separation and characterization of an igg2 antibody containing a cyclic imide in cdr1 of light chain...
TRANSCRIPT

Separation and Characterization of an IgG2Antibody Containing a Cyclic Imide in CDR1 ofLight Chain by Hydrophobic InteractionChromatography and Mass Spectrometry
John Valliere-Douglass, Laura Jones, Diana Shpektor, Paul Kodama, Alison Wallace, Alain Balland,Robert Bailey, and Yuling Zhang*
Process and Analytical Sciences, Amgen Inc., Seattle, Washington 98119
Hydrophobic interaction chromatography (HIC) was usedto separate populations of recombinant IgG2 antibody thatwere created as a result of prolonged incubation at40 °C. Antibody was separated by HIC into three majorand seven minor fractions. All but one fraction wascomposed of antibody with distinct chemical modificationsthat resulted from exposure to elevated temperature. Theresults of intact and reduced mass analysis as well aspeptide map data derived from the three major HICfractions indicated that the antibody was being chromato-graphically separated into populations containing a suc-cinimidyl intermediate in complementarity determiningregion 1 (CDR1) on zero, one, and two light chain arms.Lower level species purified by HIC were analyzed byintact and reduced mass analysis and laser-inducedfluorescence capillary electrophoresis (CE-LIF) and con-sisted of an antibody that was clipped in four differentplaces in the heavy chain as well as misfolded andaggregated antibody. The potency of the recombinantantibody containing a succinimidyl intermediate on zero,one, and two light chain arms was analyzed by LANCEbinding assay and a cell based in vitro bioassay, and theoccurrence of this modification on one or both light chainarms was associated with a reduction in the bindingaffinity of the molecule to the target by approximately 10%.We show that HIC has the unique ability as a first steppurification method to separate populations of antibodywhich are covalently modified under stability programs.The method conditions that have been developed for theHIC assay are ideal for purifying antibodies with labilemodifications for the purpose of further characterization.
HIC separates populations of molecules in order of increasinghydrophobicity. Molecules bind to a weakly hydrophobic station-ary phase in the presence of high concentrations of salt and desorbinto the mobile phase as the concentration of salt decreases. Incontrast to reversed phase chromatography, HIC is typicallycarried out under highly polar column loading conditions whichtend to protect the native structure of the molecule of interest.
HIC is widely used in a protein purification setting for isolatingtherapeutic antibodies and proteins;1-4 however, it is seldom usedas a direct characterization technique. The routine use of liquidchromatography-mass spectrometry (LC-MS) for the analysisof recombinantly produced molecules has largely displaced classicoffline chromatography techniques for molecule characterizationpurposes. LC-MS of intact and reduced molecules has greatlysimplified the characterization of heterogeneity evident in popula-tions of molecules.5-8 Digestion of monoclonal proteins withendoproteases followed by reverse phase separation and tandemmass spectrometric identification of the resulting peptides enablessources of molecule heterogeneity to be pinpointed to an aminoacid site with relative ease.
Although reversed-phase chromatography is generally ac-cepted as a more resolving chromatographic method than HICfor the separation of peptides and small proteins, it is of limiteduse for the separation and subsequent characterization of largebiomolecules. Reversed phase methods have recently beendeveloped for the separation of heterogeneous populations of IgG2antibodies, but the method conditions necessary for the recoveryof these species are detrimental to native structure.5 In contrastto reverse phase chromatography, hydrogen-deuterium exchangeperformed on proteins that are adsorbed on HIC media havedemonstrated that no significant change in protein structure wasevident using traditional HIC solvents.9,10 Recovery of chromato-graphically separated recombinant proteins in the native form is
* Corresponding author. Yuling Zhang, 1201 Amgen Ct. West, Seattle, WA98119.
(1) Shukla, A. A.; Peterson, J.; Sorge, L.; Lewis, P.; Thomas, S.; Waugh, S.Biotechnol. Prog. 2002, 18, 556-64.
(2) Wood, D. C.; Mathis, K. J.; Joy, W. D.; Minnerly, J. C.; Pegg, L. E.; Welply,J. K. Biotechnol. Appl. Biochem. 2003, 37, 31-8.
(3) Sunasara, K. M.; Xia, F.; Gronke, R. S.; Cramer, S. M. Biotechnol. Bioeng.2003, 82, 330-9.
(4) Graumann, K.; Ebenbichler, A. A. Chem. Eng. Technol. 2005, 28, 1398-407.
(5) Dillon, T. M.; Bondarenko, P. V.; Speed, R. M. J. Chromatogr., A 2004,1053, 299-305.
(6) Dillon, T. M.; Bondarenko, P. V.; Rehder, D. S.; Pipes, G. D.; Kleemann, G.R.; Ricci, M. S. J. Chromatogr., A 2006, 1120, 112-20.
(7) Liu, H.; Gaza-Bulseco, G.; Faldu, D.; Chumsae, C.; Sun, J. J. Pharm. Sci.2007, 1-22.
(8) Rehder, D. S.; Dillon, T. M.; Pipes, G. D.; Bondarenko, P. V. J. Chromatogr.,A 2006, 1102, 164-75.
(9) Jones, T. T.; Fernandez, E. J. Biotechnol. Bioeng. 2004, 87, 388-99.(10) Xiao, Y.; Freed, A. S.; Jones, T. T.; Makrodimitris, K.; O’Connell, J. P.;
Fernandez, E. J. Biotechnol. Bioeng. 2006, 93, 1177-89.
Anal. Chem. 2008, 80, 3168-3174
3168 Analytical Chemistry, Vol. 80, No. 9, May 1, 2008 10.1021/ac702245c CCC: $40.75 © 2008 American Chemical SocietyPublished on Web 03/21/2008

particularly important for the study of their therapeutic propertiesusing binding and cell based potency assays. Although there aresome examples of the application of HIC to separating intactmonoclonal antibody populations,11 historically, the technique ismore commonly used for the purpose of separating antibody Faband Fc subdomains resulting from digestion with papain.12,13
Whereas digestion with papain is an efficient strategy for char-acterization of fully humanized monoclonal IgG1 antibodies,14
IgG2’s are typically more resistant to papain cleavage15 and thusrequire a different approach.
HIC has great utility when used for purification of moleculepopulations which arise due to cellular processing or are createdduring stability studies.7 Assessment of monoclonal antibodystability involving incubation at elevated temperature and varia-tions in formulation pH typically leads to covalent modificationswhich increase the diversity of molecules present in a givensample. In this report we have used HIC to separate hydrophobicvariants that are created in a recombinant IgG2 molecule whichwas incubated at 40 °C for 12 weeks. Ten distinct hydrophobicsubpopulations were isolated, most of which were the result oftemperature induced degradation. The HIC pools were analyzedby intact and reduced molecule mass spectrometry, peptide map,CE-LIF, SEC, LANCE binding assay, and in vitro bioassay. Of thehydrophobic variants separated by HIC, the main products oftemperature degradation were found to be antibody populationswhich contained a cyclic imide from aspartic acid in complemen-tarity determining region 1 (CDR1) on one or both light chainarms (Figure 1). We also show that HIC is an efficient method toseparate other modifications including populations containing clipsto the heavy chain and covalent aggregates.
MATERIALS AND METHODSMaterials. Recombinant IgG2 antibody produced in CHO cells
was purified according to standard manufacturing procedures andformulated in acetate buffer at pH 5. The antibody was degradedby incubation at 40 °C for 12 weeks in the dark. Any reagentsused for subsequent experiments that are not explicitly describedwere of the highest obtainable purity.
HIC Fractionation. Preparative HIC purification of IgG2hydrophobic variants was carried out on an Akta Explorer (GE,Fairfield, CT) equipped with two 7.8 mm × 75 mm Dionex ProPacHIC-10 columns (Dionex, Sunnyvale, CA) connected in series toincrease the resolution of closely eluting species. The compositionof HPLC buffer A was 2.0 M ammonium acetate pH 5.2, 7%acetonitrile and was prepared from a 4.5 M stock acetic acidsolution that was adjusted to pH 5.2 with ammonium hydroxide.The composition of buffer B was 20 mM ammonium acetate pH5.2, 7% acetonitrile and was prepared by diluting buffer A 1:100 in7% acetonitrile and adjusting the pH to 5.2 with ammoniumhydroxide as needed. An amount of 15-30 mg of protein wasdissolved in 2 M ammonium acetate pH 5.2, 7% acetonitrile andbound to the columns previously equilibrated in buffer A at 1.5mL/min. The column was washed with buffer A for 9 min, andprotein was subsequently eluted with a linear gradient to 20 mMammonium acetate pH 5.2, 7% acetonitrile over 90 min at a flowrate of 1.5 mL/min. The separations were monitored by absor-bance at 280 nm, and the collection of 1.5 mL fractions com-menced 11 min after the injection. The column pressure decreasedfrom 8 MPa at the start of the run to 5.5 MPa at the end of thegradient in 100% buffer B. Subsequent injections were carried outafter the column pressure returned to 8 MPa in the presence of100% buffer A. Selected fractions were reanalyzed on a Waters2690 HPLC and 474 fluorescence detector (Waters, Milford, MA)with two 4.6 mm × 100 mm Dionex ProPac HIC-10 columnsconnected in series. The analytical separations were monitoredby excitation at 280 nm and emission at 340 nm. Fractions wereconservatively pooled to ensure maximal purity and concentratedand buffer exchanged to 20 mM sodium acetate pH 5.2 bycentrifugation in Millipore centriprep 15 mL filter units with a 30kDa molecular weight cutoff (MWCO) (Millipore, Bilerica, MA).
(11) Kwong, M. Y.; Harris, R. J. Protein Sci. 1994, 3, 147-9.(12) Cacia, J.; Keck, R.; Presta, L. G.; Frenz, J. Biochemistry 1996, 35, 1897-
903.(13) Wakankar, A. A.; Borchardt, R. T.; Eigenbrot, C.; Shia, S.; Wang, Y. J.; Shire,
S. J.; Liu, J. L. Biochemistry 2007, 46, 1534-44.(14) Yan, B.; Valliere-Douglass, J.; Brady, L.; Steen, S.; Han, M.; Pace, D.; Elliott,
S.; Yates, Z.; Han, Y.; Balland, A.; Wang, W.; Pettit, D. J. Chromatogr., A2007.
(15) Snigurowicz, J.; Powiertowska-Rezmer, M. Arch. Immunol. Ther. Exp. 1980,28, 265-73.
Figure 1. Formation of a covalent bond between the aspartic acidside chain carbonyl (1) and the amino-terminal nitrogen (5) of thesubsequent amino acid in the sequence results in the conversion ofaspartic acid into a cyclic imide (succinimide). Hydrolysis of thesuccinimide carbonyl at position 1 results in the regeneration ofaspartic acid, and hydrolysis at the carboxy-terminal carbonyl (4)yields iso-aspartic acid.
Figure 2. HIC comparison of IgG2 starting material and IgG2 afterincubation at 40 °C for 3 months. Temperature stress results in thecreation of several hydrophobic variants which are separable by HIC.Regions of the chromatogram corresponding to purified fractions1-10 are indicated.
Analytical Chemistry, Vol. 80, No. 9, May 1, 2008 3169

Peptide Map. The HIC fractions below 2 mg/mL wereconcentrated using Millipore centricon spin filters with a 5 kDaMWCO. HIC fractions were digested with trypsin, which wasadded at a 1:6.7 (w:w) ratio of enzyme to antibody in 20 mMsodium acetate pH 5.2. The digest was carried out at 37 °C for 18h and then stopped by adding trifluoroacetic acid (TFA, Pierce,Rockford IL) to a final concentration of 0.27% (w:v). The peptidemap was carried out by injecting 33.3 µg of digested material ontoan Agilent HP1200 HPLC equipped with a Phenomenex C5column of dimensions 2.1 mm × 250 mm (Phenomonex, Torrance,CA). Mobile phase buffer A consisted of 0.12% trifluoroacetic acidin water and buffer B consisted of 80% acetonitrile, 0.11%trifluoroacetic acid in water. The peptides were eluted with a lineargradient to 60% B after an initial 10 min hold at 1% B. The columnwas washed by increasing the organic from 60% to 100% B in 10min and holding at 100% B for an additional 2 min. The columnwas then brought to the initial conditions of 1% B in 1 min andre-equilibrated in the initial conditions for 40 min prior to the nextinjection. Online mass spectral data was acquired with a ThermoLTQ XL ion trap mass spectrometer (Thermo, Waltham, MA)which was set to acquire one survey scan and perform MS2 andMS3 in the data dependent manner for seven scans prior toacquiring another survey scan.
Intact and Reduced Molecule Mass Spectrometry. PurifiedHIC fractions were reduced by incubation at 55 °C in the presenceof 4 M guanidine HCl, 50 mM sodium acetate pH 5.0, and 50 mMtris[2-carboxyethyl] phosphine (TCEP, Pierce, Rockford, IL).Reduced and intact molecules were desalted prior to mass analysisby analytical SEC.16 Briefly, 4 µg of reduced antibody or 10 µg ofintact antibody were injected onto a polyhydroxyethyl aspartamidecolumn (PolyLC, The Nest Group, Southborough, MA) in 0.1%formic acid at a flow rate of 0.1 mL/min, and 98% acetonitrile, 2%formic acid was introduced postcolumn and mixed to the HPLCeluent entering the electrospray ionization (ESI) source. The mass
of the desalted reduced or intact antibody was determined withan Agilent LC-MSD time-of-flight instrument (Agilent, SantaClara, CA). The acquisition of quality intact mass data was highlydependent upon capillary voltage, declustering potential, andoctupole RF parameters, and settings of 5000, 415, and 300 V,respectively, were determined to be optimal. Capillary voltage anddeclustering potential were reduced to 4000 and 250 V, respec-tively, for reduced mass samples. Raw MS data were deconvolutedwith Agilent MassHunter Qualitative Analysis software versionB.01.02 build 1.2.122.1 patch 3 or Agilent Protein Deconvolutionsoftware version 1.0.0.1.
Laser Induced Fluorescence-Capillary Electrophoresis.Prior to labeling with the fluorophore 3-(2-furoyl)-quinoline-2-carboxaldehyde (FQ), all samples were brought to a concentrationof 1 mg/mL and buffer exchanged to 5 mM sodium acetate pH5.2 using Pierce Zeba microspin columns (Pierce, Rockford, IL).
(16) Brady, L.; Valliere-Douglass, J.; Martinez, T.; Balland, A. J. Am. Soc. Mass.Spectrom. in press.
Figure 3. Comparison of MS/MS data obtained from the fragmentation of unmodified L3 peptide (A) and L3 peptide containing a succinimidylin place of Asp (B). The mass shift of -18 Da corresponding to the loss of water from an Asp side chain is evident in the prominent y ion seriesstarting at y8 and progressing through y11 in panel B.
Figure 4. Peptide map UV trace of the L3 peptide derived fromunstressed antibody and purified HIC fractions that were digestedunder acidic conditions with trypsin. Peptide L3 succinimide 30 isenriched in HIC fractions 7 and 8 relative to HIC fraction 6 andunstressed antibody. The levels of isomerized (iso-Asp) L3 observedin HIC fractions 7 and 8 are likely the result of succinimide hydrolysisoccurring during digestion.
3170 Analytical Chemistry, Vol. 80, No. 9, May 1, 2008

Samples were labeled with FQ and analyzed according to Michelset al.17
SE Chromatography. An amount of 10 µg of antibody dilutedin formulation buffer described above was loaded onto two TosohTSK G3000 SWxl columns connected in series along with a SWxlguard column (Tosoh, Tokyo, Japan) attached before the analyticalcolumns. A Waters Alliance 2695 HPLC was used to pass mobilephase containing 50 mM sodium phosphate pH 7.3, 100 mM NaCl,and 10% ethanol through the columns at a flow rate of 0.5 mL/min. Eluting species were detected by a Waters 474 scanningfluorescence detector by excitation at 280 nm and emission at340 nm.
Receptor Phosphorylation Bioassay. PC-3 cells, a humanprostate carcinoma cell line, were obtained from American TypeCulture Collection Services (ATCC, Manassas, VA). The cells weremaintained in RPMI1640 medium with Glutamax containing 10%fetal bovine serum (FBS) at 37 °C with 10% CO2 and 90% air in ahumidified incubator. PC-3 cells were serum starved in a low-glucose medium for 24 h prior to experiment. The dose of ligandused is equivalent to the IC90 of the maximum induction of thereceptor phosphorylation for 20 000 cells per well serum starvedPC-3 cells. Reference standard, control, and test samples that weretitrated and incubated with ligand spike were then transferred toan ELISA plate coated with anti-phosphotyrosine antibody (4G10,Upstate Biotechnology Inc., Lake Placid, NY). Serum starved PC-3cells were added to the coated ELISA plate and incubated for 5-10min at 37 °C. Lysis buffer was applied followed by a freeze-thawcycle to expose the phosphorylated receptor intracellular domain,which is then captured out with the coated 4G10 anti-phospho-tyrosine antibody. Wells were washed and biotinylated anti-receptor antibody is added and incubated. Following another wash,the peroxidase-conjugated polystreptavidin was added for detec-tion. After the final wash, a chromogen solution was added to thewells and incubated for color development. Acid was then addedto arrest color development, and the optical density (OD) of eachwell was measured with a spectrophotometer. The OD values areinversely proportional to the amount of ligand bound by thereference standard and test samples. The biological activity of thetest sample is determined by comparing the response of testsample to the reference standard. Results are the average ( thestandard deviation (SD) of at least three assays.
LANCE Receptor/Ligand Inhibition Assay. Control anti-body or test samples were incubated with biotin-ligand, Europiumconjugated receptor (Eu-receptor), and streptavidin-allophycocya-nin (SA-APC) in 0.1% BSA Tris buffer, pH 7.6, in a half-area 96well black plates for 1 to 3 h protected from light. Binding inducedmolecular proximity of Europium and APC allows fluorescentresonance energy transfer, which was detected with a homoge-neous time-resolved fluorescence (HTRF) plate reader. Neutral-izing IgG2 antibody inhibits biotin-ligand binding to the Eu-receptor and prevents energy transfer, thus decreasing fluorescenceoutput. The amount of antibody present is inversely related tothe amount of fluorescence, and the test sample activity isdetermined by comparing the antibody test sample response tothe antibody reference standard.
RESULTSHIC Fractionation. The HIC method that is described in this
report is substantially different from conventional HIC assays.Recovery of antibody from the HIC column was very poor in anaqueous, ammonium sulfate based elution system but improveddramatically when acetonitrile was added to the mobile phase.This result was not unexpected because binding of the solute tothe stationary phase in an HIC system is encouraged in a highlypolar mobile phase. Addition of a relatively nonpolar solvent, e.g.,acetonitrile, to the mobile phase decreases its polarity. A less polarmobile phase attenuates the binding of protein to the resin andimproves recovery of relatively hydrophobic proteins. Furtherimprovement was obtained when ammonium acetate was usedas the elution salt. Relative to ammonium sulfate, ammoniumacetate is a weaker lyotropic salt and thus destabilizes hydrophobiccontacts between the HIC column and the antibody resulting inan earlier elution time. It was important to decrease the retentiontime of the unmodified antibody because temperature stress ledto the creation of relatively more hydrophobic populations (Figure2). On the basis of an HIC comparison of temperature stressedand unstressed antibody, 10 distinct fractions were identified andisolated from the preparative fractionation of temperature de-graded antibody by HIC (Figure 2).
Peptide Map. Typical sample preparation techniques forpeptide map involve reduction and alkylation of cysteine residuesprior to digestion with endoproteases. Reduction of disulfidebridges is carried out by adding a reducing agent and incubatingat elevated temperature in the presence of alkaline pH buffer anda chaotrope. Initial peptide map data derived from temperaturestressed antibody indicated that a succinimidyl modificationpreviously documented for this molecule18 was being hydrolyzedat alkaline pH during sample preparation to iso-Asp and Asp (datanot shown). The nonreduced low pH trypsin peptide map wasdeveloped to preserve the succinimide to facilitate confirmationof the modification site. Recently a new method for quantitationof succinimides has been proposed involving hydrolysis of thecyclic imide with an isotopic label.19 Whereas this method mayprovide better quantitation of succinimide content, we believe thatthe nonreduced, low pH peptide map is suitable for directidentification of the succinimidyl site
Peptide map analysis of the purified HIC fractions 6-8indicated that the succinimide modification at the Asp residue ofthe third light chain tryptic peptide (Figure 3) was being enrichedin the later eluting HIC fractions. It should be noted however thatthe acidic pH peptide map data obtained from HIC fractions 7and 8 indicated the presence of L3 isomerization at the sameresidue as the succinimide (Figure 4, Table 1). This result wasnot expected as molecules with an iso-Asp residue typically eluteearlier than the succinimide containing species by HIC.12,20,21 Intheir study of isomerization of model peptides with large degreeof freedom, Stephenson and Clarke have reported that hydrolysis
(17) Michels, D. A.; Brady, L. J.; Guo, A.; Balland, A. Anal. Chem. 2007, 79,5963-71.
(18) Chu, G. C.; Chelius, D.; Xiao, G.; Khor, H. K.; Coulibaly, S.; Bondarenko, P.V. Pharm. Res. 2007, 24, 1145-56.
(19) Xiao, G.; Bondarenko, P. V.; Jacob, J.; Chu, G. C.; Chelius, D. Anal. Chem.2007, 79, 2714-21.
(20) Di, D. A.; Ciardiello, M. A.; de, N. M.; Piccoli, R.; Mazzarella, L.; D’Alessio,G. J. Biol. Chem. 1993, 268, 4745-51.
(21) Harris, R. J.; Kabakoff, B.; Macchi, F. D.; Shen, F. J.; Kwong, M.; Andya, J.D.; Shire, S. J.; Bjork, N.; Totpal, K.; Chen, A. B. J. Chromatogr., B: Biomed.Sci. Appl. 2001, 752, 233-45.
Analytical Chemistry, Vol. 80, No. 9, May 1, 2008 3171

of succinimide intermediates results in a conversion to iso-Aspand Asp at a 3:1 ratio.22 A succinimide may be quite stable due tothe secondary and tertiary constraints imposed by the intactmolecule but may become more labile after proteolytic digestionas the degree of freedom increases. In our experimental results,we are seeing increasing iso-Asp at light chain position 30 in thelater eluting HIC fractions. This result supports the hypothesisthat the levels of iso-Asp observed here are likely due to methodartifacts and are not present in the undigested sample. If theassumption is made that the L3 iso-Asp observed in the peptidemap data of HIC fractions 7 and 8 is the result of succinimidylhydrolysis, then the level of succinimide present in HIC fractions7 and 8 are is greater than 50% for HIC fraction 7 and 85% forHIC fraction 8.
Intact and Reduced Mass Analysis of HIC PurifiedAntibody Populations. HIC purified fraction 6 had a retentiontime similar to that for the main component in unstressedantibody. Intact mass data derived from HIC fraction 6 indicatedthat it was similar in composition to unstressed material basedon agreement in mass (Figure 5). HIC fractions 7 and 8 were 16and 32 Da lower in mass, respectively, than HIC fraction 6.Reduction under acidic conditions with TCEP localized the lossin mass in HIC fractions 7 and 8 to the light chain, which wasconsistent with the succinimide site identification (ID) determinedby the acidic pH peptide map (Figure 3). The distribution of thesuccinimide in the reduced mass data was consistent with theoccurrence of this modification on one light chain in HIC fraction7 and both light chains in HIC fraction 8.
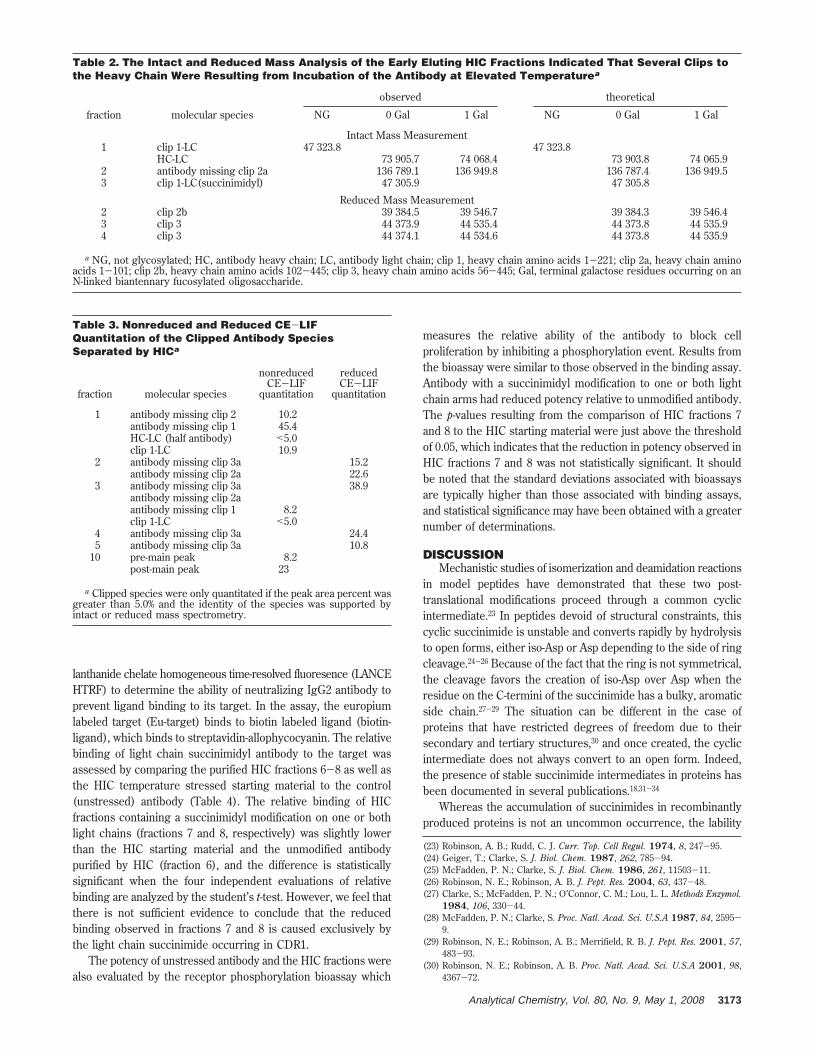
HIC fractions 1-4 contained primarily antibody with clips tothe heavy chain at four sites (Table 2). Fraction 1 consisted ofantibody with clips to the heavy chain at residue 221, which is aflanking residue to the first cysteine in the hinge region. Half(1/2) antibody and antibody with a clip at a serine residue atposition 423 are also present at lower levels. The major degradantobserved in fraction 2 was antibody with a heavy chain clip atposition 102. Clip populations in fraction 3 consisted of antibodywhich was clipped at residue 221 which also contained a succin-imidyl light chain, thus causing a retention time shift from fraction1 to fraction 3. Antibody with a clip to the heavy chain at position56 was also observed. The clip at heavy chain position 102 wasalso observed in fraction 4 except that one of the light chains ofthis species was modified to a succinimide. No significant
modifications were observed for fraction 5 by intact mass analysis,and the succinimidyl composition of fraction 9 was similar to thatof the presumptively double arm modified fraction 8 by intact andreduced mass analysis. The charge envelope obtained for wholemass analysis of fraction 10 indicated that there was dimer presentin this species, but the data quality was not sufficient to assign adeconvoluted mass. The light chain succinimide composition asdetermined by reduced mass analysis was approximately 50%.
Intact and Reduced Molecule CE-LIF. The discovery of fourdistinct heavy chain clip species which would reduce the mass ofthe intact molecule by 3-47 kDa necessitated the use of a high-resolution size based separation method for quantitation ofdegraded antibody. Laser induced fluorescence-capillary electro-phoresis was used for this purpose due to the high resolutionand low sample amounts required for analysis.17 Whereas the mainHIC fractions corresponding to intact antibody with 0-2 succin-imidyl light chains were not clipped or aggregated to anysignificant extent as indicated by CE-LIF, the clips contained inthe remaining HIC fractions varied from a high value of greaterthan 70% in HIC fraction 1 to a low value of 8.2% in fraction 10(Table 3). The resolution of nonreduced CE-LIF was not sufficientto resolve clips which removed 2527 and 6033 Da from the heavychain; however, the 6033 Da loss seen primarily in HIC fraction3 was resolved in the reduced CE-LIF heavy chain profile. Thedimer which was seen in the intact mass analysis of HIC fraction10 was confirmed by the nonreduced CE-LIF profile for thisspecies. Nonreduced CE-LIF indicated that fraction 10 contained23% aggregated material. The aggregate level determined fromSEC was 31.4%. The difference between the CE-LIF basedquantitation and SEC quantitation for this fraction is probably dueto the dissociation of noncovalent aggregate into monomericspecies in the CE-LIF sample buffer which contains SDS.
Assessment of Molecule Binding and Potency. The LANCEbinding assay is a competitive inhibition assay. The assay uses(22) Stephenson, R. C.; Clarke, S. J. Biol. Chem. 1989, 264, 6164-70.
Table 1. Peptide Map UV Quantitation of TrypticPeptide L3 Isolated from Unstressed Antibody and HICFractions Derived from Temperature StressedAntibodya
sampleL3 iso-Asp 30
(%)L3 Asp 30
(%)L3 Succinimide 30
(%)
unstressedantibody 1.8 95.3 2.9
HIC fraction 6 2.7 90.6 6.7HIC fraction 7 6.3 49.9 43.8HIC fraction 8 10.1 18.2 71.7
a The enrichment of L3 iso-Asp in fractions 7 and 8 indicates thatthere may be some hydrolysis of the cyclic imide occurring subsequentto the initial isolation by HIC.
Figure 5. The intact mass of unstressed antibody is shown in panelA. The consecutive decrease in mass of approximately 16 Da for HICpurified fraction 7 (panel C) and 32 Da for fraction 8 (panel D) relativeto fraction 6 (panel B) is evident and indicates the presence of asuccinimidyl intermediate on zero, one, and two arms of the antibodyfor HIC fractions 6, 7, and 8, respectively. Reanalysis of the antibodyfragments reduced under acidic conditions indicated that the succin-imidyl intermediate was on the antibody light chain and that themodified populations were enriched in HIC fractions 7 and 8 (panelsG and H, respectively) relative to the unstressed antibody and fraction6 (panels E and F, respectively).
3172 Analytical Chemistry, Vol. 80, No. 9, May 1, 2008
lanthanide chelate homogeneous time-resolved fluoresence (LANCEHTRF) to determine the ability of neutralizing IgG2 antibody toprevent ligand binding to its target. In the assay, the europiumlabeled target (Eu-target) binds to biotin labeled ligand (biotin-ligand), which binds to streptavidin-allophycocyanin. The relativebinding of light chain succinimidyl antibody to the target wasassessed by comparing the purified HIC fractions 6-8 as well asthe HIC temperature stressed starting material to the control(unstressed) antibody (Table 4). The relative binding of HICfractions containing a succinimidyl modification on one or bothlight chains (fractions 7 and 8, respectively) was slightly lowerthan the HIC starting material and the unmodified antibodypurified by HIC (fraction 6), and the difference is statisticallysignificant when the four independent evaluations of relativebinding are analyzed by the student’s t-test. However, we feel thatthere is not sufficient evidence to conclude that the reducedbinding observed in fractions 7 and 8 is caused exclusively bythe light chain succinimide occurring in CDR1.
The potency of unstressed antibody and the HIC fractions werealso evaluated by the receptor phosphorylation bioassay which
measures the relative ability of the antibody to block cellproliferation by inhibiting a phosphorylation event. Results fromthe bioassay were similar to those observed in the binding assay.Antibody with a succinimidyl modification to one or both lightchain arms had reduced potency relative to unmodified antibody.The p-values resulting from the comparison of HIC fractions 7and 8 to the HIC starting material were just above the thresholdof 0.05, which indicates that the reduction in potency observed inHIC fractions 7 and 8 was not statistically significant. It shouldbe noted that the standard deviations associated with bioassaysare typically higher than those associated with binding assays,and statistical significance may have been obtained with a greaternumber of determinations.
DISCUSSIONMechanistic studies of isomerization and deamidation reactions
in model peptides have demonstrated that these two post-translational modifications proceed through a common cyclicintermediate.23 In peptides devoid of structural constraints, thiscyclic succinimide is unstable and converts rapidly by hydrolysisto open forms, either iso-Asp or Asp depending to the side of ringcleavage.24-26 Because of the fact that the ring is not symmetrical,the cleavage favors the creation of iso-Asp over Asp when theresidue on the C-termini of the succinimide has a bulky, aromaticside chain.27-29 The situation can be different in the case ofproteins that have restricted degrees of freedom due to theirsecondary and tertiary structures,30 and once created, the cyclicintermediate does not always convert to an open form. Indeed,the presence of stable succinimide intermediates in proteins hasbeen documented in several publications.18,31-34
Whereas the accumulation of succinimides in recombinantlyproduced proteins is not an uncommon occurrence, the lability
(23) Robinson, A. B.; Rudd, C. J. Curr. Top. Cell Regul. 1974, 8, 247-95.(24) Geiger, T.; Clarke, S. J. Biol. Chem. 1987, 262, 785-94.(25) McFadden, P. N.; Clarke, S. J. Biol. Chem. 1986, 261, 11503-11.(26) Robinson, N. E.; Robinson, A. B. J. Pept. Res. 2004, 63, 437-48.(27) Clarke, S.; McFadden, P. N.; O’Connor, C. M.; Lou, L. L. Methods Enzymol.
1984, 106, 330-44.(28) McFadden, P. N.; Clarke, S. Proc. Natl. Acad. Sci. U.S.A 1987, 84, 2595-
9.(29) Robinson, N. E.; Robinson, A. B.; Merrifield, R. B. J. Pept. Res. 2001, 57,
483-93.(30) Robinson, N. E.; Robinson, A. B. Proc. Natl. Acad. Sci. U.S.A 2001, 98,
4367-72.
Table 2. The Intact and Reduced Mass Analysis of the Early Eluting HIC Fractions Indicated That Several Clips tothe Heavy Chain Were Resulting from Incubation of the Antibody at Elevated Temperaturea
observed theoretical
fraction molecular species NG 0 Gal 1 Gal NG 0 Gal 1 Gal
Intact Mass Measurement1 clip 1-LC 47 323.8 47 323.8
HC-LC 73 905.7 74 068.4 73 903.8 74 065.92 antibody missing clip 2a 136 789.1 136 949.8 136 787.4 136 949.53 clip 1-LC(succinimidyl) 47 305.9 47 305.8
Reduced Mass Measurement2 clip 2b 39 384.5 39 546.7 39 384.3 39 546.43 clip 3 44 373.9 44 535.4 44 373.8 44 535.94 clip 3 44 374.1 44 534.6 44 373.8 44 535.9
a NG, not glycosylated; HC, antibody heavy chain; LC, antibody light chain; clip 1, heavy chain amino acids 1-221; clip 2a, heavy chain aminoacids 1-101; clip 2b, heavy chain amino acids 102-445; clip 3, heavy chain amino acids 56-445; Gal, terminal galactose residues occurring on anN-linked biantennary fucosylated oligosaccharide.
Table 3. Nonreduced and Reduced CE-LIFQuantitation of the Clipped Antibody SpeciesSeparated by HICa
fraction molecular species
nonreducedCE-LIF
quantitation
reducedCE-LIF
quantitation
1 antibody missing clip 2 10.2antibody missing clip 1 45.4HC-LC (half antibody) <5.0clip 1-LC 10.9
2 antibody missing clip 3a 15.2antibody missing clip 2a 22.6
3 antibody missing clip 3a 38.9antibody missing clip 2aantibody missing clip 1 8.2clip 1-LC <5.0
4 antibody missing clip 3a 24.45 antibody missing clip 3a 10.8
10 pre-main peak 8.2post-main peak 23
a Clipped species were only quantitated if the peak area percent wasgreater than 5.0% and the identity of the species was supported byintact or reduced mass spectrometry.
Analytical Chemistry, Vol. 80, No. 9, May 1, 2008 3173

of this species under conditions typically used for analytical assaysmakes detection problematic. In the case of the antibody whichis the subject of this report, it has been well documented thatexposure to buffers at pH 8 or greater leads to rapid hydrolysisof the light chain succinimide18 and that addition of denaturantsfurther increases the rate of hydrolysis. It thus follows that thekey to any analytical method which is used for detection ofsuccinimides should either be carried out at low pH in nondena-turing conditions or should tag (hydrolyze) the positional site ofthe succinimide with an isotopic label so that information aboutthe site of modification can be ascertained by subsequent peptidemap analysis.19 HIC carried out under mildly acidic conditions iswell suited for the analysis of succinimides. The modification ofAsp to a succinimide results in a marked increase in hydrophobic-ity which is key to the HIC separation mechanism. Whereas cationexchange chromatography may also be applied in this situationbecause the formation of the succinimide results in the loss of anegative charge and thus the modified species elutes as a basicvariant, it may not be an ideal separation technique. Recombinantantibodies typically contain charge isoforms such as variableprocessing of C-terminal lysine residues35 and pyroglutamic acidfrom glutamine on the N-terminus of the heavy chain36,37 whichare not the product of stress induced degradation but whichnevertheless elute as basic and acidic charge variants, respectively,
by CEX. An antibody modified with a succinimide will elute inthe basic region of a CEX separation but will also potentiallycoelute with populations that do not contain succinimides but docontain C-terminal lysine or N-terminal glutamine on the heavychain. This problem is avoided by using HIC because thetechnique is relatively insensitive to charge variants which arethe result of variable processing during cell culture.
Antibodies are inherently complex molecules due to theirbivalent structure. When considering the effects of a covalentmodification on molecule potency, the effect of the modificationwhen it occurs on one arm of the antibody needs to be differenti-ated from the effect to potency when both arms are modified. Thiscan only be ascertained if the unmodified molecules’ populationscan be separated from the singly and doubly modified populationsin an intact, folded state. Analysis of the HIC purified species thatcontain the succinimidyl modification to one light chain arm ofthe antibody by the LANCE binding assay indicates that thispopulation has slightly lower binding affinity for the target whencompared to unmodified antibody. Interestingly, LANCE bindingdata for molecules which are modified on both light chain armsindicate that this species has the same reduction in affinity as theone arm modified population. We were able to optimize thechromatographic conditions to preserve the highly labile modifica-tion for subsequent analysis by orthogonal techniques andassessment of binding and potency. The power of this methodresides in its unique ability to separate intact molecules into purepopulations which contain the modification on zero, one, and twolight chain arms, providing an efficient tool for a detailedunderstanding of the degradation pathway.
Received for review October 30, 2007. Accepted February6, 2008.
AC702245C
(31) Bischoff, R.; Lepage, P.; Jaquinod, M.; Cauet, G.; Acker-Klein, M.; Clesse,D.; Laporte, M.; Bayol, A.; Van Dorsselaer, A.; Roitsch, C. Biochemistry1993, 32, 725-34.
(32) Teshima, G.; Stults, J. T.; Ling, V.; Canova-Davis, E. J. Biol. Chem. 1991,266, 13544-7.
(33) Tomizawa, H.; Yamada, H.; Ueda, T.; Imoto, T. Biochemistry 1994, 33, 8770-4.
(34) Violand, B. N.; Schlittler, M. R.; Kolodziej, E. W.; Toren, P. C.; Cabonce, M.A.; Siegel, N. R.; Duffin, K. L.; Zobel, J. F.; Smith, C. E.; Tou, J. S. ProteinSci. 1992, 1, 1634-41.
(35) Harris, R. J. J. Chromatogr., A 1995, 705, 129-34.(36) Jones, G. H. Biochemistry 1974, 13, 855-60.(37) Stott, D. I.; Munro, A. J. Biochem. J. 1972, 128, 1221-7.
Table 4. Assessment of Molecule Potency by the LANCE Receptor Ligand Binding Assay and the ReceptorPhosphorylation Bioassay Indicated That There Is a Slight Drop in Binding and Potency Associated with theSuccinimidyl Modification of the Light Chain Asp Residue at Position 30a
LANCE receptor ligandbinding inhibition assay
receptor phosphorylationbioassay
sample mean SD % CV t test mean SD % CV t test
unstressd IgG2 110 4 4 *p-value<0.05
105 0 0 *p-value<0.05
stressed IgG2 101 5 5 0.007 102 8 7 0.302HIC Fr 6 102 5 5 0.084 108 12 11 0.156HIC Fr 7 92 5 5 0.002 94 12 13 0.09HIC Fr 8 90 6 6 0.001 93 12 12 0.05
a The LANCE binding assay p-value associated with the difference in binding of fractions 7 and 8 relative to the HIC starting material (stressedIgG2) was statistically significant. Although the receptor phosphorylation bioassay also showed that fractions 7 and 8 had reduced potency relativeto the HIC starting material, these measurements were not found to be statistically significant as assessed by their associated p-value.
3174 Analytical Chemistry, Vol. 80, No. 9, May 1, 2008