therapeutic targeting of the il-6 trans-signalling/mtorc1 ... · lung tissues from emphysema...
TRANSCRIPT

Therapeutic targeting of the IL-6 trans-signalling/mTORC1 axis in pulmonary
emphysema
Saleela M. Ruwanpura1,2, Louise McLeod1,2, Lovisa F. Dousha3, Huei J. Seow3, Sultan
Alhayyani1,2, Michelle D. Tate1,2, Virginie Deswaerte1,2, Gavin D. Brooks1,2, Steven
Bozinovski3,4, Martin McDonald5, Christoph Garbers6, Paul T. King1,5, Philip G. Bardin1,5,
Ross Vlahos3,4, Stefan Rose-John6, Gary P. Anderson3 and Brendan J. Jenkins1,2*
1Centre for Innate Immunity and Infectious Diseases, Hudson Institute of Medical Research,
27-31 Wright Street, Clayton, Victoria, 3168, Australia.
2Department of Molecular Translational Science, Faculty of Medicine, Nursing and Health
Sciences, Monash University, Clayton, Victoria, 3800, Australia.
3Lung Health Research Centre, Department of Pharmacology and Therapeutics, The
University of Melbourne, Parkville, Victoria, 3050, Australia.
4School of Health and Biomedical Sciences, RMIT University, Bundoora, Victoria, 3083,
Australia.
5Monash Lung and Sleep, Monash Medical Centre, Victoria, 3168, Australia.
6Institute of Biochemistry, Christian-Albrechts-University, Olshausenstrasse 40, D-24098
Kiel, Germany.
*To whom correspondence should be addressed: Brendan J. Jenkins, Centre for Innate
Immunity and Infectious Diseases, Hudson Institute of Medical Research, 27-31 Wright
Street, Clayton, Victoria, 3168, Australia.
Tel +61 3 8572 2740; E-mail [email protected]

Author contributions: S.M.R designed and performed experiments, analysed data, prepared
the figures and co-wrote the manuscript. L.M. performed mouse experiments, cell culture,
immunohistochemistry and molecular experiments. L.F.D. and H.J.S. performed smoke and
lung function studies. M.D.T. and V.D. performed cell culture and flow cytometry analyses,
respectively, and S.A. and G.D.B. assisted with immunohistochemistry. P.B. and P.T.K.
provided human lung biopsies, and M.M. provided human sera. G.P.A. assisted with the
experimental design of smoke-related studies and editing of the manuscript. R.V. and S.B.
analyzed data, and assisted with experimental design of smoke-related studies and editing of
the manuscript. S.R-J. provided mouse strains and novel reagents to target IL-6 trans-
signalling, and assisted with the experimental design and editing of the manuscript. B.J.J.
conceived, designed and supervised the research, and co-wrote the manuscript.
Sources of support: This work was supported by a research grant from the National Health
and Medical Research Council of Australia (NHMRC) awarded to B.J.J., as well as the
Operational Infrastructure Support Program by the Victorian Government of Australia. S.M.R.
is supported by a NHMRC Post-doctoral Training Fellowship. The work of S.R-J. was
supported by the Deutsche Forschungsgemeinschaft Bonn, Germany (SFB877, project A1 and
the Cluster of Excellence 'Inflammation at Interfaces'). B.J.J. was supported by a Senior
Medical Research Fellowship awarded by the Sylvia and Charles Viertel Charitable
Foundation, and is currently supported by an NHMRC Senior Research Fellowship. S.R-J. is
an inventor of patents owned by CONARIS Research Institute, which develops the sgp130Fc
protein together with Ferring Pharmaceuticals, and he has stock ownership in CONARIS. No
conflicts of interest, financial or otherwise, are declared by all other authors.
Running title: IL-6 trans-signalling/mTORC1 axis in emphysema

Descriptor number: 9.19
Word count: 3723
At a Glance Commentary:
Scientific Knowledge on the Subject
Emphysema is a major pathological component of COPD, and there is an urgent need for new
intervention strategies against this disease. The potent immunomodulatory cytokine IL-6 is a
prominent biomarker for emphysema. However, IL-6 is highly pleiotropic, and targeting IL-6
has been problematic since both pathogenic and beneficial (homeostatic) actions are blocked
concurrently by conventional anti-IL-6 antibodies. It has thus proven difficult to exploit IL-6
as a therapeutic modality in human emphysema.
What this Study Adds to the Field
Our work addresses a new area of IL-6 biology that exploits understanding differential modes
of IL-6 signalling, and their applicability to promoting the molecular pathogenesis of
emphysema. Specifically, IL-6 utilizes two distinct signalling modes via the gp130 signal-
transducing receptor subunit to regulate many pathophysiological responses, namely classical
signalling through the membrane-bound IL-6 receptor (IL-6R) subunit, and trans-signalling
via a naturally-occurring soluble IL-6R. In a novel combinatorial approach comparing the
lungs of emphysematous patients and mouse models for spontaneous (gp130F/F) and
experimentally-induced (cigarette smoke exposure) emphysema, we have elucidated the
specific contribution of IL-6 trans-signalling to emphysema. The knowledge generated by our

findings is crucial for the future design and clinical application of novel therapeutics against
emphysema.
"This article has an online data supplement, which is accessible from this issue's table of
content online at www.atsjournals.org"

1
ABSTRACT
Rationale: The potent immunomodulatory cytokine interleukin (IL)-6 is consistently
upregulated in human lungs with emphysema, and in mouse emphysema models; however,
the mechanism(s) by which IL-6 promotes emphysema remains obscure. IL-6 signals using
two distinct modes, classical signalling via its membrane-bound IL-6 receptor (mIL-6R), and
trans-signalling via a naturally-occurring soluble IL-6R (sIL-6R).
Objectives: To identify whether IL-6 trans-signalling and/or classical signalling contribute to
the pathogenesis of emphysema.
Methods: We utilized the gp130F/F genetic mouse model for spontaneous emphysema, and
cigarette smoke-induced emphysema models. Emphysema in mice was quantified by various
methods including in vivo lung function and stereology, and TUNEL assay was employed to
assess alveolar cell apoptosis. In mouse and human lung tissues, the expression level and
location of IL-6 signalling-related genes and proteins were measured, and the levels of IL-6
and related proteins in sera from emphysematous mice and patients were also assessed.
Measurements and Main Results: Lung tissues from emphysema patients, as well as from
spontaneous and cigarette smoke-induced emphysema mouse models, were characterized by
excessive production of sIL-6R. Genetic blockade of IL-6 trans-signalling in emphysema
mouse models, and therapy with the IL-6 trans-signalling antagonist sgp130Fc, ameliorated
emphysema by suppressing augmented alveolar type II cell apoptosis. Furthermore, IL-6
trans-signalling-driven emphysematous changes in the lung correlated with mammalian target
of rapamycin complex 1 (mTORC1) hyper-activation, and treatment of emphysema mouse
models with the mTORC1 inhibitor rapamycin attenuated emphysematous changes.
Conclusions: Collectively, our data reveal that specific targeting of IL-6 trans-signalling may
represent a novel treatment strategy for emphysema.
Abstract word count: 250

2
Key words: pulmonary emphysema, interleukin-6, mammalian target of rapamycin complex
1, mouse models, trans-signalling

3
INTRODUCTION
Pulmonary emphysema is the major component of Chronic Obstructive Pulmonary Disease
(COPD), a condition predicted to be the third-leading cause of death worldwide by 2020 (1).
The primary cause of emphysema is cigarette smoke (CS), which triggers a deregulated
immune response in the lung that drives the destructive loss of alveolar cells and airspace
enlargement (2). Despite these observations, the molecular basis by which key modulators of
the immune system promote emphysema remains unclear. In this regard, increased expression
of the immunomodulatory cytokine interleukin(IL)-6 is a common feature of emphysema, and
increased IL-6 expression and IL6 gene polymorphisms correlate with rapid lung function
decline and increased disease risk in smokers (3-6). Furthermore, elevated IL-6 expression in
various mouse emphysema models has been linked to disease pathogenesis (7-11).
IL-6 utilizes two distinct signalling modes, both dependent upon the essential and
ubiquitously-expressed gp130 cytokine receptor signal-transducing β-subunit, to regulate
many pathophysiological responses. IL-6 classical signalling occurs via its membrane-bound
(m) IL-6 receptor (mIL-6R) (12), and IL-6 trans-signalling (IL-6TS) via a naturally-occurring
soluble (s) IL-6R (13) produced either by alternative splicing of IL6R mRNA or proteolytic
cleavage of mIL-6R, the latter involving metalloproteases ADAM10 and/or ADAM17 (14).
Notably, IL-6TS enables a large array of mIL-6R- cell types to respond to IL-6 (12), thus
increasing the overall IL-6 signalling spectrum. The predominant signalling pathway of either
mode is JAK/STAT3, with other pathways including PI3K/AKT, mTORC1, and ERK/MAPK
(15, 16).
The broad array of cell types responsive to IL-6TS underpins its role as the likely
pathogenic IL-6 signalling mode in various inflammatory conditions such as arthritis, asthma,
Crohn’s disease, and sepsis (17-22), as well as numerous cancers (23-25). However, despite
these observations, the causal involvement of specific IL-6 signalling modes in emphysema is

4
unknown. We have previously reported the spontaneous development of IL-6-driven alveolar
cell apoptosis leading to emphysema in gp130F/F mice (11), which have been engineered to
display IL-6/gp130-dependent hyper-activation of the JAK-mediated STAT and mTORC1
pathways, in the absence of SHP2-mediated ERK MAPK and PI3K/AKT signalling. Here, we
reveal in both gp130F/F and CS-induced emphysema models that the specific blockade of IL-
6TS with the antagonist sgp130Fc (26) suppresses alveolar type II (ATII) cell apoptosis and
disease pathogenesis. Furthermore, we uncover a novel role for mTORC1 hyper-activation in
facilitating IL-6TS-driven emphysematous changes in the lung. Notably, the IL-
6TS/mTORC1 axis was also augmented in patients with emphysema. Collectively, our
observations provide the rationale for the clinical evaluation of IL-6TS as a new target for the
treatment of emphysema.

5
METHODS
Mice and treatments
The gp130F/F and gp130F/F:Il6-/- mice have been previously reported (11), and
gp130F/F:sgp130Fctg/tg mice were generated using sgp130Fc transgenic mice (27). Gp130F/F
mice aged 3 months were intraperitoneally (i.p) administered with either sgp130Fc (once
weekly, low dose of 0.5mg/kg) (21), PBS (once weekly), rapamycin (3 times a week, 4mg/kg)
(36) or vehicle (0.8% DMSO, 5.2% PEG-400, 5.2% Tween-20, 3 times a week), over 12
weeks. For acute and chronic CS models (details are provided in the online data supplement),
mice were concurrently administered with sgp130Fc (0.5mg/kg) or PBS once over 4 days and
once weekly over 12 weeks, respectively. Rapamycin (4mg/kg) or vehicle was administered
either twice over 4 days (acute CS) or 3 times a week over 12 weeks (chronic CS). All mice
were housed under specific pathogen-free conditions. Experiments were approved by the
Monash University Animal Ethics Monash Medical Centre “A” Committee“.
Human lung biopsy and serum samples
Details are provided in an online data supplement.
RNA isolation and gene expression analysis
Details are provided in an online data supplement.
Protein extraction, ELISA and immunoblotting
Details are provided in an online data supplement.
Immunohistochemistry and immunofluorescence
Details are provided in an online data supplement.

6
Stereology and mean linear intercept (MLI) analyses
Mouse lungs were perfusion fixed by instillation of 1.5% glutaraldehyde/1.5% formaldehyde
in 0.04M phosphate-buffered saline, following which tissue blocks were prepared by
processing and embedding in glycol methacrylate, as previously described (11). Lung
stereology was performed on methylene blue-stained lung tissue sections using computer-
assisted newCAST software (version 2.14; Visiopharm, Hørsholm, Denmark (7, 11). Airspace
enlargement was quantified by the MLI technique on H&E-stained lung sections (7).
Lung function analyses
The assessment of lung function on anesthetized mice was performed using the flexiVent
system (SCIREQ, Montreal, Canada) (7, 11).
Statistical analyses
Statistical analyses were performed using GraphPad Prism for Windows version 6.0.
D'Agostino and Pearson omnibus K2 normality tests were performed for all data. Paired t-
tests were used to analyze normally distributed data among 2 sample groups, and Mann-
Whitney tests for non-normally distributed data or smaller data sets. One-way analysis of
variance (ANOVA) was used to assess differences between 3 or more groups for normally
distributed data, and Kruskal-Wallis tests for non-normally distributed data or smaller data
sets, along with appropriate post-tests. Linear regression was performed for correlations
between given markers in moderate emphysema patient groups only. Data are expressed as
the mean ± standard error of the mean (SEM), and P < 0.05 was considered statistically
significant.

7
RESULTS
Augmented expression of IL-6TS components in gp130F/F emphysematous mouse lungs
To delineate the mode of IL-6 signalling associated with IL-6-driven emphysema, we initially
assessed whether key IL-6TS components were deregulated in emphysematous lungs of 6
month old (mo) gp130F/F mice. While gene expression levels for Il6rα (11) and Adam10 were
normal, Il6 (11) and Adam17 were significantly increased in gp130F/F mouse lungs (Fig. 1A).
IL-6 protein levels were also significantly increased in the lungs and bronchoalveolar lavage
fluid (BALF) of gp130F/F compared to gp130+/+ control wild-type mice (Fig. 1B, Fig. E1A).
Since sgp130 protein levels were unchanged, the high sIL-6R levels in lungs and BALF of
gp130F/F mice (Fig. 1B, Fig. E1A) suggest augmented IL-6TS and its possible role in the
emphysema phenotype of gp130F/F mice.
Transgenic over-expression of the IL-6TS antagonist sgp130Fc in gp130F/F mice
prevents the onset of emphysema
To define a causative role for IL-6TS in the pathogenesis of emphysema in gp130F/F mice, we
crossed gp130F/F mice with sgp130Fc transgenic mice (27) to generate gp130F/F:sgp130Fctg/tg
mice over-expressing sgp130Fc, a fusion protein of recombinant sgp130 and the Fc region of
human IgG1 which acts as a potent specific inhibitor of IL-6TS-driven pathologies (21, 24).
Histological evaluation of 6mo gp130F/F:sgp130Fctg/tg mice revealed normal alveolar lung
architecture compared to enlargement of the distal air spaces and destruction of alveoli,
measured as alveolar mean linear intercept (MLI), that were observed for gp130F/F mice (Fig.
1C, Fig. E1B). Consistent with these histological observations, the elevated static compliance
and lung volume, due to destruction of elastic fibres in emphysematous lungs, that are a
feature of gp130F/F mice (11) were also normal in gp130F/F:sgp130Fctg/tg mice (Fig. 1D, E). In
further support of these findings, stereology indicated that the volume fractions of air space

8
and alveolar septal tissue, and the surface density within lung parenchyma, were similar
between gp130+/+, gp130+/+:sgp130Fctg/tg and gp130F/F:sgp130Fctg/tg mice, while these
parameters were significantly elevated in gp130F/F mice (Table E1). The serum and lung
expression levels of the acute phase response (APR) protein serum amyloid A, which is
regulated by IL-6 classical signalling, remained elevated and unchanged in
gp130F/F:sgp130Fctg/tg compared to gp130F/F mice (Fig. E1C), thus indicating that IL-6
classical signalling was unaffected in gp130F/F:sgp130Fctg/tg mice. Furthermore, the absence
of emphysema in gp130F/F:sgp130Fctg/tg mice was associated with a significant reduction in
the lung expression levels of IL-6 and sIL-6R back to those observed in
gp130+/+:sgp130Fctg/tg and gp130+/+ mice (Fig. E1D, E). Collectively, these data reveal that
IL-6TS drives the development of emphysema in gp130F/F mice.
IL-6TS promotes apoptosis-driven emphysema in gp130F/F mice
Since apoptosis is a key process associated with the development of human emphysema and
several disease models, including IL-6/gp130-driven emphysema in gp130F/F mice (11, 28,
29), we compared the extent of apoptotic TUNEL-stained cells in the lungs of 6mo mice. The
number of TUNEL-stained alveolar cells in gp130F/F:sgp130Fctg/tg mice was significantly
reduced compared to gp130F/F mice, and was comparable to gp130+/+ and
gp130+/+:sgp130Fctg/tg control mice (Fig. 2A) (7, 11). The alveolar epithelium covers >99%
of the internal surface area of the lung and is mainly composed of ATI and ATII cells, the
latter of which uniquely synthesize and secrete surfactants, including the biomarker surfactant
protein C (SPC) which prevents small airway closure and alveolar collapse, thereby
maintaining normal lung structure (30, 31). Notably, the number of SPC-positive cells in the
lungs of gp130F/F mice was significantly reduced compared to gp130+/+ mice, whereas SPC-
positive cell numbers remained elevated in the lungs of gp130F/F:sgp130Fctg/tg mice (Fig. 2B).

9
Dual immunofluorescence staining with cleaved caspase-3 (marker of apoptosis) and SPC
further confirmed that there was a significant increase in the number of apoptotic SPC-
positive ATII cells in gp130F/F mice compared to gp130+/+, gp130+/+:sgp130Fctg/tg and
gp130F/F:sgp130Fctg/tg mice (Fig. 2C), while augmented apoptosis was not detected for ATI
(podoplanin-positive) and endothelial (CD31-positive) cells (Fig. E2). Collectively, these data
suggest that IL-6TS promotes type II cells to undergo apoptosis during emphysema.
Therapeutic blockade of IL-6TS with sgp130Fc prevents alveolar cell death and
emphysema in gp130F/F mice
To determine whether IL-6TS can serve as a bona fide therapeutic target for emphysema, we
next explored whether suppression of IL-6TS upon administration with sgp130Fc would
prevent emphysema development in gp130F/F mice from 3 months of age onwards. At the
completion of 12 weeks treatment with sgp130Fc, the lungs of gp130F/F mice were
comparable to those of untreated gp130+/+ mice, with fully-preserved lung morphology (Fig.
3A), static compliance (Fig. 3B), lung volume (Fig. 3C), stereological parameters (Table E1)
and the number of apoptotic cells (Fig. 3D). Taken together these data demonstrate that
therapeutic targeting of IL-6TS can prevent emphysema development in gp130F/F mice.
Acute CS exposure of wild-type mice augments expression of IL-6TS components which
is associated with elevated alveolar cell apoptosis
Since CS is the major trigger of emphysema in humans, we next assessed whether our
mechanistic findings for spontaneous IL-6TS-driven emphysema are relevant to CS-induced
lung disease. Upon an initial exposure of 4-6 week old gp130+/+ mice to an acute (4 day) CS
model, mRNA levels for Il6 (11) and Adam17 were significantly increased, while those for
Il6rα and Adam10 remained normal, compared to non-CS-exposed control mice (Fig. 4A).

10
Acute CS exposure also significantly augmented IL-6 and sIL-6R, but not sgp130, protein
levels (Fig. 4B). By contrast, the lungs of gp130+/+:sgp130Fctg/tg mice exposed to CS showed
no changes in Il6 and Adam17 mRNA levels (Fig. E3A), nor sIL-6R and sgp130 levels (IL-6
levels remained undetectable) (Fig. E3B). The response of gp130+/+ mouse lungs to CS
exposure was confirmed by CS-induced increases in whole lung mRNA levels of
inflammatory mediators Cxcl2, Tnfα and Ccl2 (32, 33), which were ameliorated in either CS-
exposed gp130+/+:sgp130Fctg/tg mice or CS-exposed gp130+/+ mice concurrently administered
once with sgp130Fc (Fig. E3C). Moreover, while acute CS exposure of gp130+/+ mice also
augmented numbers of TUNEL-positive apoptotic alveolar cells (Fig. 4C) (11), no such
increases were observed in acute CS-exposed gp130+/+:sgp130Fctg/tg mice and CS-exposed
gp130+/+ mice treated with sgp130Fc (Fig. 4C).
Inhibition of IL-6TS with sgp130Fc suppresses alveolar cell apoptosis and
emphysematous changes induced by chronic CS exposure of wild-type mice
To determine the relevance of IL-6TS to CS-induced emphysema development, we next
utilized a chronic (12 week) CS-induced emphysema model. Over 12 weeks, we observed
significant increases in numerous parameters indicative of emphysema, namely static
compliance, lung volume and MLI, in CS-exposed gp130+/+ mice compared to their age-
matched non-CS-exposed counterparts (Fig. 4D-F). By contrast, chronic CS-exposed mice
displayed significant reductions in all of these emphysematous parameters upon inhibition of
IL-6TS with sgp130Fc (Fig. 4D-F). Importantly, these findings correlated with our data
demonstrating that chronic CS exposure increased TUNEL-positive cell numbers in the lungs
of gp130+/+ mice compared to non-CS-exposed mice, and this was significantly reduced
(~40%) in response to sgp130Fc treatment (Fig. 4G).

11
Collectively, these data in CS-exposed wild-type mice reveal that therapeutic and
genetic blockade of IL-6TS suppresses augmented expression of inflammatory mediators and
alveolar cell death, leading to amelioration of emphysema, therefore validating the relevance
of IL-6TS to the pathogenesis of CS-induced emphysema.
IL-6TS in the lungs of emphysematous gp130F/F mice is associated with hyper-activation
of the mTORC1 pathway
Our data here indicating that suppression of IL-6TS in mouse emphysema models prevents
emphysema implies that IL-6TS-driven emphysematous changes in the lung are a
consequence of increased activation of a gp130-dependent signalling pathway. We have
previously eliminated a role for IL-6-driven hyper-activation of STAT3 and STAT1 in
promoting emphysema in gp130F/F mice (7). In addition, such a pathway is unlikely to be
either SHP2-mediated ERK MAPK or PI3K/AKT, since gp130/SHP2-mediated activation of
these pathways has been abolished in gp130F/F mice (34). Another candidate pathway is
rapamycin-sensitive mTORC1, which is a large multi-protein complex comprising the
serine/threonine kinase mTOR subunit that is activated in response to diverse stimuli, such as
cytokines (35). Hyper-activation of mTORC1 is observed in many diseases, including
pulmonary fibrosis, which highlights its ability to regulate various cellular processes such as
proliferation and apoptosis (35, 36).
We observed a significant increase in the number of cells expressing phosphorylated
ribosomal S6 kinase (P-rpS6), a key mTORC1 substrate (35), in the lungs of gp130F/F mice
compared to emphysema-free gp130+/+ and gp130F/F:Il6-/- mice, the latter deficient in IL-6 (11)
(Fig. 5A). Dual immunofluorescence staining confirmed that P-rpS6 expression co-localized
to SPC-positive ATII cells (Fig. E4A), but not CD45-positive inflammatory cells (Fig. E4B).
Immunoblot analyses also indicated that P-rpS6 protein levels, as well as P-mTOR (ser2448)

12
levels, were significantly reduced in gp130F/F:Il6-/- compared to gp130F/F lungs (Fig. 5B, Fig.
E4C). Furthermore, we demonstrated activation of the IL-6TS/mTORC1 axis in cultured
gp130F/F primary ATII cells stimulated with the potent IL-6TS agonist Hyper-IL-6 (12) (Fig.
E5). Moreover, both the sgp130Fc-mediated genetic and therapeutic blockade of IL-6TS in
gp130F/F mice significantly reduced P-rpS6-positive cell numbers in the lung compared to
their respective controls (Fig. 5C, D, Fig. E6A, B). Therefore, these data support the notion
that IL-6TS augments activation of the mTORC1 pathway in the lungs of emphysematous
gp130F/F mice.
The therapeutic blockade of the mTORC1 pathway in rapamycin-treated gp130F/F mice
suppresses the development of emphysema
To specifically delineate a role for increased mTORC1 activity in IL-6TS-driven emphysema,
we explored whether mTORC1 pathway blockade with the specific mTORC1 inhibitor,
rapamycin, could alleviate emphysema development in gp130F/F and CS-induced emphysema
models. Examination of lung morphology, static compliance, lung volumes and various
stereological parameters revealed that emphysematous changes (Fig. 5E-G, Table E1), and
also TUNEL- and P-rpS6-positive cell numbers (Fig. 5H, I, Fig. E6C, D), were significantly
reduced in the lungs of gp130F/F mice following 12 weeks of rapamycin treatment compared
to vehicle-treated gp130F/F control mice.
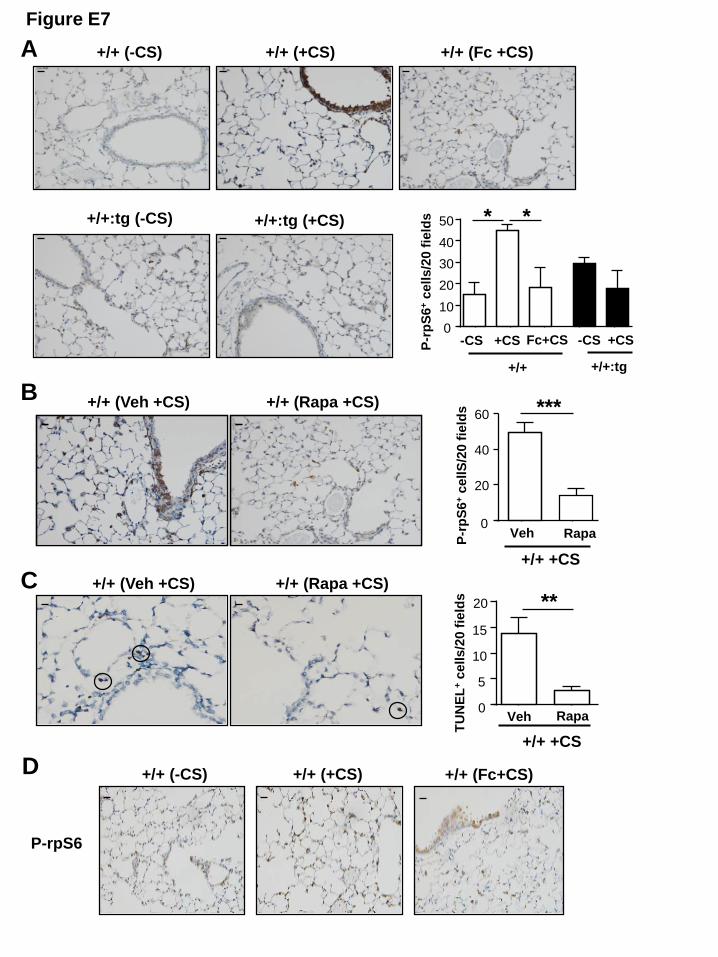
We also observed increased numbers of P-rpS6-positive cells in the lungs of acute CS-
exposed gp130+/+ mice compared to their non-CS-exposed controls (Fig. E7A). However, P-
rpS6-positive cell numbers were significantly reduced in CS-exposed gp130+/+ mice
concurrently treated with sgp130Fc compared to CS-exposed gp130+/+ mice treated with
vehicle (Fig. E7A). Furthermore, the genetic blockade of IL-6TS in gp130+/+:sgp130Fctg/tg
mice prevented any increase in P-rpS6-positive cell numbers in response to CS exposure (Fig.

13
E7A). The numbers of P-rpS6- and TUNEL-positive cells were significantly reduced in acute
CS-exposed gp130+/+ mice administered with rapamycin compared to their CS-exposed
controls that received vehicle (Fig. E7B, C). Moreover, chronic CS exposure increased P-
rpS6-positive cell numbers in the lungs of gp130+/+ mice compared to their non-CS-exposed
counterparts, and this was significantly reduced by ~45% in response to sgp130Fc treatment
(Fig. 5J, Fig. E7D). Collectively, these data strongly suggest that IL-6TS promotes mTORC1
pathway hyper-activation during emphysema.
Upregulation of IL-6TS/mTORC1 components correlates with human emphysema
To translate our emphysematous mouse model findings to human disease, we first assessed
the expression of IL-6TS-related genes in human lung tissues from disease-free and
emphysema patients as defined by pulmonary lung function tests (gas exchange, FEV1; Table
E2). The gene expression level for IL6, as well as both IL-6 and sIL-6R protein levels, were
significantly increased in lungs of emphysema patients compared to emphysema-free controls
(Fig. 6A, B). In addition, ADAM17 gene expression was elevated 2-fold in emphysema
patients compared to disease-free individuals (Fig. 6A), and ADAM17 protein levels
significantly correlated with elevated sIL-6R levels observed in emphysema patients (Fig. 6C,
D). Furthermore, we observed a significant positive correlation between sIL-6R levels and the
number of cleaved caspase-3-positive cells in the lungs of emphysema patients (Fig. 6E, F),
thus further supporting IL-6TS-induced apoptosis in human emphysema.
We next investigated the activation status of the mTORC1 pathway, and immunoblot
analyses indicated that P-rpS6 protein levels were significantly increased in lung lysates from
emphysema patients (Fig. 6G). Furthermore, regression analyses of IL-6 and P-rpS6 levels
showed significant positive correlations with sIL-6R in emphysema patients (Fig. 6H).
Notably, circulating levels of IL-6TS components, namely IL-6 and sIL-6R, were also up-

14
regulated in the serum of emphysema patients compared to healthy individuals (Fig. 6I).
Overall these clinical data are consistent with our in vivo findings and provide translational
support for a pathological role for the IL-6TS/mTORC1 axis in human emphysema.

15
DISCUSSION
Despite the established link between IL-6 and the pathogenesis of emphysema and other lung
diseases (1-5, 11), little information is known regarding the identity of the mode of IL-6
signalling and downstream pathways which drive disease pathogenesis. For instance, mouse
asthma models and clinical data indicate increased sIL-6R levels in BALF and airway smooth
muscle which may be of relevance to airway remodelling (22, 37). In addition, blocking
global IL-6 signalling via neutralizing antibodies or genetic ablation of IL-6 during chronic
stages of adenosine-mediated lung injury and asthma provide the benefit of halting airway
remodelling processes such as fibrosis. However, the above studies only hypothesize
involvement of IL-6TS rather than IL-6 classical signalling in driving lung disease (38, 39).
Here, we define for the first time that hyper-activation of the endogenous IL-6TS/mTORC1
axis in the lung augments ATII cell apoptosis during emphysema. Furthermore, our
discoveries of augmented expression and activation of IL-6TS and mTORC1 pathway
components, respectively, in the lung of patients with emphysema, together with elevated
levels of IL-6TS components (e.g. sIL-6R) in the serum of emphysema patients, have
considerable translational potential for biomarker discovery and early disease detection, as
well as patient stratification for potential responders who may gain benefit from selective
therapies against the IL-6TS/mTORC1 axis.
While our data reveal that mTORC1 hyper-activation is associated with elevated
alveolar cell apoptosis, mTORC1 is also a negative regulator of autophagy, a process which
has been implicated in emphysematous lung destruction. However, we have previously
reported that IL-6-driven emphysematous changes in the lung (which are associated with
elevated mTORC1 activation) are independent of autophagy, and for that matter inflammation
(7). Furthermore, considering that mTORC1 is activated by a vast range of stimuli (e.g.
cytokines, growth factors, stress, oxygen, nutrients) and regulates a plethora of cellular

16
responses in the lung in both a cell-type and stimulus-dependent manner (35, 40), our current
findings suggest that mTORC1 activation in the alveolar epithelium specifically by IL-6
(unlike other stimuli) does not affect autophagy and/or inflammation.
Hyper-activation of mTORC1 can promote IL-6TS by upregulating sIL-6R levels in
vivo (41), which not only supports mTORC1 being an integral component of the IL-6TS
network (at least in the lung), but also suggests a feed-forward regulatory loop involving IL-
6TS and mTORC1. A role for mTORC1 in promoting emphysema is also supported by the
demonstration that peripheral blood mononuclear cells from COPD patients and CS extract-
stimulated human monocytes display increased mTOR activity, which is associated with
corticosteroid resistance in COPD patients (42). However, in contrast to our current findings,
expression of the CS-induced stress response protein, RTP801, can increase septal cell death
leading to pulmonary injury and emphysema via inhibition of mTORC1 and NF-κB activation
(43). Since the upstream activators of mTORC1 in the above study were not documented,
they may differ from IL-6 which could cause differences in the kinetics of mTORC1
activation and subsequently the net signal output and cellular response within the lung. In this
regard, mTORC1 can exhibit pro- and anti-apoptotic activities (35), and although the ability
of hyper-activated mTORC1 to promote apoptosis has been linked to a negative feedback
loop which suppresses the AKT survival pathway (35, 44), AKT activation in the lungs of
gp130F/F mice is unaltered (data not shown). Collectively, these observations not only
highlight the importance of identifying the molecular drivers and cellular context of mTORC1
activity during disease pathogenesis prior to the potential application of therapeutics, but also
warrant investigations into the existence of alternate signalling pathways (e.g. ERK MAPK)
and target genes (e.g. p53, Bcl-2, Nox4, Bad) which can promote the pro-apoptotic actions of
mTORC1 (45) during emphysema.

17
Current strategies aimed at targeting IL-6 in the clinic have largely resulted from earlier
experimentally-induced mouse models for arthritis and inflammatory bowel diseases,
whereby antibody–mediated neutralization of IL-6R or IL-6 suppressed disease development
(46). In this respect, the humanized neutralizing IL-6R monoclonal antibody, tocilizumab, has
shown considerable promise in clinical trials in the management of rheumatoid arthritis (47).
However, tocilizumab inhibits the mIL-6R mediated pathway, which most likely accounts for
the increased incidence of infection associated with its use due to suppressing the “immune-
protective” IL-6 classical signalling pathway. Indeed, studies using IL-6 knock-out mice have
suggested that IL-6 (classical) signalling protects against mucosal ulcerations from bacterial
pathogens, such as Citrobacter rodentium infection (48). Such studies highlight how, under
certain situations, blocking IL-6 (classical) signalling may have detrimental consequences,
and also imply that other modes of IL-6 signalling promote chronic inflammatory responses
associated with disease pathogenesis.
The specific IL-6TS antagonist sgp130Fc used in our current study can exhibit
significant therapeutic activity in IL-6TS-driven animal models of inflammation,
autoimmunity and cancer, with very low immunogenicity and ~7 day in vivo half-life, as well
as no interference with IL-6 classical signalling events (18, 21, 49). Notably, blocking IL-6TS
with sgp130Fc can inhibit sIL-6R production by macrophages contributing to attenuation of
idiopathic pulmonary fibrosis (50), which is consistent with our findings that sgp130Fc
suppressed augmented levels of sIL-6R in the lungs of gp130F/F and CS-induced emphysema
models. Moreover, the efficacy of sgp130Fc, which will undergo future phase II clinical trials
in Europe for Crohn’s Disease, at suppressing emphysematous changes in the lungs of both
models provides translational implications directly to preventatives for human emphysema.
From a therapeutic perspective the selective blockade of IL-6TS, and lung-directed mTORC1
suppression, offer promising new strategies to ameliorate emphysema (and COPD), and our

18
current findings will fast-track further pre-clinical testing of sgp130Fc and the development of
humanized anti-IL-6R mAbs which target IL-6TS for clinical use in emphysema.

19
REFERENCES
1. Barnes PJ, Shapiro SD, Pauwels RA. Chronic obstructive pulmonary disease: molecular
and cellular mechanisms. Eur Respir J 2003; 22: 672-688.
2. Lokke A, Lange P, Scharling H, Fabricius P, Vestbo J. Developing COPD: a 25 year follow
up study of the general population. Thorax 2006; 61: 935-939.
3. Song W, Zhao J, Li Z. Interleukin-6 in bronchoalveolar lavage fluid from patients with
COPD. Chin Med J (Engl) 2001; 114: 1140-1142.
4. Bozinovski S, Hutchinson A, Thompson M, Macgregor L, Black J, Giannakis E, Karlsson
AS, Silvestrini R, Smallwood D, Vlahos R, Irving LB, Anderson GP. Serum amyloid
a is a biomarker of acute exacerbations of chronic obstructive pulmonary disease. Am
J Respir Crit Care Med 2008; 177: 269-278.
5. He JQ, Foreman MG, Shumansky K, Zhang X, Akhabir L, Sin DD, Man SF, DeMeo DL,
Litonjua AA, Silverman EK, Connett JE, Anthonisen NR, Wise RA, Pare PD,
Sandford AJ. Associations of IL6 polymorphisms with lung function decline and
COPD. Thorax 2009; 64: 698-704.
6. Donaldson GC, Seemungal TA, Patel IS, Bhowmik A, Wilkinson TM, Hurst JR,
Maccallum PK, Wedzicha JA. Airway and systemic inflammation and decline in lung
function in patients with COPD. Chest 2005; 128: 1995-2004.
7. Ruwanpura SM, McLeod L, Miller A, Jones J, Vlahos R, Ramm G, Longano A, Bardin PG,
Bozinovski S, Anderson GP, Jenkins BJ. Deregulated Stat3 signaling dissociates
pulmonary inflammation from emphysema in gp130 mutant mice. Am J Physiol Lung
Cell Mol Physiol 2012; 302: L627-639.
8. Kuhn C, Homer RJ, Zhu Z, Ward N, Flavell RA, Geba GP, Elias JA. Airway
hyperresponsiveness and airway obstruction in transgenic mice. Morphologic

20
correlates in mice overexpressing interleukin (IL)-11 and IL-6 in the lung. Am J
Respir Cell Mol Biol 2000; 22: 289-295.
9. Pauwels NS, Bracke K, Maes T, Pilette C, Joos GF, Brusselle, GG. The role of
interleukin-6 in pulmonary and systemic manifestations in a murine model of chronic
obstructive pulmonary disease. Exp Lung Res 2010; 36: 469-483.
10. Tasaka S, Inoue K, Miyamoto K, Nakano Y, Kamata H, Shinoda H, Hasegawa N,
Miyasho T, Satoh M, Takano H, Ishizaka A. Role of interleukin-6 in elastase-induced
lung inflammatory changes in mice. Exp Lung Res 2010; 36: 362-372.
11. Ruwanpura SM, McLeod L, Miller A, Jones J, Bozinovski S, Vlahos R, Ernst M, Armes J,
Bardin PG, Anderson GP, Jenkins BJ. Interleukin-6 promotes pulmonary emphysema
associated with apoptosis in mice. Am J Respir Cell Mol Biol 2011; 45: 720-730.
12. Peters M, Muller AM, Rose-John S. Interleukin-6 and soluble interleukin-6 receptor:
direct stimulation of gp130 and hematopoiesis. Blood 1998; 92: 3495-3504.
13. Scheller J, Rose-John S. Interleukin-6 and its receptor: from bench to bedside. Med Micro
and immuno; 2006; 195: 173-183.
14. Garbers C Janner N, Chalaris A, Moss ML, Floss DM, Meyer D, Koch-Nolte F, Rose-
John S, Scheller J. Species specificity of ADAM10 and ADAM17 proteins in
interleukin-6 (IL-6) trans-signaling and novel role of ADAM10 in inducible IL-6
receptor shedding. J Biol Chem 2011; 286: 14804-14811.
15. Heinrich P, Behrmann, I, Haan,S, Hermanns, HM, Muller-Newen, G, Schaper, F.
Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J
2003; 374: 1-20.
16. Thiem S, Pierce T, Palmieri M, Putoczki TL, Buchert M, Preaudet A, Farid RO, Love C,
Catimel B, Lei Z, Rozen S, Gopalakrishnan V, Schaper F, Hallek M, Boussioutas A,

21
Tan P, Jarnicki A, Ernst M. mTORC1 inhibition restricts inflammation-associated
gastrointestinal tumorigenesis in mice. J Clin Invest 2013; 123: 767-781.
17. Kallen KJ. The role of transsignalling via the agonistic soluble IL-6 receptor in human
diseases. Biochim Biophys Acta 2002; 1592: 323-343.
18. Nowell MA, Richards PJ, Horiuchi S, Yamamoto N, Rose-John S, Topley N, Williams
AS, Jones SA. Soluble IL-6 receptor governs IL-6 activity in experimental arthritis:
blockade of arthritis severity by soluble glycoprotein 130. J Immunol 2003; 171:
3202-3209.
19. Greenhill CJ, Rose-John S, Lissilaa R, Ferlin W, Ernst M, Hertzog PJ, Mansell A, Jenkins
BJ IL-6 trans-signaling modulates TLR4-dependent inflammatory responses via
STAT3. J Immunol 2011; 186: 1199-1208.
20. Schuett H, Oestreich R, Waetzig GH, Annema W, Luchtefeld M, Hillmer A. et al. .
Transsignaling of interleukin-6 crucially contributes to atherosclerosis in mice.
Arterioscler Thromb Vasc Biol 2012; 32: 281-290
21. Nowell MA, Williams AS, Carty SA, Scheller J, Hayes AJ, Jones GW, Richards PJ, Slinn
S, Ernst M, Jenkins BJ, Topley N, Rose-John S, Jones SA. Therapeutic targeting of
IL-6 trans signaling counteracts STAT3 control of experimental inflammatory arthritis.
J Immunol 2009; 182: 613-622.
22. Doganci A, Eigenbrod T, Krug N, De Sanctis GT, Hausding M, Erpenbeck VJ, Haddad el
B, Lehr HA, Schmitt E, Bopp T, Kallen KJ, Herz U, Schmitt S, Luft C, Hecht O,
Hohlfeld JM, Ito H, Nishimoto N, Yoshizaki K, Kishimoto T, Rose-John S, Renz H,
Neurath MF, Galle PR, Finotto S. The IL-6R alpha chain controls lung CD4+CD25+
Treg development and function during allergic airway inflammation in vivo. J Clin
Invest 2005; 115: 313-325.

22
23. Lesina M, Kurkowski M, Ludes K, Rose-John S, Treiber M, Klöppel G, Yoshimura A,
Reindl W, Sipos B, Akira S, Schmid RM, Algül H. Stat3/Socs3 activation by IL-6
transsignaling promotes progression of pancreatic intraepithelial neoplasia and
development of pancreatic cancer. Cancer Cell 2011; 19: 456-469.
24. Becker C, Fantini MC, Schramm C, Lehr HA, Wirtz S, Nikolaev A, Burg J, Strand S,
Kiesslich R, Huber S, Ito H, Nishimoto N, Yoshizaki K, Kishimoto T, Galle PR,
Blessing M, Rose-John S, Neurath MF. TGF-beta suppresses tumor progression in
colon cancer by inhibition of IL-6 trans-signaling. Immunity 2004; 21: 491-501.
25. Lo CW, Chen MW, Hsiao M, Wang S, Chen CA, Hsiao SM, Chang JS, Lai TC, Rose-
John S, Kuo ML, Wei LH. IL-6 trans-signaling in formation and progression of
malignant ascites in ovarian cancer. Cancer Res 2011; 71: 424-434.
26. Jostock T, Mullberg J, Ozbek S, Atreya R, Blinn G, Voltz N, Fischer M, Neurath MF,
Rose-John S. Soluble gp130 is the natural inhibitor of soluble interleukin-6 receptor
transsignaling responses. Eur J Biochem 2001; 268: 160-167.
27. Rabe B, Chalaris A, May U, Waetzig GH, Seegert D, Williams AS, Jones SA, Rose-John
S, Scheller J. Transgenic blockade of interleukin 6 transsignaling abrogates
inflammation. Blood 2008; 111: 1021-1028.
28. Aoshiba K, Yokohori N, Nagai A. Alveolar wall apoptosis causes lung destruction and
emphysematous changes. Am J Respir Cell Mol Biol 2003; 28: 555-562.
29. Demedts IK, Demoor T, Bracke KR, Joos GF, Brusselle GG. Role of apoptosis in the
pathogenesis of COPD and pulmonary emphysema. Respir Res 2006; 7: 53.
30. Zhao YX, Andoh A, Shimada M, Takaya H, Hata K, Fujiyama Y, Bamda T. Secretion of
complement components of the alternative pathway (C3 and factor B) by the human
alveolar type II epithelial cell line A549. Inter J Mol Med 2000; 5: 415-419.

23
31. Strunk RC, Eidlen DM, Mason RJ. Pulmonary alveolar type II epithelial cells synthesize
and secrete proteins of the classical and alternative complement pathways. J Clin
Invest 1988; 81: 1419-1426.
32. Thatcher TH, McHugh NA, Egan RW, Chapman RW, Hey JA, Turner CK, Redonnet MR,
Seweryniak KE, Sime PJ, Phipps RP. Role of CXCR2 in cigarette smoke-induced
lung inflammation. Am J Physiol Lung Cell Mol Physiol 2005; 289: 322-328.
33. Vlahos R, Bozinovski S, Jones JE, Powell J, Gras J, Lilja A, Hansen MJ, Gualano RC,
Irving L, Anderson GP. Differential protease, innate immunity, and NF-kappaB
induction profiles during lung inflammation induced by subchronic cigarette smoke
exposure in mice. Am J Physiol Lung Cell Mol Physiol 2006; 290: L931-945.
34. Tebbutt NC, Giraud AS, Inglese M, Jenkins B, Waring P, Clay FJ, Malki S, Alderman
BM, Grail D, Hollande F, Heath JK, Ernst M. Reciprocal regulation of gastrointestinal
homeostasis by SHP2 and STAT-mediated trefoil gene activation in gp130 mutant
mice. Nat Med 2002; 8: 1089-1097.
35. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012; 149:
274-293.
36. Korfhagen TR, Le Cras TD, Davidson CR, Schmidt SM, Ikegami M, Whitsett JA, Hardie
WD. Rapamycin prevents transforming growth factor-alpha-induced pulmonary
fibrosis. Am J Respir Cell Mol Biol 2009; 41: 562-572.
37. Ammit AJ, Moir LM, Oliver BG, Hughes JM, Alkhouri H, Ge Q, Burgess JK, Black JL,
Roth M. Effect of IL-6 trans-signaling on the pro-remodeling phenotype of airway
smooth muscle. A J Physio Lung Cell Mol Physio 2007; 292: L199-206.
38. Pedroza M, Schneider DJ, Karmouty-Quintana H, Coote J, Shaw S, Corrigan R, Molina
JG, Alcorn JL, Galas D, Gelinas R, Blackburn MR. Interleukin-6 contributes to

24
inflammation and remodeling in a model of adenosine mediated lung injury. PloS One
2011; 6: e22667.
39. Finotto S, Eigenbrod T, Karwot R, Boross I, Doganci A, Ito H, Nishimoto N, Yoshizaki K,
Kishimoto T, Rose-John S, Galle PR, Neurath MF. Local blockade of IL-6R signaling
induces lung CD4+ T cell apoptosis in a murine model of asthma via regulatory T
cells. Inter Immuno 2007; 19: 685-693.
40. Hu Y, Liu J, Wu YF, Lou J, Mao YY, Shen HH, Chen ZH. mTOR and autophagy in
regulation of acute lung injury: a review and perspective. Microbes Infect
2014;16:727-34
41. Garbers C, Kuck F, Aparicio-Siegmund S, Konzak K, Kessenbrock M, Sommerfeld A,
Haussinger D, Lang PA, Brenner D, Mak TW, Rose-John S, Essmann F, Schulze-
Osthoff K, Piekorz RP, Scheller J. Cellular senescence or EGFR signaling induces
Interleukin 6 (IL-6) receptor expression controlled by mammalian target of rapamycin
(mTOR). Cell cycle 2013; 12: 3421-3432.
42. Mitani A, Ito K, Vuppusetty C, Barnes PJ, Mercado N. Restoration of Corticosteroid
Sensitivity in Chronic Obstructive Pulmonary Disease by Inhibition of Mammalian
Target of Rapamycin. Am J Respir Crit Care Med 2016 193: 143-153.
43. Yoshida T, Mett I, Bhunia AK, Bowman J, Perez M, Zhang L, Gandjeva A, Zhen L,
Chukwueke U, Mao T, Richter A, Brown E, Ashush H, Notkin N, Gelfand A,
Thimmulappa RK, Rangasamy T, Sussan T, Cosgrove G, Mouded M, Shapiro SD,
Petrache I, Biswal S, Feinstein E, Tuder RM. Rtp801, a suppressor of mTOR signaling,
is an essential mediator of cigarette smoke-induced pulmonary injury and emphysema.
Nat Med 2010; 16: 767-773.

25
44. Kato H, Nakajima S, Saito Y, Takahashi S, Katoh R, Kitamura M. mTORC1 serves ER
stress-triggered apoptosis via selective activation of the IRE1-JNK pathway. Cell
Death and Diff 2012; 19: 310-320.
45. Das R, Xu S, Nguyen TT, Quan X, Choi SK, Kim SJ, Lee EY, Cha SK, Park KS.
Transforming growth factor β1-induced apoptosis in podocytes via the extracellular
signal-regulated kinase-mammalian target of rapamycin complex 1-NADPH oxidase 4
axis. J Biol Chem 2015; 290: 30830-30842.
46. Atreya R, Mudter J, Finotto S, Mullberg J, Jostock T, Wirtz S, Schutz M, Bartsch B,
Holtmann M, Becker C, Strand D, Czaja J, Schlaak JF, Lehr HA, Autschbach F,
Schurmann G, Nishimoto N, Yoshizaki K, Ito H, Kishimoto T, Galle PR, Rose-John S,
Neurath MF. Blockade of interleukin 6 trans signaling suppresses T-cell resistance
against apoptosis in chronic intestinal inflammation: evidence in crohn disease and
experimental colitis in vivo. Nat Med 2000; 6: 583-588.
47. Choy EH, Isenberg DA, Garrood T, Farrow S, Ioannou Y, Bird H, Cheung N, Williams B,
Hazleman B, Price R, Yoshizaki K, Nishimoto N, Kishimoto T, Panayi GS.
Therapeutic benefit of blocking interleukin-6 activity with an anti-interleukin-6
receptor monoclonal antibody in rheumatoid arthritis: a randomized, double-blind,
placebo-controlled, dose-escalation trial. Arthritis Rheum 2002; 46: 3143-3150.
48. Dann SM, Spehlmann ME, Hammond DC, Iimura M, Hase K, Choi LJ, Hanson E,
Eckmann L. IL-6-dependent mucosal protection prevents establishment of a microbial
niche for attaching/effacing lesion-forming enteric bacterial pathogens. J Immunol
2008; 180: 6816-6826.
49. Rose-John S, Waetzig GH, Scheller J, Grotzinger J, Seegert D. The IL-6/sIL-6R complex
as a novel target for therapeutic approaches. Expert Opin Ther Targets 2007; 11: 613-
624.

26
50. Le TT, Karmouty-Quintana H, Melicoff E, Le TT, Weng T, Chen NY, Pedroza M, Zhou
Y, Davies J, Philip K, Molina J, Luo F, George AT, Garcia-Morales LJ, Bunge RR,
Bruckner BA, Loebe M, Seethamraju H, Agarwal SK, Blackburn MR. Blockade of
IL-6 trans signaling attenuates pulmonary fibrosis. J Immunol 2014; 193: 3755-3768.

27
FIGURE LEGENDS
Figure 1 Augmented expression of IL-6TS components promotes emphysema in
gp130F/F mice. Q-PCR analyses of (A) Adam17 and Adam10 gene expression were performed
on cDNA derived from total RNA prepared from lung tissue of 6 month old (mo) gp130+/+
(+/+), gp130+/+:sgp130Fctg/tg (+/+:tg), gp130F/F (F/F) and gp130F/F:sgp130Fctg/tg (F/F:tg) mice.
Expression data are shown following normalization for 18S expression, and are presented as
the mean ± SEM from triplicate analyses from n = 5 mice per genotype. (B) IL-6, sIL-6R and
sgp130 protein concentrations in lung lysates as determined by ELISA. n = 5 mice per
genotype. (C) Representative methylene blue-stained cross-sections of lungs from 6mo +/+,
+/+:tg, F/F and F/F:tg mice. Arrows indicate airspace enlargement. Scale bars = 100μm. (D)
Static compliance as determined by flexiVent lung function analysis, and (E) lung volume per
body total weight (grams), of the above 6mo mice. Data are expressed as mean ± SEM. n =
10 mice per genotype. *P< 0.05, **P< 0.01, ***P< 0.001 and ****P< 0.0001.
Figure 2 IL-6TS-mediated apoptosis of ATII cells leads to the development of
emphysema. Stereological quantification of the number of (A) TUNEL- and (B) SPC-
positive ATII cells, and corresponding representative photomicrographs showing positive
cells in cross-sections of lungs in 6 month old (mo) gp130+/+ (+/+), gp130+/+:sgp130Fctg/tg
(+/+:tg), gp130F/F (F/F) and gp130F/F:sgp130Fctg/tg (F/F:tg) mice. Scale bars = 25μm. (C)
Representative confocal immunofluorescence photomicrographs of cells positive for either
caspase-3 alone (green), SPC alone (red), or both caspase-3 and SPC (yellow, white circles)
in cross-sections of the lung in 6mo F/F mice. The graph depicts stereological quantification
of dual labelled caspase-3- and SPC-positive cells in the lungs of 6mo mice of the indicated
genotypes. Scale bars = 10μm. Data are expressed as the mean ± SEM. n = 5 mice per
genotype. *P< 0.05 and **P< 0.01.

28
Figure 3 Therapeutic blockade of IL-6TS prevents emphysema development in
gp130F/F mice. (A) Representative methylene blue-stained cross-sections (arrows indicate
airspace enlargement), (B) static compliance and (C) lung volume, of lungs from 6 month old
(mo) gp130+/+ (+/+, n = 8) and gp130F/F (F/F, n = 8) mice, and 3mo F/F mice treated with
sgp130Fc inhibitor for 12 weeks (F/F+sgp130Fc, n = 5). Scale bars = 100μm. (D)
Stereological quantification of the number of TUNEL-positive cells, and corresponding
representative photomicrographs showing positive cells in cross-sections of lungs, from +/+
(n = 8), F/F (n = 8) and F/F+sgp130Fc (n = 5) mice. Data are expressed as the mean ± SEM.
*P< 0.05, **P< 0.01 and ***P< 0.001.
Figure 4 Blockade of IL-6TS with sgp130Fc suppresses apoptosis induced by CS
exposure to wild-type mice. (A) Q-PCR expression analyses of the indicated genes were
performed on lung cDNA, and (B) protein expression analyses of the indicated proteins were
performed on lung lysates, from 1 month old (mo) gp130+/+ (+/+) mice with (+) or without (-)
CS exposure for 4 days. Data are expressed as the mean ± SEM. n = 5 mice per genotype.
*P< 0.05. (C) Stereological quantification of the number of TUNEL-positive cells, and
corresponding representative photomicrographs showing positive cells in cross-sections of
lungs, from the following mouse groups; +/+ (- CS), +/+ (+ CS), +/+ concurrently treated
with sgp130Fc (+/+ (Fc + CS)) and gp130+/+:sgp130Fctg/tg (+/+:tg) mice with or without CS
exposure for 4 days. (D) Static compliance, (E) lung volume, (F) mean linear intercept, and
(G) stereological quantification of the number of TUNEL-positive cells and corresponding
representative photomicrographs showing positive cells in cross-sections of the lungs from
1mo gp130+/+ mice (+/+) treated with or without sgp130Fc inhibitor or CS for 12 weeks. Data
are expressed as the mean ± SEM. n = 5 mice per genotype. *P< 0.05, **P< 0.01, ***P<
0.001 and ****P< 0.0001.

29
Figure 5 IL-6-induced mTORC1 pathway hyper-activation promotes emphysematous
changes in the lungs of mice. (A) Stereological quantification of the number of P-rpS6-
stained cells, and corresponding representative photomicrographs showing positive cells in
cross-sections of lungs, from 6 month old (mo) gp130+/+ (+/+), gp130F/F (F/F) and F/F:Il6-/-
mice. n = 5 mice per genotype. *P< 0.05. (B) Immunoblotting with indicated antibodies on
whole lung lysates from 6mo F/F and F/F:Il6-/- mice. Each lane represents tissue from an
individual mouse. Densitometric quantification of P-rpS6 and total S6 protein levels per
genotype was performed and normalized against Actin protein levels present in each sample.
Data are presented as the mean fold induction ± SEM from n = 7 mice per genotype.
Stereological quantification of the number of P-rpS6-positive cells in the lungs of (C) 6
month old (mo) +/+, gp130+/+:sgp130Fctg/tg (+/+:tg), F/F and gp130F/F:sgp130Fctg/tg (F/F:tg)
mice and (D) 3mo F/F mice treated with sgp130Fc inhibitor for 12 weeks (F/F + Fc). n = 5
mice per group. *P< 0.05, **P< 0.01. (E) Representative methylene blue-stained cross-
sections of lungs from F/F mice (aged 1 month) that were treated with or without rapamycin
for 12 weeks. Arrows indicate airspace enlargement. Scale bars = 100μm. (F-I) Static
compliance (F), lung volumes (G), and the stereological quantification of the number of
TUNEL-positive cells (H) and P-rpS6-positive cells (I) in the lungs of F/F mice treated with
or without rapamycin for 12 weeks. n = 4 mice per group. *P< 0.05 and **P< 0.01. (J)
Stereological quantification of the number of P-rpS6-positive cells in the lungs of gp130+/+
mice (+/+) aged 1 month treated with or without sgp130Fc inhibitor or CS for 12 weeks. n =
4 mice per group. **P< 0.01 and ***P< 0.001.
Figure 6 Augmented levels and correlations of IL-6TS components in human
emphysema tissue and serum. (A) Q-PCR gene expression analyses of IL6 and ADAM17
were performed on human lung cDNA from emphysema patients (Emph, n = 20) or

30
emphysema-free individuals (Ctl, n = 10). Expression data are shown following
normalization for 18S expression, and are presented as the mean ± SEM from triplicate
analyses. (B) IL-6 and sIL-6R protein concentrations in lung lysates as determined by ELISA.
Data are expressed as the mean ± SEM. (C) Immunoblotting with antibodies against
ADAM17 and Actin on whole lung lysates from Emph and Ctl groups. Each lane represents
tissue from an individual. (D) Linear correlation between sIL-6R and ADAM17 (normalized
to Actin following densitometry) protein levels from individuals with emphysema (n = 9). (E)
Representative cross-section of the lung from an emphysema patient displaying cleaved
caspase-3-positive cells. Arrows indicate positive cells. Scale bar = 100μm. (F) The number
of cleaved caspase-3-positive cells per 20 high power fields in the lungs of individuals with
emphysema (n = 9) were measured, and then linear correlation was performed between sIL-
6R levels and the number of cleaved caspase-3-positive cells. (G) Immunoblotting with the
indicated antibodies on lung lysates from Emph and Ctl groups, and each lane represents
tissue from an individual. Densitometric quantification of P-rpS6 protein levels per group was
performed and normalized against Actin protein levels present in each sample. Data are
presented as the mean fold induction ± SEM. (H) Linear correlation between IL-6 and P-rpS6
protein levels compared to sIL-6R protein levels in lung tissue lysates from individuals with
emphysema (n = 20). (I) Protein levels of IL-6 and sIL-6R in the serum of Emph and Ctl
groups, as determined by ELISA. *P< 0.05 and **P< 0.05 versus Ctl group.

1
Therapeutic targeting of the IL-6 trans-signalling/mTORC1 axis in pulmonary
emphysema
Saleela M. Ruwanpura, Louise McLeod, Lovisa F. Dousha, Huei J. Seow, Sultan Alhayyani,
Michelle D. Tate, Virginie Deswaerte, Gavin D. Brooks, Steven Bozinovski, Martin
McDonald, Christoph Garbers, Paul T. King, Philip G. Bardin, Ross Vlahos, Stefan Rose-John,
Gary P. Anderson and Brendan J. Jenkins
"Online Data Supplement"

2
METHODS
Human lung biopsy and serum samples
Lung tissue from resection surgery for treatment of a solitary peripheral carcinoma was
collected from patients with evidence of emphysema, as defined by pulmonary function tests
(gas exchange, FEV1) (Table E2). Tissue from the subpleural parenchyma avoiding tumor-
bearing areas was either snap frozen for molecular analyses or formalin fixed by immersion
and paraffin embedded for histological analyses.
For serum collection, emphysema patients (n = 17) with moderate to severe stable disease
were defined by a TLCO (% of predicted post-oxygen transfer into the blood which determines
emphysema severity) of 36.2 ± 10.4 and FEV1 (% of predicted post-bronchodilator as a
measure of airflow obstruction which determines COPD severity) of 38.2 ± 12.7. These
patients also had radiological assessments such as high resolution computerized tomography
(CT) of the chest to verify emphysema. Control serum samples (n = 6) were donated by healthy
individuals with no emphysema. Lung tissue and serum were collected from individuals upon
written informed consent, and studies were approved by the Monash Health Human Research
Ethics Committee.
RNA isolation and gene expression analysis
Total RNA extraction and the preparation of cDNA for quantitative RT-PCR (Q-PCR)
expression analyses of individual genes were performed as previously described (1, 2).
Sequence information for primers against mouse genes 18s, Il-6, Il-6ra, Cxcl2, Ccl2 and Tnfa,
as well as the human gene 18S, have been previously published (1). Primer sequences for the
mouse gene Adam17 are; Adam17 forward 5’-GGCAACTCCAGGGTGGACGAAGGA-3’,
Adam17 reverse 5’-ATCTTCAGCATCTCCTGGTGGGGG-3’. Sequences for human primers
are as follows; IL-6R forward 5’-AAAGCTGGGCAGGTTGGTG-3’, IL-6R reverse 5’-

3
AGCTTGTGCAGAGGTGTTGAG-3’; ADAM17 forward 5’-
GAAGTGCCAGGAGGCGATTA-3’, ADAM17 reverse 5’-
GGGCACTCACTGCTATTACC-3’. Adam10/ADAM10 forward 5’-
CCTGAGCTCCTGAGGAAAA-3’ and Adam10/ADAM10 reverse 5’-
TGAGCAATCACAGCTTCTCG-3’ primer sequences were used for both mouse and human
genes.
Protein extraction, ELISA and immunoblotting
Total protein lysates were prepared from snap-frozen lung tissues (1, 2) and BALF, and were
subjected to ELISA. The mouse IL-6 and human IL-6 ELISA sets were purchased from BD
Sciences, (Franklin Lakes, NJ), while sIL-6R and sgp130 ELISA sets were purchased from R
& D systems (Minneapolis, MN). The SAA ELISA set was purchased from Life technologies
(Grand Island, NY). All ELISA sets were used as per the manufacturer’s instructions.
Immunoblotting of P-rpS6 (Ser240/244, Cell Signaling Technology (CST), Denver, MA), rpS6
(CST), P-mTOR (Ser2448, CST), mTOR (CST) and ADAM17 (Millipore, Temecula, CA)
were performed on lung lysates, and protein bands were visualized using the Odyssey Infrared
Imaging System (LI-COR, Lincoln, NE) and quantified using the Image J program (nih.gov).
Antibody against actin was purchased from Sigma-Aldrich (St. Louis, MO).
Immunohistochemistry and immunofluorescence
Apoptosis was determined by the terminal deoxynucleotidyl transferase (tdT)-mediated dUDP
nick-end labeling (TUNEL) technique using an ApopTag Peroxidase In Situ Apoptosis
Detection kit (Millipore, Billerica, MA). Immunohistochemistry with antibodies against SPC
(Santa Cruz Biotechnology, Dallas, Texas), P-rpS6 (Ser240/244, CST) and cleaved caspase-3

4
(CST) were performed as previously described (1). Stereological techniques were applied to
determine the number positively-stained alveolar cells.
Dual immunofluorescence staining was performed on paraffin-embedded lung tissues
using primary antibodies conjugated with fluorescent dyes or Alexa Fluor® secondary
antibodies. Briefly, lung sections were antigen retrieved by microwave in 1mM
Ethylenediaminetetraacetic acid (EDTA, pH8.0) buffer, following which they were blocked
with universal CAS block (Invitrogen, Carlsbad, CA) for 1 hour. Antigens were detected with
cleaved caspase-3 antibody conjugated with Alexa Fluor® 488 (CST), podoplanin antibody
conjugated with Alexa Fluor® 594 (BioLegend, San Diego, CA), CD31 antibody conjugated
with Alexa Fluor® 647 (BioLegend), P-rpS6 antibody conjugated with Alexa Fluor® 647
(CST), and SPC (Santa Cruz Biotechnology) and CD45 (BD BioSciences, Franklin Lakes, NJ)
primary antibodies with Alexa 488/546-conjugated donkey anti-goat and Alexa 488-
conjugated goat anti-rabbit secondary antibodies, respectively (Invitrogen). Slides were
examined under a Nikon confocal microscope and analyzed for red and green fluorescence.
Stereological techniques were applied to determine the number of dual stained cells.
Cigarette smoke (CS) exposure
Mice aged 4-6 weeks were subjected to whole body CS exposure for 4 days (acute) (1). This
smoke exposure protocol caused acute inflammation with no acute lung injury. For chronic CS
exposure, mice were subjected to whole body CS exposure over a 12 week period as per the
protocol described for acute CS exposure.
Primary lung cell cultures, and flow cytometry
Mouse primary alveolar type II (ATII) epithelial cells were prepared as previously described
(3). Following the digestion of lungs, single cell suspensions were prepared and then incubated

5
with purified CD45 antibody (BD Biosciences) and epithelial cells negatively enriched with
BioMag goat anti-rat immunoglobulin-coupled magnetic beads (Qiagen, Hilden, Germany).
The negative selection for ATI epithelial cells was performed by incubation of podoplanin-
coated petri dishes for 30 minutes at 370C. Cell purity was routinely assessed by epithelial cell
morphology and flow cytometry analysis with anti-EpCAM, anti-podoplanin and anti-SPC
(ATII) antibodies. Cells were cultured on collagen-coated (MP Biomedicals, Santa Ana, CA)
plates and stimulated with 100ng/ml Hyper-IL-6, an IL-6/soluble IL-6R fusion protein (4) over
the indicated times. Monolayers were detached with either 3mM EDTA for flow cytometry
analysis with Annexin-V (BD Biosciences) and 7-AAD to examine the level of apoptosis, or
RIPA buffer for immunoblot analyses of mTORC1 activity. Cells were analysed on a FACS-
CantoII flow cytometer (BD Biosciences) and data was performed using FACSDiva software
(BD Biosciences).

6
REFERENCES
1. Ruwanpura SM, McLeod L, Miller A, Jones J, Bozinovski S, Vlahos R, Ernst M, Armes J,
Bardin PG, Anderson GP, Jenkins BJ. Interleukin-6 promotes pulmonary emphysema
associated with apoptosis in mice. Am J Respir Cell Mol Biol 2011; 45: 720-730.
2. Ruwanpura SM, McLeod L, Miller A, Jones J, Vlahos R, Ramm G, Longano A, Bardin PG,
Bozinovski S, Anderson GP, Jenkins BJ. Deregulated Stat3 signaling dissociates
pulmonary inflammation from emphysema in gp130 mutant mice. Am J Physiol Lung
Cell Mol Physiol 2012; 302: L627-639.
3. Londrigan SL, Tate MD, Job ER, Moffat JM, Wakim LM, Gonelli CA, Purcell DF, Brooks
AG, Villadangos JA, Reading PC, Mintern JD. Endogenous murine BST-2/Tetherin is
not a major restriction factor of Influenza A virus infection. PLOS One 2015; 10:
e0142925.
4. Peters M, Muller AM, Rose-John S. Interleukin-6 and soluble interleukin-6 receptor: direct
stimulation of gp130 and hematopoiesis. Blood 1998; 92: 3495-3504.

7
SUPPLEMENTARY FIGURE LEGENDS
Figure E1: Expression of IL-6 signalling components in emphysematous gp130F/F and
emphysema-free gp130F/F:sgp130Fctg/tg mice. (A) IL-6, sIL-6R and sgp130 protein
concentrations were determined by ELISA in the BALF of 6 month old (mo) gp130+/+ (+/+)
and gp130F/F (F/F) mice. (B) Mean linear intercept of +/+, gp130+/+:sgp130Fctg/tg (+/+:tg), F/F
and gp130F/F:sgp130Fctg/tg (F/F:tg) mice aged 6 months. (C) Serum amyloid A (SAA) levels
in the serum and lungs of the indicated 6mo mice. Data are expressed as mean ± SEM. n = 5
mice per genotype. *P< 0.05, **P< 0.01 and ***P< 0.001. ND = not within assay detectable
level (assumed 0 for statistical analyses).
Figure E2: IL-6TS-mediated apoptosis is not observed in alveolar type I (ATI) and
endothelial cells from gp130F/F mice. Representative confocal immunofluorescence
photomicrographs of cells stained for (A) cleaved caspase-3 (green) and podoplanin (red, ATI
cell marker) either alone or together, or (B) cleaved caspase-3 (green) and CD31 (red,
endothelial cell marker) either alone or together, in cross-sections of lungs from 6mo gp130F/F
(F/F) mice. In both (A) and (B), sections are also stained with the nuclear marker 4',6-
diamidino-2-phenylindole (DAPI; blue) as indicated. White circles indicate examples of (A)
podoplanin-positive cells (cell membrane) and (B) CD31-positive cells (cell junction in cell
membrane) with cleaved caspase-3 (cytoplasm), while green circles (A and B) indicate
examples of cleaved caspase-3 alone cells. Scale bars = 40μm. The graphs depict stereological
quantification of dual labelled caspase-3/podoplanin- or caspase-3/CD31-positive cells in the
lungs of 6mo mice of the indicated genotypes. Data are expressed as the mean ± SEM. n = 5
mice per genotype.

8
Figure E3: Therapeutic and genetic blockade of IL-6 trans-signalling in CS-exposed wild-
type mice suppresses expression of inflammatory mediators. (A) Q-PCR expression
analyses of the indicated genes were performed on lung cDNA, and (B) ELISAs for the
indicated proteins were performed on lung lysates, from 1 month old gp130+/+:sgp130Fctg/tg
(+/+:tg) mice with (+) or without (-) cigarette smoke (CS) exposure. (C) Q-PCR expression
analyses of inflammatory mediators, Cxcl2, Tnfa and Ccl2 were performed on lung cDNA from
gp130+/+ (+/+) mice without CS (+/+ -CS), with CS (+/+ +CS), or concurrently treated with
sgp130Fc and CS (+/+ Fc+CS), as well as +/+:tg mice with/without CS exposure. Data are
expressed for n = 3 mice per genotype. *P< 0.05 and ** P< 0.01.
Figure E4: Therapeutic and genetic blockade of IL-6TS in gp130F/F mice suppresses
mTORC1 pathway activation in alveolar epithelial type II (ATII) cells. (A and B)
Representative confocal immunofluorescence photomicrographs of cells positive for either P-
rpS6 alone (red, A and B), SPC alone (green, A), CD45 alone (green, B) or both P-rpS6 and
SPC (yellow, white circles, A) in cross-sections of lungs from 6mo gp130F/F (F/F) mice. Scale
bars = 10μm. (C) Immunoblotting with indicated antibodies on whole lung lysates from 6mo
F/F and F/F:Il6-/- mice. Each lane represents tissue from an individual mouse. Densitometric
quantification of P-mTOR (Ser2448) protein levels per genotype was performed and
normalized against Actin protein levels present in each sample. Data are presented as the mean
expression ± SEM from n = 5 mice per genotype. *P< 0.05.
Figure E5: Activation of the IL-6TS/mTORC1 axis in cultured gp130F/F primary alveolar
type II (ATII) epithelial cells stimulated with the IL-6TS agonist Hyper-IL-6. (A)
Immunoblotting with indicated antibodies on lysates from cultured gp130F/F (F/F) ATII cells
following stimulation with Hyper-IL-6 (10ng/ml) over the indicated time points. (B)

9
Representative histograms of flow cytometric analyses of Annexin-V and 7AAD staining
showing percentages of Annexin-V-positive/7AAD-negative apoptotic ATII cells from wild-
type (+/+) and F/F mice.
Figure E6: Blockade of mTORC1 using rapamycin prevents alveolar cell death in
gp130F/F mice with hyper-activated IL-6TS/mTORC1 axis. (A and B) Representative
photomicrographs of P-rpS6-positive cells in cross-sections of lungs from (A) 6mo gp130+/+
(+/+), gp130+/+:sgp130Fctg/tg (+/+:tg), F/F and F/F:tg mice, and (B) 3mo +/+ and F/F mice, and
F/F mice treated with sgp130Fc inhibitor for 12 weeks (F/F + Fc). (C and D) Representative
photomicrographs showing (C) TUNEL-positive cells and (D) P-rpS6-positive cells, in cross-
sections of lungs from gp130F/F (F/F) mice aged 6 months, and 3 month old F/F mice treated
with rapamycin for 12 weeks (F/F + Rapa). Scale bars = 100μm.
Figure E7: Hyper-activation of the IL-6TS/mTORC1 pathway correlates with
augmented cellular apoptosis in the lungs of CS-exposed wild-type mice. (A) Stereological
quantification of the number of P-rpS6-positive cells, and corresponding representative
photomicrographs showing positive cells in a cross-section of lungs from +/+ -CS, +/+ +CS,
and +/+ mice concurrently treated with CS and sgp130Fc (+/+ Fc+CS), as well as
gp130+/+:sgp130Fctg/tg (+/+:tg) mice with or without CS exposure. (B) Stereological
quantification of the number of P-rpS6-positive cells, and corresponding representative
photomicrographs showing positive cells in a cross-section of lungs from +/+ mice
concurrently treated with CS and either vehicle (Veh) or rapamycin (Rapa). (C) Stereological
quantification of the number of TUNEL-positive cells, and corresponding representative
photomicrographs showing positive cells in a cross-section of lungs from +/+ mice
concurrently treated with CS and either Veh or Rapa. Data are expressed as mean ± SEM. n =

10
4 mice per genotype. Scale bars = 100μm. *P< 0.05, **P< 0.01 and ***P< 0.001. (D)
Representative photomicrographs showing P-rpS6-positive cells in cross-sections of the lungs
from gp130+/+ mice (+/+) aged 1 month treated with or without sgp130Fc inhibitor or CS for
12 weeks.

+/+ F/F +/+:tg F/F:tg F/F + sgp130Fc
F/F + Rapa
Vv (par/lung) (%)
87.9(±1.3)
93.9(±2.8)
88.3(±1.7)
91.2(±4.1)
91.9(±1.8)
93.2(±3.3)
Vv (airsp/par) (%)
68.5(±1.0)
80.3###
(±0.6)71.6****(±0.9)
69.4****(±1.5)
71.4****(±0.9)
71.0***(±1.3)
V(airsp/lung) (cm3)
59.6(±2.6)
87.9###
(±2.6)41.6****(±1.6)
55.7****(±5.1)
54.5****(±4.8)
52.3****(±4.4)
Vv (sep/par) (%)
23.2(±1.5)
14.9###
(±0.8)23.9****(±0.9)
23.05***(±0.9)
24.3****(±0.8)
24.0***(±1.0)
V (sep/lung) (cm3)
20.0(±1.0)
16.4#
(±1.1)18.9
(±0.6)18.3
(±1.2)18.5
(±1.5)17.6
(±0.7)
Sv (sep/par) (1/cm)
651(±39)
567#
(±26)623*(±48)
634*(±25)
624*(±21)
639*(±23)
S (sep/lung) (cm2)
538(±28)
583(±34)
501(±19)
511(±6)
499(±56)
535(±38)
Table E1: Comparative stereological analyses of lungs from the indicated mice. Data areexpressed as the mean ± SEM. n = 5 mice per genotype per age group. #P < 0.05 and ###P <0.001 versus age-matched +/+ mice. *P < 0.05, ***P < 0.001 and ****P < 0.0001 versus age-matched F/F mice. Vv = volume fraction; par = parenchyma; air = air space; sep = septal tissue;Sv = surface density; S = surface area.

Table E2: Clinical characteristics of the patients. Data are expressed as the mean ± SEM.FEV1 denotes forced expiratory volume in one second.
Characteristics No emphysema (n=10)
Moderate emphysema (n=20)
Sex (no. of patients)Male
Female55
128
Smoking historyPack years 10 ± 5 42 ± 9Age (years) 69 ± 5 71 ± 2Diagnosis Lung cancer (tissues were
obtained from non-cancer area)
Lung cancer (tissues were obtained from non-cancer
area)FEV1 (% of predicted post)-
COPD status 110.1 ± 4.0 88.5 ± 6.3
TLCO (gas transfer, % predicted post)-emphysema
status
92.8 ± 4.9 51.1 ± 2.0

0
20
40
60
+/+ +/+: tg
F/F F/F: tg
* **
µm
C
A
B
0
5
10
15
00.51.01.52.0
2.5
0
5
10
15
20sIL-6R sgp130IL-6
pg/m
l
pg/m
l
pg/m
l
* *
SAAng
/ml
F/F F/F:tg
Serum
ND ND+/+ F/F +/+ F/F +/+ F/F
0
200
400
600
800
1000
F/F F/F:tg
Lung
Figure E1
Mean linear intercept

A
Figure E2
B
Cleaved Casp-3 + Podoplanin Cleaved Casp-3
Podoplanin Dapi
0
2
4
6
+/+ +/+: tg
F/F F/F: tg
Cleaved Casp-3+/Podoplanin+ cells/
20 fields
Cleaved Casp-3 + CD31 Cleaved Casp-3
CD31 Dapi
0
1
2
3
4
+/+ +/+: tg
F/F F/F: tg
Cleaved Casp-3+/CD31+
cells/20 fields

A
+CS
+/+:tg
0
10
20
30
40
50 ** Ccl2
-CS +CS Fc+CS
+/+
-CS +CS+/+:tg
Rel
ativ
e Ex
pres
sion
-CS +CS Fc+CS
+/+
-CS
C
Rel
ativ
e Ex
pres
sion
Tnfa
0
5
10
15
20 ** *
-CS +CS Fc+CS+/+
-CS +CS+/+:tg
Rel
ativ
e Ex
pres
sion
0
1
2
3
4 sgp130
ng/m
l
-CS +CS
+/+:tg
0
0.2
0.4
0.6
0.8
1.0Il6
-CS +CS
+/+:tg
Rel
ativ
e Ex
pres
sion
10
15
-CS +CS
+/+:tg
sIL-6R
0
5pg/m
l
Cxcl2
0
50
100
150 **
**
***
0
0.2
0.4
0.6
0.8
1.0Adam17
-CS +CS
+/+:tg
B
Figure E3
Rel
ativ
e Ex
pres
sion

Figure E4
A
C F/F F/F:Il6-/-
P-mTOR (ser2448)
mTOR
ActinF/F F/F:Il6-/-
Rat
io to
Act
in
CD45+ P-rpS6
CD45 P-rpS6
SPC+ P-rpS6
SPC P-rpS6
0
0.5
1.0
1.5
P-mTOR (ser2448)
*
B
F/F
F/F
F/F
F/F

Figure E5
F/F
Hyper-IL-6 (hours)
0 0.5 1 3
Actin
S6
P-rpS6
A
B
Cou
nt
Annexin-V-positive/7AAD-negative
F/F+/+
35.2% 45.3%

C F/F + RapaF/F
F/F + RapaF/F
Figure E6
TUNEL
P-rpS6
+/+ F/F F/F + Fc
P-rpS6
P-rpS6
+/+ +/+:tg F/F:tgF/FA
B
D
A
P-rp
S6+
cells
/20
field
s
+/+ (-CS) +/+ (+CS) +/+ (Fc +CS)
+/+:tg (-CS) +/+:tg (+CS)
-CS0
1020304050
+CS Fc+CS
+/+
-CS +CS
+/+:tg
* *
+/+ (Veh +CS) +/+ (Rapa +CS)
Rapa
+/+ +CSVeh
0
20
40
60 ***
P-rp
S6+
cellS
/20
field
s
+/+ +CS
0
5
10
15
20
Veh Rapa
**+/+ (Veh +CS) +/+ (Rapa +CS)
TUN
EL+
cells
/20
field
s
B
C
Figure E7
+/+ (-CS) +/+ (Fc+CS)+/+ (+CS)
P-rpS6
D