polymer
ligand and metal ion is relatively weak and substrate coordinate with
high frequencv."
(iiif Chain decomposition of substrate proceeds rapidly because
concentration of catalytic site is locally enhanced.fJh9
The decomposition of Hz02 have been studied as a catalase model
rather than as a catalyst of practical use. Polymer-metal complexes showed
much higher catalytic activity for the decomposition of H202 than the
corresponding monomeric analogous."R
The catalybc activity of Fe(1II)-triethylene tetramine (trien) complex
was found to be lo7 times higher than that of ferric aqueous in the
decomposition of H202.70 The mechanism was proposed where the OOH- ion
coordinates to ferric ions as a bidentateligand, resulting in the cleavage of the
(0-0) bond with the evolution of oxygen.

Poly(P-diketone)-Cu complex and -CH- CH2-
poly(hydroxamic acid)Cu complex showed higher
catalytic activity than the monomeric catalyst.71 I
NH \
The high activity of polymer complexes is // ascribed to the weak coordination bonds between
\ the polymeric ligands and metal ions. A higher
"\ /" molecular weight of the polymer ligand gave higher /""\ activity. Polymethacrylate acetone-Cu, polyglycine- \ P Cu72 and ferric complexes of polyacrylic acidn also \
NH- //
C showed h g h catalytic activity for the decomposition I of H202.
-CH2- CH- '
It was postulated based on the action of catalase that Hz02 should be
activated prior to being decomposed by the enzyme's hemine.74 According to
this mechanism, one carbonyl and one amino group of the apoenzyme were
believed to play a role of the cofactor. These groups forms hydrogen bonds
with one molecule of HzOz which is thereby activated and the second
molecule of Hz02 forms a complex with the first one giving rise to the fission
of 0-0 bond. As for the model of such mechanism the decomposition of
H202 by F++ complex of poly(acrylic acid), which is partly amidated by
diethylene triamine (diene) was studied."

Potfmwmefsl Complexes as Ca!@sts 11 (>> 'd
H.. - P A - - 4
2.E. Polymer-metal Complexes as Heterogeneous Biologically Active Catalysts
When a homogeneous catalyst such as a transition metal complex,
transition metal or an enzyme is covalently bound or strongly absorbed on a
polymer matrlx we have a polymer-supported catalyst. The polymer can be
organic or inorganic in nature and be soluble or insoluble in the reaction
medium. Insoluble supports are preferred because the main purpose of
supporting a catalyst is to be able to recover and recycle it easily. The
advantages apart from recovery and &use are: (a) isolation of catalytic sites
and prevention of agglomeration leading to inactivation, and (b) coordinate
unsaturation introduced by the polymeric matrix resulting in enhanced
specificity.
There are three distinct stages in the preparation of a supported
catalyst. The first is the choice of suitable support material. The second is the
introduction of suitable functional groups on the support which would
interact with the catalytic species in the loading process. The choice of such
functional groups depends to a large extent on the catalyst chosen. The
incorporation of the catalyst on the support is usually carried out under mild

conditions so as to retain the activity of the catalyst and finally, the extent of
the loading acheved must be ascertained by chemical analysis.
Metal ions have an important role in the activity of bioinorganic
materials in whch metal ions are bound to proteins, nucleic acids and related
ligands. Metalloenzymes are generally formed between a polypeptide and
metal ion. The coordination structure of the complex, the conformation of the
polypeptide which is dependent on the sequence of the amino acids in the
polypeptide, stiffness of the backbone and the interaction between the
pendant groups cause specificities in metalloenzymes.76 The presence of
many kind of b~ological substances having coordinating ability around the
metal ion and the occurrence of reaction by metalloenzymes via the mixed
complex with the substrate requires to study of fundamental coordination
chemistry of mixed ligand complexes in order to elucidate the coordination
behaviour of blood plasma, the complex formation of Mg(II), Cu(II), Mn(II),
Fe(III), Co(II), Zn(II) and Pb(II) ions with amino acids and other organic acids
are s i m ~ l a t e d . ~
In metallic enzymes, the large protein ligand decides the character and
the activity of the central metal ions by providing a selective binding site for
the substrate. This selectivity is generated by the long chain of apoproteins.
A distorted coordination structure was developed for the complex by the
hydrophobic environment created by the protein molecule surrounding the
central metal ion. In blue-copper proteins the coordination geometry of the
central copper ion is not square planar but shows a distorted tetrahedral
structure which is caused by steric strain of the peptide chain. The
coordination structure of its oxidised and reduced states are very similar due
to the rigid stereostructure around the Cu(1I) ion. Such a rigid structure is
retained by a specific configuration of the polypeptide chain. The Cu(1I) ion
itself is surrounded by hydrophobic groups. Such a distorted structure and
hydrophobic environment caused by the polypeptide chain are largely
responsible for the anomalous characteristics of blue-copper proteins.

The chem~cal approach to peptide synthesis has yielded impressive
successes in recent years in the preparation of several biologically active
pep tide^.'^ With the development of new reagents and techniques the
synthesis of most small peptides has been placed within easy reach.
Insoluble polymeric supports enable separations of synthetic reactions
mixtures by simple filtrati0n.~9 They permit use of large excess of reagents in
peptide synthesis to drive the coupling.reactions to completionsU and provide
easy separation of troublesome byproducts from reactions mixtures. Another
advantage of polvmeric supports sometimes cited is "site isolation" by which
supposed that the rigid polymer chains prevent polymer-bound species from
reacting with one another, thus favouring either intramolecular reaction or
reactions with reagents in solution rather than interchain reactions within the
polymer m a t r i ~ . ~ '
Considerable attention has been focused on the preparation of
immobilised enzymes in the past decades, and many different kinds of
support matrices and techniques for enzyme immobilisation have been
d e v e l ~ p e d . ~ ~ - ~ ~ Immobilised enzyme systems can have a wide range of
potential applications in enzyme engineering, among which are enzyme
bioreactors and enzyme electrodes. Such systems have several advantages .. .
over the free enzyme including their ease of recovery and reusability,
operational simplicity and thermal and storage stability. Among the support
matrices used, polvmeric materials have been used extensively since they can
have various functional groups and can be easily modified chemically.
The immobilisation of enzyme on a macromolecular support can in
principle mimc the above conditions to some extent. The irnmobilisation of
enzymes on insoluble polymeric supports facilitates their recovery from the
reaction mixture, but it can also slow down enzyme deactivation and make
possible the exploitation of local concentration of active site.%,S9 The
characteristic effect during immobilisation of enzymes on crosslinked
~nsoluble polymer~c support is the steric effect imposed by the three-

dimensional crosslinked matrix.*,9' This often reduces the overall activity of
the enzyme. The influence of three-dimensional macromolecular matrix on
enzyme activity can be reduced by incorporating a spacer arm between
polymer matrix and en~yme.~z The immobilisation also provides an
opportumty of reutilisation. The long term stability and simple recovery of
immobilised enzymes guarantee their multiple reuse. The autodigestion of
proteolytic enzymes often leads to inactivation. In such a system,
immobilisation Increases stability as the covalent binding of enzyme
molecules to the carrier can prevent their inactivation with each other.
2.F. Macromolecular Effects on Catalytic Activities of Polymer-metal Complexes
2.F.7 Porosity and surface area
During the last few decades, heterogeneous catalytic processes have
been developed to such an extent that they constitute the main process routes
to the bulk of the output of the chemical industry. The pore-size and surface
area of the polymers varied with the nature and extent of crosslinking agent.
Optimum crosslinked polyme~ic ligands should have large surface area and
average pore-radius. Considerable insight . . has been obtained on the chemical . r
characteristics of catalytically active species and it has been well correlated
that the reaction rate increases with the exposed area of the catalytic species.
In order to bbtain reasonable reaction rates, such area has to be increased to
an adqua& level per unit volume and this is possible only by making the
catalyst htghly porous and spreading the active species over the entire surface
whch would be largely in theinterior of the catalyst pellet. Thus the internal
areas are accessible to the reactants by diffusion only.
In the case of catalytic reaction taking place in the interior surfaces of a
porous catalyst, frequently, gradients in reactant concentration and
temperature may be present inside the catalyst making the interpretation of
experimental or operational data difficult The development of a catalyst

formulation should therefore take into account not only by the chemical
nature and dispersion of the active species but the design of the catalyst
should also consider factors such as the average pore-size, the variation in
pore-size and adequate mechanical strength.
The characteristics of the pore structure also influence greatly the
response of catalysts to poisons. In general, poisons are chemical constituents
which are very strongly absorbed by the active catalytic surface. Certain
catalytic transformations are associated with consecutive reactions or side
reactions and a catalyst with high selectivity is essential to minimise
undesired products. In the hydrogenation of butadiene to butane, and the
oxidation of o-xylene to anhydride, further degradative reactions can occur
on the primary product which is the desired one. However, the diffusion
limitation during regeneration can be minimised by having macro-micro
combinations of pore-sizes. Such a catalyst will have a network of larger
sized pores which have a lower active surface but facilitate rapid diffusion of
the gas inside catalytic part as well as a network of smaller sized pores of
much shorter d~mensions which incorporate the bulk of the active catalvtic
species.
LF.2 Natum of support and extent of cmsslinking
The complexation behaviour of ligand function supported on polymer
is generally Wfeqent from the corresponding low molecular weight
.. analogous.y3 This variation is dependent on the polymer matrix to which the
ligand function is attached." Complexing groups in a macromolecular ligand
may adopt conformation different from that required by the preferred
stereochemistry of a given metal ion. Functional groups are strategically
distributed over a macromolecule so that both the more favourable and the
less favourable conformation will occur. The characteristic factors like matrix
structure, matrix configuration, matrix nature, spacer between the crosslinked
matrix and the iigand function and extent of crosslinking has a significant

effect on complexahon. The complexation characteristics of insoluble
macromolecules are strongly influenced by the nature and extent of
crosslinking in the polymer l i g a ~ ~ d . ~ ~ A high degree of crosslinking results in
a low degree of a metal ion intake and a lower stability of the resulting metal
complex by mak~ng the polymer chain more rigid. In the case of poly(4-vinyl
pyridine)-Cu complexes with increasing divinyl benzene content, conversion
of planar coordination centres to tetrahedral ones occurs. This change in
stereostructure results from an increase in steric hindrance to complex
formation wlth a polymeric ligand.
2.F.3 Spacer groups
The complexing power of macromolecular ligand depends upon the
arrangement of functional groups relative to the main chain. The shorter
distance between them, the lower the efficiency of complex formation because
of steric hndrance. By introducing a spacer to polymer matrix the less
reactive functional groups can make more reactive cause high catalytic
activity.
Functional groups directly attached to the backbone chain of high
polymers are often less reactive than if these groups were not attached to a
polymer. Ths feduced reactivity is caused by steric hindrance or by the fact
that a 1 or part of the polymer chains interferes with the reactivity of the
individual Gnctional group. Spacer groups could be designed to be flexible
or stiff, a most important capability to design macromolecular architecture by
modern polymer synthesis. The commonly used spacers are hexamethylene
dimnine,% polyethylene glycol" and a,0-dibromoalkane.9~ If the functional
groups initially present in the polymer are not sufficiently isolated, the spaces
may become doubly coupled with the polymer and is effectively lost. Such
reactions have been used to study site isolation.

Polymer-mefd Complexes as Celaph 37
References
C. W. Wharton, Int. J. Biol. Macromol., 1, 3 (1979).
I. M. Klotz, Adu. Chem. Phys., 90, 691 (1978); Y. Imanishi, in Bioactiue Polymeric Systems (Eds., C:. G. Gelelein and C. E. Carraher Jr.), Plenum Press, NewYork, 1985, p. 435.
M. E. Vollpin, G. N. Novodarova and E. M. Kolosova, Inorg. Clleirr. Acta, 5, 21 (1981).
B. Pispisa and A. Palleschi, Macromolecules, 19,904 (1986).
A. Akelah and D. C. Sherrington, Chem. Rev., 81,557 (1981).
Y . Shai, K. A. Jacobson and Patchornik, 1. Anr. Cherern. Soc., 107, 4249 (1985).
N. K. Mathur, C. K. Narang and R. E. Williams, Polymers as Aids in Organic Chemistry, Academic Press, New York, 1980.
H. Bruner and J. C. Bailar Jr., Inurg. Chem., 12,1465 (1973).
K. Kaneda, M. Terasawa, T. Imanaka and S. TeranisIu, Cllem. Lett., 1005 (1975).
T. Nosawa, M. Hatano and S. Kanbara, Kagaka Zasshi, 72,369 (1969).
T. Nosawa, Y. Nose, M. Hatano and S. Kanbara, Makrort~ol. Clletir., 73, 112 (1968).
: C. U. Pittman and Q. J. Ng, I. Organometallic Cllein., 140,187 (1977).
G. Gubitosa and H. H. Brintzinger, J. Organometallic Chers., 140, 187 (1977).
Y. Nakamura and H. Hirani, Chem. Lett., 1197 (1976).
J . E. Katon, inorganic Semiconducting Polyiners, Marcel Dekker, New York, 1970.
O i : , .K. Geoff, I . H. Simon and P. Nikki, Analyst, 117, 1243 (1992).
W. Kuhn, A. Kamel and D. H. Walten, Cl~iinia (Ztcrich), 12, 123 (1958).
J. Lingen and J. Yan, React. Polyrrz., 1, 54 (1992).
D. Lindsay, D. C. Sherrington, J. A..Greig and R. D. Hancock, React. Polym., 12, 75 (1990).
L. Xingyin, S. Zhixing, Z. Wengqn, Z. Guangyao, Xijun and Fresensius, 1. Anal. Chenz., 344(6), 2525 (1992).
M. Polymbo, A. Cosami, M. Terbojerieh and E. Pegson, 1. Am. Cllert~, Soc., 99, 939 (1977).
L. Bamaszal, J. C. Watson and J. C. Kendrew, J. Mol. Biol., 12, 130 (1965).

Paiymermde' Complexes as CatiJvsfs 38
Y. Kurimara, K. Yamada, E. Tsuchida and M. Kaneko, 1. Polyrlr. Sci., Polynr. Clletrl. Ed., 9, 3521 (1971).
N. Hojo and H. Shirai, N i p n Kagaku. Xuishi, 1316 (1972).
E. Collision, F. 5. Dainton, F.R. S.B., Mile, S. Tazuke and P. R. Smith, Nature, 193,26 (1963).
S. Tazuke and S. Okamma, J. Polym. Sci. Polynr. Chenl., 4, 141, 2461 (1966).
I. 0 . Hurtwell and J. C. Bailer Jr., J. Am. Chem. Soc., 92, 1284 (1970).
R. W. Kluiber and J. W. Lewis, 1. Anr. Chem. Soc., 82,5777 (1960).
A. Akelah, Br. Polym. J., 107 (1981).
W. Leutsch, W. Broser, W. Biedermann and H. Guichetel, Z. Nnturforsch, 48,136 (1957).
W. F. Amon and K. W. Kane, U. S. Pat, 2,505,085 (1950).
D. L. Wertz and J. L. Tywoll, 1. Inorg. Nucl. Chenl., 36, 3713 (1974).
A. Epsteln and B. S. Wilde, 1. Chem. Phys., 32,324 (1960).
A. Warshawsky, Angm. Makrotf~ol. Chem., 171 (1983).
M. Sato, Y. Inaki, K. Kondo and K. Takemoto, 1. Polytti. Sci. P O ~ ~ I I I . Cllerrr. Ed., 15, 2059 (1977).
I. P. Mathew and M. Srinivasan, Polym. Intern., 24,249 (1991).
T. Nozawa, Y. Akimoto.~,and. M. Hateno, Makrorlrol. Cllem., 155, 21 (1972).
C. U. Pithnan Jr. and L. 11. Smith, 1. Anr. Cbern. Soc., 97, 4774 (1975).
V. S. Pschezhetsyi, S.G. Ikryannikov, T. A. Kuzuetsova and V. A. Kabanov, 1. Polyrtz. Sci. Polynz. Chem. Ed., 14, 2595 (1976).
T. Ouchi, T. Nishmura and M. Imoto, 1. Polyrrr. Sri., Polynr. Cllerrr. Ed., 14, 2695 (1976).
R. V. Synder, R. J. AngeIici and R. B. Meek, 1. Ani. CIrerr/. Soc., 94, 2660 'a972).
M. Sato, K. Kondo and K. Takemoto, 1. Biol. Clleirr., 179, 601 (1978); M. Sato, Y. Inaki, K. Kondo and K. Takemoto, 1. Poly111. Sci., 15, 2059 (1977).
R. C. W. Welch and H. F. Rase, Ind. Eng. Cheirl. Fundnnl., 8, 389 (1969).
Y. Karusu, W. Storck and G. Manecke, Makronrol. Clre/rz., 176, 3185 (1975).
L. D. Rollman, J. Am. Chem. Soc., 97,2132 (1975).
H. Bruner and J. C. Bailer Jr., Inorg. Chem., 12,1965 (1973).

W. D. Bonds Jr., C. H. Brubaker, C. H. E. S. Chandrasekharan, C. Gibbons, R. H. Grubbs and L. C. Kroll, J . Awl. C k m . Soc., 97, 2128 (1975).
C. U. Pittman Jr. and L. R. Smith, J. Am. C l ~ t n . Soc., 97, 1749 (1975).
H. Hirai, Y. Nakao and N. Toshima, Polym. Prepr. Jpn., 25,2553 (1976).
H. Hirai, Y. Nakao and N. Toshima, Chenr. Lett., 545 (1978).
H. Kamogawa, J . Polytn. Sci., B 10, 711 (1972); Kamogawa, 1. Polym. Sn., A-l,12, 2317 (1974).
T. Nozawa, M. Halano and S. Kanbara, Koggo Kagaku Zasshl, 72, 373 (1969).
Y. Mor~guch, Bull. Chem. Soc. Jpn., 37,2656 (1966).
R. Breslaw and L. E. Overman, J. Am. Chem. Soc., 92, 1075 (1970).
W. 0. Haag and D. D. Whitehurst, Ges. Ofin, 1,800,379 (1969).
H. Ara~, ? Kaneko and T. Kunugi, Chetn. Lett., 265 (1975).
G. 0. Evans, C. U. Pittman Jr., R. McMillan, R. T. Beach and R. Jones, 1. Organorr~etal, Clretn., 67,295 (1974).
T. Ouch, 'T. Nishimura and M. Imeto, J. Polym. Sci., Al, 14,2695 (1976).
K. Takemoto, T. Takata and Y. Inaki, J. Polynr. Sci., Al, 10,1061 (1972).
M. Imoto, K. Ree and T. Ouchi, Makromol. C l ~ m . , 167, 353 (1973); M. Imoto, Y. Iki, Y. Kawqbata, andM. Kinoshinta, Makrotnol. Clrenr., 140,281 (1970).
T. P. Dang and H. B. Kagan, J. Am. Clznl. Soc., 95,8295 (1973).
E . Tsuchida, H. Nishikawa and E. Terada, Eur. Polynr. I., 12, 611 (1976).
T. Nozawa, M. Hateno, M. Kanbara and S. Koggo, Kagaku Znsslli, 72, 369 (1969).
T. Nozawa, Y. Nose, M. ~a teno 'and S. Kanbara, Mdconzol. Cltenr., 112, 73 (1968); 'T. Nozawa, Y. Nose, M. Hateno and S. Kanbara, Makrotnol Chern., 115, 10 (1968).
T. Sasaki and T. Matsunaga, Bull. Chetn. Soc. Jpn., 41,2440 (1968).
Y. Nose, M. Hateno and M. Kanbara, Makromol. Chetrr., 98,136 (1966).
S. L. Davgdova, V. A. Barahanov, N. A. Dlate and V. A. Kargin, Vysokomol Soed., A10,1004 (1968).
A. T. Kapancha, V. S. Pachezhetski and V. A. Kabanov, Vysokonrol. Soed., B11, 5 (1969).
J. H. Wang, I. AITI. Chem. Soc., 77,4715 (1955).

T. Nozawa, Y. Nose, M. IIatano and S. Kanbara, Makromol. C I ~ E I ~ I . , 112, 73 (1968).
H. Sigel and G. Blaner, Helu. Chim. Acta, 51,1246 (1968).
A. 'I'. Kapanchyan, V. S. Pachezhetski and V. A. Kabanov, Vysokonrol. Swd., B11, 5 (1969).
P. Jones and A. Saggett, Biochem. J., 110,621 (1968).
V. S. Pshezhetskyi, S. G. Ikruyannikov, T. A. Kuzuetsova and V. A. Kabanov, 1. Polym. Sci., Al, 14,2595 (1976).
G. L.. Eichhoru and Y. A. Shin, I. Am. Chem. Soc., 90,7323 (1968).
P. M. May, P. W. Lindler and D. R. Milliams, 1. Chern. Soc. Dalton. Trans., 588 (1977).
V. du Vigneaud, C. Ressler, J. M. Swan, C. W. Roberts, P. G. Katsoyannis and G. Gordon, 1. Am. Chenz. Soc., 75,4879 (1953).
G . Manecke and P. Renter, Pure Appl. Chern., 51,2313 (1979).
B. W. Erickson and PCB. Merrifield Jr., in The Proteins, 3rd edn., Vol. 2, Academic Press, New !%k, 1976, p. 255.
M. A. Krans and A. Patchor&, fir. I. Chern., 17,298 (1978).
R. A. Messing (Ed.), in ImnlobiliSed Enzyntes for lndristrinl Reactions, Academic Press, New York, 1975.
K. Imai, T. Shorni, K. Uchida and M. Miya, Biotechnol. Bioeng., 28, 198 (1986). . . . . . .
W. H. Seonten, Methods Enzynlol., 135, 30 (1987).
F. N. Kolisis and D. Thomas, Biotecl1nol. Biwng., 30, 160 (1987).
Y. Yokoyama, A. Tanioka and K. Miyasaka, 1. Menrbr. Sci., 38, 223 (1988).
M. F. Chaplin and C. Bucke, Enzyrne Technology, Cambridge University Press, New York, 1990.
C. K. Cotton, M. I. Nemet and R. Y. K. Yang. Alclle Syrrrp. Ser. No. 773, 74, 8 (1978).
P. A. Ramachandran, R. Krishna and C. H. Panchal, 1. Appl. CIIEIII. Biotecil., 26, 214 (1976).
R. Goldman, L. Goldstein and E. Katchalski, in Biocherrrical Aspects of Reactions on Solid Supports (Ed., G. R. Stark), Academic Press, New York, 1971. p. 27.
W . H . Dalv, Makrornol. Chenr. Suppl., 2, 3 (1979).
H. Molinan, F. Montanari and P. Tundo, Cbert~. Comnrrcn., 639 (1977).

92. G. M. Maneche and H. Heller, Makromol. Chenr., 55, 51 (1962).
93. H. P. Gregor, Angew. Chem., 64,34 (1962).
94. V. N . R. Pillai and 8. Mathew, Indian 1. Technol., 31, 302 (1993).
95. H. Egawa, T. Nonalen and M. Nakayama, Konbuski. Knko., 32, 86 (1983).
96. M. Tomo~, E. Ogawa, Y. Hosokka and H. Kakiuchi, I. Poly~n. Sci., Pnrt A, Polyrrr. Chem., 20, 3015 (1982).
97. E. Tsucluda and Y. Kurimura, Macronlol. Chem. Rapid Con~~irun., 2, 621 (1981).
98. J. 1. Crowley and H. Rapport, Acc. Chenr. Res., 9,135 (1976).