thalassemia in sardinia causedby nonsense...
TRANSCRIPT

Thalassemia in Sardinia Is Caused by a Nonsense
Mutation
RIcHARD F. TRECARTIN, STEPHENA. LIEBHABER, JUDY C. CHANG,KATHLEENY. LEE, and YUETWAI KAN, Howard Hughes Medical InstituteLaboratory and Department of Medicine, University of California,San Francisco, California 94143
MARIOFURBETTA, ANTONELLAANGIUS, and ANTONIOCAo, 2nd Pediatric Clinic,University of Calgliari, Sardinia, Italy
A B S T RA C T We report the characterization of amolecular lesion of (8 thalassemia in Sardinia. Betathalassemia in this area is predominantly the (80 typewith low levels of,8-globin mRNA.Translation assay ofthis messenger RNA in a cell-free system showed (-globin chain synthesis only with the addition of anamber (UAG) suppressor transfer RNA. Double-stranded complementary DNAprepared from recticu-locyte mRNAfrom a Sardinian patient was cloned in abacterial plasmid and a,(3-globin complementary DNAcontaining clone was isolated and sequenced. At theposition corresponding to amino acid number 39, asingle nucleotide mutation converted a glutaminecodon (CAG) to an amber termination codon (UAG). Wepreviously reported an amber nonsense mutation atamino acid 17 as a cause of Chinese (80 thalassemia.Thus, i80 thalassemia in Sardinia represents the secondexample of a nonsense mutation, and we predict thatother ,80 thalassemias with mutations at various pointsalong the ,(-globin chain will be found to form a discretesubgroup of 10 thalassemia. These experiments furtherillustrate the heterogeneity of lesions that lead todefective globin chain synthesis in 8 thalassemia.
INTRODUCTION
The molecular mechanisms that lead to defective (3-globin chain synthesis in the 3 thalassemia syndromesare extremely heterogeneous (1-3). Deletions ofvarious segments of DNAon chromosome 11 in theregion of the (3-globin gene complex (4) produce thephenotypes of 80 thalassemia (5, 6), 50,80 thalassemia,
Address correspondence to Dr. Kan, University ofCalifornia.
Received for publication 30 March 1981 and in revisedform 22 May 1981.
1012 J. Clin. Invest. © The American Society for
hereditary persistence of fetal hemoglobin (7-12), andy8(8 thalassemia (13, 14). The more commontypes of(8thalassemia, however, are not the result of gene dele-tions. The nondeletion thalassemias, which are usu-ally accompanied by elevated hemoglobin A2 levels,are divided into two broad categories: +3+ thalassemia,in which (8-globin chain synthesis is reduced butdetectable, and (83 thalassemia, in which no (l-globinchains are produced (15, 16). The molecular defectsin these forms of ( thalassemia are currently underinvestigation with molecular cloning, DNAsequenceanalysis, and functional assays. Recent studies sug-gest that some (8+ thalassemia may be due to defectiveprocessing of the primary (3-globin messenger RNA(mRNA) transcript into mature mRNA(17, 18).
The molecular bases of (0 thalassemia are alsoheterogeneous. In some individuals, (-globin mRNAsynthesis is absent; in others, a variable quantity isdemonstrable in the reticulocyte (19). Previously weinvestigated the lesion in the mRNAfrom a Chinesepatient affected by (30 thalassemia where (3-globinmRNAwas detectable (20, 21), and found the defect tobe a nonsense mutation that caused prematuretermination of the ,8-globin chain (22). In this study weinvestigated thalassemia on the island of Sardinia (23),where (0 thalassemia, the predominant molecular form,is characterized by low levels of (8-globin mRNA. Wecloned the complementary DNA(cDNA), isolated andsequenced a (3-globin cDNAclone, and found that thelesion was also due to a nonsense mutation, although ina different position from that in the Chinese. This studyfurther illustrates the heterogeneity of the lesions thatproduce 8 thalassemia.
METHODSRNAand DNApreparation and analysis. Previous studies
have identified 331 patients with /80 thalassemia in Sardinia
Clinical Investigation, Inc. * 0021-9738/81/10/1012/06 $1.00J -~ ' -~~ J7V October 191 1-0 2- 101- 1-7I
Volumne 68 October 1981 1012-1017

(23). Weprepared poly (A+) RNAfrom 20 patients and cellularDNAfrom 10 patients, as previously described (21, 24). RNAfrom 20 patients and DNAfrom five patients was quantitatedby solution hybridization with cDNA enriched in a- and 18-globin sequences as previously described (20, 24). GenomicDNAfrom 10 patients was digested with the enzymes Hpa I,Bam HI, Pst I, and Eco RI (Bethesda Research Laboratories,Gaithersburg, Md.) and analyzed by Southern blotting. Theglobin-gene containing fragments were identified by 32P_labeled cDNA or by nick-translated f8-globin cDNA asdescribed (25).
Cell-free synthesis. Poly(A+) RNAfrom five patients waspooled and the ,B-globin mRNAwas enriched by selectiveadsorption to f3-globin cDNA covalently bound to cellulose.The f3-globin cDNA recombinant plasmid (pJW 102) (26)was covalently bound to diazotyzed cellulose by the methodof Noyes and Stark (27), the only modification being theuse of 200 mMborate buffer at pH 6.0. A small amount ofradioactive 83-globin cDNAwas used to monitor the couplingefficiency, which was found to be 1.8 ,g of pJW 102/mg ofcellulose. Poly(A+) RNA (200 ng) was hybridized to 5 mg(3-globin cDNAcellulose in a 65-,ul hybridization buffer con-taining 60% formamide (vol/vol), 150 mMNaCl, 25 mMTris-HC1 (pH 7.5), 1.0 mMEDTA, 0.1 g/100 ml SDS, and 0.25mg/ml wheat germ transfer RNA (tRNA), with constant in-version at 37°C for 44 h. The cellulose was then washedtwice with the 100-,ul hybridization mixture and twice withthe mixture without tRNA at 37°C. The bound mRNAwas eluted at 65°C in two 50-.ld aliquots of hybridizationbuffer. The RNAwas precipitated in 70% (vol/vol) ethanol at-20°C after adding 20 yg/ml deacylated wheat germ tRNAand 200 mMpotassium acetate (pH 7.5).
The ability of the enriched /3-globin mRNAto direct proteinsynthesis was assayed in a micrococcal nuclease-treatedrabbit reticulocyte lysate system (28) in the presence or ab-sence of a yeast (Saccharomyces cerevisiae) serine-in-serting amber suppressor tRNA (29). A 25-,ul reticulocytelysate mixture containing 50 uCi of [35S]methionine (Amer-sham Corp., Arlington Heights, Ill.; 1,000 Ci/mmol) with orwithout the addition of 120 ,ug/ml yeast suppressor tRNAwas incubated at 30°C for 90 min. 5 mg of hemoglobin con-taining equal amounts of lysate from adult and cord bloodwas added as carrier, globin was prepared by acid acetoneprecipitation, and the globin chains were separated on car-boxymethyl cellulose in 8 M urea as described (30). The/3-globin polypeptide recovered from the column was di-gested with trypsin and the [35S]methionine-containing /3-globin peptides were identified by high voltage electro-phoresis and autoradiography, as previously described (29).
cDNApreparation. Total RNAextracted from the reticulo-cytes of one of the Sardinian subjects with ,3 thalassemia wasused as a template for cDNAsynthesis. Single-stranded cDNAwas synthesized for 3 h in a 200-,ul reaction mixture con-taining 390 Atg reticulocyte RNA, 5,000 pmol dT8GC primer,400 ,uM each of deoxyATP (dATP), deoxyguanosine triphos-phate (dGTP), and thymidine 5'-triphosphate (TTP), and100 mMa-[32P]deoxycytidine triphosphate (dCTP) diluted tosp act 2 Ci/mmol), 228 U avian myeloblastosis virus (AMV)reverse transcriptase, 50 mMTris-HCI (pH 8.3), 10 mMMgCl2,2 mMdithiothreitol, and 25 mMKC1. The reaction mixture wasextracted with phenol, and the large molecular weight prod-ucts were separated from unincorporated nucleotide triphos-phates by chromatography on a Sephadex G-50 column in abuffer containing 100 mMNaCl, 10 mMTris-HCl (pH 7.5), and5 mMEDTA, and precipitated in 70% ethanol. RNA washydrolyzed by dissolving the pellet in 25 ,ul of 0.6 N NaOHand by incubating the mixture at 37°C for 3 h. The base wasneutralized to pH 7.0 with an equivalent amount of acetic
acid followed by the addition of 2 MTris-HCl (pH 7.5) to afinal concentration of 150 mMTris-HCl. The sample wasprecipitated in 70% ethanol. About 1 ug of single-strandedcDNAwas obtained.
Oligo deoxycytidine monophosphate (dCMP) was addedto the 3' end of the cDNA (dC tailing) to provide a templatefor priming the synthesis of the second DNA strand (31).This was done by incubating the single-stranded cDNAwith275 uM a-[32P]dCTP (sp act 4 Ci/mmol) and 35 U terminaldeoxynucleotidyl transferase (P-L Biochemicals, Inc., Mil-waukee, Wis.) in a 35-Al mixture containing 140 mMpotas-sium cocadylate, 30 mMTris-HCl (pH 7.5), 1 mMcobaltchloride, and 100 mMdithiothreitol for 18 min at roomtemperature. An average of 33 dCMPresidues was added tothe 3' end of the cDNA. The reaction was extracted withphenol and precipitated in ethanol.
To synthesize the complementary strand, the oligo dC-tailed single-stranded cDNAwas incubated for 2.5 h at roomtemperature with 2 nmol dG12_18,, 500 uM each of dATP,dGTP, and TTP, 100 ,uM a-[32P]dCTP (sp act 10 Ci/mmol),and 54 UAMVreverse transcriptase in 40 ,u of the same bufferused to synthesize the first strand. The mixture was ex-tracted with phenol, fractionated on a Sephadex G-75 column,and the resulting double-stranded cDNA was precipitated inethanol. A dC tail was added to the 3' end of the double-stranded cDNAby incubation with terminal deoxynucleotidyltransferase and dCTP for 15 min under the tailing conditionsgiven above. An average of 130 dCMPresidues was addedto each end.
Preparation of the plasmid vector. 5 ,ug of the plasmidpBR 322 was linearized by digestion with Pst I and dG tailswere added by incubating the cleaved plasmid for 3 h at roomtemperature in the described tailing buffer containing 100,uM a-[32P]dGTP (New England Nuclear, Boston, Mass.) di-luted to sp act 24 Ci/mmol), 0.1 ,ug/,ul gelatin, and 35 U ter-minal deoxynucleotidyl transferase, with a second additionof 15 U TDT at 37°C for 15 h. An average of six guanosine5'-monophosphate residues was added to each 3' end.
Equimolar amounts (0.074 pmol each) of the double-stranded cDNA (dC-tailed) and the pBR 322 (dG-tailed) wereannealed to each other by incubating 200 ,ug cDNA with 28,ug pBR 322 in a 200-,ul mixture containing 100 mMNaCl,1 mMTris-HCl (pH 7.5), and 0.1 mMEDTA. The reac-tion mixture was heated to 70°C and allowed to cool slowlyto 42°C over -30 h, and then to room temperature overnightand stored at 4°C.
Transformation and isolation of the f3cDNA clone.Escherichia coli of the strain X 1776 was prepared for trans-formation according to previously described methods (32,33). 200 ,ul of the above-mentioned annealing mixture wasincubated with 400 1.l of calcium-treated X 1776 at 4°C for1 h. 600 ,ul L broth was added, and the entire mixture washeated for 3 min at 42°C and incubated at 37°C for 30 min.The mixture was grown at 37°C on L broth agar plates sup-plemented with diaminopimelic acid (100 ,g/ml), thymidine(840 ,ug/ml), and tetracycline (15 ,g/ml). Colonies appearedover the next 2 d.
Colonies were screened by in situ filter hybridizationwith a nick-translated ,8-globin cDNAprobe excised from the/3 cDNA-containing recombinant plasmid pNC 1-71. Filterswere hybridized and washed under conditions similar tothose previously reported (34), with the final wash at 55°Cfor 2 h to decrease cross hybridization of the /8-globin probewith y- and 8-globin sequences.
Colonies that hybridized strongly with the 83-cDNA probe
I Liebhaber, S. A. Unpublished observation.
/3 Thalassemia in Sardinia 1013

were grown in 25 ml culture and plasmid was prepared froma clarified lysate by banding on a CsCl gradient. 1 jAg ofeach plasmid was digested with Pst I and the recombinant(pSAR 6) with the largest insert (-600 base pairs) was usedto transform the host HblOl, grown in M-1 medium toOD630 0.77, and amplified with 150 ,ug/ml chloramphenicol.The plasmid supercoil was isolated from a clarified lysate.
DNAsequencing. The complete sequence of the (-globincDNA insert was determined by the Maxam and Gilbertmethod (35), with a modification in the A reaction (31). DNAfragments generated by restriction endonuclease digestionwere either labeled at the free 5' ends with yy-[32P]ATP andpolynucleotide kinase after dephosphorylation with bac-terial alkaline phosphatase, or at the 3' ends with E. coliDNApolymerase I (Klenow Fragment) and the appropriatea-[32P]deoxynucleotide triphosphate (2-3000 Ci/mm, Amer-sham Corp.).
RESULTS
/3-globin mRNAand 3-globin gene quantitation. Theproportion of a- to ,B-globin mRNAin the reticulocytesof 20 Sardinian patients with (3 thalassemia wasmeasured by cDNA-RNA hybridization using cDNAprobes enriched in a- and 83-globin sequences. Arepresentative hybridization analysis is shown in Fig.1. Analysis of the 1/2 Rot value (the concentration ofRNAwhere 50% of the radioactive probe is annealed)demonstrated an average (8/a ratio of 0.054 (range0.043-0.062), which indicates that the concentration of,8-globin mRNAis -5% that of the a-globin mRNA.Solution hybridization of the labeled cDNAto cellularDNAshowed that the DNAfrom these Sardinian pa-tients contained the full complement of f8-globingenes. The structure of these 3-globin genes was nor-mal by Southern blot analysis; the sizes of 8-globingene-containing fragments generated by cleavage withEco RI, BamHI, Pst I, and Hpa I were identical to nor-mal controls. (The DNAdata are not shown.)
Analysis of the function of the /3-globin mRNA. Wepreviously showed that the nonsense mutation respon-
c
4
wzz4tI-zwC-)wa.
FIGIretic0,ccDN
sible for the nonfunctional /3-globin mRNAin a Chinesepatient could be supressed in in vitro translationsystems by adding a yeast suppressor tRNA. To analyzethe function of the (3-globin mRNAin Sardinian (30thalassemia, we pooled the mRNAfrom five subjectsand enriched the ,8-globin mRNAsequences by selec-tive adsorption of reticulocyte poly(A+) mRNAto,8-globin cDNA cellulose. The /8-globin-enrichedmRNAdirected both y- and a-globin chain synthesis.When the yeast serine inserting amber suppressortRNA was added, a small but definite radioactivitypeak in the /8-globin chain region was clearly dis-cernible (Fig. 2). We pooled the [35S]methionine-labeled globin in this fraction, digested it with trypsin,and separated the peptides by high voltage electro-phoresis. We identified the methionine-containingpeptide with,S-T5 as previously described (29) (resultof peptide analysis is not shown).
In our previously reported Chinese ( thalassemiapatient, the normal codon for the 17th amino acid AAG(lysine) is changed to the amber termination codonUAG (22). Placing a serine in this position with aserine inserting amber suppressor tRNA produces amore acidic globin chain, which elutes earlier than the(3A chain on chromatography (29). In the Sardinianpatient, inserting serine in a UAGcodon produced a,8-globin chain with the same electrophoretic chargeas the normal /3A. This indicates that the / Sardiniamutation affects a codon for a neutral amino acid andis therefore different from the 3 Chinese mutation.
A single nucleotide mutation affecting codons forthe neutral amino acids leucine, serine, glutamine,tyrosine, or tryptophan can produce a UAGcodon.
0OD
0li
C-
TUBE NUMBER
"'0 \ FIGURE2 Carboxymethyl cellulose chromatography of glo-bin chains synthesized in a cell-free system with mRNA
-!a o enriched in /3-globin sequences. The two experiments per--~ ~-~-~--formed with (solid lines) and without (interrupted lines) sup-,)-°2 3 pressor tRNA are superimposed on each other. The OD
Rot3-2-10 2 3
(A280) markers are shown above the radioactive peaks. In theLog Rot inset, the radioactivity scale was expanded 10-fold to show thatJRE 1 Rate of annealing of a- and f3-globin cDNA to an authentic /8-globin peak appeared over the backgroundulocyte RNA from a Sardinian 3 thalassemia patient. radioactivity in response to the addition of the suppressor32P-labeled a-globin cDNA; 0, 3H-labeled ,8-globin tRNA. The a-globin peaks, which were unaffected by addition[A. of suppressor tRNA, are not shown.
1014 Trecartin, Liebhaber, Chang, Lee, Kan, Furbetta, Angius, and Cao
However, the leucine codon (UUG) and the serinecodon (UCG) are not used in the human /8-globinmRNA(36). Therefore the /30 Sardinia mutation prob-ably affects one of the three other amino acids found ineight positions in the coding region (Trp in positions15 and 17; Tyr in positions 35, 130, and 145; and Glnin positions 39, 127 and 131). The very low level of /8mRNAmade direct determination of 8-globin se-quences from the RNA impractical. We thereforecloned cDNA prepared from the reticulocyte mRNAto obtain enough material for sequencing.
Cloning of the cDNA. Weprimed synthesis of thefirst strand with dT8GCand added oligo dC to the 3' endof the cDNA. By using oligo dG to prime the synthesisof the second strand, we avoided creating a hairpinconnection between the two strands. The hairpin con-nection would necessitate SI cleavage that could over-digest and shorten the cDNA. The double-strandedcDNA was again dC-tailed and annealed to dG-tailedpBR 322. The recombinant plasmids were used totransform X 1776.
238 recombinants were screened with nick-trans-lated /8-globin cDNA. Three recombinants whichhybridized strongly to the / cDNA probe (Fig. 3)were analyzed by restriction endonuclease digestionwith the enzyme Mbo II, which cleaves y- and 8-, butnot ,/-globin cDNA sequences. One clone containingan insert -600 base pairs in length and resistant todigestion by Mbo II was grown up in large quantityfor further analysis.
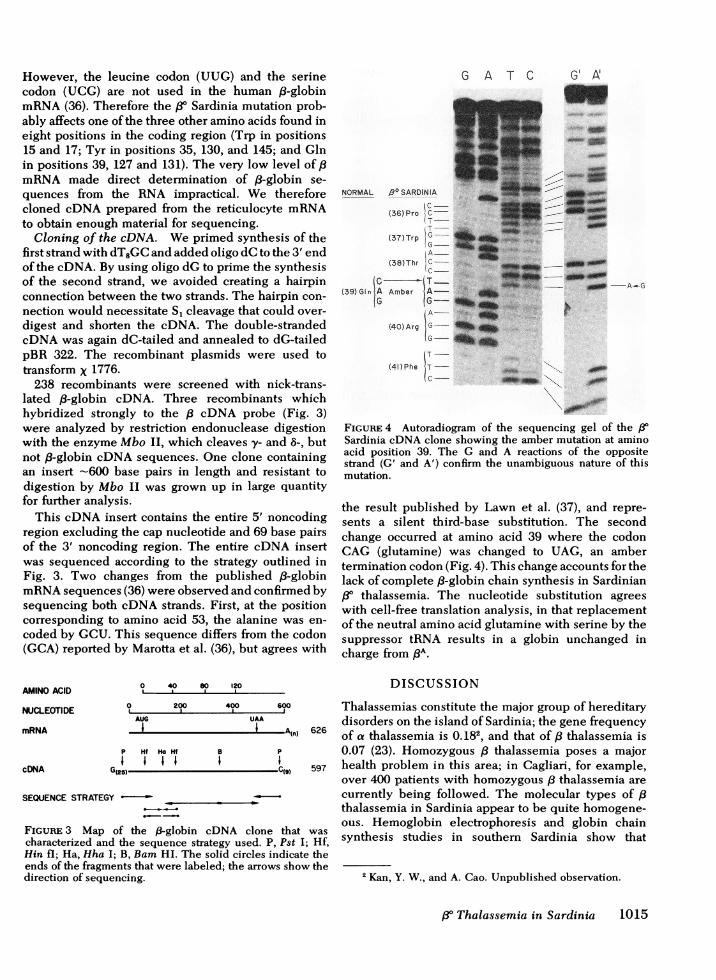
This cDNA insert contains the entire 5' noncodingregion excluding the cap nucleotide and 69 base pairsof the 3' noncoding region. The entire cDNA insertwas sequenced according to the strategy outlined inFig. 3. Two changes from the published f8-globinmRNAsequences (36) were observed and confirmed bysequencing both cDNA strands. First, at the positioncorresponding to amino acid 53, the alanine was en-coded by GCU. This sequence differs from the codon(GCA) reported by Marotta et al. (36), but agrees with
G A T C G_ V
! am.I_
"as-/ -RI_5- 0-.
NO__wmaw4-_
NORMAL /3 SARDINIA
~C-(36) Pro C=
(37)Trp G-
A(38)Thr jC--
C
(39) Gin A Amber A-G G-
A--
(40) Arg u-
I()T-
(41) phe T--C-
FIGURE 4 Autoradiogram of the sequencing gel of the /3Sardinia eDNAclone showing the amber mutation at aminoacid position 39. The G and A reactions of the oppositestrand (G' and A') confirm the unambiguous nature of thismutation.
the result published by Lawn et al. (37), and repre-sents a silent third-base substitution. The secondchange occurred at amino acid 39 where the codonCAG (glutamine) was changed to UAG, an ambertermination codon (Fig. 4). This change accounts for thelack of complete ,8-globin chain synthesis in Sardinian/3° thalassemia. The nucleotide substitution agreeswith cell-free translation analysis, in that replacementof the neutral amino acid glutamine with serine by thesuppressor tRNA results in a globin unchanged incharge from PAA.
AMINO ACID
NUCLEOTIDE
mRNA
0 40 80 120a I I
0 200 400 600
AUG UAAI I As,n)
P Hf Ho f
I i I IcDNA
B p
r-
SEQUENCESTRATEGY '--
FIGURE 3 Map of the /8-globin cDNA clone that wascharacterized and the sequence strategy used. P, Pst I; Hf,Hin fI; Ha, Hha I; B, Bam HI. The solid circles indicate theends of the fragments that were labeled; the arrows show thedirection of sequencing.
DISCUSSION
Thalassemias constitute the major group of hereditarydisorders on the island of Sardinia; the gene frequencyof a thalassemia is 0.182, and that of 3 thalassemia is0.07 (23). Homozygous /8 thalassemia poses a majorhealth problem in this area; in Cagliari, for example,over 400 patients with homozygous /8 thalassemia arecurrently being followed. The molecular types of /3thalassemia in Sardinia appear to be quite homogene-ous. Hemoglobin electrophoresis and globin chainsynthesis studies in southern Sardinia show that
2 Kan, Y. W., and A. Cao. Unpublished observation.
/W Thalassemia in Sardinia 1015
I
.1, 4000Oft-low '40ow
dwgoomp.=, = -am 400
Om-,+ 4gogO A-G

virtually all cases are the 130 type (23). The few patientswith fB+ thalassemia either have Sicilian parentage orare from northern Sardinia, which once had Greekgenetic influence. Genetic and biochemical studiesindicate that ,l thalassemia in Sardinia most likelyarose as a single mutational event (38). Our study showsthat this mutation is produced by a single nucleotidechange at codon number 39, where CAGis changedto the termination codon UAG. In accordance withpreviously proposed terminology, this mutant isdesignated either 839CAG-4UAG or 139UAG.
This is the second example of a nonsense mutationfound in humans. A review of the nucleotide sequenceof ,-globin mRNAshowed a single nucleotide muta-tion in the coding region could result in terminationcodons in 29 positions. Hence, one would expect tofind a subgroup of , thalassemia that is due todifferent nonsense mutations.
In both Sardinians and Chinese with nonsensemutations, there is a decreased amount of 18-globinmRNAin the reticulocyte. It has been postulated thatthe lack of ribosomal binding to a large section of themRNAmay expose it to nuclease degradation (22, 29).But we do not know why the 8/a globin mRNAratio is lower in the Sardinian with a termination codonat amino acid position number 39 than in the Chinesewith the termination codon at position 19. Other factors,such as the effect of the mutation on secondary mRNAstructure, could influence the mRNAlevel in thereticulocyte. Studies of termination codon mutations atother positions on the 13-globin chain may explain themechanism of mRNAdegradation.
Low level mRNAhybridization of the 18-globincDNAprobe in 1 thalassemia has often been ascribedto a cross-reaction with 8-globin mRNA(19). Thisstudy further emphasizes that a low mRNAlevel doesnot rule out the presence of a 1-globin mRNA.
,30 thalassemia comprises a heterogeneous groupof disorders. In addition to nonsense mutations, otherpossibilities include defective mRNAtranscriptionresulting in the absence of 13-globin mRNAsynthesis,and abnormal processing causing incomplete degra-dation of 8-globin mRNAin the nucleus. Our studyillustrates the efficacy of the suppressor tRNA assayas a means of distinguishing the nonsense mutationfrom other causes.
ACKNOWLEDGMENTS
We thank Julie Bailey for performing some of the liquidhybridization experiments, Dr. Ray Gesteland and NormaWells for providing the suppressor tRNA, the Office of Pro-gram Resources and Logistics (Viral Cancer Program, ViralOncology, Division of Cancer Cause and Prevention, NationalCancer Institute, Bethesda, Md.) for the reverse transcriptase,and Jennifer Gampell for editorial comments.
This investigation was supported in part by grants from theNational Institutes of Health (AM 16666, HL 24137), the
March of Dimes/Birth Defects Foundation, AssessoratoIngiene e Sanita Regione Sarde, and Centro NationaleRicerche subprogetto finalizzato "Malattie Ereditarie dell'Eritrocita.
REFERENCES1. Weatherall, D. J., and J. B. Clegg. 1979. Recent develop-
ments in the molecular genetics of human hemoglobin.Cell. 16: 467-479.
2. Forget, B. G. 1979. Molecular genetics of human hemo-globin synthesis. Ann. Intern. Med. 91: 605-616.
3. Bank, A., J. G. Mears, and F. Ramirez. 1980. Disordersof human hemoglobin. Science (Wash. D. C.) 207:486-493.
4. Maniatis T., E. F. Fritsch, J. Lauer, and R. M. Lawn. 1980.The molecular genetics of human hemoglobins. Annu.Rev. Genet. 14: 145-178.
5. Orkin, S. H., R. Kolodner, A. Michaelson, and R. Husson.1980. Cloning and direct examination of a structurallyabnormal human t3° thalassemia globin gene. Proc. Natl.Acad. Sci. U. S. A. 77: 3558-3562.
6. Flavell, R. A., R. Bernards, J. M. Kooter, E. de Boer, P. F.Little, G. Annison, and R. Williamson. 1979. The struc-ture of the human 13-globin gene in 13 thalassemia.Nucleic Acids Res. 6: 2749-2760.
7. Kan, Y. W., J. P. Holland, A. M. Dozy, S. Charache, andH. H. Kazazian. 1975. Deletion of the ,3-globin structuregene in hereditary persistance of fetal hemoglobin.Nature (Lond.) 258: 162-163.
8. Forget, B. G., D. G. Hillman, H. Lazarus, E. F. Barell,E. J. Benz, Jr., C. T. Caskey, T. H. J. Huisman, W. A.Schroeder, and D. Housman. 1976. Absence of mes-senger RNAand gene DNA for beta globin chains inhereditary persistence of fetal hemoglobin. Cell 7:323-329.
9. Mears, J. G., F. Ramirez, D. Leibowitz, F. Nakamura, A.Bloom, F. Konotey-Ahula, and A. Bank. 1978. Changesin restricted human cellular DNAfragments containingglobin gene sequences in thalassemias and relateddisorders. Proc. Natl. Acad. Sci. U. S. A. 75: 1222-1226.
10. Ottolenghi, S., B. Giglioni, P. Comi, A. M. Gianni, E.Polli, C. T. Acquaye, J. H. Oldham, and G. Masera. 1979.Globin gene deletion in HPFH, 80,30 thalassemia and HbLepore disease. Nature (Lond.). 278: 654-657.
11. Fritsch, E. F., R. M. Lawn, and T. Maniatis. 1979.Characterization of deletions which affect the expressionof fetal globin genes in man. Nature (Lond.) 279:598-603.
12. Orkin, S. H., B. P. Alter, and C. Altay. 1979. Deletion ofthe Ay-globin gene in Gy_81 thalassemia. J. Clin. Invest.64: 866-869.
13. van der Ploeg, L. H. T., A. Konings, M. Oort, D. Roos, L.Bemini, and R. A. Flavell. 1980. y-,B thalassemia studiesshowing that deletion of the y and 8 genes influences,B-globin gene expression in man. Nature (Lond.).283: 637-642.
14. Orkin, S. H., S. C. Goff, and D. G. Nathan. 1981. Hetero-geneity of DNA deletion in y8,-thalassemia. J. Clin.Invest. 67: 878-884.
15. Weatherall, D. J. and J. B. Clegg. 1972. The ThalassemiaSyndromes. Blackwell Scientific Publications, Oxford.2nd ed.
16. Bunn, H. F., B. G. Forget, and H. M. Ranney. 1977. Hu-man Hemoglobins. W. B. Saunders Co., Philadelphia.
17. Kantor J. A., P. H. Tumer, and A. W. Nienhuis. 1980.Beta+ thalassemia: mutations which affect processing ofthe fl-globin mRNAprecursor. Cell 21: 149-157.
1016 Trecartin, Liebhaber, Chang, Lee, Kan, Furbetta, Angius, and Cao

18. Maquat, L. E., A. J. Kinniburgh, L. R. Beach, G. R. Honig,J. Lazerson, W. B. Ershler, and J. Ross. 1980. Processing ofhuman f8-globin mRNAprecursor to mRNAis defectivein three patients with (+-thalassemia. Proc. Natl.Acad. Sci. U. S. A. 77: 4287-4291.
19. Benz, E. J., Jr., B. J. Forget, D. G. Hillman, M. Cohen-Solal, J. Pritchard, and C. Cavallesco. 1978. Variability inthe amount of ,8-globin mRNAin 13' thalassemia. Cell 14:299-312.
20. Kan, Y. W., J. P. Holland, A. M. Dozy, and H. E. Varmus.1975. Demonstration of non-functional /&globin mRNAin homozygous P3o thalassemia. Proc. Natl. Acad. Sci.U. S. A. 72: 5140-5144.
21. Temple, G. F., J. C. Chang, and Y. W. Kan. 1977.Authentic /8-globin mRNAsequences in homozygous,l3 thalassemia. Proc. Natl. Acad. Sci. U. S. A. 74:3047-3051.
22. Chang, J. C., and Y. W. Kan. 1979. /3" thalassemia: anonsense mutation in man. Proc. Natl. Acad. Sci. U. S. A.76: 2886-2889.
23. Cao, A., M. Furbetta, A. Ximenes, A. Angius, C. Rosatelli,T. Tuveri, M. T. Scalas, A. M. Falchi, L. Maccioni, M. A.Melis, and R. Gallanello. 1981. 3 thalassemia types insouthern Sardinia. J. Med. Genet. 18: 196-198.
24. Kan, Y. W., A. M. Dozy, H. E. Varmus, J. M. Taylor, J. P.Holland, L. E. Lie-Injo, J. Ganesan, and D. Todd. 1975.Deletion of a-globin genes in haemoglobin H diseasedemonstrates multiple a-globin structural loci. Nature(Lond.). 255: 255-256.
25. Goossens, M. and Y. W. Kan. DNAanalysis in the diag-nosis of hemoglobin disorders. 1981. Methods En-zymol. In press.
26. Wilson, J. T., L. B. Wilson, J. K. deRiel, L. Villa-Komaroff,A. Efstratiadis, B. G. Forget, and S. M. Weissman. 1978.Insertion of synthetic copies of human globin genes intobacterial plasmids. Nucleic Acids Res. 5: 563-581.
27. Noyes, B. E., and G. R. Stark. 1975. Nucleic acid hybridiza-tion using DNAcovalently coupled to cellulose. Cell. 5:301-310.
28. Pelham, H. R. B., and R. J. Jackson. 1976. An efficientmRNA-dependent translation system from reticulocytelysates. Eur. J. Biochem. 67: 247-256.
29. Chang, J. C., G. F. Temple, R. F. Trecartin, and Y. W.Kan. 1979. Suppression of the nonsense mutation inhomozygous ,3" thalassemia. Nature (Lond.). 281: 602-603.
30. Kan, Y. W., E. Schwartz, and D. G. Nathan. 1968. Globinchain synthesis in the a thalassemia syndromes. J. Clin.Invest. 47: 2515-2522.
31. Cooke, N. W., D. Coit, R. I. Weiner, J. D. Baxter, and J.Martial. 1980. Structure of cloned DNAcomplementaryto rat prolactin messenger RNA. J. Biol. Chem. 255:6502-6510.
32. Dagert, M., and S. D. Ehrlick. 1979. Prolonged incubationin calcium chloride improves the competence of Esche-richia coli cells. Gene. 6: 23-28.
33. Norgard, M. V., K. Keem, and J. J. Monahan. 1978. Fac-tors affecting the transformation of Escherichia colistrain X 1776 by pBR 332 plasmid DNA. Gene (Amst.). 3:279-292.
34. Kan, Y. W., and A. M. Dozy. 1978. Polymorphism ofDNAsequence adjacent to the human ,8-globin structuralgene: relationship to sickle mutation. Proc. Natl. Acad.Sci. U. S. A. 75: 5631-5635.
35. Maxam, A. M., and W. Gilbert. 1977. A new method forsequencing DNA. Proc. Natl. Acad. Sci. U. S. A. 74:560-564.
36. Marotta, C. A., J. T. Wilson, B. G. Forget, and S. M.Weissman. 1977. Human ,3-globin messenger RNA. III.Nucleotide sequences derived from complementaryDNA. J. Biol. Chem. 252: 5040-5043.
37. Lawn, R. M., A. Efstratiadis, C. O'Connell, and T. Mania-tis. 1980. The nucleotide sequence of the human /8-globin gene. Cell. 21: 647-651.
38. Kan, Y. W., K. Y. Lee, M. Furbetta, A. Angius, and A.Cao. 1980. Polymorphism in DNAsequences in the /3-globin region. Application to prenatal diagnosis of /thalassemia in Sardinia. N. Engl. J. Med. 302: 185-188.
/3 Thalassemia in Sardinia 1017