stanley iyadurai, msc phd md assistant professor of
TRANSCRIPT

Immune Neuropathies Stanley Iyadurai, MSc PhD MD Assistant Professor of Neurology, Neuromuscular Specialist, OSU, Columbus, OH
August 28, 2015

Case
• 32-year old woman
• 1.5 year history of numbness/tingling/weakness
• Difficulty walking
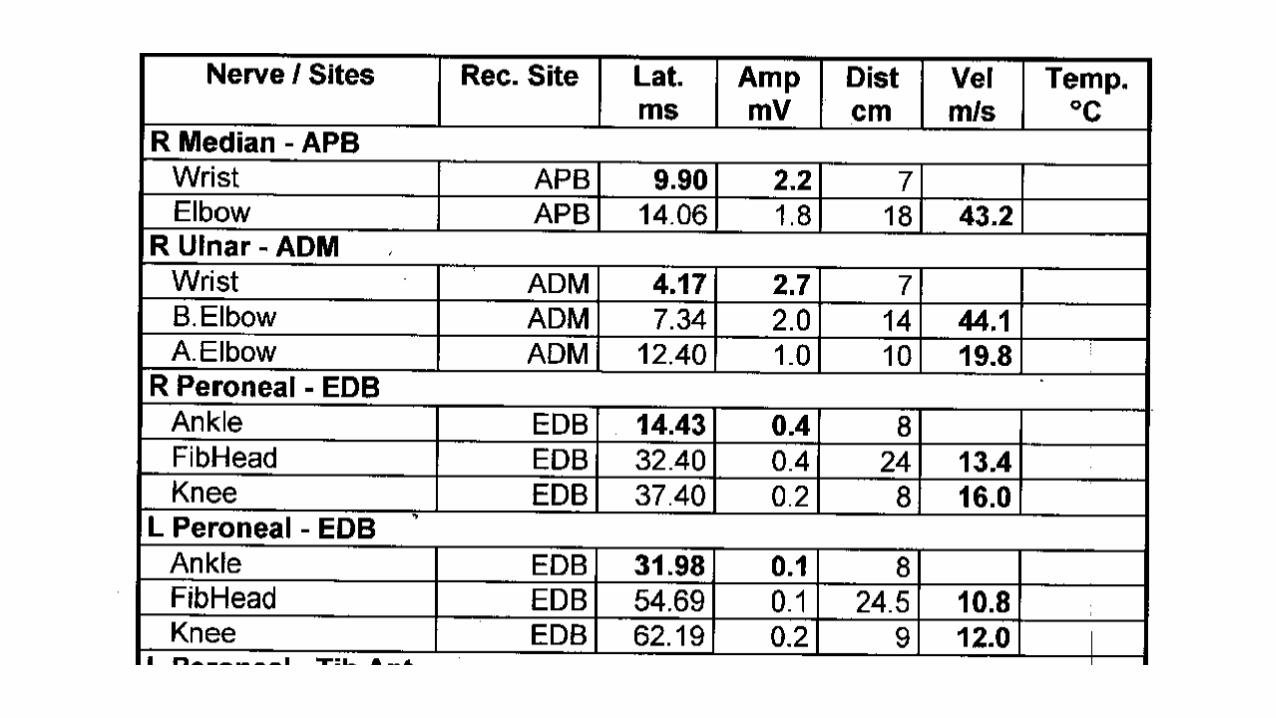
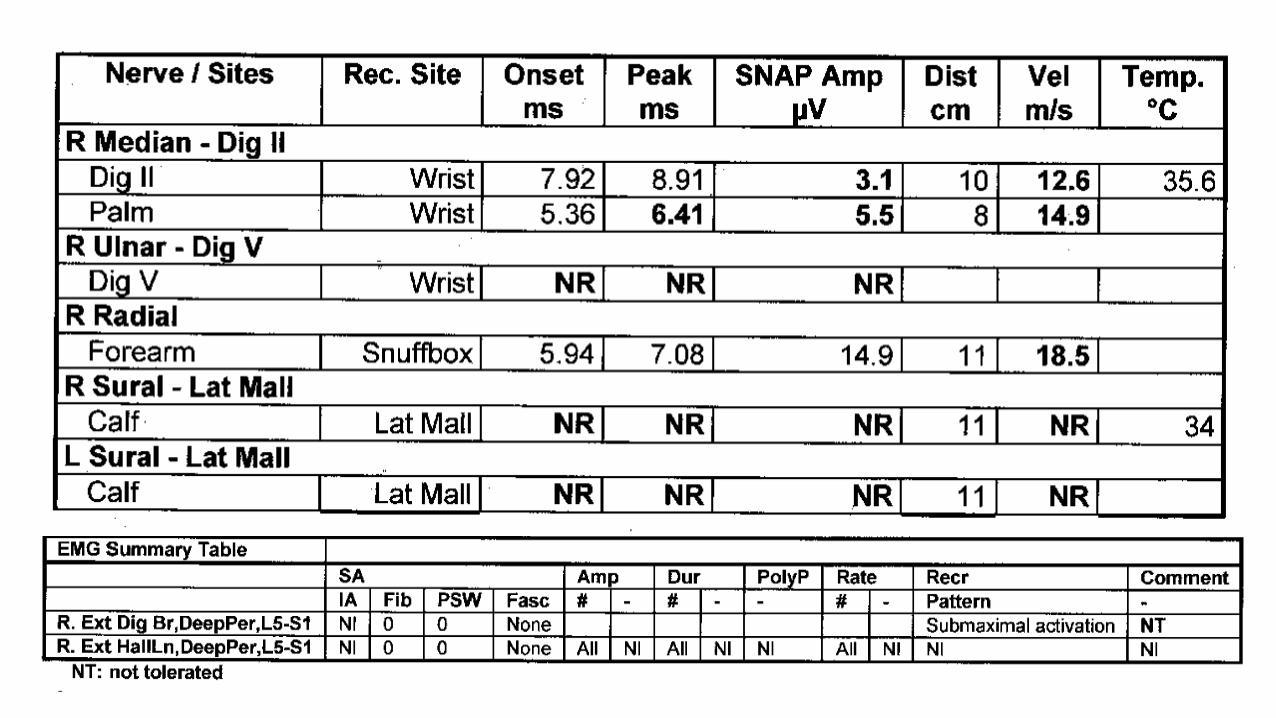
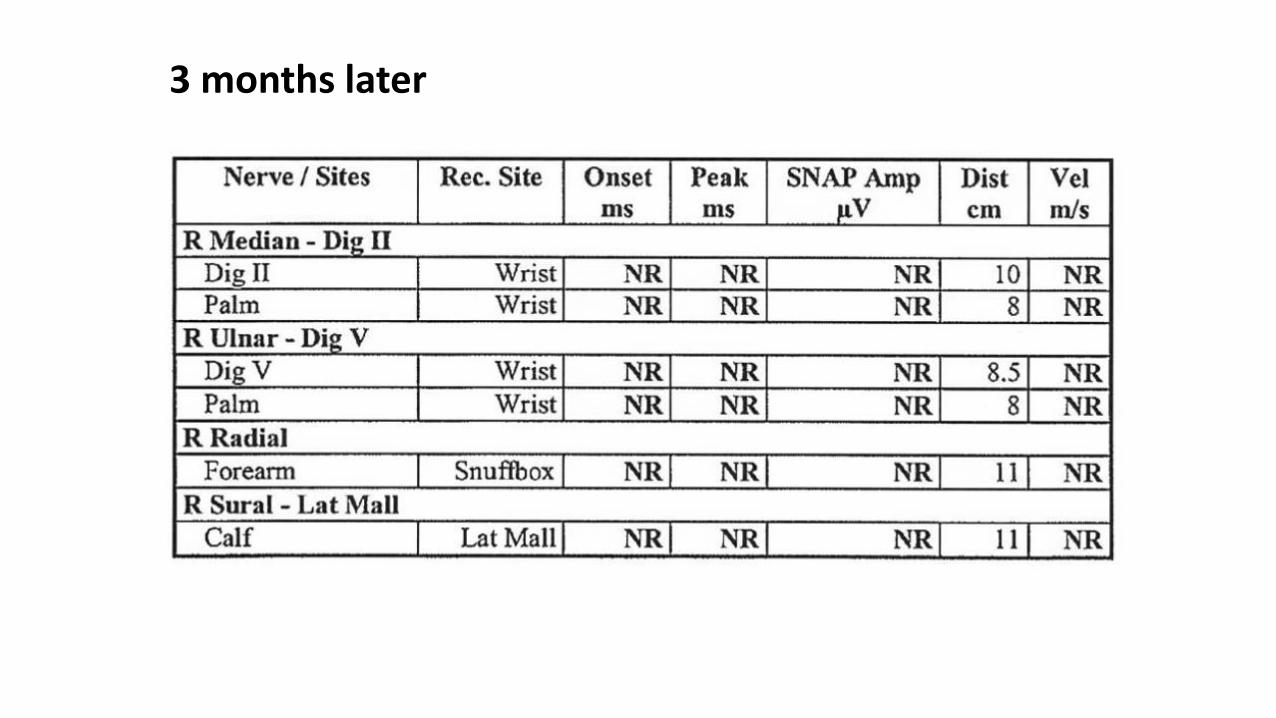
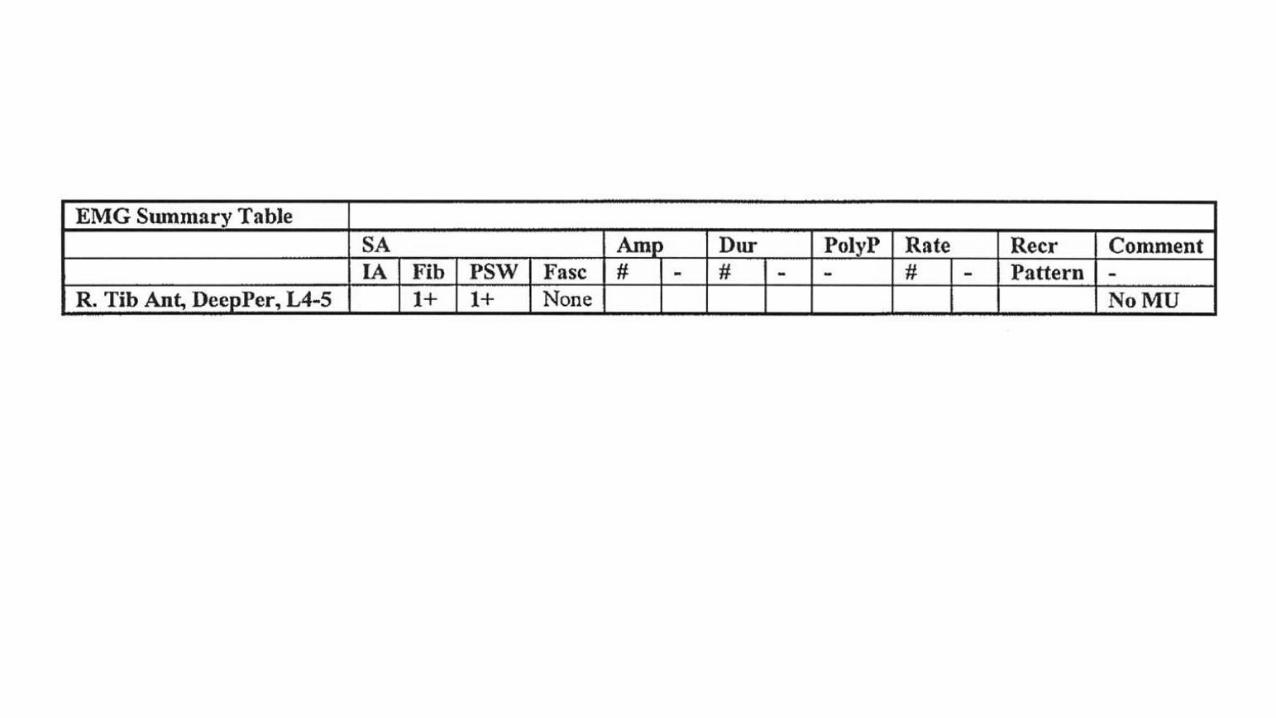
3 months later


Inflammatory or Immune?
Not Clear
Inflammatory Cells – present or not?
Immune Components Deposited?
Immune-mediated Attack?
Involves Complement?
Involves Antibodies?
Inflammatory Markers are Absent?
Treat Inflammation/Use Immunomodulation?

Types
• Guillain-Barre Syndrome and variants
• CIDP and variants
• Multifocal Motor Neuropathy
• Paraneoplastic Neuropathies
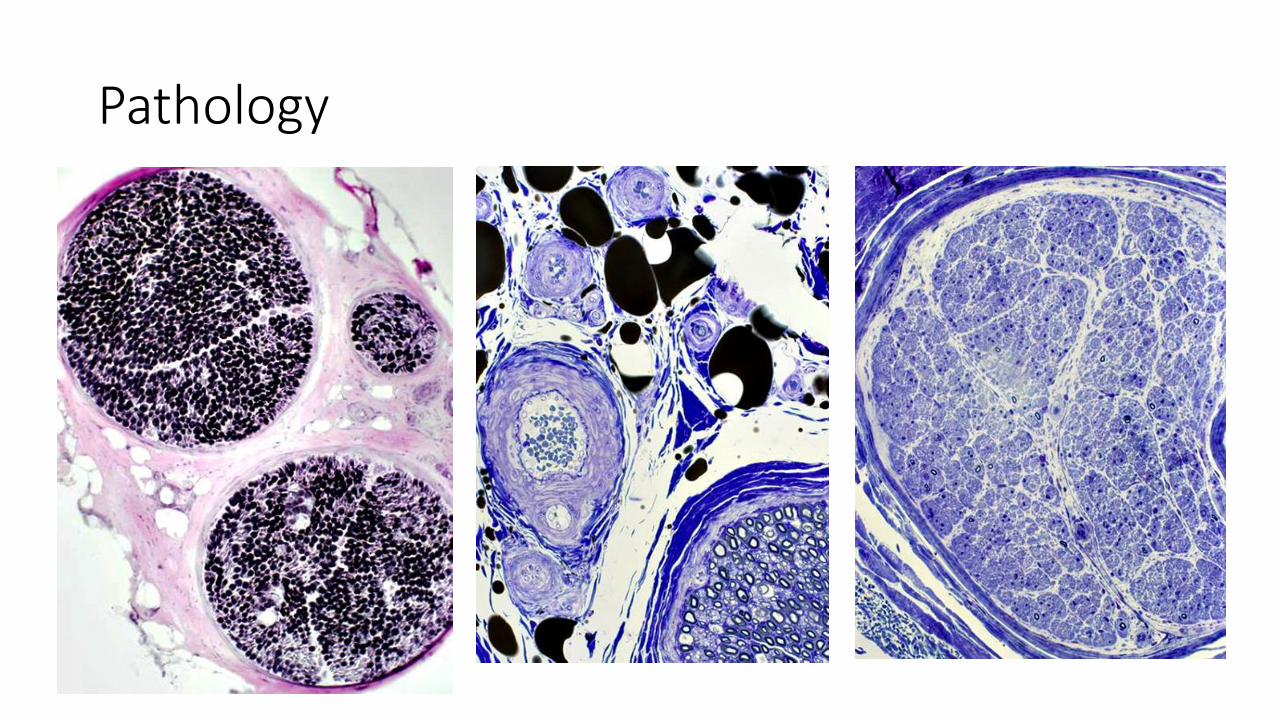
Pathology
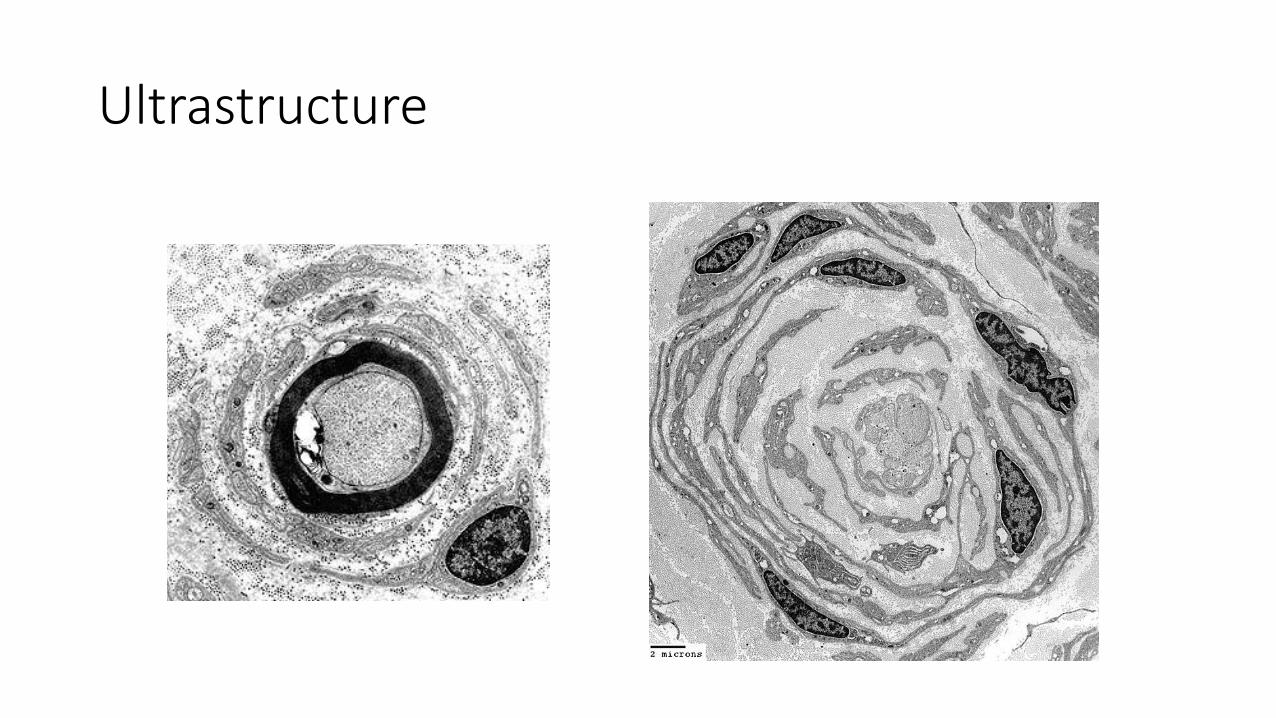
Ultrastructure
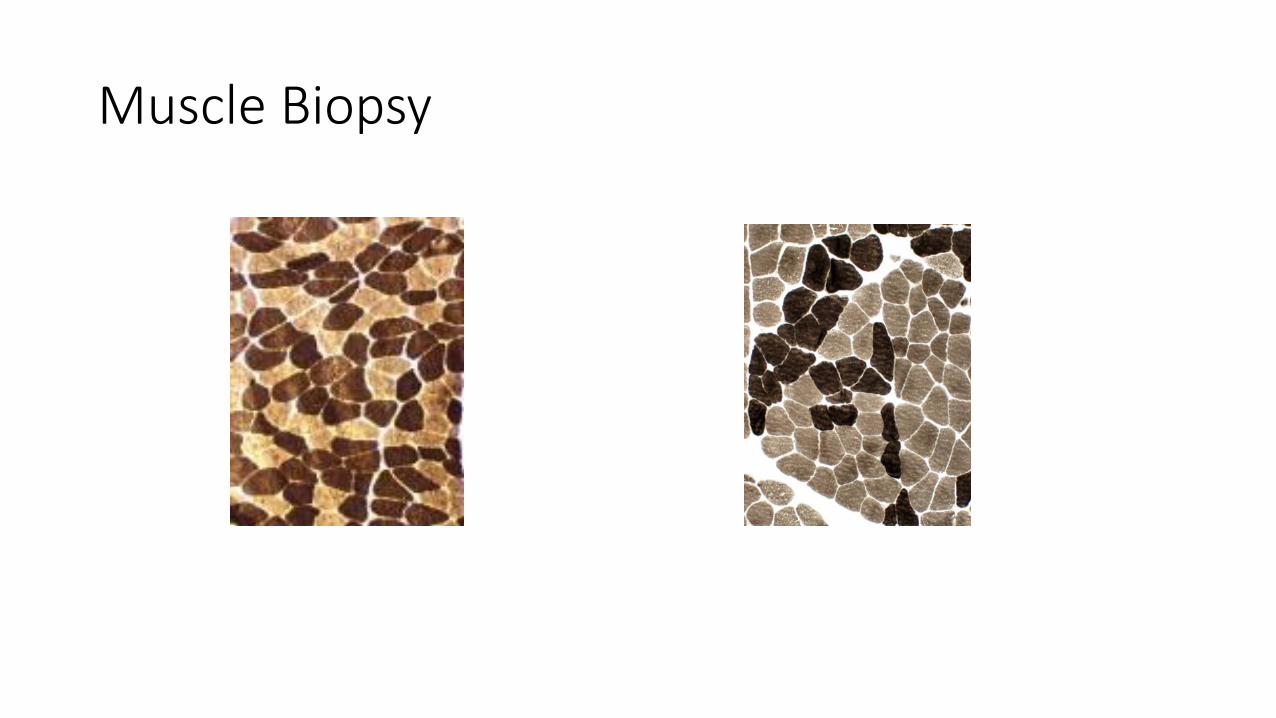
Muscle Biopsy

Guillain-Barre Syndrome
AIDP to Pure Motor, Sensory Motor, Bulbar and Miller-Fisher Syndrome
Hypothesis of Gangliosides mimicry of Campylobacter jejuni lipopolysaccharides
Ascending Weakness, Areflexia
Facial Weaknes
Increased CSF Protein
Acute Demyelinating Syndrome

Diagnosis
• Ascending Paralysis
• Areflexia
• Dysautonomia
• Nadir at 4-6 weeks
• Increased CSF Protein
• Demyelinating Velocities on EMG
• Conduction blocks
• Absent sensory responses
• Denervation – Fibs/Positive Sharp Waves

Motor-Sensory GBS
75% of GBS in Western Countries
Paresthesias
Facial Involvement
Autonomic Dysfunction
CMV infection in 20%

Pure Motor GBS
20% of GBS in Western Countries
(Paresthesias)
Distal Weakness in Extremities
Cranial Nerves, Respiratory Muscles Spared
C. jejuni 65%
IgG against GM1 abs - 40%

Miller-Fisher Variant
3% of GBS in Western Countries
Extraocular Muscle weakness, Ptosis
Paralysis of Sphincter Pupillae
Facial Weakness, Bulbar Involvement
Ataxia (50%)
IgG against GQ1b in 90%

GBS Treatment
IVIg, PE
For IgG against GM1 patients, IVIG > PE
Steroids not helpful – Harmful effects on denervated muscle or inhibit macrophage repair processes?

CIDP
• Previously known as “steroid-responsive neuropathy”.
• It affects 1 to 7.7 per 100,000 population
• Age of onset: 2 to 72 years of age with more prevalence in the fifth decade

CIDP
• chronic, monophasic/progressive or relapsing-remitting disease
• Characterized by proximal and distal weakness, in addition to sensory symptoms for more than 2 months
• On exam, hyporeflexia/areflexia is diffuse and it is usually a common finding on exam
• Objective sensory loss of 1 or more sensory modalities is present in 86% of cases

CIDP
Autoimmune!
Progressive or Relapsing/remitting
Proximal and Distal, Symmetric Weakness
Sensory Involvement + Areflexia; Variants
8 Weeks to Reach Nadir
EMG – Prolonged DL, Slowed NCV, Delayed or Absent F-waves, Conduction Block, Temporal Dispersion
CSF with Increased Protein
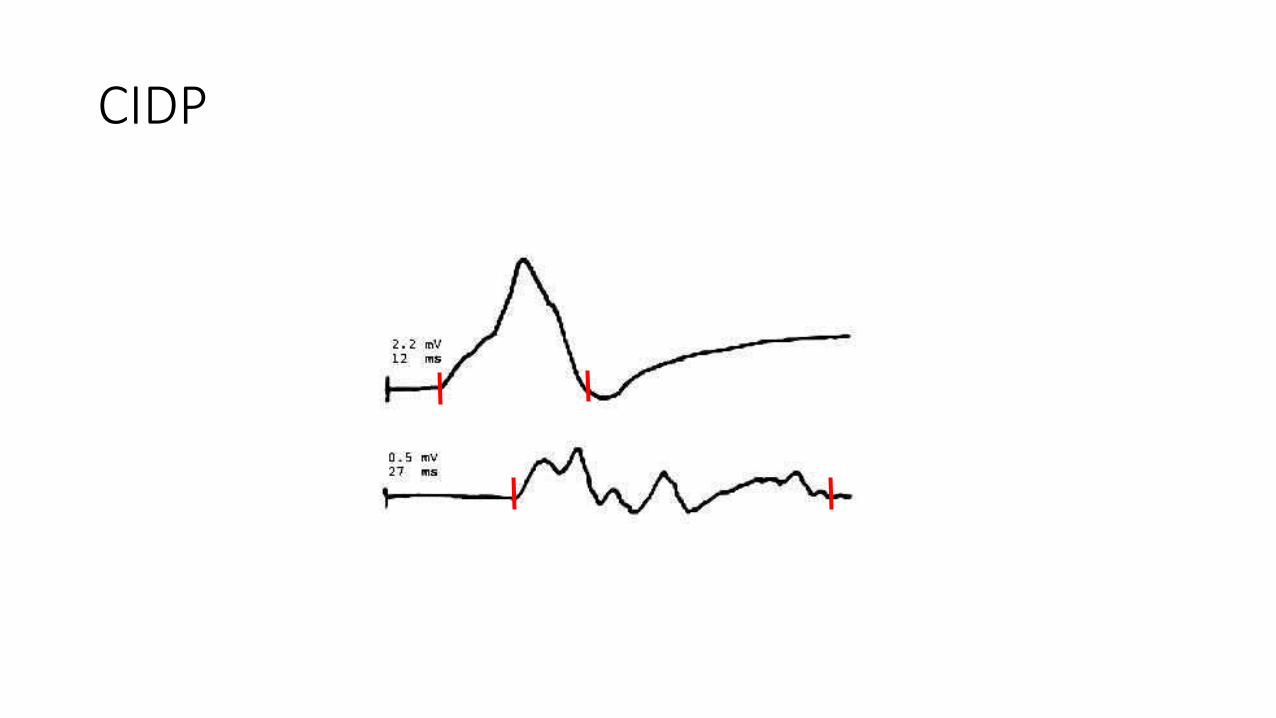
CIDP

CIDP Diagnosis
• Clinical Picture
• Cerebrospinal fluid
• Electrodiagnostic studies
• Nerve or Nerve and Muscle biopsy

CIDP Variants
• Typical CIDP constitutes about 50 % of cases of CIDP
• The rest constitutes “CIDP variants”

Major CIDP variants:
• Distal Acquired Demyelinating symmetric polyneuropathy (DADS)
• Lewis-Summer syndrome or Multifocal Acquired Demyelinating Sensory and Motor Neuropathy (MADSAM)
• Sensory predominant CIDP
• Motor predominant CIDP
• Focal CIDP

CIDP - Treatment
Prednisone, IVIg, PE
Long-term Treatment

Multifocal Motor Neuropathy
IgM against GM-1 antibodies (85%)
Pure Motor disease
Focal weakness
Involvement of subsegments of terminal nerves
Treatment with IVIg, rituximab
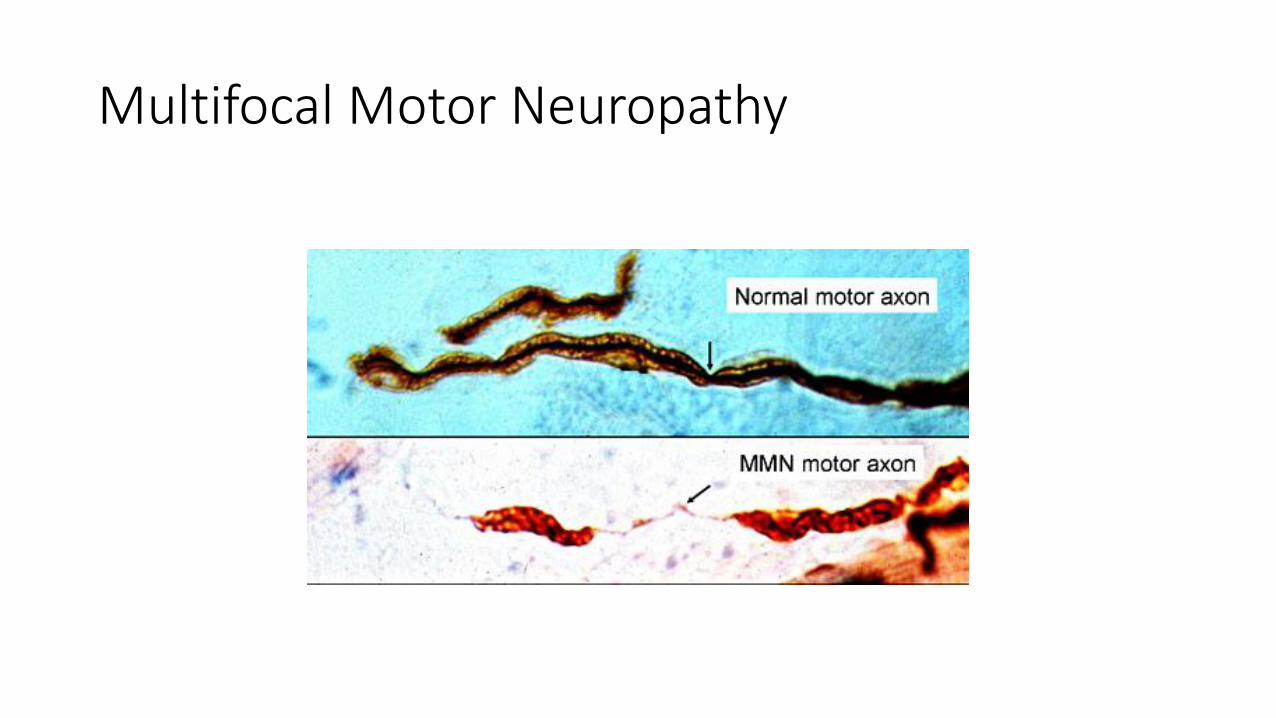
Multifocal Motor Neuropathy

Multifocal Motor Neuropathy

Distal Acquired Demyelinating symmetric polyneuropathy (DADS) • Accounts 2 to 17% of CIDP cases
• Distal form of CIDP
• It is characterized mainly by sensory symptoms, although abnormal motor nerve conduction studies in form of demyelinating disease can be observed.
• CSF shows elevated protein in 86% of cases
• In 2/3 of cases, an abnormal monoclonal protein (M protein) on serum protein electrophoresis or IFE is found, and in 2/3 of those, antibody against Myelin-Associated Glycoprotein (MAG)
• Poor response to Immunotherapy, and treatment is mostly supportive

MAG Neuropathy

Lewis-Summer syndrome or Multifocal Acquired Demyelinating Sensory and Motor Neuropathy (MADSAM) • It accounts 6-15% of CIDP cases
• It is a distal form of CIDP
• Males are commonly affected in the fifth decade
• Upper extremities are affected mostly (Lower extremities are involved in 38% of cases
• Mistaken as entrapment neuropathies at median and ulnar nerves (wrist and elbow respectively).
• Cranial nerve involvement including II, III, IV and V has been reported

MADSAM
• Main electrodiagnostic feature is conduction block (importance to have proximal stimulation at Erb’s point)
• CSF protein is normal or mildly elevated
• Intravenous Immunoglobulin is the first line treatment

Sensory Predominant CIDP
• It accounts for 6-12% of CIDP
• Symptoms are sensory and include the following: numbness and tingling in “stocking-glove distribution”, neuropathic pain, or proprioception deficit leading to sensory ataxia.
• Strength is usually normal
• On electrodiagnostic studies, demyelinating changes are seen in motor as well in sensory nerves
• Treatment if clinically disabled

Motor CIDP
• Pure Motor form of CIDP

Focal CIDP
• Only in specific location

Other Dysimmune Neuropathies
• Anti-Hu syndrome • Treat cancer
• Anti-SS-A, SS-B • Hydroxychloroquine
• MGUS (monoclonal gammopathy of unknown significance) • Plasma Exchange, cyclophosphamide
• POEMS (Polyneuropathy, organomegaly, endocrinopathy, M-protein, Skin changes) • Plasma Exchange, cyclophosphamide, Stem cell transplant?
• GALOP (Gait disorder, autoantibody, Late-onset, Polyneuropathy) • ??
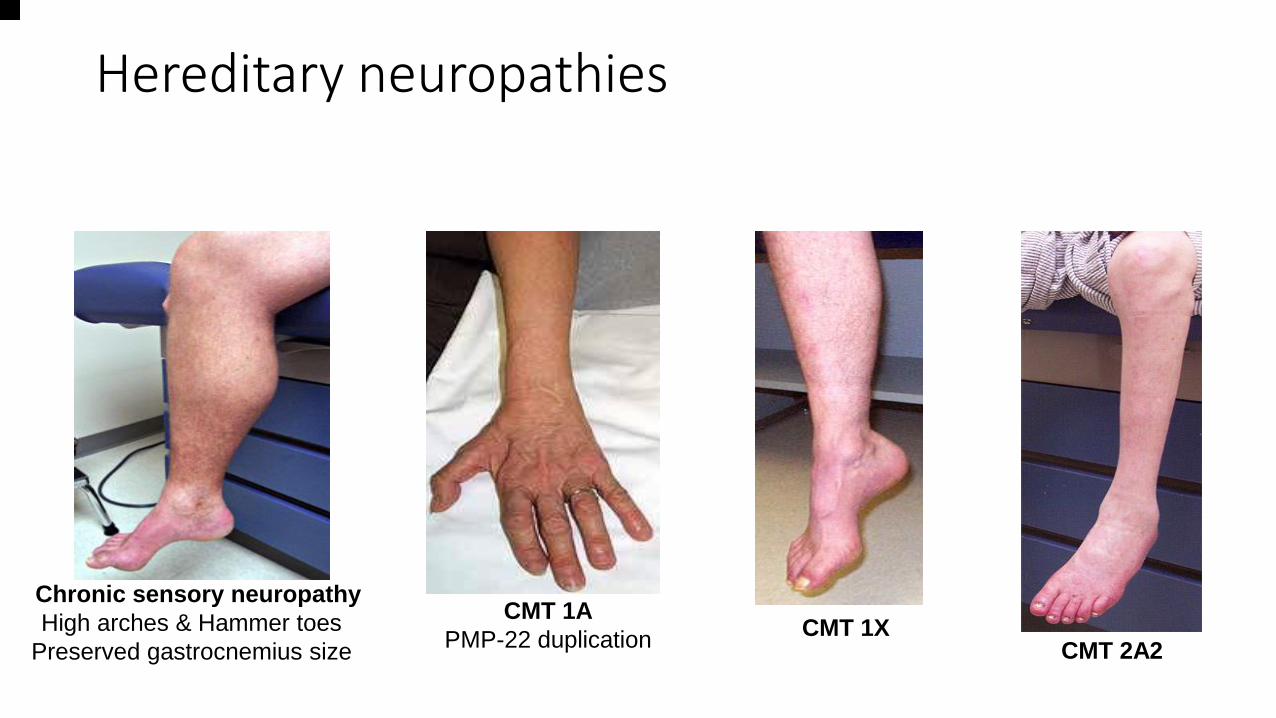
Hereditary neuropathies
Chronic sensory neuropathy
High arches & Hammer toes
Preserved gastrocnemius size
CMT 1A
PMP-22 duplication CMT 1X
CMT 2A2