synonyms nature of ald different forms symptoms gene location
TRANSCRIPT

SYNONYMS
NATURE OF ALD
DIFFERENT FORMS
SYMPTOMSGENE LOCATION

AMN
Addison Disease with Cerebral Sclerosis
Addison-Schilder Disease
Adrenomyeloneuropathy
Adult Onset ALD
Bronze Schilder’s Disease
Encephalitis Periaxilais Diffusa
Flatau-Schilder’s Disease
Melanodermic Leukodystophy
Myelinoclastic Diffuse Sclerosis
Schilder Disease
Schilder Encephalitis
Slewering-Creutzfeldt Disease
Sudanophilic Leukodystrophy,ADL

Adrenoleukodystrophy (ALD) is a rare, genetic disorder characterized by the breakdown or loss of the myelin sheath surrounding nerve cells in the brain and progressive disfunction of the adrenal gland.
People with ALD accumulate high levels of saturated, very long chain fatty acids in their brain and adrenal cortex because the fatty acids are not broken down by an enzyme in the normal manner.
The first report of a patient with X-chromosomal linked adrenoleukodystrophy (X-ALD) was published in the medical literature in 1923.
The name adrenoleukodystrophy was introduced by Michael Blaw in 1970, and is defined as a serious progressive, genetic disorder, which affects the adrenal glands and the white matter of the nervous system. It was first recognized in 1923 and has been known as Schilder's disease and sudanophilic leukodystrophy. Blaw coined the name adrenoleukodystrophy; adreno refers to the adrenal glands; leuko refers to the white matter of the brain, and dystrophy means imperfect growth or development.

There are several forms of ALD. Onset of the classic childhood form, which is the most severe and affects only boys, may occur between ages 4 and 10. Features of this form may include visual loss, learning disabilities, seizures, dysarthria (poorly articulated speech), dysphagia (difficulty swallowing), deafness, disturbances of gait and coordination, fatigue, intermittent vomiting, melanoderma (increased skin pigmentation), and progressive dementia.
The most common symptoms are usually behavioral changes such as abnormal withdrawal or aggression, poor memory, and poor school performance. In the milder adult-onset form, which typically begins between
ages 21 and 35, symptoms may include leg stiffness, progressive spastic paraparesis (stiffness, weakness and/or paralysis) of the lower extremities, and ataxia. Although adult-onset ALD progresses more slowly than the classic childhood form, it can also result in deterioration of brain function.
Another form of ALD is occasionally seen in women who are carriers of the disorder. Symptoms are mild and may include spastic paraparesis of the lower limbs, ataxia, hypertonia (excessive muscle tone), mild peripheral neuropathy, and urinary problems. Neonatal ALD affects both male and female newborns. Symptoms may include mental retardation, facial abnormalities, seizures, retinal degeneration, hypotonia (low muscle tone), heptomegaly (enlarged liver), and adrenal dysfunction. This form is usually quickly
progressive.

X-ALD is found in about 1 in 21.000 new born males and approximately 1 in 14.000 new born females are carrier for this disease.
The overall frequency for X-ALD is about 1 in 17.000 life births.
asymptomatic
mild myelopathy
moderate to severe myeloneuropathy
cerebral involvement
clinically evident adrenocortical insufficiency
asymptomatic or presymptomatic
childhood cerebral ALD
adolescent cerebral ALD
adult cerebral ALD
adrenomyeloneuropathy
Addison-only phenotype

-ALD is an X-linked disorder, which means it affects only males and is transmitted by a female carrier. Such disorders are referred to as "X-linked" since the genetic abnormality involves the X-chromosome.
X-ALD is a peroxisomal storage disease whereby abnormal function of peroxisomes leads to the accumulation of very long-chain fatty acids (VLCFA) in tissues of the body, especially the brain and the adrenal glands.
This gene is located on the X-chromosome (it’s official name is the ABCD1 gene).
•Chromosome Xq28; Recessive

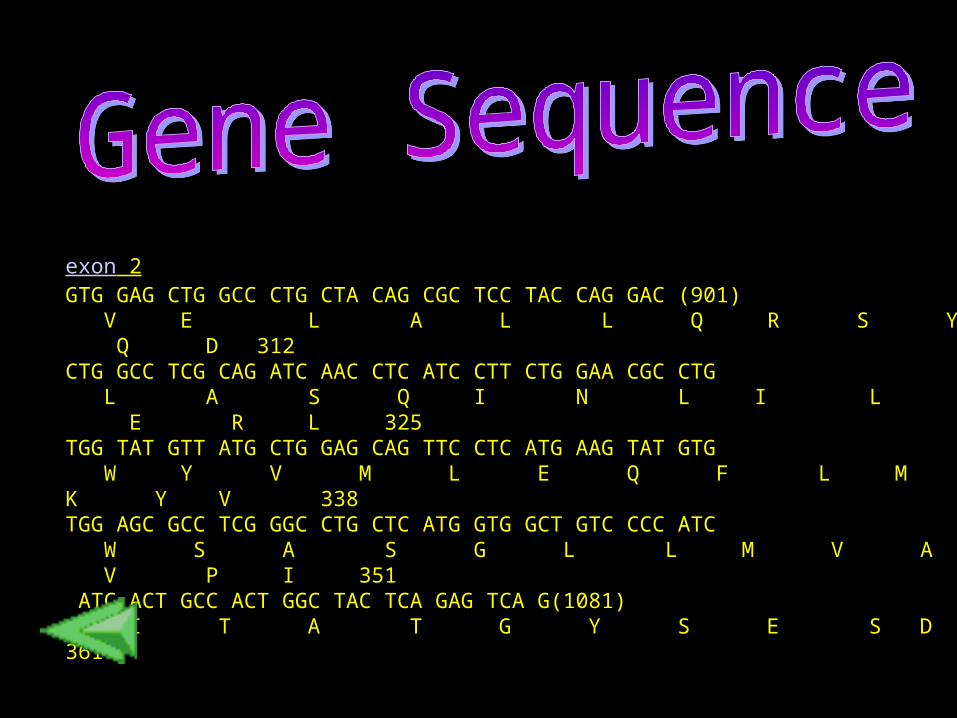
exon 2 GTG GAG CTG GCC CTG CTA CAG CGC TCC TAC CAG GAC (901) V E L A L L Q R S Y Q D 312CTG GCC TCG CAG ATC AAC CTC ATC CTT CTG GAA CGC CTG L A S Q I N L I L L E R L 325 TGG TAT GTT ATG CTG GAG CAG TTC CTC ATG AAG TAT GTG W Y V M L E Q F L M K Y V 338 TGG AGC GCC TCG GGC CTG CTC ATG GTG GCT GTC CCC ATC W S A S G L L M V A V P I 351 ATC ACT GCC ACT GGC TAC TCA GAG TCA G(1081) I T A T G Y S E S D 361

ALLELIC VARIANTS
Selected examples
Allelic variants are given a 10 digit number: the 6-digit number of the parent locus followed by a decimal point and a unique 4-digit variant number.
Note that for most gene loci, only selected mutations are included as specific subentries. Criteria for inclusion include the first mutation to be discovered, high population frequency, distinctive phenotype, historic significance, unusual mechanism of mutation, unusual pathogenetic mechanism, and distinctive inheritance (e.g., dominant with some mutations, recessive with other mutations in the same gene).
Most of the allelic variants represent disease-producing mutations. A few polymorphisms are included, many of which show a positive statistical correlation with particular common disorders.
Oligodendrocytes (red), the myelin-producing cells of the brain, shown here in tissue culture in association with astrocytes (green).

Mutation typeTotal number of
mutationsNucleotide substitutions (missense / nonsense)
151
Nucleotide substitutions (splicing) 12
Nucleotide substitutions (regulatory) 0Small deletions
38
Small insertions 14
Small indels 4
Gross deletions 12
Gross insertions & duplications 0Complex rearrangements (including inversions)
1
Repeat variations 0TOTAL 232

•0001:ADRENOLEUKODYSTROPHY
•Mutation : ABCD1, GLU291LYS
•0002 : ADRENOLEUKODYSTROPHY
•Mutation : ABCD1, PRO484ARG
•0003 : ADRENOLEUKODYSTROPHY
•Mutation : ABCD1, IVS6AS, A-G, -2, FS546TER
•0004 : ADRENOLEUKODYSTROPHY
•Mutation : ABCD1, IVS8AS, G-A, -10, 8-BP INS, FS638TER
•0005 : ADRENOMYELONEUROPATHY
•Mutation : ABCD1, ARG389GLY
•0006 : ADRENOLEUKODYSTROPHY
•Mutation : ABCD1, ASN148SER
•0011 : ADRENOMYELONEUROPATHY
•Mutation : ABCD1, ARG464TER
•0012 : ADRENOLEUKODYSTROPHY
•Mutation : ABCD1, 2-BP DEL, FS, TER
•0013 : ADRENOLEUKODYSTROPHY
•Mutation : ABCD1, GLU477TER
•0014 : ADRENOLEUKODYSTROPHY
•Mutation : ABCD1, SER515PHE
•0015 : ADRENOLEUKODYSTROPHY
•Mutation : ABCD1, 1-BP DEL, 1937C, FS557TER
•0007 : ADRENOLEUKODYSTROPHY
•Mutation : ABCD1, TYR174ASP
•0008 : ADRENOLEUKODYSTROPHY
•Mutation : ABCD1, GLY266ARG
•0009 : ADRENOLEUKODYSTROPHY
•Mutation : ABCD1, ARG401GLN
•0010 : ADRENOLEUKODYSTROPHY
•Mutation : ABCD1, ARG418TRP


Proteins (According to SWISS-PROT and/or MIPS)
ALD_HUMAN•Size: 745 amino acids; 82908 Da •Function: PROBABLE TRANSPORTER. THE NUCLEOTIDE-BINDING FOLD ACTS AS AN ATP-BINDING SUBUNIT WITHATPASE ACTIVITY.
•Subunit: CAN FORM HOMO- AND HETERODIMERS WITH ABCD2/ALDR AND ABCD3/PMP70. DIMERIZATION IS NECESSARYTO FORM AN ACTIVE TRANSPORTER.
•Subcellular location: Integral membrane protein. Peroxisomal.
•Similarity: BELONGS TO THE ABC TRANSPORTER FAMILY. ALD SUBFAMILY.
MIPS Pedant Viewer: 34833
REFSEQ proteins: NP_000024.2

There are currently two different techniques for getting disease-free stem cells into an ALD patient: a bone marrow transplant (BMT) or an umbilical cord blood transplant (UCBT). Both bone marrow and umbilical cord blood are rich in stem cells. By transplanting healthy donor stem cells into an ALD patient, ALD progression may be halted and in some cases reversed. There are many similarities between bone marrow transplants and umbilical cord blood transplants. The goal of both types of transplants is to get healthy stem cells which produce a functioning ALD protein which is lacking in an ALD patient.
Physical therapy
Psychological support
Special education
The picture above shows the makeup of oleic and erucic acids and how they combine to make "Lorenzo's Oil."
Other treatment includes:

There are two distinct advantages to gene therapy within the context of treating ALD.
1. The chemotherapy and/or radiation that is required to allow the donor cells to engraft, or become accepted by the host would no longer be such a dangerous issue. The cells that are transplanted into the patient after gene correction are autologous, in other words, they come from the patient himself. Therefore, all issues related to the discordance between host and donor tissues, and the subsequent medical complications that ensue, would be significantly lessened.
2. Many ALD individuals are not eligible for a BMT or UCBT due to lack of a suitable donor. Due to the autologous nature of the treatment, each person serves as his own donor and recipient. Matching donors and recipients will no longer be an issue.
The goal of successful gene therapy would yield the positive results of BMT with significantly decreased morbidity and mortality. In addition, no patient would be turned away due to lack of a match. In summary, a safer treatment that could be offered to a much larger group of patients.

4 Phenylbuterate is a drug that is currently used for a variety of disorders, and has shown some promise as far as treating ALD. Dr. Hugo Moser is currently proposing a clinical trial. However, because he must apply for funding in the form of a grant, which must be submitted, reviewed, etc., these trials will not take place in the immediate future. If Dr. Moser had ready access to funding, he need only apply for an IRB, a minor task, and the trial could proceed. Thus it is important for The Stop ALD Foundation to have an infrastructure in place that can properly and efficiently evaluate these types of proposals, take the necessary action, and provide the necessary resources -- whatever they may be.
Other Possible Therapies: 4 Phenylbuterate – A Case in Point

National Institution of Neurological Disorder and stroke (NINDS) supports research on genetic disorders. The aim of this research is to find ways to prevent, treat, and cure diseases.
Much research is being done in different areas. People are working on ways of remyelinating nerves (The Myelin Project, founded by Augusto and the late Michaela Odone (the parents of Lorenzo Odone, for whom Lorenzo's Oil is named)). As well, research is being conducted into various gene therapies, but those
are years away from human trials.
More recently, all the transporters related to ALD protein have been found in the yeast Saccharomyces cerevisiae, and a mouse model for the human disease has been developed. These and other molecular biology approaches should further our understanding of ALD and hasten our progress towards effective therapies.