synaptic localization - pennsylvania state university
TRANSCRIPT

The Pennsylvania State University
The Graduate School
Eberly College of Science
AN INVESTIGATION OF THE DOMAINS OF THE GAMMA TWO SUBUNIT OF
GABAA RECEPTORS, GEPHYRIN AND COLLYBISTIN REQUIRED FOR
SYNAPTIC LOCALIZATION
A Thesis in
Biochemistry, Microbiology, and Molecular Biology
by
Melissa J. Alldred
© 2005 Melissa J. Alldred
Submitted in Partial Fulfillment of the Requirements
for the Degree of
Doctor of Philosophy
August 2005

The thesis of Melissa J. Alldred was reviewed and approved* by the following:
Bernhard Lüscher Associate Professor of Biology, Biochemistry and Molecular Biology Thesis Advisor Chair of Committee
Graham Thomas Associate Professor of Biology, Biochemistry and Molecular Biology
B. Franklin Pugh Associate Professor of Biochemistry and Molecular Biology
Douglas Cavener Head of the Department of Biology, Professor of Biology
Randen Patterson Assistant Professor of Biology
Robert Schlegel Professor of Biochemistry and Molecular Biology Head of the Department of Biochemistry and Molecular Biology
*Signatures are on file in the Graduate School

iii
ABSTRACT
Gamma-aminobutyric acid (GABA) type A receptors are heteropentameric
ligand-gated Cl--channels that mediate the majority of fast inhibitory
neurotransmission in the brain. GABAA receptor clustering at postsynaptic sites is
critical for the function of inhibitory synapse. Moreover, changes in synaptic
receptor concentration are believed to contribute to functional plasticity of
neurons. Typical postsynaptic GABAA receptor subtypes are composed of α, β,
and γ2 subunits and they are co-localized at synapses with the putative clustering
protein gephyrin, which is thought to link GABAA receptors to the cytoskeleton.
The multifunctional protein gephyrin represents a major component of the
subsynaptic protein scaffold of inhibitory synapses and provides an interface for
interaction with diverse other postsynaptic proteins including components of the
microtubule and actin cytoskleton, as well as the GDP-GTP exchange factor
collybistin, which is implicated in postsynaptic deposition of gephyrin.
Analysis of γ2 subunit-deficient mice and neurons revealed that the γ2
subunit is essential for postsynaptic clustering of GABAA receptors and gephyrin,
but largely dispensable for expression of functional GABA-gated chloride
channels at the cell surface (Essrich et al., 1998). Interaction of the γ2 subunit
with diverse putative trafficking proteins of GABAA receptors, such as GABARAP
and GODZ, further suggests that this subunit acts as an important determinant of
postsynaptic receptor concentration. Nevertheless, the mechanism by which the

iv
γ2 subunit contributes to accumulation of GABAA receptors at synapses is poorly
understood.
The main objective of this doctoral thesis was to determine the subunit
domain(s) of the γ2 subunit that are essential (i) for proper trafficking and
localization of postsynaptic GABAA receptors (ii) for recruitment of gephyrin to
postsynaptic GABAA receptors and (iii) for normal inhibitory synaptic function of
postsynaptic GABAA receptors in γ2 subunit-deficient neurons. Surprisingly, the
fourth transmembrane domain (TM4) of the γ2 subunit was found to be sufficient
for postsynaptic localization of GABAA receptors. However, the cytoplasmic loop
domain is required in addition to TM4 for recruitment of gephyrin to postsynaptic
GABAA receptors and for restoration of inhibitory synaptic function. These
experiments point to a novel mechanism in subcellular targeting of ligand-gated
ion channels and clearly dissociate postsynaptic GABAA receptor targeting
mechanisms from interaction with gephyrin.
As part of a collaborative project with Dr. Harvey’s group at UC London, I
was involved in mapping the protein-protein interaction domains between
gephyrin and the GTP exchange factor collybistin to further elucidate their roles
in synaptic localization and anchoring of GABAA receptors at the postsynaptic
membrane. These experiments revealed that proper localization of gephyrin
requires both the plextrin homology domain of collybistin and the collybistin
binding sequence in gephyrin. Additionally, a single point mutation in the SH3
domain of collybistin known to underlie an atypical form of hyperekplexia in
humans was shown to result in mislocalization of gephyrin and postsynaptic

v
GABAA receptors in transfected neurons. These experiments for the first time
showed an essential function of collybistin in formation of GABAergic inhibitory
synapses
Finally, preliminary results addressing the role of palmitoylation of the γ2
subunit of GABAA receptors with respect to postsynaptic localization of GABAA
receptors suggest that proper localization of GABAA receptors can occur
independently of palmitoylation. Rather than proper localization, palmitoylation is
therefore implicated in regulating the stability of GABAA receptors in the plasma
membrane. Preliminary experiments also addressed potential functional
redundancy between different members of the GABAA receptor-associated
protein (GABARAP) family of γ2 subunit binding proteins implicated in trafficking
of GABAA receptors. The results suggest that different members of the
GABARAP family of proteins are functionally redundant with respect to
interaction with GABAA receptors, gephyrin and N-maleimide-sensitive factor
(NSF)

vi
TABLE OF CONTENTS
LIST OF FIGURES......................................................................................... ix
LIST OF TABLES........................................................................................... xi
ABBREVIATIONS ......................................................................................... xii
ACKNOWLEDGEMENTS .............................................................................. xiv
CHAPTER 1. INTRODUCTION..................................................................... 1 1.1 Structure and Molecular Diversity of GABAA Receptors............... 1 1.2 Receptor Assembly ...................................................................... 4 1.3 Membrane localization of GABAA receptors ................................. 5 1.4 The γ2 subunit for receptor localization and efficacy .................... 6 1.5 Distribution and Function of GABAA Receptors ............................ 8 1.6 Synaptic verses extrasynaptic receptors and their function.......... 10 1.7 GABAA Receptors and their effect on brain function .................... 12 1.8 Postsynaptic proteins localized at GABAergic synapses.............. 13 1.8.1 Gephyrin ......................................................................... 13 1.8.2 Structure of Gephyrin...................................................... 15 1.8.3 Gephyrin-interacting proteins.......................................... 18 1.8.4 Dystrophin-Glycoprotein complex ................................... 20 1.9 Regulation and Modulation of GABAA Receptors at synapses ..... 21 1.9.1 Proteins associated with GABAA receptors..................... 21 1.10 Lateral diffusion of GABAA receptors on plasma membrane ...... 24 1.11 Endocytosis of GABAA receptors................................................ 24 1.12 Modulation of GABAA Receptors ................................................ 26 1.13 Aim of Study ................................................................................ 28 CHAPTER 2. MATERIALS AND METHODS ................................................. 30
2.1 Mouse lines utilized ...................................................................... 30 2.2 Sequencing .................................................................................. 30 2.3 Preparation of plasmids................................................................ 30
2.3.1 Generation of chimeric plasmid constructs ..................... 31 2.3.2 Generation of cysteine mutant constructs....................... 32 2.3.3 Generation of TAC/IL-2 constructs ................................. 35 2.3.4 Generation of GST-fusion constructs.............................. 36 2.3.5 Generation of GFP-GABARAP-L1 fusion constructs ...... 37
2.4 Growth of GST fusion proteins ..................................................... 38 2.5 Protein extracts ............................................................................ 38 2.6 Western blot analyses .................................................................. 39 2.7 Tissue culture and transfection..................................................... 40
2.7.1 Neuronal tissue culture .................................................. 41 2.7.2 Neuronal transfections for immunohistochemistry .......... 43

vii
2.8 Immunofluorescence analyses ..................................................... 44 2.9 Quantitation of immunofluorescent staining.................................. 47 2.10 Electrophysiology ....................................................................... 49 2.11 Brain extracts ............................................................................. 51 2.12 Dialysis of brain membrane extracts .......................................... 52
2.13 GST pull down assays................................................................ 52 2.14 Generation of antisera ............................................................... 53 2.15 Analysis of Antisera.................................................................... 55 CHAPTER 3. RESULTS I: DISTINCT γ2 SUBUNIT DOMAINS MEDIATE
CLUSTERING AND SYNAPTIC FUNCTION OF POSTSYNAPTIC GABAA RECEPTORS AND GEPHYRIN..... 56
3.1 Aim of Study ................................................................................. 56 3.2 Results .......................................................................................... 56 3.2.1 Generation and transfection of GFP-γ2........................... 56 3.2.2 Surface localization of GFP-γ2 subunit ........................... 60 3.2.3 Design and functional characterization of chimeric constructs ................................................................................. 62 3.2.4 Cellular distribution of chimeric subunits......................... 63 3.2.5 Receptor domains required for postsynaptic localization 68 3.2.6 Recruitment of gephyrin to GABAA receptors ................. 73 3.2.7 Assessment of inhibitory synaptic clustering................... 76 3.2.8 Rescue of inhibitory synaptic function............................. 77 CHAPTER 4. RESULTS II: GEPHYRIN, COLLYBISTIN AND THEIR EFFECT
ON PROPER LOCALIZATION................................................ 81 4.1 Aim of Study ................................................................................. 81 4.2 Results ......................................................................................... 81 4.2.1 Functional analyses of collybistin isoforms ..................... 82 4.2.2 Collybistin mutation underlying hyperekplexia ................ 86 4.2.3 Functional Analysis of CB3SH3+G55A mutation ............... 87 4.2.4 Gephyrin domains required for collybistin interaction ..... 91 CHAPTER 5. RESULTS III: UNPUBLISHED RESULTS............................... 95 5.1 Aim 1 ............................................................................................. 95 5.2 Results Aim 1 ............................................................................... 95 5.2.1 Generation and cellular localization of chimeric constructs in heterologous cells...................................... 95 5.2.2 Localization of chimeras in neurons................................ 100 5.3 Aim 2 ............................................................................................ 102 5.4 Results Aim 2 ............................................................................... 102 5.5 Aim 3 ............................................................................................ 107 5.6 Results .......................................................................................... 107 5.6.1 Antibody generation for GABARAP and homologs......... 107 5.6.2 Redundant interactions of GABARAP and homologs ..... 108

viii
5.6.3 Subcellular localization of GABARAP-L1........................ 110 5.6.4 GABARAP-L1 localization in neurons............................. 112 CHAPTER 6. DISCUSSION........................................................................... 114 6.1 GABAA receptor domains required for postsynaptic localization .. 115 6.2 IL-2 α subunit linked to γ2 and α2 sequences for membrane
localization.................................................................................... 122 6.3 Collybistin as a determinant of gephyrin clustering ...................... 123 6.3.1 Deletions of collybistin domains...................................... 124 6.3.2 G55A mutation and consequences................................. 125 6.4 Gephyrin domain for collybistin interaction ................................... 126 6.5 Mutation of cysteine residues within γ2 subunit ............................ 126 6.6 GABA receptor associated protein L1 .......................................... 128 6.7 Outlook......................................................................................... 130 6.7.1 Long-term application of this research............................ 132 BIBLIOGRAPHY ............................................................................................ 136 APPENDIX: LIST OF PUBLICATIONS ......................................................... 156

ix
LIST OF FIGURES
Figure 1.1 Schematic representation of GABAA receptor................ 3 Figure 1.2 Schematic representation of the postsynaptic scaffold
at a GABAergic synapse................................................ 17 Figure 1.3 Membrane localization of GABAA receptors................... 25 Figure 2.1 Sequence comparison of GABAA receptor subunits ...... 33 Figure 3.1 Restoration of postsynaptic GABAA receptors and
gephyrin clusters in γ2-/- neurons by transfection of GFP-tagged γ2 subunit .................................................. 59
Figure 3.2 Surface expression of GFP-γ2 and 9E10γ2 comparison in 293T cells................................................................... 61
Figure 3.3 Schematic representation of chimeric subunit constructs and analysis of their expression following transfection into 293T cells................................................................ 64
Figure 3.4 Analysis of surface expression of chimeric subunit constructs transfected into 293T cells............................ 67
Figure 3.5 GABA dose-response curves of GABAA receptors containing chimeric subunits expressed in 293T cells ... 67
Figure 3.6 Restoration of postsynaptic GABAA receptor clusters in γ2-/- neurons transfected with chimeric γ2/α2 subunit constructs....................................................................... 70
Figure 3.7 Quantitative analyses of postsynaptic clusters formed by chimeric constructs transfected into γ2-/- neurons...... 72 Figure 3.8 Recruitment of gephyrin to GABAA receptor clusters ..... 75 Figure 3.9 Functional analyses of 9E10α−γ−α and 9E10γ−γ−α constructs in wildtype neurons. ...................................... 80 Figure 3.10 Rescue of mIPSCs in γ2-/- neurons requires both the major intracellular loop and the fourth transmembrane
domain of the γ2 subunit ................................................ 80 Figure 4.1: Schematic representation of collybistin and gephyrin
construct ........................................................................ 83 Figure 4.2 Functional collybistin is required for accumulation of
gephyrin in dendritic clusters.......................................... 85 Figure 4.3 Mutation within CB3SH3+ in patient with hyperekplexia and epilepsy................................................................... 88

x
Figure 4.4 Mutation in CB3SH3+ results in loss of synaptic GABAA receptors........................................................................ 90
Figure 4.5 Disruption of the collybistin binding site on gephyrin prevents accumulation of gephyrin at postsynaptic sites ............................................................................... 94
Figure 5.21.1 Schematic representation of IL-2 α fusion constructs .... 97 Figure 5.2.2 Differential surface targeting in the presence of GABAA
receptor subunits ........................................................... 97 Figure 5.2.3 Differential surface targeting of IL-2/γ2 and IL-2/α2γ2 fusion constructs ............................................................ 99 Figure 5.2.4 IL-2 fusion constructs do not localize to synapse in
neurons.......................................................................... 101 Figure 5.4.1 Schematic representation of cysteine substituted γ2 subunit constructs .......................................................... 103 Figure 5.4.2 Localization of cysteine mutants in γ2-/- neurons............ 104 Figure 5.4.3 Localization of cysteine mutants in wild-type neurons ... 106 Figure 5.6.1 Antisera specificity for GABARAP, GABARAP-L1 and
GATE-16........................................................................ 109 Figure 5.6.2 Protein-protein interactions of GABARAP family
members........................................................................ 111 Figure 5.6.3 Subcellular localization of GABARAP-L1 in HEK 293T
cells................................................................................ 113 Figure 5.6.4 GFP-GABARAP-L1 localization in neurons.................... 113 Figure 6.1 Sequence alignments of nACh and GABAA receptor TM4 domains ................................................................. 118 Figure 6.2 Structural features of the γ2 TM4 domain ...................... 118

xi
LIST OF TABLES
Table 2.1 Primers Utilized for Generation of Chimeric Plasmid Constructs.......................................................................... 34
Table 2.2 Primers for Cysteine Mutants ............................................. 35 Table 2.3 Rabbits used for Immunization Protocol ............................. 54 Table 2.4 Immunization Protocol for GATE-16 ................................... 54 Table 2.5 Immunization Protocol for GABARAP and GABARAP-L1 .. 55

xii
ABBREVIATIONS
5-HT3 5-hydroxytrytamine 3 ACh acetylecholine AP2 clathrin adaptor protein 2 APV 2-amino-5-phosphonovaleric acid BSA bovine serum albumin BZ benzodiazepine CaMKII calcium calmodulin-dependent protein kinase II cDNA complementary DNA CNS central nervous system CNQX 6-cyano-7-nitroquinoxaline-2,3-dione Cy3 carboxymethylindocyanine DIV days in vitro DLC1/2 dynein light chains 1 and 2 DH Dbl homology domain DMEM Dulbecco’s modified Eagle’s medium DPC dystrophin-associated protein complex DTT 1, 4 dithio-DL-threitol ECL enhanced chemiluminescence EDTA ethylenediaminetetraacetic acid EGFP enhanced green fluorescent protein FBS fetal bovine serum GA glutathione agarose GABA γ-aminobutyric acid GABARAP GABAA receptor-associated protein GAD glutamic acid decarboxylase GATE-16 Golgi-associated ATPase enhancer protein-16 kDa GEF guanine nucleotide exchange factor GFP green fluorescent protein GlyR glycine receptor GODZ Golgi-specific DHHC zinc finger domain protein GST glutathione S-transferase HBSS Hank’s buffered salt solution HEPES 4-(2-hydroxyethyl)-piperazine-1-ethane sulfonic acid HRP horseradish peroxidase IL-2 interleukin-2 alpha subunit IPTG Isopropyl-Beta-d-Thiogalactopyranoside IR immunoreactivity KCC2 potassium/chloride cotransporter 2 LC3 light chain 3 MEM modified Eagle’s medium Mena mammalian enabled mIPSC miniature inhibitory postsynaptic current mRNA messenger ribonucleic acid

xiii
MW molecular weight NA numerical aperature nAChR nicotinic acetylcholine receptor NB-A Neurobasal A medium NMDA N-methyl D-aspartate NSF N-ethylmaleimide-sensitive factor NT neurotransmitter PAGE polyacrylamide gel electrophoresis PBS phosphate buffered saline PCR polymerase chain reaction PH plextrin homology PKA protein kinase A PKC protein kinase C PMFS Phenylmethanesulfonyl Fluoride PRIP1/2 phospholipase C-related catalytically inactive protein 1 and 2 RT room temperature SEM standard error of mean SDS sodium dodecyl sulphate SH3 src Homology domain 3 SynGAP synaptic GTPase activating protein TM transmembrane domain Tris tris(hydroxymethyl) aminoethane TTX tetrodotoxin VASP vasodilator stimulated phosphoprotein VIAAT vesicular inhibitory amino acid transporter WT wildtype

xiv
ACKNOWLEDGEMENTS
I would like to give my thanks and appreciation to the following people:
First, I would like to express my gratitude to my thesis advisor, Dr. Bernhard Lüscher. Without his support, guidance and vast scientific knowledge that he has shared with me, I would never have been able to complete this work. I would also like to gratefully acknowledge Dr. Cheryl Keller, who has given me both scientific advice and encouragement all along the way. Additionally, I thank Michelle Martin and Sue Lingenfelter for all of their help with tissue culture and their willingness to lend an ear when things got rough. I would like to thank Kristin Harvey, Robert Harvey, Sep Mulder-Rosi and Gong Chen for their collaborations. I would like to thank Rob Lyon for help with the GATE-16 antisera, Jodi Stewart for help in generating the GST fusion constructs and Laura Snyder for her work with the IL-2 constructs. In addition, I would like to give special thanks to my family and friends for all of their support during my graduate career Finally, my lab mates, Cheryl Keller, Claude Schweizer, Michelle Martin, Scott DiLoreto, Clint Earnheart, Sue Lingenfelter, Cheng Fang, Xu Yuan, Shoko Masuda and Hal Wrigley, deserve thanks for their help and support along the way,
Figures 3.1, 3.3-3.10 and 6.1 are reproduced from The Journal of Neuroscience, 2005, vol.25 (3), by copyright permission from The Journal of Neuroscience. The complete citation is: Alldred, M.J, J. Mulder-Rosi, S.E. Lingenfelter, G. Chen, B. Lüscher (2005). Distinct γ2 Subunit Domains Mediate Clustering and Synaptic Function of Postsynaptic GABAA Receptors and Gephyrin. J. Neurosci. 25(3): 594-603.
Figures 4.1-4.4 are reproduced from The Journal of Neuroscience,
2004, vol.24 (25), by copyright permission from The Journal of Neuroscience. The complete citation is: Harvey, K., I.C. Dunguid, M.J. Alldred, S.E. Beatty, H. Ward, N.H. Keep, S.E. Lingenfelter, B.R. Pearce, J. Lundgren, M.J. Owens, T.G. Smart, B. Lüscher, M.I. Rees, R.J. Harvey (2004). The GDP-GTP Exchange Factor Collybistin: An Essential Determinant of Neuronal Gephyrin Clustering. J. Neurosci. 24(25): 5816-5826.

CHAPTER 1
INTRODUCTION
The A-type γ-aminobutyric acid (GABAA) receptors are heteropentameric
ligand-gated chloride channels that mediate the majority of fast inhibitory
neurotransmission in the brain. These receptors bind the neurotransmitter GABA,
which is released from GABAergic terminals. For efficient synaptic transmission,
GABAA receptors need to be localized at postsynaptic sites apposed to these
terminals. Modulation of the expression, cellular distribution and function of
GABAA receptors has profound effects on GABAergic transmission and neural
excitability. Therefore, it is important to understand the mechanisms that
modulate the concentration and function of these receptors at synapses.
1.1 Structure and Molecular Diversity of GABAA Receptors
GABAA receptors are members of the superfamily of ligand-gated ion
channels, which also includes the nicotinic acetylcholine (nACh), glycine, and 5-
hydroxytrytamine 3 (5-HT3) receptors (Lynch, 2004). These receptors share the
same basic structural features, consisting of five membrane-spanning subunits
arranged around a central ion-conducting pore (Fig. 1.1 A) (Nayeem et al., 1994).
Each subunit is composed of a large extracellular N-terminal domain, followed by
four transmembrane (TM) domains, with a large cytoplasmic loop between TM3
and TM4, and a short extracellular C-terminal domain (Fig. 1.1 B). Hydropathy

2
plotting predicts an α helical arrangement for all four transmembrane domains,
which has been confirmed by crystal structure analyses of the nACh receptor
(Miyazawa et al., 1999; Miyazawa et al., 2003). The intracellular loop region of
these receptor subunits is a poorly conserved domain that often contains multiple
binding sites for putative trafficking and synaptic scaffolding proteins and
phosphorylation sites for serine/threonine and tyrosine kinases. The C-terminal
region faces the extracellular space, but in most cases is predicted to barely
extrude from the membrane (reviewed by Luscher and Keller, 2004).
In mammals, GABAA receptor subunits are encoded by 19 known subunit
genes and, based on homology, the corresponding subunits can be grouped into
eight distinct subunit classes (α 1−6, β 1−3, γ 1−3, δ ,π, θ, ρ 1−3, ε) (Simon et al.,
2004). Additionally, some subunits exist as alternately spliced variants, such as
the γ2S and γ2L subunits, which differ by eight amino acids within the
cytoplasmic loop region. A 70-80% homology is present within each subunit class
and this identity decreases to approximately 30-40% between subunit classes
(reviewed in Macdonald and Olsen, 1994). As expected, the transmembrane
domains display the largest degree of homology, whereas the extracellular and
intracellular regions are more divergent. While genes encoding the
α, β, γ, δ, σ, θ, ε and π subunits are principally expressed in the brain, the
expression of genes encoding the ρ1-3 subunits is largely limited to the retina.
When expressed in heterologous cells, the ρ subunits form homomeric receptors

3
Figure 1.1: Schematic representation of a GABAA receptor and an individual subunit. A. GABAA receptors represent heteropentameric ion channels and are commonly composed of two α, two β and one γ2 subunit. Upon binding of GABA, the channel opens allowing for the influx Cl- ions into the cell. B. Each subunit of GABAA receptors shares the same structural topology, with a large N-terminal extracellular domain, four transmembrane domains, a short C-terminal extracellular tail and a large intracellular domain between transmembrane domains three and four.

4
with a distinct pharmacology, which is why ρ subunit-containing receptors are
often referred to as GABAC receptors (Bormann and Melzig, 2000). They are not
discussed further here.
1.2 Receptor Assembly
The large number of GABAA receptor subunits likely gives rise to a much
larger number of pentameric receptor subtypes, many as of yet undefined.
However, the number of GABAA receptor subtypes expressed in brain is limited
by the expression patterns of the subunits, which are regulated both spatially and
temporally in the brain (reviewed by Fritschy and Mohler, 1995), and by rules that
govern subunit assembly into functional receptors (reviewed by Barnes, 2001;
Kittler et al., 2002; Luscher and Keller 2004). While studies in non-neuronal cell
types have shown that α and β subunits are able to form functional receptors on
the membrane surface that are modulated by barbiturates and steroids,
coexpression of γ 1−3, δ, ε, π, or θ subunits is required to form receptors that
mimic the electrophysiological and pharmacological parameters of native
receptors (reviewed by Whiting et al., 1999). The current established consensus,
based on diverse experimental approaches, indicates that the majority of GABAA
receptors are composed of two α, two β and one γ2 subunit (Chang et al., 1996;
Tretter et al., 1997; Farrar et al., 1999; Knight et al., 2000; Baumann et al., 2001).
The γ1 and γ3 subunits, while functionally similar to the γ2 subunit, are thought to
be part of minor populations of GABAA receptors in restricted brain regions

5
(Laurie et al., 1992; Wisden et al., 1992). Similarly, the δ, ε, and θ subunits
exhibit a highly restricted expression pattern in the brain and likely form minor
receptor subtypes (Laurie et al., 1992; Wisden et al., 1992; Sieghart et al., 1999;
Steiger and Russek, 2004).
1.3 Membrane localization of GABAA receptors
While the majority of GABAA receptors found in the brain contain 2α, 2β
and the γ2 subunit, studies in heterologous cells indicate GABAA receptors can
localize to the membrane surface with less stringent criteria. Analyses of GABAA
receptor subtype surface expression have been influential in determining the
minimal subunits required for membrane localization. Heterologous expression
of α, β and γ subunits has shown that both αβ and αβγ subunit combinations can
produce functional receptors that are expressed at the cell surface (Macdonald
and Olsen, 1994; Connolly et al., 1999a). While none of the α subunits alone
have proven to localize to the membrane surface as homomeric receptors
(Connolly et al., 1996b; Connolly et al., 1996a), homomeric β3 and β1 receptors
can localize to the membrane surface (Connolly et al., 1996a; Krishek et al.,
1996; Wooltorton et al., 1997). By comparison, the γ2S subunit appears to be
able to localize to the cell surface in the absence of additional subunits in
heterologous cells, although electrophysiological and sucrose gradient studies
indicate the γ2S subunit cannot form functional homomeric receptors (Connolly et
al., 1996a; Connolly et al., 1999a). Thus far, single subunit expression of the β2,

6
γ1, γ2L or γ3 subunit does not result in surface expression in heterologous cells
(Connolly et al., 1996b; Connolly et al., 1996a; Connolly et al., 1999a). While
homomeric receptors can apparently localize to the membrane surface in
heterologous cells, (Kittler et al., 2000) there is no evidence for the existence of
homomeric GABAA receptors in vivo and overexpression studies have not looked
at homomeric subunit expression in subunit-deficient transgenic mice, likely
indicating that the ability to localize to the membrane surface is not the only
criteria necessary in vivo for GABAA receptor expression.
1.4 The γ2 subunit for receptor localization and efficacy
The γ2 subunit of GABAA receptors has generated much excitement in the
last decade, as more has been discovered about the unique properties of γ2
subunit-containing GABAA receptors. Mice that had the γ2 subunit gene knocked
out mice were originally generated to investigate the role of benzodiazepine
binding in the brain (Günther et al., 1995). While this study did show that
ablation of the γ2 subunit reduced the binding of flumazenil (a benzodiazepine)
by 94 %, with only a small decrease (22%) of total GABAA receptors, the loss of
the γ2 subunit resulted in an early postnatal lethal phenotype with a maximal life
expectancy of 18 days. Although, immunohistochemical studies indicated that the
γ2 subunit was not required for expression, trafficking to the membrane or
surface localization of GABAA receptors (Günther et al., 1995), further studies
demonstrated that the loss of the γ2 subunit resulted in the absence of

7
postsynaptic clustering of GABAA receptors (Essrich et al., 1998). Consistent
with these findings, γ2 subunit-deficient neurons showed an almost complete lack
of GABAA receptor-mediated miniature inhibitory postsynaptic currents (mIPSCs)
(Essrich et al., 1998; Alldred et al., 2005).
While the γ2 and γ3 subunits share 64% identity at the amino acid level,
unlike the γ2 subunit, the γ3 subunit has very restricted expression in the adult
brain. Overexpression of the γ3 subunit in γ2 subunit-deficient neurons showed
that the γ3 subunit could partly restore postsynaptic localization of GABAA
receptors, even in regions of the brain where the endogenous γ3 subunit is not
normally expressed (Baer et al., 1999). Additionally, the γ3 subunit was able to
restore mIPSCs in these neurons to levels comparable to receptors containing
the γ2 subunit (Baer et al., 1999). However, overexpression of the γ3 subunit
could not rescue the lethal phenotype seen in γ2 subunit-deficient mice, possibly
due to insufficient expression of the transgene or functional differences between
the two types of subunits. Overexpression studies utilizing either the γ2S or γ2L
splice variant alone in the γ2-/- mice showed a complete rescue of the γ2-/- lethal
phenotype (Baer et al., 2000). This study indicated that either splice variant is
sufficient to fulfill the fundamental functions essential for postnatal life (Baer et
al., 2000). Homanics et al. (1999) confirmed the ability of the γ2S subunit to
replicate all functions fundamental for postnatal life by the generation of knock
out mice lacking the γ2L-specifici mini exon. In addition to the molecular and
pharmacological deficits seen in the γ2 subunit deficient mice (Günther et al.,

8
1995; Essrich et al., 1998), behavioral phenotypes due to the underlying the
molecular deficits were studied in heterozygous γ2 mutant mice (Crestani et al.,
1999). These mice showed a reduction in postsynaptic GABAA receptors most
notably in the hippocampus, cortex and dentate gyrus, which correlated with a
mixture of single channel conductance levels corresponding to those found in
recombinant receptors composed of αβ or αβγ2 subunits. This molecular
phenotype was manifested behaviorally in a trait anxiety-like phenotype (Crestani
et al., 1999),
The above-mentioned studies demonstrated a critically important role of
the γ2 subunit for the development of GABAergic synapses. Further analyses of
the γ2 subunit in functionally mature neurons using conditional knockout mice
(Schweizer et al., 2003) revealed that the γ2 subunit is not only critical for
localization of postsynaptic GABAA receptors during initial formation of synapses
in developing neurons, but also for the maintenance of GABAA receptors at
mature synapses.
1.5 Distribution and Function of GABAA Receptors
The binding of GABA to GABAA receptors can induce both tonic and
phasic inhibition in the mammalian brain (Mody et al., 1994). GABAA receptors
mediate ‘fast’ synaptic transmission through the release of GABA from the
synaptic bouton of an activated cell. In the presynaptic terminal, the
neurotransmitter (NT) is packaged into synaptic vesicles and the vesicles dock at
the presynaptic membrane (reviewed by Lin and Scheller, 2000). Upon

9
stimulation of the presynaptic cell, Ca2+ enters the synaptic terminal thereby
triggering the fusion of docked NT-containing vesicles with the terminal
membrane and the release of NT into the synaptic cleft (reviewed by Zucker,
1996; Chen et al., 2001; Rizo and Sudhof, 2002). The neurotransmitter then
binds to receptors localized in the postsynaptic membrane, which then triggers
the opening of the receptor-intrinsic ion channel and the influx of Cl- ions into the
cell, thus hyperpolarizing the postsynaptic cell (Rabow et al., 1995). The number
of postsynaptic receptors determines the amplitude of the postsynaptic current
(Kittler et al., 2000). Hyperpolarization of the membrane reduces the likelihood
that nearby depolarizing inputs to the same cell, generated by excitatory receptor
activation, can reach threshold for activation of voltage-gated Na+ channels
required for the generation of an action potential. Alternatively, GABAA receptors
can increase the membrane conductance and thereby directly interfere with
glutamate induced depolarization (reviewed by Luscher and Keller, 2004).
In immature neurons, the chloride equilibrium potential tends to be more
positive than the resting membrane potential and, in this situation, activation of
GABAA receptors results in efflux of Cl-, causing depolarization (reviewed by
Ben-Ari, 2002). GABAA receptor-mediated membrane depolarization is thought to
activate voltage-gated Ca2+ channels, which in turn is thought to deliver the signal
for induction of transcription for the KKC2 cotransporter (Ganguly et al., 2001;
Hubner et al., 2001; Nabekura et al., 2002). This activation of the KKC2
cotransporter is followed by a switch in the chloride equilibrium potential during
maturation of the neurons (Plotkin et al., 1997; Kakazu et al., 1999; Rivera et al.,

10
1999). To date, all evidence suggests that activation of GABAA receptors and
presynaptic inputs are not required for the development of inhibitory synapses
(Rao et al., 2000; Verhage et al., 2000; Gally and Bessereau, 2003).
In non-glutamatergic autaptic neurons, GABAA receptors mislocalize
apposed to glutamatergic presynaptic terminals, indicating GABAA receptor
function is not required for receptor clustering and that glutamatergic terminals
release a signal required for postsynaptic differentiation similar to the signal
released form GABAergic terminals (Rao et al., 2000; Brunig et al., 2002; Christie
et al., 2002). Additionally, mice with targeted deletions in the synaptic vesicle
machinery are able to develop synapses that are morphologically normal
(Verhage et al., 2000; Varoqueaux et al., 2002), indicating that functional GABAA
receptors are not required for development of inhibitory synapses. This
observation is in stark contrast to findings with glycinergic synapses, where
glycine-receptor mediated depolarization is required for targeting of glycine
receptors to the developing inhibitory synapse (Kirsch and Betz, 1998; Levi et al.,
1998).
1.6 Synaptic verses extrasynaptic receptors and their function
GABAA receptor subunits have specific subcellular localization patterns
and this is essential for normal brain function. Differential expression of these
subunits within neurons can modify the current seen by the postsynaptic cell.
Critically important for this modulation is the γ2 subunit, which is required for
synaptic localization of GABAA receptors. Loss of the γ2 subunit, seen in γ2

11
subunit-deficient neurons, showed drastically reduced postsynaptic clustering for
both the α1 and α2 subunits, as seen by the loss of punctate immunoreactivity
(IR) for these subunits in the cortex and cerebellum (Essrich et al., 1998). In
contrast, in the peripheral nervous system, immunohistochemical analysis of
cellular localization of the β2/3 subunit on dorsal root ganglion neurons, which
lack synaptic membrane specializations, appeared normal (Günther et al., 1995),
indicating the γ2 subunit is specifically required for synaptic localization of GABAA
receptors in neurons, but not for membrane localization of GABAA receptors.
Although the γ2 subunit is essential for postsynaptic clustering of GABAA
receptors, the γ2 subunit containing GABAA receptors can also localize
extrasynaptically, seen by diffuse distribution in somato-dendritic membranes
(Fritschy et al., 1998). GABAA receptor subtypes that incorporate the α4, α6 or
the δ subunit are exclusively extrasynaptic, where they mediate tonic inhibition
(Fritschy et al., 2003; Luscher and Keller, 2004; Petrini et al., 2004; Sun et al.,
2004; Mangan et al., 2005). These extrasynaptic receptors are believed to
mediate the tonic inhibition in response to low levels of ambient GABA. The δ
and γ2 subunits appear to compete for assembly with the same α and β subunits,
indicating that their relative expression levels can affect the ratio of synaptic
verses extrasynaptic receptors (Tretter et al., 2001; Peng et al., 2002; Mangan et
al., 2005).

12
1.7 GABAA Receptors and their effect on brain function
The binding of different drug classes to the receptors can modulate
GABAA receptor efficacy. Two types of endogenous modulators are
benzodiazepines, which occur naturally in mammals, and neurosteroids, which
are progesterone-derived neuromodulators. In addition, exogenous compounds
such as benzodiazepines, barbiturates, neurosteroids, volatile anesthetics and
ethanol can be utilized as modulators of GABAA receptor function. The clinical
effects of barbiturates were discovered in the early 1900’s and was quickly
followed by the discovery of major side effects associated with clinical use, such
as toxicity and dependence (Nemeroff, 2003). Due to these complications, this
drug class is rarely used today as a modulator of GABAA receptor function.
Benzodiazepines have replaced barbiturates and can be used as anxiolytics,
sedatives, muscle relaxants and antiepileptics. This drug class binds to the
extracellular region at the α and γ interface. Benzodiazepines bind at modulatory
sites different from GABA, however, this binding can alter the efficacy of GABA
binding (reviewed by Sigel and Buhr, 1997). Modulation by neurosteroids has
been documented from the 1940s, however, much is still unknown about this
drug class. While the effect of neurosteroids on GABAA receptor modulation has
been extensively documented, the GABAA receptor binding pocket for the
steroids is still unknown (Lambert et al., 2003).
In addition to drug interactions altering GABAA receptor function,
mutations within the subunits can result in modifications of the GABAA receptor
conductance, assembly, expression, stability, trafficking or localization, any of

13
which can have drastic effects on normal brain function. Several studies have
looked at the physical manifestation of disease states based on mutations in
GABAA receptor subunits. Different mutations within the gene encoding the β3
subunit have been linked to Angelman Syndrome and autism (Nurmi et al., 2001;
Silva et al., 2002). The γ2 subunit gene has mutations associated with familial
idiopathic epilepsies (Cohen et al., 2002; Sancar and Czajkowski, 2004) and
mutations within the α5 subunit gene have been linked with bipolar disorder
(Papadimitriou et al., 2001). In addition to these mutations or deficits that result in
mental disorders, there have also been several studies on the outcome of
modulation of GABAA receptor function in rodent model systems, including effects
on anxiety disorders such as post-traumatic stress disorder (Anagnostaras et al.,
1999), schizophrenia (Wassef et al., 2003) and on alcohol dependence (Mehta
and Ticku, 2005). The association of GABAA receptor defects with such
disorders makes it critically important to understand the mechanisms that
modulate receptor expression, function and correct cellular localization.
1.8 Postsynaptic proteins localized at GABAergic synapses
1.8.1 Gephyrin
Efficient synaptic transmission requires the postsynaptic localization of
neurotransmitter receptors. However, the mechanism for localization and
stabilization of GABAA receptors at postsynaptic sites is poorly understood.
Several studies suggest that gephyrin is required for the postsynaptic localization
of at least a subset of GABAA receptors (Fig. 1.2) (Essrich et al., 1998; Kneussel

14
et al., 1999; Levi et al., 2004). Gephyrin is a 93 kDa tubulin-binding protein that
was originally discovered as a component of affinity purified glycine receptor
(GlyR) complexes (Kirsch et al., 1993). Gephyrin anchors GlyRs at postsynaptic
membranes by direct interaction with a 20 amino acid stretch in the intracellular
loop of the GlyRβ subunit (Meyer et al., 1995). Additionally, gephyrin is known to
bind polymerized tubulin and is essential for postsynaptic clustering of GlyRs in
spinal cord, retinal and hippocampal neurons (Prior et al., 1992; Kirsch et al.,
1993; Todd et al., 1996; Zucker, 1998; Meier et al., 2000; Levi et al., 2004).
Mice lacking the γ2 subunit and subsequently, postsynaptic GABAA
receptors revealed prominent loss of postsynaptic gephyrin as a direct
consequence of the γ2 gene deletion (Essrich et al., 1998). Gephyrin antisense
treatment of wild type hippocampal neurons displayed a simultaneous reduction
of gephyrin and GABAA receptor punctate staining. These results demonstrated
that gephyrin was not only required for clustering of GlyRs, but also of GABAA
receptors. The essential role gephyrin plays in GABAA receptor clustering at
postsynaptic sites was confirmed by analysis of gephyrin knock out (geph-/-) mice
(Kneussel et al., 1999). However, while a significant reduction in the number of
α2, α3 and γ2 subunit-containing GABAA receptors was seen in the geph-/- mice
(Fischer et al., 2000; Kneussel et al., 2001), Kneussel et al., (2001) showed no
differences in the number of α1 and α5 subunit containing GABAA receptors. A
recent study by Levi et al., (2004) investigated further the localization of GABAA
receptors in geph-/- mice and showed that a small percentage of α2- or γ2-

15
containing receptors can localize to the synapse. These studies suggest that
GABAA receptors are able to localize to synapses in a manner independent of
gephyrin, but that gephyrin might be required for aggregation and possibly
stabilization of GABAA receptors and other postsynaptic components at the
synaptic membrane (Kneussel et al., 2001; Levi et al., 2004).
1.8.2 Structure of Gephyrin
Gephyrin is widely expressed in the rat and human brain (Prior et al.,
1992; Kirsch et al., 1993; Rees et al., 2003). The 93 kDa isoform of gephyrin
contains an N-terminal domain closely homologous to MogA and a C-terminal
domain closely related to MoeA, two bacterial proteins involved in molybdenum
cofactor biosynthesis (Feng et al., 1998). The linker region between these two
domains contains a stretch of 14 amino acids implicated in binding of gephyrin to
microtubules (Ramming et al., 2000). Gephyrin is thought to form a hexagonal
lattice through a trimeric interaction at the N-terminal domain and a dimer at the
C-terminus. X-ray crystal structure analysis of the N-terminal domain indicates
that a parallel beta pleated sheet with 6 beta strands is surrounded by 7-8 alpha
helices (Schwarz et al., 2001; Sola et al., 2001). The trimeric interface contains 3
points of interaction and different splice variants can produce three variants of
this N-terminal domain, which could modify these sites of interaction. The C-
terminal region of gephyrin including the MoeA domain, crystallized as four
domains, with a dimer forming in solution. The I, III and IV domains are required
for dimerization to occur (Xiang et al., 2001). Xiang et al., also give a convincing

16
model for how these interactions at the N- and C-terminal regions of gephyrin
might contribute to a hexagonal lattice structure. This hexagonal lattice is
believed to serve as a subsynaptic scaffold with which receptors and other
postsynaptic proteins could associate.
Gephyrin has a variety of functions in both neuronal tissues, as discussed
here, as well as in non-neuronal tissues. This functional diversity has generated
speculation about the molecular basis of this array of functions. Several studies
have revealed multiple alternative splice variants of gephyrin that could
potentially give rise to functionally distinct isoforms. Initial analysis of rat brain
transcripts identified four N-terminal splicing cassettes (Prior et al., 1992). A
more comprehensive analysis revealed that the gephyrin gene comprises at least
27 exons in the human gene (Rees et al., 2003) and 29 exons in mouse
(Ramming et al., 2000). Alternative splicing of five of these exons gives rise to 11
distinct gephyrin isoforms (Ramming et al., 2000; Rees et al., 2003).
Furthermore, alternatively spliced isoforms of gephyrin may underlie differential
recruitment of GABAA and glycine receptors to different types of inhibitory
synapses (Meier and Grantyn, 2004).

17
Figure 1.2 Schematic representation of the postsynaptic scaffold at a GABAergic
synapse. Localization of GABAA receptors to postsynaptic sites requires the γ2 subunit. Gephyrin is proposed to form a hexagonal lattice structure, where it is able to bind many additional subsynaptic proteins such as Dlc1/2, profilin, and Mena/VASP, which are thought to link gephyrin to cytoskeletal microtubules and microfilaments. Collybistin is proposed to traffic gephyrin and associated proteins via the exchange of GDP-GTP on Cdc42 and actin cytoskeletal rearrangement. In addition, the dystrophin-dystroglycan protein complex, which includes α-dystroglycan, β-dystroglycan, dystrophin and syntrophin localizes to a subset of GABAergic postsynaptic sites. Finally, neuroligin-2 has recently been shown to localize to postsynaptic GABAA receptor sites and interact with neurexin on the presynaptic terminal. While these proteins form a complex network at inhibitory synapses, to date, none have been shown to directly interact with any GABAA receptor subunits. Thus, the mechanism for targeting and maintenance of GABAA receptors at the synapse is still unclear.

18
1.8.3 Gephyrin-interacting proteins
Gephyrin interacts directly with a number of cytoskeletal and cytoplasmic
proteins, including tubulin (Prior et al., 1992), profilin (Mammoto et al., 1998),
dynein light chains 1 and 2 (Dlc1/2) (Fuhrmann et al., 2002), Mena/VASP
(Giesemann et al., 2003) and collybistin (Kins et al., 2000). Tubulin, profilin and
Mena/VASP have all been associated with microtubule/microfilament systems,
suggesting a role for gephyrin in the anchoring of postsynaptic proteins with the
cytoskeleton. Dlc1/2 have been described as light chains of cytoplasmic dynein,
which is a large protein complex that mediates the movement of proteins along
microtubule tracts. The interaction between Dlc1/2 and gephyrin was confirmed
further by colocalization assays in neurons, showing an enrichment of Dlc1/2 at
inhibitory synapses, suggesting a role for these proteins in the subcellular
localization of gephyrin (Fig. 1.2) (Fuhrmann et al., 2002).
Collybistin is a member of the guanine nucleotide exchange factor (GEF)
family of proteins that catalyze the GTP-GDP exchange on Rho family GTPases.
Collybistin specifically activates the small GTPase Cdc42, which is known to play
a role in actin cytoskeleton reorganization. Recent studies have shown that
human and rat collybistin can exist in three isoforms, collybistin 1, collybistin 2
and collybistin 3 (CB1/2/3), that differ in their C-termini (Harvey et al., 2004).
Two of these isoforms (CB2 and CB3) can exist as two different splice variants
differing by the presence or absence of an N-terminal src homology 3 (SH3)
domain (Harvey et al., 2004). All the isoforms share the same basic structure,
with an N-terminal domain, which may include an SH3 binding region, followed

19
by a Dbl homology (DH) domain, also called the RhoGEF domain. Further
downstream, there is a plextrin homology (PH) domain, followed by variable C-
terminal region that distinguishes the three isoforms (Kins et al., 2000; Harvey et
al., 2004) . The first isoform of collybistin, CB1, exhibits very limited expression.
The most abundant isoforms of collybistin in rat and human were found to be
CB2SH3+ and CB3SH3+, indicating that regulation of collybistin activity might come
through this domain (Harvey et al., 2004). However, the CB2 isoform lacking the
SH3 domain, (CB2SH3-) has been shown to induce the formation of
submembraneous gephyrin clusters that can recruit GlyRs to the membrane in
heterologous cells (Kins et al., 2000; Harvey et al., 2004), indicating that the
gephyrin collybistin interaction with inhibitory receptors can be modified by the
SH3 domain. The interaction between collybistin and gephyrin is direct and
occurs via polar amino acid residues within the C-terminal half of the linker region
between the N-terminal domain and the DH domain of collybistin (Grosskreutz et
al., 2001). However, little is known about the collybistin-binding domain on
gephyrin. It would be interesting to determine this domain, as this interaction
could be involved in the clustering mechanism of GABAA receptors at synapses.
However, as all attempts to show interaction between GABAA receptor subunits
and gephyrin have failed, the mechanism for GABAA receptor localization to
synapses likely requires additional unknown protein interactions (reviewed by
Luscher and Fritschy, 2001).

20
1.8.4 Dystrophin-Glycoprotein complex
A second complex of proteins has been shown to colocalize specifically
with a subset of GABAergic synapses is the dystrophin-glycoprotein complex
(Fig. 1.2) (Knuesel et al., 1999). This protein complex is specifically expressed
by neurons in the neocortex, hippocampus, and cerebellum. Mice devoid of
dystrophin (mdx) exhibit altered synaptic clustering of GABAA receptors (Knuesel
et al., 1999). Recent studies have shown that the dystrophin-associated protein
complex (DPC), which includes syntrophin and β-dystroglycan, is exclusively
localized opposite GABAergic terminals (Brunig et al., 2002). In glutamatergic
neurons that lack GABAergic innervation, GABAA receptors and gephyrin are
frequently mislocalized to sites apposed to glutamate terminals. Interestingly,
dystrophin and the DPC are never mislocalized to glutamatergic sites, even in
neurons lacking GABAergic synapses. In neurons innervated by a single
GABAergic axon, dystrophin and the DPC are localized opposite GABAergic
boutons, and never seen with GABAA receptors and gephyrin clusters that are
mislocalized. In addition, dystrophin and the DPC are localized to GABAergic
sites even in γ2 subunit-deficient neurons where postsynaptic GABAA receptors
and gephyrin are largely absent (Brunig et al., 2002). However, an additional
study found that accumulation of dystrophin and the DPC at synapses is
dependent on another member of the DPC, β−dystroglycan. The β-dystroglycan
protein, but not full-length dystrophin, was found to be required for the
localization of the DPC to GABAergic postsynaptic sites. However, neither β-
dystroglycan nor dystrophin were required for GABAergic synaptic differentiation,

21
nor accumulation of GABAA receptors and gephyrin to synaptic sites (Levi et al.,
2002). This suggests that β−dystroglycan is critical for the localization of
dystrophin and the DPC to the postsynaptic membrane. The findings by both
Levi et al., (2002.) and Brünig et al., (2002) indicate that the DPC localizes to the
postsynaptic membrane by a GABAA receptor and gephyrin-independent
mechanism. The data propose the existence of presynaptic factors that
contribute to postsynaptic aggregation of dystrophin and of the DPC across from
GABAergic terminals, however, as yet none have been found.
1.9 Regulation and Modulation of GABAA Receptors at synapses
1.9.1 Proteins associated with GABAA receptors
The mechanism that underlies GABAA receptor clustering and targeting to
the postsynaptic membrane has yet to be elucidated. However, many recently
discovered proteins have been implicated in the trafficking and targeting of
GABAA receptors to the membrane and to postsynaptic specializations (Fig. 1.3).
One such protein is GABAA receptor associated protein (GABARAP), which was
shown by yeast two hybrid screening to interact with the intracellular loop of the
γ2 subunit of GABAA receptors (Wang et al., 1999). This 13.9 kDa protein has
homology to light chain 3 (LC3) of MAP 1A/B, a tubulin binding protein (Mann
and Hammarback, 1994), and to Golgi-associated ATPase enhancer protein of
16 kDa (GATE-16) (Kneussel et al., 2000; Sagiv et al., 2000). GATE-16 is a
soluble transport factor that has been shown to bind to N-ethylmaleimide-
sensitive factor (NSF), a vesicle transport factor, and vesicle-soluble NSF

22
attachment protein receptors (v-SNARES), (Sagiv et al., 2000). In vitro assays
indicate that GABARAP can bind to microtubules, NSF and gephyrin, giving
further evidence for a role in membrane trafficking of GABAA receptors. Previous
work has shown that neuronal colocalization of GABARAP with gephyrin and
GABAA receptors at inhibitory synapses was not significant (Kneussel et al.,
2000; Kittler et al., 2001) and the majority of GABARAP localized to the Golgi
complex and other vesicular bodies (Kneussel et al., 2000; Kittler et al., 2001).
However, a recent report indicated that a small percentage of GABARAP did
localize to proximal dendrite regions with γ2 subunit-containing GABAA receptors
apposed to synaptic contacts (Leil et al., 2004). This latest study indicates a
critical role for GABARAP in receptor translocalization from the Golgi to the cell
membrane (Leil et al., 2004).
In addition to GABARAP, several other proteins have been identified that
interact with GABAA receptor subunits to modify GABAA receptor localization.
The proteins discussed here are limited to those that interact directly with the γ2
subunit, and do not reflect the proteins that modify receptor localization
exclusively by interaction with α or β subunits. AP2 interacts with the intracellular
loops of β and γ subunits and has been shown to decrease surface expression of
receptors and mediate endocytosis through clathrin-coated pits (Kittler et al.,
2000; Hering et al., 2003). Recently, Golgi-specific DHHC zinc finger domain
protein (GODZ) has been shown to interact with and palmitoylate the γ2 subunit,
and is thought to regulate trafficking of γ subunit-containing GABAA receptors

23
(Keller et al., 2004). In addition, phospholipase C-related catalytically inactive
protein 1 and 2 (PRIP-1 and -2) modulate surface expression of γ2 subunit-
containing GABAA receptors by competing with GABARAP for subunit binding
(Kanematsu et al., 2002; Uji et al., 2002). In contrast to modulating surface
localization, calcineurin modulates receptor activity by binding to the γ2 subunit
after NMDA driven Ca2+ influx, which results in dephosphorylation of γ2 subunit
and induces long-term depression of GABA mediated IPSCs (Wang et al., 2003).
In the absence of PKC phosphorylation on the β2, β3, or γ2 subunits, endocytosis
of GABAA receptors is stimulated, resulting in a decrease in the number of
GABAA receptors on the membrane surface (Connolly et al., 1999b; Kittler et al.,
2000). By comparison, phosphorylation can also modulate receptor activity, for
example Src phosphorylation of the intracellular loop of the γ2 subunit results in
receptor activity modulation (Brandon et al., 2001). In addition, other proteins
can modulate surface expression without phosphorylation, insulin and brain
derived neurotrophic factor are both extracellular signals that act to increase and
decrease surface expression respectively (reviewed in Luscher and Keller,
2004). However, none of the proteins mentioned have been shown to colocalize
with GABAA receptors at the postsynaptic membrane, suggesting the γ2 subunit
interacts with additional as yet undefined proteins.

24
1.10 Lateral diffusion of GABAA receptors on plasma membrane
Accumulation of GABAA receptors at postsynaptic sites is important to
mediate effective synaptic transmission (Fig. 1.3). There are several regulatory
mechanisms that can modify receptor number at synapses, including proteins
that modify endo- and exo-cytosis rates and lateral diffusion of receptors to
synaptic sites. Lateral mobility of receptors on the plasma membrane allows for
rapid modification of synaptic signal intensity. A recent study by Jacob et al.,
(SJ. Moss, personal communication) looked at surface expression of pHluorin-
tagged GABAA receptor subunits to compare the movement of receptors within
synapses and those at extrasynaptic sites. They found that β3 and γ2 subunit-
containing receptors at extrasynaptic spaces diffuse more rapidly than those
found at synapses. Additionally, inhibition of gephyrin expression by plasmid-
based RNAi leads to a dramatic increase in dendritic mobility of GABAA
receptors. This result is consistent with previous work done on glycine receptors
in which beads were attached to receptors via antibody interactions and receptor-
bead movement was measured (Meier et al., 2001). Both these studies suggest
that synaptic efficacy can be modulated through lateral movements of GABAA
receptors.
1.11 Endocytosis of GABAA receptors
While the above study has improved the knowledge on lateral
diffusion of GABAA receptors, the mechanism is still poorly understood.
However, lateral mobility is not the only mechanism to modify synaptic efficacy.

25
Figure 1.3: Membrane localization of GABAA receptors. Receptor concentration at
postsynaptic sites is important for efficient signal transmission from cell to cell. The balance of receptors localized synaptically and extrasynaptically is regulated by a multitude of processes, including. regulatory mechanisms that modulate the receptor concentration, such as exocytosis of receptors released from the Golgi and clathrin mediated endocytosis and recycling of receptors. Stability of the proteins at the synapse by interaction with clustering and anchoring proteins also plays an important role in receptor concentration.

26
There have been many studies on the mechanism for endocytosis, exocytosis
and recycling of GABAA receptors to modulate synaptic efficacy. Work by Kittler
et al., (2002) has shown GABAA receptors can be localized to clathrin coated
vesicles implicating GABAA receptor endocytosis occurs via clathrin coated pits.
Additionally, this work found that β and γ subunits interacted directly with the
clathrin adaptor protein AP2 (Kittler et al., 2002). Blockage of clathrin-mediated
endocytotic pathway has resulted in the absence of GABAA receptor
internalization (Kittler et al., 2002; Hering et al., 2003); indicating endocytosis can
modulate the receptor number on the membrane surface.
1.12 Modulation of GABAA Receptors
Modulation of the function of GABAA receptors can be done by two
independent means, first by modulating the ligand binding affinity utilizing
endogenous or exogenous compounds and second by modulating the subunit
structure by phosphorylation. There are a variety of drug targets that can bind to
extracellular epitopes on the subunits, as well as changes in phosphorylation
states of different subunits. Modulation of GABAA receptor function can be
demonstrated by differential phosphorylation of specific GABAA receptor
subunits. Specificity of the phosphorylation targets will allow for the differential
modulation of different subsets of GABAA receptors containing different subunits.
Diverse phosphorylation sites have been identified on the β1-3 and γ2 subunits.
The β1-3 subunits all contain conserved serine resides which can be differentially
phosphorylated. The Ser408/Ser409 residues of β3 subunit are phosphorylated by

27
PKA, resulting in the potentiation of the GABA response. However, the Ser410 in
the β2 subunit cannot be phosphorylated by PKA, and phosphorylation of the
Ser408 on β1 results in inhibition of the GABA response (reviewed by Luscher and
Keller, 2004). In addition to PKA-mediated phosphorylation, there are various
other enzymes that can phosphorylate and dephosphorylate the β subunits to
modulate GABAA receptor function. Some enzymes that can affect
phosphorylation states are PKC, which can phosphorylate the β3 subunit
(Brandon et al., 2002), Src, which modulates tyrosine phosphorylation
(Valenzuela et al., 1995; Wan et al., 1997), and PP2A, which can
dephosphorylate β3 (Jovanovic et al., 2004).
The γ2 subunit contains multiple residues for phosphorylation, and the γ2L
splice variant contains an additional phosphorylation site. The serine/threonine
kinases that modulate GABAA receptor function are PKC and CaMKII. These
kinases can phosphorylate Ser327, however, the kinase that basally
phosphorylates Ser327 in vivo is unknown (McDonald, 1994; Wang et al., 2003).
In addition to Src phosphorylation, phosphorylation by the Src homolog Fyn can
modulate GABAergic transmission by altering the expression of functional
GABAA receptors. While it is known that phosphorylation by Fyn is required for
this modulation, the molecular substrate is unknown (Boehm et al., 2004). Much
evidence has been shown for the phosphorylation-induced modulation of GABAA
receptors; however, more work will be required to understand fully the effect of
this in model systems.

28
1.13 Aim of Study
The aim of this PhD thesis was to identify the domain(s) of the γ2 subunit
required for synaptic localization and to further study proteins known to modify
GABAA receptor postsynaptic localization. Previous studies have shown that the
γ2 subunit is critical for postsynaptic localization of GABAA receptors and that
loss of the γ2 subunit also results in loss of gephyrin localization to inhibitory
postsynaptic sites (Essrich et al., 1998). The γ2 subunit has also been shown to
be required not only for the localization of GABAA receptors to inhibitory
synapses, but also for the maintenance of the receptors at mature synapses
(Schweizer et al., 2003). However, no studies have shown the direct interaction
between these two proteins. Indeed, further studies show a subset of γ2 subunit-
containing receptors is able to localize to synapses in the absence of gephyrin.
This suggests that the domain of the γ2 subunit required for postsynaptic GABAA
receptor localization may be different from that of gephyrin colocalization.
In particular, the following specific questions were addressed in the current study:
1. Can we identify the domain(s) of the γ2 subunit required for postsynaptic
localization, are these domain(s) also responsible for colocalization of
GABAA receptors with gephyrin at inhibitory synapses and is association
of GABAA receptors essential for proper function of inhibitory synapses?
2. What effect does mutating the interaction domains of gephyrin and
collybistin have on gephyrin and GABAA receptor localization in neurons?

29
3. What effect does modifying cysteine residues implicated in palmitoylation
of the γ2 subunit have on their localization?
4. Can we determine the ability of GABARAP-L1 to interact with cytoskeletal
proteins and localize to synapses with GABAA receptors?

30
CHAPTER 2
MATERIALS AND METHODS
2.1 Mouse lines utilized
Mice with a targeted disruption of the γ2 subunit gene (γ2-/+) have been
previously described (Günther et al., 1995; Essrich et al., 1998) and maintained
on an inbred 129SvJ background (Crestani et al., 1999). All offspring were
genotyped by PCR amplification of DNA from tail biopsies, using primers specific
for the wildtype and mutant γ2 locus (Both: upper 5’ CTCTCCATCGCTAAGAA
TGTTCGGGAAGT 3’; WT: lower 5’ GCTGACAAAATAATGCAGGGTGCCATA
CTC 3’, Mutant: lower 5’ATGCTCCAG ACTGCCTTGGGAAAAGC 3’.
2.2 Sequencing
All sequencing reactions were done utilizing either the Penn State Nucleic
Acid Facility (University Park, PA, 16802) or Macrogen Inc. (Seoul, Korea).
2.3 Preparation of plasmids
Plasmids were transformed into electrocompetent XL-1 Blue E. coli cells
and colonies containing the plasmids were grown in Luria broth with appropriate
antibiotics. Plasmids transfected into cortical neuron cultures were prepared
utilizing Sigma’s GenElute Endotoxin-Free plasmid kits (Sigma, St Louis, MO).
All other plasmid applications utilized Eppendorf’s plasmid kits (Eppendorf,

31
Hamburg, Germany). Plasmid concentration and purity was determined by UV
spectrophotometry.
2.3.1 Generation of chimeric plasmid constructs
The mouse γ2S (γ2) subunit cDNA (Connolly et al., 1999a), including 51
nucleotides of untranslated leader and 33 nucleotides of 3’-untranslated mRNA,
was cloned into pEGFP-N (Clontech, Paolo Alto, CA), substituting the γ2 cDNA
for EGFP (GFP) (Cormack et al., 1996). An oligonucleotide (5’-CAAAA ACTAA
TATCA GAAGA AGACC TAACT AGT-3’) encoding the nine amino acid 9E10
myc epitope and an adjacent Spe I site (QKLISQQDL-TS) was inserted between
amino acids four and five of the mature γ2 polypeptide by site-directed
mutagenesis. A GFP-tagged version of this 9E10γ2 construct (GFP−γ2) was
constructed by PCR amplification of the EGFP open reading frame of pEGFP-N
using Spe I site adaptor primers (Table 1, A-B) and insertion of this fragment into
the Spe I site downstream of the 9E10 tag of 9E10γ2 (Table 1, C-F). Towards
construction of chimeric subunits containing portions of the mouse γ2 and either
the rat α2 (Benson et al., 1998) or mouse β2 subunit (Malherbe et al., 1990), the
nucleotide sequences flanking the cytoplasmic loop region (amino acids 318 –
404; HYFVSNR….RIAKMDS) of the γ2 polypeptide in the 9E10γ2 construct were
subjected to site-directed mutagenesis to introduce silent Eco 0109 I and Eco NI
restriction sites (primers, Table 1, G-L). PCR-generated fragments derived from
the α2 or β2 subunit cDNA that were homologous to the γ2 subunit domains to

32
be exchanged were amplified using adapter primers (Table 2.1; α2 primers, M-R;
β2 primers, S-T) that contained the matching restriction sites and inserted into
the restriction enzyme digested 9E10-tagged γ2 subunit backbone, thereby
replacing the corresponding γ2 subunit fragment (Table 2.1). Thus, the 5’
untranslated sequences, as well as the leader peptide and the 15 N-terminal
amino acids including the epitope tag and Spe I site of the mature polypeptides,
are identical for all these 9E10-tagged subunit constructs. The constructs lacked
GABAA receptor subunit-derived 3’-untranslated sequences except for 33
nucleotides in constructs that contained the γ2 transmembrane domain four
(TM4) region. All constructs were verified by sequencing. The expression
vectors for untagged α2, β2 and β3 subunits have been described (Malherbe et
al., 1990; Benson et al., 1998)(Fig. 2.1).
2.3.2 Generation of cysteine mutant constructs
To determine the effect that cysteine residues, both within the cytoplasmic
loop and within TM4, have on the cellular localization of the γ2 subunit, each
cysteine residue within this region was mutated by site-directed mutagenesis.
The mouse γ2 subunit within the pRK5 backbone was modified to incorporate
mutations within the cytoplasmic or TM4 region. PCR primers were utilized to
generate point mutations by sequential PCR steps (Table 2.2). Initial PCR
incorporated the cysteine mutations on both 5’ and 3’ fragments of the
cytoplasmic loop, which overlapped by 12-20 bp. This was followed by nested

33
γ2S -38 MSSPNTWSIG SSVYSPVFSQ KMTLWILLLL SLYPGFTSQK SDDDYEDYAS 12 α2 -28 ---------- ----MRTKLS TCNVWFPLLV LLVWNPARLV LANIQEDEAK 8 β2 -24 ---------- --MWRVRKRG YFGIWSFPLI IAAVCAQS-- -----VNDPS 7 β3 -25 ---------- --MWGFAGGR LFGIFSAPVL VAVVCCAQS- -----VNDPG 7 γ2S NKTWVLTPKV PEGDVTVILN NLLEGYDNKL RPDIGVKPTL IHTDMYVNSI GPVNAINMEY 72 α2 NNITIFT--- ------RILD RLLDGYDNRL RPGLGDSITE VFTNIYVTSF GPVSDTDMEY 50 β2 NMSLVKET-- --------VD RLLKGYDIRL RPDFGGPPVA VGMNIDIASI DMVSEVNMDY 47 β3 NMSFVKET-- --------VD KLLKGYDIRL RPDFGGPPVC VGMNIDIASI DMVSEVNMDY 47 γ2S TIDIFFAQTW YDRRLKFNST IKVLRLNSNM VGKIWIPDTF FRNSKKADAH WITTPNRMLR 132 α2 TIDVFFRQKW KDERLKFKGP MNILRLNNSM ASKIWTPDTF FHNGKKSVAH NMTMPNKLLR 110 β2 TLTMYFQQAW RDKRLSYNVI PLNLTLDNRV ADQLWVPDTY FLNDKKSFVH GVTVKNRMIR 107 β3 TLTMYFQQYW RDKRLAYSGI PLNLTLDNRV ADQLWVPDTY FLNDKKSFVH GVTVKNRMIR 107 γ2S IWNDGRVLYT LRLTIDAECQ LQLHNFPMDE HSCPLEFSSY GYPREEIVYQ WKRSSVEVGD 192 α2 IQDDGTLLYT MRLTVQAECP MHLEDFPMDA HSCPLKFGSY AYTTSEVTYI WTYNPSDSVQ 170 β2 LHPDGTVLYG LRITTTAACM MDLRRYPLDE QNCTLEIESY GYTTDDIEFY WRGDDNAVTG 167 β3 LHPDGTVLYG LRITTTAACM MDLRRYPLDE QNCTLEIESY GYTTDDIEFY WRGGDKAVTG 167 γ2S TR--SWRLYQ FSFVGLRNTT EVVKTTSGDY VVMSVYFDLS RRMGYFTIQT YIPCTLIVVL 250 α2 VAPDGSRLNQ YDLLGQSIGK ETIKSSTGEY TVMTAHFHLK RKIGYFVIQT YLPCIMTVIL 230 β2 VTK--IELPQ FSIVDYKLIT KKVVFSTGSY PRLSLSFKLK RNIGYFILQT YMPSILITIL 225 β3 VER--IELPQ FSIVEHRLVS RNVVFATGAY PRLSLSFRLK RNIGYFILQT YMPSIMITIL 225 γ2S SWVSFWINKD AVPARTSLGI TTVLTMTTLS TIARKSLPKV SYVTAMDLFV SVCFIFVFSA 310 α2 SQVSFWLNRE SVPARTVFGV TTVLTMTTLS ISARNSLPKV AYATAMDWFI AVCYAFVFSA 290 β2 SWVSFWINYD ASAARVALGI TTVLTMTTIN THLRETLPKI PYVKAIDMYL MGCFVFVFMA 285 β3 SWVSFWINYD ASAARVALGI TTVLTMTTIN THLRETLPKI PYVKAIDMYL MGCFVFVFLA 285 γ2S LVEYGTLHYF VSNRKPSKDK DKKKKNPAPT ID-------- IRPRSATIQM NNATHLQERD 362 α2 LIEFATVNYF TKRGWAWDGK SVVNDKKKEK GS-------- VMIQNNAYAV AVANYAPNLS 352 β2 LLEYALVNYI FFGRGPQRQK KAAEKAANAN NEKMRLDVNK MDPHENILLS TLEIKNEMAT 345 β3 LLEYAFVNYI FFGRGPQRQK KLAEKTAKAK NDRSKSEINR VDAHGNILLA PMDVHNEMN- 344 γ2S EEYGYECLDG KDCASFFCCF EDCRTGAWRH GRIHIR---- ---------- ---------- 398 α2 KDPVLSTIS- -KSATTPEPN KKPENKPAEA KKTFNS---- ---------- ---------- 386 β2 SEAVMGLGDP RSTMLAYDAS SIQYRKAGLP RHSFGRNALE RHVAQKKSRL RRRASQLKIT 404 β3 -EVAGSVGDT RNSAISFDNS GIQYRKQSMP KEGHGRYMGD RSIPHKKTHL RRRSSQLKIK 403 γ2S ------IAKM DSYARIFFPT AFCLFNLVYW VSYLYL---- ---- 428 α2 ------VSKI DRMSRIVFPV LFGTFNLVYW ATYLNREPVL GVSP 423 β2 IPDLTDVNAI DRWSRIFFPV VFSFFNIVYW LYYVN------ --- 449 β3 IPDLTDVNAI DRWSRIVFPF TFSLFNLVYW LYYVN------ --- 448
Figure 2.1 Sequence comparison of GABAA receptor subunits. Shown here is the mouse
γ2S subunit protein sequence with rat α2, β2, and β3 protein sequences with proposed membrane spanning domains indicated by gray shading. Amino acid sequence numbering starts at the putative N-terminal residue of the subunit, with presumed signal peptide sequences indicated by negative numbering. Yellow highlighted amino acids indicate differences in amino acids between rat and mouse protein sequences. Note that for the γ2S and β2 sequence that no amino acids differ in the mature protein from rat to mouse.

34
Table 2.1: Primers utilized for generation of chimeric plasmid constructs.
A
5’ primer insert GFP with Spe I site flanking
CAC CGG TCG CCA CCA CTA GTA TGG TGA GCA AGG GC
B 3' primer binding GFP with Spe I site flanking
GTC GCG GCC GCT TTA ACT AGT CTT GTA CAGCTC G
C 5' primer inserting Spe I site flanking 9E10 epitope
CTA ATA TCA GAA GAA GAC CTA ACT AGT GAT GAC TAT GAA GAT TAC GC
D 3' primer binding EcoR V site CGA GGA TAT CCA TAA CTG GAG E 5' primer in EGFP plasmid upstream
of start codon CGC TCG AGG CCA CCA TGA GTT CGC CAA ATA CAT G
F 3' primer inserting Spe I site flanking 9E10 epitope in γ2 sequence
GCG TAA TCT TCA TAG TCA TCA CTA GTT AGG TCT TCT TCT GAT ATT AG
G 5' primer binding to Spe I site/ 5th a.a. of γ2 sequence
CCC ACT AGT GAT GAC TAT GAA GAT TAC GCT TCT A
H 3' primer inserting Eco0109 I at 5' end of loop
GCT GAC AAA ATA ATG CAG GGT CCC ATA CTC CAC C
I 5' primer inserting Eco0109 I at 5' end of loop
GGT GGA GTA TGG GAC CCT GCA TTA TTT TGT CAG C
J 3' primer within vector 3' of γ2 sequence
GTA TGG CTG ATT ATG ATC TAG AGT CGC GGC
K 3' primer inserting EcoN I at 3' end of loop sequence
GGT AGG GAA GAA GAT CCT AGC ATA GGA G
L 5' primer inserting EcoN I at 3' end of loop sequence
CTC CTA TGC TAG GAT CTT CTT CCC TAC C
M 3' primer inserting extracellular/ TM1-3 of α2 with Eco0109 I site flanking
AAG GGT CCC AAA TTC AAT TAA GGC AGA GAA CAC
N 5' primer inserting extracellular/TM1-3 of α2 with Spe I site flanking
GGC ACT AGT AAC ATC CAA GAA GAT GAG GC
O 5' primer inserting α2 cytoplasmic loop region with Eco0109 I site flanking
AAG GGA CCC TTA ATT ACT TCA CGA AAA GAG G
P 3' primer 3' end of α2 sequence with Apa I site flanking
CGC GGG CCC TCA AGG ACT AAC CCC TAA TAC AGG C
Q 5' primer inserting α2 cytoplasmic loop region with Eco0109 I site flanking
CGG CCT AGC ATA GGT GTC GAT TTT GCT GAC AC
R 3' primer 3' end of α2 sequence with Apa I site flanking
GGG CCT ATG CTA GGA TAG TGT TCC CGG TTC TGT TTG G
S 3' primer inserting β2 extracellular/ TM1-3 including Eco0109 I site
GGG CCT AGC ATA GGA ATC AAT GGC ATT CAC
T 5' primer inserting β2 extracellular/ TM1-3 with Spe I site flanking
GGG ACT AGT GAC CCT AGT AAT ATG TCG CTG G

35
PCR utilizing the 5’ and 3’ fragments of the cytoplasmic loop incorporating the
cysteine to alanine mutations as template, to generate a new cytoplasmic loop
that contains the mutation of interest. The same concept was applied to the
cysteine mutation within TM4. (see Chapter 5, Fig. 5.2.1 for schematic).
Table 2.2: Primers for Cysteine mutations
Primer name: Sequence (5’-3’) Cysteine* Modified:
g2cysala1upp GCTCTGGATGGCAAGGACTG C369A g2cysala1low CTTGCCATCCAGAGCCTCATAGCCATATTCTT C369A g2cysala2upp GCTGCCAGTTTCTTCTGCTGT C375A g2cysala2low CAGCAGAAGAAACTGGCAGCGTCCTTGCCATC C375A g2cysala3+4upp GCCGCTTTTGAAGATTGCCGAACA C380/381A g2cysala3+4low GCAATCTTCAAAAGCGGCGAAGAAACTGGCA C380/381A GAGTCCTTGCCATC C385A Upper TGT TTT GAA GAT GCC CGA ACA GGA G C385A C385A Lower ATCTTCAAAACAGCAGAAGAAACTGGCACAGTCCTT C385A C404A Lower CAAGATTGAACAGGCAAAGGCGGTAGGGAAGAA C415A C404A Upper GCCTTGTTCAATCTTGTTTACTGGGTC C415A 464Eco0109 I sense GGTGGAGTATGGGACCCTGCATTATTTTGTCAGC Start of loop 3’-5’ end γ2 loop GGTAGGGAAGAAGATCCTAGCATAGGAG End of loop
* numbering of amino acid residues refers to the amino acid sequence of the mouse γ2 subunit described in Fig. 2.1. (Kofuji et al., 1991)
2.3.3 Generation of TAC/IL-2 constructs
The IL-2 α subunit fusion protein with the cytoplasmic loop and C-terminal
region of the GABAA receptor γ2 subunit was generated via directed PCR,
utilizing specific restriction enzyme sites within the polylinker region of the IL-2
plasmid (Standley et al., 2000) (gift of M. Ehlers, Duke University Medical Center,
Durham, NC). The IL-2 α subunit, also called the TAC subunit of the IL-2
receptor, contained the full-length human IL-2 α sequence including 44-bp of
upstream 5’ UTR. The IL-2/γ2 fusion construct was generated by linking the 3’

36
region of the γ2 subunit incorporating the intracellular loop from amino acid 309
(Fig. 2.1) to the C-terminus (including 58-bp of downstream 3’ UTR), to the 3’ end
of the IL-2 α. This γ2 subunit fragment was inserted into the C-terminal Hind III
site at amino acid 268 of the IL-2 α subunit, 6-bp upstream from the stop codon,
and an Xba I site from the vector multicloning site. The γ2 insert was generated
by PCR of a mouse γ2S vector, containing the full-length γ2S sequence, as
template (Connolly et al., 1999a), with the 5’ primer GTC CAA GCT TAT TTT
GTC AGC AAC and the chimeric downstream primer J (Table 1). A similar
construct containing the α2 loop sequence with the γ2 TM4, IL-2/α2γ2, was made
likewise via PCR, inserting the α2 sequence from amino acid 306 to 389 of the
putative α2 loop sequence (Benson et al., 1998), then the C-terminal region of
the γ2 subunit from amino acid 395 to C-terminal, including 58-bp of 3’ UTR (Fig.
2.1 for α2 and γ2 sequences). One of the chimeric vectors containing the α2
loop with the γ2 TM domain was utilized as template, with the 5’ primer GTC GAA
GCT TAC TTC ACG AAA AGA and the chimeric downstream primer J.
2.3.4 Generation of GST-fusion constructs
A GST-γ2 fusion construct was made using the γ2 cytoplasmic loop
sequence from the pSOS- γ2S vector (rat 309-396 a.a.) (Keller et al., 2004), and
inserting it into the pGEX-4T-1 (Amersham Pharmacia, Piscataway, NJ), via a 5’
BamH I site that was cleaved and filled with Klenow polymerase and a 3’ Not I
site (Amersham Pharmacia). This γ2 intracellular loop was inserted into the

37
pGEX-4T-1 vector Sma I and Not I sites, generating a GST-fusion construct with
GST and γ2 intracellular loop separated by 11 amino acids containing a thrombin
cleavage site.
The pGEX-4T-1 plasmid was utilized as GST control. The GST fusion
construct for GABARAP (Wang et al., 1999) was generated by cloning a RT-PCR
fragment flanked by EcoR I and Xho I into the multi-cloning sites of pGEX-4T-1,
generating a GST fusion of the full-length GABARAP (117 a.a.) with a 15 amino
acid spacer, again containing a thrombin cleavage site. The GST-GABARAP-L1
(117 a.a.) (Xin et al., 2001) and GATE-16 (117 a.a.) (Sagiv et al., 2000) were
likewise inserted into the pGEX-4T-1 by cloning RT-PCR fragments flanked by
EcoR I and EcoR V sites, inserting into the multi-cloning sites at EcoR I and Sma
I restriction sites, both with 15 amino acid spacers with the thrombin cleavage
site. (J. Stewart and B. Lüscher previously generated these three fusion
constructs.)
2.3.5 Generation of GFP-GABARAP-L1 fusion constructs
To generate a GFP-GABARAP-L1 construct, the pEGFP-C3 (Clontech)
vector was digested with EcoR I and Sma I, and full-length GABARAP-L1 (117
a.a.), flanked by EcoR I and EcoR V sites, was ligated into the vector. This
generated a GFP fusion protein with 13 amino acids between GFP and
GABARAP-L1 (a.a. 1-117) followed by 9 amino acids of the polylinker from
pEGFP-C1 prior to the stop codon.

38
2.4 Growth of GST fusion proteins
GST plasmids were transformed into electrocompetent BL-21 E. coli cells
and colonies containing the plasmids were grown on LB agar plates with
ampicillin antibiotic (Sigma, LB-Amp; 100 µg/ml). Starter cultures of single
colonies were picked and grown in LB-Amp broth (5 ml) at 37 ºC overnight, then
transferred to 500 ml of LB-Amp broth and grown to OD600 of ~ 0.6 at 37 ºC.
IPTG (100 µM) was added to induce expression of GST and GST-fusion proteins
and cultures were grown for 30 min. to 1 hr at 28 ºC. Cells were harvested by
centrifugation at 4 ºC and pellets were resuspended in 5 ml MT-PBS +10 %
glycerol (150 mM NaCl, 16 mM Na2HPO4, 4 mM NaH2PO4, 10 % v/v glycerol, pH
7.3). Cells were sonicated and Triton X-100 was added to 1 % final
concentration. Cellular debris was centrifuged and supernatant utilized for GST
assays.
2.5 Protein extracts
Transfected human embryonic kidney (HEK) 293T cells (American Type
Culture Collection, Manassas, VA) were extracted in 10 mM Tris-HCl, pH 7.4,
150 mM NaCl, 1 mM EDTA, 0.2 % Triton X-100, 1 µg/ml antipain, 1 µg/ml
pepstatin-A, 1 µg/ml leupeptin, and 0.5 mM PMSF. The extracts were cleared by
centrifugation (10,000 x g, 5 min) and the supernatant stored at –80 ºC.
Expression of chimeric proteins was assessed by analysis of supernatants on a
10 % SDS-PAGE gel (40 µg protein per lane) followed by western blot.

39
2.6 Western blot analyses
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE)
was performed according to Laemmli (1970). Samples of 4 µg GST-fusion
protein diluted with 2x SDS-loading dye (20 mM Tris-HCl pH 6.8, 200 mM DTT, 2
% SDS, 20 % glycerol, 0.2 % bromophenol blue) were heated by boiling for 5
min. Proteins were separated on appropriate percentage polyacrylamide
minigels (90 x 60 x 1.5 mm) at 175 volts in electrophoresis buffer (25 mM Tris-Cl,
192 mM glycine, 0.1 % SDS) on a Proteon 2 or 3 system (BioRad, Hercules,
CA). After SDS-PAGE, nitrocellulose (0.2 µm, Bio-Rad), blotting paper (VWR)
and gels were equilibrated in transfer buffer (39 mM glycine, 48 mM Tris-Cl, 0.04
% SDS, 20 % methanol) and proteins were transferred to nitrocellulose
membranes using a semi-dry electro-blotting apparatus (Bio-Rad) for 60 min at
15 volts.
For immunodetections, nitrocellulose membranes were blocked for 2 hrs
at room temperature in TBST (10 mM Tris-Cl pH 8.0, 150 mM NaCl, 0.05 %
Tween-20) containing 5 % non-fat dry milk powder. Membranes were incubated
with primary antibodies [mouse anti-9E10 (S. Lingenfelter, 1:50), goat anti-
gephyrin (Santa Cruz Biologicals, Santa Cruz, CA 1:250), mouse anti-gephyrin
(BD Transduction Labs, San Jose, CA, 1:250), rabbit anti-GABARAPL1 (rabbit
#6, K. Baer, 1:3000), rabbit anti-α1 (Gift D. Benke, 1:500), mouse anti-β tubulin
I/II (Sigma, 1:1000), mouse anti-NSF (Exalpha, Maynard, MA 1:2000)] overnight
at 4 ºC in TBST/5 % milk Membranes were washed in RIPEA buffer (20 mM Tris-
Cl pH 7.5, 60 mM NaCl, 2 mM EDTA, 0.4 % SDS, 0.4 % Triton X-100, 0.4 %

40
deoxycholate) for 15 min., then washed four times for 10 min. in TBST.
Membranes were reblocked for 30 min. in TBST/5 % milk, then horseradish
peroxidase labeled secondary antibodies (HRP α-goat, 1:10,000; HRP α-guinea
pig, 1:5000; HRP α-rabbit, 1:5000; HRP α-mouse, 1:5000; all from Amersham
Pharmacia) were applied for 2 hrs at RT to detect the proteins. Membranes
were rewashed as above, the membranes were incubated with enhanced
chemiluminescence reagents (ECL Plus, Amersham Pharmacia), and visualized
by exposure to x-ray film (Hyperfilm ECL, Amersham Pharmacia).
For detection of total protein loaded and purity of samples, Commassie
staining of SDS-PAGE gels was performed. SDS-PAGE gels were incubated in
Commassie stain (50 % methanol, 0.05 % Commassie Brilliant Blue, 10 % glacial
acetic acid) for up to 1 hr at room temperature. Gels were destained for 1 hr to
overnight in destaining solution (50 % methanol, 10 % glacial acetic acid, 40 %
MilliQ UV purified H2O; Sambrook et al., 1989).
2.7 Tissue culture and transfection
HEK 293T and HeLa cells were maintained in DMEM (Invitrogen),
supplemented with 10 % fetal bovine serum (FBS) at 37 ºC in 5 % CO2. For
protein expression assays, cells were seeded onto 60 mm dishes and allowed to
reach 70 % confluency. They were transfected with a mixture of plasmids
harboring cDNAs containing the indicated chimeric construct (10 µg) and GABAA
receptor α2 and β3 subunits (5 µg each), using the standard CaPO4 transfection
method (Chen and Okayama, 1987). For surface expression assays, cells were

41
seeded onto poly-L-lysine-coated coverslips, and analogously transfected with
the construct of interest (4 µg each) alone or together with a mixture of GABAA
receptor α2 and β3 subunits (4 µg each). IL-2 fusion constructs were also
subjected to cotransfection with the γ2S subunit (Kofuji et al., 1991). The cells
were harvested or analyzed by immunofluorescent staining 36 - 48 hr after
addition of the DNA precipitate.
2.7.1 Neuronal tissue culture
Cortical neurons were cultured from γ2 subunit deficient embryonic day
14.5 embryos, generated by crossing of γ2+/- mice on a 129SvJ inbred
background (Essrich et al., 1998). Cortical hemispheres were collected in
Phosphate Buffered Saline (PBS) containing 5.5 mM glucose. Tissue was treated
with papain (0.5 mg/ml) and DNase I (10 µg/ml, both from Sigma, St. Louis, MO)
in PBS/Glu/BSA (PBS Invitrogen, Carlsbad, CA) with 10 mM glucose and 1
mg/ml bovine serum albumin (Fraction V, Sigma) for 15 min at RT. Tissue was
then dissociated by trituration with a fire-polished Pasteur pipette and an equal
volume of DMEM, supplemented with 2% FBS and buffered with HEPES (DMEM
with 2% FBS, 10 mM HEPES, 0.5 mM Glutamax I, 100 units/ml
penicillin/streptomycin; all from Invitrogen) was utilized to stop the papain
reaction. Cells were centrifuged at 900 x g for 5 min. and supernatant was
discarded. Cells were resuspended in MEM/2 %FBS (MEM with 2% FBS, 2 mM
Glutamax I, 1 mM pyruvic acid, 0.52 % glucose, 100 units/ml

42
penicillin/streptomycin; all Invitrogen) and counted. Cells were diluted to cell
plating concentration with MEM/10% FBS (MEM with 10% FBS, 2mM Glutamax
I, 1 mM pyruvic acid, 0.52 % glucose, 100 units/ml penicillin/streptomycin) and
plated on poly-L-lysine-coated glass cover slips (22 x 22 mm) at 4 x 104 cells/cm2
in an atmosphere of 10% CO2 at 37 º C. After 60 min., the medium was replaced
with fresh MEM/10 % FBS. The genotype of cultures was determined using PCR
as follows: tail biopsies (3 mm) were incubated for 30 min at 55 º C in 25 µl lysis
buffer (200 mM NaCl, 5 mM EDTA, 0.2 % SDS, 100 mM Tris-HCl, pH 8.5) and 1
% of the supernatant volume was used for PCR using standard conditions with
the primers 5’-CATCT CCATC GCTAA GAATG TTCGG GAAGT-3’ combined
with either 5’-GCTGA CAAAA TAATG CAGGG TGCCA TACTC-3’ to amplify the
wildtype γ2 locus or the primer 5’-ATGCT CCAGA CTGCC TTGGG AAAAG C-3’
to amplify the mutant γ2 locus. The 24 hr old cultures with the desired genotypes
were turned over onto a glial feeder layer in a 35 mm Petri dish containing
Neurobasal-A supplemented with B27 (Invitrogen), in an atmosphere of 10% CO2
(Banker and Goslin, 1998).
Feeder cells were prepared from cortices of newborn rat pups as
described previously (Banker and Goslin, 1998). Briefly, 2-3 day old postnatal
rat pups were euthanized and their cortices placed into cold Hanks’s Buffered
Salt Solution (1X HBSS [Invitrogen], 1 mM HEPES, 100 units/ml
penicillin/streptomycin). Cortices were minced, centrifuged and supernatant was
removed, then the tissue was incubated in 1.5 mL each of 2.5 % trypsin and 1 %
DNase for 15 min. at 37 ºC. Tissue was triturated with a fire-polished pipette and

43
passed through a 72 µm nylon filter. Cells were centrifuged and resuspended in
glial medium (MEM, 0.6 % glucose, 100 units/ml penicillin/streptomycin, 10 %
FBS, 2 mM Glutamax I) and incubated at 37 ºC in 5 % CO2. After cells reached
confluency, they were split 1:3 and then grown to confluency again, after which
they were plated on 35-mm dishes. At 70-80% confluency, their growth was
stopped with poly-uridine (35 µg uridine, 15 µg fluoro-deoxyuridine per dish, both
Sigma) and media was changed to neuronal media (NB-A with 1x B27, 2 mM
Glutamax I, 100 units/ml penicillin/streptomycin).
2.7.2 Neuronal transfections for immunohistochemistry
Neuron cultures were maintained without medium change for 18 days in
vitro (Cormack et al., 1996) and then transferred into new Petri dishes containing
Neurobasal A/B27 supplemented with 1 µM 6-cyano-7-nitoquinoxaline-2,3-dione
(CNQX) and 100 µM 2-amino-5-phosphonovaleric acid (APV) (Sigma, St. Louis,
MO), with the cells facing up. They were transfected with 8 µg of either epitope-
tagged GABAA receptor subunit chimeric constructs, GFP-tagged gephyrin
constructs, epitope-tagged collybistin constructs, GFP-tagged GABARAP-L1 or
IL-2 fusion constructs, using a CaPO4 transfection kit (BD Biosciences). In some
experiments, which tested chimeric constructs that failed to cluster, we
cotransfected 100 ng pEGFP-C1 (BD Biosciences, Palo Alto, CA) to
unambiguously identify transfected cells. In neurons identified by positive
chimeric puncta, GFP was cotransfected with chimeric construct in 80-90 % of

44
transfected cells. The DNA precipitate (200 µl) was prepared according to the
instructions of the kit manufacturer, allowed to precipitate for 15 min, added onto
the cells for 45 min, and the cover slips were then returned to the original dishes
containing original media. Neurons were processed for immunofluorescent
analysis at 21 DIV.
2.8 Immunofluorescence analyses
For labeling of GABAA receptor subunits expressed in the plasma
membrane of HEK 293T cells, the cells were washed three times with PBS (136
mM NaCl, 1.75 mM NaH2PO4, 8.25 mM Na2HPO4), fixed in 4%
paraformaldehyde in 150 mM sodium phosphate buffer pH 7.4 for 12 min without
permeabilization, and incubated overnight at 4 ºC with rabbit anti-myc (Medical
and Biological Labs, Woburn, MA, 1:1000), and guinea pig anti-α2 (gift of J.M.
Fritschy, University of Zurich, Switzerland, 1:700). After primary antibody
incubation, coverslips were washed in PBS (3 washes/10 min per wash), then
secondary antibody was added and cultures were incubated for 45 min. at room
temperature followed by repetition of wash steps. Coverslips were mounted
utilizing mounting solution (50% v/v glycerol, 50% 0.1 M NaHCO3 pH 7.4) and
stored at 4 ºC until ready for imaging.
To label the surface and internal receptor localization of the IL-2 chimeric
receptors, the HEK 293T or HeLa cells were stained as follows: the cells were
washed briefly in live staining buffer (2 mM CaCl2; 2 mM MgCl2; I µM glycine; 30
mM glucose; 25 mM HEPES, pH 8.0; 5 mM KCl; 119 mM NaCl; 0.5 µM TTX;

45
solution then pHed to 7.4), then incubated at 8-10 ºC for 1 hr (Covance, Berkeley,
CA, mouse anti-IL-2, 1:1000), then washed with the live staining buffer (3
washes/10 min. each). The secondary antibody (Molecular Probes, AlexaFluoro
488 anti-mouse, 1:500) was then added for 30 min. at 8-10 ºC, after which the
washes were repeated. To visualize the internal receptors, the cells were then
moved to room temperature, fixed in 4 % paraformaldehyde for 12 min. and
permeabilized for 4 min. with 0.2 % Triton X-100 in PBS containing 10 % donkey
serum. The cells were then reincubated with primary antibody (mouse anti-IL-2,
1:1000 Covance) in PBS with 15 % donkey serum for 90 min. at RT, followed by
PBS washes (4 washes/8 min. per wash). Secondary antibody, utilizing a
different fluorophore (Cy3 anti-mouse, 1:500, Jackson Immunoresearch Labs),
was incubated for 45 min. at RT and washes were repeated. Coverslips were
then mounted onto slides and stored at 4 ºC until viewing.
Neurons used for immunofluorescence studies were washed three times
in PBS, fixed in 4 % paraformaldehyde for 12 min, and permeabilized for 4 min
with 0.2 % Triton X-100 in PBS containing 10 % donkey serum. After a brief
wash in PBS, cells were incubated with primary antibody overnight at 40 C using
the following dilutions: rabbit anti-myc (Medical and Biological Labs, 1:500),
guinea pig anti-α2 (1:2000) and mouse anti-gephyrin mAb 7a (gift of H. Betz,
Max Plank Institute, Frankfurt, Germany, 1:500), mouse anti-IL-2 (Covance,
1:1000), GABAA receptor antibody guinea pig anti-γ2 (1:1500, Gift of J.M.
Fritschy), mouse anti-gephyrin mAb 7a (used for GFP-gephyrin and collybistin
images, 1:100, mAb7a; Alexis Biochemicals, San Diego, CA), or mAb GAD-6

46
(0.5 µg/ml; Developmental Studies Hybridoma Bank, University of Iowa, IA). For
detection of primary antibodies, AlexaFluoro 488-conjugated goat anti-rabbit,
AlexaFluoro 647-conjugated goat anti-mouse or rabbit (Molecular Probes,
Eugene, OR), or Cy3 donkey anti-mouse or guinea pig (Jackson
ImmunoResearch, West Grove, PA) were used as appropriate. Note that for all
GFP-gephyrin and epitope-tagged collybistin transfected cells, 0.15 % saponin
was substituted for 0.2 % Triton X-100 in permeabilization. As saponin is a non-
permanent detergent, 0.15 % saponin was added to all solutions following
permeabilization. Also note, for experiments using EGFP-gephyrin constructs,
mAb7a is assumed to recognize both native and recombinant gephyrin.
Fluorescent images were captured using a Zeiss Axiophot2 microscope
equipped with a 40 x 1.3 NA objective and an ORCA-100 video camera linked to
an OpenLab imaging system (Improvision, Lexington, MA). Digital gray scale
images were pseudo-colored using green, red and blue for the sequentially
recorded fluorescences. Images of cells that had been co-transfected with trace
amounts of GFP were developed using AlexaFluoro 647 as a secondary antibody
to reveal immunoreactivity of the chimeric constructs and then pseudo-colored
green to match the color of images that had been developed with AlexaFluoro
488. Images were adjusted for contrast using OpenLab and assembled into
figure palettes using Adobe Photoshop.

47
2.9 Quantitation of immunofluorescent staining
For semiquantitative analyses of GABAA receptor clusters seen by
chimeric transfections, digitized microscopic images were recorded from cells
that were innervated by GABAergic axons, as judged by glutamic acid
decarboxylase staining. Two properly innervated dendritic segments of 40 µm in
length were selected from each of 13 - 18 different cells transfected in three or
more independent experiments. Immunoreactive puncta stained for the 9E10
epitope were automatically selected using OpenLab imaging software. Receptor
clusters were defined as immunofluorescent puncta within the region of interest
that exceeded a fluorescence intensity threshold that was 2-fold greater than the
diffuse fluorescence measured on the shaft of the same dendrite and fit a target
size range of 0.2 to 2 µm in diameter. To determine the percentage of 9E10-
immunoreactive puncta at synapses, the fraction of puncta that were maximally
one pixel apart from punctate GAD immunoreactivity was declared to be
postsynaptic. Puncta determined to be postsynaptic by this method were
selected to compute the average size (i.e. area) of postsynaptic GABAA receptor
clusters. 9E10-immunoreactive puncta were selected in dendritic segments of 40
µm in length, using the intensity and size limitations indicated above. The fraction
of puncta that exhibited at least one pixel overlap with punctate gephyrin
immunoreactivity was defined to be colocalized with gephyrin (n = 13 - 26 cells
per construct). Transfected neurons that did not show any punctate gephyrin
immunoreactivity were excluded from the analysis to ensure that cells that were
not viable or did not express gephyrin were excluded from analysis. Statistical

48
comparisons were performed using ANOVA one-way comparison with Dunnett’s
post-test (Instat software).
To quantify the effects of transfecting collybistin and gephyrin constructs,
either wild-type or mutant, into neurons, two dendritic segments of 40 µm in
length were selected per cell (n = 10-12 transfected cells per construct) that
exhibited low overall background staining for gephyrin (mAb7a) or the GABAA
receptor γ2 subunit. Collybistin transfected cells were quantitated for either
gephyrin or GABAA receptor clusters, seen by mAb7a or γ2 IR, respectively.
Quantitation of GFP-gephyrin, GFP-gephyrin A4 and A5 measured the total
amount of gephyrin puncta, seen by mAb7a IR. The segments exhibiting low
background staining were analyzed within the region of interest to select
immunoreactive puncta using a threshold of 2-fold greater fluorescence intensity
than the diffuse fluorescence on dendritic shafts. Using this threshold, the
number of puncta, over the 40 µm segment per cell and within a size range of 0.2
µm to 2 µm in diameter, was determined to compute the average size of gephyrin
or GABAA receptor clusters. Synaptic GABAA receptor clusters were determined
by close apposition (maximally 1 pixel length apart) to GABAergic innervation
(GAD-IR). Each segment was averaged per cell and analysis was performed
using OpenLab imaging software with Microsoft Excel (Seattle, WA). Student’s t-
test was used for all statistical comparisons of immunofluorescence.

49
2.10 Electrophysiology
To determine the GABA dose response curves of recombinant GABAA
receptors, HEK 293T cells were plated onto poly-L-lysine-coated glass coverslips
(12 mm diameter) and transfected 24 to 36 hrs later as described above using
200 ng of pEGFP-C1 (Clontech, Palo Alto, CA) and 1 µg each of the α2 and β3
expression vectors supplemented with 1 µg chimeric construct as indicated in the
figures. Transfected cells were identified by EGFP fluorescence 24 - 48 h after
transfection and membrane currents were recorded in the whole-cell mode with
the membrane potential clamped at -60 mV using a Multiclamp 700A amplifier
(Axon Instruments, Foster City, CA). Borosilicate glass pipettes (Harvard
Apparatus, Holliston, MA) were fire polished for a final resistance of 2-6 MΩ. The
recording chamber was perfused continuously with a bath solution containing
128 mM NaCl, 30 mM D-glucose, 25 mM HEPES, 5 mM KCl, 2 mM CaCl2, and 1
mM MgCl2 (adjusted to pH 7.4 using NaOH). The pipette solution contained 147
mM KCl (or CsCl), 5 mM disodium phosphocreatine, 2 mM EGTA, 10 mM
HEPES, 2 mM MgATP, and 0.3 mM Na2GTP (pH 7.4, adjusted with KOH). GABA
solutions were prepared daily from stock solution containing 100 mM GABA
(Acros Organics, Geel, Belgium). Series resistances were typically 10 - 25 MΩ
with a membrane resistance mostly in the range of 250 - 800 MΩ. Data were
acquired using PCLAMP 8 software, sampled at 10 kHz and filtered at 1 - 2 kHz
and analyzed using CLAMPFIT 8/9 software (Axon Instruments). GABA-induced
currents were normalized to fractions of the maximum current recorded in the
same cell under the same conditions. Using SigmaPlot 8.0 (SPSS Inc., Chicago

50
Il), dose-response values of each cell were fitted to the equation I = Imax/[1 +
(EC50/A)n], where A is the GABA concentration, EC50 is the concentration of
GABA eliciting a half maximal current amplitude, Imax is the maximal current
amplitude, I is the measured current amplitude, and n is the Hill coefficient.
Curves determined separately for 3 - 10 cells expressing the same
subunit/chimera combination were averaged to yield the corresponding EC50
value. The EC50 values of different subunit/chimera combinations were compared
using a two-tailed t-test and are expressed as mean ± standard error of
measurement.
For analysis of mIPSCs, cortical neurons were cultured and transfected at
16 DIV using 8 µg cDNA per construct and 100 ng pEGFP-C1 as described
above. Twenty-four to 48 h later, miniature synaptic currents were recorded in
the whole-cell voltage-clamp mode. Holding potential was set at -70 mV.
Miniature inhibitory postsynaptic currents were recorded in the presence of 200
nM tetrodotoxin (TTX) (Sigma) and 10 µM CNQX (Tocris Cookson, Ellisvile, MO).
The GABAergic nature of these events was confirmed by blocking with 40 µM
bicuculline (Tocris). Miniature events were analyzed using MiniAnalysis software
(Synaptosoft, Decatur, GA) and inspected visually for accuracy. The average
amplitude and frequency of mini events (n = 6 - 15 neurons per construct or
genotype) were compared using a two-tailed Student t-test.

51
2.11 Brain extracts
Crude Brain extract was generated utilizing a modified protocol from
Kneussel and Olsen (2001). Briefly, adult mouse brains were buffered in 10x wet
brain weight of homogenization buffer (100 mM NaCl, 10 mM Tris-Cl pH 7.5, 5
mM EDTA, 10 mM MgCl2, 0.5 % NP-40, 1 % Triton X-100, 1 mM PMSF, and 10
µg/ml each antipain, leupeptin, pepstatin-A and aprotinin). Brains were
incubated at 4 ºC for 60 min then homogenized utilizing Polytron homogenizer
(Brinkmann Instruments, Eppendorf) and centrifuged at 10,000 g for 30 min at 4
ºC. Supernatant was collected and utilized fresh or frozen at –80 ºC until use.
To generate brain membrane extract (modified from Kristin Baer, 1999),
rat or mouse brains were dissected and immediately transferred into ice-cold
sucrose buffer (0.32 M Sucrose, 5 mM EDTA pH 8.0, 0.2 mM PMSF, 10 mM
Tris-acetate pH 7.4, 0.05 % NaN3). Brain tissue was homogenized utilizing a
Polytron homogenizer at top speed within 1 volume sucrose buffer for 1 min, and
sucrose buffer was then added to 10 times volume of homogenate. The
suspension was centrifuged at 1,500 x g for 15 min. at 4 ºC, and the supernatant
was transferred to fresh tube. The supernatant was then centrifuged at 20,000 x
g for 20 min. at 4 ºC. The membrane pellet was washed three times with sucrose
buffer, homogenizing between each wash (5 ml sucrose buffer/brain) and
centrifuged at 15,000 x g for 20 min. at 4 ºC. Final membrane pellets were
resuspended in 1 mL sucrose buffer per brain with protease inhibitors, pepstatin-
A 1 µg/ml, antipain 1 µg/ml, leupeptin 1 µg/ml, aprotinin 1 µg/ml and PMSF 1

52
mM. Protein concentration was determined by Bradford protein assay (Bio-Rad)
according to manufacturer’s instructions.
2.12 Dialysis of brain membrane extracts
Brain membrane extracts were diluted in an equal volume of crude brain
homogenization buffer and dialyzed overnight. Dialysis tubing of MW 6,000 to
8,000 kD (Spectrum Laboratories, Inc., Racho Dominguez, CA) was utilized
buffered in 500 ml of crude brain homogenization buffer with fresh protease
inhibitors at 4 ºC. Once dialyzed, protein concentration was determined by
Bradford and aliquots were utilized fresh or frozen at –80 ºC.
2.13 GST pull down assays
Glutathione agarose (GA) beads were prepared according to the
manufacturer’s specifications (Sigma). GA beads were then washed in column
(BioRad Micro Bio-Spin columns) and equilibrated with 3 washes of MTPBS +10
% glycerol (10x bed volume/wash). GST-fusion proteins (20 µg) were added to
the column in 2-4 aliquots with each aliquot bound on a rotator at 4 ºC for 15 min,
then centrifuged (750 x g). Unbound protein was washed out with MTPBS +10 %
glycerol (2 washes, 10x bed volume/wash). GA beads with bound GST-fusion
protein were transferred to 15 ml tube and 20-30 mg of either crude brain extract
or dialyzed brain membrane extract was incubated for 4 hrs at 4 ºC with fresh
protease inhibitors added. Protein bound beads were centrifuged back into a
column and washed extensively with homogenization buffer (for GST-GABARAP,

53
GST-GABARAP-L1 and GST-GATE-16 constructs, increased NP-40 to 1 % and
Triton X-100 to 2 % in wash buffer). GA beads with fusion complexes bound
were then removed to fresh Eppendorf tube and 2x SDS-Loading dye (125 mM
Tris-Cl pH 6.8, 20 % glycerol, 4.6 % SDS, 2 mM β-mercaptoethanol, 0.002 %
bromophenol blue) was added to solubilize proteins.
2.14 Generation of antisera
To generate polyclonal antisera against GABARAP and homologues, full-
length proteins were generated as follows to inoculate rabbits. GST-fusion
proteins for GABARAP, GABARAP-L1 and GATE-16 were generated and bound
to glutathione agarose beads as above, with the bed volume increased to 200 µl
in 10 ml columns (BioRad Poly-prep) binding 1-2 mg of GST fusion protein.
GST-fusion protein bound to GA beads (Sigma) were washed twice with 20x bed
volumes of 1 % Triton X-100 in PBS and centrifuged (750 x g) to remove wash
solution. GA beads were then washed in GST wash buffer (50 mM Tris-Cl pH
7.5, 150 mM NaCl), then washed in 20 volumes of thrombin cleavage buffer (50
mM Tris-Cl pH 7.5, 150 mM NaCl, 2.5 mM CaCl2). After centrifugation to remove
buffer, beads were resuspended in 1 bed volume of thrombin cleavage buffer
and thrombin (0.4 mg/ml final concentration) was added and the mixture
incubated on rotator for 4 hrs at RT. Cleaved proteins were eluted from column
by washing 3 times with 1 bed volume of GST wash buffer. GST was eluted from
columns utilizing GST elution buffer (50 mM Tris-Cl, pH 8.0 with 5 mM reduced
glutathione). Cleavage and purity of proteins was determined by SDS-PAGE

54
stained with Commassie (Sambrook et al., 1989). Cleaved protein amounts were
determined by Bradford assay (Bio-Rad).
Once purified full-length proteins for GABARAP and homologues were
generated, immunization and bleeding of rabbits was done by Cocalico
Biologicals, Inc (Table 2.3). For the first immunization, 100 µg of antigen mixed
with Complete Freud’s Adjuvant was utilized and for all boosters done at 14, 21
and 49 days, 50 µg of antigen was mixed with Incomplete Freund’s Adjuvant
(Tables 2.4, 2.5). Pre-immune sera were taken prior to the first injections.
Table 2.3: Rabbits used for Immunization protocol Injected protein Identity number of Rabbits
GATE-16 PSU58, PSU59 GABARAP-L1 PSU62, PSU63 GABARAP PSU60, PSU61
Table 2.4: Immunization protocol for GATE-16 Date Procedure Performed with Animals Volume
05/31/00 Pre-Bleed PSU58, PSU59 5 ml, 6 ml 05/31/00 Initial Inoculation PSU58, PSU59 100µg antigen 06/14/00 First Boost PSU58, PSU59 50 µg antigen 06/21/00 Second Boost PSU58, PSU59 50 µg antigen 07/05/00 First Test Bleed PSU58, PSU59 7 ml, 7 ml 07/19/00 Third Boost PSU58, PSU59 50 µg antigen 07/26/00 Second Test Bleed PSU58, PSU59 6 ml, 6 ml 08/21/00 Fourth Boost PSU58, PSU59 50 µg antigen 08/28/00 Production Bleed 1 PSU58, PSU59 26 ml, 22 ml 09/11/00 Production Bleed 2 PSU58, PSU59 21 ml, 14 ml 09/18/00 Fifth Boost PSU58, PSU59 50 µg antigen 09/28/00 Exsanguination PSU58, PSU59 92 ml, 80 ml

55
Table 2.5: Immunization protocol for GABARAP, GABARAP-L1 Date Procedure Performed with Animals Volume
08/23/00 Pre-Bleed PSU60,61; PSU62,63 4,8 ml; 8,8 ml 08/23/00 Initial Inoculation PSU60,61; PSU62,63 100 µg antigen 09/06/00 First Boost PSU60,61; PSU62,63 50 µg antigen 09/13/00 Second Boost PSU60,61; PSU62,63 50 µg antigen 09/27/00 First Test Bleed PSU60,61; PSU62,63 7,7 ml; 7,7 ml 10/11/00 Third Boost PSU60,61; PSU62,63 50 µg antigen 10/18/00 Second Test Bleed PSU60,61; PSU62,63 7,6 ml; 7,7 ml 10/30/00 Exsanguination PSU60,61; PSU62,63 91,87 ml; 73,74 ml The test bleeds were taken at day 35 and day 56, then production bleeds
were taken after examination of test bleeds. Sera were aliquoted into 500 µl or 1
ml aliquots and stored at –80 ºC.
2.15 Analysis of Antisera
Purified antiserum was tested for binding to recombinant and endogenous
proteins by SDS-PAGE and Western analysis. Both purified recombinant
protein, and rat and mouse brain extracts (both crude and brain membrane) were
loaded onto 15 % SDS-PAGE and western analysis was done with different
antibody concentrations. In addition to determining the specificity of the antisera
for each homolog, each antiserum was tested with purified recombinant
GABARAP, GABARAP-L1 and GATE-16 proteins.

56
CHAPTER 3: RESULTS I
DISTINCT γ2 SUBUNIT DOMAINS MEDIATE CLUSTERING AND SYNAPTIC FUNCTION OF POSTSYNAPTIC GABAA RECEPTORS AND GEPHYRIN
3.1 Aim of Study
The γ2 subunit of GABAA receptors is essential for the clustering of
GABAA receptors at synapses and for proper function of inhibitory synapses. In
addition, the γ2 subunit recruits the subsynaptic scaffold protein gephyrin to
inhibitory synapses. This synaptic localization of GABAA receptors to the
synapse is critically important for fast inhibitory neurotransmission. While the γ2
subunit has been identified as a critical component for postsynaptic localization,
the mechanism and the molecular components required for this localization are
unknown. To elucidate the role of the γ2 subunit in clustering of GABAA receptors
and gephyrin and in enabling inhibitory synaptic function, we set out to map the
subunit domain(s) of the γ2 subunit required for synaptic localization of GABAA
receptors and for recruitment of gephyrin to synapses.
3.2 Results
3.2.1 Generation and transfection of GFP-γ2
Previous work done in cortical neurons and brain slices has shown that in
the absence of the γ2 subunit, GABAA receptors are unable to localize to

57
postsynaptic sites (Essrich et al., 1998). To ensure this phenotype was the same
when utilizing different culture conditions, immunohistochemical analysis was
performed on mature wildtype and γ2-subunit deficient neurons (γ2-/-). As
expected, punctate immunoreactivity for the γ2 subunit was completely lost in γ2-/-
neurons, whereas innervation indicated by immunoreactivity for the GABAergic
presynaptic terminal marker, glutamic acid decarboxylase (GAD)(Fig 3.1, A, B)
was unaffected. To confirm that loss of the γ2 subunit results in loss of
postsynaptic GABAA receptors, we further analyzed the cellular distribution of the
α2 subunit and the postsynaptic scaffold protein gephyrin, which are normally
colocalized with punctate γ2 subunit IR. Consistent with previous results obtained
using mixed glial neuron cultures (Essrich et al., 1998), these experiments
confirmed that loss of the γ2 subunit result in almost complete loss of
immunoreactive puncta for the α2 subunit and gephyrin (Fig. 3.1 C, D). Studies
by Schweitzer et al., (2003) have shown that the γ2 subunit is not only required
for GABAA receptor clustering during synaptogenesis when GABAA receptors are
presumably excitatory (Chen et al., 1996), but that the γ2 subunit is also required
for maintenance of receptors at mature inhibitory synapses. Consistent with this
observation, introducing the γ2 subunit into mature neurons allows rescue of
postsynaptic structure under conditions where GABAergic transmission is
inhibitory, indicating that GABA-mediated membrane depolarization is not a
prerequisite for γ2 subunit-dependent GABAA receptor clustering (Schweizer et
al., 2003).

58
A fusion construct consisting of green fluorescent protein (GFP) and the γ2
subunit (GFP-γ2) was generated to determine whether transfection of this subunit
into γ2-/- neurons would restore clustering of postsynaptic GABAA receptor in γ2
subunit-deficient neurons. The transfected GFP-γ2 produced a punctate pattern
of fluorescence similar to the punctate staining of the γ2 subunit in WT neurons.
Moreover, transfection of GFP-γ2 restored the punctate staining of the γ2 and α2
subunits suggesting that the transfected GFP-γ2 subunit rescued the formation of
receptors containing the α2 subunit. These receptors were localized at
postsynaptic sites as indicated by the perfect apposition of GFP fluorescence
and α2 immunoreactive puncta across from immunoreactive puncta for
presynaptic GAD (Fig. 3.1 E). Additionally, restoration of postsynaptic GABAA
receptors was associated with re-emergence of punctate gephyrin
immunoreactivity that was colocalized with postsynaptic GABAA receptors (Fig.
3.1 F), These results support the view that gephyrin is recruited to postsynaptic
sites by γ2-subunit containing GABAA receptors, rather than vice-versa.
Moreover, the findings suggest that transfection of γ2-/- neurons with γ2 subunit-
derived constructs provides a means to determine the subunit domains required
for proper postsynaptic targeting of GABAA receptors and for recruitment of
gephyrin to postsynaptic sites, without interference by the endogenous γ2
subunit.

59
Figure 3.1 Restoration of postsynaptic GABAA receptors and gephyrin clusters in γ2-/-
neurons by transfection of GFP-tagged γ2 subunit. A - D. Cortical neurons cultured from embryonic day 14.5 γ2+/+ (A, C) or γ2--/- embryos (B, D)(20 DIV) were double stained with antibodies specific for (A, B) the γ2 subunit (shown in green) and GAD (red) or (C, D) the α2 subunit (blue) and gephyrin (red); colocalization is shown in yellow (A, B) and magenta (C, D), respectively. Note the dramatic loss of punctate staining for the γ2 and α2 subunits as well as gephyrin in γ2-/- neurons, whereas presynaptic GAD staining is unchanged. E, F. Cortical neurons cultured from γ2-/- embryos where transfected at 18 DIV with GFP-tagged γ2 subunit (GFPγ2). They were fixed, permeabilized and processed for immunofluorescent staining at 21 DIV with an antiserum specific for the GABAA receptor α2 subunit (blue) and antibodies specific for either (E) GAD (red) indicating GABAergic terminals or (F) gephyrin (red), as indicated. Boxed dendritic segments are shown enlarged in the panels on the right for either GFPγ2 alone (green) or as merged images. Colocalization in the merged enlargement of the cell in (A) representing GFPγ2 (green) and the α2 subunit (blue) is shown in cyan blue and colocalization of GFPγ2 and GAD (red) is shown in yellow. Colocalization in the merged enlargement of the cell in (B) between gephyrin and the α2 subunit is shown in magenta and colocalization of GFPγ2 and gephyrin is shown in yellow. Scale bars, 10 µm.

60
3.2.2 Surface localization of the GFP-γ2 subunit
In neurons, the GFP-γ2 subunit had very low transfection efficiency
indicating that the neurons may have difficultly in either expressing this fusion
construct or in the proper postsynaptic localization of this construct. To
determine the ability of GFP-γ2 to efficiently express, localize and insert into the
plasma membrane, the GFP-γ2 subunit, which had a 9E10 epitope tag directly
downstream of GFP, was co-expressed with α2 and β3 untagged subunits and
visualized for surface expression in HEK 293T cells. Surface expression was
visualized by surface labeling of fixed, unpermeabilized cells with antibodies
directed against the α2 subunit and the myc epitope on the GFP-γ2 fusion
construct. While the GFP-γ2 subunit was able to efficiently express in these
cells, as seen by both GFP fluorescence and myc staining (mAb) (Fig. 3.2 A, in
green), surface labeling of the extracellular myc epitope of GFP-γ2 (rabbit anti-
myc, blue) was barely detectable, indicating that a major portion of the GFP-γ2
subunit did not reach the cell surface (Fig. 3.2 A’’). Unlike the GFP-γ2 subunit,
untagged GABAA receptor subunits efficiently targeted to the membrane surface,
as seen by the α2-IR detected on non-permeabilized cells (Fig. 3.2 A’). In
contrast, the 9E10 epitope-tagged γ2 subunit, lacking the GFP fusion protein at
the N-terminus was able to efficiently localize to the membrane surface (rabbit
anti-myc, blue, Fig. 3.2 B’’’) and colocalize with α2 subunit immunoreactivity at
the cell surface (Fig 3.2 B’’). Importantly the majority of the 9E10γ2 subunit was
localized to the membrane surface as evidenced by the low level of intracellular

61
Figure 3.2 Surface expression of GFP-γ2 and 9E10γ2 comparison in 293T cells. HEK 293T
cells were transfected with either GFP-γ2 (A) or 9E10γ2 (B), and α2 and β3 subunits and examined for surface expression of the γ2 subunit and colocalization with alpha and beta subunits. A. Transfected GFP-γ2 showed high expression levels seen by GFP fluorescence (green), however, the GFP-γ2 subunit could not efficiently localize to the membrane surface as seen by immunofluorescent staining of non-permeabilized cells against the 9E10 epitope (rabbit α myc) found directly downstream of GFP (blue). Efficient targeting of GABAA receptors to the membrane surface was seen by surface labeling of the α2 subunit utilizing an antiserum against an extracellular epitope (red). B. By comparison, the 9E10γ2 subunit allows for efficient localization to the membrane surface as seen by the high intensity of surface labeling (blue, rabbit α myc) and this 9E10γ2 subunit was able to colocalize with surface α2-IR (red, guinea pig α α2). Little of the 9E10γ2 subunit was trapped intracellularly, as seen by total cell labeling with a mouse α myc antibody after permeabilization (green).

62
9E10γ2 immunoreactivity (myc mAb, green, Fig. 3.2 B). Compared to 9E10γ2,
transfection of GFP-γ2 would therefore severely compromise the trafficking and
immunofluorescent detection of recombinant receptors containing transfected γ2
subunit constructs.
3.2.3 Design and functional characterization of chimeric subunit
constructs
Towards mapping the γ2 subunit domains required for proper localization
of GABAA receptors, we generated a series of chimeric constructs in which
different extracellular, transmembrane and intracellular domains of the γ2 subunit
were replaced with homologous domains derived from the α2 subunit or the β2
subunit (Fig.3.3). The α2 subunit was chosen for construction of γ2/α2 chimeric
subunits because it is normally expressed almost exclusively at postsynaptic
sites yet strictly dependent on the γ2 subunit and gephyrin for postsynaptic
localization (Fig. 3.1 A-D) (Essrich et al., 1998; Schweizer et al., 2003). We
adopted a tripartite nomenclature to describe these constructs (for example
9E10α-γ-γ), with the first term indicating the subunit origin of the epitope-tagged
extracellular domain together with the first three transmembrane domains, the
second term indicating the major cytoplasmic loop domain between TM3 and
TM4, and the third term indicating the origin of the TM4 domain and the short C-
terminal tail (Fig. 3.3 B). In addition to these α2/γ2 chimeric constructs, the result
of one construct (9E10α-α-γ) was verified with an analogous construct in which the

63
α2 subunit portion was replaced with the corresponding domain derived from the
β2 subunit (9E10β-β-γ). Sequences from the β2 rather than the β1 or β3 subunit
were used because, unlike the β1 and β3 subunit, the β2 subunit requires
assembly with an α subunit for surface expression in heterologous cells (Sigel et
al., 1989; Connolly et al., 1996; Krishek et al., 1996; Wooltorton et al., 1997). As
noted above, to maximize insertion into the plasma membrane of recombinant
subunit constructs we relied on the 9E10 epitope rather than GFP as a tag to
monitor expression of transfected constructs. In addition to the vector backbone,
non-coding sequences at the 5’ end including the leader peptide, the first four
amino acids and the 9E10 epitope were kept the same in all constructs to
minimize potential differences in expression levels. Save for maximally 32
nucleotides downstream of the translational stop signal, all GABAA receptor
subunit-derived 3’ untranslated mRNA sequences were deleted to avoid
sequences that might affect dendritic targeting of transcripts. Proper expression
of all constructs was assessed following co-transfection with α2 and β3 subunits
into HEK 293T cells. Western blot analyses of whole cell extracts of transfected
cells indicated that the chimeric constructs gave rise to stable polypeptides of the
expected mobility when analyzed by SDS-PAGE and were expressed at similar
steady state levels (Fig. 3.3 C).
3.2.4 Cellular distribution of chimeric subunits
To determine the ability of these chimeric subunits to properly form
receptors and localize to the membrane surface, we compared their cellular

64
Figure 3.3 Schematic representation of chimeric subunit constructs and analysis of their expression following transfection into 293T cells. A. Schematic representation of the myc 9E10 epitope-tagged γ2 subunit with the position of silent restriction sites inserted flanking the large cytoplasmic loop domain. The γ2 subunit translational open reading frame is shown as a gray box, with N- and C-termini indicated. Short black lines above this box indicate the positions of the four transmembrane domains. The 9E10 epitope tag and an adjacent Spe I site are inserted between the 4th and 5th amino acid of the open reading frame. B. Schematic representation of chimeric constructs with the � 2 subunit-derived portion shown in gray and the α2 subunit-derived portion shown in black. The nomenclature for chimeric construct indicated underneath each drawing is explained in the text. C. Western blot analysis of chimeric constructs cotransfected with α2 and β2 subunits into HEK 293T cells. Equal amounts of protein (40 µg) were loaded on the gel and the blot was developed using an antiserum raised against the 9E10 myc epitope. The ubiquitous protein band running as a band of approximately 60-kDa represents endogenous myc and serves as a gel loading control. The constructs and subunits transfected in each lane are indicated.

65
distribution in the presence and absence of the α2 and β3 subunits in HEK 293T
cells. Previous studies have shown that the γ2S subunit alone is able to form
homomeric receptors that can localize to the membrane surface in heterologous
cells (Connolly et al., 1999). However, only low levels of surface expression can
be seen for the α2 subunit, which is thought to be due to formation of
heteromeric receptors utilizing endogenous receptor subunits expressed in HEK
cells. To visualize the surface expression, immunofluorescent staining of non-
permeabilized HEK293T cells was performed for all transfected constructs. In
the absence of α/β the only construct that was able to efficiently localize to the
membrane surface was the γ2 subunit, as expected (Connolly et al., 1999) (Fig.
3.4 A). In contrast, none of the chimeric constructs, nor the α2 subunit were able
to efficiently reach the cell surface when expressed alone (Fig. 3.4 B-H).
However, when coexpressed with α2 and either β2 or β3 subunits, all chimeric
constructs were able to localize on the plasma membrane (Fig 3.4 A’-H’).
Surface expression of α subunits is known to require coassembly with β subunits
into heteromeric ion channels (Connolly et al., 1999 and data not shown).
Therefore, the data indicate that all the chimeric constructs are able to assemble
with the α2 and or β2/3 subunits, thereby forming heteromeric complexes that
are efficiently inserted into the plasma membrane.
To confirm the ability of chimeric subunits to form functional receptors on
the membrane surface of HEK 293T cells with α and β subunits,
electrophysiological analysis was performed (collaboration with G. Chen and J.

66
Mulder-Rosi). The GABA efficacy of receptors found in γ2 subunit-deficient
neurons is significantly below that of wildtype receptors (Günther et al., 1995;
Essrich et al., 1998) consistent with reduced single channel conductance of γ2
subunit-deficient GABAA receptors (Crestani et al., 1999; Lorez et al., 2000) and
studies of recombinant receptors indicating that the γ2 subunit contributes to
normal GABAA receptor channel function (Verdoorn et al., 1990; Angelotti and
Macdonald, 1993). Therefore, in order to determine whether the chimeric
constructs could contribute to functional GABA-gated ion channels, we used
whole cell patch clamp analyses of HEK 293T cells transfected with either α2
and β3 subunits alone or together with different chimeric constructs. GABA dose
response curves were recorded to evaluate differences in the GABA efficacy of
putative receptors (EC50 = GABA concentration resulting in half maximal GABA-
evoked currents) (Fig. 3.5 A, B). On co-expression with α2β3 subunits, the
9E10α−γ−γ, 9E10γ−α−γ, 9E10α-α-γ and 9E10γ-γ-α constructs produced channels with
GABA EC50 values significantly lower than the value observed for α2β3 receptors
[EC50 (α2β3) = 48.9 ± 4.5 µM (s.e.m.), (α2β3 + 9E10α-γ-γ = 26.2 ± 1.9 µM, p ≤
0.01; (α2β3 + 9E10γ-α-γ = 21.0 ± 2.7 µM, p ≤ 0.001; (α2β3 + 9E10α-α-γ = 18.4 ± 1.6
µM, p ≤ 0.001; (α2β3 + 9E10 γ-γ-α = 16.7 ± 1.9 µM, p ≤ 0.01] and comparable or
even lower than the value for the α2β3 + 9E10γ-γ-γ subunit combination (EC50 =
25.6 ± 3.9 µM). Similar results were obtained when the β2 subunit was
substituted for β3 and irrespective of whether the chimeric constructs were
transfected at a 1:1 ratio with α2 and β3 subunits or at a 10-fold excess (data not

67
Figure 3.4 Analysis of surface expression of chimeric subunit constructs transfected into
293T cells. The 9E10-tagged constructs indicated were transfected either alone (A - H) or together with α2 and β3 subunits (A’ - H’). Non-permeabilized cells were stained with antibody specific for the 9E10-tagged construct indicated (A - H) or double labeled for this construct and the α2 subunit (A’ - H’). Both antibodies are directed against N-terminal epitopes and selectively recognize subunits inserted into plasma membrane. The cells were developed with fluorescent secondary antibody for imaging. Note the efficient expression of the γ2 subunit (9E10γ-γ-γ) in the plasma membrane independent of whether it is expressed alone (A) or together with α2 and β3 subunits (A’). In contrast, efficient incorporation of the α2 subunit (B, B’) or chimeric constructs (C - H, C’ - H’) depends on coexpression of the α2 or β3 subunit or both.
Figure 3.5 GABA dose-response curves of GABAA receptors containing chimeric subunits
expressed in 293T cells. A, B. The α2 and β3 subunits were transfected either alone or in combination with (A) the 9E10γ−γ−γ, 9E10α−γ−α or 9E10γ−γ−α constructs or (B) in combination with the 9E10α−γ−γ, 9E10γ−α−γ or 9E10α−α−γ constructs. GABA-evoked whole-cell currents were normalized to the maximal responses obtained at 1 - 5 mM GABA application. For each concentration tested, the data were averaged from 3 - 11 cells

68
shown). In contrast, the 9E10α-γ-α construct co-expressed with α2 and β3
subunits produced less responsive receptors with an EC50 value significantly
greater than that of α2β3 receptors [EC50 (α2β3+ 9E10α-γ-α) = 107.9 ± 17.2, p <
0.05] (Fig. 3.5 B). Taken together, the GABA dose response curves of
recombinant receptors containing α/γ subunit chimeric constructs confirm and
extend the conclusions from immunofluorescent analyses and show that the
9E10α-γ-γ, 9E10γ-α-γ, 9E10γ-γ-α, 9E10α-α-γ, and 9E10α-γ-α constructs can each
assemble with α and/or β subunits and contribute to the formation of GABA-
gated ion channels that are functionally distinct from channels produced by α2
and β3 subunits alone.
3.2.5 Receptor domains required for postsynaptic localization
In order to determine the γ2 subunit domains required for trafficking and
accumulation of GABAA receptors at postsynaptic sites, each of the constructs
was transfected into cultured cortical neurons derived from γ2-/- embryos at 18
DIV and the cultures processed for immunofluorescence analyses three days
later. Whereas immunoreactivity for the 9E10γ-γ-γ construct (anti-myc antiserum)
was found to accumulate at membrane sites apposed to GAD-positive
GABAergic terminals, essentially no punctate staining was evident for the 9E10α-
α-α construct, as expected (Fig. 3.6, A, B). This confirms that the
overexpression of any subunit into the γ2-/- neurons does not result in
accumulation at postsynaptic sites, and indicates that a γ2-specific signal is

69
required for postsynaptic localization. To narrow down the region required for
this localization, chimeric constructs containing portions of the α2 sequence
inserted into the γ2 subunit backbone were transfected into γ2-/- neurons and
examined for their subcellular localization. The 9E10α-γ-γ construct revealed
punctate staining apposed to GAD-positive terminals similar to 9E10γ-γ-γ,
indicating that the extracellular domain and the first three transmembrane
domains of the γ2 subunit are dispensable for postsynaptic localization (Fig. 3.6
C). Surprisingly, the 9E10α-α-γ construct accumulated at postsynaptic sites similar
to 9E10γ-γ-γ, suggesting that clustering and postsynaptic localization of γ2 subunit-
containing GABAA receptors can be mediated by the TM4 domain of the γ2
subunit and that the cytoplasmic domain of γ2 subunit is dispensable (Fig. 3.6 D).
Consistent with this notion, the 9E10γ-α-γ construct (Fig. 3.6 E) was clustered and
localized to postsynaptic sites similar to the 9E10α-α-γ construct. However, both
the 9E10α-γ-α and 9E10γ-γ-α constructs (Fig. 3.6 F, G) failed to cluster at post-
synaptic sites. These constructs had limited, mainly diffuse expression in
dendrites. To further corroborate these findings, we analyzed whether the TM4
region of the γ2 subunit could function similarly when tested in the context of the
β2 subunit backbone (9E10β-β-γ). While the 9E10β-β-γ expression level was
comparatively low in HEK293T cells, this construct produced immunoreactive
puncta that were apposed to GAD-positive terminals (Fig. 3.6 H), consistent with
the previous work indicating the TM4 of the γ2 subunit is sufficient for

70
Figure 3.6 Restoration of postsynaptic GABAA receptor clusters in γ2-/- neurons transfected with chimeric γ2/α2 subunit constructs. A – H. Cortical neurons isolated from γ2-/- embryos were transfected at 18 DIV with (9E10) epitope-tagged constructs indicated and processed for immunofluorescence analysis at 21 DIV for the 9E10 epitope (shown in green) and presynaptic GAD (red). Shown are merged images with colocalization represented in yellow. Boxed dendritic segments of the images are shown enlarged in separate panels below each image. Note the faithful formation of clusters revealed in the form of punctate staining for the 9E10γ-γ-γ (A), 9E10α-γ-γ (C), 9E10α-α-γ (D), γ-α-γ (E) and β−β−γ (H) constructs that were closely apposed to presynaptic GAD, whereas the 9E10α-α-α (B), 9E10α-γ-α (F) and 9E10γ-γ-α (G) constructs failed to form clusters and showed no juxtaposition to GAD immunoreactivity. Arrows point to clusters apposed to GAD, arrowheads indicate 9E10 immunoreactivity that is diffuse or punctate but not apposed to GAD immunoreactivity. Scale bar is 10 µm per image and 5 µm per dendritic enlargement.

71
postsynaptic localization of GABAA receptors. To confirm our visual impression,
cells with proper GABAergic innervation (n = 13 - 18 cells for each construct)
were subjected to semiquantitative analyses using automatic detection of
immunofluorescent puncta above a set fluorescence intensity threshold (see
Material and Methods). The total number of clusters per dendritic segment of 40
µm detected was examined first for each of the constructs. The 9E10α-γ-γ (9.87 ±
0.67), 9E10α-α-γ (9.53 ± 0.81), 9E10γ-α-γ (7.68 ± 0.57) and 9E10β−β−γ � � � ± 1.40)
constructs were indistinguishable from values determined for the 9E10γ-γ-γ (8.5 ±
0.66 ) (Fig. 3.7 A). Moreover, the percentage of these clusters that were
apposed to presynaptic GAD immunoreactivity were indistinguishable from
corresponding values observed for the γ2 subunit (87.9% ± 1.65 s.e.m.) with the
9E10α-γ-γ with 73.8% ± 5.56, the 9E10α-α-γ with 84.8% ± 3.67, the 9E10γ-α-
γ with, 88.6% ± 3.07 and the 9E10β−β−γ with 76.6% ± 5.71 (Fig. 3.7 B, C). In
contrast, significantly fewer clusters were detected for the 9E10α-α-α (4.38% ±
0.85 ; p<0.01)., 9E10α-γ-α (4.92% ± 0.93; p<0.05) and 9E10γ-γ-α (491% ± 0.73;
p<0.05) constructs (Fig. 3.7 A) and almost none of these clusters were apposed
to GAD 9E10α-α-α (12.94% ± 14.8, p<0.001), 9E10α-γ-α (14.95% ± 13.8, p<0.001)
and 9E10γ-γ-α (17.69% ± 17.63, p<0.001)(Fig. 3.7 B) indicating that the clusters
seen represented extrasynaptic receptors. The area of the synaptic puncta was
indistinguishable for the synaptic constructs with the 9E10γ-γ-γ having an average
area of 1.53 µm ± 0.139, the 9E10α-γ-γ having 1.30 µm ± 0.136, the 9E10α-α-
γ, 1.274 µm ± 0.153, the 9E10γ-α-γ 1.29 µm ± 0.095 and the 9E10β−β−γ 1.18 µm ±

72
Figure 3.7 Quantitative analyses of postsynaptic clusters formed by chimeric constructs
transfected into γ2-/- neurons. Cortical cultures isolated from γ2-/- embryos were transfected and double labeled for the 9E10 epitope tag of the transfected construct and for the GABAergic terminal marker GAD as shown in Fig. 5. Immunoreactive puncta on dendritic segments that were innervated by a GABAergic axon as revealed by GAD immunoreactivity were quantified from digitized video images as described in Material and Methods. A. The average number of 9E10 immunoreactive puncta per 40 µm dendritic segment determined for each of the chimeric constructs including 9E10α-α-α was compared to the value determined for the γ2 subunit (9E10γ-γ-γ). Note the similar number of puncta observed for the 9E10γ-γ-γ, 9E10α-γ-γ, 9E10α-α-γ, 9E10γ-α-γ, and 9E10β-β-γ constructs (n = 13, 15, 19, 16, 10 cells, respectively). In contrast, the number of puncta observed for the 9E10α-γ-α (n = 14) and 9E10γ-γ-α (n = 16) constructs was similar to that observed for the α2 subunit (9E10α-α-α, n = 17) and greatly reduced compared to the γ2 subunit (9E10γ-γ-γ). B. For comparison of the number of clusters localized to postsynaptic sites, the fraction of puncta apposed to punctate GAD immunoreactivity was determined for each chimeric construct and compared to the value determined for 9E10γ-γ-γ. Note that the same constructs that showed a high propensity to cluster in (A) similar to the γ2 subunit are also indistinguishable from the γ2 subunit in that they result in a high percentage of clusters that are postsynaptic. In contrast, the percentage of immunoreactive puncta for the 9E10α-γ-α and 9E10γ-γ-α constructs and the α2 subunit (9E10α-α-α) are significantly reduced compared to the value for the γ2 subunit. C. The average size of postsynaptic clusters induced by the 9E10α-γ-γ, 9E10α-α-γ, 9E10γ-α-γ, ανδ 9E10β-β-γ constructs is indistinguishable from the value observed for the γ2 subunit (9E10γ-γ-γ). Error bars, standard error; ∗,∗∗, ∗∗∗, p < 0.05, 0.01, 0.001.

73
0.171. However, the area of the synaptic puncta for the negative chimeric
subunits could not be accurately measured due to the low number of synaptic
puncta. In summary, the TM4 domain of the γ2 subunit is required and sufficient
to deliver αβγ2 receptors to dendritic sites apposed to GABAergic terminals.
Conversely, the γ2 subunit major cytoplasmic loop domain is neither sufficient nor
required for postsynaptic clustering.
3.2.6 Recruitment of gephyrin to GABAA receptors
Postsynaptic GABAA receptors are invariably colocalized with the
subsynaptic scaffold protein gephyrin (Sassoe-Pognetto et al.; Cabot et al., 1995;
Sassoé-Pognetto et al., 1995; Giustetto et al., 1998; Sassoe-Pognetto et al.,
2000). However, the function of gephyrin with respect to clustering and targeting
of GABAA receptors remains ill-defined. In the case of the closely related glycine
receptors, interaction of receptors and gephyrin is mediated by the major
cytoplasmic loop domain of the receptor β subunit and this interaction is believed
to mediate clustering and postsynaptic anchoring of glycine receptors in the
postsynaptic membrane (Prior et al., 1992; Meyer et al., 1995; Kneussel et al.,
1999). To address whether the same region of the γ2 subunit that localizes the
receptors to the synapse also can recruit gephyrin to the synapse, all of the
constructs shown to induce postsynaptic clusters above (9E10γ-γ-γ, 9E10α-γ-γ, 9E10γ-
α-γ, 9E10α-α-γ, and 9E10β-β-γ ) were analyzed with respect to their ability to induce
colocalization with gephyrin (Fig. 3.8, A-D). Indeed, following transfection into γ2-

74 /- cortical neurons all of the constructs that formed postsynaptic clusters in Fig.
3.6 also resulted in recruitment and clustering of gephyrin as evident by
colocalization of punctate immunoreactivity for the transfected constructs (anti-
myc antiserum) and endogenous gephyrin (mAb7a). However, whereas the
9E10α-γ-γ construct recruited gephyrin as effectively as the γ2 subunit (9E10γ-γ-γ) as
determined by the percentage of 9E10 immunoreactive puncta colocalized with
punctate gephyrin immunoreactivity [9E10γ-γ-γ, 73.8 ± 4.1 (s.e.m.); 9E10α-γ-γ, 83.7 ±
3.9, p > 0.05], a significantly lower fraction of the 9E10γ-α-γ, 9E10α-α-γ and 9E10β-β-γ
clusters were colocalized with gephyrin compared to 9E10γ-γ-γ (9E10γ-α-γ, 37.1±
6.1, p < 0.01; 9E10α-α-γ, 30.8 ± 7.0, p < 0.01; 9E10β-β-γ, 45.2 ± 7.6, p < 0.01; n = 10
- 26 cells per construct) (Fig. 3.8 E). Less efficient recruitment of gephyrin by the
9E10γ-α-γ, 9E10α-α-γ � � � 9E10β-β-γ constructs was reflected by a significant
number of immunoreactive puncta for these constructs that were not colocalized
with gephyrin immunoreactivity (Fig. 3.8 C, D). Moreover, cells transfected with
the 9E10γ-α-γ or 9E10α-α-γ or 9E10β-β-γ constructs more often than the 9E10γ-γ-γ and
9E10α−γ-γ constructs failed to recruit gephyrin entirely (data not shown). The low
percentage colocalization given for these constructs in Fig. 3.8 F are likely to be
overestimated because transfected cells that lacked gephyrin immunoreactivity
were excluded from quantitation. Thus, it appears that both the TM4 and the
major cytoplasmic loop region of the γ2 subunit contribute to GABAA receptor-
mediated recruitment of gephyrin to postsynaptic sites. It is possible that the
cytoplasmic sequences from the β or α subunits also contributes to the residual

75
Figure 3.8 Recruitment of gephyrin to GABAA receptor clusters. The γ2 subunit (A) and the three other γ2 subunit TM4-containing chimeric constructs shown in Figure 5 to form clusters (B, 9E10α−γ−γ; C, 9E10γ−α−γ; D, 9E10α−α−γ, � , 9E10β-β-γ) were transfected into γ2-/- neurons and analyzed for colocalization of punctate 9E10 immunoreactivity (green) with endogenous gephyrin (red). In addition, analysis of the diffusely expressed construct 9E10α−γ−α (F), which is not concentrated at synapses (Fig. 3.6 F), is shown for comparison. Boxed dendritic segments are shown enlarged in color-separated and merged panels underneath each image with arrows pointing to colocalized clusters (yellow) and arrowheads indicating punctate 9E10 immunoreactivity that was not colocalized (green). Note the similar high degree of colocalization seen for the γ2 subunit (A) and for the 9E10α-γ-γ construct (B) with only few 9E10-immunoreactive puncta that were not colocalized with gephyrin (green puncta in the merged enlargement). In contrast, only a fraction of the 9E10 immunoreactive puncta that were formed by the 9E10γ-α-γ, 9E10α-α-γ, � � � 9E10β-β-γ constructs were colocalized with gephyrin (C-E). This visual impression was confirmed by quantitative analyses (G). The percentage of 9E10α-γ-γ puncta colocalized with gephyrin was similar to values seen with the γ2 subunit. In contrast, the percentage of 9E10γ-α-γ, 9E10α-α-γ, or 9E10β-β-γ puncta colocalized was significantly reduced compared to the γ2 subunit, consistent with the notion that GABAA receptors can form clusters in the absence of gephyrin. Colocalization of 9E10α−γ−α and gephyrin (F) could not be quantified because expression of this construct was mostly diffuse. Scale bar is 10 µm per image and 5 µm per dendritic enlargement.

76
recruitment of gephyrin in the presence of the γ2 subunit TM4 domain alone,
which enables the TM4 domain to contribute to limited recruitment of gephyrin
independent of other γ2 subunit sequences. Alternatively, the few gephyrin
clusters seen in the absence of the γ2 cytoplasmic loop region may represent
gephyrin associated with γ3 subunit containing receptors. Regardless, the
increase in efficiency of recruitment by the γ2 subunit cytoplasmic loop domain is
strictly dependent upon the γ2 subunit TM4 domain.
3.2.7 Assessment of inhibitory synaptic clustering
The finding that the cytoplasmic portion of the γ2 subunit was unable to
induce clustering of GABAA receptors is surprising, given that postsynaptic
targeting of the closely related glycine receptors, is mediated by interaction of
gephyrin with the cytoplasmic domain of the receptor β subunit. It is possible that
constructs that contain the γ2 subunit cytoplasmic domain in the absence of the
TM4 domain would interfere with synapse formation in a dominant negative
fashion. To address this possibility, we transfected the 9E10γ-γ-γ, 9E10α-γ-α and
9E10γ-γ-α constructs into wildtype neurons and addressed whether they would
interfere with postsynaptic clustering of endogenous gephyrin (Fig. 3.9). The γ2
subunit (9E10γ-γ-γ) formed immunoreactive puncta that were colocalized with
gephyrin clusters and juxtaposed to GABAergic sites as expected (Fig. 3.9 A).
Similar to observations made in γ2-/- neurons, the 9E10α-γ-α and 9E10γ-γ-α
constructs were diffusely expressed in dendrites of wildtype neurons and they did

77
not interfere with clustering and postsynaptic localization of endogenous gephyrin
(Fig. 3.9 B, C). Thus, failure of these constructs to cluster and to accumulate at
postsynaptic sites in γ2-/- neurons is unlikely to be due to negative effects of
these constructs on mechanisms involved in synapse formation. To confirm that
these constructs did not have negative effects on mechanisms involved in
synapse formation, we also transfected these dysfunctional constructs into γ2-/-
neurons and examined them for proper formation of immunoreactive puncta
apposed to glutamatergic sites. Immunohistochemical analysis was performed
utilizing an antibody against SynGap, a protein component of the postsynaptic
density of excitatory synapses (Vazquez et al., 2004). The γ2-/- neurons were
transfected with either the positive control 9E10γ−γ−γ, or the dysfunctional chimeric
constructs 9E10α−γ−α, 9E10γ−γ−α or 9E10α−α−α. None of these chimeric constructs
showed overt differences in the immunoreactive puncta for SynGAP compared to
neurons transfected 9E10γ−γ−γ control construct (data not shown). These
experiments confirmed that the 9E10α−γ−α, 9E10γ−γ−α failed to cluster at
postsynaptic due to lack of proper targeting signals rather than due to dominant
negative or toxic effects of the artificial polypeptides.
3.2.8 Rescue of inhibitory synaptic function
Efficient insertion of chimeric constructs into the plasma membrane of
293T cells requires coexpression of α and β subunits (Fig. 3.4), suggesting that
these constructs assemble into heteromeric complexes. To test whether chimeric

78
constructs can assemble with endogenous α and β subunits and restore the
function of GABAergic inhibitory synapses in γ2-/- neurons, we engaged in a
collaboration with J. Mulder-Rosi and G. Chen to record miniature inhibitory
postsynaptic currents (mIPSCs) in transfected neurons. The frequency of
mIPSCs recorded from γ2-/- compared to wildtype cortical neurons (DIV 17 - 22)
was greatly reduced as previously shown (Essrich et al., 1998). Upon
transfection of γ2-/- neurons with the γ2 subunit (9E10γ-γ-γ) or with the 9E10α-γ-γ
chimeric construct, the mIPSC frequency was restored to values similar to the
wildtype control, consistent with restoration of postsynaptic localization and
function of GABAA receptors (Fig. 3.10 A, B). Furthermore, the 9E10γ-γ-α and
9E10α-γ-α constructs both failed to restore mIPSCs, consistent with the notion that
the γ2 subunit TM4 is required for postsynaptic clustering of GABAA receptors
and gephyrin.
Unexpectedly, however, the 9E10γ-α-γ and 9E10α-α-γ constructs failed to
restore mIPSCs upon transfection into γ2-/- neurons although they formed
clusters at postsynaptic sites similar to the 9E10γ-γ-γ and 9E10α-γ-γ constructs. The
very low mIPSC frequency observed for the 9E10γ-α-γ and 9E10α-α-γ constructs
expressed in γ2-/- neurons was similar to that recorded from untransfected γ2-/-
neurons (Fig. 3.10 A, B). Moreover, the low mIPSC frequency of 9E10γ-α-γ or
9E10α-α-γ transfected cells was mirrored in a significantly lower amplitude of rare
miniature currents detected compared to values seen with the 9E10γ-γ-γ construct
(9E10γ-γ-γ, 34.4 ± 3.1 pA, n = 10 cells showing minis; 9E10γ-α-γ, 16.4 ± 1.7 pA, n =

79
8, p < 0.001; 9E10α-α-γ, 16.2 ±1.0 pA, n = 3, p < 0.001; Students t-test). Not
surprisingly, the number of γ2-/- neurons transfected with the 9E10γ-γ-α or 9E10α-γ-α
constructs that revealed any mIPSCs at all were greatly reduced compared to
9E10γ-γ-γ transfected γ2-/- neurons (not shown). The amplitudes of rare synaptic
events detected in a minority of the 9E10γ-α-γ or 9E10α-α-γ transfected cells did not
differ significantly from the amplitude of mIPSCs sporadically detected in a
subset of untransfected γ2-/- neurons (13.5 ± 2.5 pA, n = 5, p > 0.3 for
comparison to both, 9E10γ-α-γ and 9E10α-α-γ) confirming that these two constructs
did not rescue synaptic function. Thus, reminiscent of the structural prerequisites
for efficient recruitment of gephyrin, restoration of mIPSCs requires both the γ2
subunit TM4 and cytoplasmic loop domains. Conversely, the data suggest that
postsynaptic accumulation of GABAA receptors alone is not sufficient to ensure
synaptic function.

80
Figure 3.9 Functional analyses of 9E10α-γ-α and 9E10γ-γ-α constructs in wildtype neurons. The
9E10γ-γ-γ, 9E10α-γ-α, and 9E10γ-γ-α construct which ailed to form clusters on transfection into γ2-/- neurons were transfected into wildtype neurons and double labeled for the 9E10 epitope tagged construct and endogenous gephyrin to test for negative effects on formation of gephyrin clusters. A. The 9E10γ-γ-γ construct formed clusters colocalized with gephyrin as expected. The 9E10α-γ-α (B) and 9E10γ-γ-α (C) constructs failed to cluster but endogenous gephyrin clusters remained unaffected, indicating that failure of these constructs to accumulate at synapses was not associated with dominant negative effects on synapse formation. Scale bars, 10 µm.
Figure 3.10 Rescue of mIPSCs in γ2-/- neurons requires both the major intracellular loop
and the fourth transmembrane domain of the γ2 subunit. A. Representative traces are shown of mIPSCs recorded from γ2+/+ and γ2-/- neurons and of γ2 /- neurons transfected with the γ2 subunit (9E10γ-γ-γ) or chimeric subunits as indicated. B. Summary of data showing mIPSC frequencies confirming that transfection of either 9E10γ-γ-γ or 9E10α-γ-γ restored the function of inhibitory synaptic transmission in γ2-/- neurons to values similar to those found in wildtype neurons. Note that none of the 9E10γ-γ-α, 9E10γ-α-γ, 9E10α-γ-α and 9E10α-α-γ constructs were able to restore mIPSCs of γ2-/- neurons, indicating that both the γ2 subunit cytoplasmic loop and TM4 domain are required for restoration of synaptic function in γ2-/- neurons. Error bars, standard error; ∗∗∗, p < 0.001.

81
CHAPTER 4: RESULTS II
GEPHYRIN, COLLYBISTIN AND THEIR ROLE IN PROPER LOCALIZATION OF GABAA RECEPTORS
4.1 Aim of Study
Collybistin, a GTP-GDP exchange factor, is known to interact with and
translocate gephyrin to sub-membranous clusters. It has been postulated that
collybistin initiates local remodeling of the subsynaptic cytoskeleton of inhibitory
synapses (Kneussel and Betz, 2000). However, very little is currently known
about the mechanism and downstream effects of the collybistin-gephyrin
interaction. We joined a multi-laboratory collaborative effort to examine the
protein domains that mediate interaction between gephyrin and collybistin, and to
assess the functional consequences of a naturally occurring mutation in
collybistin found in a patient suffering from hyperekplexia and epilepsy, in which I
examined the phenotypes of the isoforms and mutants when transfected into
neurons. The consequences of this mutation were analyzed with respect to its
effects on trafficking and postsynaptic clustering of gephyrin and postsynaptic
GABAA receptors.
4.2 Results
Collybistin (CB) is cdc42-directed GTP exchange factor with RhoGEF
(also known as dbl or DH) and PH domains (Fig. 4.1 A). The protein was

82
originally identified as gephyrin binding protein and is also known as hPEM-2
(Reid et al., 1999). Initially two rat isoforms of collybistin were described, that
differ by the presence (SH3+, CB1) or absence (SH3-, CB2) of an SH3 domain in
the N-terminal sequence and a coiled-coiled domain at the C-terminus of the
CB1 isoform (Kins et al., 2000). Using heterologous expression in HEK 293 cells,
the SH3 domain-lacking variant CB2 was shown to promote submembrane
clustering of gephyrin and surface expression of recombinant glycine receptors in
a gephyrin-dependent manner (Kins et al., 2000). However, more detailed
analyses of collybistin cDNAs from three different species (rat, mouse and
human) revealed the existence of three distinct alternatively spliced main forms
of collybistin termed CB1, CB2 and CB3 (Harvey et al., 2004). Each of these
isoforms can again exist as two alternate splice variants distinguished by the
presence (SH3+) or absence (SH3-) of an N-terminal SH3 domain. RT-PCR
analyses of rodent spinal cord and brain tissues revealed that CB2SH3+ and
CB3SH3+ were the predominant isoforms expressed in these species.
4.2.1 Functional analyses of collybistin isoforms
To determine the cellular localization of the major collybistin isoforms in
neurons, mouse cortical wild-type neurons were transfected with myc epitope-
tagged collybistin constructs (Fig. 4.1 A). Both myc-CB2SH3- and myc-CB2SH3
were diffusely distributed throughout the soma and dendrites of transfected cells
as seen by the diffuse immunoreactivity (Fig. 4.2 A, B). We further assessed
whether overexpression of either of these isoforms had any effect on gephyrin

83
Figure 4.1: Schematic representation of collybistin and gephyrin constructs. Amino acid numbers are indicated above each construct. A. Collybistin isoforms, CB2 and CB3 are most abundant in rat and human. CB2 exists in two isoforms, differing in an N-terminal SH3 domain (SH3+, SH3-). Differences in C-terminal domains are indicated by grey (CB3) or black (CB2). Mutant collybistin constructs were generated by deleting either the RhoGEF domain or the PH domain. Each collybistin isoform contains an N-terminal 9E10 epitope tag. B. Schematic representation of gephyrin and gephyrin mutants with MogA and MoeA domains in light grey. Linker regions are denoted by dark grey. GFP was linked to the N-terminus of each gephyrin construct.

84
clusters. However, the average number of gephyrin clusters [myc-CB2SH3+: 9.96
± 0.64 (s.e.m.); myc-CB2SH3- : 9.86 ± 0.68 clusters per 40 µm segment of
dendrite, Fig. 4.2 E], and the average gephyrin cluster area [myc-CB2SH3+: 1.60 ±
0.25 µm2 , myc-CB2SH3-: 1.39 ± 0.14 µm2 for the (n = 11 neurons per construct)]
were indistinguishable in cells transfected with the two constructs.
To search for collybistin domain(s) that might be required for normal
synaptic localization of gephyrin, putative dominant negative constructs were
generated which lacked either the RhoGEF domain (amino acids 114-293, myc-
CB2SH3- Δ RhoGEF) or the PH domain (amino acids 326-432, myc-CB2SH3- Δ PH)
(Fig. 4.1 A). The myc-CB2SH3- Δ RhoGEF construct showed no overt effect on
gephyrin localization (Fig. 4.2 D). This is consistent with data from HEK 293
cells, in which lack of the RhoGEF domain abolished gephyrin-collybistin
interaction (Harvey et al., 2004) and thus could not affect gephyrin trafficking
despite likely being functionally inactive. However, the myc-CB2SH3- Δ RhoGEF
construct yielded a noticeably lower percentage of transfected cells than any
other collybistin construct, consistent with a possible toxic effect (data not
shown).
Overexpression of the collybistin construct lacking the PH domain (myc-
CB2SH3- Δ PH) resulted in the formation of large aggregates of gephyrin in the
soma and dendrites of neurons associated with a dramatic loss of punctate
gephyrin staining [2.1 ± 1.13 clusters per 40 µm dendrite segment (n = 10

85
Figu
re 4
.2
Func
tiona
l col
lybi
stin
is r
equi
red
for
accu
mul
atio
n of
gep
hyrin
in d
endr
itic
clus
ters
. Exp
ress
ion
of m
yc-ta
gged
C
B2SH
3- (
A)
and
CB2
SH3+
(B
) is
ofor
ms
in t
rans
fect
ed m
ouse
cor
tical
neu
rons
rev
eals
a d
iffus
e di
strib
utio
n th
roug
hout
the
cell
som
a an
d de
ndrit
es w
ith n
o ef
fect
on
geph
yrin
pun
cta.
In
trans
fect
ed c
ells
, CB2
SH3-Δ
PH a
cts
as
a do
min
ant-n
egat
ive
fact
or,
com
petin
g w
ith e
ndog
enou
s co
llybi
stin
for
bin
ding
site
s on
gep
hyrin
(C
). G
ephy
rin
stai
ning
usi
ng m
Ab7a
indi
cate
s a
loss
of s
ynap
tic g
ephy
rin c
lust
ers
and
accu
mul
atio
n of
end
ogen
ous
geph
yrin
in li
pid
raft-
like
stru
ctur
es i
n th
e pr
oxim
al d
endr
ites.
Th
is c
an b
e cl
early
see
n in
den
driti
c en
larg
emen
t. H
owev
er,
the
CB2
SH3-Δ
Rho
GE
F m
utan
t doe
s no
t sho
w a
ny a
ccum
ulat
ion
of g
ephy
rin in
intra
cellu
lar
aggr
egat
es, i
nste
ad s
how
ing
norm
al p
unct
ate
geph
yrin
(D
). E
. Q
uant
itatio
n of
CB2
SH3-
, C
B2 S
H3+
and
CB2
SH3-Δ
PH s
how
s a
sign
ifica
nt
decr
ease
in th
e am
ount
of g
ephy
rin p
unct
a fo
r the
mut
ant c
onst
ruct
(p<0
.001
***
). S
cale
bar
s 10
µm
.

86
neurons)], compared to either myc-CB2SH3+ or myc-CB2SH3- (9.96 ± 0.64 and
9.86 ± 0.68 clusters respectively; n=11 neurons; p<0.001; Student’s t-test)(Fig.
4.2 C). In contrast, the distribution of myc-CB2SH3- Δ PH itself was not different
from that of the control construct, myc-CB2SH3-. The data indicate that the PH
domain is critically important for normal functioning of collybistin and for
postsynaptic targeting of gephyrin. As collybistin constructs lacking the PH
domain are still able to interact with gephyrin, the ablation of this domain appears
to have a dominant negative effect on gephyrin localization.
4.2.2 Collybistin mutation underlying hyperekplexia
Hereditary hyperekplexia is a rare neurological disorder that is
characterized by an increased startle response and neonatal hypertonia (Zhou et
al., 2002). Most patients diagnosed with hyperekplexia have mutations within
either the GlyR α1 (Rees et al., 2001) or β (Rees et al., 2002) subunit, indicating
that a deficit in glycine receptors is often causal for this disease. However, not all
patients have mutations within these genes indicating that mutations in other
genes that may or may not be associated with glycine receptor function could
result in manifestation of a similar disease phenotype. Genetic analysis of a
patient who suffered from hereditary hyperekplexia but lacked mutations in the
GlyR subunit genes or in gephyrin revealed a missense mutation in ARHGEF9,
the human gene encoding collybistin (Harvey et al., 2004). This mutation
(G164C) was predicted to result in a glycine to alanine switch at position 55
within the SH3 domain. Because CB3SH3+ is the only isoform with detectable

87
expression levels in human brain and spinal cord, this mutation is likely to affect
most or all functions of collybistin, perhaps by disrupting the trafficking of glycine
or GABAA receptors or both.
4.2.3 Functional analysis of the CB3SH3+G55A mutation
To investigate the consequences of this mutation in neurons, myc-tagged
versions of CB3SH3+ and CB3SH3+G55A were transfected into cultured cortical
neurons and subjected to immunofluorescent analysis of the transfected
collybistin construct as well as endogenous gephyrin and GABAA receptors. Like
the CB2 constructs, myc-CB3SH3+ was diffusely expressed within the soma and
dendrites of neurons (Fig. 4.3 A) and as expected, did not seem to affect the
punctate distribution of gephyrin (Fig. 4.3 A, D). In stark contrast, myc-
CB3SH3+G55A was confined to large intracellular aggregates in the soma and
proximal dendrites of transfected neurons (Fig. 4.3 B, C). In cells that seemed to
express very large amounts of the transfected myc-CB3SH3+G55A, the mutant
protein was more strongly confined to large somatic aggregates that were often
colocalized with gephyrin aggregates (Fig. 4.3 C). Indeed, semiquantitative
analyses revealed that immunoreactive puncta for gephyrin seen in myc-
CB3SH3+G55A-transfected neurons (1.55 ± 0.72 clusters per 40 µm segment)
were greatly reduced compared to myc-CB3SH3+ transfected cells (11.9 ± 4.44,
n= 11 neurons, p<0.001; Student’s t-test).

88
Figu
re 4
.3
Mut
atio
n w
ithin
CB
3 SH
3+ i
n pa
tient
with
hyp
erek
plex
ia a
nd e
pile
psy.
Tr
ansf
ectio
n of
mou
se
corti
cal n
euro
ns w
ith C
B3SH
3+ o
r CB3
SH
3+G
55A
wer
e ex
amin
ed w
ith a
ntib
odie
s sp
ecifi
c fo
r the
myc
ta
g of
CB3
SH3+
, CB
3 SH
3+G
55A
and
gep
hyrin
(mAb
7a).
Whe
reas
wild
-type
col
lybi
stin
(CB3
SH3+
, A,
gree
n) i
s ex
pres
sed
thro
ugho
ut s
oma
and
dend
rites
, th
e C
B3SH
3+G
55A
form
s la
rge
som
atic
and
de
ndrit
ic a
ggre
gate
s (B
, hig
h le
vels
of e
xpre
ssio
n, g
reen
), an
d sh
ows
a lo
ss o
f gep
hyrin
pun
cta
(B’,
red)
. R
emai
ning
red
pun
cta
(rep
rese
nt g
ephy
rin c
lust
ers
from
non
-tran
sfec
ted
cells
in
the
sam
e cu
lture
. W
hen
expr
esse
d at
low
er l
evel
s th
e C
B3SH
3+G
55A
mut
ant
(C,
gree
n) s
how
s a
tight
as
soci
atio
n w
ith g
ephy
rin (
C’,
red)
in c
lust
ers
conf
ined
to p
roxi
mal
den
drite
s (C
’’, c
oloc
aliz
ed y
ello
w).
D, q
uant
itatio
n of
gep
hyrin
pun
cta
show
ed a
sig
nific
ant d
ecre
ase
in th
e am
ount
of g
ephy
rin c
lust
ers
in
CB3
SH3+
G55
A tr
ansf
ecte
d ce
lls c
ompa
red
to C
B3SH
3+ (*
** p
<0.0
01).
Sca
le b
ars
10 �
m.

89
The ability of the myc-CB3SH3+G55A mutant to trap gephyrin intracellularly,
together with the notion that gephyrin is required for postsynaptic clustering of
glycine receptors (Kirsch et al., 1993; Feng et al., 1998) and α2 subunit-
containing GABAA receptors (Essrich et al. 1998, Kneussel et al 1999) suggests
that a patient harboring this mutation in the ARHGEF9 gene would suffer from a
major deficit in inhibitory synaptic transmission. To examine the role of collybistin
in trafficking of α2 and/or γ2 subunit-containing GABAA receptors, we examined
the cellular localization of GABAA receptor in myc-CB3SH3+ and myc-
CB3SH3+G55A-transfected neurons. Neurons transfected with myc-CB3SH3+ or
myc-CB3SH3+G55A (Fig. 4.4 A, B; myc immunoreactivity) were stained for the γ2
subunit of GABAA receptors (Fig. 4.3 A’, B’) and for the presynaptic terminal
marker GAD (A’’, B’’). Whereas staining for the γ2 subunit and GAD revealed
proper apposition of GABAA receptors and presynaptic terminals in myc-CB3SH3+
transfected cells (Fig. 4.3 A’’’) as expected, neurons transfected with myc-
CB3SH3+G55A showed significantly fewer immunoreactive puncta for the γ2
subunit (Fig. 4.3 B’’’, 2.08 ± 1.57 clusters per 40 µm segment) compared to myc-
CB3SH3+, (10.04 ± 1.57, n= 12 neurons, p<0.001; Student’s t-test). This deficit in
postsynaptic GABAA receptor localization associated with loss of punctate
dendritic gephyrin indicates a major deficit in the localization of inhibitory
neurotransmitter receptors in the patient that suffered from this mutation. A
similar deficit is expected to occur with respect to clustering of postsynaptic

90
Figu
re 4
.4
Mut
atio
n in
CB
3 SH
3+ r
esul
ts in
loss
of
syna
ptic
GA
BA
A r
ecep
tors
. Tr
iple
sta
inin
g of
neu
rons
tran
sfec
ted
with
eith
er
CB3
SH3+
or
CB
3 SH
3+G
55A
with
ant
ibod
ies
agai
nst
the
myc
tag
of
CB2
SH3+
(gre
en),
pres
ynap
tic G
AD (
blue
) an
d th
e G
AB
AA
rece
ptor
or γ2
sub
unit
(red
). A
. C
B3SH
3+-tr
ansf
ecte
d m
ouse
cor
tical
neu
rons
GAB
AA
rece
ptor
clu
ster
s ar
e ju
xtap
osed
to
GAD
-pos
itive
te
rmin
als.
B
.
By c
ontra
st,
in n
euro
ns e
xpre
ssin
g C
B3SH
3+G
55A,
GA
BA A
rec
epto
r im
mun
orea
ctiv
ity is
con
fined
to th
e ce
ll so
ma
(red
), an
d la
rge
colly
bist
in a
ggre
gate
s (g
reen
) ar
e ob
serv
ed t
hrou
ghou
t the
de
ndrit
es.
Not
ably
, th
ese
colly
bist
in a
ggre
gate
s ar
e no
t ju
xtap
osed
to
pres
ynap
tic G
ABA
ergi
c te
rmin
als,
whi
ch a
re
unch
ange
d (b
lue)
. Q
uant
itativ
e an
alys
is o
f γ2
sub
unit
imm
unof
luor
esce
nce
(C)
indi
cate
s a
sign
ifica
nt l
oss
of G
ABA
A
rece
ptor
clu
ster
s (*
**, p
<0.0
01; n
=12
cells
per
con
stru
ct)
for
the
CB3
SH3+
G55
A m
utan
t com
pare
d w
ith C
B3SH
3+.
Scal
e ba
rs 1
0 µ
m.

91
glycine receptors. Together the glycinergic and GABAergic deficits could cause
the clinical symptoms seen in the patient.
4.2.4 Gephyrin domains required for collybistin interaction
The gephyrin interaction domain in collybistin has been previously
mapped to the linker region between the SH3 and the DH domains of collybistin
(Grosskreutz et al., 2001). To further elucidate the gephyrin domains required for
interaction with collybistin, the essential amino acids required for collybistin
binding were determined by yeast two-hybrid assays (Harvey et al., 2004). These
experiments revealed that the gephyrin domain required for interaction with
collybistin is distinct from the domain required for interaction with the glycine
receptor β subunit. Specifically, the collybistin binding site was mapped to the
boundary of the linker region and the MoeA domain of gephyrin (amino acids 305
and 323).
The critical amino acids were further narrowed by alanine scanning
mutations in that region and revealed two constructs that failed to interact with
collybistin (EGFP-GephA4, EGFP-GephA5) (Fig. 4.1 B). The functional relevance
of collybistin-gephyrin interaction was then analyzed by transfection of these
constructs into neurons. Cortical neurons were transfected either with EGFP
tagged gephyrin or with the alanine scanning mutants EGFP-GephA4 (a.a. 320-
324, PFPLT, at boundary of linker region and MoeA domain replaced with
alanines) and EGFP-GephA5 (a.a. 325 - 329, SMDKA, at beginning of MoeA
domain replaced with alanines). Expression of wild-type gephyrin, EGFP-

92
gephyrin, revealed a punctate pattern of GFP fluorescence throughout the
dendritic segments, and the immunoreactive puncta were apposed to presynaptic
GAD, as expected. Immunohistochemical analysis utilizing an antibody against
gephyrin (mAb7a), which reacts with both transfected and endogenous gephyrin,
showed perfect concordance of gephyrin immunoreactivity and EGFP
fluorescence, indicating that transfected EGFP-tagged gephyrin localized
indistinguishably from endogenous gephyrin (Fig. 4.5 A).
In contrast to wildtype EGFP-gephyrin, the EGFP-GephA4 construct
accumulated in large aggregates in the cell soma (Fig. 4.5 B) and dendrites of
transfected cortical neurons (Fig. 4.5 C). Compared to EGFP-gephyrin (9.77 ±
2.74 clusters per 40 µm segment per cell, n = 13 neurons), the number of
clusters observed for EGFP-GephA4 was greatly reduced (Fig. 4.5 I; 2.0 ± 1.95
clusters per 40 µm dendrite, n = 12 neurons, p<0.001; Student’s t-test). Unlike
EGFP-gephyrin clusters (Fig. 4.5 E), the larger aggregates formed by EGFP-
GephA4 were not localized to postsynaptic sites as indicated by the lack of
apposition to presynaptic GAD (Fig. 4.5 F, G).
The mutant gephyrin EGFP-GephA5 construct resulted in a concurrent
deficit in localization of endogenous gephyrin (Fig. 4.5 D). While this deficit was
not as severe as that seen for EGFP-GephA4, EGFP-GephA5 nevertheless
produced significantly fewer clusters (Fig. 4.5 I; 4.63 ± 2.01 clusters, n = 12
neurons, p < 0.001; Student’s T-test) than wildtype EGFP-gephyrin. Compared to
EGFP-GephA4, which resulted in an almost complete loss of dendritic gephyrin
puncta, EGFP-GephA5 formed a few large dendritic aggregates and appeared to

93
trap endogenous gephyrin at these sites. Unlike EGFP-GephA4, transfected
EGFP-GephA5 at least partially colocalized with GAD immunoreactivity,
indicating that this construct could partially localize to postsynaptic sites (Fig. 4.5
H). This observation might indicate that the EGFP-GephA5 construct can
coassemble with endogenous gephyrin, and that the protein therefore might be
transported to synapses passively by interaction with endogenous gephyrin.

94
Figu
re 4
.5 D
isru
ptio
n of
the
col
lybi
stin
bin
ding
site
on
geph
yrin
pre
vent
s ac
cum
ulat
ion
of g
ephy
rin a
t po
stsy
napt
ic s
ites.
EG
FP-ta
gged
gep
hyrin
(A,E
) and
the
alan
ine
scan
ning
mut
ants
Gep
h-A4
(B,C
,F,G
) and
Gep
h-A
5 (D
,H) w
ere
trans
fect
ed
into
cul
ture
d co
rtica
l neu
rons
and
the
cells
sta
ined
with
ant
ibod
ies
spec
ific
for g
ephy
rin (m
Ab7a
, A-D
) or
GAD
(mAb
GAD
-6,
E-H
) sh
own
in r
ed. T
wo
exam
ples
of c
ells
are
sho
wn
for
Gep
h-A
4 re
pres
entin
g co
mpa
rativ
ely
low
(B,
F) a
nd h
igh
(C,G
) le
vels
of
expr
essi
on a
nd c
orre
spon
ding
agg
rega
te p
heno
type
s. N
ote
the
perfe
ct c
oloc
aliz
atio
n of
tra
nsfe
cted
EG
FP-
geph
yrin
flu
ores
cenc
e (g
reen
) an
d im
mun
oflu
ores
cenc
e fo
r en
doge
nous
gep
hyrin
(A)
. Th
e fe
w r
emai
ning
red
pun
cta
indi
cate
end
ogen
ous
geph
yrin
exp
ress
ed o
n de
ndrit
es o
f no
n-tra
nsfe
cted
nei
ghbo
ring
cells
tha
t ar
e th
eref
ore
not
colo
caliz
ed w
ith E
GFP
-gep
hyrin
. EG
FP-g
ephy
rin is
loca
lized
at p
osts
ynap
tic s
ites
juxt
apos
ed to
pre
syna
ptic
GAD
(E,
with
cl
ose
appo
sitio
n in
yel
low
). M
utan
ts G
eph-
A4 a
nd G
eph-
A5 fo
rm la
rge
aggr
egat
es in
the
som
a an
d de
ndrit
es, w
hich
are
no
long
er ju
xtap
osed
to G
AB
Aerg
ic te
rmin
als
(F-H
). En
doge
nous
gep
hyrin
has
bee
n tra
pped
in th
ese
extra
syna
ptic
gep
hyrin
m
utan
t ag
greg
ates
, w
hich
the
refo
re a
ppea
r ye
llow
(B
-D).
Qua
ntita
tive
anal
ysis
of
mAb
7a s
tain
ing
indi
cate
s a
stas
tical
ly
sign
ifica
nt lo
ss o
f gep
hyrin
clu
ster
s (I,
***
,* p<
0.00
1,0.
05, n
= 1
2-13
cel
ls p
er c
onst
ruct
) fo
r th
e EG
FP-G
ephA
4 an
d EG
FP-
Gep
hA5
mut
ants
com
pare
d w
ith E
GFP
-gep
hyrin
. Sc
ale
bar,
10 µ
m.

95
CHAPTER 5
RESULTS III: UNPUBLISHED DATA
5.1 Aim 1
Previous work has shown that the TM4 domain of the γ2 subunit is
sufficient for postsynaptic localization of GABAA receptors, and that the
intracellular loop, while not required for localization, is necessary for recruitment
of gephyrin and formation of functional receptors. Here, we further addressed
whether the cytoplasmic loop and TM4 region of the γ2 subunit could traffic to
inhibitory synapses independent of assembly with other subunits
5.2 Results Aim 1
5.2.1 Generation and cellular localization of chimeric constructs in
heterologous cells
The fragment containing the cytoplasmic and TM4 domains of the γ2
subunit was tested as fusion constructs with the IL2 α subunit of the interleukin
receptor. The IL-2α subunit has previously been shown to efficiently translocate
to the plasma membrane and has previously been used in chimeric reporter
constructs to map trafficking signals present in the cytoplasmic portion of NMDA
receptor subunits (Standley et al., 2000). A similar approach was utilized here,
where the IL-2 α portion of the IL2α/γ2 chimeric reporter molecule should provide
a signal sequence and first transmembrane domain of this fusion protein suitable
for proper orientation of the γ2 subunit C-terminal fragment in the plasma

96
membrane. Different fusion constructs were analyzed for their ability to target to
the plasma membrane and to localize to synapses. A first IL-2/γ2 chimeric
construct was generated by fusing the C-terminal portion of the mouse γ2 subunit
containing the entire cytoplasmic loop and TM4 domains (amino acids 317-428,
Figure 2.1) to the C-terminal region of the IL-2 α subunit (see Material and
Methods). In addition, a second fusion construct contained the IL-2 α subunit
fragment fused to the cytoplasmic region of the α2 subunit (amino acids 307 -
391, Fig 2.1) followed by the γ2 TM4 domain (amino acids 405 – 428, Fig 2.1)
(Fig. 5.2.1). Chimeric constructs were then tested in HEK 293T cells using
immunofluorescent staining of live cells to determine the extent of translocation
of the different constructs to the plasma membrane. Total protein expression (in
intracellular compartments and plasma membrane) was visualized by staining of
the same cells following permeabilization utilizing a second secondary antibody
linked to a different fluorophore. The IL-2 α subunit was found to be localized at
the membrane surface as expected (Fig. 5.2.2 A) (Bonifacino et al., 1990). In
contrast, the IL-2/γ2 chimeric construct was mostly retained intracellularly (Fig.
5.2.2 B), with less than 10% of the transfected cells showing significant surface
expression (n = 29). However, on cotransfection with α2 and β3 subunits,
expression of the IL-2/γ2 construct in the plasma membrane was significantly
increased with approximately 70% of the cells showing significant surface
expression (n = 26) (Fig. 5.2.2 C). Thus, the γ2 subunit fragment contains an

97
Figure 5.2.1 Schematic representation of IL-2 α- γ 2 subunit fusion constructs. The IL-2 α subunit full-length protein (A) was C-terminally fused 2 a.a. upstream of stop codon to either the γ2 subunit intracellular loop and TM4 domains (B) or with the intracellular domain of α2 linked to the TM4 domain of the γ2 subunit (C) of GABAA receptors.
Figure 5.2.2 Differential surface targeting in the presence of GABAA receptor subunits. HEK 293T cells were transfected with the IL-2 α (A), IL-2/γ2 (B) and IL-2/γ2 with α2 and β3 GABAA receptor subunits (C). Surface expression was examined by live staining utilizing antibody against an extracellular epitope of IL-2. Cells were then fixed, permeabilized and stained for total expression. The IL-2 subunit (A) efficiently targets to the membrane surface, however, the IL-2/γ2 fusion construct requires α and β subunits to efficiently target to the membrane surface (B, C). Scale bar 5 µm.

98
intracellular retention signal that interferes with trafficking of the γ2 subunit to the
plasma membrane and this block can be released by interaction with α and/or β
subunits.
To confirm this finding, the subcellular localization of the IL-2/γ2 construct
as well as the 2nd construct (IL-2/α2γ2) containing the α2 intracellular domain and
the γ2 TM4 domain were similarly analyzed, this time by transfection into HeLa
cells. Similar to HEK 293 cells, this cell line has been demonstrated to allow
faithful trafficking to the plasma membrane of IL-2 α fusion proteins (Standley et
al., 2000; Roche et al., 2001; Scott et al., 2001). As in transfected HEK 293T
cells, the IL-2 α subunit efficiently trafficked to the cell surface, whereas efficient
expression of the IL-2/γ2 fusion construct required coexpression of α2 and β3
subunits (Fig. 5.2.3 A-C). Compared to IL-2/γ2, the IL-2/α2γ2 construct appeared
to exhibit a generally higher level of expression upon transfection into HeLa cells.
This increase in expression may in part contribute to the notion that the IL-2/α2γ2
fusion construct showed higher expression in the plasma membrane than the IL-
2 α/γ2 construct, independent of whether it was coexpressed with α2 and β3
subunits or not (Fig. 5.2.3 D, E). Consistent with this interpretation, a
comparatively large amount of this IL-2/α2γ2 protein remained localized to
intracellular compartments, even when the protein was co-expressed with α and
β subunits. Final interpretation of these experiments will require careful
quantitation of the relative fluorescence intensities at the cell surface and in

99
Figure 5.2.3 Differential surface targeting of IL-2/γ2 and IL-2/α2γ2 fusion constructs. HeLa cells were transfected with different IL2α – GABAA receptor subunit fusion constructs in the presence (A, B, D) or absence of cotransfected α2 and β 3 subunits (C, E) and processed for immunofluorescent double labeling before and after fixation and permeabilization of cells, respectively. Cell surface labeling of the IL2-α subunit before permeabilization is shown in green and total expression determined following fixation and permeabilization using an antibody raised against the same epitope is shown in red. Merged images show surface expression in yellow. Note that the Il-2 α construct efficiently targets to the cell surface membrane (A). In contrast, IL-2/γ2 does not efficiently target to the membrane surface in the absence of other GABAA receptor subunits (B; surface fluorescence (green) appeared to be less that 20% of total signal (red)). However, cotransfection of the IL-2/γ2 construct with α and β subunits (C) increased the percentage of IL-2/γ2 localized to the membrane surface, The IL-2/α2γ2 efficiently targets to the membrane of HeLa cells both in the absence (D) and presence (E) of α and β subunits. Scale bar 5 µm.

100
intracellular compartments. The preliminary result that the IL-2/γ2 and IL-2/α2γ2
chimeras were able to localize to the membrane surface in the presence of the
α2 and β3 subunits suggests that the C-terminal region alone may assemble with
α and/or β subunits and that this interaction facilitates integration of a
heteromeric complex into the plasma membrane.
5.2.2 Localization of chimeras in neurons
To further analyze the ability of the γ2 cytoplasmic loop and TM4 domains
to properly localize in the absence of additional domains, IL-2/γ2 and IL-
2/α2γ were analyzed by transfection into γ2-/- neurons. The IL-2 α construct
mainly localized to the soma of γ2-/- neurons with some of the protein localized to
immunofluorescent puncta in neurites (Fig. 5.2.4 A). However, these puncta were
not apposed to synaptic terminals visualized with an antibody against vesicular
inhibitory amino acid transporter (VIAAT). The cellular expression patterns of the
IL-2/γ2 and IL-2/α2γ2 constructs were similar to that of the IL-2 α control
construct (Fig. 5.2.4 B, C), indicating that the γ2 cytoplasmic and TM4 domains
alone are not sufficient to localize to post synaptic sites.

101
Figure 5.2.4 IL-2 fusion constructs do not localize to synapse in neurons. When
transfected into DIV 18 γ2-/- mouse cortical neurons and examined for synaptic localization, the IL-2 α construct localizes to the soma and forms puncta along neurites not apposed to inhibitory synapses (A). Both the IL-2/γ2 (B) and IL-2/α2γ2 (C) show the same staining pattern in neurons as that seen by the IL-2 subunit alone. Scale bars 10 µm.

102
5.3 Aim 2
The γ2 subunit cytoplasmic loop region is subject to palmitoylation by GODZ
(Keller et al 2004). To address the functional significance of this reversible
posttranslational modification for postsynaptic targeting of GABAA receptors, we
generated a series of cysteine to alanine substituted γ2 subunit constructs and
determined their localization following transfection into γ2 subunit-deficient and
WT neurons.
5.4 Results Aim 2
Several constructs with single or multiple cysteine to alanine or serine
substitutions within the cytoplasmic loop and TM4 of the γ2S subunit were
generated (Fig. 5.4.1) and analyzed in γ2-/- neurons as described earlier (Fig.
3.6). The 9E10γ2S subunit was able to properly localize to the synapse as
expected (Fig. 5.4.2 A) (Alldred et al., 2005). In addition, when cysteine mutant
constructs were transfected into γ2 subunit-deficient neurons, all mutant subunits
were able to localize to the synapse (Fig. 5.4.2 B-E), confirming that these
cysteine residues are not critical for postsynaptic localization of γ2 subunit-
containing GABAA receptors. Preliminary quantification indicates that there is no
difference in number or size of synaptic puncta compared to controls.
While the ability of the cysteine mutant constructs to localize to synapses
in the absence of endogenous γ2 subunit indicated that these mutations were not
critical for synaptic localization, the ability of these mutants to compete with

103
Figure 5.4.1 Schematic representation of cysteine substituted γ2 subunit constructs.
The vector backbone of these constructs was identical to that of the γ−γ−γ construct in Fig. 3.3. The C-terminal half of the cytoplasmic loop region is represented with amino acid number referring to the γ2 subunit sequence shown in Fig. 2.1. The position of the GODZ and GABARAP binding sites are indicated. * = N-terminal myc tag inserted between amino acids 4 and 5 of the mature polypeptide

104
Figure 5.4.2 Localization of cysteine mutant γ2 subunit constructs transfected into in
γ2-/- neurons. A-E, Cortical neurons isolated from γ2-/- embryos were transfected at 18 DIV with the 9E10 epitope-tagged cysteine mutant γ2 subunit constructs indicated and processed for immunofluorescence analysis at 21 DIV of the 9E10 epitope (shown in green) and presynaptic GAD (red), with colocalization represented in yellow in merged images. Boxed dendritic segments of each cell are shown enlarged in separate panels beside each image. Note the faithful formation of clusters revealed in the form of immunoreactive puncta apposed to GAD immunoreactive varicosities for all constructs. Scale bar 10 µm.

105
endogenous γ2 was unknown. Therefore, duplicate transfections and
immunohistochemical analysis of these mutants into WT neurons was
undertaken. The 9E10γ2S subunit was able to properly localize to the synapse as
expected (Fig. 5.4.3 A). In addition, all constructs tested were able to target to
GABAergic synapses, as seen in the γ2-/- neurons. Specifically, the C369A (Fig.
5.4.3 B), the C375A (Fig. 5.4.3 C), the C385A (Fig.5.4.3 D) and the C415A (Fig.
5.4.3 E) were all able to localize to the synapse apposed to GAD-IR and
preliminary quantification confirms there was no significant differences for the
cysteine mutant constructs. However, it should be noted that for the C385A
mutant subunit, very few cells showed punctate IR, indicating that there could be
a difference in the ability of this mutant subunit to localize to traffic to dendrites or
to the membrane. These results are in apparent conflict with results published by
Rathenberg et al. (2004) showing a significant reduction in surface expression
and postsynaptic clustering of C-A substituted γ2 subunit construct. Possible
reasons for this discrepancy are discussed in Chapter 6, section 5.

106
Figure 5.4.3 Localization of cysteine mutants in wild-type neurons. A-E, Cortical
neurons isolated from wild-type γ2+/+ embryos were transfected at 18 DIV with (9E10) epitope-tagged cysteine mutant constructs indicated and processed for immunofluorescence analysis at 21 DIV for the 9E10 epitope (shown in green) and presynaptic GAD (red), with colocalization represented in yellow in merged images. Boxed dendritic segments of the images are shown enlarged in separate panels beside each image. Note the faithful formation of clusters revealed in the form of punctate staining for the 9E10-γ2 and each of the cysteine mutant constructs. Scale bar 10 µm.

107
5.5 Aim 3
Recent work addressing the function of GABARAP strongly suggests that
this protein contributes to surface expression of γ2 subunit containing GABAA
receptors (Leil et al., 2004). In addition to the γ2 subunit, GABARAP interacts
with microtubules (Wang et al., 1999; Wang and Olsen, 2000), the vesicular
trafficking factor NSF (Kittler et al., 2001), and gephyrin (Kneussel et al., 2000).
GABARAP is member of small family of proteins including GATE-16 (Sagiv et al.,
2000), GABARAP-L1 (also known as GEC1) (Vernier-Magnin et al., 2001; Xin et
al., 2001) and the more distantly related light chain 3 (LC3) of microtubule
associated proteins (Mann and Hammarback, 1994). In order to address the
degree of functional redundancy among these proteins we here determined their
ability to interact with GABAA receptors and other cytoskeletal proteins previously
shown to interact with GABARAP. In addition, the ability of GABARAP-L1 to
colocalize with GABAA receptors was examined following cotransfection into
heterologous cells and in neurons.
5.6 Results
5.6.1 Antibody generation for GABARAP and homologs
To date no antibodies are available that distinguish between GABARAP
and GABARAP-L1 (Mansuy et al., 2004). Antibodies specifically recognizing
individual homologs are essential for further analysis of these proteins and their
role in GABAA receptor trafficking. Full-length GABARAP, GABARAP-L1 and
GATE-16 proteins were generated from cleaved GST fusion proteins and utilized

108
for antibody production in rabbits (see Materials and Methods). Antisera were
tested for specificity to recombinant homologs, as well as crude brain extract and
brain membrane extract. The crude antisera raised against the GATE-16 protein
(rabbits 58 and 59) were able to detect recombinant GABARAP, GABARAP-L1
and GATE-16 expressed in E. coli , but failed to detect endogenous proteins from
brain extracts (Fig. 5.6.1 A, B). Similarly, affinity purified antibodies failed to
detect native proteins in brain extracts (data not shown). The antisera for
GABARAP (rabbits 60 and 61) and GABARAP-L1 (rabbits 62 and 63; not shown)
showed affinity for recombinant GABARAP, GABARAP-L1 and GATE-16, but
were unable to detect endogenous proteins, similar to the antisera for GATE-16
(Fig. 5.6.1 C-E). While all three homologs contain the same amino acid number,
differences in mobility seen here are likely due to differences in post-translational
modifications.
5.6.2 Protein-protein interactions of GABARAP and homologs
To determine possible functional redundance of GABARAP family proteins
we performed glutathione-S-transferase (GST) pull-down assays using agarose
beads charged with GST-GABARAP, GST-GABARAP-L1 or GST-GATE-16.
Bound proteins were eluted and analyzed by western blot using antibodies
specific for gephyrin, the � 1 subunit of GABA� receptors, tubulin (α/β subunits of
tubulin I/II), NSF, and GABARAP-L1 (Fig. 5.6.2 A-D). The results indicate that all
three members can interact with each of the GABARAP binding proteins thereby

109
Figure 5.6.1 Antisera specificity for GABARAP, GABARAP-L1 and GATE-16 were tested on crude brain extract (lane 1), recombinant GABARAP (10 ng) (lane 2), GABARAP-L1 (10 ng, lane 3) and GATE-16 (10 ng, lane 4). The position of the 15-kD molecular weight marker is indicated on the left of panels. Note that the crude antisera against GATE-16 (A, rabbit 58; B, rabbit 59) recognized all recombinant proteins but failed to detect endogenous proteins. Similarly, crude antisera against GABARAP (C, rabbit 60; D, rabbit 61) and GABARAP-L1 (E, rabbit 62; rabbit 63 not shown) only recognized recombinant proteins, with no specificity for any one homolog. Upper arrow in each panel indicates GABARAP and GABARAP-L1, lower arrow indicates GATE-16. Other bands are degradation products.

110
confirming known interactions for GABARAP (Wang et al., 1999; Kneussel et al.,
2000; Kittler et al., 2001) and expanding on published results for GABARAP-L1
(Mansuy et al., 2004) and GATE-16 (Sagiv et al., 2000). In addition, interaction
between GABARAP and GABARAP-L1 was confirmed (Mansuy et al., 2004),
and self-interaction of GABARAP-L1 and interaction with GATE-16 was shown
(Fig. 5.6.2 E). Taken together, this data suggest extensive functional
redundancy between GABARAP family proteins and that these proteins form a
complex.
5.6.3 Subcellular localization of GABARAP-L1
A GFP-fusion protein linking GFP to the N-terminus of GABARAP-L1 was
generated to test for colocalization of GABARAP-L1 and GABAA receptors. GFP-
GABARAP-L1 was cotransfected with α2β3γ2 subunit GABAA receptors into HEK
293T cells and the GABAA receptors aggregated with a γ2 subunit specific
antiserum. No colocalization was detected between aggregated GABAA
receptors and GFP-GABARAP-L1 at the plasma membrane consistent with
previous evidence from GATE-16 (Sagiv et al., 2000) and GABARAP (Kneussel
et al., 2000), suggesting that GABARAP-L1 is localized to intracellular
compartments (Fig. 5.6.3 A-C). This data contradicts earlier claims that
GABARAP proteins might contribute to clustering of GABAA receptors (Wang et
al., 1999), and is more compatible with more recent data showing that GABARAP
proteins contributes to intracellular trafficking of GABAA receptors (Leil et al.,
2004).
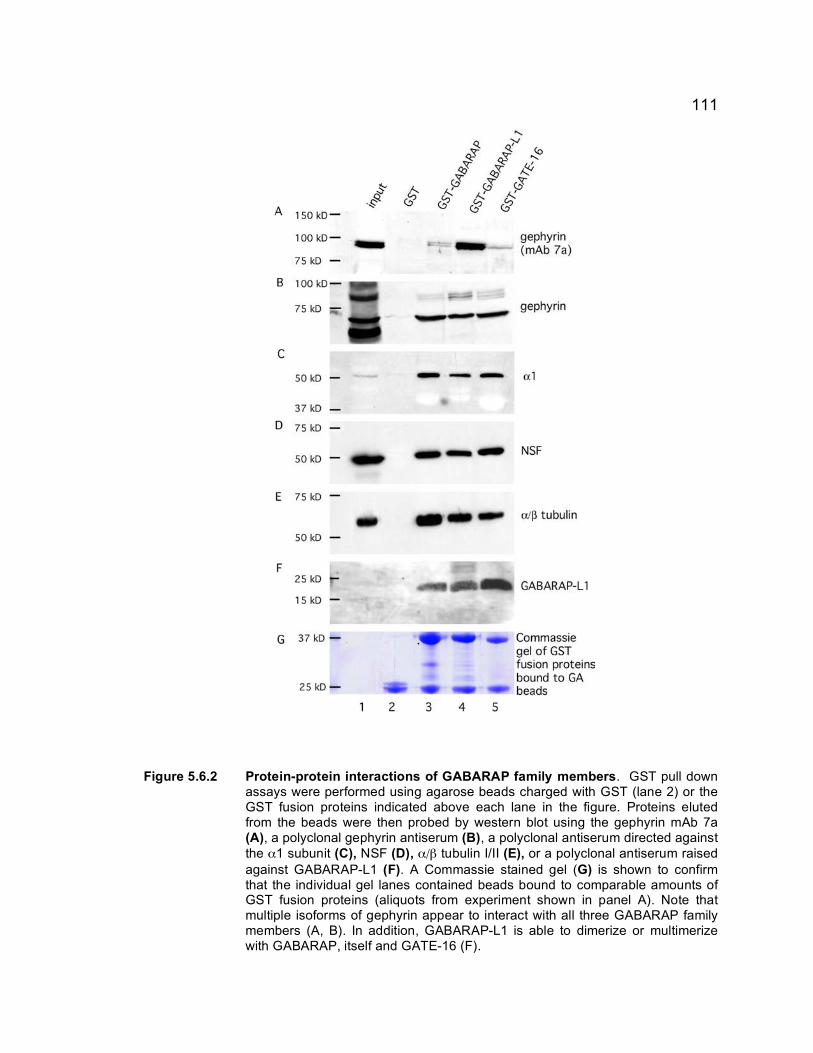
111
Figure 5.6.2 Protein-protein interactions of GABARAP family members. GST pull down assays were performed using agarose beads charged with GST (lane 2) or the GST fusion proteins indicated above each lane in the figure. Proteins eluted from the beads were then probed by western blot using the gephyrin mAb 7a (A), a polyclonal gephyrin antiserum (B), a polyclonal antiserum directed against the α1 subunit (C), NSF (D), α/β tubulin I/II (E), or a polyclonal antiserum raised against GABARAP-L1 (F). A Commassie stained gel (G) is shown to confirm that the individual gel lanes contained beads bound to comparable amounts of GST fusion proteins (aliquots from experiment shown in panel A). Note that multiple isoforms of gephyrin appear to interact with all three GABARAP family members (A, B). In addition, GABARAP-L1 is able to dimerize or multimerize with GABARAP, itself and GATE-16 (F).

112
5.6.4 GABARAP-L1 localization in neurons
To examine the subcellular localization of GABARAP-L1 in neurons and
determine the ability of GFP-GABARAP-L1 to interact with endogenous GABA�
receptors, GFP-GABARAP-L1 was transfected into wild-type cortical neurons.
GFP-GABARAP-L1 localized mainly to the soma, with a small portion showing a
punctate distribution along dendrites. A subset of GFP-GABARAP-L1 puncta
localized with α2-IR puncta on presumed dendrites indicating a portion of the
GABARAP-L1 colocalized with GABAA receptors in neurons (Fig. 5.6.4). This
finding mimics what was seen for GABARAP (Kittler et al., 2001; Leil et al.,
2004), suggesting these proteins could have functional redundancy or could both
localize with intracellular GABAA receptors in neurons. This colocalization, taken
with the GST pull down assays, implies that GABARAP-L1 is involved in
trafficking of the receptors to the synapse.

113
Figure 5.6.3 Subcellular localization of GABARAP-L1 in HEK 293T cells. Control GFP
construct transfected together with GABAA receptor subunits, α2, β3 and γ2 show no specificity and does colocalize with the γ2 subunit (A). GFP-GABARAP-L1 (B and C) shows intracellular fluorescence with little to no colocalization with the γ2 subunit of GABAA receptors.
Figure 5.6.4 GFP-GABARAP-L1 localization in neurons. GFP-GABARAP-L1 (shown in
green) was transfected into DIV18 wild-type cortical neurons and the cells fixed and stained three days later using an antiserum specific for the α2 subunit of GABAA receptors (shown in red). Colocalization between GFP-GABARAP-L1 and α2-subunit containing GABAA receptors is seen in yellow in merged images. Insets show enlarged dendritic segments indicated by boxed areas. Arrows indicate colocalization, whereas arrowheads indicate α2 containing GABAA receptors that did not colocalize with GABARAP-L1. Scale bar 10 µm.

114
CHAPTER 6
DISCUSSION
The γ2 subunit is critical for postsynaptic localization of GABAA receptors
and gephyrin (Essrich et al., 1998; Schweizer et al., 2003; Alldred et al., 2005).
However, while gephyrin is known to colocalize with GABAA receptors at
inhibitory postsynaptic sites (Essrich et al 1998, Sassoe-Pognetto et al 2000,
Brünig et al 2002), in vitro binding experiments designed to show direct
interaction of gephyrin with GABAA receptors have invariably failed (Meyer et al.,
1995; Kannenberg et al., 1997). Moreover, gephyrin is required for postsynaptic
clustering of only a subset of GABAA receptor subtypes (Essrich et al., 1998;
Kneussel et al., 2001; Levi et al., 2004). Proteins other than GABAA receptors
that are localized to the inhibitory synapse have not been shown to interact
directly with GABAA receptor subunits. To gain further insights into mechanisms
that contribute to postsynaptic localization of GABAA receptors, we determined
that the subunit domain of the γ2 subunit required for synaptic localization. We
also attempted to map the interaction domains between the subsynaptic scaffold
protein gephyrin and the GTP exchange factor collybistin, in collaboration with
Robert Harvey and Mark Rees. Indeed, we established that collybistin exerts an
essential role in trafficking of postsynaptic GABAA receptors. Finally, we
addressed functional redundancy of GABARAP family proteins with respect to
interactions with GABAA receptors and postsynaptic proteins.

115
6.1 GABAA receptor subunit domains required for postsynaptic localization
The C-terminal domain including the TM4 domain of the γ2 subunit is
necessary and sufficient for postsynaptic localization of GABAA receptors.
Surprisingly, the γ2 intracellular domain is dispensable for postsynaptic
localization, but essential for the recruitment of gephyrin to GABAA receptor
clusters and is required for restoration of mIPSCs in γ2-/- neurons. These results
indicate that there may be independent mechanisms for the localization of
receptors to synaptic sites and restoration of synaptic activity. Moreover, the
correlation between gephyrin localization to GABAA receptor clusters and
restoration of synaptic activity suggests that gephyrin may be essential for
synaptic function of GABAA receptors. Thus, rather than serving to stabilize or
anchor GABAA receptors in the postsynaptic plasma membrane, gephyrin might
provide a scaffold for an endocytic recycling machinery localized to a subsynaptic
compartment underneath the postsynaptic membrane. This compartment would
be tailored for reinsertion of endocytosed γ2 subunit containing GABAA receptors
into the postsynaptic membrane. It is possible that recruitment of gephyrin and
restoration of synaptic activity in γ2-/- neurons are mechanistically linked. The
ability of gephyrin to partially localize to synapses in the absence of the γ2
subunit intracellular loop domain may indicate an inactive endocytic recycling
apparatus, as it did not contribute to synaptic activity. Previous work by Essrich
et al., (1998) demonstrated that loss of the γ2 subunit resulted in a dramatic but
incomplete loss in gephyrin clusters. Baer et al. (1999) demonstrated that the γ3

116
subunit can partially substitute for the γ2 subunit in γ2-/- neurons. Thus, it is
unclear whether this partial recruitment of gephyrin seen in γ2-/- neuron upon
transfection of 9E10γ−α−γ and 9E10α−α−γ (Fig. 3.8) represents (i) partial
colocalization with receptors composed of α and β subunits (ii) low levels of
endogenous γ3 subunit, or (iii) partial recruitment of gephyrin dependent on the
transfected 9E10γ−α−γ and 9E10α−α−γ constructs. Irrespective of the answer to
these questions, association of GABAA receptors with gephyrin appears to be
indirect and probably involves other proteins (Meyer et al 1995, Kannenberg et al
1997). Alternatively, the requirement for the cytoplasmic loop for gephyrin
colocalization may indicate that there are protein-protein interactions with the
cytoplasmic loop domain, which directly affects the localization of gephyrin.
Gephyrin, in turn could affect synaptic activity of GABAA receptors by stabilizing
the receptors at synaptic sites.
Interaction of the γ2 intracellular loop with itself and other proteins (Wang
et al., 1999; Kittler et al., 2000; Nymann-Andersen et al., 2002; Keller et al.,
2004; Terunuma et al., 2004) suggested a mechanism for the synaptic
localization of GABAA receptors, similar to the presumed mechanisms for the
closely related glycine and NMDA receptors. However, glycine and NMDA
receptors rely on direct protein-protein interactions of the cytoplasmic subunit
domains with the subsynaptic protein scaffold containing gephyrin (reviewed by
Kirsch et al 1996) or with PDZ domain-containing proteins such as PSD-95
(Kornau et al 1997), respectively. Therefore, it appears that the mechanism for

117
synaptic localization of GABAA receptors is fundamentally different from glycine
and glutamate receptor types.
We have identified an important function of the γ2 subunit TM4 domain in
accumulation of GABAA receptors at postsynaptic sites. A precedent exists for
the function of the TM4 domain in nACh receptors, which are structurally similar
to GABAA receptors. The nACh receptors are postulated to rely on the TM4
domain of the α and γ subunits to interact with the lipid bilayer through
cholesterol/sphignolipid-rich vesicles or microdomains in the plasma membrane
(lipid rafts) (Blanton and Cohen, 1994; de Almeida et al., 2004; Pediconi et al.,
2004). However, the identity between the TM4 domains of GABAA receptor γ2
subunit and α or γ subunits of nACh receptors is surprisingly low, with 17%
identity for the α subunit and 5% for the γ subunit (Fig. 6.1). Nevertheless, nACh,
AMPA and GABAA receptors have all been shown to colocalize with lipid raft
markers and treatment of neurons with cholesterol disrupting agents effects the
postsynaptic clustering of AMPA, GABAA and nACh receptors (Bruses et al.,
2001; Hering et al., 2003; Pediconi et al., 2004). Thus, the mechanism for GABAA
receptor localization might involve selective association of the γ2 TM4 domain
with lipid rafts of the plasma membrane or cholesterol-rich vesicular membranes
in a subsynaptic compartment. Whereas the transmembrane domain regions of
GABAA receptor subunits are highly conserved across all subunit classes, it is
interesting to note that among the α1/2, β2/3 and γ2 subunit TM4 domains six
amino acid residues are uniquely present in γ2. Intriguingly, five out of six of

118
% identity with γ2
GABAA γ2TM4 1 YARIFFPTAFCLFNLVYWVSYLYL GABAA α2TM4 1 MSRIVFPVLFGTFNLVYWATYL-- 59% GABAA β3TM4 1 WSRIVFPFTFSLFNLVYWLYYV-- 59% nACh α TM4 1 ---RIFLWVFILVCILGTAG---- 17% nACh γ TM 4 1 ----CFLAMLSLF-ICGTAGIF-- 5%
Figure 6.1 Sequence alignments of mouse nACh and GABAA receptor TM4 domains. Single
amino acid codes in red indicate identical amino acids, while amino acids indicated in blue indicate conservative changes in amino acids. Sequences were aligned using ClustalW sequence alignment program with Boxshade. Percentages at right indicate % identity to the γ2 TM4 of GABAA receptors. Nicotinic ACh receptor subunit α (Yu et al., 1986) and γ (Boulter et al., 1986) TM4 domains were predicted utilizing PredictProtein (http://cubic.bioc.columbia.edu/predictprotein/submit_def.html).
Figure 6.2 Structural features of the γ2 TM4 domain. A. Alignment of the C-terminal
domains of major GABAA receptor subunits know to constitute postsynaptic GABAA receptors reveals seven amino acids that are uniquely present in the γ2 subunit TM4 domain (shown as white text on black background). B. A helical wheel representation of the putative α-helix of the γ2 subunit TM4 domain predicts that amino acids uniquely present in the γ2 subunit TM4 domain map to two narrow faces of the putative TM4 α-helix, suggesting potential contact sites that are uniquely present in this subunit. Amino acids in (B) are denoted in single letter code with numeric subscripts indicating the position of the amino acid in the transmembrane domain, as shown in (A). Amino acids that are unique for the γ2 subunit TM4 domain are shown as white text on black background.

119
these unique amino acids map to two discrete faces of the presumed TM4 α-
helix of the γ2 subunit (Fig. 6.2). This finding suggests that the interaction(s) that
contribute to proper localization are likely mediated by these unique amino acids,
either as a unique interface with the lipid bilayer or by interaction with another
integral membrane protein.
There are several integral membrane proteins that localize to the
postsynaptic membrane of GABAergic synapses. The DPC is so far the only
complex known to localize to GABAergic synapses independently of postsynaptic
GABAA receptors (Brunig et al., 2002; Levi et al., 2002). However, this complex
accumulates at synapses much later during development than GABAA receptors,
and is detected at only a subset of GABAergic synapses. This indicates that the
DPC is unlikely to be responsible for the synaptic localization of GABAA receptors
(Brunig et al., 2002; Levi et al., 2002). Another candidate, neuroligin-2 is
recruited to postsynaptic sites by neurexin expressed on presynaptic GABAergic
terminals. It appears to provide an essential signal that induces postsynaptic
differentiation of GABAergic synapses and the matching apposition of pre and
postsynaptic elements of GABAergic synapses (Graf et al., 2004; Varoqueaux et
al., 2004). Four different neuroligins have been identified to date. Neuroligin-1
was determined to be a postsynaptic cell adhesion molecule that was specifically
localized to excitatory synapses (Song et al., 1999), and later shown to trigger
presynaptic development of glutamatergic synapses (Scheiffele et al., 2000).
However, more recent evidence suggests different neuroligins act in concert to
modulate the number and size of excitatory (neuroligin-1) (Prange et al., 2004)

120
and inhibitory (neuroligin-2) (Graf et al., 2004; Varoqueaux et al., 2004; Chih et
al., 2005) synapse formation. While neuroligin-2 is implicated in inhibitory
synaptic development and colocalizes with GABAA receptors at synapses in both
developing and mature neurons (Varoqueaux et al., 2004; Graf et al, 2004),
current evidence suggests that the interaction between neuroligin-2 and gephyrin
or GABAA receptors is indirect (Graf et al., 2004). A critical question is whether
neuroligin-2 can localize to GABAergic synapses independently of gephyrin and
GABAA receptors, acting as an autonomous signal for postsynaptic
differentiation, or whether neuroligin acts in concert with GABAA receptors and
gephyrin to localize to proper membrane sites. Acute depletion of neuroligin-1, 2,
3 by RNAi leads to significant, but incomplete loss of GABAergic and
glutamatergic synapses, suggesting that other unrelated synapse-inducing
molecules can partly compensate for neuroligins (Chih et al. 2005).
A recent report by Van Rijnsoever et al, (2005) suggests that postsynaptic
GABAA receptor clusters identified by immunofluorescent techniques are
localized in a sub-membranous intracellular compartment rather than in the
plasma membrane. This study also suggests that GABAA receptors localized to
the plasma membrane surface do not, cluster at synapses. However, it is
possible that there could be two distinct mechanisms of synaptic localization for
GABAA receptors, one that localizes receptors to a subsynaptic pool and one for
receptors localized at the membrane surface. Localization of GABAA receptors to
either the subsynaptic site or the plasma membrane could involve a synaptic
targeting protein or by the unique lipid composition of the membrane(s) at the

121
synapse. Thus, the data supplied by Van Rijnsoever et al. (2005) in combination
with our own data, indicates that GABAA receptors are localized to a sub-plasma
membrane compartment via the C-terminal TM4 domain, and that the
intracellular loop region of the γ2 subunit is required for the insertion of GABAA
receptors into the plasma membrane. In support of this mechanism, Connelly et
al. (1999 a,b) found that the γ2 subunit plays a critical role in intracellular
trafficking of endocytic recycling of GABAA receptors expressed in heterologous
cells (Connolly et al., 1999a; Connolly et al., 1999b).
Physical interaction of the γ2 subunit cytoplasmic domain with the
phosphatase calcineurin is implicated in NMDA receptor-dependent functional
plasticity of inhibitory synapses (Lu et al., 2000; Wang et al., 2003). Moreover,
the γ2 subunit cytoplasmic loop domain mediates interaction with the GABAA
receptor trafficking factor GABARAP (Wang et al., 1999) and the palmitoyl
transferase GODZ (Keller et al., 2004). Whereas neither of these proteins have
been detected consistently at synapses, palmitoylation of cysteines in the γ2
subunit cytoplasmic loop domain appears to ensure the trafficking of GABAA
receptors to the cell surface and synapses (Keller et al., 2004; Rathenberg et al.,
2004). Furthermore, the γ� subunit contains a binding site for the clathrin adaptor
AP2 and clathrin-mediated endocytosis of GABAA receptors has been shown to
limit the amplitude of mIPSCs in cultured neurons (Kittler et al., 2000).
The findings presented in this Ph.D. thesis clearly indicate that the TM4
domain of the γ2 subunit is sufficient for postsynaptic localization of GABAA

122
receptors. The electrophysiological and immunohistochemical data presented in
Chapter 3, along with the data by Van Rijnsoever et al., (2005), support the
interpretation that the γ2 subunit cytoplasmic domain contributes to normal
endocytosis and recycling of GABAA receptors and thereby maintains the steady
state receptor concentration in the postsynaptic plasma membrane. While we
have made much progress in the understanding of domains required for
postsynaptic localization and gephyrin interaction, it remains unclear how
different γ2 subunit binding proteins and gephyrin contribute to synaptic
localization of GABAA receptors and to synaptic function
6.2 IL-2 α subunit linked to γ2 and α2 sequences for membrane
localization
The observation that plasma membrane translocation of the γ2 subunitC-
terminal region is linked to IL-2α requires coexpression of α and β subunits (Figs.
5.1.2, 5.1.3). This suggests that the γ2 subunit C-terminal region contains a
cytoplasmic retention signal that is masked by coassembly with α and β subunits.
Intracellular retention is also observed with neuronal GABAA receptors when
cysteines in the cytoplasmic loop region/ GODZ binding site of the γ2 subunit are
mutated to alanine (Rathenberg et al., 2004), whereas chimeric subunits that
lacked the γ2 subunit cytoplasmic loop region seemed to be trafficked to
synapses normally (Alldred et al., 2005). The simplest interpretation would
indicate that palmitoylation of the γ2 subunit releases intracellular retention

123
mediated by the γ2 subunit cytoplasmic region and that intracellular retention is
not observed when the corresponding sequence is absent. Increased expression
in the plasma membrane of IL-2α/α2γ2 compared to the IL-2α/γ2 construct and
tested in the absence of α and β subunits indicates that the intracellular retention
signal is contained within the γ2 subunit cytoplasmic loop region. Consistent with
the existence of an intracellular retention signal within the γ2 subunit, previous
analyses in heterologous cells has shown that, in contrast to αβ and αβγ
receptors, αγ or βγ subunit combinations fail to express at the membrane surface
(Connolly et al., 1999; Kittler et al., 2000).
A report by Meier et al (2004) indicates that GFP linked to either the γ2S
or γ2L intracellular loops alone have differential expression when transfected into
neurons. This report shows that in spinal cord WT neurons, the GFP-γ2L
construct was able to efficiently localize to synaptic sites, whereas the GFP-γ2S
construct could not. This suggests that the 8 amino acid difference in the
cytoplasmic loop region, which contains a phosphorylation site, is critical for
synaptic localization of the γ2 loop sequence in the absence of any additional
sequence. In conjunction with our results, it indicates that the intracellular
retention signal we see in the γ2S cytoplasmic loop sequence could be masked
by this phosphorylation.
6.3 Collybistin as a determinant of gephyrin clustering

124
The results in Chapter 4 indicate that collybistin is a critical factor for
proper localization of gephyrin and GABAA receptors. Specifically, the PH
domain of collybistin is crucial for gephyrin localization, while the MoeA domain
of gephyrin is required for interaction with collybistin. In addition, a naturally
occurring point mutant form of CB3 containing a glycine to alanine substitution in
the SH3 domain of CB3, known to cause symptoms of hyperekplexia and
epilepsy in a patient, acts as a dominant negative protein that suppresses the
postsynaptic clustering of gephyrin and GABAA receptors upon transfection into
cultured neurons. Thus, these experiments establish an essential function for
collybistin in clustering of postsynaptic clustering of gephyrin and GABAA
receptors and probably glycine receptors as well.
6.3.1 Deletions of collybistin domains
The CB2SH3-ΔPH mutant resulted in accumulation of endogenous gephyrin
in proximal dendrites and a significant reduction in number of gephyrin clusters.
This suggests that collybistin is involved in dendritic transport of gephyrin to
inhibitory synapses. However, it is unclear how the PH domain contributes to the
function of collybistin and whether this mutation disrupts PH domain-dependent
membrane phosphoinositide interaction or the binding of unknown other
protein(s) to collybistin. The dominant negative effect of CB2SH3-Δ RhoGEF
indicates that absence of the RhoGEF domain also interferes with the function of
collybistin. Whereas this protein was largely deficient in submembrane targeting
of gephyrin in HEK 293 cells (Harvey et al., 2004), a small percentage of

125
gephyrin was able to localize to the dendrites of transfected neurons. Thus while
CB2SH3-Δ RhoGEF could not completely abolish the function of endogenous
collybistin, it nevertheless appears to act as a dominant negative protein. In
summary, the results indicate that both the RhoGEF domain and the plextrin
homology domain of collybistin are essential for gephyrin interaction and
localization, with the PH domain essential for proper localization of gephyrin and
likely required for GABAA receptor clustering at synapses.
6.3.2 G55A mutation and consequences
Overexpression of CB3SH3+ G55A resulted in a dramatic reduction in the
number of postsynaptic gephyrin and GABAA receptors, indicating that this
mutant acts as a dominant negative protein for collybistin function. In addition,
this result appears to explain the severe hyperekplexia and epilepsy phenotypes
observed in a patient carrying this X-linked mutation, and illustrates the
importance of collybistin for postsynaptic localization of gephyrin and the
associated inhibitory receptors. While recent studies have shown binding of
ligands to SH3 domains (Douangamath et al., 2002; Groemping et al., 2003; Liu
et al., 2003) and this could lead to interference in the binding surface, it is more
likely that the mutation here of the conserved glycine residue affected the ability
to fold the SH3 domain. However, the perturbation of the expression pattern of
the CB3SH3+ G55A mutant in neurons suggests that this mutation causes a
complex deficit in dendritic trafficking not only of gephyrin and GABAA receptors,
but also of the collybistin mutant itself.

126
6.4 Gephyrin domain that interacts with collybistin
The binding site for collybistin was localized to the interface of the linker
region and the MoeA domain in gephyrin. Transfection of gephyrin constructs
with mutations in this region resulted in a significant reduction in immunoreactive
puncta for both the transfected and endogenous gephyrin. This study identified
critical sequences for gephyrin-collybistin interactions and that these interactions
were essential for normal collybistin-mediated trafficking and clustering of
gephyrin and GABAA receptors to postsynaptic sites.
6.5 Mutation of cysteine residues within γ2 subunit
Similar experiments by Rathenberg et al., (2004) performed in wildtype
neurons utilizing different transfection protocol suggests that cysteine-alanine
substitution in γ2 subunit constructs are less efficiently translocated to the plasma
membrane than the endogenous γ2 subunit. However, when replicating these
experiments with different transfection protocol, all of the cysteine mutant
constructs tested in our hands were indistinguishable from the control γ2 subunit
construct, with no statistically significant differences in size or number of puncta
in preliminary quantitation (Fig. 5.2.2 and Fig. 5.2.3). However, in our hands,
analysis was limited to neurons that showed at least some punctate staining,
which could explain why partial deficits in surface expression or postsynaptic
targeting might have escaped detection in our assays.

127
Rathenberg et al. (2004) utilized a long-term expression of these mutant
constructs, which resulted in reduced surface expression compared to wildtype
constructs. Such differences would not necessarily be apparent following acute
short-term overexpression. It is likely that under these conditions long term
deficits in receptor trafficking would be amplified, whereas short-term expression
in γ2-/- or WT cells may allow for assembly and synaptic localization of the
transfected mutant construct. In addition, Rathenberg et al. used constructs
tagged with GFP at the N-terminus of the γ2 subunit, which we have shown to be
less efficiently inserted into the plasma membrane than those with a smaller
epitope tag (our own unpublished observations; Fig 3.2). Taken together, the
conditions used by Rathenberg et al. might be better suited to pick up subtle
deficits in surface expression of mutant γ2 subunit constructs. Future
experiments therefore should redress this issue with a non-biased method for
quantification of surface expression and include analyses of mutant constructs
with multiple substitutions of cysteines such as the C380/381A and C357S and
C380/381A constructs. One way to facilitate quantification would be to co-
transfect these constructs with minute amounts of GFP to determine the
percentage of transfected cells that show synaptic puncta.
Independent of the experimental approach used, the partial ability of
cysteine mutant γ2 subunit constructs to localize to inhibitory synapses strongly
suggests that palmitoylation of the γ2 subunit is not critical for the postsynaptic
localization of GABAA receptors. Instead, the long-term deficits in postsynaptic
localization and cluster formation observed by Rathenberg et al. suggest that

128
palmitoylation contributes to proper localization in more subtle ways. It will be
interesting to determine the extent to which cysteine mutant γ2 constructs are
localized in the plasma membrane surface and to determine their rate of
endocytosis and recycling to the plasma membrane. It is also possible that
palmitoylation is important for receptor recycling, rather than simply required for
membrane translocation.
6.6 GABA receptor associated protein L1
We found that the GABARAP-L1 homolog was indistinguishable from
GABARAP with respect to interaction with GABAA receptors, gephyrin, NSF and
with respect to partial colocalization with GABAA receptors in neurons. Together
with the high degree of sequence conservation between these two proteins, our
data suggests that these two proteins are functionally redundant. While GATE-
16 is more distantly related, and showed less robust interaction with gephyrin, it
similarly interacted with GABAA receptors, tubulin and NSF, indicating that
GABARAP, GABARAP-L1 and GATE-16 likely have similar sites for protein
interactions. A genetic deletion of one of these homologs would allow the study
of the effects on GABAA receptor localization. These homologs may all bind to
the same proteins, or alternatively they could form a multi-protein complex. A
second possibility is that interaction between the homologs is required to traffic
GABAA receptors to the membrane surface. Indeed, if a gene deletion
generating a knock out phenotype of one protein would result in the loss of
postsynaptic GABAA receptors, this would indicate these proteins do not have

129
functional redundancy and would point towards complex formation for the
trafficking of GABAA receptors. While a single knockout would allow us to
examine the effect of a single homolog, further studies with a double or triple
knock out would allow us to determine whether GABARAP family of proteins
together contribute to clustering of GABAA receptors.

130
6.7 Outlook
The research completed here has helped further our understanding of
GABAA receptor mediated neurotransmission, detailing it as a complex and
dynamic process controlled by multiple mechanisms. These studies have
demonstrated that the domain of the γ2 subunit is required for postsynaptic
localization of GABAA receptors and identify additional domains required for
gephyrin recruitment and synaptic function. The mechanism for postsynaptic
GABAA receptor clustering was determined to require the TM4 domain of the γ2
subunit, invalidating previous hypotheses, which suggested that GABAA receptor
channel activation, or interactions with known clustering proteins, were required
for synaptic localization. The work presented here, in combination with recent
studies by Van Rijnsoever et al. (2005), suggest that aggregation of GABAA
receptors at synapses is localized to intracellular pools, suggests that there could
be separate mechanisms for receptor localization to postsynaptic sites,
membrane insertion, and function of receptors. Future studies should therefore
concentrate on elucidating these two mechanisms, and identify the role of
proteins involved in each mechanism. However, while proteins have been
identified that interact with the cytoplasmic loop domain of the γ2 subunit and
have putative roles in trafficking of receptors to the membrane and synapses, no
proteins have been shown to interact with the transmembrane domains of the γ2
subunit. A search for novel proteins should focus on proteins that interact with
the TM4 domain, as it is critical for our understanding of synaptic localization and

131
stabilization of GABAA receptors at synaptic sites. It is possible that such proteins
will contribute to synaptic localization of GABAA receptors.
Additionally, work done to elucidate the gephyrin-collybistin interaction and
requirement of these proteins for synaptic localization of GABAA receptors is
necessary to understand the function of these proteins and their effect on
maintenance of GABAA receptors at synaptic sites. The collaboration detailed in
Chapter 4 determined that the gephyrin binding site on collybistin, and
demonstrated the essential role this interaction plays on proper localization of
both gephyrin and GABAA receptors (Harvey et al., 2004). However, further work
to clarify the function of the different collybistin isoforms and the consequences of
mutation or deletion of collybistin on gephyrin, glycine and GABAA receptors will
allow us to further understand the role of collybistin in the mechanisms of
localization and stabilization of synaptic inhibitory receptors. In addition, the
consequences of mutations that alter the function of collybistin and the effects of
these mutations on downstream protein-protein interactions, which may manifest
as neurological disorders, should be carefully considered. Not surprisingly, as
collybistin is a member of a larger family of functionally similar proteins,
collybistin mutant mice are apparently viable and do not show the perinatal lethal
phenotypes of gephyrin or γ2 subunit KO mice (H. Betz, personal
communication).
The role of other proteins known to bind to the γ2 subunit of GABAA
receptors needs to be elucidated. Several proteins known to interact with the
cytoplasmic loop domain, such as GODZ and GABARAP, do not localize directly

132
with GABAA receptors at postsynaptic sites, and are believed to be trafficking
factors for the receptors to synaptic sites. However, with the results here, it is
clear that the cytoplasmic loop is not responsible for synaptic localization of the
GABAA receptors; rather, this domain contributes to gephyrin recruitment and
function of synapses. Therefore understanding the mechanism of action for
these proteins may provide a better understanding for their role in endocytic
recycling of GABAA receptors. The notion that the γ2 cytoplasmic loop is
dispensable suggests that palmitoylation per se is not the signal for localization
of receptors to synapses. However, palmitoylation appears to be essential for
surface expression of γ2 subunit containing GABAA receptors (Rathenberg et al
2004). Further work examining the ability of the γ2 subunit, containing multiple
cysteine mutations, to properly localize to synaptic sites in the presence or
absence of endogenous γ2 should be performed to determine the mechanism of
action for palmitoylation. .
6.7.1 Long term applications of this research
Understanding the mechanisms of trafficking of GABAA receptors is critical
for the further understanding of disease states caused by mislocalization or
deficits in recycling of receptors. Long-term changes in synaptic efficacy brought
about by activity dependent changes are thought to be critical for learning,
memory formation, neuronal development and neuronal excitability in the CNS.
This long-term plasticity can be induced by either long-term potentiation (LTP),
an increase in synaptic strength, or long-term depression (LTD), a decrease in

133
synaptic strength. Studies examining LTD of GABAergic synapses have
suggested that long lasting modifications of synaptic strength can be induced by
LTD, and in fact, repeated electrical stimulations in hippocampal slices have
resulted in epileptiform activity (reviewed in Giasara et al., 2002). Recent work by
Maguire et al. (2005), demonstrates cyclic changes in extrasynaptic GABAA
receptors, which have resulted in altered seizure susceptibility and anxiety. This
study suggests that certain epilepsy phenotypes are possibly due to deficits in
regulatory mechanisms, such as trafficking and recycling, of δ subunit containing
GABAA receptors (Maguire et al., 2005). Finally, a study involved in examining
ischemia in the hippocampus demonstrates that prolonged ischemia induces
release of GABA by exocytosis and followed by GAT-1 transporter reversal,
leading to an increase in Cl- influx into the postsynaptic cell. This increase in Cl-
entry through GABAA receptor channels is thought to promote potentially
neurotoxic cell swelling. This study suggests that the timing of GABA application
can induce a neuroprotective or neurotoxic effect (Allen et al., 2004). These
studies also demonstrate that modifications of cellular mechanisms can alter
either receptor localization, recycling of receptors, or presynaptic release of
neurotransmitters. This could induce long-term changes that may result in
neurological deficits. By elucidating the mechanisms of receptor trafficking and
recycling, researchers can devise novel therapeutic approaches for the treatment
of diseases affected by modulation of GABAA receptors.
According to the epilepsy foundation, over 180,000 new cases of epilepsy,
of which over 20 different types of seizure disorders are characterized, are

134
diagnosed each year in the United States (www.epilepsyfoundationsewi.org/). At
the molecular level, it has been well documented that mutations within the γ2
subunit of GABAA receptors interfere with normal trafficking of GABAA receptors,
leading to a dramatic deficit in γ2 subunit-containing GABAA receptors, which
results in epileptogenic phenotypes (Cohen et al., 2002; Sancar and Czajkowski,
2004; Hales et al., 2005). In addition, rodent stroke models also have a decrease
in GABAA receptor expression in area surrounding the lesion, which leads to an
increase in neuronal excitability and leads to epileptic seizures in 5-20% of
patients after stroke (Schiene et al., 1996). Therefore, the study of GABAA
receptor localization, proteins involved in this localization and stabilization on the
postsynaptic membrane, with particular emphasis on the γ2 subunit, is critically
important for elucidating the mechanisms of action of epilepsy and may help
understand of excitotoxicity observed following ischemia.
The research presented here gives researchers a basis to investigate
clinically important drug targets for epilepsy, stroke and hyperekplexia patients.
In addition, the γ2 subunit TM4 domain can be examined for novel protein-protein
interactions. These proteins could be substrates for clinically important drugs or
could be utilized for gene therapies. Moreover, the identification of these
proteins could result in the discovery of genetically inheritable mutations,
resulting in awareness and prevention of seizures. As the majority of epilepsies
manifest in children, and can cause mild to severe brain damage, the ability to
accurately predict and treat these patients could result in enormous benefits.
Certainly, the continuation of basic research to elucidate the mechanisms of

135
GABAA receptor localization is needed to understand the complex interactions
between GABAA receptors. Understanding the variety of cytoskeletal and
membrane proteins that interact with these receptors is imperative for both
clinical research and patient therapies.

136
BIBLIOGRAPHY
Alldred MJ, Mulder-Rosi J, Lingenfelter SE, Chen G, Luscher B (2005). Distinct gamma2 subunit domains mediate clustering and synaptic function of postsynaptic GABAA receptors and gephyrin. J Neurosci 25:594-603.
Allen NJ, Rossi, DJ, Attwell D (2004). Sequential release of GABA by exocytosis
and reversed uptake leads to neuronal swelling in simulated ischemia of hippocampal slices. J. Neurosci. 24: 3837-3849
Anagnostaras SG, Craske MG, Fanselow MS (1999). Anxiety: at the intersection
of genes and experience. Nature Neurosci 2:780-782. Angelotti TP, Macdonald RL (1993). Assembly of GABAA receptor subunits: α1β1
and α1β1γ2S subunits produce unique ion channels with dissimilar single channel properties. J Neurosci 13:1429-1440.
Baer K, Essrich C, Benson JA, Benke D, Bluethmann H, Fritschy J-M, Lüscher B (1999). Postsynaptic clustering of GABAA receptors by the γ3 subunit in vivo. PNAS 96:12860-12865.
Baer K, Essrich C, Balsiger S, Wick M, Harris RA, Sieghart W, Benson J,
Fritschy J-M, Lüscher B (2000). Rescue of γ2 subunit-deficient mice by transgenic overexpression of the γ2sS or γ2L ubunit. Soc Neurosci Abst 25:1709.
Banker G, Goslin K (1998). Culturing nerve cells, 2nd Edition. Cambridge, Mass.: MIT Press.
Barnes EM, Jr. (2001). Assembly and intracellular trafficking of GABAA receptors. Int Rev Neurobiol 48:1-29.
Baumann SW, Baur R, Sigel E (2001). Subunit arrangement of gamma-aminobutyric acid type A receptors. JBC 276:36275-36280.

137
Ben-Ari Y (2002). Excitatory actions of gaba during development: the nature of the nurture. Nat Rev Neurosci 3:728-739.
Benson JA, Low K, Keist R, Mohler H, Rudolph U (1998). Pharmacology of recombinant gamma-aminobutyric acidA receptors rendered diazepam-insensitive by point-mutated alpha-subunits. FEBS 431:400-404.
Blanton MP, Cohen JB. (1994). Identifying the lipid-protein interface of the Torpedo nicotinic acetylcholine receptor: secondary structure implications. Biochemistry 33:2859-2872.
Biederer Sara Y, Mozhayeva M, Atasoy D, Liu X, Kavalali ET, Sudhof TC (2002). SynCAM, a Synaptic Adhesion Molecule That Drives Synapse Assembly, Science 297: 1525-1531
Boehm SL, 2nd, Peden L, Harris RA, Blednov YA (2004). Deletion of the fyn-kinase gene alters sensitivity to GABAergic drugs: dependence on beta2/beta3 GABAA receptor subunits. J Pharmacol Exp Ther 309:1154-1159.
Bonifacino JS, Cosson P, Klausner RD (1990). Colocalized transmembrane determinants for ER degradation and subunit assembly explain the intracellular fate of TCR chains. Cell 63:503-513.
Bormann H, Melzig MF (2000). Inhibition of metallopeptidases by flavonoids and related compounds. Pharmazie 55:129-132.
Boulter J, Evans K, Goldman D, Martin G, Treco D, Heinemann S, Patrick J. (1986). Isolation of a cDNA clone coding for a possible neural nicotinic acetylcholine receptor alpha-subunit. Nature 319:368-374.
Brandon NJ, Jovanovic JN, Smart TG, Moss SJ (2002). Receptor for activated C
kinase-1 facilitates protein kinase C-dependent phosphorylation and functional modulation of GABA(A) receptors with the activation of G-protein-coupled receptors. J Neurosci 22:6353-6361.

138
Brandon NJ, Delmas P, Hill J, Smart TG, Moss SJ (2001). Constitutive tyrosine phosphorylation of the GABA(A) receptor gamma 2 subunit in rat brain. Neuropharmacol 41:745-752.
Brunig I, Suter A, Knuesel I, Luscher B, Fritschy JM (2002). GABAergic terminals are required for postsynaptic clustering of dystrophin but not of GABA(A) receptors and gephyrin. J Neurosci 22:4805-4813.
Bruses JL, Chauvet N, Rutishauser U. (2001). Membrane lipid rafts are necessary for the maintenance of the (alpha)7 nicotinic acetylcholine receptor in somatic spines of ciliary neurons. J Neurosci 21:504-512.
Cabot JB, Bushnell A, Alessi V, Mendell NR (1995). Postsynaptic gephyrin immunoreactivity exhibits a nearly one-to-one correspondence with γ-aminobutyric acid-like immunogold-labeled synaptic inputs to sympathetic preganglionic neurons. JCN 356:418-432.
Chang Y, Wang R, Barot S, Weiss DS (1996). Stoichiometry of a recombinant GABAA receptor. J Neurosci 16:5415-5424.
Chen C, Okayama H (1987). High-efficiency transformation of mammalian cells by plasmid DNA. Mol Cell Biol 7:2745-2752.
Chen G, Trombley P, van del Pol A (1996). Excitatory actions of GABA in developing rat hypothalamic neurones. J of Physio 494.2:451-464.
Chen Z, Xin YR, Jiang Y, Jiang JX (2001). Cloning a novel mouse Gabarapl2 cDNA and its characterization. Acta Pharmacol Sin 22:751-755.
Chih B, Engelman H, Scheiffele P (2005). Control of excitatory and inhibitory synapse formation by neuroligins. Science 307:1324-1328.
Christie SB, Miralles CP, De Blas AL (2002). GABAergic innervation organizes synaptic and extrasynaptic GABAA receptor clustering in cultured hippocampal neurons. J Neurosci 22:684-697.

139
Cohen I, Navarro V, Clemenceau S, Baulac M, Miles R (2002). On the origin of interictal activity in human temporal lobe epilepsy in vitro. Science 298:1418-1421.
Connolly CN, Wooltorton JR, Smart TG, Moss SJ (1996a). Subcellular localization of γ-aminobutyric acid type A receptors is determined by receptor β subunits. Proc Natl Acad Sci U S A 93:9899-9904.
Connolly CN, Krishek BJ, McDonald BJ, Smart TG, Moss SJ (1996b). Assembly and cell surface expression of heteromeric and homomeric γ-aminobutyric acid type A receptors. JBC 271:89-96.
Connolly CN, Uren JM, Thomas P, Gorrie GH, Gibson A, Smart TG, Moss SJ (1999a). Subcellular localization and endocytosis of homomeric gamma2 subunit splice variants of gamma-aminobutyric acid type A receptors. MCN 13:259-271.
Connolly CN, Kittler JT, Thomas P, Uren JM, Brandon NJ, Smart TG, Moss SJ (1999b). Cell surface stability of gamma-aminobutyric acid type A receptors. Dependence on protein kinase C activity and subunit composition. JBC 274:36565-36572.
Cormack BP, Valdivia RH, Falkow S (1996). FACS-optimized mutants of the green fluorescent protein (GFP). Gene 173:33-38.
Crestani F, Lorez M, Baer K, Essrich C, Benke D, Laurent JP, Belzung C, Fritschy JM, Lüscher B, Mohler H (1999). Decreased GABAA-receptor clustering results in enhanced anxiety and a bias for threat cues. Nature Neurosci 2:833-839.
de Almeida RF, Loura LM, Prieto M, Watts A, Fedorov A, Barrantes FJ. (2004). Cholesterol modulates the organization of the gammaM4 transmembrane domain of the muscle nicotinic acetylcholine receptor. Biophys J 86:2261-2272.
Douangamath A, Filipp FV, Klein AT, Barnett P, Zou P, Voorn-Brouwer T, Vega MC, Mayans OM, Sattler M, Distel B, Wilmanns M. (2002). Topography for independent binding of alpha-helical and PPII-helical ligands to a peroxisomal SH3 domain. Mol Cell 10:1007-1017.

140
Essrich C, Lorez M, Benson J, Fritschy J-M, Lüscher B (1998). Postsynaptic clustering of major GABAA receptor subtypes requires the γ2 subunit and gephyrin. Nature Neurosci 1: 563-571.
Farrar SJ, Whiting PJ, Bonnert TP, McKernan RM (1999). Stoichiometry of a ligand-gated ion channel determined by fluorescence energy transfer. JBC 274:10100-10104.
Feng G, Tintrup H, Kirsch J, Nichol MC, Kuhse J, Betz H, Sanes JR (1998). Dual requirement for gephyrin in glycine receptor clustering and molybdoenzyme activity. Science 282:1321-1324.
Fischer F, Kneussel M, Tintrup H, Haverkamp S, Rauen T, Betz H, Wassle H (2000). Reduced synaptic clustering of GABA and glycine receptors in the retina of the gephyrin null mutant mouse. J Comp Neurol 427:634-648.
Fritschy J-M, Mohler H (1995). GABAA receptor heterogeneity in the adult rat brain: differential regional and cellular distribution of seven major subunits. JCN 359:154-194.
Fritschy JM, Johnson DK, Mohler H, Rudolph U (1998). Independent assembly and subcellular targeting of GABAA-receptor subtypes demonstrated in mouse hippocampal and olfactory neurons in vivo. Neurosci lett 249:99-102.
Fritschy JM, Schweizer C, Brunig I, Luscher B (2003). Pre- and post-synaptic mechanisms regulating the clustering of type A gamma-aminobutyric acid receptors (GABAA receptors). Biochem Soc Trans 31:889-892.
Fuhrmann JC, Kins S, Rostaing P, El Far O, Kirsch J, Sheng M, Triller A, Betz H, Kneussel M (2002). Gephyrin interacts with Dynein light chains 1 and 2, components of motor protein complexes. J Neurosci 22:5393-5402.
Gally C, Bessereau JL (2003). GABA is dispensable for the formation of junctional GABA receptor clusters in Caenorhabditis elegans. J Neurosci 23:2591-2599.

141
Ganguly K, Schinder AF, Wong ST, Poo M (2001). GABA itself promotes the developmental switch of neuronal GABAergic responses from excitation to inhibition. Cell 105:521-532.
Giasara JL, Caillard O, Ben-Ari Y (2002). Long-term plasticity at GABAergic and glycinergic synapses: mechanisms and functional significance. Trends Neurosci. 25:564-570.
Giesemann T, Schwarz G, Nawrotzki R, Berhorster K, Rothkegel M, Schluter K, Schrader N, Schindelin H, Mendel RR, Kirsch J, Jockusch BM (2003). Complex formation between the postsynaptic scaffolding protein gephyrin, profilin, and Mena: a possible link to the microfilament system. J Neurosci 23:8330-8339.
Giustetto M, Kirsch J, Fritschy JM, Cantino D, Sassoe-Pognetto M (1998). Localization of the clustering protein gephyrin at GABAergic synapses in the main olfactory bulb of the rat. J Comp Neurol 395:231-244.
Graf ER, Zhang X, Jin SX, Linhoff MW, Craig AM. (2004). Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell 119:1013-1026.
Groemping Y, Lapouge K, Smerdon SJ, Rittinger K. (2003). Molecular basis of phosphorylation-induced activation of the NADPH oxidase. Cell 113:343-355.
Grosskreutz Y, Hermann A, Kins S, Fuhrmann JC, Betz H, Kneussel M (2001). Identification of a gephyrin-binding motif in the GDP/GTP exchange factor collybistin. Biol Chem 382:1455-1462.
Günther U, Benson J, Benke D, Fritschy J-M, Reyes G, Knoflach F, Crestani F, Aguzzi A, Arigoni M, Lang Y, Bluethmann H, Mohler H, Lüscher B (1995). Benzodiazepine-insensitive mice generated by targeted disruption of the γ2 subunit gene of GABAA receptors. PNAS 92:7749-7753.
Hales TG, Tang H, Bollan KA, Johnson SJ, King DP, McDonald NA, Cheng A, Connolly CN (2005). The epilepsy mutation, γ2(R43Q) disrupts a highly conserved inter-subunit contact site, perturbing the biogenesis of GABAA receptors. MCN 29:120-127.

142
Harvey K, Duguid IC, Alldred MJ, Beatty SE, Ward H, Keep NH, Lingenfelter SE, Pearce BR, Lundgren J, Owen MJ, Smart TG, Luscher B, Rees MI, Harvey RJ (2004). The GDP-GTP exchange factor collybistin: an essential determinant of neuronal gephyrin clustering. J Neurosci 24:5816-5826.
Hering H, Lin CC, Sheng M (2003). Lipid rafts in the maintenance of synapses, dendritic spines, and surface AMPA receptor stability. J Neurosci 23:3262-3271.
Homanics GE, Harrison NL, Quinlan JJ, Krasowski MD, Rick CE, de Blas AL, Mehta AK, Kist F, Mihalek RM, Aul JJ, Firestone LL (1999). Normal electrophysiological and behavioral responses to ethanol in mice lacking the long splice variant of the gamma2 subunit of the gamma- aminobutyrate type A receptor. Neuropharmacol 38:253-265.
Hubner RA, Kubota E, Casley DJ, Johnston CI, Burrell LM (2001). In-vitro and in-vivo inhibition of rat neutral endopeptidase and angiotensin converting enzyme with the vasopeptidase inhibitor gemopatrilat. J Hypertens 19:941-946.
Jacob, TC, Bogdanov, YD, Kittler, JT, Haydon, PG, Moss SJ (2005). Gephyrin regulates the cell surface dynamics of synaptic GABAA receptors. (personal communication)
Jovanovic JN, Thomas P, Kittler JT, Smart TG, Moss SJ (2004). Brain-derived neurotrophic factor modulates fast synaptic inhibition by regulating GABA(A) receptor phosphorylation, activity, and cell-surface stability. J Neurosci 24:522-530.
Kakazu Y, Akaike N, Komiyama S, Nabekura J (1999). Regulation of intracellular chloride by cotransporters in developing lateral superior olive neurons. J Neurosci 19:2843-2851.
Kanematsu T, Jang IS, Yamaguchi T, Nagahama H, Yoshimura K, Hidaka K, Matsuda M, Takeuchi H, Misumi Y, Nakayama K, Yamamoto T, Akaike N, Hirata M, Nakayama K (2002). Role of the PLC-related, catalytically inactive protein p130 in GABA(A) receptor function. EMBO 21:1004-1011.

143
Kannenberg K, Baur R, Sigel E (1997). Proteins associated with a1-subunit-containing GABAA receptors from bovine brain. J Neurochem. 68:1352-1360
Keller CA, Yuan X, Panzanelli P, Martin ML, Alldred M, Sassoe-Pognetto M, Luscher B (2004). The gamma2 subunit of GABA(A) receptors is a substrate for palmitoylation by GODZ. J Neurosci 24:5881-5891.
Kins S, Betz H, Kirsch J (2000). Collybistin, a newly identified brain specific GEF induces submembrane clustering of gephyrin. Nature Neurosci 3:22-29.
Kirsch J, Betz H (1998). Glycine-receptor activation is required for receptor clustering in spinal neurons. Nature 392:717-720.
Kirsch J, Kuhse J, Betz H. (1995). Targeting of glycine receptor subunits to gephyrin-rich domains in transfected human embryonic kidney cells. Mol Cell Neurosci 6:450-461.
Kirsch J, Meyer G, Betz H (1996). Synaptic targeting of ionotropic neurotransmitter receptors. Mol. Cell Neurosci. 8:93-98
Kirsch J, Wolters I, Triller A, Betz H (1993). Gephyrin antisense oligonucleotides prevent glycine receptor clustering in spinal neurons. Nature 366:745-748.
Kittler JT, McAinsh K, Moss SJ (2002). Mechanisms of GABAA receptor assembly and trafficking: implications for the modulation of inhibitory neurotransmission. Mol Neurobiol 26:251-268.
Kittler JT, Wang J, Connolly CN, Vicini S, Smart TG, Moss SJ (2000). Analysis of GABAA receptor assembly in mammalian cell lines and hippocampal neurons using gamma 2 subunit green fluorescent protein chimeras. MCN 16:440-452.
Kittler JT, Rostaing P, Schiavo G, Fritschy JM, Olsen R, Triller A, Moss SJ (2001). The subcellular distribution of GABARAP and its ability to interact with NSF suggest a role for this protein in the intracellular transport of GABA(A) receptors. MCN 18:13-25.

144
Kneussel M, Betz H (2000). Receptors, gephyrin and gephyrin-associated proteins: novel insights into the assembly of inhibitory postsynaptic membrane specializations. J Physiol 525 Pt 1:1-9.
Kneussel M, Hermann A, Kirsch J, Betz H (1999). Hydrophobic interactions mediate binding of the glycine receptor beta-subunit to gephyrin. J Neurochem 72:1323-1326.
Kneussel M, Brandstatter JH, Gasnier B, Feng G, Sanes JR, Betz H (2001). Gephyrin-independent clustering of postsynaptic GABA(A) receptor subtypes. MCN 17:973-982.
Kneussel M, Haverkamp S, Fuhrmann JC, Wang H, Wassle H, Olsen RW, Betz H (2000). The gamma-aminobutyric acid type A receptor (GABAAR)-associated protein GABARAP interacts with gephyrin but is not involved in receptor anchoring at the synapse. Proc Natl Acad Sci U S A 97:8594-8599.
Knight AR, Stephenson FA, Tallman JF, Ramabahdran TV (2000). Monospecific antibodies as probes for the stoichiometry of recombinant GABA(A) receptors. Receptors Channels 7:213-226.
Knuesel I, Mastrocola M, Zuellig RA, Bornhauser B, Schaub MC, Fritschy JM (1999). Altered synaptic clusterin of GABAA receptors in mice lacking dystrophin (mdx mice). Eur J Neurosci in press.
Kofuji P, Wang JB, Moss SJ, Huganir RL, Burt DR (1991). Generation of two forms of the γ-aminobutyric acid receptor γ2-subunit in mice by alternative splicing. J Neurochem 56:713-715.
Kornau HC, Seeburg PH, Kennedy MB (1997). Interaction of ion channels and receptors with PDZ domain proteins. Curr Opin Neurobiol 7(3):368-73
Krishek BJ, Moss SJ, Smart TG (1996). Homomeric beta 1 gamma-aminobutyric acid A receptor-ion channels: evaluation of pharmacological and physiological properties. Mol Pharmacol 49:494-504.

145
Lambert JJ, Belelli D, Peden DR, Vardy AW, Peters JA (2003). Neurosteroid modulation of GABAA receptors. Prog Neurobiol 71:67-80.
Lamelli, UK. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680-685.
Laurie DJ, Seeburg PH, Wisden W (1992). The distribution of 13 GABAA receptor subunit mRNAs in the rat brain. II. Olfactory bulb and cerebellum. J Neurosci 12:1063-1076.
Leil TA, Chen ZW, Chang CS, Olsen RW (2004). GABAA receptor-associated protein traffics GABAA receptors to the plasma membrane in neurons. J Neurosci 24:11429-11438.
Levi S, Vannier C, Triller A (1998). Strychnine-sensitive stabilization of postsynaptic glycine receptor clusters. J Cell Science 111:335-345.
Levi S, Logan SM, Tovar KR, Craig AM (2004). Gephyrin is critical for glycine receptor clustering but not for the formation of functional GABAergic synapses in hippocampal neurons. J Neurosci 24:207-217.
Levi S, Grady RM, Henry MD, Campbell KP, Sanes JR, Craig AM (2002). Dystroglycan is selectively associated with inhibitory GABAergic synapses but is dispensable for their differentiation. J Neurosci 22:4274-4285.
Lin RC, Scheller RH (2000). Mechanisms of synaptic vesicle exocytosis. Annu Rev Cell Dev Biol 16:19-49.
Liu Q, Berry D, Nash P, Pawson T, McGlade CJ, Li SS. (2003). Structural basis for specific binding of the Gads SH3 domain to an RxxK motif-containing SLP-76 peptide: a novel mode of peptide recognition. Mol Cell 11:471-481.
Lorez M, Benke D, Luscher B, Mohler H, Benson JA (2000). Single-channel properties of neuronal GABAA-receptors lacking the gamma2 subunit. J Physiol 527:11-31.

146
Lu YM, Mansuy IM, Kandel ER, Roder J. (2000). Calcineurin-mediated LTD of GABAergic inhibition underlies the increased excitability of CA1 neurons associated with LTP. Neuron 26:197-205.
Luscher B, Fritschy JM (2001). Subcellular localization and regulation of GABAA receptors and associated proteins. Int Rev Neurobiol 48:31-64.
Luscher B, Keller CA (2004). Regulation of GABAA receptor trafficking, channel activity, and functional plasticity of inhibitory synapses. Pharmacol Ther 102:195-221.
Lynch JW (2004). Molecular structure and function of the glycine receptor chloride channel. Physiol Rev 84:1051-1095.
Macdonald RL, Olsen RW (1994). GABAA receptor channels. Ann Rev Neurosci 17:569-602.
Maguire JL, Stell BM, Rafizadeh M, Mody I (2005). Ovarian cycle-linked changes in GABAA receptors mediating tonic inhibition alter seizure susceptibility and anxiety. Nat. Neurosci. 8: 797-804.
Malherbe P, Draguhn A, Multhaup G, Beyreuther K, Mohler H (1990). GABAA-receptor expressed from rat brain alpha- and beta-subunit cDNAs displays potentiation by benzodiazepine receptor ligands. Mol Brain Res 8:199-208.
Mammoto A, Sasaki T, Asakura T, Hotta I, Imamura H, Takahashi K, Matsuura Y, Shirao T, Takai Y (1998). Interactions of drebrin and gephyrin with profilin. Biochem Biophys Res Commun 243:86-89.
Mangan PS, Sun C, Carpenter M, Goodkin HP, Sieghart W, Kapur J (2005). Cultured Hippocampal Pyramidal Neurons Express Two Kinds of GABAA Receptors. Mol Pharmacol 67:775-788.
Mann SS, Hammarback JA (1994). Molecular characterizationof light chain 3. LBC 269:1149-11497.

147
Mansuy V, Boireau W, Fraichard A, Schlick JL, Jouvenot M, Delage-Mourroux R. (2004). GEC1, a protein related to GABARAP, interacts with tubulin and GABA(A) receptor. Biochem Biophys Res Commun 325:639-648.
McDonald JD (1994). Producing mouse genetic models for human diseases. In: Gene therapeutics. Methods and applications of diretct gene transfer (Wolff JA, ed), pp 26-39. Boston, Basel, Berlin: Birkhäuser.
Mehta AK, Ticku MK (2005). Effect of chronic administration of ethanol on GABAA receptor assemblies derived from alpha2-, alpha3-, beta2- and gamma2-subunits in the rat cerebral cortex. Brain Res 1031:134-137.
Meier J, Grantyn R (2004). A gephyrin-related mechanism restraining glycine receptor anchoring at GABAergic synapses. J Neurosci 24:1398-1405.
Meier J, Meunier-Durmort C, Forest C, Triller A, Vannier C (2000). Formation of glycine receptor clusters and their accumulation at synapses. J Cell Sci 113 (Pt 15):2783-2795.
Meier J, Vannier C, Serge A, Triller A, Choquet D (2001). Fast and reversible trapping of surface glycine receptors by gephyrin. Nat Neurosci 4:253-260.
Meyer G, Kirsch J, Betz H, Langosch D (1995). Identification of a gephyrin binding motif on the glycine receptor β subunit. Neuron 15:563-572.
Miyazawa A, Fujiyoshi Y, Unwin N (2003). Structure and gating mechanism of the acetylcholine receptor pore. Nature 423:949-955.
Miyazawa A, Fujiyoshi Y, Stowell M, Unwin N (1999). Nicotinic acetylcholine receptor at 4.6 A resolution: transverse tunnels in the channel wall. J Mol Biol 288:765-786.
Mody I, Koninck Y, Otis TS, Soltesz I (1994). Bridging the cleft at GABA synapses in the brain. TINS 17:517-525.

148
Nabekura J, Ueno T, Okabe A, Furuta A, Iwaki T, Shimizu-Okabe C, Fukuda A, Akaike N (2002). Reduction of KCC2 expression and GABAA receptor-mediated excitation after in vivo axonal injury. J Neurosci 22:4412-4417.
Nayeem N, Green TP, Martin IL, Barnard EA (1994). Quaternary structure of the native GABAA receptor determined by electron microscopic image analysis. J Neurochem 62:815-818.
Nemeroff CB (2003). Anxiolytics: past, present, and future agents. J Clin Psychiatry 64 Suppl 3:3-6.
Nurmi EL, Bradford Y, Chen Y, Hall J, Arnone B, Gardiner MB, Hutcheson HB, Gilbert JR, Pericak-Vance MA, Copeland-Yates SA, Michaelis RC, Wassink TH, Santangelo SL, Sheffield VC, Piven J, Folstein SE, Haines JL, Sutcliffe JS (2001). Linkage disequilibrium at the Angelman syndrome gene UBE3A in autism families. Genomics 77:105-113.
Nymann-Andersen J, Sawyer GW, Olsen RW. (2002). Interaction between GABAA receptor subunit intracellular loops: implications for higher order complex formation. J Neurochem 83:1164-1171.
Papadimitriou GN, Dikeos DG, Karadima G, Avramopoulos D, Daskalopoulou EG, Stefanis CN (2001). GABA-A receptor beta3 and alpha5 subunit gene cluster on chromosome 15q11-q13 and bipolar disorder: a genetic association study. Am J Med Genet 105:317-320.
Pediconi MF, Gallegos CE, De Los Santos EB, Barrantes FJ. (2004). Metabolic cholesterol depletion hinders cell-surface trafficking of the nicotinic acetylcholine receptor. Neuroscience 128:239-249.
Peng Z, Hauer B, Mihalek RM, Homanics GE, Sieghart W, Olsen RW, Houser CR (2002). GABA(A) receptor changes in delta subunit-deficient mice: altered expression of alpha4 and gamma2 subunits in the forebrain. J Comp Neurol 446:179-197.
Petrini EM, Marchionni I, Zacchi P, Sieghart W, Cherubini E (2004). Clustering of extrasynaptic GABA(A) receptors modulates tonic inhibition in cultured hippocampal neurons. JBC 279:45833-45843.

149
Plotkin MD, Snyder EY, Hebert SC, Delpire E (1997). Expression of the Na-K-2Cl cotransporter is developmentally regulated in postnatal rat brains: a possible mechanism underlying GABA's excitatory role in immature brain. J Neurobiol 33:781-795.
Prange O, Wong TP, Gerrow K, Wnag YT, El-Husseini A (2004). A balance between excitatory and inhibitory synapses is controlled by PSD-95 and neuroligin. PNAS 101(38):13915-13920.
Prior P, Schmitt B, Grenningloh G, Pribilla I, Multhaup G, Beyreuther K, Maulet Y, Werner P, Langosh D, Kirsch J, Betz H (1992). Primary structure and alternative splice variants of gephyrin, a putative glycine receptor-tubulin linker protein. Neuron 8:1161-1170.
Rabow LE, Russek SJ, Farb DH (1995). From ion currents to genomic analysis: recent advances in GABAA receptor research. Synapse 21:189-274.
Ramming M, Kins S, Werner N, Hermann A, Betz H, Kirsch J (2000). Diversity and phylogeny of gephyrin: tissue-specific splice variants, gene structure, and sequence similarities to molybdenum cofactor-synthesizing and cytoskeleton-associated proteins. Proc Natl Acad Sci U S A 97:10266-10271.
Rao A, Cha EM, Craig AM (2000). Mismatched appositions of presynaptic and postsynaptic components in isolated hippocampal neurons. J Neurosci 20:8344-8353.
Rathenberg J, Kittler JT, Moss SJ. (2004). Palmitoylation regulates the clustering and cell surface stability of GABAA receptors. Mol. Cell. Neurosci. 26:251-257.
Rees MI, Harvey K, Ward H, White JH, Evans L, Duguid IC, Hsu CC, Coleman SL, Miller J, Baer K, Waldvogel HJ, Gibbon F, Smart TG, Owen MJ, Harvey RJ, Snell RG (2003). Isoform heterogeneity of the human gephyrin gene (GPHN), binding domains to the glycine receptor, and mutation analysis in hyperekplexia. JBC 278:24688-24696.
Rees MI, Lewis TM, Kwok JB, Mortier GR, Govaert P, Snell RG, Schofield PR, Owen MJ (2002). Hyperekplexia associated with compound heterozygote

150
mutations in the beta-subunit of the human inhibitory glycine receptor (GLRB). Hum Mol Genet 11:853-860.
Rees MI, Lewis TM, Vafa B, Ferrie C, Corry P, Muntoni F, Jungbluth H, Stephenson JB, Kerr M, Snell RG, Schofield PR, Owen MJ (2001). Compound heterozygosity and nonsense mutations in the alpha(1)-subunit of the inhibitory glycine receptor in hyperekplexia. Hum Genet 109:267-270.
Reid T, Bathoorn A, Ahmadian MR, Collard JG (1999). Identification and characterization of hPEM-2, a guanine nucleotide exchange factor specific for Cdc42. JBC 274:33587-33593.
Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K (1999). The K+/Cl- co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 397:251-255.
Rizo J, Sudhof TC (2002). Snares and Munc18 in synaptic vesicle fusion. Nat Rev Neurosci 3:641-653.
Roche KW, Standley S, McCallum J, Dune Ly C, Ehlers MD, Wenthold RJ (2001). Molecular determinants of NMDA receptor internalization. Nat Neurosci 4:794-802.
Sagiv Y, Legesse-Miller A, Porat A, Elazar Z (2000). GATE-16, a membrane transport modulator, interacts with NSF and the Golgi v-SNARE GOS-28. EMBO Journal 19:1494-1504.
Sambrook J, Fritsch EF, Maniatis T, eds (1989). Molecular Cloning, a laboratory manual., 2nd Edition. Cold Spring Harbor: Cold Spring Harbor Laboratory Press.
Sancar F, Czajkowski C (2004). A GABAA receptor mutation linked to human epilepsy (gamma2R43Q) impairs cell surface expression of alphabetagamma receptors. JBC 279:47034-47039.

151
Sassoe-Pognetto M, Panzanelli P, Sieghart W, Fritschy JM (2000). Colocalization of multiple GABA-A receptor subtypes with gephyrin at postsynaptic sites. the Journal of Compartive Neurology 420:481-498.
Sassoe-Pognetto M, Giustetto M, Panzanelli P, Cantino D, Kirsch.J., Fritschy JM (1999). Postsynaptic colocalizationof gephyrin and GABA-A receptors. Annals New York Academy of Sciences:693-696.
Sassoé-Pognetto M, Kirsch J, Grünert U, Greferath U, Fritschy JM, Mohler H, Betz H, Wässle H (1995). Colocalization of gephyrin and GABAA-receptor subunits in the rat retina. JCN 357:1-14.
Scheiffele P, Fan J, Choih J, Fetter T, Serafini T (2000). Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons Cell 101:657-669.
Scheine K, Bruehl C, Zilles K, Qü M, Hagemann G, Kraemer M, Witte OW (1996). Neuronal hyperexcitability and reduction of GABAA-receptor expression in the surround of cerebral photothrombosis. J Cereb Blood Flow Metab. 16: 906-914
Schwarz G, Schrader N, Mendel RR, Hecht HJ, Schindelin H (2001). Crystal structures of human gephyrin and plant Cnx1 G domains: comparative analysis and functional implications. J Mol Biol 312:405-418.
Schweizer C, Balsiger S, Bluethmann H, Mansuy IM, Fritschy JM, Mohler H, Luscher B (2003). The gamma 2 subunit of GABA(A) receptors is required for maintenance of receptors at mature synapses. MCN 24:442-450.
Scott DB, Blanpied TA, Swanson GT, Zhang C, Ehlers MD (2001). An NMDA receptor ER retention signal regulated by phosphorylation and alternative splicing. J Neurosci 21:3063-3072.
Sieghart W, Fuchs K, Tretter V, Ebert V, Jechlinger M, Hoger H, Adamiker D (1999). Structure and subunit composition of GABA(A) receptors. Neurochem Int 34:379-385.

152
Sigel E, Baur R, Malherbe P, Mohler H (1989). The rat beta 1-subunit of the GABAA receptor forms a picrotoxin-sensitive anion channel open in the absence of GABA. FEBS 257:377-379.
Sigel E, Buhr A (1997). The benzodiazepine binding site of GABAA receptors. TIPS 18:425-429.
Silva AE, Vayego-Lourenco SA, Fett-Conte AC, Goloni-Bertollo EM, Varella-Garcia M (2002). Tetrasomy 15q11-q13 identified by fluorescence in situ hybridization in a patient with autistic disorder. Arq Neuropsiquiatr 60:290-294.
Simon J, Wakimoto H, Fujita N, Lalande M, Barnard EA (2004). Analysis of the set of GABA(A) receptor genes in the human genome. JBC 279:41422-41435.
Sola M, Kneussel M, Heck IS, Betz H, Weissenhorn W (2001). X-ray crystal structure of the trimeric N-terminal domain of gephyrin. JBC 276:25294-25301.
Song JY, Ichtchenko K, Sudhof TC, Brose N (1999). Neuroligin 1 is a postsynaptic cell-adhesion molecule of excitatory synapses. PNAS 96:1100-1105.
Standley S, Roche KW, McCallum J, Sans N, Wenthold RJ (2000). PDZ domain suppression of an ER retention signal in NMDA receptor NR1 splice variants. Neuron 28:887-898.
Steiger JL, Russek SJ (2004). GABAA receptors: building the bridge between subunit mRNAs, their promoters, and cognate transcription factors. Pharmacol Ther 101:259-281.
Sun C, Sieghart W, Kapur J (2004). Distribution of alpha1, alpha4, gamma2, and delta subunits of GABAA receptors in hippocampal granule cells. Brain Res 1029:207-216.
Terunuma M, Jang IS, Ha SH, Kittler JT, Kanematsu T, Jovanovic JN, Nakayama KI, Akaike N, Ryu SH, Moss SJ, Hirata M. (2004). GABAA receptor

153
phospho-dependent modulation is regulated by phospholipase C-related inactive protein type 1, a novel protein phosphatase 1 anchoring protein. J Neurosci 24:7074-7084.
Todd AJ, Watt C, Spike RC, Sieghart W (1996). Colocalization of GABA, glycine and their receptors at synapses in the rat spinal cord. J Neurosci 16:974-982.
Tretter V, Ehya N, Fuchs K, Sieghart W (1997). Stoichiometry and assembly of a recombinant GABAA receptor subtype. J Neurosci 17:2728-2737.
Tretter V, Hauer B, Nusser Z, Mihalek RM, Hoger H, Homanics GE, Somogyi P, Sieghart W (2001). Targeted disruption of the GABA(A) receptor delta subunit gene leads to an up-regulation of gamma 2 subunit-containing receptors in cerebellar granule cells. JBC 276:10532-10538.
Uji A, Matsuda M, Kukita T, Maeda K, Kanematsu T, Hirata M (2002). Molecules interacting with PRIP-2, a novel Ins(1,4,5)P3 binding protein type 2: Comparison with PRIP-1. Life Sci 72:443-453.
Yu L, LaPolla RJ, Davidson N. (1986). Mouse muscle nicotinic acetylcholine receptor gamma subunit: cDNA sequence and gene expression. Nucleic Acids Res 14:3539-3555.
Valenzuela CF, Machu TK, McKernan RM, Whiting P, VanRenterghem BB, McManaman JL, Brozowski SJ, Smith GB, Olsen RW, Harris RA (1995). Tyrosine kinase phosphorylation of GABAA receptors. Brain Res Mol Brain Res 31:165-172.
van Rijnsoever C, Sidler C, Fritschy JM. (2005). Internalized GABA-receptor subunits are transferred to an intracellular pool associated with the postsynaptic density. Eur J Neurosci 21:327-338.
Varoqueaux F, Jamain S, Brose N (2004) Neuroligin 2 is exclusively localized to inhibitory synapses. Eur J Cell Biol 83:449-456.
Varoqueaux F, Sigler A, Rhee JS, Brose N, Enk C, Reim K, Rosenmund C (2002). Total arrest of spontaneous and evoked synaptic transmission but

154
normal synaptogenesis in the absence of Munc13-mediated vesicle priming. Proc Natl Acad Sci U S A 99:9037-9042.
Vazquez LE, Chen HJ, Sokalova I, Knuesel I, Kennedy, MB (2004). SynGAP regulates spine formation. J Neurosci. 24(40):8862-8872.
Verdoorn TA, Draguhn A, Ymer S, Seeburg PH, Sakmann B (1990). Functional properties of recombinant rat GABAA receptors depend upon subunit composition. Neuron 4:919-928.
Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, Toonen RF, Hammer RE, van den Berg TK, Missler M, Geuze HJ, Sudhof TC (2000). Synaptic assembly of the brain in the absence of neurotrasmitter secretion. Science 287:864-869.
Vernier-Magnin S, Muller S, Sallot M, Radom J, Musard JF, Adami P, Dulieu P, Remy-Martin JP, Jouvenot M, Fraichard A (2001). A novel early estrogen-regulated gene gec1 encodes a protein related to GABARAP. Biochem Biophys Res Commun 284:118-125.
Wan Q, Man HY, Braunton J, Wang W, Salter MW, Becker L, Wang YT (1997). Modulation of GABAA receptor function by tyrosine phosphorylation of β subunits. J Neuroscience 17:5062-5069.
Wang H, Bedford FK, Brandon NJ, Moss SJ, Olsen RW (1999). GABAA-receptor-associated protein links GABAA receptors and the cytoskeleton. Nature 397:69-72.
Wang J, Liu S, Haditsch U, Tu W, Cochrane K, Ahmadian G, Tran L, Paw J, Wang Y, Mansuy I, Salter MM, Lu YM (2003). Interaction of calcineurin and type-A GABA receptor gamma 2 subunits produces long-term depression at CA1 inhibitory synapses. J Neurosci 23:826-836.
Wang H, Olsen RW (2000). Binding of the GABA(A) receptor-associated protein (GABARAP) to microtubules and microfilaments suggests involvement of the cytoskeleton in GABARAPGABA(A) receptor interaction. J Neurochem 75:644-655.

155
Wassef A, Baker J, Kochan LD (2003). GABA and schizophrenia: a review of basic science and clinical studies. J Clin Psychopharmacol 23:601-640.
Whiting PJ, Bonnert TP, McKernan RM, Farrar S, Le Bourdelles B, Heavens RP, Smith DW, Hewson L, Rigby MR, Sirinathsinghji DJ, Thompson SA, Wafford KA (1999). Molecular and functional diversity of the expanding GABA-A receptor gene family. Ann N Y Acad Sci 868:645-653.
Wisden W, Laurie DJ, Monyer H, Seeburg PH (1992). The distribution of 13 GABAA receptor subunit mRNAs in the rat brain. I. Telencephalon, diencephalon, mesencephalon. J Neurosci 12:1040-1062.
Wooltorton JR, Moss SJ, Smart TG (1997). Pharmacological and physiological characterization of murine homomeric beta3 GABA(A) receptors. Eur J Neurosci 9:2225-2235.
Xiang S, Nichols J, Rajagopalan KV, Schindelin H (2001). The crystal structure of Escherichia coli MoeA and its relationship to the multifunctional protein gephyrin. Structure (Camb) 9:299-310.
Xin Y, Yu L, Chen Z, Zheng L, Fu Q, Jiang J, Zhang P, Gong R, Zhao S (2001). Cloning, expression patterns, and chromosome localization of three human and two mouse homologues of GABA(A) receptor-associated protein. Genomics 74:408-413.
Zucker CL (1998). Localization of gephyrin and glycine receptor subunit immunoreactivity in the rabbit retina. Vis Neurosci 15:389-395.
Zucker RS (1996). Exocytosis: a molecular and physiological perspective. Neuron 17:1049-1055.
Zhou L, Chillag KL, Nigro MA (2002). Hyperekplexia: a treatable neurogenetic
disease. Brain Dev 24:669-674.

156
APPENDIX:
LIST OF PUBLICATIONS Alldred, MJ, Mulder-Rosi J, Lingenfelter SE, Chen G, Luscher B (2005) Distinct
gamma2 subunit domains mediate clustering and synaptic function of postsynaptic GABAA receptors and gephyrin. J Neurosci 25:594-603.
Keller CA, Yuan X, Panzanelli P, Martin ML, Alldred M, Sassoe-Pognetto M,
Luscher B (2004) The gamma2 subunit of GABA(A) receptors is a substrate for palmitoylation by GODZ. J Neurosci 24:5881-5891.
Harvey K, Duguid IC, Alldred MJ, Beatty SE, Ward H, Keep NH, Lingenfelter
SE, Pearce BR, Lundgren J, Owen MJ, Smart TG, Luscher B, Rees MI, Harvey RJ (2004) The GDP-GTP exchange factor collybistin: an essential determinant of neuronal gephyrin clustering. J Neurosci 24:5816-5826.

VITA
Melissa J. Alldred
Education2005 Ph.D. The Pennsylvania State University BMMB1999 B.S. Cook College, Rutgers University BiotechnologySpring1997- University of Queensland, Australia study abroad
Professional Experience04/98 - 08/99 Research Assistant, Cook College Extension Program,08/99 - present Graduate Research Assistant, The Pennsylvania State
University
PublicationsAlldred, M.J., J. Mulder-Rosi, S.E. Lingenfelter, G. Chen, and B. Lüscher
(2005). Distinct γ2 subunit domains mediate clustering and synaptic function ofpostsynaptic GABAA receptors and gephyrin. J. Neurosci., 25 (3): 594-603
Harvey, K., I. C. Duguid†, M. J. Alldred†, S. E. Beatty, H. Ward, N. H.Keep, S. E. Lingenfelter, B. R. Pearce, J. Lundgren, M. J. Owen, T. G. Smart, B.Lüscher, M. I. Rees and R. J. Harvey. The GDP-GTP exchange factor collybistin:An essential determinant of neuronal gephyrin clustering. J. Neurosci. 24, 5816-5826 †authors contributed equally to work
Cheryl A. Keller, X. Yuan, P. Panzanelli, M. L. Martin, M. Alldred,M.Sassoè-Pognetto, and B. Lüscher. The γ2 subunit of GABAA receptors is asubstrate for palmitoylation by GODZ. J. Neurosci. 24: 5881-5891
Oral PresentationsAn investigation of the domains of the γ2 subunit of GABAA receptors,
gephyrin and collybistin required for synaptic localization. Ph.D. Defenseseminar, Department of Biochemistry, Microbiology and Molecular Biology,Pennsylvania State University. May 18th, 2005
Protein domains implicated in synaptic targeting and clustering of GABAA
receptors. Department of Biology, Neuroscience Seminar Series, PennsylvaniaState University. June, 23, 2004.
Protein domains implicated in synaptic targeting and clustering of GABAA
receptors. Department of Biochemistry, Microbiology and Molecular BiologyResearch Forum, Pennsylvania State University. April 3rd, 2003.
The role of the γ2 subunit and interacting proteins in the clustering ofGABAA receptors Department of Biochemistry, Microbiology and MolecularBiology Research Forum, Pennsylvania State University. Oct. 18th 2001.