sdf-1 modulates bcl-xl - the journal of biological … modulates bcl-x l, noxa, and bak to induce...
TRANSCRIPT

SDF-1 modulates Bcl-XL, Noxa, and Bak to induce apoptosis
1
SDF-1/CXCR4 signaling induces apoptosis in acute myeloid leukemia cells via regulation of the Bcl-2 family members Bcl-XL, Noxa, and Bak
Kimberly N. Kremer1, Kevin L. Peterson2, Paula A. Schneider2, X. Wei Meng,2 Haiming Dai,2 Allan D. Hess3, B. Douglas Smith3, Christie Rodriguez-Ramirez1, Judith E. Karp3, Scott H. Kaufmann2,4, and
Karen E. Hedin1*
Departments of 1Immunology, 2Oncology and 4Molecular Pharmacology & Experimental Therapeutics, Mayo Clinic College of Medicine, Mayo Clinic, Rochester, MN 55905 and 3Sidney Kimmel Cancer
Center at Johns Hopkins, Baltimore, Maryland 21287
*Running Title: SDF-1 modulates Bcl-XL, Noxa, and Bak to induce apoptosis Direct correspondence to: Dr. Karen E. Hedin, Mayo Clinic, Department of Immunology, Guggenheim Building 3rd floor, 200 First Street Southwest, Rochester, MN 55905; PH: 507-284-6441; FAX: 507-284-4957; email: [email protected] Keywords: AML, CXCR4, SDF-1, CXCL12, apoptosis CAPSULE Background: The chemokine receptor CXCR4 plays a role in AML. Result: SDF-1, the ligand of CXCR4, induces apoptosis in AML cell lines and patient samples via modulation of Bcl-2 family members. Conclusion: SDF-1 induces apoptosis of AML cells via upregulation of Bak and Noxa and downregulation of Bcl-XL. Significance: SDF-1/CXCR4 signaling could induce AML cell apoptosis if bone marrow survival cues can be disrupted.
The CXCR4 chemokine receptor promotes survival of many different cell types. Here, we describe a previously unsuspected role for CXCR4 as a potent inducer of apoptosis in acute myeloid leukemia (AML) cell lines and a subset of clinical AML samples. We show that SDF-1, the sole ligand for CXCR4, induces the expected migration and ERK activation in the KG1a AML cell line transiently overexpressing CXCR4, but ERK activation did not lead to survival. Instead, SDF-1 treatment led via a CXCR4-dependent mechanism to apoptosis, as evidenced by increased annexin V staining, condensation of chromatin, and cleavage of both procaspase-3 and PARP. This SDF-1-induced death pathway was partially inhibited
by hypoxia, which is often found in the bone marrow of AML patients. SDF-1-induced apoptosis was inhibited by dominant negative procaspase-9 but not by inhibition of caspase-8 activation, implicating the intrinsic apoptotic pathway. Further analysis showed that this pathway was activated by multiple mechanisms, including upregulation of Bak at the level of mRNA and protein, stabilization of the Bak activator Noxa, and downregulation of anti-apoptotic Bcl-XL. Furthermore, adjusting expression levels of Bak, Bcl-XL, or Noxa individually altered the level of apoptosis in AML cells, suggesting that the combined modulation of these family members by SDF-1 coordinates their interplay to produce apoptosis. Thus, rather than mediating survival, SDF-1 may be a means to induce apoptosis of CXCR4-expressing AML cells directly in the SDF-1-rich bone marrow microenvironment if the survival cues of the bone marrow are disrupted. The bone marrow microenvironment provides a variety of signals that promote the survival of acute myeloid leukemia (AML) cells. Both the endosteal and vascular niches provide an environment that supports normal hematopoietic stem cells as well as leukemic stem cells. A variety of cytokines, chemokines, and integrins
http://www.jbc.org/cgi/doi/10.1074/jbc.M113.449926The latest version is at JBC Papers in Press. Published on June 24, 2013 as Manuscript M113.449926
Copyright 2013 by The American Society for Biochemistry and Molecular Biology, Inc.
by guest on June 9, 2018http://w
ww
.jbc.org/D
ownloaded from

SDF-1 modulates Bcl-XL, Noxa, and Bak to induce apoptosis
2
present in these niches mediate both the homing and survival signals necessary to propagate leukemia. In addition, the expansion of hypoxic areas during the development of leukemia leads to additional signals that support the proliferation and survival of AML cells (1, 2). Determining which of these signaling events are critical for the survival of AML cells will help identify a strategy to disrupt the protection that the bone marrow microenvironment provides for AML cells.
CXCR4 is a G-protein-coupled chemokine receptor that signals in response to its sole physiological ligand, SDF-1 (also known as CXCL12). CXCR4 has been characterized as a key player in the development of AML due to the abundant expression of SDF-1 within the bone marrow microenvironment. CXCR4 expression is essential for enabling AML cells to home to the bone marrow (1-3). Conversely, the CXCR4 antagonist AMD3100 causes mobilization of cells from the protective bone marrow microenvironment. This agent has been approved by the Food and Drug Administration for treatment of lymphoma and multiple myeloma in combination with chemotherapeutics to target the displaced cancer cells. In contrast, AMD3100 has not thus far been approved for treatment of AML (4). Although AMD3100 does mobilize a majority of AML cells from the bone marrow, the leukemic stem cells appear to resist mobilization, remain in the bone marrow and contribute to the relapse seen in the murine models (5-9). These unexpected effects indicate the need to better understand the role of CXCR4 in AML.
In addition to initiating migration and ERK activation, CXCR4 has also been shown to contribute to apoptosis in a variety of cell types. In response to prolonged exposure to SDF-1, CXCR4 has been shown to induce apoptosis in T cells and colorectal carcinoma cells (10-12). Moreoever, lymphocytes, neurons, breast cancer cells, endothelial cells, and cardiomyocytes all undergo apoptosis in response to CXCR4 binding to gp120, the envelope glycoprotein of HIV-1 (13-16).
Previous studies have suggested that CXCR4 can activate death signaling via either the extrinsic or intrinsic pathway. The extrinsic pathway is triggered when ligands on initiator cells bind to death receptors on target cells, leading to caspase-8 activation and downstream apoptotic events in
those target cells. The intrinsic apoptotic pathway is activated by changes that tip the balance between anti- and pro-apoptotic members of the Bcl-2 family. Upon activation, the effector members of this family, Bak and Bax, oligomerize and permeabilize the outer mitochondrial membrane, resulting in the release of cytochrome C, activation of procaspase-9 and subsequent caspase-mediated cleavages. The effectors are in turn regulated by the two other classes of Bcl-2 family members. The BH3-only containing pro-apoptotic members, which include Bim, Puma, Bid and Noxa, promote apoptosis by directly binding and inducing the oligomerizaton of Bak and/or Bax or by neutralizing the anti-apoptotic members. The anti-apoptotic family members directly bind and antagonize Bak and Bax or sequester the BH3-only family members to prevent interactions with the effectors (17-19). In AML cells, various reagents have been described to induce apoptosis via altering levels of different Bcl-2 family members, suggesting that AML cells are sensitive to regulation of Bcl-2 family proteins (20-23).
Many studies analyzing the role of CXCR4 in AML have focused on inhibiting CXCR4 signaling by utilizing AMD3100 or other CXCR4 antagonists. Based on these prior studies, we set out to determine the signaling mechanisms involved in SDF-1-mediated survival of AML cells. However, we found that SDF-1 did not promote AML cell survival, but instead induced apoptosis of AML cells expressing CXCR4. Here, we characterize a novel SDF-1-mediated apoptosis pathway in AML cells and provide evidence suggesting that this intrinsic apoptotic mechanism is normally suppressed within the bone marrow microenvironment. These results describe a new role for SDF-1/CXCR4 signaling in AML cells and indicate that SDF-1/CXCR4 functions in AML are more complex than previously appreciated. EXPERIMENTAL PROCEDURES Materials. Reagents were obtained from the following suppliers: the broad spectrum caspase inhibitor Q-VD-OPh (SM Biochemicals, Anaheim, CA); Calcein-AM (Invitrogen, Carlsbad, VA); SDF-1 (R&D Systems, Minneapolis, MN); pertussis toxin and control toxin (pertussis toxin B-oligomer) (Millipore, Billerica, MA); and
by guest on June 9, 2018http://w
ww
.jbc.org/D
ownloaded from

SDF-1 modulates Bcl-XL, Noxa, and Bak to induce apoptosis
3
Hoechst 33258, cytarabine, cycloheximide, and AMD3100 (Sigma-Aldrich, St. Louis, MO). CCX704 and CCX711 were kind gifts from ChemoCentryx, Mountain View, CA. PD325901 and CI-1040 (Parke Davis, Ann Arbor, MI) were kind gifts from Judith Siebolt.
Antibodies for immunoblotting or flow cytometry were obtained from the following suppliers: rabbit anti-Bak (Millipore); rabbit polyclonal antibodies to Bax, Bim, Bcl-XL, Mcl-1 and procaspase-9 (Cell Signaling Technology, Danvers, MA); murine monoclonal anti-Bcl-2 (Dako, Carpenteria, CA); murine monoclonal anti-Noxa (Enzo Life Sciences, Farmingdale, NY); murine monoclonal anti-GFP (Clontech, Mountain View, CA); murine monoclonal anti-actin (Novus Biologicals, Littleton, CO); rabbit anti-ERK (Santa Cruz Biotechnology, Dallas, Texas); murine monoclonal antibodies to CXCR4, CXCR7, and EGFR (R&D Systems); and APC-conjugated annexin-V, Alexafluor-647-conjugated antibody to cleaved PARP, and an active caspase-3 apoptosis kit (BD Biosciences, San Jose, CA).
Cells. After informed consent was obtained, samples of bone marrow were harvested from AML patients prior to chemotherapy according to an IRB-approved protocol. Following sedimentation on a Ficoll-Paque (1.077 g/cm3) step gradient (24), mononuclear cells were recovered and cultured in Medium A (RPMI 1640 supplemented with 10% FCS, 2 mM L-glutamine, 1 mM sodium pyruvate, non-essential amino acids, and 5.5 μM 2-ME) at a density of 1 million cells per ml. The AML cell lines, KG1a and U937, and the E6 Jurkat acute T cell leukemia line (ATCC, Manassas, VA), were cultured in RPMI supplemented with 10% FCS at a cell density of 0.6-1.0 million cells per ml. HeLa cells (ATCC) were cultured to confluency in RPMI supplemented with 10% FCS.
To generate CXCR4-expressing KG1a, U937, and E6 Jurkat cells, we transiently-transfected cells with a plasmid encoding a CXCR4-YFP fluorescent fusion protein (25) using a BTX 830 square wave electroporator at 240 V for 10 ms, incubated for 16-18 hrs to allow expression of the transgene, and treated as indicated in the various figures. The transfection efficiencies of KG1a and U937 with control YFP empty vector were typically 10-20% and 10-15%, respectively. The
E6 Jurkat T cell line was transfected as previously described (25). Flow cytometric data were gated to compare cells expressing similar levels of YFP. To stably suppress either Bak or Bim protein expression, KG1a cells were transduced with lentiviral particles that target the sequences CCCATTCACTACAGGTGAA for Bak or GACCGAGAAGGTAGACAATTGC for Bim in pSIH-H1, respectively (26, 27). Following transfection of plasmids encoding shRNA or empty vector into HEK293 cells along with the packaging plasmids psPAX2 and pMD2.G, supernatants were collected and filtered through 0.45 µm filters. After viral infection on two successive days, KG1a cells were cultured in medium containing 2 µg/ml puromycin for a week. Bak- and Bim-specific antibodies were used to assay Bak and Bim expression via western blot. To transiently suppress Noxa, Bak, or Bim protein expression, KG1a or Jurkat cells were transiently transfected as indicated above with the following siRNAs from Ambion (Austin, TX): Control siRNA (Silencer® Negative Control #1); Noxa siRNA: sense 5’GGAGAUUUGGAGACAAACUTT3’ and antisense 5’AGUUUGUCUCCAAAUCUCCTG3’; Bak siRNA: sense: GUACGAAGAUUCUUCAAAUTT and antisense: AUUUGAAGAAUCUUCGUACCA; and Bim siRNA: sense 5’GACCGAGAAGGUAGACAAUTT3’ and antisense 5’AUUGUCUACCUUCUCGGUCTT3’. The cells were cultured for 6-7 hr prior to addition of SDF-1. Depletion of Noxa, Bak, and Bim was confirmed by immunoblotting with specific antibodies using lysates prepared 24 hr after transfection.
Tert-immortalized bone marrow derived stromal cells (BMSC) were a kind gift of Dario Campana (St. Jude Children’s Research Hospital, Memphis, TN)(28, 29). BMSC were cultured in RPMI supplemented with 10% FCS, 2 mM L-glutamine, and 1 µM hydrocortisone prior to addition of AML isolates, at which time the coculture was maintained in Medium A.
Assaying CXCR4 levels. Clinical AML isolates were cultured for 16-18 hr in Medium A, then
by guest on June 9, 2018http://w
ww
.jbc.org/D
ownloaded from

SDF-1 modulates Bcl-XL, Noxa, and Bak to induce apoptosis
4
assayed for CXCR4 cell surface expression by flow cytometry (30) after staining with a CXCR4 monoclonal antibody. For assaying total cellular CXCR4 levels, cells were incubated at 37 °C for 10 min in BD Phosflow Lyse/Fix buffer, sedimented, permeabilized on ice for 30 min with BD Perm Buffer III, washed with BD stain buffer (BD Biosciences), stained with either CXCR4 mAb or a control, anti-EGFR mAb, for 30 min at room temperature, and assayed by flow cytometry. Where indicated, the KG1a cells were treated + 5 x 10-8 M SDF-1 for 20 min prior to staining for CXCR4.
Assays of migration and active ERK. KG1a cells were transfected with either CXCR4-YFP or YFP empty vector, and cultured for 18 hr prior to the migration assay or ERK activation assay. For the migration assay, the cells were stained with calcein-AM and assayed for migration as described in (31). Where indicated, either 10 µM PD325901, 10 µM CI-1040 or DMSO (vehicle) was added 16-18 hr prior to addition of SDF-1; or either 100 ng/ml pertussis toxin or its control toxin was added 4 hr prior to addition of SDF-1. For analysis of active phospho-ERK, cells were stimulated + 5 x 10-8 M SDF-1 and assayed by flow cytometry (31). Flow cytometric data was gated to compare ERK phosphorylation in cells expressing similar levels of YFP.
Apoptosis Assays. For assays measuring apoptosis, the indicated cells were cultured at 0.25 x 106 cells/ml in the presence or absence of 1.3 x 10-8 M SDF-1 or 6 ng/ml of CH.11 agonistic anti-Fas antibody (Millipore) for 16-18 hr and then assayed for apoptosis using one of the techniques listed below. Where indicated, 30 µM AMD3100, 1 µM CCX704, 1 µM CCX771, 10 µM PD325901, 10 µM CI-1040 was added 1 hr prior to addition of SDF-1; 100 ng/ml pertussis toxin or its control toxin was added 4 hr prior to addition of SDF-1; cells were placed in a hypoxia chamber at 37°C, 5% CO2, and 1% O2 for 16-18 hr prior to the annexin V staining; cells were incubated for 2 hr under hypoxic or normoxic conditions and then treated with 1 µM cytarabine for 16-18 hr in the indicated oxygen conditions; cells were cotransfected with CXCR4-YFP and a plasmid encoding the viral c-FLIP homolog MC159 (a
kind gift from Greg Gores, Mayo Clinic, Rochester, MN), a dominant-negative caspase-9/EGFP fusion protein (obtained from Emad Alnemri, Thomas Jefferson University, Philadelphia, PA), human Noxa bearing L29A and D34A mutations (Noxa-2A) (32) fused to EGFP or S-peptided-tagged-Bcl-XL; or cells were transfected with GFP-Bak or GFP-Noxa. MC159 expression was confirmed via semi-quantitative RT-PCR with the following primers: MC159 Forward: ATGTCCGACTCCAAGGAGGTCCCTAG; MC159 Reverse: TCAAGTCGTTTGCTCGGGGCTGTCGCTG; GAPDH Forward: GGCAAATTCCATGGCACCGTCAGG; and GAPDH Reverse: GGAGGCATTGCTGATGATCCTGAGG.
For detecting cell surface phosphatidylserine, cells were stained with APC-conjugated annexin-V as described (33). To detect fragmented nuclei, cells were washed with PBS, resuspended in 3:1 methanol:acetic acid, incubated at room temperature for 1 hr, centrifuged, and mounted in 50% glycerol/50% 0.1 M Tris-HCl (pH 7.4) containing 1 µg/ml Hoechst 33258 (34). The cells were then visualized 16 hr later with Hoechst/DAPI filters. Cleaved PARP and active caspase-3 were assayed by flow microfluorimetry in YFP positive cells using an Alexafluor-647-conjugated antibody to cleaved PARP and an active caspase-3 apoptosis kit, respectively, according to the suppliers’ instructions.
Apoptosis assay with clinical AML isolates. AML isolates were cultured in Medium A for 1-2 hours prior to plating on BMSC. The isolates were plated at a density of 0.25 x 106 cells/ml onto a confluent layer of BMSC for 1 hr and then 5 x 10-8 M SDF-1 was added for 16-18 hr in Medium A. Annexin V staining was performed as described above.
Detecting Bcl-2 family members by qRT-PCR and western blot. For the quantitative RT-PCR (qRT-PCR) and the immunoblots of Bcl-2 family members, KG1a cells were transfected with CXCR4-YFP, treated with 5 µM Q-VD-OPh, cultured for 16-18 hr + SDF-1, and then sorted for the brightest 12% of YFP-expressing cells. The qRT-PCR was performed in triplicate using 100
by guest on June 9, 2018http://w
ww
.jbc.org/D
ownloaded from

SDF-1 modulates Bcl-XL, Noxa, and Bak to induce apoptosis
5
ng RNA and TaqMan One-Step RT-PCR Master Mix (Applied Biosystems) per the supplier's instructions. Using GAPDH (4352934E), Bak (Hs00832876_g1), Bax (Hs00180269_m1), Bcl-2 (Hs00608023_m1), Bcl2A1 (Hs00187845_m1), Bcl-XL (Hs00236329_m1), Bim (Hs00197982_m1), Mcl-1 (Hs03043898_m1), Noxa (Hs00560402_m1), Bmf (Hs00372937_m1), and BNIP3 (Hs00969291_m1) probe sets, the PCR was performed as previously described (35). For detection of protein levels of the Bcl-2 family members by western blot, the sorted cells were sedimented at 200 x g for 10 min, washed once with ice-cold RPMI 1640 medium containing 10 mM HEPES (pH 7.4 at 4°C) and prepared for electrophoresis as described (36).
Analyzing Noxa stability. KG1a cells were cotransfected with CXCR4-YFP and Noxa2A-GFP, cultured for 16 hr with the caspase inhibitor Q-VD-OPh in the presence or absence of SDF-1, then treated with 25 µg/ml cycloheximide for the indicated time, fixed with paraformaldehyde, and analyzed via flow microfluorimetry for Noxa2A-GFP expression in gated CXCR4-YFP positive cells. The amount of Noxa2A-GFP remaining after the indicated cycloheximide treatment was determined as a percent of the Noxa2A-GFP present at the 0 hr time point. RESULTS CXCR4 is expressed at variable levels on AML cells. In initial experiments, we observed that CXCR4 is expressed at varying levels on the cell surface of primary AML cells from patient bone marrow (Fig. 1A). We also confirmed that CXCR4 is expressed intracellularly in patient samples, as indicated by the increased signal observed upon permeabilizing the plasma membrane prior to CXCR4 staining (Fig. 1B). Patient data shown in Figs. 1A&B are representative of all patients analyzed during the study period (Supplementary Table 1). These results confirm previous findings that CXCR4 is expressed variably on the cell surface of AML isolates, and that CXCR4 is maintained intracellularly at high levels in most AML isolates (9, 37, 38).
To begin to characterize the signaling pathways initiated by SDF-1/CXCR4 signaling in AML cells, we utilized the human M0 AML cell
line KG1a , which has been extensively studied as a model of human AML. KG1a cells lack CXCR4 expression on the cell surface (Fig. 1C) (9, 37, 39), however, KG1a do express CXCR4 that is maintained inside the cell as shown by CXCR4 staining following permeabilization (Fig. 1C and (9)). To analyze the effects of SDF-1 treatment on KG1a, we transiently overexpressed CXCR4 at sufficiently high levels to achieve cell-surface expression. A CXCR4-YFP fluorescent fusion protein was used for this purpose in order to permit the unambiguous identification of individual, transfected cells expressing similar levels of CXCR4 via flow cytometry. Here and below, we refer to these CXCR4-expressing KG1a cells as “KG1a-CXCR4”. Control cells consisted of KG1a cells transiently-transfected with a YFP-encoding vector in the same experiment. Compared to cells transiently transfected with a control vector, the KG1a-CXCR4 displayed elevated cell-surface levels of CXCR4 (Fig. 1C). As expected the level of CXCR4 on the cell surface of KG1a-CXCR4 cells was downregulated in response to SDF-1 (Fig. 1C).
SDF-1 signaling induces migration and ERK activation in the KG1a-CXCR4. SDF-1/CXCR4 signaling is critical for homing of AML cells to the SDF-1-rich bone marrow microenvironment where they are protected from chemotherapeutic agents (1, 2, 40). Consistent with these results, transient CXCR4 expression enabled SDF-1 induced migration of KG1a cells, in contrast to KG1a cells transfected with a vector control plasmid (p < 0.05, Fig. 2A).
Next we assessed activation of the ERK mitogen-activated protein kinase, a key initiator of SDF-1-induced survival pathways in a variety of cell types (41, 42). Transfected cells were treated with SDF-1 for the indicated times, and levels of active, phosphorylated ERK in individual cells were assayed by flow microfluorimetry. KG1a-CXCR4 cells responded to SDF-1 treatment by significantly increasing levels of active, phosphorylated ERK at 2 and 5 min, with this response declining at 8 min (p < 0.05, Fig. 2B&C). In contrast, there was no change in ERK phosphorylation in cells expressing empty vector. In summary, KG1a cells transiently expressing CXCR4 are able to migrate and increase mitogen-activated protein kinase signaling in response to
by guest on June 9, 2018http://w
ww
.jbc.org/D
ownloaded from

SDF-1 modulates Bcl-XL, Noxa, and Bak to induce apoptosis
6
SDF-1. These results are consistent with a previous report that similarly analyzed KG1a cells overexpressing CXCR4 (40).
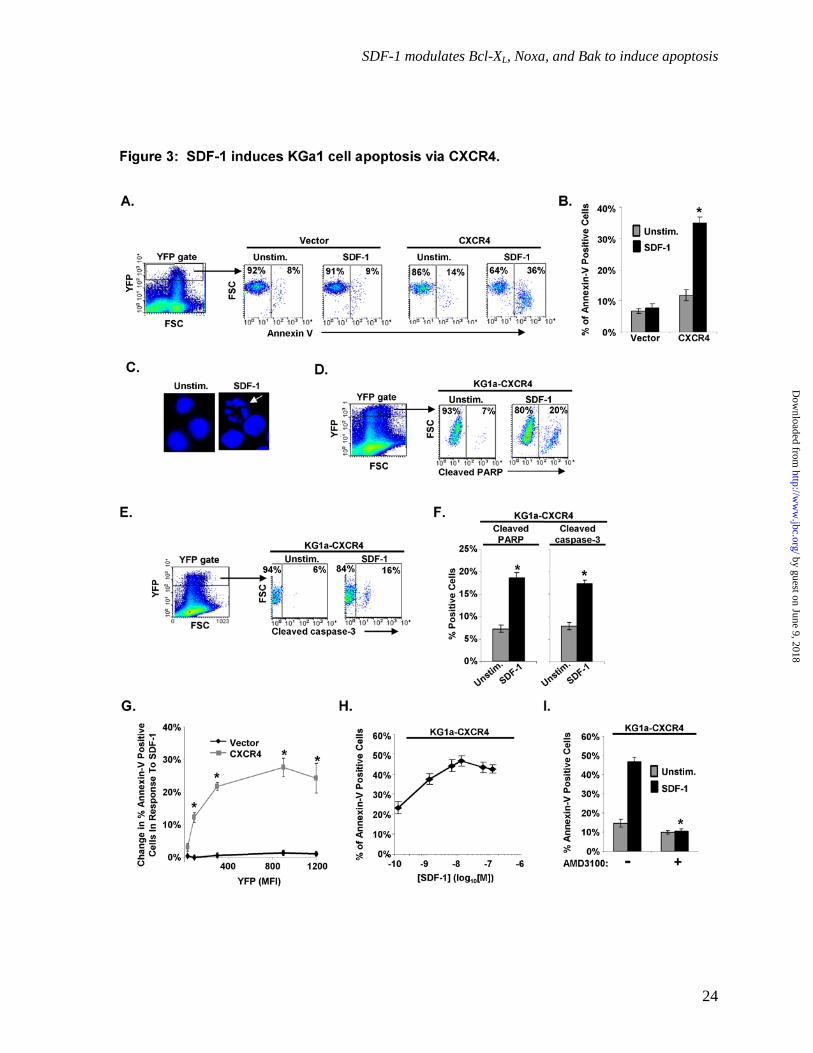
SDF-1 induces KG1a cell apoptosis via CXCR4. Because KG1a-CXCR4 cells undergo SDF-1-induced migration and ERK activation, we anticipated that prolonged SDF-1 treatment would protect KG1a-CXCR4 cells from death. To assess survival, we assayed annexin-V staining on KG1a-CXCR4 cells following 16-18 hr of SDF-1 treatment. Surprisingly, SDF-1 treatment resulted in a 3-fold increase in the percentage of annexin-V positive KG1a-CXCR4 cells compared to unstimulated cells (p < 0.05, Fig. 3A&B). In contrast, cells transfected with the YFP vector control plasmid did not show a significant increase in annexin-V positive cells in response to SDF-1.
To rule out the possibility that increased annexin-V staining was a result of plasma membrane reorganization without apoptosis, as has been observed in mitogen-stimulated lymphocytes (43, 44) we stained SDF-1-treated cells with Hoechst 33258, a dye which allows visualization of nuclear morphological changes. As shown in Fig. 3C, KG1a-CXCR4 cells treated with SDF-1 displayed fragmented nuclei, a hallmark of apoptosis. Utilizing fluorochrome-conjugated antibodies specific for cleaved PARP and cleaved caspase-3, we also observed that SDF-1 treatment induced a significant increase in cleaved PARP and cleaved procaspase-3 in KG1a-CXCR4 cells (p < 0 .05, Fig. 3D-F).
To confirm that SDF-1 was causing apoptosis via CXCR4, we examined the relationship between levels of cell surface CXCR4 and annexin V binding. Our results demonstrate that increased CXCR4 on the cell surface, as indicated by YFP fluorescence, led to a significantly increased percentage of annexin-V positive cells following SDF-1 treatment (p < 0.05, Fig. 3G). Similarly, the percentage of apoptotic cells also increased in a dose-dependent manner with increasing amounts of SDF-1 (Fig. 3H). The concentration of SDF-1 that achieves half maximal increase in apoptosis is approximately 4.1 x 10-9 M, a result fully consistent with SDF-1 inducing apoptosis by binding to CXCR4 (25, 45). In addition, use of the CXCR4-specific antagonist, AMD3100, prevented SDF-1-induced apoptosis in KG1a-CXCR4 cells (p < 0.05, Fig. 3I). Collectively,
these results show that SDF-1 induces apoptosis in KG1a-CXCR4 cells via a mechanism requiring CXCR4.
SDF-1/CXCR4 induces apoptosis in the U937 AML cell line and in a subset of clinical AML isolates. To ensure that the apoptosis induced by SDF-1 was not unique to KG1a cells, we utilized U937, an M2 AML cell line. U937 cells express low levels of CXCR4 on the cell surface (9), lower even than many clinical AML isolates. Accordingly, we transiently-transfected U937 with CXCR4-YFP and gated on YFP positive cells, which we designated U937-CXCR4 cells (Fig. 4A). In contrast to U937 cells transfected with the vector control plasmid, SDF-1 significantly increased the percentage of annexin-V positive U937-CXCR4 cells (p < 0.05, Fig. 4B).
In addition, we obtained a number of clinical AML isolates and assayed for the ability of SDF-1 to induce apoptosis via endogenously expressed CXCR4. Interestingly, SDF-1 treatment increased the percentage of annexin-V positive cells in three of 10 isolates (Fig. 4C). Samples from patients 4 and 10, which had the highest CXCR4 cell surface expression, also displayed a substantial increase in SDF-1-induced apoptosis, suggesting that higher CXCR4 levels may correlate with increased apoptosis in response to SDF-1 (Fig. 1A&4C). Unfortunately, we were unable to assess CXCR4 cell surface levels in the sample from patient 3 due to the limited number of cells obtained. Together, the results in Figs. 1-4 indicate that SDF-1/CXCR4 signaling can induce apoptosis in CXCR4-expressing AML cell lines and also in a subset of clinical AML isolates expressing high levels of endogenous cell surface CXCR4.
Hypoxia protects CXCR4-expressing AML cell lines from apoptosis. Because SDF-1 is abundant in the bone marrow microenvironment, it was unclear how CXCR4-expressing AML cells could thrive in this environment if they are sensitive to SDF-1-induced cell death. We hypothesized that an environmental signal must override or inhibit SDF-1-induced apoptosis. One possible inhibitory signal may derive from areas of hypoxia in the bone marrow microenvironment. Expansion of hypoxic regions has been noted to occur during the progression of leukemia, with leukemic cells adapting and then proliferating in these hypoxic
by guest on June 9, 2018http://w
ww
.jbc.org/D
ownloaded from

SDF-1 modulates Bcl-XL, Noxa, and Bak to induce apoptosis
7
regions (2). When KG1a-CXCR4 cells were exposed to SDF-1 in an environment of 1% oxygen, significantly fewer cells underwent apoptosis as compared to KG1a-CXCR4 cells treated with SDF-1 in ambient oxygen as measured by staining for either Annexin-V or cleaved PARP (p < 0.05, Fig. 5A&B). Although SDF-1 was still able to induce an increase in apoptosis under hypoxic conditions, the overall level of apoptosis in SDF-1 stimulated cells under hypoxic conditions was low compared to normoxic conditions. These results suggest that hypoxia provides signals to protect AML cells from apoptosis. This protective effect of hypoxia on KG1a-CXCR4 cells treated with SDF-1 was not due to reduced CXCR4 cell surface levels (Fig. 5C). Similarly, both the unstimulated and SDF-1-treated U937-CXCR4 cells were significantly protected from apoptosis by the hypoxic environment (p < 0.05, Fig. 5D&E), again without any reduction in cell surface CXCR4. Indeed, the U937-CXCR4 cells significantly increased levels of cell-surface CXCR4 upon exposure to hypoxia (p < 0.05, Fig. 5F). To determine if hypoxia protects AML cells from apoptosis induced by a different treatment, we exposed KG1a cells to cytarabine, currently the most effective single agent for treatment of AML (46), and measured apoptosis under normoxic and hypoxic conditions. Compared to normoxic conditions, cytarabine-treated cells maintained in a hypoxic environment exhibited significantly less apoptosis (p < 0.05, Fig. 5G). These results suggest that hypoxia could provide a survival signal that allows AML cells to thrive in the bone marrow microenvironment, despite the presence of apoptosis-inducing agents, including SDF-1.
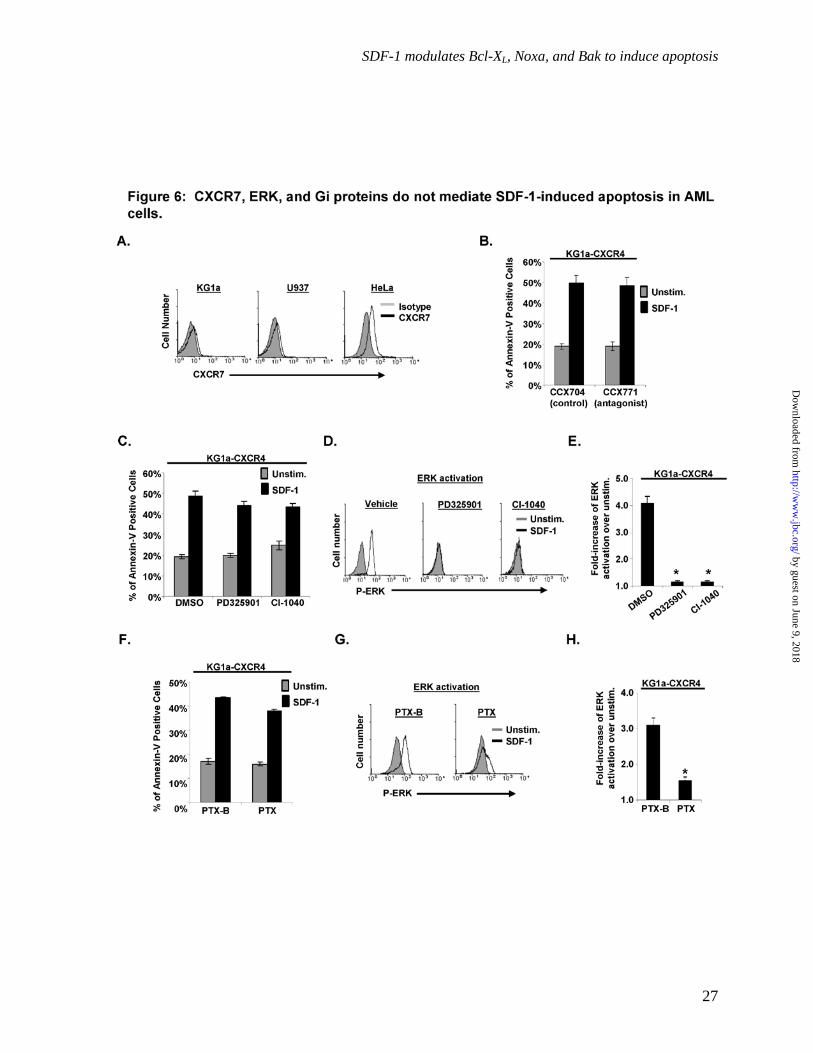
CXCR7, ERK activation, and Gi proteins do not mediate SDF-1-induced apoptosis in AML cells. To further confirm that SDF-1 was inducing apoptosis via CXCR4, we assessed the role of CXCR7 (47-50), the only other chemokine receptor known to bind SDF-1. CXCR7 was not detectably expressed on the cell surface of KG1a or U937 but was expressed on HeLa cells (Fig. 6A). Furthermore, treatment of the KG1a-CXCR4 cells with the CXCR7 antagonist, CCX771, did not inhibit the SDF-1-induced apoptosis compared to the KG1a-CXCR4 cells treated with the control compound, CCX704 (p > 0.05, Fig. 6B). Thus,
CXCR7 does not mediate SDF-1-induced apoptosis in AML cells.
ERK activation, which often mediates survival signals, has also been reported to induce apoptosis in some cell types (51-53). To determine whether the SDF-1-induced ERK activation demonstrated in Fig. 2B & C contributes to SDF-1-induced apoptosis, we exposed cells to the MEK inhibitors PD325901 or CI-1040 before SDF-1 treatment. As shown in Fig. 6C, pretreating KG1a-CXCR4 cells with these MEK inhibitors did not substantially inhibit SDF-1-induced apoptosis, even though the MEK inhibitors did effectively abolish ERK activation in response to SDF-1 (p < 0.5, Fig. 6D&E).
Signaling by Gi-type G proteins has previously been shown to induce apoptosis in hepatocytes and neutrophils (54, 55), and CXCR4 is able to signal through Gi-type G proteins in response to SDF-1 (25, 48, 56). To address whether CXCR4 utilizes Gi-type G proteins to induce apoptosis, we treated KG1a-CXCR4 cells with pertussis toxin (PTX), an exotoxin that inhibits Gi-type G proteins. PTX did not substantially inhibit SDF-1-induced apoptosis in KG1a-CXCR4 cells as compared to cells treated with the control toxin PTX-B (p < 0.5, Fig. 6F). However, PTX did significantly inhibit SDF-1-induced ERK activation in KG1a-CXCR4 cells (p < 0.05, Fig. 6G&H). Together, the results in Fig. 6 indicate that SDF-1/CXCR4-induced apoptosis is not mediated by pathways that require signaling by CXCR7, ERK, or Gi-type G proteins.
Requirement for the caspase-9/intrinsic death pathway, but not the caspase-8/extrinsic pathway, for SDF-1/CXCR4-mediated apoptosis. To further characterize the apoptosis induced by SDF-1/CXCR4 signaling in AML cells, we first determined whether the intrinsic or extrinsic apoptotic pathway was utilized. The Molluscum contagiosum virus MC159 protein inhibits caspase-8-dependent death receptor pathways, including those mediated by Fas, tumor necrosis factor (TNF), and TNF-related apoptosis inducing ligant (TRAIL) (57). MC159 expression (Fig. 7E) failed to protect KG1a-CXCR4 cells from SDF-1-induced apoptosis (Fig. 7A) while, as expected, MC159 expression significantly inhibited the apoptosis of Jurkat cells in response to a Fas agonist (CH.11) (p < 0.05, Fig. 7B) (58),
by guest on June 9, 2018http://w
ww
.jbc.org/D
ownloaded from

SDF-1 modulates Bcl-XL, Noxa, and Bak to induce apoptosis
8
demonstrating the ability of this construct to inhibit the extrinsic apoptotic pathway. In contrast, expression of a dominant-negative Caspase-9 (DN-Casp-9-GFP) in KG1a-CXCR4 cells significantly diminished SDF-1-induced apoptosis (p < 0.05, Fig. 7A&D). Similarly, U937-CXCR4 cells coexpressing DN-Casp-9-GFP displayed significantly decreased levels of apoptosis compared to U937-CXCR4 cells coexpressing the control vector or MC159 (p < 0.05, Fig. 7C-E). Collectively these results are consistent with the view that SDF-1/CXCR4 signaling induces apoptosis in KG1a and U937 cells via a caspase-9-dependent apoptotic pathway.
Role of SDF-1-mediated Bak upregulation in SDF-1/CXCR4-mediated apoptosis. To evaluate potential mechanisms for SDF-1-induced activation of the intrinsic pathway, we assessed the effect of SDF-1 on levels of mRNA encoding the anti-apoptotic Bcl-2 family members Bcl-2, Bcl-2A1, Bcl-XL, and Mcl-1, as well as the pro-apoptotic Bcl-2 family members Bim, Noxa, Bmf, BNIP3, Bak, and Bax in KG1a-CXCR4 cells. As shown in Figure 8A, only Bak mRNA levels in KG1a-CXCR4 cells were substantially altered by SDF-1 treatment (p < 0.05). Consistent with these results, western blotting showed that SDF-1 also upregulates Bak at the protein level in KG1a-CXCR4 cells (Fig. 8B).
We next determined whether increased expression of Bak alone was sufficient to induce apoptosis. Transiently expressing a plasmid encoding GFP-Bak in KG1a cells resulted in a dramatic decrease in the number of GFP positive cells compared to the control GFP vector (Fig. 8C). Treating the GFP-Bak expressing cells with the caspase inhibitor Q-VD-OPh resulted in a greater number of GFP positive cells (Fig. 8C), suggesting that GFP-Bak is inducing apoptosis before expression levels get very high. Indeed, ~93% of GFP-Bak expressing cells were annexin-V positive in the absence of Q-VD-OPh (Fig. 8D&E).
To further confirm the role of Bak in SDF-1-induced apoptosis, we created KG1a cells that had Bak stably knocked down by shRNA to ~30 % of control levels (Fig. 8F). Compared to the KG1a-vector shRNA control cells expressing CXCR4-YFP, KG1a-shBak cells expressing CXCR4 exhibited significantly less apoptosis after
treatment with SDF-1 (p < 0.05, Fig. 8G). As a control, despite an 85% reduction in Bim, a KG1a-shBim cell line expressing CXCR4 had similar levels of SDF-1-induced apoptosis as the KG1a-vector shRNA control cells (Fig. 8H&I). These results indicate that Bak is required to mediate SDF-1-induced apoptosis and that SDF-1-mediated upregulation of Bak is sufficient to induce apoptosis in CXCR4-expressing AML cells.
SDF-1-induced downregulation of Bcl-XL and upregulation of Noxa contribute to apoptosis of AML cells. To assess the potential role of SDF-1-induced changes in other Bcl-2 family members, we assayed the protein levels of Bcl-XL, Bcl-2, Mcl-1, Noxa and Bax in KG1a-CXCR4 cells treated with SDF-1. As shown in Fig. 9A, Noxa expression increased and Bcl-XL expression decreased in response to SDF-1 treatment, while Bcl-2, Mcl-1 and Bax remained unchanged. Noxa is a pro-apoptotic Bcl-2 family member that, upon binding to Bak increases apoptosis, while Bcl-XL is an anti-apoptotic family member that inhibits apoptosis upon binding Bak (32, 59, 60).
To analyze the potential contribution of Bcl-XL downregulation on SDF-1-induced death, we evaluated whether forced overexpresson of Bcl-XL could protect the cells. As anticipated, fewer KG1a-CXCR4 cells expressing S peptide-tagged Bcl-XL bound annexin-V compared to KG1a-CXCR4 cells expressing the vector control (p < 0.05, Fig. 9B&C). Thus, in combination with Bak upregulation, SDF-1 utilizes Bcl-XL downregulation as an additional mechanism to mediate apoptosis in AML cells.
To determine whether increased expression of pro-apoptotic Noxa is sufficient to increase apoptosis in AML cells, we transiently expressed a plasmid encoding GFP-Noxa in KG1a cells (Fig. 9D). Similar to transient expression of GFP-Bak (Fig. 8C&D), treatment with Q-VD-OPh increased the number of cells expressing GFP-Noxa compared to the untreated cells (Fig. 9E), indicating that induction of apoptosis limits how much Noxa can be expressed in these cells. Likewise, approximately 92% of GFP-Noxa expressing cells were annexin-V positive in the absence of Q-VD-OPh (p < 0.05, Fig. 9F&G), indicating that increased expression of Noxa can mediate apoptosis in KG1a cells.
by guest on June 9, 2018http://w
ww
.jbc.org/D
ownloaded from

SDF-1 modulates Bcl-XL, Noxa, and Bak to induce apoptosis
9
To address the mechanism by which SDF-1 increases Noxa expression, we analyzed the stability of Noxa protein in the cells stimulated with SDF-1. Because the elevated levels of endogenous Noxa that arise in response to SDF-1 rapidly cause the apoptosis of these cells (Fig. 9F&G), we measured turnover of Noxa2A-GFP, a fluorescent fusion protein of Noxa that has amino acids L29 and D34 mutated to inhibit its interaction with Bak and ability to cause apoptosis (32). SDF-1 treatment significantly increased the stability of Noxa2A-GFP in KG1a-CXCR4 cells as compared to unstimulated cells (p < 0.05, Fig. 9H).
To assess whether the increase in Noxa protein expression contributes to apoptosis in response to SDF-1, we decreased Noxa expression via siRNA (Fig. 9I). Noxa depletion resulted in a significant decrease in SDF-1-induced apoptosis as well as in the basal level of apoptosis (p < 0.05, Fig. 9J). Thus, SDF-1 treatment increases Noxa expression and thereby apoptosis in AML cells, via a mechanism that involves increased stability of the Noxa protein.
To determine whether these results were unique to AML cells, we examined the effect of SDF-1 treatment after CXCR4 expression in Jurkat T-cell ALL cells. Interestingly, an SDF-1-induced apoptosis pathway involving Noxa and Bak that is similar to the SDF-1-induced apoptosis pathway in AML cells was discerned. Jurkat cells endogenously express high levels of CXCR4 and transiently transfecting Jurkat cells with CXCR4 (creating Jurkat-CXCR4 cells) further elevated expression of CXCR4 (Fig. S1A). As shown if Fig. S1B, SDF-1 did not increase apoptosis in Jurkat cells transiently transfected with a control vector. In contrast, SDF-1 dramatically increased the percentage of Jurkat-CXCR4 cells undergoing apoptosis (p < 0.05, Fig. S1B). Thus, increasing CXCR4 levels above endogenous levels permits SDF-1 treatment to drive apoptosis in Jurkat cells. To assess whether SDF-1 utilizes similar mechanisms to induce apoptosis in Jurkat cells as in AML cells, we used siRNA to knock down Noxa, Bak, and Bim (Fig. S1C). In contrast to cells depleted of Bim, depletion of either Noxa or Bak prevented SDF-1-induced apoptosis in Jurkat-CXCR4 cells (Fig. S1D) as it did in AML cells (Figs. 8 & 9). Thus, high levels of CXCR4 may be
capable of mediating apoptosis in other cell types in addition to AML cells.
Collectively, the results in Figs. 8 and 9 indicate that SDF-1 potently induces the apoptosis of KG1a AML cells by a multifactorial intrinsic mechanism involving upregulation of the apoptosis effector Bak and BH3-only protein Noxa with the simultaneous downregulation of the anti-apoptotic family member Bcl-XL.
DISCUSSION
Although the initial response rate to current AML therapies is a promising 60-70 %, relapse and death within a few years of initial treatment continues to be a frequent outcome. Relapse is most often mediated by resistance to therapy that is likely driven by the difficulty of eradicating the leukemia stem cells residing within the protected bone marrow microenvironment. Chemotherapeutics are not as effective at targeting the AML cells present in the bone marrow (1, 2, 61). AMD3100, the CXCR4 antagonist, showed initial promise in removing a large number of AML cells from the bone marrow to the blood stream where they could then be targeted by the chemotherapeutics. More recent evidence, however, indicates that AMD3100 may have a more limited benefit in AML because CXCR4 appears to only be required for initial homing of AML cells to the bone marrow, not for their retention within the bone marrow (1, 9, 62, 63). Thus, targeting leukemic stem cells within the bone marrow microenvironment may be a more effective strategy for eliminating these cells.
We show here for the first time that SDF-1 treatment initiates an intrinsic apoptotic pathway in AML cells expressing CXCR4. Essential features of this pathway (Fig. 10) include upregulation of Bak expression at the mRNA and protein level as well as increasing the stability and protein expression of the Bak activator Noxa (32) and downregulation of the Bak inhibitor Bcl-XL (59, 60). As summarized below, the effects of a variety of pathway perturbations, including treatment with AMD3100; forced alterations in expression of Bak, Noxa and Bcl-XL; and treatment with dn-procaspase 9 or caspase inhibitors, are consistent with this model.
We characterized the SDF-1-mediated apoptotic process utilizing the AML cell lines KG1a and U937 as well as AML isolates from
by guest on June 9, 2018http://w
ww
.jbc.org/D
ownloaded from

SDF-1 modulates Bcl-XL, Noxa, and Bak to induce apoptosis
10
patients. Transient expression of CXCR4 on KG1a resulted in the anticipated migration and ERK activation in response to short treatments with SDF-1 (Fig. 2) as has been previously characterized for SDF-1/CXCR4 signaling (40). Surprisingly, ERK activation in response to SDF-1 did not promote the survival of KG1a cells expressing CXCR4. Instead, an 18 hr SDF-1 treatment triggered apoptosis, as indicated by phosphatidylserine exposure on the cell surface, nuclear fragmentation, activation of caspase-3, and cleavage of PARP (Fig. 3).
We also show here that SDF-1 induced apoptosis in 3 of 10 clinical AML isolates, while the level of apoptosis remained largely unchanged in the other patient isolates (Fig. 4C). Our additional studies confirmed that cell surface expression of CXCR4 varied widely among clinical AML isolates. Importantly, both of the AML isolates with high levels of CXCR4 had an increase in apoptosis in response to SDF-1 (cf. Figs. 1A and 4C). Unfortunately, the third clinical sample that exhibited SDF-1-induced apoptosis had too few cells to assay CXCR4 levels. Nonetheless, the correlation between high cell surface CXCR4 levels and SDF-1-induced apoptosis in the samples where both assays could be performed likely indicates that a subset of AMLs, i.e., the set with high cell surface CXCR4 expression, is sensitive to SDF-1-induced apoptosis. Further analysis of clinical samples will be required to determine whether SDF-1 sensitive phenotype tracks with any other clinicopathological parameter. Meanwhile, we also showed that CXCR4 was retained inside the cell at relatively high levels in all of the AML isolates assayed, in agreement with previous studies (9, 37). Retention of pools of intracellular CXCR4 has been described in a variety of cancers, however, the mechanisms utilized to prevent transport to the cell surface have not been described (64-66). In view of the relationship between cell surface CXCR4 expression and SDF-1-induced apoptosis, strategies to force trafficking of CXCR4 to the cell surface could alter the survival of these cancer cells.
Our studies showed that SDF-1 mediates apoptosis via a pathway that involves modulation of Bak, Noxa and Bcl-XL as well as the initiator caspase of the intrinsic pathway, procaspase-9 (Fig. 10). We further showed that this pathway
does not require signaling by CXCR7, ERK activation, or Gi-type G proteins. To characterize the mechanism of this SDF-1-mediated apoptosis, we analyzed the SDF-1-induced changes in protein expression levels of Bcl-2 family proteins as well as used procaspase-8 and procaspase-9 inhibitors. Further support for our model comes from the ability of Bak downregulation, Noxa downregulation, or Bcl-XL overexpression to protect the cells from SDF-1-induced apoptosis. We previously demonstrated that Noxa can directly bind and oligomerize Bak (32). Utilizing Noxa2A-GFP, a form of Noxa that cannot bind Bak and kill the cells, we show here that SDF-1 increases the half-life of Noxa as a means to mediate apoptosis. Conversely, we and others have shown that Bcl-XL has the ability to bind and neutralize active Bak (59, 60). Thus, our results together indicate that SDF-1 treatment affects three related proteins in the mitochondrial apoptotic pathway in a concerted fashion to mediate apoptosis.
If SDF-1 can induce apoptosis in CXCR4-expressing AML cells, how can AML cells survive in the SDF-1-rich environment of the bone marrow? A variety of cancers have co-opted hypoxia-driven signaling pathways in order to resist the death initiated by apoptosis-inducing agents, via a large variety of mechanisms including hypoxia-inducible factors (HIFs), p53, and regulation of the Bcl-2 family members (67, 68). We show here that hypoxic conditions significantly decrease the level of apoptosis seen in AML cells in either the absence or presence of apoptosis-inducing agents such as SDF-1 or cytarbine (Fig. 5). Thus, hypoxia provides a survival signal that can override apoptosis-inducing signals in AML cells. These observations highlight the importance of our ongoing studies of SDF-1-induced apoptosis in AML cells in hopes of understanding how its inhibition in the SDF-1-rich bone marrow microenvironment can be overcome.
In summary, the present results indicate that SDF-1 is not a factor utilized by the bone marrow microenvironment to protect AML cells. Instead, SDF-1 may be a means to induce robust apoptosis in CXCR4-expressing AML cells directly in the bone marrow microenvironment, particularly if the survival cues of the SDF-1-rich bone marrow can be disrupted. Further characterization of this
by guest on June 9, 2018http://w
ww
.jbc.org/D
ownloaded from

SDF-1 modulates Bcl-XL, Noxa, and Bak to induce apoptosis
11
novel pathway may provide valuable insights into targets that may be modulated to enhance the killing of AML cells.
by guest on June 9, 2018http://w
ww
.jbc.org/D
ownloaded from

SDF-1 modulates Bcl-XL, Noxa, and Bak to induce apoptosis
12
REFERENCES 1. Doan, P. L., and J. P. Chute. 2012. The vascular niche: home for normal and malignant
hematopoietic stem cells. Leukemia 26:54-62. 2. Konopleva, M. Y., and C. T. Jordan. 2011. Leukemia Stem Cells and Microenvironment:
Biology and Therapeutic Targeting. J Clin Oncol 29:591-599. 3. Ceradini, D. J., A. R. Kulkarni, M. J. Callaghan, O. M. Tepper, N. Bastidas, M. E.
Kleinman, J. M. Capla, R. D. Galiano, J. P. Levine, and G. C. Gurtner. 2004. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med 10:858-864.
4. Pusic, I., and J. F. DiPersio. 2010. Update on clinical experience with AMD3100, an SDF-1/CXCL12-CXCR4 inhibitor, in mobilization of hematopoietic stem and progenitor cells. Curr Opin Hematol 17:319-326.
5. Clements, D., L. J. Markwick, N. Puri, and S. R. Johnson. 2010. Role of the CXCR4/CXCL12 axis in lymphangioleiomyomatosis and angiomyolipoma. J Immunol 185:1812-1821.
6. Monaco, G., M. Konopleva, M. Munsell, C. Leysath, R. Y. Wang, C. E. Jackson, M. Korbling, E. Estey, J. Belmont, and M. Andreeff. 2004. Engraftment of acute myeloid leukemia in NOD/SCID mice is independent of CXCR4 and predicts poor patient survival. Stem Cells 22:188-201.
7. Stolzel, F., M. Wermke, C. Rollig, C. Thiede, U. Platzbecker, and M. Bornhauser. 2010. Mobilization of PML/RARalpha negative peripheral blood stem cells with a combination of G-CSF and CXCR4 blockade in relapsed acute promyelocytic leukemia pre-treated with arsenic trioxide. Haematologica 95:171-172.
8. Zeng, Z., Y. X. Shi, I. J. Samudio, R. Y. Wang, X. Ling, O. Frolova, M. Levis, J. B. Rubin, R. R. Negrin, E. H. Estey, S. Konoplev, M. Andreeff, and M. Konopleva. 2009. Targeting the leukemia microenvironment by CXCR4 inhibition overcomes resistance to kinase inhibitors and chemotherapy in AML. Blood 113:6215-6224.
9. Tavor, S., I. Petit, S. Porozov, A. Avigdor, A. Dar, L. Leider-Trejo, N. Shemtov, V. Deutsch, E. Naparstek, A. Nagler, and T. Lapidot. 2004. CXCR4 regulates migration and development of human acute myelogenous leukemia stem cells in transplanted NOD/SCID mice. Cancer Res 64:2817-2824.
10. Colamussi, M. L., P. Secchiero, A. Gonelli, M. Marchisio, G. Zauli, and S. Capitani. 2001. Stromal derived factor-1 alpha (SDF-1 alpha) induces CD4+ T cell apoptosis via the functional up-regulation of the Fas (CD95)/Fas ligand (CD95L) pathway. J Leukoc Biol 69:263-270.
11. Herbein, G., U. Mahlknecht, F. Batliwalla, P. Gregersen, T. Pappas, J. Butler, W. A. O'Brien, and E. Verdin. 1998. Apoptosis of CD8+ T cells is mediated by macrophages through interaction of HIV gp120 with chemokine receptor CXCR4. Nature 395:189-194.
12. Drury, L. J., M. K. Wendt, and M. B. Dwinell. 2010. CXCL12 chemokine expression and secretion regulates colorectal carcinoma cell anoikis through Bim-mediated intrinsic apoptosis. PLoS One 5:e12895.
13. Castedo, M., J. L. Perfettini, K. Andreau, T. Roumier, M. Piacentini, and G. Kroemer. 2003. Mitochondrial apoptosis induced by the HIV-1 envelope. Ann N Y Acad Sci 1010:19-28.
by guest on June 9, 2018http://w
ww
.jbc.org/D
ownloaded from

SDF-1 modulates Bcl-XL, Noxa, and Bak to induce apoptosis
13
14. Ullrich, C. K., J. E. Groopman, and R. K. Ganju. 2000. HIV-1 gp120- and gp160-induced apoptosis in cultured endothelial cells is mediated by caspases. Blood 96:1438-1442.
15. Endo, M., A. Inatsu, K. Hashimoto, N. Takamune, S. Shoji, and S. Misumi. 2008. Human immunodeficiency virus-induced apoptosis of human breast cancer cells via CXCR4 is mediated by the viral envelope protein but does not require CD4. Curr HIV Res 6:34-42.
16. Lusso, P. 2006. HIV and the chemokine system: 10 years later. EMBO J 25:447-456. 17. Kaufmann, S. H., and W. C. Earnshaw. 2000. Induction of apoptosis by cancer
chemotherapy. Exp Cell Res 256:42-49. 18. Shamas-Din, A., H. Brahmbhatt, B. Leber, and D. W. Andrews. 2011. BH3-only
proteins: Orchestrators of apoptosis. Biochim Biophys Acta 1813:508-520. 19. Opferman, J. T. 2008. Apoptosis in the development of the immune system. Cell Death
Differ 15:234-242. 20. Rahmani, M., M. M. Aust, E. Attkisson, D. C. Williams, Jr., A. Ferreira-Gonzalez, and S.
Grant. 2012. Inhibition of Bcl-2 antiapoptotic members by obatoclax potently enhances sorafenib-induced apoptosis in human myeloid leukemia cells through a Bim-dependent process. Blood 119:6089-6098.
21. Schimmer, A. D. 2007. Novel therapies targeting the apoptosis pathway for the treatment of acute myeloid leukemia. Curr Treat Options Oncol 8:277-286.
22. Fathi, A. T., S. Grant, and J. E. Karp. 2010. Exploiting cellular pathways to develop new treatment strategies for AML. Cancer Treat Rev 36:142-150.
23. Konopleva, M., M. Milella, P. Ruvolo, J. C. Watts, M. R. Ricciardi, B. Korchin, T. McQueen, W. Bornmann, T. Tsao, P. Bergamo, D. H. Mak, W. Chen, J. McCubrey, A. Tafuri, and M. Andreeff. 2012. MEK inhibition enhances ABT-737-induced leukemia cell apoptosis via prevention of ERK-activated MCL-1 induction and modulation of MCL-1/BIM complex. Leukemia 26:778-787.
24. English, D., and B. R. Andersen. 1974. Single-step separation of red blood cells. Granulocytes and mononuclear leukocytes on discontinuous density gradients of Ficoll-Hypaque. J Immunol Methods 5:249-252.
25. Kumar, A., T. D. Humphreys, K. N. Kremer, P. S. Bramati, L. Bradfield, C. E. Edgar, and K. E. Hedin. 2006. CXCR4 physically associates with the T cell receptor to signal in T cells. Immunity 25:213-224.
26. Kepp, O., K. Rajalingam, S. Kimmig, and T. Rudel. 2007. Bak and Bax are non-redundant during infection- and DNA damage-induced apoptosis. EMBO J 26:825-834.
27. Huang, S., K. Okumura, and F. A. Sinicrope. 2009. BH3 mimetic obatoclax enhances TRAIL-mediated apoptosis in human pancreatic cancer cells. Clin Cancer Res 15:150-159.
28. Iwamoto, S., K. Mihara, J. R. Downing, C. H. Pui, and D. Campana. 2007. Mesenchymal cells regulate the response of acute lymphoblastic leukemia cells to asparaginase. J Clin Invest 117:1049-1057.
29. Mihara, K., C. Imai, E. Coustan-Smith, J. S. Dome, M. Dominici, E. Vanin, and D. Campana. 2003. Development and functional characterization of human bone marrow mesenchymal cells immortalized by enforced expression of telomerase. Br J Haematol 120:846-849.
30. Kumar, A., K. N. Kremer, D. Dominguez, M. Tadi, and K. E. Hedin. 2011. Galpha13 and Rho mediate endosomal trafficking of CXCR4 into Rab11+ vesicles upon stromal cell-derived factor-1 stimulation. J Immunol 186:951-958.
by guest on June 9, 2018http://w
ww
.jbc.org/D
ownloaded from

SDF-1 modulates Bcl-XL, Noxa, and Bak to induce apoptosis
14
31. Kremer, K. N., I. C. Clift, A. G. Miamen, A. O. Bamidele, N. X. Qian, T. D. Humphreys, and K. E. Hedin. 2011. Stromal cell-derived factor-1 signaling via the CXCR4-TCR heterodimer requires phospholipase C-beta3 and phospholipase C-gamma1 for distinct cellular responses. J Immunol 187:1440-1447.
32. Dai, H., A. Smith, X. W. Meng, P. A. Schneider, Y. P. Pang, and S. H. Kaufmann. 2011. Transient binding of an activator BH3 domain to the Bak BH3-binding groove initiates Bak oligomerization. J Cell Biol 194:39-48.
33. Lee, S. H., X. W. Meng, K. S. Flatten, D. A. Loegering, and S. H. Kaufmann. 2013. Phosphatidylserine exposure during apoptosis reflects bidirectional trafficking between plasma membrane and cytoplasm. Cell Death Differ 20:64-76.
34. Meng, X. W., M. P. Heldebrant, and S. H. Kaufmann. 2002. Phorbol 12-myristate 13-acetate inhibits death receptor-mediated apoptosis in Jurkat cells by disrupting recruitment of Fas-associated polypeptide with death domain. J Biol Chem 277:3776-3783.
35. Gupta, M., A. E. Hendrickson, S. S. Yun, J. J. Han, P. A. Schneider, B. D. Koh, M. J. Stenson, L. E. Wellik, J. C. Shing, K. L. Peterson, K. S. Flatten, A. D. Hess, B. D. Smith, J. E. Karp, S. Barr, T. E. Witzig, and S. H. Kaufmann. 2012. Dual mTORC1/mTORC2 inhibition diminishes Akt activation and induces Puma-dependent apoptosis in lymphoid malignancies. Blood 119:476-487.
36. Kaufmann, S. H., S. Okret, A. C. Wikstrom, J. A. Gustafsson, and J. H. Shaper. 1986. Binding of the glucocorticoid receptor to the rat liver nuclear matrix. The role of disulfide bond formation. J Biol Chem 261:11962-11967.
37. Mohle, R., F. Bautz, S. Rafii, M. A. Moore, W. Brugger, and L. Kanz. 1998. The chemokine receptor CXCR-4 is expressed on CD34+ hematopoietic progenitors and leukemic cells and mediates transendothelial migration induced by stromal cell-derived factor-1. Blood 91:4523-4530.
38. Spinello, I., M. T. Quaranta, R. Riccioni, V. Riti, L. Pasquini, A. Boe, E. Pelosi, A. Vitale, R. Foa, U. Testa, and C. Labbaye. 2011. MicroRNA-146a and AMD3100, two ways to control CXCR4 expression in acute myeloid leukemias. Blood Cancer J 1:e26.
39. Charrad, R. S., Z. Gadhoum, J. Qi, A. Glachant, M. Allouche, C. Jasmin, C. Chomienne, and F. Smadja-Joffe. 2002. Effects of anti-CD44 monoclonal antibodies on differentiation and apoptosis of human myeloid leukemia cell lines. Blood 99:290-299.
40. van Buul, J. D., C. Voermans, J. van Gelderen, E. C. Anthony, C. E. van der Schoot, and P. L. Hordijk. 2003. Leukocyte-endothelium interaction promotes SDF-1-dependent polarization of CXCR4. J Biol Chem 278:30302-30310.
41. Mendoza, M. C., E. E. Er, and J. Blenis. 2011. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci 36:320-328.
42. Teicher, B. A., and S. P. Fricker. 2010. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin Cancer Res 16:2927-2931.
43. Elliott, J. I., A. Sardini, J. C. Cooper, D. R. Alexander, S. Davanture, G. Chimini, and C. F. Higgins. 2006. Phosphatidylserine exposure in B lymphocytes: a role for lipid packing. Blood 108:1611-1617.
44. Fischer, K., S. Voelkl, J. Berger, R. Andreesen, T. Pomorski, and A. Mackensen. 2006. Antigen recognition induces phosphatidylserine exposure on the cell surface of human CD8+ T cells. Blood 108:4094-4101.
by guest on June 9, 2018http://w
ww
.jbc.org/D
ownloaded from

SDF-1 modulates Bcl-XL, Noxa, and Bak to induce apoptosis
15
45. Hesselgesser, J., M. Liang, J. Hoxie, M. Greenberg, L. F. Brass, M. J. Orsini, D. Taub, and R. Horuk. 1998. Identification and characterization of the CXCR4 chemokine receptor in human T cell lines: ligand binding, biological activity, and HIV-1 infectivity. J Immunol 160:877-883.
46. Tallman, M. S., D. G. Gilliland, and J. M. Rowe. 2005. Drug therapy for acute myeloid leukemia. Blood 106:1154-1163.
47. Kumar, R., V. Tripathi, M. Ahmad, N. Nath, R. A. Mir, S. S. Chauhan, and K. Luthra. 2012. CXCR7 mediated Gialpha independent activation of ERK and Akt promotes cell survival and chemotaxis in T cells. Cell Immunol 272:230-241.
48. Sanchez-Martin, L., P. Sanchez-Mateos, and C. Cabanas. 2013. CXCR7 impact on CXCL12 biology and disease. Trends Mol Med 19:12-22.
49. Tarnowski, M., R. Liu, M. Wysoczynski, J. Ratajczak, M. Kucia, and M. Z. Ratajczak. 2010. CXCR7: a new SDF-1-binding receptor in contrast to normal CD34(+) progenitors is functional and is expressed at higher level in human malignant hematopoietic cells. Eur J Haematol 85:472-483.
50. Zabel, B. A., Y. Wang, S. Lewen, R. D. Berahovich, M. E. Penfold, P. Zhang, J. Powers, B. C. Summers, Z. Miao, B. Zhao, A. Jalili, A. Janowska-Wieczorek, J. C. Jaen, and T. J. Schall. 2009. Elucidation of CXCR7-mediated signaling events and inhibition of CXCR4-mediated tumor cell transendothelial migration by CXCR7 ligands. J Immunol 183:3204-3211.
51. Cagnol, S., and J. C. Chambard. 2010. ERK and cell death: mechanisms of ERK-induced cell death--apoptosis, autophagy and senescence. FEBS J 277:2-21.
52. Tang, D., D. Wu, A. Hirao, J. M. Lahti, L. Liu, B. Mazza, V. J. Kidd, T. W. Mak, and A. J. Ingram. 2002. ERK activation mediates cell cycle arrest and apoptosis after DNA damage independently of p53. J Biol Chem 277:12710-12717.
53. Zhuang, S., and R. G. Schnellmann. 2006. A death-promoting role for extracellular signal-regulated kinase. J Pharmacol Exp Ther 319:991-997.
54. Karimian, G., M. Buist-Homan, K. N. Faber, and H. Moshage. 2012. Pertussis toxin, an inhibitor of G(alphai) PCR, inhibits bile acid- and cytokine-induced apoptosis in primary rat hepatocytes. PLoS One 7:e43156.
55. Kostylina, G., D. Simon, M. F. Fey, S. Yousefi, and H. U. Simon. 2008. Neutrophil apoptosis mediated by nicotinic acid receptors (GPR109A). Cell Death Differ 15:134-142.
56. Kremer, K. N., A. Kumar, and K. E. Hedin. 2011. G alpha i2 and ZAP-70 mediate RasGRP1 membrane localization and activation of SDF-1-induced T cell functions. J Immunol 187:3177-3185.
57. Garvey, T. L., J. Bertin, R. M. Siegel, G. H. Wang, M. J. Lenardo, and J. I. Cohen. 2002. Binding of FADD and caspase-8 to molluscum contagiosum virus MC159 v-FLIP is not sufficient for its antiapoptotic function. J Virol 76:697-706.
58. Meng, X. W., K. L. Peterson, H. Dai, P. Schneider, S. H. Lee, J. S. Zhang, A. Koenig, S. Bronk, D. D. Billadeau, G. J. Gores, and S. H. Kaufmann. 2011. High cell surface death receptor expression determines type I versus type II signaling. J Biol Chem 286:35823-35833.
59. Willis, S. N., L. Chen, G. Dewson, A. Wei, E. Naik, J. I. Fletcher, J. M. Adams, and D. C. Huang. 2005. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev 19:1294-1305.
by guest on June 9, 2018http://w
ww
.jbc.org/D
ownloaded from

SDF-1 modulates Bcl-XL, Noxa, and Bak to induce apoptosis
16
60. Dai, H., X. W. Meng, S. H. Lee, P. A. Schneider, and S. H. Kaufmann. 2009. Context-dependent Bcl-2/Bak interactions regulate lymphoid cell apoptosis. J Biol Chem 284:18311-18322.
61. Estey, E. H. 2012. How to manage high-risk acute myeloid leukemia. Leukemia 26:861-869.
62. Matsunaga, T., N. Takemoto, T. Sato, R. Takimoto, I. Tanaka, A. Fujimi, T. Akiyama, H. Kuroda, Y. Kawano, M. Kobune, J. Kato, Y. Hirayama, S. Sakamaki, K. Kohda, K. Miyake, and Y. Niitsu. 2003. Interaction between leukemic-cell VLA-4 and stromal fibronectin is a decisive factor for minimal residual disease of acute myelogenous leukemia. Nat Med 9:1158-1165.
63. Jin, L., K. J. Hope, Q. Zhai, F. Smadja-Joffe, and J. E. Dick. 2006. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med 12:1167-1174.
64. Yao, X., L. Zhou, S. Han, and Y. Chen. 2011. High expression of CXCR4 and CXCR7 predicts poor survival in gallbladder cancer. J Int Med Res 39:1253-1264.
65. Sacanna, E., T. Ibrahim, M. Gaudio, L. Mercatali, E. Scarpi, W. Zoli, P. Serra, S. Bravaccini, R. Ricci, L. Serra, and D. Amadori. 2011. The role of CXCR4 in the prediction of bone metastases from breast cancer: a pilot study. Oncology 80:225-231.
66. Speetjens, F. M., G. J. Liefers, C. J. Korbee, W. E. Mesker, C. J. van de Velde, R. L. van Vlierberghe, H. Morreau, R. A. Tollenaar, and P. J. Kuppen. 2009. Nuclear localization of CXCR4 determines prognosis for colorectal cancer patients. Cancer Microenviron 2:1-7.
67. Majmundar, A. J., W. J. Wong, and M. C. Simon. 2010. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell 40:294-309.
68. Sermeus, A., M. Genin, A. Maincent, M. Fransolet, A. Notte, L. Leclere, H. Riquier, T. Arnould, and C. Michiels. 2012. Hypoxia-induced modulation of apoptosis and BCL-2 family proteins in different cancer cell types. PLoS One 7:e47519.
by guest on June 9, 2018http://w
ww
.jbc.org/D
ownloaded from

SDF-1 modulates Bcl-XL, Noxa, and Bak to induce apoptosis
17
FOOTNOTES *This work was supported by the Mayo Foundation, the Joanne G. and Gary N. Owen Fund in Immunology Research, the Alma B. Stevenson Endowment Fund for Medical Research, and the National Institutes of Health Grants RO1 CA166741 and U02 CA70095. The abbreviations used are: PARP, poly(ADP-ribose) polymerase; SDF-1, stromal cell-derived factor-1. ACKNOWLEDGEMENTS We greatly acknowledge the gifts of reagents from D. Campana, G.J. Gores, E. Alnemri, and S. Huang; discussions with H. Dai and H. Ding; assistance of personnel in the Mayo Clinic Flow Cytometry Core Facility and Johns Hopkins Oncology Center SAC Lab; and the diligence of numerous clinical colleagues in providing the AML specimens used in this study.
by guest on June 9, 2018http://w
ww
.jbc.org/D
ownloaded from

SDF-1 modulates Bcl-XL, Noxa, and Bak to induce apoptosis
18
FIGURE LEGENDS Fig. 1: CXCR4 is expressed at variable levels on AML cells. A) Bone marrow isolates were harvested from AML patients prior to chemotherapy. Patient samples were cultured for 16-18 hr before levels of CXCR4 protein on the cell surface were analyzed via incubation of intact cells with anti-CXCR4 mAb followed by flow microfluorimetry. Results from nine different patient isolates are shown. B) To assay intracellular CXCR4 protein levels, cells from AML isolates were fixed, permeabilized, and then incubated with either a CXCR4 antibody or a control antibody prior to flow microfluorimetry. Results of a representative isolate is shown, n = 8. C) Either endogenous, cell surface CXCR4 (left panel) or endogenous intracellular CXCR4 (middle panel) were assayed for the human AML cell line, KG1a; representative results are shown, n = 3. Right panel: Cell surface CXCR4 levels of KG1a cells transfected with either a control plasmid (filled histogram), or a plasmid encoding the CXCR4-YFP fluorescent fusion protein (open histograms) are shown, either before ("Unstim.") or after 20 min stimulation with SDF-1 ("SDF-1") that induced CXCR4 internalization; representative results from one experiment are shown; n = 3.
Fig. 2: SDF-1 signaling induces migration and ERK activation in the KG1a-CXCR4. KG1a cells were transfected with either a vector control plasmid or CXCR4-YFP as in Fig. 1C. A) Transfected cells were assayed for migration in response to SDF-1. Each point denotes the mean % increase over basal migration in response to the indicated concentration of SDF-1 + S.E.M., n = 3. *, significantly different from KG1a cells transfected with the vector control plasmid, p < 0.05. B) Transfected cells were assayed for active, phosphorylated ERK in response to treatment with SDF-1 for the indicated times in YFP positive cells; representative flow cytometric plots from one experiment are shown. C) Summary of the results of three independent experiments performed as in B), each point denotes the average geometric mean of active, phosphorylated ERK + S.E.M. *, indicates significantly different from KG1a cells transfected with the vector control plasmid and treated with SDF-1, p < 0.05.
Fig. 3: SDF-1 induces KG1a cell apoptosis via CXCR4. KG1a cells transfected with either the vector control plasmid or CXCR4-YFP were cultured + SDF-1 for 16-18 hr. A) Cells were stained with APC-conjugated annexin V and assayed via flow microfluorimetry for apoptosis as indicated by annexin V binding to the cell surface. Gating as shown was used to compare apoptosis among cells expressing similar high levels of either CXCR4-YFP or YFP (vector control). B) Summary of results from three independent experiments done as in A). Each bar denotes the % of cells positive for annexin V + S.E.M. *, significantly different from KG1a cells transfected with the vector control plasmid and treated with SDF-1, p < 0.05. C) Apoptosis of SDF-1-treated cells was confirmed by confocal microscopic imaging of Hoechst 33258 stained nuclei. A representative image is shown; the arrow denotes nuclear fragmentation typically associated with apoptosis. D&E) Cells were fixed and permeabilized, then stained with an antibody specific for either cleaved PARP or for cleaved (active) caspase-3 and analyzed by flow microfluorimetry. Gating as shown was used to analyze cells expressing similar levels of CXCR4-YFP. F) Results of multiple independent experiments performed as in D&E. Each bar denotes the % of cells with each of the indicators of apoptosis + S.E.M., n = 3; *, significantly different from unstimulated cells, p < 0.05. G) Cells were analyzed for the correlation between apoptosis as assayed by annexin V binding and expression of either CXCR4-YFP or YFP (vector control). Each point denotes the mean change in % of cells positive for annexin V in response to SDF-1 + S.E.M., n = 3. *, significantly different from KG1a cells transfected with the vector control plasmid and treated with SDF-1, p < 0.05. H) Results of multiple experiments as in A), in which the concentration of SDF-1 was varied as indicated prior to assay of annexin V binding. Each point denotes the mean % of cells positive for annexin V among cells expressing similar levels of CXCR4-YPF + S.E.M., n = 3. I) The indicated cells were treated with 30 µM AMD3100 (a CXCR4 antagonist) prior to stimulation with SDF-1. Following SDF-1 treatment the cells were assayed for annexin V binding as in A. Each bar denotes the mean % of cells positive for annexin V + S.E.M., n = 3. *, significantly different from untreated KG1a cells transfected with CXCR4-YFP and stimulated with SDF-1, p < 0.05.
by guest on June 9, 2018http://w
ww
.jbc.org/D
ownloaded from

SDF-1 modulates Bcl-XL, Noxa, and Bak to induce apoptosis
19
Fig. 4: SDF-1/CXCR4 induces apoptosis in the U937 AML cell line and in a subset of clinical AML isolates. A) The AML cell line U937 was transfected with either a vector control plasmid (YPF) or CXCR4-YFP, cultured for 16-18 hr, stained with an APC-conjugated CXCR4 antibody, and assayed for CXCR4 expression by flow microfluorimetry while gating on cells expressing similar levels of CXCR4-YFP or YFP. A representative flow cytometric plot is shown, n = 3. B) Cells were transfected as in A) and assayed for annexin V positive cells as in Fig. 3A. *, significantly different from unstimulated CXCR4 transfected cells, p < 0.05. C) Clinical AML isolates were cultured for 1-2 hr and then plated onto a confluent layer of BMSC, cultured for 1 hr, and then treated + 5 x 10-8 M SDF-1 for 16-18 hr. Isolates were then stained with APC-conjugated annexin V and assayed for apoptosis by flow microfluorimetry as in Fig. 3A. Results for 10 different patients are shown.
Fig. 5: Hypoxia protects CXCR4-expressing AML cell lines from apoptosis. Either KG1a or U937 cells were transiently transfected with CXCR4-YFP to generate KG1a-CXCR4 or U937-CXCR4 cells, respectively. Cells were stimulated with SDF-1 for 16-18 hr in ambient (normoxic) or 1% oxygen (hypoxic) conditions, as indicated. A&D) Apoptosis was assayed by annexin V binding as in Fig. 3A. *, significantly different from unstimulated cells in normoxic conditions, p < 0.05; **, significantly different from SDF-1 treated cells in normoxic conditions, p < 0.05; n = 3. B&E) Apoptosis was assayed by detection of cleaved PARP as in Fig. 3F. *, significantly different from unstimulated cells in normoxic conditions, p < 0.05; **, significantly different from SDF-1 treated cells in normoxic conditions, p < 0.05; n = 3. C&F) CXCR4 cell surface levels were assayed by flow microfluorimetry on the same unstimulated cells used for experiments as in A&D. *, significantly different from cell culture in normoxic conditions, p < 0.05, n = 3. G) Untransfected KG1a cells were incubated in hypoxic or normoxic conditions for 2 hr and then treated with 1 µM cytarabine for 18 hr in normoxic or hypoxic conditions prior to analysis of annexin V binding. *, significantly different from cytarabine-treated cells in normoxic conditions, p < 0.05; n = 3.
Fig. 6: CXCR7, ERK activation, and Gi proteins do not mediate SDF-1-induced apoptosis in AML cells. A) CXCR7 cell surface levels were assayed by flow microfluorimetry on the indicated cell lines; representative results from one experiment are shown; n = 3. B&C&F) KG1a-CXCR4 cells were pretreated with CCX771 (CXCR7 antagonist), CCX704 (the control), PD325901, CI-1040, or the vehicle for 1 hr prior to addition of SDF-1; or with PTX-B or PTX for 4 hr prior to the addition of SDF-1. Cells were then cultured for 16-18 hours and assayed for apoptosis via annexin V binding as in Fig. 3A, n = 3. D&E) KG1a-CXCR4 cells were treated with PD325901, CI-1040, or vehicle for 16-18 hr, stimulated with SDF-1 for 2 min, and then assayed for ERK activation as in Fig. 2B&C. *, significantly different from control-treated cells, p < 0.05; n = 3. G-H) KG1a-CXCR4 cells were cultured for 16 hr, treated with PTX-B or PTX for 4 hr, stimulated with SDF-1 for 2 min, and assayed for ERK activation as in Fig. 2B&C. *, significantly different from SDF-1-stimulated control-treated cells, p < 0.05 n = 3.
Fig. 7: Requirement for the caspase-9/intrinsic death pathway, but not the caspase-8/extrinsic pathway, for SDF-1/CXCR4-mediated apoptosis. A&C) KG1a or U937 cells were transiently transfected with CXCR4-YFP together with either a control plasmid (Vector) or a plasmid encoding either MC159 or DN-Casp-9-GFP. Cells were then assayed for SDF-1-dependent apoptosis via annexin V binding as in Fig. 3A. *, significantly different from SDF-1-treated control vector, p < 0.05, n = 3. B) Jurkat T cells were transiently transfected with a plasmid encoding either a control vector or the caspase-8 inhibitor, MC159, then stimulated with agonistic anti-Fas antibody for 16-18 hr and assayed for apoptosis via annexin V binding as in Fig. 3A. *, significantly different from the CH.11-treated control vector results, p < 0.05, n = 3. D) Immunoblots of whole cell lysates from A&C), showing overexpression of DN-Casp-9-GFP as compared to endogenous Procasp-9. The same membrane was stripped and re-blotted for actin as a control. E) mRNA from cells transfected as in A-C) was assayed via RT-PCR for expression of the mRNA encoding the viral protein, MC159. GAPDH mRNA was assayed as a control, n = 3.
by guest on June 9, 2018http://w
ww
.jbc.org/D
ownloaded from

SDF-1 modulates Bcl-XL, Noxa, and Bak to induce apoptosis
20
Fig. 8: Role of SDF-1-mediated Bak upregulation in SDF-1/CXCR4-mediated apoptosis. A) KG1a cells were transfected with CXCR4-YFP and treated + SDF-1 for 16-18 hr. The cells were sorted using a flow cytometer to obtain the top 12% of YFP positive cells, and mRNA of the indicated Bcl-2 family members was assayed via qRT-PCR. Each bar denotes the mean fold increase of mRNA expression in response to SDF-1 treatment + S.E.M. as compared to unstimulated cells, following normalization of the results to the mRNA levels of GAPDH, for three independent experiments and sorts done on different days. *, significantly different from GAPDH control, p < 0.05. B) KG1a cells were transfected, treated, and sorted as in A, and Bak and ERK (control) protein expression levels were assayed in whole cell lysates via immunoblotting, n = 3. C&D) KG1a cells were transfected with either a GFP plasmid control vector or GFP-Bak, cultured for 24 hr (together with the caspase inhibitor, Q-VD-OPh, where indicated), and analyzed for GFP expression levels via flow microfluorimetry C) and annexin V positivity in these GFP expressing cells D). Representative experiments are shown. E) Summary of results from 3 independent experiments performed as in D). Each bar denotes the % of annexin V positive cells + S.E.M. *, significantly different from control vector, p < 0.05. F&H) Immunoblots showing depletion of Bak or Bim in the KG1a cell lines stably-expressing Bak shRNA or Bim shRNA, respectively; as compared to the KG1a cell line expressing control shRNAs. The same immunoblots were stripped and reprobed with actin as a control, n = 3. G&I) KG1a cells stably-expressing Bak shRNA, Bim shRNA or the shRNA control vector were transiently transfected with CXCR4-YFP, then assayed for SDF-1-dependent apoptosis as in Fig. 3A. *, significantly different from SDF-1 treated control vector, p < 0.05, n = 5 and n = 2 respectively.
Fig. 9: SDF-1-induced downregulation of Bcl-XL and upregulation of Noxa contribute to apoptosis of AML cells. A) KG1a cells were transfected, treated, and sorted as in Fig. 8A, and the protein expression levels of the indicated Bcl-2 family members were assayed via immunoblot. The same immunoblots were stripped and reprobed with actin as a control, n = 3. . B) KG1a cells were transfected with CXCR4-YFP and either a control vector or S-peptide-tagged-Bcl-XL (labeled S-tagged-Bcl-XL) and assayed for Bcl-XL and actin (control) protein levels via immunoblot. C) KG1a cells were transfected as in B), cultured for 6 hr, treated with SDF-1 for 18 hr, and then assayed for annexin V positivity in YFP expressing cells. Results from 3 independent experiments are shown. Each bar denotes the mean % of cells positive for annexin V + S.E.M. *, significantly different from control vector, p < 0.05. D-F) KG1a cells were transfected with either a plasmid encoding GFP or GFP-Noxa, cultured for 24 hr (together with the caspase inhibitor, Q-VD-OPh, where indicated), and assayed for D) expression levels via western blot, E) GFP expression via flow microfluorimetry and F) annexin V positivity in GFP expressing cells. Representative experiments are shown. G) Summary of results from 3 independent experiments performed as in F). Each bar denotes the % of annexin V positive cells + S.E.M. *, significantly different from control vector, p < 0.05. H) KG1a cells were cotransfected with CXCR4-YFP and Noxa2A-GFP, cultured for 16 hr in the presence or absence of SDF-1, then treated with cycloheximide for the indicated time, fixed, and analyzed via flow microfluorimetry for Noxa2A-GFP expression in CXCR4-YFP positive cells. Each point denotes the % of Noxa2A-GFP expression as compared to the 0 hr. *, significantly different from vehicle control, p < 0.05, n = 3. Inset: Immunoblot of the indicated whole cell lysates confirming expression of Noxa2A-GFP compared to actin (control). I) Immunoblots of whole cell lysates from KG1a transfected with CXCR4-YFP and an siRNA for Noxa or a control siRNA, cultured for 24 hr, and blotted for Noxa or actin (control). J) KG1a cells were transfected as in I), cultured for 6-7 hr prior to the addition of SDF-1, then cultured +/- SDF-1 for an additional 16 hr, and assayed for annexin V positive cells as in Fig. 3A). Each bar denotes the % of annexin V positive cells + S.E.M. *, significantly different from SDF-1-stimulated cells treated with the control siRNA, p < 0.05.
Fig. 10: Proposed model of SDF-1-induced apoptosis. SDF-1-induced CXCR4 activation induces Noxa stabilization, Bak upregulation and Bcl-XL downregulation. Collectively, these lead to Bax/Bak activation followed by activation of caspase-9 and caspase-3. Consistent with this model, increased
by guest on June 9, 2018http://w
ww
.jbc.org/D
ownloaded from

SDF-1 modulates Bcl-XL, Noxa, and Bak to induce apoptosis
21
expression of Noxa or Bak is shown to induce apoptosis in these cells and Bcl-XL expression, Bak shRNA, or Noxa siRNA is shown to inhibit SDF-1-induced apoptosis. Likewise, AMD3100 and DN-caspase 9 inhibit apoptosis, providing further support for the involvement of CXCR4 and the caspase 9-mediated intrinsic pathway in this process.
by guest on June 9, 2018http://w
ww
.jbc.org/D
ownloaded from

SDF-1 modulates Bcl-XL, Noxa, and Bak to induce apoptosis
22
by guest on June 9, 2018http://w
ww
.jbc.org/D
ownloaded from

SDF-1 modulates Bcl-XL, Noxa, and Bak to induce apoptosis
23
by guest on June 9, 2018http://w
ww
.jbc.org/D
ownloaded from
SDF-1 modulates Bcl-XL, Noxa, and Bak to induce apoptosis
24
by guest on June 9, 2018http://w
ww
.jbc.org/D
ownloaded from

SDF-1 modulates Bcl-XL, Noxa, and Bak to induce apoptosis
25
by guest on June 9, 2018http://w
ww
.jbc.org/D
ownloaded from

SDF-1 modulates Bcl-XL, Noxa, and Bak to induce apoptosis
26
by guest on June 9, 2018http://w
ww
.jbc.org/D
ownloaded from
SDF-1 modulates Bcl-XL, Noxa, and Bak to induce apoptosis
27
by guest on June 9, 2018http://w
ww
.jbc.org/D
ownloaded from

SDF-1 modulates Bcl-XL, Noxa, and Bak to induce apoptosis
28
by guest on June 9, 2018http://w
ww
.jbc.org/D
ownloaded from

SDF-1 modulates Bcl-XL, Noxa, and Bak to induce apoptosis
29
by guest on June 9, 2018http://w
ww
.jbc.org/D
ownloaded from

SDF-1 modulates Bcl-XL, Noxa, and Bak to induce apoptosis
30
by guest on June 9, 2018http://w
ww
.jbc.org/D
ownloaded from

SDF-1 modulates Bcl-XL, Noxa, and Bak to induce apoptosis
31
by guest on June 9, 2018http://w
ww
.jbc.org/D
ownloaded from

Kaufmann and Karen E. HedinAllan D. Hess, B. Douglas Smith, Christie Rodriguez-Ramirez, Judith E. Karp, Scott H.
Kimberly N. Kremer, Kevin L. Peterson, Paula A. Schneider, X. Wei Meng, Haiming Dai,regulation of the Bcl-2 family members Bcl-XL, Noxa, and Bak
SDF-1/CXCR4 signaling induces apoptosis in acute myeloid leukemia cells via
published online June 24, 2013J. Biol. Chem.
10.1074/jbc.M113.449926Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
Supplemental material:
http://www.jbc.org/content/suppl/2013/06/24/M113.449926.DC1
by guest on June 9, 2018http://w
ww
.jbc.org/D
ownloaded from