polymerase chain reaction-aided genomic sequencing of an x
TRANSCRIPT

Proc. Nati. Acad. Sci. USAVol. 87, pp. 8252-8256, November 1990Genetics
Polymerase chain reaction-aided genomic sequencing of an Xchromosome-linked CpG island: Methylation patternssuggest clonal inheritance, CpG site autonomy, andan explanation of activity state stability
(DNA methylation/5-azacytidine/PGKI gene/gene regulation/cell memory)
G. P. PFEIFER*, S. D. STEIGERWALD*, R. S. HANSENt, S. M. GARTLERt, AND A. D. RIGGS**Molecular Biology Section, Beckman Research Institute of the City of Hope, Duarte, CA 91010; and tDepartments of Genetics and Medicine and the Centerfor Inherited Diseases, University of Washington, Seattle, WA 98195
Contributed by S. M. Gartler, July 20, 1990
ABSTRACT The 5' region of the gene encoding human Xchromosome-linked phosphoglycerate kinase 1 (PGKI) is apromoter-containing CpG island known to be methylated at 119of 121 CpG dinucleotides in a 450-base-pair region on theinactive human X chromosome in the hamster-human cell lineX8-6T2. Here we report the use of polymerase chain reaction-aided genomic sequencing to determine the complete methyl-ation pattern of this region in clones derived from X8-6T2 cellsafter treatment with the methylation inhibitor 5-azacytidine. Wefind (i) a clone showing full expression of human phosphogly-cerate kinase is fully unmethylated in this region; (ii) clones notexpressing human phosphoglycerate kinase remain methylatedat =50% ofCpG sites, with a pattern ofinterspersed methylated(M) and unmethylated (U) sites different for each clone; (iN)singles, defined as M-U-M or U-M-U, are common; and (iv) afew CpG sites are partially methylated. The data are interpretedaccording to a model of multiple, autonomous CpG sites, andestimates are made for two key parameters, maintenance effi-ciency (Em 99.9% per site per generation) and de novomethylation efficiency (Ed 5%). These parameter values andthe hypothesis that several independent sites must be unmeth-ylated for transcription can explain the stable maintenance ofXchromosome inactivation. We also consider how the activeregion is kept free of methylation and suggest that transcriptioninhibits methylation by decreasing Em so that methylationcannot be maintained. Thus, multiple CpG sites, independentwith respect to a dynamic methylation system, can stabilize twoalternative states of methylation and transcription.
Inheritance of DNA methylation patterns is generally ob-served in tissue culture, consistent with the maintenancemethylase concept (1, 2) and the in vitro observed preferenceof DNA methyltransferase for hemimethylated sites (3-5).However, little is known about the in vivo ratio of mainte-nance to de novo methylation. Methylation maintenanceprobably is a key part of the maintenance of X chromosomeinactivation, a phenomenon where one has extremely stable,clonally heritable differentiation of identical DNA se-quences. Studies on X chromosome inactivation have, infact, provided strong evidence that cytosine methylation isone of the mechanisms used by mammalian cells to aid cellmemory (6) and to maintain genetic silence during develop-ment (7-12). For X chromosome-linked genes, a strongcorrelation exists between the inactive state and hyperme-thylation of methylation-sensitive restriction sites (usuallyHpa II) in the 5' region (13-17). Most housekeeping genes,including those on the X chromosome, have 5'-associated
G+C-rich regions, termed CpG islands, which are up to10-fold enriched for CpG dinucleotides (18). Autosomal CpGislands are characteristically unmethylated, but, in contrast,several X chromosome-linked CpG islands are highly meth-ylated on the inactive X chromosome (Xi) (13-17). When thestabilizing effect ofDNA methylation is not present (19) or isperturbed by 5-azacytidine (SzC) treatment, reactivation ofgenes on the inactive X is frequently observed (20, 21).DNA methylation information at every cytosine can be
determined by genomic sequencing (22), but technical prob-lems have limited its application, especially for G+C-richregions. However, the recent development of genomic se-quencing procedures (23-25) that use a ligation-mediatedpolymerase chain reaction (LMPCR) procedure has greatlyincreased reproducibility, specificity, and sensitivity. Thefirst step of LMPCR-aided genomic sequencing is base-specific chemical cleavage of DNA, generating 5'-phos-phorylated molecules. Next, primer extension of a gene-specific oligonucleotide (primer 1) generates molecules thathave a blunt end on one side. Linkers are ligated to the bluntends, and then an exponential PCR amplification of thelinker-ligated fragments is done by using the longer oligonu-cleotide of the linker (linker-primer) and a second gene-specific primer (primer 2). This method provides high-qualitysequence ladders suitable for methylation and protein foot-print analysis (23-25). Here, we have used LMPCR-aidedgenomic sequencing to determine cytosine methylation inboth strands of 62 CpG sites (information obtained for 121CpG dinucleotides) of the human PGK-associated CpG-richisland in human-hamster hybrid cell lines that were clonedafter 5zC treatment.
MATERIALS AND METHODSHuman-hamster hybrid cells containing either an activehuman X chromosome (Xa) (cell line Y162-11C) or an Xi (cellline X8-6T2 and clonal derivatives) were cultivated as de-scribed (16), and DNA was prepared (24). Chemical cleavage,LMPCR, sequence gel electrophoresis, and hybridizationwere as described (24, 25). A complete protocol is availableupon request. Hydrazine-treated DNA had an average frag-ment length of 100-200 bases, and 1-2 ,g was used in theLMPCR reactions. Eight primer sets were used (25), and PCRamplification was for 15-18 cycles. Hybridization probes
Abbreviations: 5zC, 5-azacytidine; HPRT, hypoxanthine guaninephosphoribosyltransferase; LMPCR, ligation-mediated polymerasechain reaction; PGK1, X chromosome-linked phosphoglycerate ki-nase 1 enzyme; M, methylated CpG; U, unmethylated CpG; P,partially methylated CpG; Xa, active X chromosome; Xi, inactive Xchromosome.
8252
The publication costs of this article were defrayed in part by page chargepayment. This article must therefore be hereby marked "advertisement"in accordance with 18 U.S.C. §1734 solely to indicate this fact.

Proc. Natl. Acad. Sci. USA 87 (1990) 8253
were cDNA fragments derived from in vitro synthesizedRNA, as described (24, 26).
RESULTSExperimental System, Cell Lines, and Genomic Sequencing
Procedure. Fig. 1 outlines the cell lines and experimentalsystem. Hamster-human hybrid cells containing a human Xiwere treated for 24 hr with 5zC, allowed to recover, clonedwith selection for activity of the human HPRTgene, and thengrown forDNA extraction and enzyme assays (16). Note thatno selection was applied for human PGKI; selection was onlyfor HPRT, which is X chromosome-linked but distant fromPGK1. Xi reactivation after 5zC treatment usually is piece-meal (27), a fact confirmed by the finding that only 20% of thereactivant clones express the human X-linked phosphoglyc-erate kinase enzyme (PGK1) (16). DNA for genomic sequenc-ing was from cloned reactivants grown for at least 30 gener-ations without additional subcloning.
RepresentativeDNA sequence ladders obtained by LMPCR-aided genomic sequencing are shown in Fig. 2. Because5-methylcytosine does not react with hydrazine in high salt,cytosine methylation is indicated by the reduction of acytosine band to the background level. As a consequence ofthe linker-ligation step, every PCR-amplified molecule con-tains the same primer target sequences (linker-specific primerand gene-specific primer), so amplification efficiencies aresimilar, although not identical, for each fragment. The ratioof nearby bands in the same sequence ladder is an intrinsicfunction of the sequence and is quite reproducible (23-25,28); thus, the percent methylation at each CpG can beestimated from the band ratios compared with the control,unmethylated DNA lane. Fig. 2 shows control lanes of totalHeLa DNA, which is unmethylated at the PGK island (25),and cytosine reaction lanes ofDNA from the hamster-humancell lines. Cell line Y162-11C contains a human Xa, whereascell line X8-6T2 contains an Xi. Cell lines 15A, SACD, and111-9 are derivative clones, obtained from X8-6T2 after 5zCtreatment but which are human PGK1-negative. Cell lineV-2B is a similar derivative clone but is expressing high levelsof human PGK1.
Methylation of the Parental Lines. The Xi in X8-6T2 andnormal human lymphocytes was known to be highly meth-ylated (25, 29). Fig. 3B shows that the human Xi in X8-6T2is fully methylated at 117 of the 121 CpGs dinucleotidesanalyzed. In this manuscript, "site" refers to a double-stranded CpG site, and "CpG dinucleotide" refers to a singlestrand. Only one site at -260 base pairs (bp) escapes meth-ylation. One CpG dinucleotide on the lower strand at position-7 gave no methylation information because a strong band,perhaps caused by a nick or premature termination of Ther-mus aquaticus (Taq) polymerase, appeared across all lanes ofthe sequencing gel for both Xa and Xi, even in the guanineand G+A channels. CpG sites at positions -417 and +68were analyzed in one strand only. In sharp contrast to the Xi,no methylation is seen in this 450-bp region of the Xa inY162-11C (Fig. 3A) (or normal human cells, ref. 25). Back-ground varies somewhat from band to band, apparentlydependent on sequence, but we estimate that any CpGdinucleotide methylated >20o would be recognized as par-tially methylated. Partial methylation, as used in this paper,
G ATCa -L_--321
--4-
__li__b
0 4
do
f -dd
4*qw,. -
46lb dlb.t..4a.Mf * -
lA.f S-:: W*
BG C U3 < I) (NG A TC -Uu?-)
-338
~-l -*4-^
...-
lbm^
a.,41Ff -. 4--
lbII l 40GP
O--373
-263
FIG. 2. LMPCR-aided genomic sequencing data showing meth-ylation at CpG dinucleotides in the 5' region of PGKI. Lanes G,G+A, T+C, and C are sequencing controls obtained from HeLaDNA. Only the cytosine-specific reaction is shown for the hybrid cellline DNAs. Arrows, position of methylated cytosines; P, partialmethylation site in cell lines 15A and III-9. (A) Primer set D. (B)Primer set C (25).
means that a specific site is methylated in some X chromo-somes of the culture and unmethylated in other X chromo-somes of the same culture; it does not refer to the partialmethylation seen for the entire region. With one exception(position -257 in X8-6T2 cells), partially methylated sites arenot seen in the parental cell lines, a result confirmed for tworestriction sites (Hpa II at +23 and Nar I at -343) by anearlier study (28). Most sites probably are >90% methylated.We also confirm here that methylation is only in CpG sites.
Analysis of Cells Cloned After 5zC Treatment. The reacti-vant expressing human PGK (V-2B, Fig. 3F) is completelyunmethylated. Thus, the reactivant is like a normal Xa (25).A quite different picture is seen for the PGK-negative reac-tivants. Mosaic patterns are seen (Fig. 3 C-E) with inter-spersed methylated CpG (M) and unmethylated CpG (U)sites; overall only 48% are M. Methylation is much moreextensive around the transcription start region and down-stream; 89% of the CpG sites are methylated downstream ofposition -7. It is noteworthy that all three PGK-negativeclones are methylated at five CpG sites approximately cen-tered around the transcription start point. This is the onlyregion where a cluster ofmore than two methylated cytosinesis present in all three clones, suggestive that methylation ofthis region is critical for maintaining transcriptional silence.Previous studies also indicated that demethylation of thisregion seems necessary, although probably not sufficient, forreactivation (16, 29). This region also contains a consensusbinding site for a recently discovered protein factor (HIP)
X8-6T2CHO with Hu XiHu HPRT-Hu PGK -
AzaC24 hr
Clone withselectionfor HPRT+
2 daysRecovery
FIG. 1. Outline of 5zC treatment, cloning, and analysis. Hu, human; gen, generations.
Hu PGKActivity
15A5ACD
30 111-930 gen V2B
PrepareDNA
P.DetermineMethylation ofPGK CpG Island
Genetics: Pfeifer et al.

8254 Genetics: Pfeifer et al.
AAATTCCAGGGGTTGGGGTTO GCCTTTTCCAAGGCAGCCCTC-WTTTG~iC AOGGAoCkZ
GCTGCTCTaGG8TGTTCCoGGAAM~Al9Z(A(, CCC'TCGGTC.$TACATTCT
TCA CACCATTCfAGCCACC9gATCTTCTCoTACCTTGTGcCCCCC'cA
9bTTCCTGCTC oCCCTAAGTAGGAAGGTTCCTTGgTTGC-A9g
GCAgMCCA jyCTCACTAGTACCCT1g6A gCA 5CAGGGAGCAA~~~~~~~~~~~TGGCAR&? T AcgTGcTCACCA
TGTTCCTGCwTGTTC~gATTCTGCACCCGTCGTATAC TCACTA
GCC oT6A~AATCACC&ACCTCTCTCCCCAGCTGTATTTCCAAAAT
CAATTCCAGGGGTTGGGG6TTGGCCTTTTCCAAGGCAGCCCTGGGTTTG!P¶GGGACG
%gTTCCTGCTC9 9CAAT~GAAGGTTCCTTG?~gTT~P&GCPGGAGT
GACMAAAAAGCGCA~TCTCACTAGTACCCTtcAGAGGACAG23CAGGGAGCAA
TGGCA65|2oA~tATGGGCTGTGGCCMT C G IR&
AGAGCAGGC AAG6V26TGP6GAGJRVGT TAGTGTGCGCCC
TGTTCCTGCCPG TTTCGCATTCTGCAAGCCTC GGAG?9kT~CAG9GCTCCCTGTTGA AATCACC&ACCTCTCTCCCCAGCTGTATTTCCAAA ATG
C~~~
AATTCCAGGGGTTGGGGTT2OCCTTTTCCAAGGCAGCCCTGGGTTT6 CAGGGA%
GCGTTGG T6GTTCgGAAA9AG G OWo~~TG~oACATTCT
TCAiTCK jVT~tgC A JITCACC ATCT ~tCACCCTTGTGGGCCCCCegfA
9rgTTCCTOCTC9CvCCCTAA6 gGAAG6TTCCTTGPC, T%PST6C?66A?&T
GACAAA?#AGC 6ATCTCACTAGTACCT?~AGA?&ACAGtiCAGGGAGCA
TGGCA%9&CAtGGGTOWTGTCCCAATACTWCTGCTCAGCAGAW&AG6CG V6CGGGGGgGrGA6GC-GG6TGTGGtTG, GG~c
TGTTCCTGCC"T,6TTCt&CA ATCC TGA t&CAtzCG9CACTt&GCTCCCTT&TTGACAATCACC&ACCTCTCTCCCCAGCTGTATTTCCAA ATG
Proc. Natl. Acad. Sci. USA 87 (1990)
BAATTCCAGGGGTTGGGGTT6?GCCTTTTCCAAGGCAOCCCTGGGTTTGPCAGG6AG& -378
GCTGCTCTGG~t OOTT GAAIAR&P tCCCTCGGT ACATTCT -3 18
TCA~tT\~UAG?( CACCtssTCT~t&Ct&CTACCCT~oTGGGCCACT 2586 rT 1 ACTTT,6cc~p 19
t&ICTTCCTGCTCtICCCCTAA6Tt&GA^G6TTCCTT6?4ITTt?? &Tcry 9
ToCwCToCooC~T6&CoT~C~ -78
AGA6CA~tzgGAAGG6GtgT~t&GGAG~ttGTGTGG6Gt>AGTT66CC -118
GCTCCCTATTG CTCACCTCTCTCCCCAGCTGTAATCCAAA ATG +98
ig
DAATTCCAGCGGTTGGGGTTGGGCCTTTTCCAGGCAGCCCTGGTTTGitCAGGGAt,<GCTGCTCTGG 8GGTT tGAAA? _~gg28CCTGTogCTTTCAt&T(,Ct(TTtU~AGCAJTCACC? ATCTTC -OTACCTGGCCCCCCSZ&A
gTTCCTCGCT tCCCCTAAGT28GGAGGTCTTCC'TTG>I6 RCt&A T
GACAAA96AAGCgA?26TCTCACTAGTACCCT GCAGAGGACA2gCAGGGAGCAA
AGAOCA tACAAGGGCGTGGCMAGGt&CGTAGI~
AACRGCVAGG IGTGAG &GGTGTGGG R6TAGTGTGGGCCC
TGTTCCTGCC TCGCATTCTGCAAGCCTCtGAjCA?( r?( AGAOGCTCCCT?3TG W
A GAATCACCbACCTCTCTCCCCAGCTGTATTTCCAAA ATG
FAATTCCAGGGGTTGGGGTTPGGCCTTTTCCAAGGCAGCCCTGGGTTTOR AGGGAn6GCTGCTCT GGg6TTC oGAAA16 CARRR9gAcCCTGGGTC$T ACATTCT
TCA96TC0T AG? TCACCGGATCT9GcoCTACCCTTGTGGGccccGcRo
9&TTCCTGCTC9&CCCTAAGT9 AAGOTTCCTTGc9 TTZGG(GCbGART
GACAAA93AAG A CTCACTAGTACCCT9&AGA9gACAG~CAGGGAGCA
TGG6CA ffGAi TGGGCTGTGGCCAATAG6P WGTACGGfC?
AGAGCA6RG8AAGGKGGGGTGGGGAG23GGTGTGGGG2GTAGTGTGGGCCC
TGTTCCTGCC STGTTC9gATTCTGCAAGCCTCgAGgA' T9dCAGT%GCTCCCP&TTGA GATCACC&ACCTCTCTCCCCAGCTGTATTTCCAAA ATG
-378
-318
-258
-198
-138
-78
-18
+43
+98
-378
-318
-258
- 198
- 138
-78
-18
+43
+98
FIG. 3. Summary ofDNA methylation data. (A) Y162-11C cells carrying an active human X chromosome (human PGK-positive). (B) X8-6T2cells carrying an inactive human X chromosome (human PGK-negative). (C) Cell line 15A (human PGK-negative). (D) Cell line 5ACD (humanPGK-negative). (E) Cell line 111-9 (human PGK-negative). (F) Cell line V-2B (human PGK-positive). o, Unmethylated cytosine in a CpGdinucleotide; *, full methylation; *, partial methylation; +, major transcription initiation site.
that appears important in the transcription of several house-keeping genes (30). Upstream of position -7, the averagemethylation level is much less: 38% M, 52% U, and 10%partially methylated (P). Singles, defined as M-U-M orU-M-U, occur at the frequency expected for a sequentiallyrandom pattern (Table 1). However, the pattern is notcompletely random. Run test analysis (31) indicates that theupstream patterns in 15A and 5ACD clones could, indeed, berandom, but clone III-9 and the combined data are signifi-cantly different from random. Runs of length 2 are under-represented, and some statistically unlikely runs are pre-sent-e.g., a run of 12 unmethylated sites in clone l5A. Wecannot provide a certain explanation for these data, but ourworking hypothesis is as follows. DNA methyltransferase isprocessive (32) and is inactivated when it encounters a sitehaving recently incorporated 5-azadeoxycytidine in place ofcytosine (33, 34). This may lead to clusters ofhemimethylatedsites, many of which, however, will be repaired during the
2-day recovery period, in which much remethylation isknown to occur (35, 36). This "recovery" methylation couldbe, in large part, random.For estimating de novo methylation, partially methylated
sites (P, indicated as grey in Fig. 3) give useful information.A site was scored as partially methylated only when bandintensity was reproducibly above background for severalindependent analyses; also in each case the assignment wasconfirmed by data for the opposite strand. Partials are (i) 10oof sites in the upstream region, (ii) distributed differently ineach clone, (iii) only weakly clustered, if at all (three clustersof 2 of 15 total P sites), and (iv) found in highly unmethylatedas well as in highly methylated regions.
DISCUSSIONSummary of Experimental Observations. Previous work
using methylation-sensitive restriction enzymes indicatedmosaic methylation patterns in PGK-negative clones and
Proc. Natl. Acad. Sci. USA 87 (1990) 8255
Table 1. Run-length analysis for PGK-negative clones
Run Methylated Unmethylatedlength Observed Expected* Observed Expected*
1 18 22 13 132 2 8 1 83 1 3 4 54 3 1 7 3
25 3 1 5 5
Sites analyzed are upstream of position -7.*Expected number of runs of length n was calculated from theequation: Exp = 150(1 - q)2q", where 150 is the total number ofCpG sites (50 in each clone), q is the fraction of indicated methyl-ation state (0.38 for methylated or 0.62 for unmethylated). Partialswere scored as unmethylated because they probably are due tomethylation of initially unmethylated sites.
suggested that demethylation ofmuch or all of the island wasnecessary for transcriptional activity (29). To keep our basicexperimental observations from being confused with inter-pretation, they are summarized as follows: (i) the parental Xiin clone X8-6T2 is fully methylated at 117 of the 121 CpGdinucleotides analyzed in the 450-bp region containing thepromoter and transcription start site; (ii) the normal Xa inthese hybrid cells and the reactivant showing full expressionofhuman PGK are fully unmethylated in this region; (iii) brieftreatment with 5zC causes mosaic methylation patterns over30 generations later; (iv) the PGK-negative clones each have-50% ofCpG sites unmethylated; (v) singles, both M-U-M orU-M-U, are common; and (vi) partial methylation is seen atsome sites in the PGK-negative clones.Lack ofCooperativity and Pattern Maintenance. Our finding
of frequent singles is strong evidence that neither remethy-lation during the repair stage nor de novo methylation duringgrowth is highly cooperative in the upstream region. For thisreason, much ofthe analysis below assumes considerable siteautonomy. The mosaic patterns seen differ for each clone,suggesting clonal inheritance, but a question is whether theobserved patterns reflect maintenance of patterns fixed bycloning shortly after 5zC treatment or whether the patternschange quickly. Hansen and Gartler (29) did additional sub-cloning experiments that bear on this question. After sub-cloning and growth for DNA extraction (25-30 generations),analysis by methylation-sensitive restriction enzymes indi-cated that the original pattern was generally retained, butsome de novo methylation could be detected in most sub-clones. This result establishes that maintenance predomi-nates in human-hamster hybrid cells, but, in addition, denovo methylation occurs at a significant rate.
Methylation Patterns in a Growing Population. Otto andWalbot (37) appropriately considered methylation mainte-nance in a growing population and derived a recursiveequation for the special case in which methylcytosine is neverlost from the "old" strand and demethylation occurs pas-sively from failure to methylate the hemimethylated sitecreated by DNA replication. They showed that an equilib-rium methylation level will be reached that will depend on theratio of the efficiency of maintenance methylation (Em) to theefficiency ofde novo methylation (Ed). Much ofthe reasoningis directly applicable to our system, but because of recentevidence that methylation can be lost quickly and withoutDNA replication (38, 39), we present here an alternativetreatment that does not specify how, or from which strand,methylcytosines are lost.Consider a specific CpG site that has two alternative
methylation states, M or U. In a population of growing cells,
dM/dt = a-Em-M + a-Ed-U [1]
dU/dt = b-(l - Ed)-U + b.(l - Em)-M,
whereM is the number ofmethylated sites (one per cell in cellline X8-6T2), U is the number ofunmethylated sites, Em is themaintenance efficiency, Ed is the de novo methylation effi-ciency, and a and b are cell-growth rate constants. Bydefinition, the fraction methylated is
M/A = M/Total = M/(M + U). [3]
If a = b, as is likely for our system, the differential equationscan be readily solved (see Fig. 4), and at equilibrium, whendMA/dt = 0,
MA = Ed/(l + Ed - Em). [4]
If each CpG site in the CpG island of PGK is considered asan autonomous entity, then the above reasoning and equa-tions can be applied, and average methylation constants canbe estimated from the experimental data.
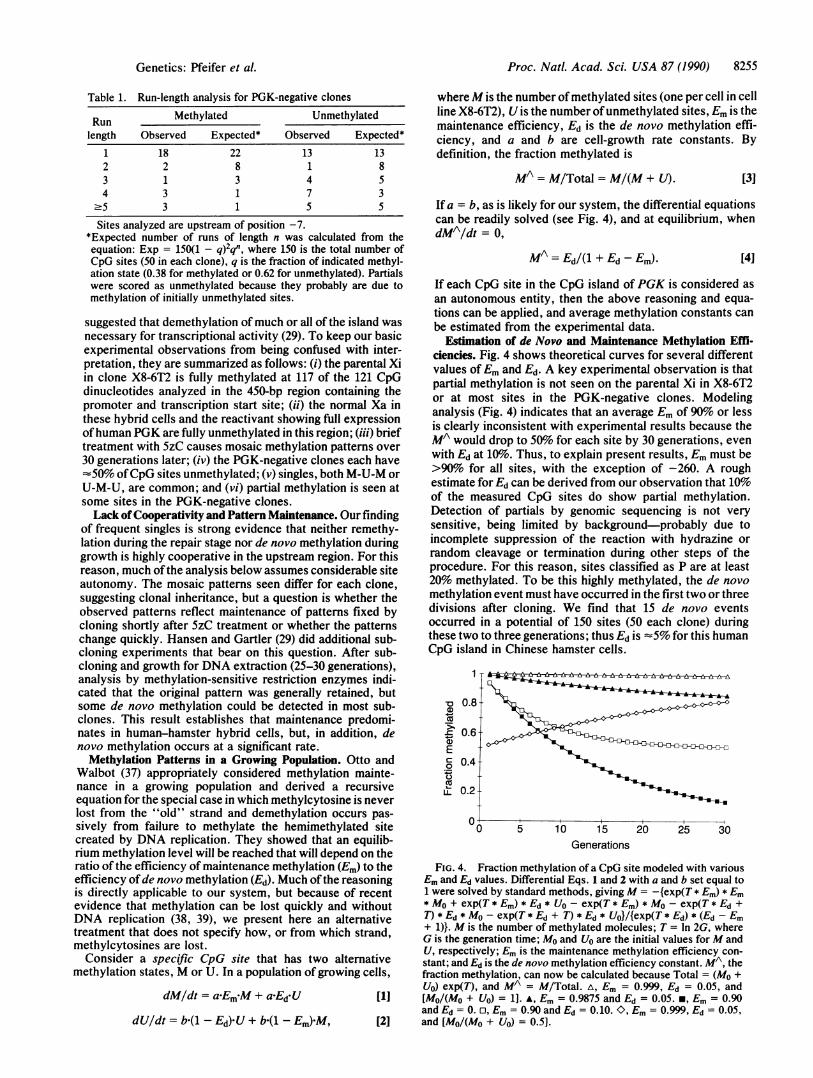
Estimation of de Novo and Maintenance Methylation Effl-ciencies. Fig. 4 shows theoretical curves for several differentvalues ofEm and Ed. A key experimental observation is thatpartial methylation is not seen on the parental Xi in X8-6T2or at most sites in the PGK-negative clones. Modelinganalysis (Fig. 4) indicates that an average Em of 90% or lessis clearly inconsistent with experimental results because theMA would drop to 50% for each site by 30 generations, evenwith Ed at 1o. Thus, to explain present results, Em must be>90%o for all sites, with the exception of -260. A roughestimate for Ed can be derived from our observation that 10%of the measured CpG sites do show partial methylation.Detection of partials by genomic sequencing is not verysensitive, being limited by background-probably due toincomplete suppression of the reaction with hydrazine orrandom cleavage or termination during other steps of theprocedure. For this reason, sites classified as P are at least20%o methylated. To be this highly methylated, the de novomethylation event must have occurred in the first two or threedivisions after cloning. We find that 15 de novo eventsoccurred in a potential of 150 sites (50 each clone) duringthese two to three generations; thus Ed is -5% for this humanCpG island in Chinese hamster cells.
s 0.86
0.6-E0 0.4--
Z_ 0.2
15Generations
FIG. 4. Fraction methylation of a CpG site modeled with variousEm and Ed values. Differential Eqs. 1 and 2 with a and b set equal to1 were solved by standard methods, giving M = -{exp(T * Em) * Em* MO + exp(T * Em) * Ed * UO - exp(T * Em) * MO - exp(T * Ed +T) * Ed * MO - exp(T * Ed + T) * Ed * UO}/{exp(T * Ed) * (Ed - Em+ 1)}. M is the number of methylated molecules; T = In 2G, whereG is the generation time; MO and U0 are the initial values for M andU, respectively; Em is the maintenance methylation efficiency con-stant; and Ed is the de novo methylation efficiency constant. MA, thefraction methylation, can now be calculated because Total = (MO +U0) exp(T), and MA = M/Total. A, Em = 0.999, Ed = 0.05, and[Mo/(Mo + Uo) = 1]. A, Em = 0.9875 and Ed = 0.05. *, Em = 0.90and Ed = 0. E, Em = 0.90 and Ed = 0.10. O, Em = 0.999, Ed = 0.05,and [Mo/(Mo + U0) = 0.5].
Genetics: Pfeifer et al.

Proc. Natl. Acad. Sci. USA 87 (1990)
Knowing Ed, one can calculate Em from Eq. 4, and Emequals 98.75% for an MA equal to 80%o (detectable incom-pleteness of methylation) and an Ed of 5%. This approach toestimation of Em is applicable for each site and thus gives aminimum estimate for the average Em of the entire region. Anearlier study (28) assayed two restriction sites in this CpGisland of X8-6T2 DNA and found them at least 98% methyl-ated; thus, by Eq. 4, Em was equal to 99.9%. The Otto-Walbot treatment (37) similarly estimates Em at 99.8%. Theefficiency of in vivo maintenance thus seems quite high. Fig.4 shows curves modeling Ed equal to 5% and Em equal to99.9%. With these parameters methylation is maintainedextremely well; moreover, when perturbed (for example, to50% methylation by 5zC treatment), a drift back toward fullmethylation occurs. Maintenance errors do not accumulate.The current values for Em and Ed are only rough estimates,but the use of this hybrid cell system for future subcloningexperiments, growth rate studies, and more accurate quan-titation ofpartials should allow testing ofthe assumptions andmore accurate determination of the intrinsic constants con-trolling the maintenance of methylation patterns.Maintenance of X Chromosome Inactivation and the Xa: Xi
Methylation Differential. The estimated values of Em and Edfor the Xi favor a highly methylated state and provide for"repair" of occasional methylation failure. The theoreticaltreatment and the estimated values are based on the hypoth-esis of multiple, independent CpG sites. If this hypothesiswere true, then the maintenance of the methylated state of aCpG island would be, in essence, explained. Note that thismodel is generally applicable to any methylation-sensitivecritical element and depends on multiple, autonomous CpGsites rather than on CpG islands per se. If transcriptionrequires that several CpG sites in a critical element(s) beunmethylated, then the stable maintenance of the inactivestate is, in large part, explained by the Em and Ed valuessuggested by current data.A key question now becomes how a sequence that is
obviously susceptible to methylation is kept free of methyl-ation on the Xa. Twenty percent of the clones treated with5zC reexpressed detectable human PGK (16), and thesehigh-level expressors are unmethylated at all restriction sites(29). We now find that a high expressor, cell line V-2B, isunmethylated at all CpG sites in this region. Current resultsmake it virtually certain that the methylation patterns seen inthe nonexpressors reflect patterns established shortly aftertreatment with 5zC. Given the almost random methylationpatterns with 50% of the sites retained as fully methylated Msites in the nonexpressors, it is highly improbable thatcomplete demethylation as seen in V-2B cells came first byrandom fluctuation of the methylation patterns. More likely,demethylation of much smaller critical regions allowed fac-tors to bind and some transcription to begin, with moreextensive demethylation following as a secondary event.Eight footprints distributed over much ofthe upstream regionhave been found on the activePGK promoter but none on theinactive promoter (25). Thus, one can imagine that a largetranscription complex, once formed, would decrease Em,either by steric hindrance ofDNA methylase or an increasein the active removal of 5-methylcytosine. A clear predictionis that Em will be less for clones expressing even low levelsof human PGK, and those clones should drift toward de-methylation and higher expression with growth and subclon-ing.High cooperativity has often been invoked as a mechanism
for maintaining all-or-none differentiated states (for review,see ref. 40). Therefore, it is of general interest that, at leastfor this system, multiple, independent CpG sites, rather thancooperativity between sites, more likely provide the founda-tion for the fail-safe maintenance of the inactive state. The
active state could be maintained by a feedback in whichtranscription interferes with the maintenance system. Asystem where methylation inhibits transcription and tran-scription inhibits methylation maintenance tends toward twoalternative stable states.
We thank Dr. Ron Shymko, City of Hope Department of Endo-crinology, for help in solving the simultaneous differential equations,and the City of Hope Department of Statistics with help in statisticalanalysis. This work was supported by National Institute of AgingGrant AGO81% to A.D.R., by National Institutes of Health GrantHD-16659 to S.M.G., and by a fellowship from the DeutscheForschungsgemeinschaft (Pf212/1-1) to G.P.P.
1. Riggs, A. D. (1975) Cytogenet. Cell Genet. 14, 9-11.2. Holliday, R. & Pugh, J. E. (1975) Science 187, 226-232.3. Gruenbaum, Y., Cedar, H. & Razin, A. (1982) Nature (London) 295,
620-622.4. Zucker, K., Riggs, A. D. & Smith, S. S. (1985)J. Cell. Biochem. 29,
337-351.5. Spiess, E., Tomassetti, A., Hernaiz-Driever, P. & Pfeifer, G. P.
(1988) Eur. J. Biochem. 177, 29-34.6. Riggs, A. D. (1989) Cell Biophys. 15, 1-13.7. Razin, A. & Riggs, A. D. (1980) Science 210, 604-610.8. Riggs, A. D. & Jones, P. A. (1983) Adv. Cancer Res. 40, 1-30.9. Doerfler, W. (1983) Annu. Rev. Biochem. 52, 93-124.
10. Holliday, R. (1987) Science 238, 163-170.11. Cedar, H. (1988) Cell 53, 3-4.12. Grunwald, S. & Pfeifer, G. P. (1989) Prog. Clin. Biochem. Med. 9,
61-103.13. Wolf, S. F., Jolly, D. J., Lunnen, K. D., Friedmann, T. & Migeon,
B. R. (1984) Proc. Natl. Acad. Sci. USA 81, 2806-2810.14. Yen, P. H., Patel, P., Chinault, T., Mohandas, T. & Shapiro, L. J.
(1984) Proc. Natl. Acad. Sci. USA 81, 1759-1763.15. Keith, D. H., Singer-Sam, J. & Riggs, A. D. (1986) Mol. Cell. Biol.
6, 4122-4125.16. Hansen, R. S., Ellis, N. A. & Gartler, S. M. (1988) Mol. Cell. Biol.
8, 4692-4699.17. Toniolo, D., Martini, G., Migeon, B. R. & Dono, R. (1988) EMBO
J. 7, 401-406.18. Bird, A. P. (1986) Nature (London) 321, 209-213.19. Kaslow, D. C. & Migeon, B. R. (1987) Proc. Natl. Acad. Sci. USA
84, 6210-6214.20. Migeon, B. R., Jan de Beur, S. & Axelman, J. (1989) Exp. Cell Res.
182, 597-609.21. Grant, S. G. & Worton, R. G. (1989) Mol. Cell. Biol. 9, 1635-1641.22. Church, G. M. & Gilbert, W. (1984) Proc. Natl. Acad. Sci. USA 81,
1991-1995.23. Mueller, P. R. & Wold, B. (1989) Science 246, 780-786.24. Pfeifer, G. P., Steigerwald, S., Mueller, P. R., Wold, B. & Riggs,
A. D. (1989) Science 246, 810-813.25. Pfeifer, G. P., Tanguay, R. L., Steigerwald, S. D. & Riggs, A. D.
(1990) Genes Dev. 4, 1277-1287.26. Weih, F., Stewart, A. F. & Schutz, G. (1988) Nucleic Acids Res. 16,
1628.27. Grant, S. G. & Chapman, V. M. (1988) Annu. Rev. Genet. 22,
199-233.28. Steigerwald, S. D., Pfeifer, G. P. & Riggs, A. D. (1990) Nucleic
Acids Res. 18, 1435-1439.29. Hansen, R. S. & Gartler, S. M. (1990) Proc. Nail. Acad. Sci. USA
87, 4174-4178.30. Means, A. L. & Farnham, P. J. (1990) Mol. Cell. Biol. 10, 653-661.31. Zar, J. H. (1984) Biostatistical Analysis (Prentice-Hall, Englewood
Cliffs, NJ), 2nd Ed., pp. 416-419.32. Drahovsky, D. & Morris, N. R. (1971) J. Mol. Biol. 57, 475-489.33. Pfeifer, G. P., Grunwald, S., Boehm, T. 1. J. & Drahovsky, D.
(1983) Biochim. Biophys. Acta 740, 323-330.34. Taylor, S. M. & Jones, P. A. (1982) J. Mol. Biol. 162, 679-693.35. Creusot, F., Acs, G. & Christman, J. K. (1982) J. Biol. Chem. 257,
2041-2048.36. Flatau, E., Gonzales, F. A., Michalowsky, L. A. & Jones, P. A.
(1984) Mol. Cell. Biol. 4, 2098-2102.37. Otto, S. P. & Walbot, V. (1990) Genetics, 124, 429-437.38. Razin, A., Szyf, M., Kafri, T., Roll, M., Giloh, H., Scarpa, S.,
Carotti, D. & Cantoni, G. L. (1986) Proc. Nail. Acad. Sci. USA 83,2827-2831.
39. Saluz, H. P., Jiricny, J. & Jost, J. P. (1986) Proc. Natl. Acad. Sci.USA 83, 7167-7171.
40. McCarrey, J. & Riggs, A. D. (1986) Proc. Nall. Acad. Sci. USA 83,679-683.
8256 Genetics: Pfeifer et al.