smad3 knockout mice exhibit a resistance to skin chemical ...knockout mice do not exhibit epithelial...
TRANSCRIPT

[CANCER RESEARCH 64, 7836–7845, November 1, 2004]
Smad3 Knockout Mice Exhibit a Resistance to Skin Chemical Carcinogenesis
Allen G. Li,1,2,3 Shi-Long Lu,1,2,3 Ming-Xiang Zhang,1,2,3 Chuxia Deng,4 and Xiao-Jing Wang1,2,3
Departments of 1Otolaryngology, 2Dermatology, and 3Cell and Developmental Biology, Oregon Health and Science University, Portland, Oregon; and 4Mammalian GeneticsSection, Genetics of Development and Disease Branch, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland
ABSTRACT
It has been shown that Smad3 exerts both tumor-suppressive and-promoting roles. To evaluate the role of Smad3 in skin carcinogenesis invivo, we applied a chemical skin carcinogenesis protocol to Smad3 knockoutmice (Smad3�/� and Smad3�/�) and wild-type littermates (Smad3�/�).Smad3�/� mice exhibited reduced papilloma formation in comparisonwith Smad3�/� mice and did not develop any squamous cell carcinomas.Further analysis revealed that Smad3 knockout mice were resistant to12-O-tetradecanoylphorbol-13-acetate (TPA)–induced epidermal hyper-proliferation. Concurrently, increased apoptosis was observed in TPA-treated Smad3�/� skin and papillomas when compared with those ofwild-type mice. Expression levels of activator protein-1 family members(c-jun, junB, junD, and c-fos) and transforming growth factor (TGF)-�were significantly lower in TPA-treated Smad3�/� skin, cultured kerati-nocytes, and papillomas, as compared with Smad3�/� controls. Smad3�/�
papillomas also exhibited reduced leukocyte infiltration, particularly areduction of tumor-associated macrophage infiltration, in comparisonwith Smad3�/� papillomas. All of these molecular and cellular alterationsalso occurred to a lesser extent in Smad3�/� mice as compared withSmad3�/� mice, suggesting a Smad3 gene dosage effect. Given thatTGF-�1 is a well-documented TPA-responsive gene and also has a potentchemotactic effect on macrophages, our study suggests that Smad3 may berequired for TPA-mediated tumor promotion through inducing TGF-�1–responsive genes, which are required for tumor promotion, and throughmediating TGF-�1–induced macrophage infiltration.
INTRODUCTION
Transforming growth factor (TGF)-� is a multifunctional cytokinethat regulates cell proliferation, apoptosis, tissue remodeling, andangiogenesis, all of which are associated with cancer development(for a recent review, see ref. 1). Smad family members are the majorintracellular mediators of TGF-� signaling (for a recent review, seeref. 1). Among the Smad family members, Smad2 and Smad3 areresponsible for TGF-� and activin signaling (2). On binding of TGF-�to its receptors, Smad2 and Smad3 are phosphorylated and subse-quently form heteromeric complexes with Smad4, which then trans-locate from the cytoplasm to the nucleus. The Smad complex func-tions in the nucleus as a transcription factor and regulates expressionof TGF-� downstream targets (2).
TGF-� signaling plays a complex role in cancer development. As apotent growth inhibitor for keratinocytes, TGF-�1 is considered toplay a tumor-suppressive role at early stages of skin carcinogenesis.For instance, loss of TGF-�1 expression results in early progression tomalignancy (3). Similarly, transgenic mice overexpressing the domi-nant negative TGF-� type II receptor (��RII) in the epidermis ex-hibited a higher susceptibility to skin chemical carcinogenesis (4, 5).
Paradoxically, TGF-�1 is overexpressed in many cancer types inhumans (6). In experimental carcinogenesis models, expression ofTGF-�1 is also induced after application of tumor promoters, such as12-O-tetradecanoylphorbol-13-acetate [TPA (7)]. Studies from trans-genic mice show that overexpression of TGF-�1 in the epidermisresults in inhibition of tumor formation at early stages but accelerationof tumor progression at later stages (8, 9). These studies suggest thatTGF-�1 has a dual role in skin carcinogenesis. It remains to bedetermined, however, whether TGF-�1 overexpression at an earlystage of skin carcinogenesis exerts any tumor promotion effect.
As the major signaling mediators of the TGF-� superfamily, Smadsmay be actively involved in mediating the effects of TGF-�1 incarcinogenesis. In vitro studies have shown that TGF-�–inducedgrowth inhibition in epithelial cells is preferentially mediated bySmad3 (10, 11) and that cyclin-dependent kinase (cdk) 2 and cdk4 canphosphorylate Smad3, thus inactivating its ability to instigate cellcycle arrest (12). Therefore, Smad3 may mediate TGF-�1–inducedgrowth inhibition during cancer development. Supporting this notion,Smad3-null keratinocytes transduced by v-rasHa undergo acceleratedmalignant conversion when they are transplanted onto nude mice (13).Further analyses revealed that the loss of Smad3 abrogates the func-tions of TGF-�1 in regulating genes critical for cell cycle control,such as p15 and c-myc (13), suggesting that Smad3 is required forTGF-�–induced growth inhibition in keratinocytes. However, Smad3knockout mice do not exhibit epithelial hyperproliferation (14), sug-gesting that the role of Smad3 in mediating TGF-�1–induced growthinhibition is dispensable in vivo. In addition, TGF-�1 overexpressionin keratinocytes in vivo induces severe skin inflammation (15, 16),whereas Smad3 knockout mice exhibit accelerated wound healingwith decreased inflammation in vivo (17). These studies suggest thatSmad3 may also mediate TGF-�1–induced skin inflammation. Be-cause inflammation plays an important role in tumor promotion (18),affecting TGF-�1–induced skin inflammation may significantly affectthe role of TGF-�1 in skin carcinogenesis. Furthermore, a recentstudy using breast cancer cell lines showed that Smad3 exhibits bothtumor-suppressive and -promoting roles (19). To evaluate the role ofSmad3 in TGF-� signaling during skin carcinogenesis in vivo, weapplied a chemical carcinogenesis protocol to Smad3 knockout mice(designated Smad3�/� and Smad3�/� hereafter), in which TGF-�1expression is induced by TPA promotion (7). Here we demonstratethat Smad3 knockout mice exhibited a resistance to skin chemicalcarcinogenesis, correlating with decreased proliferation and increasedapoptosis in Smad3 knockout keratinocytes during TPA promotionand reduced tumor-associated macrophage (TAM) infiltration inSmad3 knockout tumors. Consistently, TGF-� and activator protein(AP)-1 family members, which are critical factors for TPA-inducedtumor promotion, were not up-regulated in Smad3 knockout skin andtumors. Our results suggest that Smad3 is required for TPA-mediatedtumor promotion, possibly by regulating TGF-�–responsive genesthat are involved in tumor promotion and by mediating TGF-�1–induced inflammation.
MATERIALS AND METHODS
Skin Chemical Carcinogenesis Protocol. The Smad3 knockout mice wereproduced in a C57BL/6 � 129SVEV background (20) and backcrossed to
Received 4/14/04; revised 8/16/04; accepted 9/2/04.Grant support: National Institutes of Health grants CA87849 and CA79998 (X-J.
Wang).The costs of publication of this article were defrayed in part by the payment of page
charges. This article must therefore be hereby marked advertisement in accordance with18 U.S.C. Section 1734 solely to indicate this fact.
Note: M-X. Zhang is presently in the Department of Physiology, Baylor College ofMedicine, Houston, Texas.
Requests for reprints: Xiao-Jing Wang, 3710 SW US Veterans Hospital Road, Mailcode R&D46, Portland, OR 97239. Phone: 503-220-8262, ext. 54273; Fax: 503-402-2817;E-mail: [email protected].
©2004 American Association for Cancer Research.
7836
Research. on February 19, 2020. © 2004 American Association for Cancercancerres.aacrjournals.org Downloaded from

129SVEV for �12 generations during a 2-year period before their use in thisskin carcinogenesis study. Twelve hemizygous (Smad3�/�) breeding pairswere set up to generate mice with Smad3�/�, Smad3�/�, and Smad3�/�
genotypes. In this study, 35 Smad3�/� (15 males and 20 females), 44Smad3�/� (23 males and 21 females), and 16 Smad3�/� (7 males and 9females) mice were used. The chemical carcinogenesis protocol was applied asdescribed previously (21). Briefly, neonatal pups at day 3 were treated topi-cally with 20 �g of 7,12-dimethylbenz(a)anthracene (DMBA; dissolved in 50�L of acetone; Sigma, St. Louis, MO). TPA (5 �g; dissolved in 50 �L ofacetone; Sigma) was applied to pup skin at day 4 and day 6, and applicationwas continued twice a week for 30 weeks.
Acute 12-O-Tetradecanoylphorbol-13-acetate Treatment on AdultSkin. The dorsal skins of 6-week–old mice were shaved and treated with 5 �gof TPA (dissolved in 50 �L of acetone). Forty-eight hours after TPA treatment,mice received injection with 0.125 mg/g bromodeoxyuridine (BrdUrd; Sigma).One hour later, the mice were euthanized, and the dorsal skins were harvestedfor histologic analysis, BrdUrd labeling, immunohistochemical staining, andRNA and protein extractions.
Tissue Histology. Dissected skin and tumor samples were fixed in 10%neutral-buffered formalin at 4°C overnight, embedded in paraffin, sectioned to6-�m thickness, and stained with hematoxylin and eosin (H&E). Tumor typeswere determined by H&E analysis using the criteria described previously (22)and confirmed by Dr. C. Corless, a pathologist and the director of the CancerPathology Core facility at Oregon Health and Science University. Generally,papillomas appeared as exophytic, pedunculated proliferations consisting ofmultiple finger-like hyperkeratotic epidermal projections having an intactbasement membrane creating a distinct border from the underlying dermis.Squamous cell carcinomas (SCCs) were characterized by invasive tumor cellproliferation into the dermis in a lobular pattern and numerous mitotic figureswithin tumor lobules. SCCs were further classified into well-, moderately, andpoorly differentiated groups based on the criteria described previously (22).
Double Stain Immunofluorescence. Double stain immunofluorescencewas performed as we have described previously (23). The primary antibodiesincluded fluorescein isothiocyanate-conjugated BrdUrd (undiluted; Becton-Dickinson, Franklin Lakes, NJ), keratin 1 (K1; 1:500; ref. 24), keratin 13 (K13;1:500; ref. 24), and nuclear factor (NF)-�B p50 (1:50; Santa Cruz Biotech-nology, Santa Cruz, CA). Briefly, paraffin-embedded sections were deparaf-finized in fresh xylene and rehydrated. Antigen retrieval was performed bymicrowaving slides in 10 mmol/L sodium citrate solution for 10 minutes. Eachsection was incubated overnight at 4°C with a primary antibody diluted in PBScontaining 12% bovine serum albumin, together with a guinea pig antiserumagainst mouse keratin 14 (K14; 1:500), the latter of which highlights theepithelial compartment of the skin (9). The sections were then washed withPBS and incubated with fluorescence dye-conjugated secondary antibodies, anAlexa Fluo 488-conjugated (green) secondary antibody against the species ofthe primary antibody [1:100; Molecular Probes, Eugene, OR (except for thesections incubated with fluorescein isothiocyanate-conjugated BrdUrd anti-body)] and Alexa Fluo 594-conjugated (red) anti-guinea pig secondary anti-body (1:100; Molecular Probes). The fluorescence dye-conjugated secondaryantibodies were diluted in 10% bovine serum albumin-PBS and applied to thetissue sections at room temperature for 30 minutes. After several PBS washes,sections were visualized under a Nikon Eclipse E600W fluorescence micro-scope (Nikon, Melville, NY). The BrdUrd labeling index in the epidermis wasexpressed as the mean number of BrdUrd-positive cells per millimeter ofbasement membrane � SD.
Immunohistochemistry. To examine inflammatory cell subtypes, immu-nohistochemistry was performed on frozen sections, as we have describedpreviously (16), using primary antibodies for different leukocyte markersincluding CD45 (1:20), CD4 (1:20), and Ly-6G [1:20; ref. 25 (all from BDBiosciences, San Diego, CA)]. The BM8 antibody (1:400; BMA Biomedicals,Augst, Switzerland), which recognizes macrophages (26), was also used.Immunohistochemical detection of proliferating cell nuclear antigen (PCNA)and phosphorylated Smad2 was performed on paraffin-embedded sectionsusing a PCNA antibody (1:100; Santa Cruz Biotechnology) and a phospho-rylated Smad2 antibody (1:50; Cell Signaling, Beverly, MA). After deparaf-finization, the sections were subjected to antigen retrieval as described above,followed by incubation with 5% serum (from the species in which the sec-ondary antibody was developed) for 1 hour at room temperature. Incubationwith primary antibodies was carried out at 4°C overnight. The sections were
then sequentially incubated with biotinylated secondary antibodies (1:250) andan avidin-peroxidase reagent (Vector Laboratories, Burlingame, CA) at roomtemperature for 10 and 5 minutes, respectively. The immune complexes in thesections were visualized using diaminobenzidine (DAKO, Carpinteria, CA).Quantitative measurement of positively stained cells was performed usingMetaMorph software (Universal Imaging Corp., Burnaby, British Columbia,Canada) and expressed as the mean number of PCNA-positive cells per mm2
tumor area � SD.Apoptosis Assays. Following the manufacturer’s instructions, apoptosis
was evaluated using terminal deoxynucleotidyltransferase-mediated uridinenick end labeling (TUNEL) assay with the DeadEnd Fluorometric TUNEL kit(Promega, Madison, WI). Briefly, paraffin-embedded sections were deparaf-finized, rehydrated, and washed in 0.85% NaCl. The sections were thensequentially prefixed (in 4% formaldehyde-PBS), permeabilized (in 20 �g/mLproteinase K), and postfixed (in 4% formaldehyde-PBS). After several PBSwashes, the sections were incubated in a buffer that contained fluorescein-dUTP and recombinant terminal deoxynucleotidyltransferase at 37°C for 1hour in a humidified chamber in the dark. The reaction was then terminated in2� SSC for 15 minutes at room temperature, and the slides were washed inPBS. The slides were then immersed in a 1 �g/mL propidium iodide solutionfor 15 minutes at room temperature to counterstain the nuclei. The sampleswere then analyzed under a fluorescence microscope. Quantitative measure-ment of apoptotic cells was performed using MetaMorph software (UniversalImaging Corp.) and expressed as the mean number of apoptotic cells per mm2
tumor area � SD.Keratinocyte Culture and 12-O-Tetradecanoylphorbol-13-acetate
Treatment. Primary keratinocytes were isolated from neonatal Smad3�/�,Smad3�/�, and Smad3�/� skin and cultured in conditioned medium supple-mented with 0.05 mmol/L Ca2�, as we have described previously (27). Onreaching subconfluence, the cells were treated with TPA (100 nmol/L) for 12hours before harvesting (28).
RNA Extraction, Quantitative Reverse Transcription-PolymeraseChain Reaction, and RNase Protection Assay. Total RNA was isolatedfrom skin, chemically induced tumors, and cultured keratinocytes using RNA-zol B (Tel-Test, Friendswood, TX), as we have described previously (11), andfurther purified using a Qiagen RNeasy Mini kit (Qiagen, Valencia, CA). Thequantitative reverse transcription-polymerase chain reaction (qRT-PCR) wasachieved by combining in vitro reverse transcription with quantitative poly-merase chain reaction, which was performed in a Stratagene Mx3000P thermalcycler (Stratagene, La Jolla, CA; ref. 29). Briefly, 5 �g of RNA from eachsample were treated with DNase (Ambion, Austin, TX). The RNA was thensubjected to a reverse transcription reaction using avian myeloblastosis virusreverse transcriptase (Roche, Indianapolis, MN). The resultant cDNA productswere used as templates for quantitative polymerase chain reaction to examinethe levels of transcripts of mouse Smad3, TGF-�, and AP-1 family membersincluding c-jun, junB, junD, and c-fos using corresponding TaqMan Assays-on-Demand probes (Applied Biosystems, Foster City, CA). Tumor RNAsamples were also assayed for gene expression levels of interleukin (IL)-1�
and monocyte chemotactic protein (MCP-1). An 18S RNA probe was used asan internal control, and the data (CT values) were analyzed using StratageneMx3000P Comparative Quantitation software. The expression level of eachgene was normalized with 18S using a comparative CT (�CT) and expressed asthe difference of the CT values from a test gene (e.g., TGF-�) and 18S[CT(18S) minus CT(TGF-�)]. The relative RNA expression levels were calcu-lated using the �CT method (30), and the average results from three to fivesamples from three to five mice of each genotype are shown, with theexception that only two Smad3�/� mice developed tumors. For Smad3 geneexpression assays, the expression levels from all Smad3�/� samples were setas 1 arbitrary unit, which was used as a baseline to compare expression levelsof the same gene in samples with different Smad3 genotypes. In analyzing therelative expression levels of other genes, the expression level from oneSmad3�/� sample (unless otherwise specified) of each particular gene beinganalyzed was set as 1 arbitrary unit. To examine expression levels of variouscell cycle control genes, RNase protection assay was performed using RNaseprotection assay III kits (Ambion), as we have described previously (31). Theprobes used included multiprobe gene sets [mCC1c, mCYC-1, and mCYC-2(BD Biosciences)] and individual probes for c-myc, p15, and p21 (31).
7837
SKIN CHEMICAL CARCINOGENESIS ON Smad3 KNOCKOUT MICE
Research. on February 19, 2020. © 2004 American Association for Cancercancerres.aacrjournals.org Downloaded from

Statistics. Significant differences between the values obtained in eachassay on samples from various genotypes were determined using Student’s ttest throughout this study.
RESULTS
Smad3 Knockout Mice Exhibited Reduced Tumor Formationand Malignant Progression during Skin Chemical Carcinogene-sis. To determine the effect of loss of Smad3 on skin carcinogenesisin vivo, we applied a chemical carcinogenesis protocol to the skin ofSmad3 knockout mice (Smad3�/� and Smad3�/�). Because adultSmad3�/� mice develop a wasting syndrome (20), which could po-tentially affect the outcome of the skin carcinogenesis experiment, weapplied DMBA to Smad3 knockout mice 3 days after birth, followedby applications of TPA twice a week for 30 weeks. Before 30 weeksof age, animals of all three genotypes remained apparently healthy,with the exception of 10% to 20% lower body weights in Smad3�/�
mice in comparison with Smad3�/� and Smad3�/� mice. By 30weeks of age, the Smad3�/� mice exhibited an approximately 30%reduction in body weight (males, 22 � 3.3 g; females, 20 � 2.1 g;P � 0.01), as compared with Smad3�/� mice (males, 39 � 4.1 g;females, 37 � 2.1 g) or Smad3�/� mice (males, 36 � 3.7 g; females,34 � 4.2 g). After 30 weeks of age, the wasting syndrome in some ofthe Smad3�/� mice became dominant, resulting in a rapid decline inoverall health of these mice, as described previously (20). Therefore,tumorigenesis in mice with different Smad3 genotypes was monitoreduntil 30 weeks after DMBA initiation.
Smad3�/� mice began to develop benign papillomas 6 weeks afterDMBA initiation, and by 16 weeks, approximately 60% of Smad3�/�
mice had developed papillomas (Fig. 1A). The first papillomas onSmad3�/� mice began to develop at 11 weeks after DMBA initiation,and only 2 of 16 (�20%) Smad3�/� mice developed papillomas by16 weeks (Fig. 1A). Smad3�/� mice also exhibited reduced tumori-genesis (47% by 20 weeks; Fig. 1A) as compared with Smad3�/�
mice. Additionally, Smad3�/� mice developed significantly fewertumors as compared with Smad3�/� mice. Smad3�/� mice averaged2.5 tumors per mouse at the end of the promotion stage (30 weeks;Fig. 1B). In contrast, Smad3�/� mice only averaged 0.5 tumor permouse by the end of TPA promotion, a 5-fold reduction in comparison
with Smad3�/� mice (Fig. 1B; P � 0.01). Smad3�/� mice averaged1 tumor per mouse by 30 weeks, which was also a significantreduction in tumor number when compared with Smad3�/� mice(Fig. 1B; P � 0.01). Furthermore, 40% of the tumors that developedon Smad3�/� mice progressed to SCCs after termination of TPAtreatment (30 weeks; Fig. 1C), with an average of 0.8 � 0.04 SCCsper mouse (Fig. 1D). In contrast, all of the tumors dissected fromSmad3�/� mice at the same time point were benign papillomas (Fig.1C and D). Smad3�/� mice also exhibited a decreased frequency ofSCC development, with only 27% of Smad3�/� tumors progressingto SCC (Fig. 1C) and a reduced average number of SCCs per mouse(0.4 � 0.03; P � 0.01) in comparison with Smad3�/� mice. Histo-logically, Smad3�/� SCCs appeared more advanced and less differ-entiated than Smad3�/� SCCs, which displayed features of typicalcarcinomas in situ or well-differentiated SCCs (Fig. 2). Consistently,the Smad3�/� and Smad3�/� tumors classified as SCCs showed alack of K1 expression, a marker of benign papillomas (24), whereasall of the Smad3�/� tumors retained uniform or at least focal K1expression (Fig. 2). In contrast, strongly positive immunofluorescencestaining for K13, a malignancy marker in squamous epithelia (32),was observed in tumors classified as SCCs from both Smad3�/� andSmad3�/� mice, but not in tumors from Smad3�/� mice (Fig. 2),further confirming that the tumors from the Smad3�/� mice werebenign papillomas.
Smad3 Deletion in Keratinocytes Resulted in a Resistance to12-O-Tetradecanoylphorbol-13-acetate–Induced Epidermal Hy-perplasia and an Increased Rate of Apoptosis. To determinewhether the resistance to skin chemical carcinogenesis in Smad3knockout mice is due to the blockade of DMBA-induced initiation oftumor development, we examined tumor samples from differentSmad3 genotypes for c-rasHa mutations, which are induced by DMBAin �90% of papillomas (33). Sequence analysis for a c-rasHa genomicfragment (5) revealed that all tumor samples examined from thedifferent Smad3 genotypes possessed a c-rasHa mutation at codon 12,13, or 61 (data not shown), suggesting that loss of Smad3 likely doesnot affect DMBA initiation. Next, we analyzed whether Smad3 dele-tion contributes to a reduction of susceptibility to TPA-induced tumorpromotion by examining adult skin with different Smad3 genotypes
Fig. 1. Tumor kinetics. A, percentage of tumor-bearingmice in each group. B, average number of tumors permouse. C, percentage of mice that developed carcinoma by30 weeks after TPA treatment. None of the Smad3�/� micepapillomas converted to carcinomas. D, average number ofSCCs per mouse.
7838
SKIN CHEMICAL CARCINOGENESIS ON Smad3 KNOCKOUT MICE
Research. on February 19, 2020. © 2004 American Association for Cancercancerres.aacrjournals.org Downloaded from

48 hours after a single TPA treatment. As compared with TPA-treatedSmad3�/� skin, which was characterized by obvious epidermal hy-perplasia (8.1 � 1.6 cell layers in epidermis), Smad3�/� (4.7 � 1.2layers; P � 0.01; n 3) and Smad3�/� (2.3 � 0.4 layers; P � 0.01;n 3) skin showed a resistance to TPA-induced epidermal hyperpla-sia (Fig. 3A). Accordingly, the number of BrdUrd-labeled keratino-cytes was significantly reduced in TPA-treated Smad3 knockout skin.The number of BrdUrd-labeled keratinocytes was 189 � 23.4 per mmof epidermis in TPA-treated Smad3�/�skin, whereas it was reducedto 122 � 14.2 (P � 0.01; n 3) and 35.6 � 9.0 per mm of epidermis(P � 0.01; n 3) in Smad3�/and Smad3�/� skins, respectively (Fig.3A). In contrast to the change in epidermal proliferation, CD45immunostaining showed no difference in the degree of inflammatorycell infiltration in TPA-treated skin from three groups (Fig. 3A). Thedegree of infiltration by different leukocyte subsets, including CD4�cells, granulocytes, and macrophages, in TPA-treated skin of threegroups was also indistinguishable (data not shown). These resultssuggest that loss of Smad3 has a direct effect on keratinocyte prolif-eration during the promotion stage of skin chemical carcinogenesis.Analysis of cell proliferation in Smad3 knockout papillomas revealedthat the number of PCNA-positive cells was reduced by �50% inSmad3�/� papillomas (163 � 12.2/mm2 tumor area; P � 0.01; n 3)in comparison with Smad3�/� papillomas (389 � 28.9/mm2) at thesame stage and further reduced by 90% in Smad3�/� papillomas(37.2 � 4.5/mm2) as compared with Smad3�/� papillomas (Fig. 3B).
To determine whether Smad3 deletion alters the apoptotic process,TUNEL assay was performed on TPA-treated adult skin and papillo-mas with different Smad3 genotypes. The numbers of apoptotic cellsin the epidermis of Smad3�/� (24.8 � 3.53/mm2 epidermis; n 3)and Smad3�/� skin (19.8 � 2.65/mm2 epidermis; n 3) weresignificantly increased as compared with the number of apoptotic cellsin Smad3�/� skin (5.25 � 1.28/mm2 epidermis; n 3; P � 0.01) 48hours after TPA treatment (Fig. 3C). The apoptotic cells in thesesamples were primarily located in the suprabasal layers of the epider-mis (Fig. 3C). Similarly, apoptotic cells, located mainly in suprabasallayers, were prominent in Smad3�/� (31.3 � 2.83/mm2 tumor epi-thelia) and Smad3�/� papillomas (38.7 � 3.69/mm2) but sparse inSmad3�/� papillomas (11.6 � 1.47/mm2; n 3; P � 0.01; Fig. 3C).
To further confirm the correlation between the above-mentionedeffects and the inactivation of the Smad3 gene, we examined therelative expression levels of the Smad3 transcript by qRT-PCR. Asshown in Fig. 4A, Smad3 expression levels in nontreated skin, TPA-treated skin, and papillomas, all from Smad3�/� mice, were reducedby about 50% as compared with the levels detected in the same tissuesfrom Smad3�/� mice. Expression of Smad3 was undetectable in allsamples from Smad3�/� mice (Fig. 4A). To elucidate whether theeffect of Smad3 deletion on skin carcinogenesis involves alteredactivation of its signaling partner, Smad2, the presence of phospho-rylated Smad2 (pSmad2) was examined by immunohistochemistryusing a pSmad2 antibody. Nontreated skins from different Smad3genotypes exhibited comparable pSmad2 nuclear staining (Fig. 4B).Noticeably, TPA-treated skin from all three Smad3 genotypes exhib-ited increased pSmad2 in both the epidermis and dermis (Fig. 4B),suggesting that TPA-induced endogenous TGF-�1 activates Smad2.However, both TPA-treated skins and papillomas exhibited compara-ble pSmad2 nuclear staining among different Smad3 genotypes (Fig.4B), suggesting that the effect of Smad3 deletion on skin chemicalcarcinogenesis is independent of Smad2 activation.
Reduced Expression of Activator Protein-1 Family Membersand Transforming Growth Factor � in 12-O-Tetradecanoylphor-bol-13-acetate–Treated Skin and Tumors of Smad3 KnockoutMice. To determine the molecular mechanisms of Smad3 knockoutskin resistance to TPA-induced epidermal hyperplasia and increasedapoptotic rate, we examined expression levels of proliferative/survivalfactors that normally respond to TPA. In contrast to a previous reportshowing decreased expression of the cdk inhibitor p15 and increasedexpression of c-myc in vrasHa-transduced Smad3-null keratinocytes(13), we did not find differences among the different Smad3 geno-types in expression of TGF-�1 target genes involved in cell cyclecontrol, including the cdk inhibitors p15 and p21 and the c-myconcogene, in TPA-treated skin of each genotype (data not shown).These results suggest that either TPA itself or an endogenous mech-anism(s) is able to regulate expression of these molecules in theabsence of Smad3. We next examined the expression of major AP-1family members, which are TGF-�–responsive genes (34) that arerapidly up-regulated in response to TPA and mediate TPA-induced
Fig. 2. Histology and immunofluorescence analysesof the most advanced tumor types from each genotype.H&E examination revealed a poorly differentiatedSCC, an early-stage SCC, and a papilloma inSmad3�/�, Smad3�/�, and Smad3�/� mice, respec-tively. A K14 antibody (red) was used as a counterstainfor immunofluorescence. The bar in the top left panelrepresents 200 �m for H&E sections and 60 �m forimmunofluorescence sections.
7839
SKIN CHEMICAL CARCINOGENESIS ON Smad3 KNOCKOUT MICE
Research. on February 19, 2020. © 2004 American Association for Cancercancerres.aacrjournals.org Downloaded from

tumor promotion (35). We found that expression levels of c-jun, junB,junD, and c-fos in TPA-treated skin and papillomas were highest inSmad3�/� mice and lowest in Smad3�/� mice (Fig. 5).
Because TGF-� overexpression induced by TPA is thought to becritical for TPA-induced tumor promotion (36) and for protection oftumor cells from apoptosis (37–39), we examined expression ofTGF-� in TPA-treated skin and tumors. Expression of TGF-� wasalmost undetectable in untreated skin (data not shown) but wasinduced in Smad3�/� skin after TPA treatment (Fig. 5). However,expression of TGF-� was 8- to 10-fold lower in TPA-treatedSmad3�/� skin and almost undetectable in TPA-treated Smad3�/�
skin. A similar pattern of TGF-� expression was observed in tumorsfrom the three genotypes (Fig. 5).
To further determine whether changes in expression levels of thesegenes represent a direct effect of Smad3 deletion on keratinocytes, wecultured primary keratinocytes isolated from mice with different Smad3genotypes and treated these cells with TPA for 12 hours. Expression ofthe above-mentioned five genes was significantly lower in TPA-treatedSmad3�/� keratinocytes than in Smad3�/� cells (Fig. 5). TPA-treatedSmad3�/� keratinocytes exhibited a reduction in the expression of thesegenes by about 45% to 66% relative to Smad3�/� samples (Fig. 5),suggesting a dose-dependent effect of Smad3 inactivation on the tran-scription of these genes in keratinocytes in response to TPA.
Reduced Tumor-Associated Macrophages and Inflammation inSmad3 Knockout Papillomas. Because Smad3 can affect infiltrationof inflammatory cells (20), we examined leukocyte infiltration in
Fig. 3. Proliferation and apoptosis in TPA-treated skin and papillomas from each group. Dot-ted lines denote the epidermal–dermal junction. A,histology, BrdUrd labeling (green), and CD45staining on TPA-treated skin. Arrows point to typ-ical BrdUrd-labeled cells. A K14 antibody (red)was used as a counterstain for immunofluores-cence. Sections stained by immunohistochemistryfor CD45 were counterstained with hematoxylin.The bar in the top left panel represents 100 �m forH&E- and BrdUrd-stained sections and 40 �m forCD45-stained sections. B, PCNA staining of rep-resentative papillomas from each genotype. Thesections are counterstained with hematoxylin. Redarrows point to examples of positively stainednuclei. The bar in the top left panel represents 100�m. C, TUNEL assay on TPA-treated skin (toppanels) and papillomas (bottom panels) from eachgroup. Arrows point to representative apoptoticnuclei (green or yellowish). The bar in the top leftpanel represents 40 �m for all sections.
7840
SKIN CHEMICAL CARCINOGENESIS ON Smad3 KNOCKOUT MICE
Research. on February 19, 2020. © 2004 American Association for Cancercancerres.aacrjournals.org Downloaded from

Fig. 4. Smad3 expression and Smad2 phosphorylation. A,qRT-PCR results of Smad3 expression in 6-week–old mouseskin (skin), TPA-treated skin, and papillomas from the threedifferent genotypes. The Smad3 expression level of all sam-ples from Smad3�/� mice was set as 1 arbitrary unit. B,immunohistostaining of pSmad2. The bar in the top left panelrepresents 40 �m for all sections. Hematoxylin was used as acounterstain.
Fig. 5. Relative expression levels of AP-1 familymembers and TGF-� as determined by real-timeRT-PCR. Results from two representative samplesin each group are shown. The expression level ofeach molecule in one Smad3�/� sample was set as1 arbitrary unit. An exception was made for TGF-�expression levels in TPA-treated skins, in whichone Smad3�/� skin sample was set as 1 arbitraryunit, because TGF-� expression was not detectablein TPA-treated Smad3�/� skin.
7841
SKIN CHEMICAL CARCINOGENESIS ON Smad3 KNOCKOUT MICE
Research. on February 19, 2020. © 2004 American Association for Cancercancerres.aacrjournals.org Downloaded from
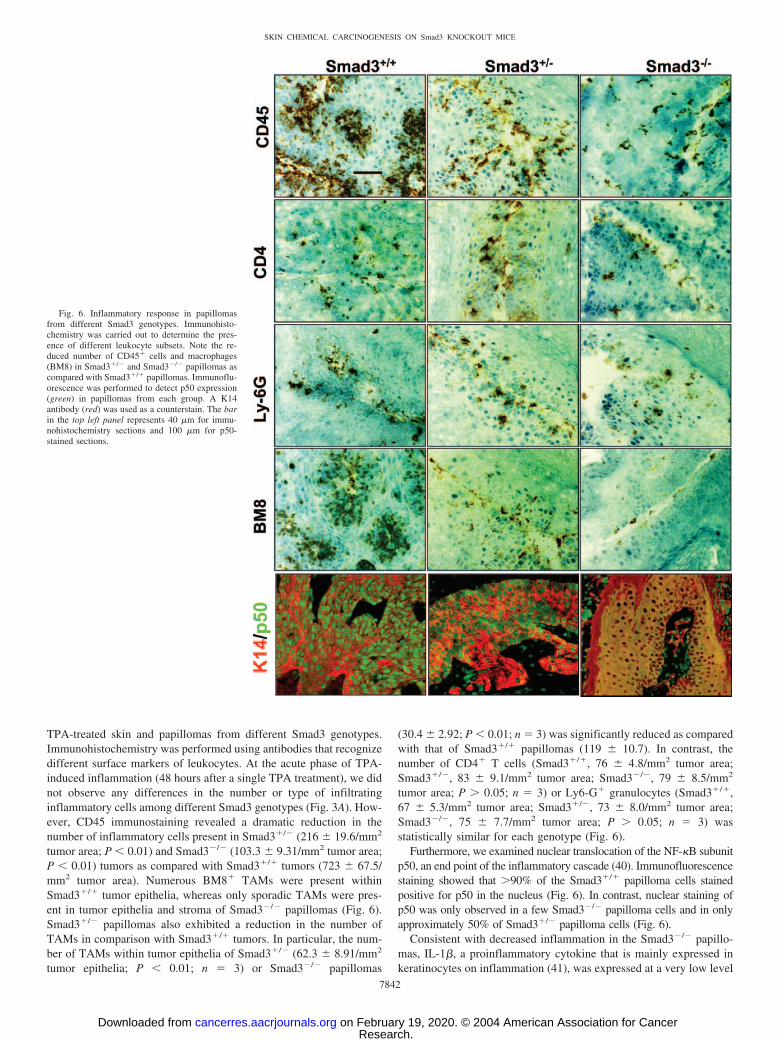
TPA-treated skin and papillomas from different Smad3 genotypes.Immunohistochemistry was performed using antibodies that recognizedifferent surface markers of leukocytes. At the acute phase of TPA-induced inflammation (48 hours after a single TPA treatment), we didnot observe any differences in the number or type of infiltratinginflammatory cells among different Smad3 genotypes (Fig. 3A). How-ever, CD45 immunostaining revealed a dramatic reduction in thenumber of inflammatory cells present in Smad3�/� (216 � 19.6/mm2
tumor area; P � 0.01) and Smad3�/� (103.3 � 9.31/mm2 tumor area;P � 0.01) tumors as compared with Smad3�/� tumors (723 � 67.5/mm2 tumor area). Numerous BM8� TAMs were present withinSmad3�/� tumor epithelia, whereas only sporadic TAMs were pres-ent in tumor epithelia and stroma of Smad3�/� papillomas (Fig. 6).Smad3�/� papillomas also exhibited a reduction in the number ofTAMs in comparison with Smad3�/� tumors. In particular, the num-ber of TAMs within tumor epithelia of Smad3�/� (62.3 � 8.91/mm2
tumor epithelia; P � 0.01; n 3) or Smad3�/� papillomas
(30.4 � 2.92; P � 0.01; n 3) was significantly reduced as comparedwith that of Smad3�/� papillomas (119 � 10.7). In contrast, thenumber of CD4� T cells (Smad3�/�, 76 � 4.8/mm2 tumor area;Smad3�/�, 83 � 9.1/mm2 tumor area; Smad3�/�, 79 � 8.5/mm2
tumor area; P � 0.05; n 3) or Ly6-G� granulocytes (Smad3�/�,67 � 5.3/mm2 tumor area; Smad3�/�, 73 � 8.0/mm2 tumor area;Smad3�/�, 75 � 7.7/mm2 tumor area; P � 0.05; n 3) wasstatistically similar for each genotype (Fig. 6).
Furthermore, we examined nuclear translocation of the NF-�B subunitp50, an end point of the inflammatory cascade (40). Immunofluorescencestaining showed that �90% of the Smad3�/� papilloma cells stainedpositive for p50 in the nucleus (Fig. 6). In contrast, nuclear staining ofp50 was only observed in a few Smad3�/� papilloma cells and in onlyapproximately 50% of Smad3�/� papilloma cells (Fig. 6).
Consistent with decreased inflammation in the Smad3�/� papillo-mas, IL-1�, a proinflammatory cytokine that is mainly expressed inkeratinocytes on inflammation (41), was expressed at a very low level
Fig. 6. Inflammatory response in papillomasfrom different Smad3 genotypes. Immunohisto-chemistry was carried out to determine the pres-ence of different leukocyte subsets. Note the re-duced number of CD45� cells and macrophages(BM8) in Smad3�/� and Smad3�/� papillomas ascompared with Smad3�/� papillomas. Immunoflu-orescence was performed to detect p50 expression(green) in papillomas from each group. A K14antibody (red) was used as a counterstain. The barin the top left panel represents 40 �m for immu-nohistochemistry sections and 100 �m for p50-stained sections.
7842
SKIN CHEMICAL CARCINOGENESIS ON Smad3 KNOCKOUT MICE
Research. on February 19, 2020. © 2004 American Association for Cancercancerres.aacrjournals.org Downloaded from

in Smad3�/� papillomas (Fig. 7). In contrast, IL-1� expression was�500-fold higher in Smad3�/� papillomas as compared withSmad3�/� papillomas (P � 0.01). IL-1� expression levels inSmad3�/� papillomas were approximately 400-fold less than IL-1�expression levels in Smad3�/� papillomas (P � 0.01; Fig. 7). Inaddition, the expression level of MCP-1, a major monocyte/macro-phage-attracting chemokine (42), was expressed at approximately8-fold higher levels in Smad3�/� papillomas than in Smad3�/�
papillomas (P � 0.01; Fig. 7), and expression of MCP-1 in Smad3�/�
papillomas was only 2-fold higher than that in Smad3�/� papillomas(P � 0.01; Fig. 7).
DISCUSSION
In this report, we have shown that mice lacking Smad3 exhibitedresistance to skin chemical carcinogenesis compared with wild-typelittermates. This result contradicts a previous report that loss of Smad3in keratinocytes accelerates malignant conversion in a tumor trans-plant model (13). Because nutritional status can greatly affect skincarcinogenesis (43), we were concerned that results from a skinchemical carcinogenesis study on adult Smad3 knockout mice, whichdevelop a wasting syndrome (20), may not accurately reflect the effectof the loss Smad3 on susceptibility to skin chemical carcinogenesis.To avoid this problem, we used neonatal mice and applied a previ-ously described chemical carcinogenesis protocol (44) that is initiated3 days after birth, when Smad3�/� pups are morphologically normal(20). By the time Smad3�/� mice began to develop papillomas (6 to15 weeks), Smad3�/� mice remained free of obvious wasting syn-drome symptoms yet demonstrated a resistance to tumorigenesis. Inaddition, Smad3�/� mice, which did not develop wasting syndromeand maintained body weight similar to wild-type littermates, showeda reduction in tumor formation and malignant conversion. Further-more, Smad3�/� and Smad3�/� skin exhibited a gene dosage-depen-dent resistance to epidermal proliferation after acute TPA treatment.In fact, Smad3 gene dosage effects during skin chemical carcinogen-esis were seen in almost every variable we analyzed on littermateswith different Smad3 genotypes. Because activation of a Smad3partner, Smad2, remained unaffected by Smad3 deletion (Fig. 4), ourresults suggest that the resistance to skin chemical carcinogenesis inSmad3 knockout mice is likely a direct effect of the loss of Smad3gene expression. Supporting our observations, a recent report shows
that Smad3�/� mice were resistant to skin chemical carcinogenesis,whereas Smad2�/� mice, with the same genetic background, exhib-ited accelerated skin carcinogenesis (45).
Lack of AP-1 Activation and TGF-� Overexpression in Smad3Knockout Keratinocytes Contributes to Resistance to TPA-In-duced Tumor Promotion. Unlike v-rasHa-transduced keratinocytes,which can progress to benign papillomas on grafting onto the athymicmouse skin (13), normal keratinocytes harboring a point mutation inthe c-rasHa gene induced in vivo by a subcarcinogenic dose of DMBAdo not typically form papillomas in normal mice (5), and furthertumor promotion is required for these cells to undergo tumorigenesis.The tumor promotion effect of TPA depends mainly on the ability ofTPA to induce keratinocyte hyperproliferation, which expands thepopulation of initiated stem cells [e.g., stem cells carrying a c-rasHa
mutation (46, 47)]. On TPA application, TGF-�1 expression is ele-vated (7). Although TGF-�1 directly inhibits keratinocyte prolifera-tion, it also activates expression of genes closely related to TPA-induced tumor promotion, e.g., the AP-1 family members (34). Smad3has been found to mediate the effect of TGF-�1 on transactivation ofc-fos, c-jun, junB, and junD by binding to their promoters (48–50). Inour current study, we show that induction of these AP-1 familymembers did not occur in TPA-treated Smad3 knockout skin andtumors or TPA-treated Smad3 knockout keratinocytes in vitro. Thus,Smad3 appears to be required for transcriptional activation of thesegenes during TPA promotion. In addition to direct transcriptionalregulation by Smad3, the AP-1 family members are also transcrip-tionally regulated on activation of epidermal growth factor receptor(51). Therefore, the lack of overexpression of TGF-�, the epidermalgrowth factor receptor ligand, in Smad3 knockout skin and tumorsduring TPA promotion may also contribute to the low levels of AP-1family members in Smad3 knockout skin and tumors. Each AP-1family member has been suggested to play an essential role in skincarcinogenesis through regulating cellular proliferation and apoptosis(52). For instance, c-fos, c-jun, and junD proteins positively regulatecell proliferation, whereas junB may negatively regulate cell prolif-eration in the presence of c-jun (52). Thus, it could be predicted thatdecreased expression of these molecules in TPA-treated skin andtumors of Smad3 knockout mice can cause a decrease in keratinocyteproliferation. With respect to apoptosis, the effects of AP-1 familymembers are somewhat contradictory and highly tissue specific. c-junknockout embryos exhibit massive apoptosis in hepatocytes, indicat-ing an antiapoptotic activity of c-jun protein. A previous study hasshown an antiapoptotic effect of jun/fos proteins on skin carcinogen-esis (53). Overall, the role of AP-1 family members in skin carcino-genesis has been at least partially attributed to their ability to promotekeratinocyte proliferation and inhibit apoptosis (52, 54). In vivo ex-periments showed that c-jun is required for the development ofpapillomas (55), whereas c-fos is required for malignant transforma-tion (56). Thus, lack of AP-1 expression/activation during TPA pro-motion in Smad3 knockout skin appears to greatly contribute toresistance to papilloma formation as well as malignant progressionduring skin chemical carcinogenesis. In the case of constant overex-pression of v-rasHa in transplanted keratinocytes (13), the tumorpromotion event can bypass TPA-induced AP-1 activation; thus, theeffect of Smad3 on AP-1 gene expression is dispensable. Under thiscircumstance, the effect of the loss of Smad3 appears to predomi-nantly abrogate TGF-�1–induced growth inhibition, resulting in ac-celerated malignant conversion.
Another growth factor that is typically rapidly induced by TPAtreatment of normal keratinocytes is TGF-�. TPA up-regulates TGF-�expression through signaling via protein kinase C (PKC)-dependentand -independent pathways (57). In the latter case, it is well-docu-mented that TGF-� is an autoinductive cytokine and that the TGF-�
Fig. 7. Relative expression levels of IL-1� and MCP-1 in papillomas as determined byqRT-PCR. The expression level in one Smad3�/� sample was set as 1 arbitrary unit.
7843
SKIN CHEMICAL CARCINOGENESIS ON Smad3 KNOCKOUT MICE
Research. on February 19, 2020. © 2004 American Association for Cancercancerres.aacrjournals.org Downloaded from

gene promoter contains a TGF-�/epidermal growth factor-responsiveelement (58). It has also been shown that TGF-�1 transcriptionallyregulates TGF-� expression (59). Because PKC is rapidly down-regulated after TPA application (60), it is reasonable to believe thatthe PKC-independent pathways are primarily responsible for thepersistent TGF-� overexpression. Although the exact mechanism bywhich Smad3 deletion blocks TGF-� overexpression remains to bedetermined, it is likely that loss of Smad3 abrogates TGF-�1–inducedTGF-� overexpression, consequently perturbing the autoinduction ofTGF-� expression. TGF-� is overexpressed in many cancer types andfunctions as a potent mitogen for cancer cell proliferation (61, 62). Inaddition, TGF-� is a critical survival factor against apoptosis forcancer cells (37, 38). Thus, persistent TGF-� overexpression isthought to be critical for the TPA-induced tumor promotion effect(36). Supporting this, TGF-�–deficient mice (Wa-1 mutant) exhibit aresistance to TPA promotion (63). Consistent with the above-men-tioned studies, lack of TPA- induced TGF-� overexpression in Smad3knockout skin and papillomas correlated with decreased proliferationand increased apoptosis in keratinocytes, which apparently contrib-uted to the resistance of Smad3 knockout mice to skin chemicalcarcinogenesis. Because TGF-� overexpression is critical for activa-tion of several signal transduction pathways, such as the ras-mitogen-activated protein kinase pathway (64), the lack of TGF-� overexpres-sion in Smad3 knockout skin/tumors may also explain thecontradictory data between our present study and a previous reportshowing accelerated malignant conversion in skin tumors that resultedfrom transplantation of Smad3-null keratinocytes transduced withv-rasHa (13). In that study, consistently high levels of v-rasHa expres-sion in keratinocytes should be able to bypass the effect of TGF-�overexpression as seen during TPA-induced tumor promotion.
Reduced Inflammation in Smad3 Knockout Skin May AlsoContribute to Resistance to Skin Carcinogenesis In vivo. Given theimportance of inflammation in cancer development (18), TPA-induced skin inflammation is expected to contribute to the tumorpromotion effect of TPA. In keratinocytes, TPA application inducesexpression of inflammatory cytokines, including IL-1 (65), which notonly induces inflammation but also stimulates keratinocyte prolifer-ation (66). In TPA-treated preneoplastic skin, we did not find differ-ences in inflammation among the different Smad3 genotypes. Thissuggests that TPA-induced inflammation may not be completelymediated by TGF-�1 overexpression, and therefore the loss of Smad3cannot attenuate TPA-induced inflammation, at least at the acutephase. At this stage, the resistance to skin carcinogenesis in Smad3knockout mice may be mainly a result of altered keratinocyte prop-erties (i.e., resistance to proliferation and increased apoptosis). How-ever, because TGF-�1 overexpression has been shown to induce skininflammation (15, 16), persistent TGF-�1 overexpression at laterstages of skin carcinogenesis may maintain a certain level of inflam-mation that is required for tumor development (18). In particular,TAMs play an important role in tumor promotion and progression(18). Thus, a reduction in TAM infiltration in Smad3 knockout tumorsmay also explain, at least in part, the resistance of these tumors tomalignant conversion. The impaired chemotactic effect of TGF-�1 onSmad3�/� monocytes (20) is possibly responsible for reduced TAMinfiltration in Smad3 knockout tumors. Supporting this notion, re-duced local inflammation associated with decreased monocyte infil-tration has also been reported in Smad3 knockout wounds as com-pared with Smad3�/� wounds (17). TGF-�1 has a potent chemotacticeffect on monocytes (67) and may induce expression of multiplechemokines in the skin (16, 68). MCP-1, which was down-regulatedin Smad3 knockout tumors, is a chemokine that attracts monocyte-macrophage infiltration to sites of inflammation and can be up-regulated on TGF-�1 overexpression in the skin (16). It is thus likely
that Smad3�/� monocytes do not respond to the chemotactic effect ofTGF-�1. Alternatively, Smad3�/� stromal cells and endothelial cellsmay affect monocyte infiltration. In either case, reduced TAM infil-tration would not occur in tumors generated in mice in which Smad3is deleted only in keratinocytes, as indicated in a previous study thatreported accelerated skin carcinogenesis of transplanted Smad3-nullkeratinocytes (13). However, because inflammation was reduced inSmad3 knockout papillomas, as evidenced by reduced IL-1� and thelack of NF-�B activation, reduced TAM infiltration in Smad3 knock-out papillomas may also be a consequence of reduced expressionlevels of inflammatory cytokines and chemokines in Smad3�/� tumorepithelia. All of the above-mentioned possibilities can be tested in thefuture by generating mice with epidermal-specific deletion of Smad3and performing carcinogenesis experiments on these mice.
In summary, the present study shows that loss of Smad3 in vivosignificantly reduced tumor formation and malignant conversion dur-ing skin chemical carcinogenesis. Our study suggests that, at earlystages of skin carcinogenesis, TGF-�1 overexpression induced byTPA may also have a tumor promotion effect via activation of AP-1family members in keratinocytes and inflammation in the stroma, bothof which may require wild-type Smad3. Our study instigates futurestudies to investigate the complex nature of the role of Smad3 incancer development and its underlying molecular mechanisms underdifferent pathological conditions.
ADDENDUM
While this article was under review, a report was published (45) showing that
Smad3�/� mice were resistant to skin chemical carcinogenesis.
REFERENCES
1. Siegel PM, Massague J. Cytostatic and apoptotic actions of TGF-beta in homeostasisand cancer. Nat Rev Cancer 2003;3:807–21.
2. Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature (Lond) 2003;425:577–84.
3. Glick AB, Lee MM, Darwiche N, et al. Targeted deletion of the TGF-beta 1 genecauses rapid progression to squamous cell carcinoma. Genes Dev 1994;8:2429–40.
4. Amendt C, Schirmacher P, Weber H, Blessing M. Expression of a dominant negativetype II TGF-beta receptor in mouse skin results in an increase in carcinoma incidenceand an acceleration of carcinoma development. Oncogene 1998;17:25–34.
5. Go C, Li P, Wang XJ. Blocking transforming growth factor beta signaling intransgenic epidermis accelerates chemical carcinogenesis: a mechanism associatedwith increased angiogenesis. Cancer Res 1999;59:2861–8.
6. Cohen MM Jr. TGF beta/Smad signaling system and its pathologic correlates. Am JMed Genet 2003;116A:1–10.
7. Patamalai B, Burow DL, Gimenez-Conti I, et al. Altered expression of transforminggrowth factor-beta 1 mRNA and protein in mouse skin carcinogenesis. Mol Carcinog1994;9:220–9.
8. Cui W, Fowlis DJ, Bryson S, et al TGFbeta1 inhibits the formation of benign skintumors, but enhances progression to invasive spindle carcinomas in transgenic mice.Cell 1996;86:531–42.
9. Weeks BH, He W, Olson KL, Wang XJ. Inducible expression of transforming growthfactor beta1 in papillomas causes rapid metastasis. Cancer Res 2001;61:7435–43.
10. Kretschmer A, Moepert K, Dames S, et al. Differential regulation of TGF-betasignaling through Smad2, Smad3 and Smad4. Oncogene 2003;22:6748–63.
11. He W, Cao T, Smith DA, Myers TE, Wang XJ. Smads mediate signaling of theTGFbeta superfamily in normal keratinocytes but are lost during skin chemicalcarcinogenesis. Oncogene 2001;20:471–83.
12. Matsuura I, Denissova NG, Wang G, et al. Cyclin-dependent kinases regulate theantiproliferative function of Smads. Nature (Lond) 2004;430:226–31.
13. Vijayachandra K, Lee J, Glick AB. Smad3 regulates senescence and malignantconversion in a mouse multistage skin carcinogenesis model. Cancer Res 2003;63:3447–52.
14. Sporn MB, Roberts AB. Transforming growth factor-beta: recent progress and newchallenges. J Cell Biol 1992;119:1017–21.
15. Liu X, Alexander V, Vijayachandra K, et al. Conditional epidermal expression ofTGFbeta 1 blocks neonatal lethality but causes a reversible hyperplasia and alopecia.Proc Natl Acad Sci USA 2001;98:9139–44.
16. Li AG, Wang D, Feng XH, Wang XJ. Latent TGFbeta1 overexpression in keratino-cytes results in a severe psoriasis-like skin disorder. EMBO J 2004;23:1770–81.
17. Ashcroft GS, Yang X, Glick AB, et al. Mice lacking Smad3 show accelerated woundhealing and an impaired local inflammatory response. Nat Cell Biol 1999;1:260–6.
18. Coussens LM, Werb Z. Inflammation and cancer. Nature (Lond) 2002;420:860–7.
7844
SKIN CHEMICAL CARCINOGENESIS ON Smad3 KNOCKOUT MICE
Research. on February 19, 2020. © 2004 American Association for Cancercancerres.aacrjournals.org Downloaded from

19. Tian F, DaCosta BS, Parks WT, et al. Reduction in Smad2/3 signaling enhancestumorigenesis but suppresses metastasis of breast cancer cell lines. Cancer Res2003;63:8284–92.
20. Yang X, Letterio JJ, Lechleider RJ, et al. Targeted disruption of SMAD3 results inimpaired mucosal immunity and diminished T cell responsiveness to TGF-beta.EMBO J 1999;18:1280–91.
21. Bottinger EP, Letterio JJ, Roberts AB. Biology of TGF-beta in knockout and trans-genic mouse models. Kidney Int 1997;51:1355–60.
22. Aldaz CM, Conti CJ, Klein-Szanto AJ, Slaga TJ. Progressive dysplasia and aneu-ploidy are hallmarks of mouse skin papillomas: relevance to malignancy. Proc NatlAcad Sci USA 1987;84:2029–32.
23. Wang XJ, Liefer KM, Tsai S, O’Malley BW, Roop DR. Development of gene-switchtransgenic mice that inducibly express transforming growth factor beta1 in theepidermis. Proc Natl Acad Sci USA 1999;96:8483–8.
24. Greenhalgh DA, Wang XJ, Rothnagel JA, et al. Transgenic mice expressing targetedHPV-18 E6 and E7 oncogenes in the epidermis develop verrucous lesions andspontaneous, rasHa-activated papillomas. Cell Growth Differ 1994;5:667–75.
25. Ledbetter JA, Herzenberg LA. Xenogeneic monoclonal antibodies to mouse lymphoiddifferentiation antigens. Immunol Rev 1979;47:63–90.
26. Malorny U, Michels E, Sorg C. A monoclonal antibody against an antigen present onmouse macrophages and absent from monocytes. Cell Tissue Res 1986;243:421–8.
27. Wang XJ, Greenhalgh DA, Bickenbach JR, et al. Expression of a dominant-negativetype II transforming growth factor beta (TGF-beta) receptor in the epidermis oftransgenic mice blocks TGF-beta-mediated growth inhibition. Proc Natl Acad SciUSA 1997;94:2386–91.
28. Rennecke J, Rehberger PA, Furstenberger G, et al. Protein-kinase-Cmu expressioncorrelates with enhanced keratinocyte proliferation in normal and neoplastic mouseepidermis and in cell culture. Int J Cancer 1999;80:98–103.
29. Lu SL, Reh D, Li AG, et al. Overexpression of transforming growth factor beta1 inhead and neck epithelia results in inflammation, angiogenesis, and epithelial hyper-proliferation. Cancer Res 2004;64:4405–10.
30. Wong MH, Huelsken J, Birchmeier W, Gordon JI. Selection of multipotent stem cellsduring morphogenesis of small intestinal crypts of Lieberkuhn is perturbed bystimulation of Lef-1/beta-catenin signaling. J Biol Chem 2002;277:15843–50.
31. Go C, He W, Zhong L, et al. Aberrant cell cycle progression contributes to theearly-stage accelerated carcinogenesis in transgenic epidermis expressing the domi-nant negative TGFbetaRII. Oncogene 2000;19:3623–31.
32. Nischt R, Roop DR, Mehrel T, et al. Aberrant expression during two-stage mouse skincarcinogenesis of a type I 47-kDa keratin, K13, normally associated with terminaldifferentiation of internal stratified epithelia. Mol Carcinog 1988;1:96–108.
33. Quintanilla M, Brown K, Ramsden M, Balmain A. Carcinogen-specific mutation andamplification of Ha-ras during mouse skin carcinogenesis. Nature (Lond) 1986;322:78–80.
34. Hocevar, AH, Howe PH. Regulation of AP-1 activity by TGF-beta. In: Howe PH,editor. Transforming growth factor-beta protocols. Totowa, NJ: Human Press; 2001.p. 97–108.
35. Hsu TC, Young MR, Cmarik J, Colburn NH. Activator protein 1 (AP-1)- and nuclearfactor kappaB (NF-kappaB)-dependent transcriptional events in carcinogenesis. FreeRadic Biol Med 2000;28:1338–48.
36. DiGiovanni J, Rho O, Xian W, Beltran L. Role of the epidermal growth factorreceptor and transforming growth factor alpha in mouse skin carcinogenesis. ProgClin Biol Res 1994;387:113–38.
37. Reinartz J, Bechtel MJ, Kramer MD. Tumor necrosis factor-alpha-induced apoptosisin a human keratinocyte cell line (HaCaT) is counteracted by transforming growthfactor-alpha. Exp Cell Res 1996;228:334–40.
38. Kanda D, Takagi H, Toyoda M, et al. Transforming growth factor alpha protectsagainst Fas-mediated liver apoptosis in mice. FEBS Lett 2002;519:11–5.
39. Amundadottir LT, Nass SJ, Berchem GJ, Johnson MD, Dickson RB. Cooperation ofTGF alpha and c-Myc in mouse mammary tumorigenesis: coordinated stimulation ofgrowth and suppression of apoptosis. Oncogene 1996;13:757–65.
40. Tak PP, Firestein GS. NF-kappaB: a key role in inflammatory diseases. J ClinInvestig 2001;107:7–11.
41. Murphy JE, Robert C, Kupper TS. Interleukin-1 and cutaneous inflammation: acrucial link between innate and acquired immunity. J Investig Dermatol 2000;114:602–8.
42. Bonifati C, Ameglio F. Cytokines in psoriasis. Int J Dermatol 1999;38:241–51.43. Liu Y, Duysen E, Yaktine AL, et al. Dietary energy restriction inhibits ERK but not
JNK or p38 activity in the epidermis of SENCAR mice. Carcinogenesis (Lond)2001;22:607–12.
44. Rodriguez-Villanueva J, Greenhalgh D, Wang XJ, et al. Human keratin-1.bcl-2transgenic mice aberrantly express keratin 6, exhibit reduced sensitivity to keratino-
cyte cell death induction, and are susceptible to skin tumor formation. Oncogene1998;16:853–63.
45. Tannehill-Gregg SH, Kusewitt DF, Rosol TJ, Weinstein M. The roles of Smad2 andSmad3 in the development of chemically induced skin tumors in mice. Vet Pathol2004;41:278–82.
46. Yuspa SH. Alterations in epidermal functions resulting from exposure to initiatorsand promoters of carcinogenesis. Curr Probl Dermatol 1983;11:227–41.
47. Kiguchi K, Beltran LM, You J, Rho O, DiGiovanni J. Elevation of transforminggrowth factor-alpha mRNA and protein expression by diverse tumor promoters inSENCAR mouse epidermis. Mol Carcinog 1995;12:225–35.
48. Verrecchia F, Tacheau C, Schorpp-Kistner M, Angel P, Mauviel A. Induction of theAP-1 members c-Jun and JunB by TGF-beta/Smad suppresses early Smad-drivengene activation. Oncogene 2001;20:2205–11.
49. Liberati NT, Datto MB, Frederick JP, et al. Smads bind directly to the Jun family ofAP-1 transcription factors. Proc Natl Acad Sci USA 1999;96:4844–9.
50. Wong C, Rougier-Chapman EM, Frederick JP, et al. Smad3-Smad4 and AP-1complexes synergize in transcriptional activation of the c-Jun promoter by transform-ing growth factor beta. Mol Cell Biol 1999;19:1821–30.
51. Jorissen RN, Walker F, Pouliot N, et al. Epidermal growth factor receptor: mecha-nisms of activation and signalling. Exp Cell Res 2003;284:31–53.
52. Shaulian E, Karin M. AP-1 as a regulator of cell life and death. Nat Cell Biol2002;4:E131–6.
53. Wach S, Schirmacher P, Protschka M, Blessing M. Overexpression of bone morpho-genetic protein-6 (BMP-6) in murine epidermis suppresses skin tumor formation byinduction of apoptosis and downregulation of fos/jun family members. Oncogene2001;20:7761–9.
54. Eferl R, Wagner EF. AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer2003;3:859–68.
55. Young MR, Li JJ, Rincon M, et al. Transgenic mice demonstrate AP-1 (activatorprotein-1) transactivation is required for tumor promotion. Proc Natl Acad Sci USA1999;96:9827–32.
56. Saez E, Rutberg SE, Mueller E, et al. c-fos is required for malignant progression ofskin tumors. Cell 1995;82:721–32.
57. Klein SB, Fisher GJ, Jensen TC, et al. Regulation of TGF-alpha expression in humankeratinocytes: PKC-dependent and -independent pathways. J Cell Physiol 1992;151:326–36.
58. Awwad R, Humphrey LE, Periyasamy B, et al. The EGF/TGFalpha response elementwithin the TGFalpha promoter consists of a multi-complex regulatory element.Oncogene 1999;18:5923–35.
59. Lynch MJ, Pelosi L, Carboni JM, et al. Transforming growth factor-beta 1 inducestransforming growth factor-alpha promoter activity and transforming growth factor-alpha secretion in the human colon adenocarcinoma cell line FET. Cancer Res1993;53:4041–7.
60. Wang XJ, Warren BS, Rupp T, Beltran LM, DiGiovanni J. Loss of mouse epidermalprotein kinase C isozyme activities following treatment with phorbol ester andnon-phorbol ester tumor promoters. Carcinogenesis (Lond) 1994;15:2795–803.
61. Sanders S, Thorgeirsson SS. Promotion of hepatocarcinogenesis by phenobarbital incmyc/TGF-alpha transgenic mice. Mol Carcinog 2000;28:168–73.
62. Wang XJ, Greenhalgh DA, Lu XR, Bickenbach JR, Roop DR. TGF alpha and v-foscooperation in transgenic mouse epidermis induces aberrant keratinocyte differenti-ation and stable, autonomous papillomas. Oncogene 1995;10:279–89.
63. Kiguchi K, Beltran L, Dubowski A, DiGiovanni J. Analysis of the ability of 12-O-tetradecanoylphorbol-13-acetate to induce epidermal hyperplasia, transforminggrowth factor-alpha, and skin tumor promotion in wa-1 mice. J Investig Dermatol1997;108:784–91.
64. Keshamouni VG, Mattingly RR, Reddy KB. Mechanism of 17-beta-estradiol-inducedErk1/2 activation in breast cancer cells. A role for HER2 AND PKC-delta. J BiolChem 2002;277:22558–65.
65. Updyke LW, Yoon HL, Chuthaputti A, Pfeifer RW, Yim GK. Induction of interleu-kin-1 and tumor necrosis factor by 12-O-tetradecanoylphorbol-13-acetate in phorbolester- sensitive (SENCAR) and resistant (B6C3F1) mice. Carcinogenesis (Lond)1989;10:1107–11.
66. Cooper KD, Hammerberg C, Baadsgaard O, et al. Interleukin-1 in human skin:dysregulation in psoriasis. J Investig Dermatol 1990;95:24S–6S.
67. Wahl SM. Transforming growth factor beta (TGF-beta) in inflammation: a cause anda cure. J Clin Immunol 1992;12:61–74.
68. Wahl SM, Allen JB, Weeks BS, Wong HL, Klotman PE. Transforming growth factorbeta enhances integrin expression and type IV collagenase secretion in humanmonocytes. Proc Natl Acad Sci USA 1993;90:4577–81.
7845
SKIN CHEMICAL CARCINOGENESIS ON Smad3 KNOCKOUT MICE
Research. on February 19, 2020. © 2004 American Association for Cancercancerres.aacrjournals.org Downloaded from

2004;64:7836-7845. Cancer Res Allen G. Li, Shi-Long Lu, Ming-Xiang Zhang, et al. CarcinogenesisSmad3 Knockout Mice Exhibit a Resistance to Skin Chemical
Updated version
http://cancerres.aacrjournals.org/content/64/21/7836
Access the most recent version of this article at:
Cited articles
http://cancerres.aacrjournals.org/content/64/21/7836.full#ref-list-1
This article cites 66 articles, 20 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/64/21/7836.full#related-urls
This article has been cited by 7 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
SubscriptionsReprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. (CCC)Click on "Request Permissions" which will take you to the Copyright Clearance Center's
.http://cancerres.aacrjournals.org/content/64/21/7836To request permission to re-use all or part of this article, use this link
Research. on February 19, 2020. © 2004 American Association for Cancercancerres.aacrjournals.org Downloaded from