sensitivitytopoly(adp-ribose)polymerase(parp)inhibition ... · may 7, 2010•volume 285•number 19...
TRANSCRIPT

Sensitivity to Poly(ADP-ribose) Polymerase (PARP) InhibitionIdentifies Ubiquitin-specific Peptidase 11 (USP11) as aRegulator of DNA Double-strand Break Repair*□S
Received for publication, January 15, 2010, and in revised form, March 12, 2010 Published, JBC Papers in Press, March 15, 2010, DOI 10.1074/jbc.M110.104745
Timothy D. Wiltshire‡, Courtney A. Lovejoy‡, Tong Wang§, Fen Xia§, Mark J. O’Connor¶, and David Cortez‡1
From the Departments of ‡Biochemistry and §Radiation Oncology, Vanderbilt University School of Medicine, Nashville, Tennessee 37232and ¶KuDOS Pharmaceuticals Ltd., 410 Cambridge Science Park, Cambridge CB4 OPE, United Kingdom
DNA damage repair and checkpoint responses preventgenome instability and provide a barrier to the development ofcancer. Inherited mutations in DNA damage response (DDR)genes such as those that encode the homologous recombination(HR) proteins BRCA1 and BRCA2 cause cancer predispositionsyndromes. PARP inhibitors are an exciting new class of tar-geted therapy for treating patients with HR repair-defectivetumors. In this study, we use an RNAi screen to identify genesthat when silenced cause synthetic lethality with the PARPinhibitor AZD2281. This screen identified the deubiquitylatingenzyme USP11 as a participant in HR repair of DNA double-strand breaks. Silencing USP11 with siRNA leads to sponta-neous DDR activation in otherwise undamaged cells andhypersensitivity to PARP inhibition, ionizing radiation, andother genotoxic stress agents. Moreover, we demonstrate thatHR repair is defective in USP11-silenced cells. Finally, therecruitment of a subset of double-strand break repair proteinsincludingRAD51 and 53BP1 to repair foci ismisregulated in theabsence of USP11 catalytic activity. Thus, our synthetic lethalapproach identified USP11 as a component of the HR double-strand break repair pathway.
Faithful DNA replication and repair is critical to maintaingenome integrity. An intact DNA damage response (DDR)2prevents mutations and gross chromosomal instability. Defectsin the DDR cause cancer predisposition syndromes, demon-strating the importance of the DDR as a barrier to tumorigen-esis (1, 2). Most cancers have defects in some part of the DDR.This difference between normal and cancer cells not onlyexplains the genetic instability associated with cancer, but alsoprovides an opportunity for therapeutic intervention. This con-cept is the basis for many genotoxic therapies where the DNA
damage is repaired in healthy cells but has lethal consequencesin cancer cells.Targeted therapy of tumors is a method of treatment that
seeks to maximize response while reducing harmful sideeffects to healthy cells. Inhibition of the DNA repair enzymepoly(ADP-ribose) polymerase 1 (PARP1) is a recently devel-oped strategy for cancer therapy that exploits DDR defects in asubset of cancers (3). PARP1 is a DNA repair enzyme that isresponsible for the sensing and repair of single-strand DNAbreaks via short-patch base-excision repair (4). Thousands ofsingle-strand breaks occur per cell per day. When a replicationfork encounters a single-strand break, the result is collapse ofthe fork and formation of a double-strand break (5). In wild-type cells, these replication-associated double-strand breaksare often repaired via homologous recombination (HR). Cellsdeficient in PARP activity have increased recombination as evi-denced by elevated rates of sister chromatid exchange (6) andincreased intranuclear foci of the RAD51 recombination pro-tein (7, 8). Cells deficient in HR proteins such as BRCA1 andBRCA2 are unable to repair these double-strand breaks effi-ciently and die (7, 8). Thus, PARP inhibitors are being success-fully used to treat HR-deficient tumors such as those found inthe breast of women with inherited mutations in BRCA1 orBRCA2 (9).PARP inhibitors also preferentially kill cells defective in
many other components of the DDR that respond to double-strand breaks including the ataxia-telangiectasia mutated(ATM) kinase (10, 11). ATM is recruited to double-strandbreaks by the Mre11-Rad50-Nbs1 (MRN) complex where itphosphorylates the histone variant H2AX on Ser-139 (�H2AX)in nucleosomes surrounding the break. H2AXphosphorylationand the MRN complex itself initiate the recruitment of manyadditional double-strand break repair and signaling proteinsincludingMDC1, BRCA1, RAD51, and 53BP1 (12–16). Assem-bly of these proteins within damage-induced foci initiates cellcycle checkpoint signaling and HR. Search for the homologoussister chromatid and strand invasion during HR requires for-mation of a RAD51 filament on the single-strandedDNA and isfacilitated by BRCA2 (17–21).Ubiquitin modification at the sites of double-strand breaks
has emerged as an essential regulator of signaling and repair.Recruitment of BRCA1 and 53BP1 requires ubiquitylation atdouble-strand break sites catalyzed by the E3 ubiquitin ligases,RNF8 andRNF168 (22–28). First,MDC1directly binds�H2AXvia a BRCT domain and is also a target of phosphorylation by
* This work was supported, in whole or in part, by NCI, National Institutes ofHealth Grants (R21CA132010 and R01CA136933) and a Susan G. Komen forthe Cure grant (to D. C.). Support for research facilities was provided by theVanderbilt Center for Molecular Toxicology (Grant P30 ES000267) and theVanderbilt-Ingram Cancer Center (Grant P30 CA068485).
□S The on-line version of this article (available at http://www.jbc.org) containssupplemental Table S1 and Fig. S1.
1 To whom correspondence should be addressed: 613 Light Hall, 2215 Gar-land Ave., Nashville, TN 37232. Tel.: 615-322-8547; Fax: 615-343-0704;E-mail: [email protected].
2 The abbreviations used are: DDR, DNA damage response; PARP, poly(ADP-ribose) polymerase; HR, homologous recombination; GFP, green fluores-cent protein; HA, hemagglutinin; WT, wild type; DSB, double-strand break;DUB, de-ubiquitylating enzymes; IR, ionizing radiation.
THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 285, NO. 19, pp. 14565–14571, May 7, 2010© 2010 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in the U.S.A.
MAY 7, 2010 • VOLUME 285 • NUMBER 19 JOURNAL OF BIOLOGICAL CHEMISTRY 14565
by guest on February 2, 2019http://w
ww
.jbc.org/D
ownloaded from

ATM (29, 30). After MDC1 is phosphorylated, it interacts withthe FHA domain of RNF8 which ubiquitylates H2A (23–25).Ubiquitylated H2A serves as an interacting partner for RNF168that further propagates the ubiquitylation of H2A and otherunknown targets at the double-strand break site (22, 27).RAP80 binds ubiquitylated H2A and along with Abraxas medi-ates accumulation of BRCA1 (31–35). Histone ubiquitylation atdouble-strand break sites is also required for recruitment of53BP1, although the precise mechanism remains unclear (22).RAD51 recruitment to double-strand breaks requires the E2enzyme Ubc13, a known interaction partner of RNF8 (26).A number of de-ubiquitylating enzymes (DUBs) also func-
tion at double-strand breaks including USP3 and BRCC36.BRCC36 is part of the RAP80-Abraxas-BRCA1 complexand antagonizes RNF8-dependent ubiquitylation to main-tain steady state levels required for appropriate signaling (31,36–38). USP3 is a chromatin-associated DUB that also antag-onizes RNF8-mediated ubiquitylation (39).Because HR deficiency causes cellular sensitivity to PARP
inhibitors, PARP inhibitor sensitivity may indicate a defect inHR repair. Indeed, RNAi screening for PARP inhibitor sensitiv-ity has identified new participants in the DDR (40, 41). In thisstudy, we used siRNA screening to identify and validate theDUB USP11 as a regulator of double-strand break repairinvolved in modulating sensitivity to DNA damage.
EXPERIMENTAL PROCEDURES
Cell Culture, siRNA, and Plasmids—U2OS cell were main-tained in Dulbecco’s modified Eagle’s medium (DMEM) with7.5% fetal bovine serum at 37 °C and 5% CO2. siRNA transfec-tions were performed with HiPerfect reagent (Qiagen) with afinal siRNA concentration of 5 nM. Target siRNA sequences(listed in supplemental Table S1) and the non-targeting controlsequence AUGAACGUGAAUUGCUCAA were purchasedfrom Dharmacon and Qiagen. Full-length cDNA for USP11(clone ID: 2961383) was obtained from Open Biosystems. Aplasmid resistant to USP11 siRNA was created by introducingwobble mutations through PCR amplification of the targetsequences. Primers usedwere as follows: USP11_6: sense: TGTT-CAGTTCAGCCATACCGATTCTATCGGTCTGGTGTTG-CGCACAGCTCGG; antisense: CCGAGCTGTGCGCAACA-CCAGACCGATAGAATCGGTATGGCTGAACTGAACA;USP11_10: sense: AGCCAGAGATGAAGAAGCGTTACTA-CGATGAAGTTGAGGCTGAGGGCTA; antisense: TAGCC-CTCAGCCTCAACTTCATCGTAGTAACGCTTCTTCAT-CTCTGGCTC. The C318S, mutation was made using thefollowing primers: sense: CAATCTGGGCAACACGAGCTT-CATGAACTCGGC; antisense: GCCGAGTTCATGAAGCT-CGTGTTGCCCAGATTG (mutated site underlined). StableU2OS cells infected with siRNA-resistant constructs of USP11were selected and maintained in 1 �g/ml of puromycin.Drug Treatment andDNADamage—PARP inhibitor (AZD2281)
was dissolved in DMSO at a concentration of 10 mM. Ionizingradiation (IR) treatment was done using a Cs137 source at a rateof 1.8 Gy/min.Viability Assay and Statistical Analysis—Reverse transfec-
tion was performed by spotting siRNA in 96-well plates fol-lowed by addition of HiPerfect reagent in Optimem media.
After 5min of incubation at room temperature, 5000 cells wereadded to each well and incubated 24 h at 37 °C. Cells were split1:4 with two plates treated with 5 �M AZD2281 and two platesas an untreated control. After 96 h of incubation, medium wasreplaced with fresh medium containing 80 �l of a 1:10 dilutionofWST-1 reagent (Roche) inDMEM. Plates were incubated 1 hat 37 °C, then assayed at 450 nm on a spectrophotometer. Sen-sitivity was calculated by normalizing absorbance readings tothe average of the non-targeting controls within each plate. Themean of two replicates for each experiment was used to calcu-late a viability ratio by dividing treated by untreated wells foreach gene. The log was calculated for these ratios and normal-ized to the non-targeting control, which represents the relativesensitivity toAZD2281.Multiple replicates of the log ratio wereused to calculate a p value using unpaired, two-tailed t test. Thesensitivity index was also calculated as previously described(42).Antibodies and Immunoblotting—Cells were lysed for 20min
on ice in 50 mM Tris, pH 7.5, 150 mMNaCl, 0.5% Igepal, 10 mM
NaF supplemented with 1 mM phenylmethylsulfonyl fluoride,20mM�-glycerophosphate, 1mMsodiumvanadate,1mMdithio-threitol, 5 �g/ml aprotinin, and 5 �g/ml leupeptin. Lysateswere cleared by centrifugation prior to Bradford protein con-centration determination (Bio-Rad). Total cellular protein wasseparated by SDS-PAGE and transferred to nitrocellulosemembranes. Protein detection was done using infrared fluores-cent-conjugated secondary antibodies on an Odyssey imagingsystem (LI-COR). Antibodies to �H2AX were purchased fromCell Signaling. RAD51 and BRCA1 antibodies were purchasedfrom EMD BioSciences. 53BP1 and USP11 antibodies werepurchased from Bethyl Laboratories. GAPDH antibody waspurchased from Millipore. HA antibody was purchased fromCovance. ORC2 antibody was obtained from BD Pharmingen.Immunofluorescence—Cells were plated on coverslips and
allowed to attach before treatment with IR. After incubation,cell were fixed in 3% paraformaldehyde and permeabilized with0.5% Triton X-100 solution before incubation with primaryantibodies. Fluorescein isothiocyanate and rhodamine red-X-conjugated secondary antibodies were obtained from JacksonImmunoresearch. Cells were visualized and foci counted on aZeiss Axioplan 2.Clonogenic Survival Assay—Sensitivity to IR was determined
by transfecting U2OS cells with non-targeting and USP11siRNA for 24 h followed by plating in 60-mm dishes at increas-ing cell densities. Treatment with 3 and 5 Gy IR was carried out72 h after siRNA knockdown. Colonies were allowed to growfor 7–10 days and stained with 2% methylene blue in a 50:50solution of methanol/water. Colonies of �50 cells werecounted, and the surviving fraction was calculated and normal-ized to untreated control.Chromosomal Homologous Recombinational Repair (HR)
Analysis—HR repair assay was carried out as previouslydescribed (43). HEK293DRGFP cells carrying a chromosomallyintegrated single copy of homologous recombinational repair(HR) substrate were used to test USP11 role in HR. DSB-in-duced HR results in restoration and expression of GFP and wasquantified by FACS. Briefly, 48 h after one repeat of transfec-tion of control or USP11-targeting siRNA, chromosomal DSBs
USP11 Regulates DNA Double-strand Break Repair
14566 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 285 • NUMBER 19 • MAY 7, 2010
by guest on February 2, 2019http://w
ww
.jbc.org/D
ownloaded from
were induced through the expression of I-SceI. 48 h later, cellswere subjected to two-color fluorescence analysis, whichrevealed the percentage of green fluorescent cells relative to thetotal viable cell number. For each analysis, 100,000 cells wereprocessed.
RESULTS
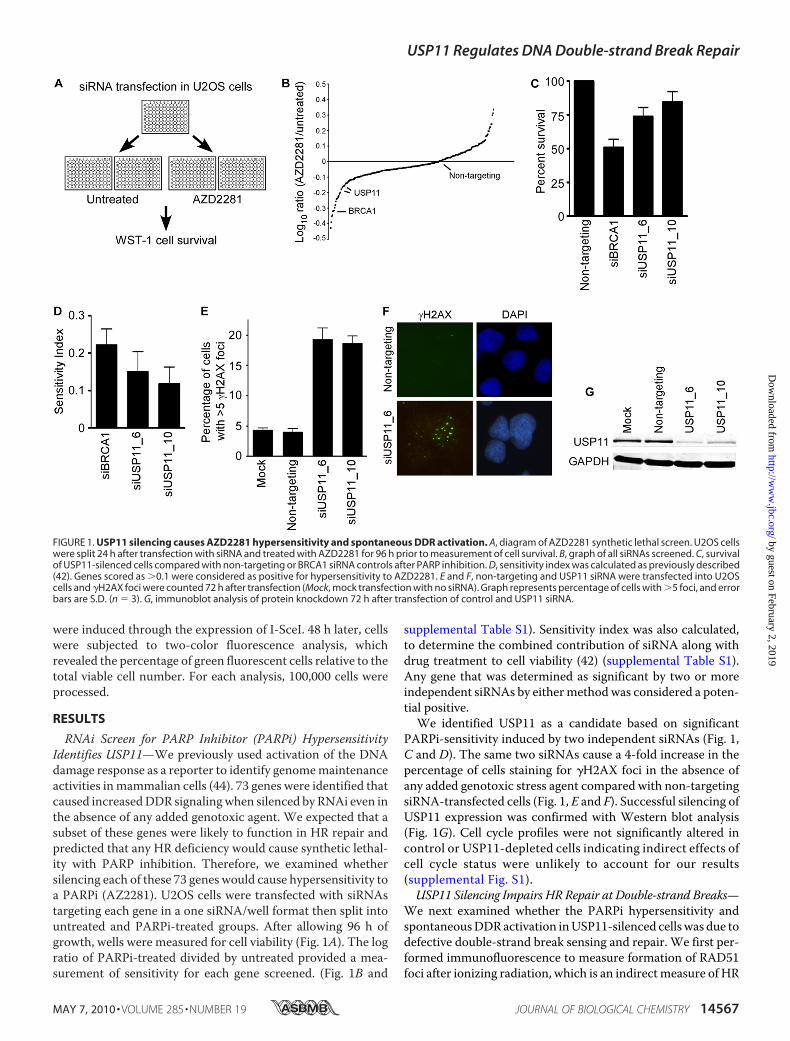
RNAi Screen for PARP Inhibitor (PARPi) HypersensitivityIdentifies USP11—We previously used activation of the DNAdamage response as a reporter to identify genomemaintenanceactivities inmammalian cells (44). 73 genes were identified thatcaused increasedDDR signalingwhen silenced by RNAi even inthe absence of any added genotoxic agent. We expected that asubset of these genes were likely to function in HR repair andpredicted that any HR deficiency would cause synthetic lethal-ity with PARP inhibition. Therefore, we examined whethersilencing each of these 73 genes would cause hypersensitivity toa PARPi (AZ2281). U2OS cells were transfected with siRNAstargeting each gene in a one siRNA/well format then split intountreated and PARPi-treated groups. After allowing 96 h ofgrowth, wells were measured for cell viability (Fig. 1A). The logratio of PARPi-treated divided by untreated provided a mea-surement of sensitivity for each gene screened. (Fig. 1B and
supplemental Table S1). Sensitivity index was also calculated,to determine the combined contribution of siRNA along withdrug treatment to cell viability (42) (supplemental Table S1).Any gene that was determined as significant by two or moreindependent siRNAs by eithermethodwas considered a poten-tial positive.We identified USP11 as a candidate based on significant
PARPi-sensitivity induced by two independent siRNAs (Fig. 1,C and D). The same two siRNAs cause a 4-fold increase in thepercentage of cells staining for �H2AX foci in the absence ofany added genotoxic stress agent compared with non-targetingsiRNA-transfected cells (Fig. 1, E and F). Successful silencing ofUSP11 expression was confirmed with Western blot analysis(Fig. 1G). Cell cycle profiles were not significantly altered incontrol or USP11-depleted cells indicating indirect effects ofcell cycle status were unlikely to account for our results(supplemental Fig. S1).USP11 Silencing Impairs HR Repair at Double-strand Breaks—
We next examined whether the PARPi hypersensitivity andspontaneousDDRactivation inUSP11-silenced cellswas due todefective double-strand break sensing and repair. We first per-formed immunofluorescence to measure formation of RAD51foci after ionizing radiation, which is an indirectmeasure of HR
FIGURE 1. USP11 silencing causes AZD2281 hypersensitivity and spontaneous DDR activation. A, diagram of AZD2281 synthetic lethal screen. U2OS cellswere split 24 h after transfection with siRNA and treated with AZD2281 for 96 h prior to measurement of cell survival. B, graph of all siRNAs screened. C, survivalof USP11-silenced cells compared with non-targeting or BRCA1 siRNA controls after PARP inhibition. D, sensitivity index was calculated as previously described(42). Genes scored as �0.1 were considered as positive for hypersensitivity to AZD2281. E and F, non-targeting and USP11 siRNA were transfected into U2OScells and �H2AX foci were counted 72 h after transfection (Mock, mock transfection with no siRNA). Graph represents percentage of cells with �5 foci, and errorbars are S.D. (n � 3). G, immunoblot analysis of protein knockdown 72 h after transfection of control and USP11 siRNA.
USP11 Regulates DNA Double-strand Break Repair
MAY 7, 2010 • VOLUME 285 • NUMBER 19 JOURNAL OF BIOLOGICAL CHEMISTRY 14567
by guest on February 2, 2019http://w
ww
.jbc.org/D
ownloaded from
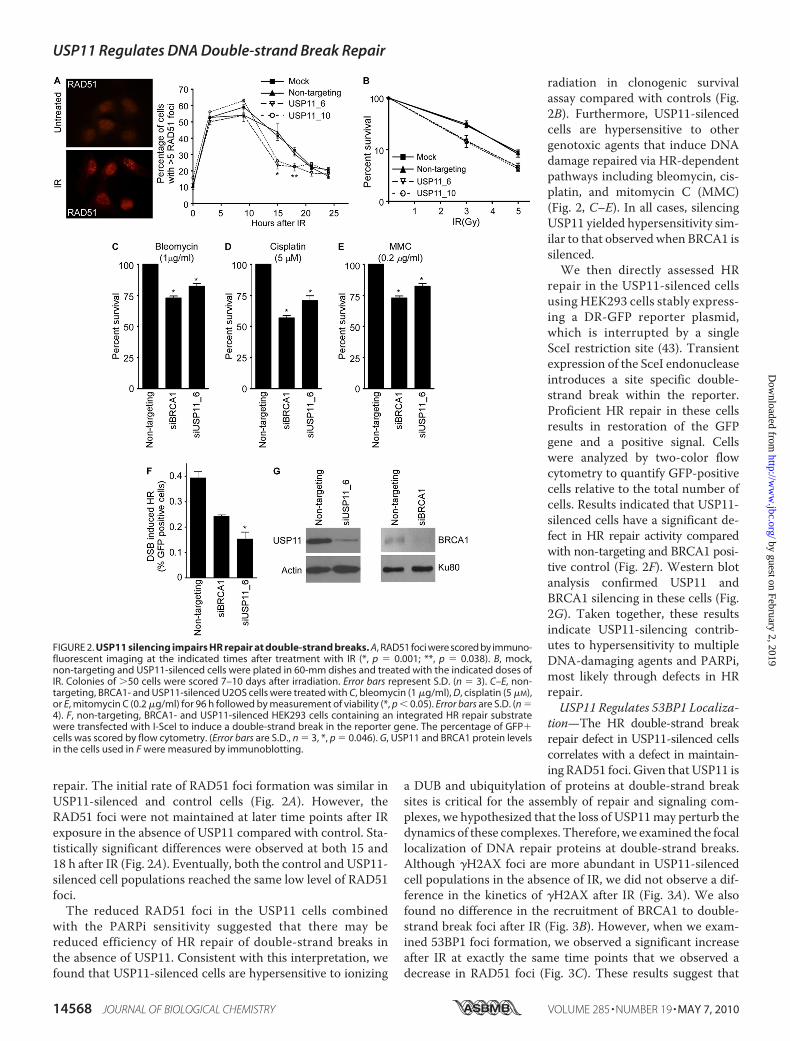
repair. The initial rate of RAD51 foci formation was similar inUSP11-silenced and control cells (Fig. 2A). However, theRAD51 foci were not maintained at later time points after IRexposure in the absence of USP11 compared with control. Sta-tistically significant differences were observed at both 15 and18 h after IR (Fig. 2A). Eventually, both the control and USP11-silenced cell populations reached the same low level of RAD51foci.The reduced RAD51 foci in the USP11 cells combined
with the PARPi sensitivity suggested that there may bereduced efficiency of HR repair of double-strand breaks inthe absence of USP11. Consistent with this interpretation, wefound that USP11-silenced cells are hypersensitive to ionizing
radiation in clonogenic survivalassay compared with controls (Fig.2B). Furthermore, USP11-silencedcells are hypersensitive to othergenotoxic agents that induce DNAdamage repaired via HR-dependentpathways including bleomycin, cis-platin, and mitomycin C (MMC)(Fig. 2, C–E). In all cases, silencingUSP11 yielded hypersensitivity sim-ilar to that observed when BRCA1 issilenced.We then directly assessed HR
repair in the USP11-silenced cellsusing HEK293 cells stably express-ing a DR-GFP reporter plasmid,which is interrupted by a singleSceI restriction site (43). Transientexpression of the SceI endonucleaseintroduces a site specific double-strand break within the reporter.Proficient HR repair in these cellsresults in restoration of the GFPgene and a positive signal. Cellswere analyzed by two-color flowcytometry to quantify GFP-positivecells relative to the total number ofcells. Results indicated that USP11-silenced cells have a significant de-fect in HR repair activity comparedwith non-targeting and BRCA1 posi-tive control (Fig. 2F). Western blotanalysis confirmed USP11 andBRCA1 silencing in these cells (Fig.2G). Taken together, these resultsindicate USP11-silencing contrib-utes to hypersensitivity to multipleDNA-damaging agents and PARPi,most likely through defects in HRrepair.USP11 Regulates 53BP1 Localiza-
tion—The HR double-strand breakrepair defect in USP11-silenced cellscorrelates with a defect in maintain-ingRAD51 foci. Given that USP11 is
a DUB and ubiquitylation of proteins at double-strand breaksites is critical for the assembly of repair and signaling com-plexes, we hypothesized that the loss of USP11may perturb thedynamics of these complexes. Therefore, we examined the focallocalization of DNA repair proteins at double-strand breaks.Although �H2AX foci are more abundant in USP11-silencedcell populations in the absence of IR, we did not observe a dif-ference in the kinetics of �H2AX after IR (Fig. 3A). We alsofound no difference in the recruitment of BRCA1 to double-strand break foci after IR (Fig. 3B). However, when we exam-ined 53BP1 foci formation, we observed a significant increaseafter IR at exactly the same time points that we observed adecrease in RAD51 foci (Fig. 3C). These results suggest that
FIGURE 2. USP11 silencing impairs HR repair at double-strand breaks. A, RAD51 foci were scored by immuno-fluorescent imaging at the indicated times after treatment with IR (*, p � 0.001; **, p � 0.038). B, mock,non-targeting and USP11-silenced cells were plated in 60-mm dishes and treated with the indicated doses ofIR. Colonies of �50 cells were scored 7–10 days after irradiation. Error bars represent S.D. (n � 3). C–E, non-targeting, BRCA1- and USP11-silenced U2OS cells were treated with C, bleomycin (1 �g/ml), D, cisplatin (5 �M),or E, mitomycin C (0.2 �g/ml) for 96 h followed by measurement of viability (*, p � 0.05). Error bars are S.D. (n �4). F, non-targeting, BRCA1- and USP11-silenced HEK293 cells containing an integrated HR repair substratewere transfected with I-SceI to induce a double-strand break in the reporter gene. The percentage of GFP�cells was scored by flow cytometry. (Error bars are S.D., n � 3, *, p � 0.046). G, USP11 and BRCA1 protein levelsin the cells used in F were measured by immunoblotting.
USP11 Regulates DNA Double-strand Break Repair
14568 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 285 • NUMBER 19 • MAY 7, 2010
by guest on February 2, 2019http://w
ww
.jbc.org/D
ownloaded from

USP11 acts upstream in the regulation of RAD51 and 53BP1but downstream of the initial �H2AX signal and independentlyof the BRCA1 localization mechanism.The DUB Activity of USP11 Is Necessary for Its Function in
Double-strand Break Repair—To gain further insights into themechanism of action of USP11 in double-strand break repairwe first sought to determine if it localized to double-strandbreak sites. Immunofluorescence localization of endogenousUSP11 did not reveal an accumulation of the protein in dam-age-induced foci. However, we did observe a significant frac-tion of USP11 on chromatin in cells both before and after treat-ment with ionizing radiation (Fig. 4A).We next asked whether the DUB activity of USP11 is
required for its function in double-strand break repair.We gen-erated stable cell lines expressing either HA-tagged wild type(WT) or catalytically inactive USP11 (C318S) that are resistantto both USP11 siRNAs because of wobble base pair changesin their target recognition sequences. These proteins wereexpressed at equal levels and unaffected by transfection with
the siRNAs (Fig. 4B). We first determined whether the sponta-neous activation of the DDR following silencing of USP11 wascomplemented by either the WT or C318S proteins. WTUSP11 efficiently complemented the �H2AX induced by bothUSP11 siRNAs (Fig. 4C). In contrast, C318S-USP11 expressionwas unable to complement this USP11-deficient phenotype(Fig. 4C). Similarly, WT but not C318S-USP11 could comple-ment the reduced RAD51 foci and increased 53BP1 foci inUSP11 siRNA-transfected cells following ionizing radiationtreatment (Fig. 4, D and E). Thus, USP11 catalytic activity isrequired for its function in the double-strand break response.
DISCUSSION
Our results demonstrate a role for USP11 in the repair ofdouble-strand breaks via homologous recombination. Deple-tion of USP11 by introduction of siRNA leads to spontaneousDNA damage in the absence of genotoxic stress. USP11-si-lenced cells are hypersensitive to IR, bleomycin, cisplatin, mito-mycin C, and PARPi treatment. They also exhibit a defect inHR directed double-strand break repair. Proper kinetic regula-tion of protein accumulation at double-strand breaks isimpaired in USP11-depleted cells. RAD51 foci are not main-tained and 53BP1 foci are increased at the same time pointsafter IR exposure. Moreover, USP11 is a chromatin-associatedprotein and its catalytic activity is required for its genomemain-tenance activities. Taken together, these results indicate thatUSP11 is a DUB that functions in the DNA damage response todouble-strand breaks.Ubiquitylation of proteins at double-strand breaks is regu-
lated by multiple ubiquitin ligases and DUBs creating a hetero-geneous landscape (45). Ubiquitin conjugation regulates therecruitment of multiple proteins including RAD51 and 53BP1through as yet undefined mechanisms (23, 26, 46). The accel-erated disappearance of RAD51 foci in USP11-silenced cellsmay indicate an inability to maintain a sufficient RAD51 fila-ment to facilitateHR repair. This could be due to changes in theresection of the double-strand breaks or perhaps inappropriateprocessing of the resected end.It is unclear whether the persistent 53BP1 foci observed at
later times after IR in USP11-silenced cells is a cause or conse-quence of the repair defect. 53BP1 recruitment is dependent onubiquitylation (47). However, 53BP1 does not bind directly toubiquitin-modified proteins. Instead, 53BP1 tudor domainsbind methylated histones. The precise role of ubiquitin isunknown. The prolonged 53BP1 foci could be caused by thepersistence of unrepaired double-strand breaks or increasedlevels of ubiquitin-conjugation at theDSB. Interestingly, 53BP1has been shown to be required forXRCC4-dependent end-join-ing repair but not required for HR (48). Furthermore, 53BP1deficiency actually increases HR repair. Thus, the increased53BP1 retention at DSBs observed in USP11-silenced cellscould be causally connected to the decrease inHR repair. In anycase, the alterations in 53BP1 and RAD51 foci support a dimin-ished capacity for HR repair in USP11-depleted cells leading toincreased spontaneous �H2AX foci, PARPi sensitivity, andreduced survival after IR.Our data indicate a requirement for the DUB activity of
USP11 in the DNA damage response, suggesting defective
FIGURE 3. USP11 regulates 53BP1 foci after IR. A–C, mock, non-targetingand USP11-silenced U2OS cells were treated with IR and allowed to recoverfor the indicated time prior to fixation and staining with antibodies to A,�H2AX; B, BRCA1; or C, 53BP1 (*, p � 0.0006; **, p � 0.074). The percentage ofcells with �5 foci were scored at each time point. Error bars are S.D. (n � 3).
USP11 Regulates DNA Double-strand Break Repair
MAY 7, 2010 • VOLUME 285 • NUMBER 19 JOURNAL OF BIOLOGICAL CHEMISTRY 14569
by guest on February 2, 2019http://w
ww
.jbc.org/D
ownloaded from

ubiquitin de-conjugation or ubiquitin turnover causes the dou-ble-strand break repair defect in USP11-silenced cells. How-ever, we cannot rule out thatmutation of the catalytically activesite of USP11 could cause disruption of protein interactions,accounting for the defects we see in our C318S mutant. Theidentification of the USP11 target(s) mediating its repair func-tion is an important future goal. USP11was previously reportedto interact with BRCA2 (49). BRCA2 is ubiquitylated in cells;however, Schoenfeld et al. found that catalytic-inactive USP11had no effect on BRCA2 ubiquitylation or protein levels. It ispossible the BRCA2 interaction provides a mechanism torecruitUSP11 to double-strand breakswhere it can act on othersubstrates.However, wewere unable to find an accumulation ofUSP11 in foci or a significant increase in its chromatin associ-ation after IR.Although the interaction of USP11 and BRCA2 suggests a
direct activity ofUSP11 at double-strand breaks, other reportedUSP11 interactions are consistent with indirect effects. Massspectrometry results indicate that USP11 interacts with a largenumber of proteins including the transcriptional elongationfactors TCEAL1 and TCEAL4, other DUBs including USP7,and the NRF2 regulatory protein KEAP1 (50). Thus, USP11may have multiple functions within the cell, and we cannotexclude the possibility that the double-strand break repairdefects are caused by indirect functions of USP11 in transcrip-tion regulation or other cellular pathways.In summary, we have used a synthetic lethal approach to
identify USP11 as a participant in the HR double-strand breakrepair pathway. PARP inhibitors are a new class of targetedtherapywith exciting clinical promise in a subset of patients (9).
USP11 status or the status of other HR-repair proteins intumors may provide biomarkers for use of PARP inhibitors.Furthermore, our approach provides an experimental model toidentify other HR proteins and potential biomarkers. The samemethodologies could also be applied to determine targets forcombination therapy with PARP inhibitors.
Acknowledgment—We thank Fei Ye for help with statistical analysis.
REFERENCES1. Kastan, M. B., and Bartek, J. (2004) Nature 432, 316–3232. Huen, M. S., and Chen, J. (2010) Trends Biochem. Sci. 35, 101–1083. Ashworth, A. (2008) J. Clin. Oncol. 26, 3785–37904. Herceg, Z., and Wang, Z. Q. (2001)Mutat. Res. 477, 97–1105. Saffhill, R., and Ockey, C. H. (1985) Chromosoma 92, 218–2246. Wang, Z.-Q., Stingl, L., Morrison, C., Jantsch, M., Los, M., Schulze-
Osthoff, K., and Wagner, E. F. (1997) Genes Dev. 11, 2347–23587. Bryant, H. E., Schultz, N., Thomas, H. D., Parker, K.M., Flower, D., Lopez,
E., Kyle, S., Meuth, M., Curtin, N. J., and Helleday, T. (2005) Nature 434,913–917
8. Farmer, H., McCabe, N., Lord, C. J., Tutt, A. N., Johnson, D. A., Richard-son, T. B., Santarosa, M., Dillon, K. J., Hickson, I., Knights, C., Martin,N.M., Jackson, S. P., Smith,G.C.M., andAshworth, A. (2005)Nature434,917–921
9. Fong, P. C., Boss, D. S., Yap, T. A., Tutt, A., Wu, P., Mergui-Roelvink, M.,Mortimer, P., Swaisland, H., Lau, A., O’Connor, M. J., Ashworth, A., Car-michael, J., Kaye, S. B., Schellens, J. H., and de Bono, J. S. (2009) N. Engl.J. Med. 361, 123–134
10. McCabe, N., Turner, N. C., Lord, C. J., Kluzek, K., Bialkowska, A., Swift, S.,Giavara, S., O’Connor, M. J., Tutt, A. N., Zdzienicka, M. Z., Smith, G. C.,and Ashworth, A. (2006) Cancer Res. 66, 8109–8115
11. Bryant, H. E., and Helleday, T. (2006) Nucleic Acids Res. 34, 1685–1691
FIGURE 4. USP11 is a chromatin-associated protein, and its DUB activity is required for its activity in double-strand break repair. A, whole cell extracts(WCE), soluble fractions (S2 and S3) and chromatin fractions (P3) from cells treated with IR and allowed to recover for the indicated times were immunoblottedfor USP11, ORC2, and GAPDH. B–E, HA-tagged wild-type (WT), and catalytic inactive (C318S) USP11 cDNAs that are insensitive to the USP11 siRNAs were stablyexpressed in U2OS cells. B, expression levels of HA-tagged proteins after non-targeting and USP11 siRNA transfections were determined by immunoblotting.C, parental-, WT-, and C318S USP11-expressing cells were transfected with the indicated siRNAs and scored for �H2AX foci in undamaged cells. D and E,parental-, WT-, and C318S-expressing cells were transfected with the indicated siRNAs, irradiated with 1 or 4 Gy of IR, allowed to recover for 15 h, then scoredfor RAD51 and 53BP1 foci by immunofluorescent imaging. Error bars in C–E are S.D. (n � 3).
USP11 Regulates DNA Double-strand Break Repair
14570 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 285 • NUMBER 19 • MAY 7, 2010
by guest on February 2, 2019http://w
ww
.jbc.org/D
ownloaded from

12. Lavin, M. F. (2008) Nat. Rev. Mol. Cell Biol. 9, 759–76913. Kitagawa, R., and Kastan, M. B. (2005) Cold Spring Harbor Symp. Quant.
Biol. 70, 99–10914. Stucki, M., and Jackson, S. P. (2006) DNA Repair 5, 534–54315. Harper, J. W., and Elledge, S. J. (2007)Mol. Cell 28, 739–74516. Yuan, J., and Chen, J. (2010) J. Biol. Chem. 285, 1097–110417. Sung, P. (1994) Science 265, 1241–124318. Baumann, P., Benson, F. E., and West, S. C. (1996) Cell 87, 757–76619. Shinohara, A., Ogawa, H., and Ogawa, T. (1992) Cell 69, 457–47020. Davies, A. A., Masson, J. Y., McIlwraith, M. J., Stasiak, A. Z., Stasiak, A.,
Venkitaraman, A. R., and West, S. C. (2001)Mol. Cell 7, 273–28221. Yuan, S. S., Lee, S. Y., Chen, G., Song, M., Tomlinson, G. E., and Lee, E. Y.
(1999) Cancer Res. 59, 3547–355122. Stewart, G. S., Panier, S., Townsend, K., Al-Hakim, A. K., Kolas, N. K.,
Miller, E. S., Nakada, S., Ylanko, J., Olivarius, S., Mendez, M., Oldreive, C.,Wildenhain, J., Tagliaferro, A., Pelletier, L., Taubenheim, N., Durandy, A.,Byrd, P. J., Stankovic, T., Taylor, A. M., and Durocher, D. (2009) Cell 136,420–434
23. Mailand, N., Bekker-Jensen, S., Faustrup, H., Melander, F., Bartek, J., Lu-kas, C., and Lukas, J. (2007) Cell 131, 887–900
24. Huen, M. S., Grant, R., Manke, I., Minn, K., Yu, X., Yaffe, M. B., and Chen,J. (2007) Cell 131, 901–914
25. Kolas, N. K., Chapman, J. R., Nakada, S., Ylanko, J., Chahwan, R., Sweeney,F. D., Panier, S., Mendez,M.,Wildenhain, J., Thomson, T.M., Pelletier, L.,Jackson, S. P., and Durocher, D. (2007) Science 318, 1637–1640
26. Zhao, G. Y., Sonoda, E., Barber, L. J., Oka, H., Murakawa, Y., Yamada, K.,Ikura, T., Wang, X., Kobayashi, M., Yamamoto, K., Boulton, S. J., andTakeda, S. (2007)Mol. Cell 25, 663–675
27. Doil, C., Mailand, N., Bekker-Jensen, S., Menard, P., Larsen, D. H., Pep-perkok, R., Ellenberg, J., Panier, S., Durocher, D., Bartek, J., Lukas, J., andLukas, C. (2009) Cell 136, 435–446
28. Wang, B., and Elledge, S. J. (2007) Proc. Natl. Acad. Sci. U. S. A. 104,20759–20763
29. Stucki, M., Clapperton, J. A., Mohammad, D., Yaffe, M. B., Smerdon, S. J.,and Jackson, S. P. (2005) Cell 123, 1213–1226
30. Stewart, G. S., Wang, B., Bignell, C. R., Taylor, A. M., and Elledge, S. J.(2003) Nature 421, 961–966
31. Sobhian, B., Shao, G., Lilli, D. R., Culhane, A. C., Moreau, L. A., Xia, B.,Livingston, D. M., and Greenberg, R. A. (2007) Science 316, 1198–1202
32. Kim, H., Chen, J., and Yu, X. (2007) Science 316, 1202–120533. Wang, B., Matsuoka, S., Ballif, B. A., Zhang, D., Smogorzewska, A., Gygi,
S. P., and Elledge, S. J. (2007) Science 316, 1194–119834. Kim, H., Huang, J., and Chen, J. (2007)Nat. Struct. Mol. Biol. 14, 710–71535. Yan, J., Kim, Y. S., Yang, X. P., Li, L. P., Liao, G., Xia, F., and Jetten, A. M.
(2007) Cancer Res. 67, 6647–665636. Chen, X., Arciero, C. A., Wang, C., Broccoli, D., and Godwin, A. K. (2006)
Cancer Res. 66, 5039–504637. Dong, Y., Hakimi, M. A., Chen, X., Kumaraswamy, E., Cooch, N. S., God-
win, A. K., and Shiekhattar, R. (2003)Mol. Cell 12, 1087–109938. Shao, G., Lilli, D. R., Patterson-Fortin, J., Coleman, K. A., Morrissey, D. E.,
andGreenberg, R. A. (2009) Proc. Natl. Acad. Sci. U. S. A. 106, 3166–317139. Nicassio, F., Corrado, N., Vissers, J. H., Areces, L. B., Bergink, S., Marteijn,
J. A., Geverts, B., Houtsmuller, A. B., Vermeulen, W., Di Fiore, P. P., andCitterio, E. (2007) Curr. Biol. 17, 1972–1977
40. Lord, C. J., McDonald, S., Swift, S., Turner, N. C., and Ashworth, A. (2008)DNA Repair 7, 2010–2019
41. Turner, N. C., Lord, C. J., Iorns, E., Brough, R., Swift, S., Elliott, R., Rayter,S., Tutt, A. N., and Ashworth, A. (2008) EMBO J. 27, 1368–1377
42. Swanton, C., Marani, M., Pardo, O., Warne, P. H., Kelly, G., Sahai, E.,Elustondo, F., Chang, J., Temple, J., Ahmed, A. A., Brenton, J. D., Down-ward, J., and Nicke, B. (2007) Cancer Cell 11, 498–512
43. Zhang, J.,Willers, H., Feng, Z., Ghosh, J. C., Kim, S.,Weaver, D. T., Chung,J. H., Powell, S. N., and Xia, F. (2004)Mol. Cell. Biol. 24, 708–718
44. Lovejoy, C. A., Xu, X., Bansbach, C. E., Glick, G. G., Zhao, R., Ye, F., Sirbu,B. M., Titus, L. C., Shyr, Y., and Cortez, D. (2009) Proc. Natl. Acad. Sci.U. S. A. 106, 19304–19309
45. Messick, T. E., and Greenberg, R. A. (2009) J. Cell Biol. 187, 319–32646. Botuyan, M. V., Lee, J., Ward, I. M., Kim, J. E., Thompson, J. R., Chen, J.,
and Mer, G. (2006) Cell 127, 1361–137347. Stewart, G. S. (2009) Cell Cycle 8, 1532–153848. Xie, A., Hartlerode, A., Stucki, M., Odate, S., Puget, N., Kwok, A., Naga-
raju, G., Yan, C., Alt, F. W., Chen, J., Jackson, S. P., and Scully, R. (2007)Mol. Cell 28, 1045–1057
49. Schoenfeld, A. R., Apgar, S., Dolios, G., Wang, R., and Aaronson, S. A.(2004)Mol. Cell. Biol. 24, 7444–7455
50. Sowa, M. E., Bennett, E. J., Gygi, S. P., and Harper, J. W. (2009) Cell 138,389–403
USP11 Regulates DNA Double-strand Break Repair
MAY 7, 2010 • VOLUME 285 • NUMBER 19 JOURNAL OF BIOLOGICAL CHEMISTRY 14571
by guest on February 2, 2019http://w
ww
.jbc.org/D
ownloaded from

and David CortezTimothy D. Wiltshire, Courtney A. Lovejoy, Tong Wang, Fen Xia, Mark J. O'Connor
Break RepairUbiquitin-specific Peptidase 11 (USP11) as a Regulator of DNA Double-strand
Sensitivity to Poly(ADP-ribose) Polymerase (PARP) Inhibition Identifies
doi: 10.1074/jbc.M110.104745 originally published online March 15, 20102010, 285:14565-14571.J. Biol. Chem.
10.1074/jbc.M110.104745Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
Supplemental material:
http://www.jbc.org/content/suppl/2010/03/15/M110.104745.DC1
http://www.jbc.org/content/285/19/14565.full.html#ref-list-1
This article cites 50 references, 19 of which can be accessed free at
by guest on February 2, 2019http://w
ww
.jbc.org/D
ownloaded from