short primer on comparative genomics today: special guest lecture 12pm, alway m108 comparative...
Post on 22-Dec-2015
214 views
TRANSCRIPT

Short Primer on Comparative Genomics
Today: Special guest lecture12pm, Alway M108 Comparative genomics of animals and plants
Adam SiepelAssistant Professor of Biological Statistics and Computational Biology Cornell University

Evolution at the DNA level
…ACGGTGCAGTTACCA…
…AC----CAGTCCACCA…
Mutation
SEQUENCE EDITS
REARRANGEMENTS
Deletion
InversionTranslocationDuplication

Orthology and Paralogy
HB HumanHB Human
WB WormWB Worm
HA1 HumanHA1 Human
HA2 HumanHA2 Human
YeastYeast
WA WormWA Worm
Orthologs:Derived by speciation
Paralogs:Everything else

Orthology, Paralogy, Inparalogs, Outparalogs

Synteny maps
Comparison of human and mouse

Synteny maps

Building synteny maps
Recommended local aligners• BLASTZ
Most accurate, especially for genes Chains local alignments
• WU-BLAST Good tradeoff of efficiency/sensitivity Best command-line options
• BLAT Fast, less sensitive Good for
• comparing very similar sequences • finding rough homology map

Index-based local alignment
Dictionary:
All words of length k (~10)
Alignment initiated between words of alignment score T
(typically T = k)
Alignment:
Ungapped extensions until score
below statistical threshold
Output:
All local alignments with score
> statistical threshold
……
……
query
DB
query
scan
Question: Using an idea from overlap detection, better way to find all local alignments between two genomes?

Local Alignments

After chaining

Chaining local alignments
1. Find local alignments
2. Chain -O(NlogN) L.I.S.
3. Restricted DP

Progressive Alignment
• When evolutionary tree is known:
Align closest first, in the order of the tree In each step, align two sequences x, y, or profiles px, py, to generate a new
alignment with associated profile presult
Weighted version: Tree edges have weights, proportional to the divergence in that edge New profile is a weighted average of two old profiles
x
w
y
z

Threaded Blockset Aligner
Human–Cow
HMR – CDRestricted AreaProfile Alignment

Reconstructing the Ancestral Mammalian Genome
Human: C
Baboon: C
Cat: C
Dog: G
C
C or G
G

Neutral Substitution Rates

Finding Conserved Elements (1)
• Binomial method 25-bp window in the human genome Binomial distribution of k matches in N bases given the neutral
probability of substitution

Finding Conserved Elements (2)
• Parsimony Method Count minimum # of mutations explaining each column Assign a probability to this parsimony score given neutral model Multiply probabilities across 25-bp window of human genome
A
CAAG
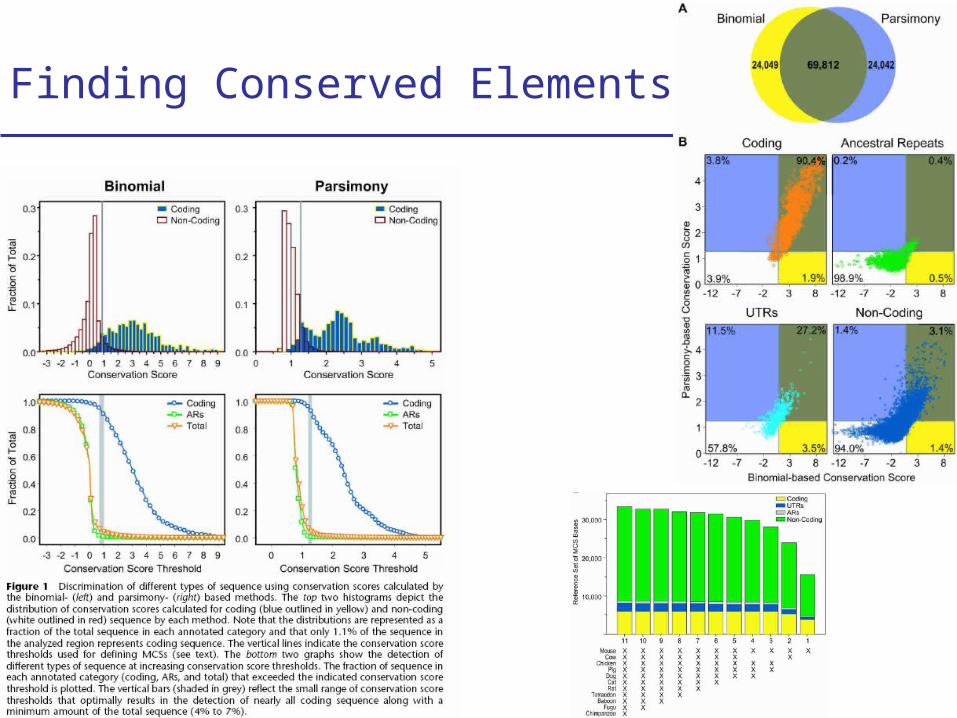
Finding Conserved Elements

Finding Conserved Elements (3)
GERP

Phylo HMMs
HMM
Phylogenetic Tree Model
Phylo HMM

Finding Conserved Elements (3)
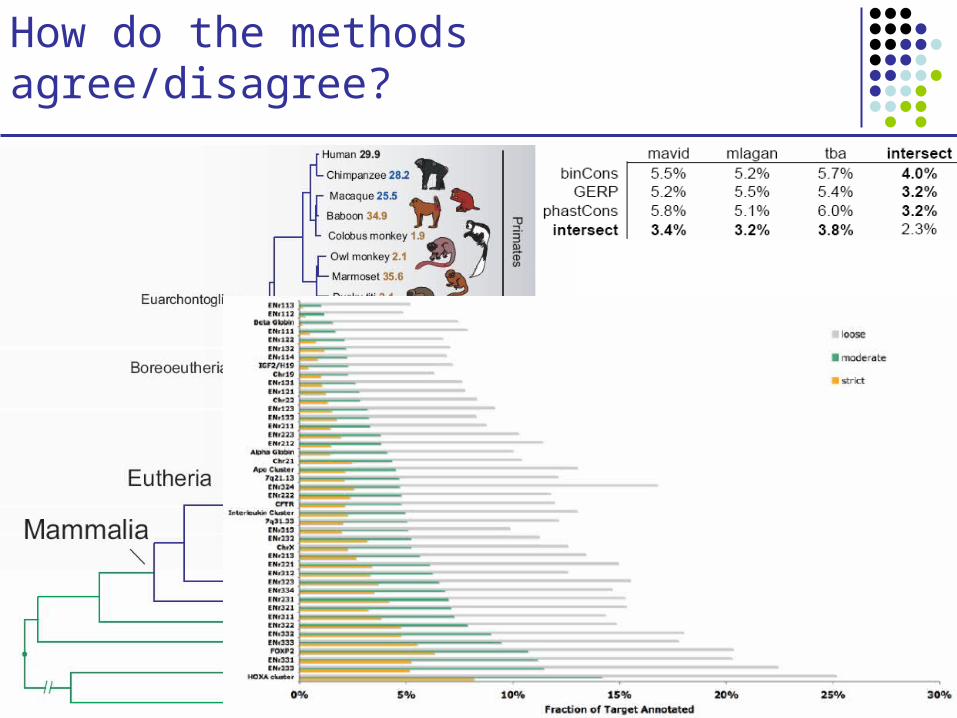
How do the methods agree/disagree?

Statistical Power to Detect Constraint
L
N
C: cutoff # mutationsD: neutral mutation rate: constraint mutation rate relative to neutral

Statistical Power to Detect Constraint
L
N
C: cutoff # mutationsD: neutral mutation rate: constraint mutation rate relative to neutral