the surface energy of a bounded electron gas
TRANSCRIPT

J. Phys. F: Metal Phys., Vol. 4, August 1974. Printed in Great Britain. Q 1974.
The surface energy of a bounded electron gas
J Harris and R 0 Jones Institut fiir Festkorperforschung der Kernforschungsanlage Julich, 5 17 Jiilich, Germany
Received 28 December 1973
Abstract. An exact expression for the exchange and correlation energy of an inhomogeneous electron gas, as defined by Hohenberg, Kohn and Sham, is derived. This expression is separated into exchange and correlation terms and a formula linking the surface exchange energy of a half space to the Kohn-Sham one electron potential follows without approxi- mation. For an infinite barrier model, the local density (Slater) approximation gives a surface exchange energy 50% greater than the exact value, a large and previously un- suspected error. An exact evaluation of the surface correlation energy is not feasible, but we argue that the dominant contribution, srising from the difference in zero point energy between bounded and unbounded systems, can beestimated using a simple model. Numerical results, not dependent on the introduction of arbitrary plasmon wavevector cutoffs, give surface correlation energies larger than Lang and Kohn (LK), who work from a local formula, by a factor of six. We discuss the consequences of our work for the agreement with experimental surface energies achieved by LK.
1. Introduction
The calculation of the surface energy of a free electron like metal. long a subject of interest. has recently become rather controversial. Early work (see Huntington 1951 or Stratton 1953) concentrated on the kinetic and electrostatic energy and it was only recently that a serious attempt was made by Lang and Kohn (LK 1970) to include exchange and correlation effects as well as the influence of the ionic lattice. In developing a general theory of the static properties of a bounded electron gas. LK employed the Hohenberg-Kohn-Sham selfconsistency scheme for calculating the density of an inhomogeneous electronic system (see Sham and Kohn 1966 and references therein). From this density. or equivalently from the one electron potential generating it. they calculated the surface energy and the work function of the electron gas Of particular interest is their conclusion that the surface energy of a bounded electron gas becomes negative for I , 5 2.4. Adding a contribution from the discrete ionic lattice. which they evaluate to first order in a pseudopotential. they were however able to reproduce remarkably well the experimental surface energies (more correctly. surface tensions) of Simple metals. which increase monotonically for decreasing I , .
More recently Craig (1972) and Schmit and Lucas (1972) suggested that both trend and magnitude of the observed surface energies can be explained by assuming the correla- tion contribution to be dominant. They assume a sharp cutoff model for the electron gas and estimate the zero point energy (ZPE) difference between bounded and unbounded systems by considering only dispersionless bulk and surface plasmons. While the details
1170

The surface energy of a bounded electron gas 1171
of this calculation have been criticized (Jonson and Srinivasan 1973. private communica- tion. Feibelman 1973. Kohn 1973, Heinrichs 1973). the basic questions as to the relative magnitude of the ZPE contribution. where it appears in the Lang-Kohn formalism. and what its consequences might be concerning agreement with experiment. have not been answered. To resolve these questions is the main aim of this paper.
We note. firstly. that the ZPE contribution is not contained at all in the LK formalism. This is most readily apparent by considering the step procedure used by Lang and Kohn for including the ionic lattice contribution (see figure 5 of LK 1970) Step I involves splitting the sample. but requiring the electron density to maintain its bulk value. If the ions are represented by a uniform positive background. this step costs no correlation energies in LK’s local density formalism. In reality. however. the correlation energy involved can be estimated using the Lifshitz formula for the force between two dielctric halfspaces (see Landau and Lifshitz 1960). But the Lifshitz formula. which also accounts for the Van der Waals forces between atoms. can be shown to contain a contribution due to the different in ZPE between joined and separated halfspaces. For sufficiently large separations, for example, Gerlach (1971) has shown that the ZPE of the surface plasmons is the dominant contribution. If this is a significant omission in LK, the agree- ment between their energies and experiment is somewhat puzzling.
The LK calculation of the surface exchange and correlation energy E;c proceeds from the local formula
Here the electron density is n(z) with bulk value E. and &n) is the exchange and correla- tion energy of a homogeneous electron gas of density n. It is immediately obvious that LK obtain no surface exchange and correlation energy unless n(z) deviates at some point from its asymptotic bulk value. That a truly local theory does not include the proper ZPE contribution may be understood as follows. A basic assumption is that. locally. the system behaves as if it were homogeneous. The normal modes of such a system there- fore include ‘local’ bulk plasmons of frequency (4nn(z))”’. but no surface plasmons since such modes could always decay into a local bulk mode. The correlation part of (1.1) therefore includes a contribution corresponding to the ZPE of the ‘local’ bulk modes. Such a term, whose physical relevance in a system of rapidly varying electron density is obscure, must be small since the ZPE of bulk plasmons is only weakly density dependent. The LK correlation energies must be correspondingly small$.
To take account of the ZPE contribution properly. we must replace ( 1.1) by a non local relation between E:, and n(z). a procedure which we carry out exactly in § 2. More pre- cisely. we derive a method whereby one could, in principle. determine E,, exactly once the exact electron density is known. We then separate E,, into exchange and correlation parts and consider the correlation energy in 9 3 For a slowly varying system. where the wavefunctions could be represented by plane waves with a position dependent wave- vector, we would reproduce ( 1 . I ) . We argue that. so far as the correlation energy is concerned. a metal surface more nearly resembles the opposite limit where the system
t Owing to the long range of the Coulomb interaction this contribution has very little to do with the way in which the electron density at the surface relaxes. Thus we expect the correlation energy to be relatively in- sensitive to this relaxation, as distinct from kinetic. exchange and electrostatic contributions which are dominated by it. :This fact was apparently first reaiiied by G Toulouse.

1172 J Harris and R 0 Jones
cuts off infinitely sharply. though no account is taken of the change in the electron wavefunctions close to the boundary. Under these circumstances our expression for EQ reduces to the simple formula previously obtained by Peuckert (1970. 1971). This formula includes the ZPE of the surface plasmons as well as contributions from the single particle excitations. No problems with cutoff wavevectors arise. Numerical evaluation leads to results for E: six times as great as the LK energies and 15-50% less than the estimates of the plasmon contribution given by Schmit and Lucas.
In $4 we examine the surface exchange energy. which we separate from the bulk contribution without approximation. This expression, whose derivation is described in appendix 2. affords an exact evaluation of E: once the electron density is known. Since our aim is to check the likely error in using the local approximation (1. 1). we consider an infinite barrier model where the numerical evaluation of the exact E: is simplified. We show that for this model the local approximation overestimates the exchange energy by some 50 %. By comparing the exchange energies of atoms as calculated via complete Hartree-Fock calculations and via the local approximation we argue that such a large and previously unsuspected error is. in fact. reasonable.
Section 5 then summarizes our results. The very large errors in the LK surface cor- relation energies are substantially cancelled in the small r, region by the equally large. but proportionally smaller. errors in the LK exchange energies. Since we have not calculated E:, for the LK density profiles. we do not compare our results with experiment. Such comparisons are often misleading. since the net surface energy is obtained from a number of large contributions. each subject to considerable uncertainty. Nevertheless. we believe that our calculations resolve the question of the ZPE contribution and its relation to the LK theory. lend indirect support to the LK density profiles. and confirm the conclusion that a contribution from the discrete ionic lattice is essential to explain the observed surface energies of low rc metals.
Throughout the paper we use Hartree units (e ( = h = m = 1, the unit of energy being 1 Hartree = 27.2 eV: vectors x are separated into components xll. z parallel to and perpendicular to the predominant surfaces of the sample : the Fermi momentum and energy are denoted k , and E, respectively.
2. Exchange and correlation energy of a bounded system
The system we consider consists of a collection of ions. represented by a fixed positive background of charge density - N ( x ) and an equal number of electrons which in the ground state give rise to a charge density n ( x ) The Hamiltonian is
I? = [dx $+(x)( - iV2)$(x) + 4 j d x S dx’$+(x)$+(x’)dx - x‘)$(x’)$(x)
- [dx {dx’ [N(x )v (x - X ‘ ) & ~ ( X ’ ~ X ’ ) - i N ( x ) v ( x - x ’ ) N ( x ‘ ) ] (2.1)
= $ + P where dx) is the Coulomb interaction. We wish to calculate the energy E = (4 1 I-? 1 I)>. where 1 I)> is the many body ground state of H. In a homogeneous system a very useful technique for determining E is to consider the Hamiltonian
I?;, z T + i v (2 .2 )
where T and P are the kinetic and potential parts of (’.1) and i is a parameter which varies from 0 to 1 . When the state I $ > represents a system which is bounded only by virtue

The surface energy of a bounded electron gas 1173
of the interactions, this scheme must be modified since the ground state of A,, is funda- mentally different from 1 $). This difficulty may be overcome by adding and subtracting from A a term
(2.3) Po = s dx v , , ,~x )$ '~x )~~x)
and considering the Hamiltonian
A, = T + Po + l.(P - Po). (2.4)
Clearly 8, = E? and Po may be so chosen that if 1 ILL) is the ground state of H,.
n,(x) = ($"~$+(X)$(X) l$O> = n ( x ) (2.5)
the exact electron density of the physical system. This property is assured if we take for v,,,lx) the Kohn-Sham effective potential. The qualitative difference between the states 1 $o) and 1 $;,) is now removed and the effect of 'switching on' the electron-electron interaction in (2.4) is minimized. The energy E is given by
(2.6) may be written
E = T + E, , + E,, (2.8)
where
T = ($ol"> (2.9) E, , = 4 J dx S dx'C(X - x ' ) (~ (x ) - N ( x ) ) ( n ( x ' ) - N ( x ' ) ) (2.10)
and
E,, = dx tieXt(x) d).(n(x) - n,(x)) J JO
- &x - x')n,(x)] (2.1 1)
Here T is the kinetic energy of an assembly of noninteracting electrons having the same density as the physical system. E,, is the total electrostatic energy of the system. and E,, is by definition the exchange and correlation energy. T and E,, are identical in form to the corresponding quantities in the Kohn-Sham functional and so. provided we use the electron densities calculated by LK, (2.9) and (2.10) give identically their kinetic and electrostatic contributions to the surface energy. We note that (2.9) does not give the

1174 J Harris and R 0 Jones
kinetic energy of the interacting system. part of which is included in the exchange and correlation energy. The final term of (2.11) includes explicitly the contribution to the energy due to the density fluctuations of the system. It may be rewritten
where xA(x . x’. io) is the density response function of a system of electrons described by the Hamiltonian Ha.
The exchange part of E,, may be isolated by evaluating the 1. integrand at I. = 0. Using (2.12) we obtain
E , = - 1 dx dx’ u(x - x’) - xo(x. x’iw) + n(x)h(x - x’) . (2.13) s s 1 This result may be cast in a more familiar form. If
( - :v2 + ~~&))(fgX) = E&X)
defines the complete and orthonormal set of eigenfunctions +i(x) of the Kohn-Sham potential. then (see. for example. Hedin and Lundqvist 1969)
The factor of two accounts for the double occupancy of each orbital Using (2.14) in (2.13) we obtain
E , = - 4 J dx 1 dx’ V(X - x’) 1 n(x)h(x - x‘)
+ 2 1 [O(EF - E ; ) - - E J ) ] ~ S ( X ) ~ ~ X ’ ) ~ : ( X ’ ) ~ ~ ( X ) } I ]
E r ’ € ,
= - a Sdx l d x ’ V ( ~ - x ’ ) ~ ~ ( x . x ’ ) / ~ (2.15)
where
(2.16)
is the one particle density matrix. Thus (2.1 3) is equivalent to evaluating the Fock energy in a Hartree-Fock calculation using the functions 4 i (x ) . This may be regarded as a definition of the exchange energy of a bounded system?. Since only one potential t’,,,(x) can generate the exact electron density. the functions $Jx) and hence the exchange energy (2.1 3) are unique.
Subtracting (2.1 3) from (2.1 1) we have for the correlation energy
(2.17)
t This is different from the exchange energy arising from a Hartree-Fock calculation. since the functions 4 i ( x ) are generally not solutions of the HF equations. In a homogeneous system, the LK and HF selfconsistent potentials are both constant and the corresponding eigenfunctions (plane waves) are identical. This is not so in general. leading to some arbitrariness in the definition of the exchange energy for inhomogeneous systems. Our choice is the most convenient for calculational purposes.

The surface energy of a bounded electrox gas 1175
where 4 ;.(x) is the electrostatic potential set up by the charge density distribution &x) + n;.(xj -. 2N(x)) and 1
fTx. x’. io) ~‘(x. x’. io) - xo(x. x’. io).
One advantage of separating E,, into E, and E , is that. once the functions 4i(x) are known, the exchange contribution can be evaluated exactly. Secondly, the final term in (2.17) represents density fluctuations due solely to the Coulomb interaction. In the present context, these are bulk and surface plasmons whose energies depend only weakly on the details of the Hamiltonian A,. In practice, this means that we may replace id in (2.1 7) by an approximation which would be unjustlfied if applied directly to x’ in (2.12).
In applying the above analysis to the case of an electron gas which is homogeneous in two spatial directions. it is useful to introduce the two dimensional Fourier transforms
(2.1 8)
where the coordinate z is the direction in which the system is inhomogeneous. Equations (2.13) and (2.17) may then be written
f ( k l l . z ) = J dxII exp( - ikil .xll)f(x)
(2.19)
x ?-(kll, z:z’. io) (2.20)
where
These equations give the exchange and correlation energy of a film of thickness L and cross sectional area A correct to order L/ J A and form the basis of the remainder of the paper.
3. Correlation energy
The computation of the correlation energy of a bulk system from (2.20) is straightforward. one method being to deform the contour of the o integral to enclose the positive real axis (see, for example. Pines 1963). A contribution to E , comes from the plasmon pole in 2’. A similar procedure for a film geometry is less trivial. but additional contributions from the surface plasmon pole in f’ are clearly expected? Since both bulk and surface plasmon energies depend only weakly on the detailed properties of the electron gas.
t A complication is that, due to the presence of the surface, ii has a cut along the entire real axis, even in the time dependent Hartree approximation.

1176 J Harris and R 0 Jones
these poles should give comparable contributions for any reasonable model. It is crucial. however. to include single particle excitations and to avoid introducing arbitrary cutoff wavevectors.
To satisfy the above requirements within a soluble model. we must represent the bounded electron gas by a system which cuts off sharply at a plane. The dynamical properties of such a system are qualitatively similar to those of a more realistic model. however. and should give a reasonable estimate of the 'dynamical' contribution to E,. contained in the second term of (2.20). The first term, which has a different origin. is expected to give a small contribution. The function (n(z) - n,(z)). which occurs in this term. is zero for 1. = 0,l and, since the density profile is relatively insensitive to changes in the surface potential. should be small for intermediate 1..
If the electron density is constant in the region 0 < z < L and zero elsewhere. we may introduce the cosine transform
nn L
q = - n = 0. 1 . . . x (3.la) f (q) = s' dz cos qzf(z) 0
with inverse
The second term in (2.20) may then be written
(3.2)
where u(kll, q) = 4n/(ki + q2) and C' implies that ( q + q') must be an even multiple of n/Lt. Assuming specular reflection of the electrons at the boundaries of the film the density response function is given by (Peuckert 1971. Griffin and Zaremba 1973)
where
and xi(kII, q, w) is the Lindhard density response function for a homogeneous eleetron gas with charge JA. Substituting (3.3) into (3 2), simplifying and performing the E. integra- tion, we obtain Peuckert's expression for the surface correlation energy of a halfspace
An outline of the main steps in going from (3.2) to (3.4). is given in appendix 1. We define the surface energy per surface particle to be
t Since we eventually take the halfspace limit, L + CO, we have excluded a term proportional to exp [ - k , , L ] which represents the coupling between the surfaces.
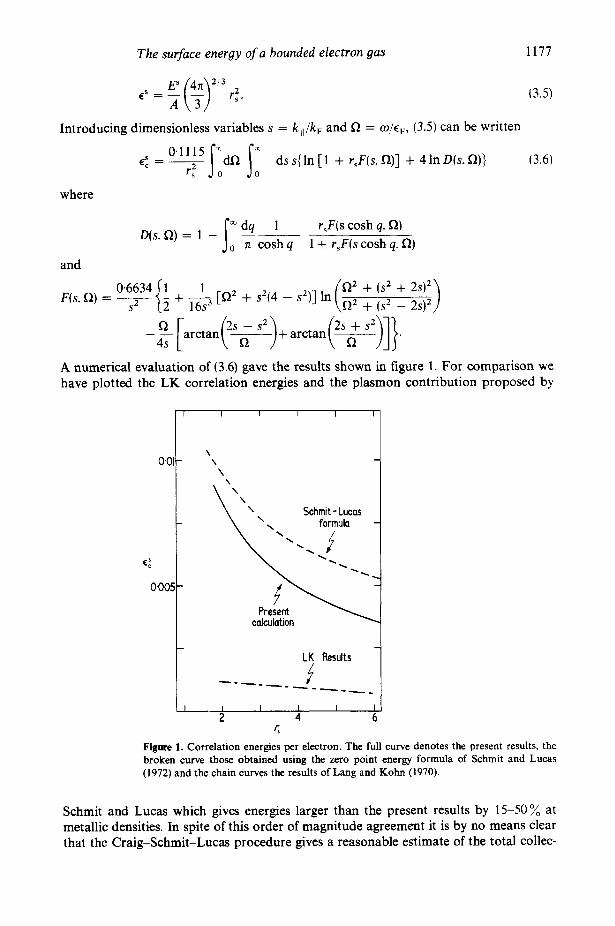
The surface energy of a bounded electron gas 1177
(3.5)
Introducing dimensionless variables s = ki l /k , and R = o / E ~ , (3.5) can be written
E: = 0.1115 J:dR J z ds s{ln [ 1 + r,F(s. R)] + 4 in D(s , R)} (3.6) r: 0
where
r ,F(s cosh q. R) 1 + r,F(s cosh q. R) n cosh q
and
- 4s [arctan?+)+ arctan?+)]}.
A numerical evaluation of (3.6) gave the results shown in figure 1. For comparison we have plotted the LK correlation energies and the plasmon contribution proposed by
\ \ - \ \
Schmit -Lucas
LK Results
4 ------------
Figme 1. Correlation energies per electron. The full curve denotes the present results, the broken curve those obtained using the zero point energy formula of Schmit and Lucas (1972) and the chain curves the results of Lang and Kohn (1970).
Schmit and Lucas which gives energies larger than the present results by 15-50 % at metallic densities. In spite of this order of magnitude agreement it is by no means clear that the Craig-Schmit-Lucas procedure gives a reasonable estimate of the total collec-

1178 J Harris and R 0 Jones
tive contribution to ( 3 6). A normal mode analysis of this formula (Griffin and Kranz 1974 private communication) suggests that the contribution arising from the bulk and surface plasmon poles shows a marked dependence on the wavevector and that its value at k , , = 0 constitutes only a small contribution to the total correlation energy. This is due to the weighting factor of kll in the integrand?.
Since our model assumes the dynamic Hartree approximation our results probably form an upper bound for 6;. Nevertheless, deficiencies in the model are unlikely to account for the factor of six by which they exceed the LK correlation energies. and we conclude that the local approximation grossly underestimates this contribution to the surface energy. It should be noted, however, that for r, N 2, our correlation energy is considerably smaller than the LK kinetic and exchange energies and roughly equal to the electrostatic contribution. Direct comparison of correlation energies and experi- ment (Craig 1972, Schmit and Lucas 1972) is therefore incorrect.
4. Exchange energy
In the local density approximation. the exchange energy per surface electron is given by
(4.1)
where x = k,z and p ( x ) is the electron density divided by the bulk density. The I, de- pendence of the density profile means that (4. l ) generally has a different rs behaviour from the bulk exchange energy per electron, - 0.458/rS. The conditions under which (4.1) may be derived from the exact expressions (2.1 3) or (2.1 5), namely when the electronic wavefunctions are plane waves with a position dependent wavevector k(x), clearly do not obtain when u,,,(z) varies by a large amount over a distance k; '. When applied to atoms, the local approximation nevertheless gives exchange energies which agree with Hartree-Fock calculations within 5-1 5 % (Tong and Sham 1966) To conclude, however. that (4.1) should exhibit a similar percentage error is incorrect. since in this case the bulk contribution, which the local theory gives exactly, has been subtracted. A better comparison could be made by subtracting from the atomic exchange energies the exchange energy of a jellium sphere having the average density of the atomic electrons. This would give a 'surface' exchange energy for the atom. analogous to (4.1) and substantially smaller than the total exchange energy. The error involved in estimating this quantity within the local approximation would therefore be considerably greater than 5-1 5 %:.
To demonstrate this point and to check the error likely to arise from using (4.1) to calculate the surface exchange energy, we work from the exact expression (2.1 3) which we separate into bulk and surface contributions as detailed in appendix 2. The final expression for E: (A2.25) is exact, once the functions c#J~(x) are known, but in practice its evaluation would involve a prodigious computation. The more limited aim of checking the probable error in (4.1) can be achieved however, by considering a model in which
t In fact, the major contribution to (3.6) comes from particle-hole rather than plasmon modes. We thank A Grifin for discussions concerning this point.
This effect is discernible in the results of Tong and Sham (1966). which show significantly worse agreement for atoms with larger surface to volume ratios.

The surface energy of a bounded electron gas 1179
This ‘infinite barrier’ model has been widely used. and more details may be found in papers by Newns (1970) and Beck and Celli (1970). Using (4.2). the expression for E: is substantially simplified and its numerical evaluation is relatively straightforward. The result ist
IB 0.0106 exact r s
compared with the local density approximation (4.1 )
IB 00164
local
(4.3)
(4.4)
Although the local approximation gives the order of magnitude of E: correctly, it over- estimates the exact surface exchange energy by approximately 50%. This error is important. since exchange is a large contribution to the surface energy. In connection with our remarks above. we note that in this model the total energy of a film (per unit volume is)
where L is the thickness and where is the bulk exchange energy per electron. It is clear that the local approximation would give very good results for E, for even relatively thin films.
There is no reason why the local approximation should be significantly better for the LK potentials than for the infinite barrier model, particularly for larger rs where the LK potential closely resembles a step. Even at rs - 2, however, the wavefunctions vary rapidly over distances comparable with the Fermi wavelength AF, so that an error of roughly the same order is to be expected. We conclude that the LK surface exchange energies overestimate the correct values by an amount approaching 50 %.
5. Conclnding remarks
We have obtained exact closed form expressions for the exchange and correlation energy of a bounded system which are directly additive to the kinetic and electrostatic terms in the Kohn-Sham functional. Using the LK density profiles, our results for the kinetic and electrostatic contributions are identical with those of LK. Correlation energies obtained from a sharp cutoff model are six times greater than those derived from a local approximation, a result attributable to the different treatment of the density fluctuations. An exact expression for the surface exchange energy has been evaluated for a simple model within which the local approximation overestimates this contribution by about 50%.
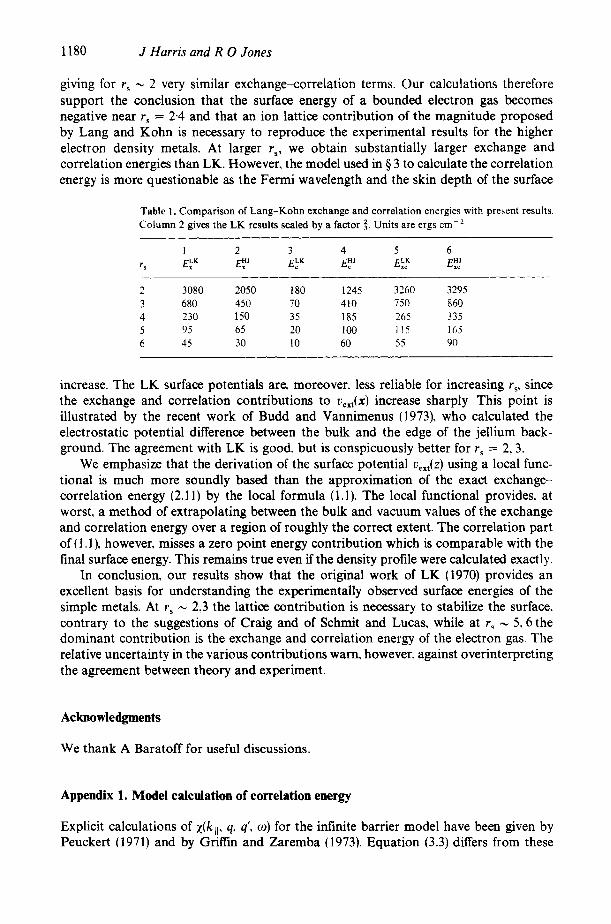
Our numerical results are summarized in table 1. Columns 1 . 3 and 5 give the LK estimates for exchange, correlation and ‘exchangesorrelation’ surface energies. Column 4 gives the correlation energies as calculated in $2, column 2 are the LK results scaled by a factor i- (corresponding to the ratio of (4.3) to (4.4)) and the final column is our resulting estimate for the exchange-correlation energy. It is particularly striking that the large discrepancy between the correlation energies EkK. E!’ is substantially cancelled. t The r, dependence arises from the fact that the only length in the problem is k;’, ie u,Jz) is rr independent
1180 J Harris and R 0 Jones
giving for r , - 2 very similar exchange-correlation terms. Our calculations therefore support the conclusion that the surface energy of a bounded electron gas becomes negative near r, = 2.4 and that an ion lattice contribution of the magnitude proposed by Lang and Kohn is necessary to reproduce the experimental results for the higher electron density metals. At larger Is, we obtain substantially larger exchange and correlation energies than LK. However. the model used in 0 3 to calculate the correlation energy is more questionable as the Fermi wavelength and the skin depth of the surface
Table 1. Comparison of Lang-Kohn exchange and correlation energies with present results. Column 2 gives the LK results scaled by a factor 3. Units are ergs cm-’
I 2 3 4 5 6 r s E k K E:’ E:K E;’ E:: E::
2 3080 2050 180 1245 3260 3295 3 680 450 70 410 750 860 4 230 150 35 185 265 335 5 95 65 20 100 I15 165 6 45 30 10 60 55 90
increase. The LK surface potentials are, moreover. less reliable for increasing r,. since the exchange and correlation contributions to u,,,(x) increase sharply This point is illustrated by the recent work of Budd and Vannimenus (1973), who calculated the electrostatic potential difference between the bulk and the edge of the jellium back- ground. The agreement with LK is good, but is conspicuously better for r , = 2 , 3 .
We emphasize that the derivation of the surface potential u,,,Cz) using a local func- tional is much more soundly based than the approximation of the exact exchange- correlation energy (2.1 1) by the local formula (1.1). The local functional provides. at worst, a method of extrapolating between the bulk and vacuum values of the exchange and correlation energy over a region of roughly the correct extent. The correlation part of (1 . I ) , however, misses a zero point energy contribution which is comparable with the final surface energy. This remains true even if the density profile were calculated exactly.
In conclusion, our results show that the original work of LK (1970) provides an excellent basis for understanding the experimentally observed surface energies of the simple metals. At rs - 2,3 the lattice contribution is necessary to stabilize the surface. contrary to the suggestions of Craig and of Schmit and Lucas, while at T , - 5,6 the dominant contribution is the exchange and correlation energy of the electron gas. The relative uncertainty in the various contributions warn, however, against overinterpreting the agreement between theory and experiment.
Acknowledgments
We thank A Baratoff for useful discussions.
Appendix 1. Model calculation of correlation energy
Explicit calculations of ~ ( k , , , q, q’, w ) for the infinite barrier model have been given by Peuckert (1971) and by Griffin and Zaremba (1973). Equation (3.3) differs from these

The surface energy of a bounded electron gas 1181
results only via the factor 1 /aq which arises from the use of cosine rather than exponential transforms. One should remark that in deriving (3.3) the interference between electron waves incident on and reflected by the surface bamer has been neglected When this effect is included, no closed expression for the density response function of the bounded system can be written down (see Newns 1970, Beck and Celli 1970). The influence of the interference terms on the correlation energy is currently being considered by Jonson and Srinivasan (private communication).
Equation (3.4) has been obtained previously by Peuckert (1970). We give our derivation because Peuckert’s is not available in the published literature. Firstly we note that
Writing (3.2) as
(Al.1)
(Al.2)
using (Al.1) to convert all sums on q to integrals, and (3.3) for i A ( k l l , q, q‘, io) we obtain.
xi$kll, 4. io) x5kll, q’, io) D”(kll, io)
X
The first term in (A1.3) gives the bulk correlation energy,
(Al.3)
Here we have used the fact that z$(kII, q. o) depends only on ki + q2 k2 The same property ensures the exact cancellation of the second and third terms in (Al.3). (Note that this depends crucially on a proper treatment of the q = 0 Fourier components.) The surface correlation energy is thus
(A1.5)

1182 J Harris and R 0 Jones
We now make use of the dynamic Hartree approximation
to perform the I integration. Following Peuckert (1970) we note that the E. integrand may be written
Thus (A1.5) reduces to
(Al.6)
Again using the fact that x; (kI l , 4, io) depends only on (kfi + q2), we arrive at equation (3.4). Use of (3.3) and (A1.l) constitutes taking the halfspace limit. so (A1.6) is the correlation energyfor a single surface.
Appendix 2. Surface exchange energy
We derive an exact expression for the surface exchange energy in terms of the occupied eigenstates of uext(z), starting from (2.19). For a film, the states $Jx) which appear in the expression (2.14) for the response function take the form
(A2.1)
where the q5,, are chosen to be real. Far from the surfaces. these functions have the asymptotic form
(A2.2)
1 q5i(x) = -exp(ikII . xll) q5,(z)
J A
#,(z) + &(z) = N , sin (k,z + 6,)
where N , is a normalization constant and the 6, are phaseshifts. The energies ei are given
(A2.3)
and we have assumed that j k : < eF + a, where is the work function. We choose the origin of z to be a distance q from the edge of the jellium background, where q is such that for every function $,(z), with k , < (2~,)"~, $,,(O) is effectively zero. We write
I : = L + 2 q (A2.4)
where L is the distance between the edges of the positive background (Note that the phaseshifts 6, will depehd on q.) The spectrum of energies for the film is obtained most easily by noting that ;b,(z) must be even or odd about z = $E. Thus
by
~i = i-(ki + kfi)
nn 26, " L: I:'
k = - - -
Using (A2.1) and (A2.3) in (2.14) we obtain
(A2.5)

The surface energy of a bounded electron gas 1183
(A2.6)
where
and
Integrating over o, we find
(A2.9)
where
[.$.,$I’ < kf - k:, with
* f k t nn 11, E Aij , /kl , . (A2.10)
Note that the n’ sum in (A2.9) is restricted to exclude states above the Fermi level, Substituting (A2.9) in (2.19), we have for the exchange energy of the film,
(A2.11)
where the matrix elements @.“,I). are defined by
@i,$ z /Im dzIm dz’ expt - kl l 1 z - z’ 1) @,,.(z, 2’). (A2.12)
Using the competeness relation for the &(z) and the expression for the electron density.
n(z) = - (kf - k:) &(z) (A2.13)
- -m
1 nr
277 ,,=
the two terms in (A2.11) may be combined to give
(A2.14)
(A2.15)

1184 J Harris and R 0 Jones
Since fi;. is zero for k , , > 2k, or k, > k,. the sums in (A2.14) are restricted to the Fermi sphere.
We now write the matrix elements e,$ as
@:!, = N;fN$ (Li$ + Mi;,) (A2.16) where
Lk,$ = Jr d r y dz' sin(k,z + 6,) sin(k,,z + dnf) exp( - k , , Iz - z'l) sin(k,z' + 6,) 0
x sin(k,.z' + 6,,) (A2.17)
(A2.18)
Here 8 denotes the asymptotic value of @. Thus M i $ contains no contribution of order L! and can be evaluated by numerical integration over a region in the neighbourhood of each surface. A straightforward but tedious calculation gives
where
(A2.19)
k , , sin 26- sin26, sin26,. k l l sin 26, sin26, 2(kfi + k?) (lk_ - - - ~ 2k,' ) - 2Cki + k ? ) ( r --
p;, = - 2kn
sin 'a,,,) - - [ (k cos 6 - - _ _
2knf k, sin 6,
Here we have used the fact that n, n' # 0 and introduced the abbreviations
k, = k, k k,, 6, 3 6, * a,,. Equation (A2.14) may now be written
+ Mi;, + i:;.].
To separate this into bulk and surface terms. we write
2kn
(A2.20)
(A2.21)
(A2.22)
(A2.23)
where lk,z is a microscopic length depending on the choice made for 17. Using (A2.5) and the Euler-Maclaurin series

The surface energy of a bounded electron gas 1185
we have our final result for the exchange energy of a film (two surfaces)
Here R = A L is the volume of the film and
(A2.25)
(A2.26)
To show that the volume term is the same as would be obtained for a homogeneous system, we can use the representation for f”,, given in (A2.9) and extend the k integrals to - 00. Variable changes then reproduce the result one obtains from (2.1 3) and (2.14) if the 4 i ( x ) are plane waves. This term gives eventually -0.458/r, per electron.
In the infinite barrier model considered in 9 4, the quantities S(k), Zb Mi!, in (A2.25) are all zero, charge neutrality gives 17 = 3n/8kF (Bardeen 1936) and
In units where kF = 1, the surface exchange energy per surface particle is then
(A2.27)
(A2.28)
This expression may be evaluated numerically, giving the result (4.3). In the three dimensional integral. a large contribution comes from small values of kll . This diffkulty was overcome by writing k = kllx and evaluating
The two dimensional integral 17a) approaches a constant as a tends to zero and
I(0) = I (a) - aZY0).
References
Bardeen J 1936 Phys. Ret.. 49 65343 Beck D E and Celli V 1970 Phys. Ret.. B 2 295540 Budd H F and Vannimenus J 1973 Phys. Rec.. Lett. 31 1218-21 Craig R A 1972 Phys. Reo. B 6 1134-42 Feibelman P J 1973 Solid S t . Commun. 13 319-21 Gerlach E 1971 Phys. Rev. B 4 3 9 3 4 Griffin A and Zaremba E 1973 Phys. Ret.. A 8 486-99 Hedin L and Lundqvist S 1969 Solid State Physics vol 23 ed F Seitz, D Turnbull and H Ehrenreich (New
Heinrichs J 1973 Solid S t . Commun. 13 1599-602 York: Academic Press) pp 1-181

1186 J Harris and R 0 Jones
Huntington H B 1951 Phys. Reo. 81 1035-9 Jonson M and Srinivasan G 1973 Phys. L e f t . 43A 427-8 Kohn W 1973 Solid St. Commun. 13 323-4 Landau L D and Lifshitz E M 1960 Electrodynamics of Confinuous Media (Oxford: Pergamon) $90 Lang N D and Kohn W 1970 Phys. Rev. B 1 455568 Newns D M 1970 Phys. Rev. B 1 3304-22 Peuckert V 1970 Dissertation Universitiit Koln
Pines D 1963 Elementary Excitations in Solids (New York: Benjamin) appendix C Schmit J and Lucas A A 1972 Solid Si. Commuii. 11 415-8 Sham L J and Kohn W 1966 Phys. Rev. 145 561-7 Stratton R 1953 Phil. Mag. 44 123646 Tong B Y and Sham L J 1966 Phys. Re t . 144 1-4 Wagner D 1966 Z . Naturf. 21a 634-42
- 1971 Z . Phys. 241 191-204