unique device identification system; proposed rule · unique device identification system; proposed...
TRANSCRIPT

CDRH200816
4160-01-P
DEPARTMENT OF HEALTH AND HUMAN SERVICESFood and Drug Administration
Unique Device Identification System; Proposed Rule
Docket No. FDA-2011-N-0090
Preliminary Regulatory Impact AnalysisInitial Regulatory Flexibility Analysis
Unfunded Mandates Reform Act Analysis
Economics StaffOffce of Planning
Offce of Policy and PlanningOffce of the Commissioner

2 CDRH200816
Table of Contents
1. Analysis ofImpacts
A. Summar of Costs
1. Costs to Domestic Labelers
2. Costs to Issuing Agencies
3. Costs to FDA to Establish and Maintain the GUDID
4. Cost to Foreign Labelers
5. Uncertainty
6. Alternatives
B. Summar of Regulatory Flexibility Analysis
C. Sumar of Benefits
D. Need for Regulation and Sumar of the Proposed Rule
E. Medical Device Manufactuing Industry Profie
1. Number of Labeler Establishments and Firms
2. Baseline Practice
F. Costs of the Proposed Rule
1. Costs for Initial Labelers
2. Costs for Repackagers and Relabelers
3. Efforts to Reduce Regulatory Burdens for Certain Low Risk Devices
4. Costs of the Proposed Rule to Labelers with Simplifying Assumptions
5. Costs of the Proposed Rule to Labelers Under FDA's Proposed Implementation
Schedule
6. Cost to Issuing Agencies
7. Cost to FDA for the GUDID

3 CDRH200816
8. Impact on Foreign Trade
9. Summar of Total Costs of the Proposed Rule
G. Analysis of the Uncertainty of Costs
H. Benefits
1. Improved Reporting of Adverse Medical Device Events
2. Improved Effciency in Removing Recalled Devices from Use
3. Reduced Device Related Medical Errors and UDI
4. The Public Health Implications of Better Device Analyses by FDA
1. Alternatives to the Proposed Regulation
1. Full UDI Requirements for Unclassified and Class I, II, and III Devices
2. A Requirement for Labeling Only
3. Apply UDI Requirements Only to Class II and III Devices.
4. A Unique Device Identifier that Includes Only Static Information
5. Apply the UDI Requirements Only to Class III Devices
6. Sumar of Alternatives
J. Small Business Impact
1. Need for the Rule and Objectives of the Rule
2. Number of Affected Small Entities
3. Description of the Reporting and Recordkeeping Burdens and Personnel Skil Levels
4. Impact ofthe Rule on Small Entities.
5. Alternatives Considered
I1. References

4 CDRH200816
L Analysis of Impacts
FDA has examined the impacts of the proposed rule under Executive Order 12866,
Executive Order 13563, the Regulatory Flexibility Act (5 U.S.C. 601-612), and the Unfuded
Mandates Reform Act of 1995 (Public Law 104-4). Executive Orders 12866 and 13563 direct
Agencies to assess all costs and benefits of available regulatory alternatives and, when regulation
is necessary, to select regulatory approaches that maximize net benefits (including potential
economic, environmental, public health and safety, and other advantages; distributive impacts;
and equity). The Agency believes that this proposed rule is a significant regulatory action under
Executive Order 12866.
The Regulatory Flexibility Act requires Agencies to analyze regulatory options that
would minimize any significant impact of a rule on small entities. Because we are uncertain
whether the proposed rule would have a significant economic impact on a substantial number of
small entities, this RIA and other sections of the preamble to the proposed rule constitute the
Agency's regulatory flexibility analysis.
Section 202(a) of the Unfuded Mandates Reform Act of 1995 requires that Agencies
prepare a written statement, which includes an assessment of anticipated costs and benefits,
before proposing "any rule that includes any Federal mandate that may result in the expenditue
by State, local, and tribal governents, in the aggregate, or by the private sector, of
$100,000,000 òr more (adjusted anually for inflation) in anyone year." The curent threshold
after adjustment for inflation is $139 milion, using the most curent (2011) Implicit Price
Deflator for the Gross Domestic Product. The estimated costs of this proposed rule would result
in a I-year expenditue that exceeds this amount.

5 CDRH200816
This proposed rule would require the label and package of medical devices to bear a
unique device identifier (UDI) and would provide for alternative placement or an exception for a
particular device or type of device. In addition, this proposed rule would require certain devices
to be directly marked with a UDI, with exceptions. Medical device records throughout the
required device recordkeeping and reporting systems would need to be modified to include the
UD1. Under this proposed rule FDA would establish the Global Unique Device Identification
Database (GUDID), a public database containing information about devices labeled with a UD1.
The proposed rule would require labelers of medical devices to submit information concerning
each device to the GUDID. In addition, the proposed rule would also establish the accreditation
requirements for agencies that may operate a system for the issuance ofUDIs and establish the
conditions for when FDA might act as an issuing agency.
A. Sumar of Costs
The detailed data for this cost analysis were developed by Eastern Research Group, Inc.
(ERG) under contract to FDA and are presented in the full report "Unique Device Identification
(UDI) for Medical Devices," 20 11 (cited in Ref.ll). We refer to this analysis below and
welcome comments on the assumptions and estimates contained in the report.
Table 1 of this document presents for each affected sector a summary of the estimated
present value and the anualized domestic costs of this proposed rule over 10 years using
discount rates of 7 percent and 3 percent. Over 10 years, the present value of the domestic costs
would be $514.0 milion using a 7 percent discount rate and $588.6 milion using a 3 percent
rate, and the anualized costs would be $68.4 milion using a 7 percent discount rate and $66.9
milion using a 3 percent discount rate.

6 CDRH200816
Table 1.--Sumar of the Estimated Regulatory Costs of the Proposed Rule (2010 dollars)!
Total Present Value of Total Anualized Costs
Affected SectorsCost over 10 years Over 10 Years
($ milion) ($ milion)
3 Percent 7 Percent 3 Percent 7 PercentDomestic Labelers $571.5 $499.4 $65.0 $66.5Issuing Agencies $1.0 $0.9 $0.1 $0.1FDA $16.1 $13.7 $1.8 $1.8
ImportsNot Not Not Not
quantified quantified quantified quantifiedTotal Domestic Cost of the Proposed
$588.6 $514.0 $66.9 $68.4Rulei Present value and annuahzed costs calculated at the beginning of the penod.
1. Costs to Domestic Labelers
The majority of the costs of this proposed rule would be incured by labelers of medical
devices. Labelers include manufacturers, reprocessors, specification developers, repackagers
and relabelers that cause a label to be applied to a medical device. The estimated present value
of the costs for domestic labelers over 10 years would be $499.4 milion at a 7 percent discount
rate and $571.5 milion at 3 percent. Over 10 years, the anualized costs for domestic labelers ~
would be $66.5 milion at a 7 percent discount rate and $65.0 milion at 3 percent. The largest
components of one-time costs would include the costs to integrate the UDI into existing
information systems, to install, test and validate barcode printing software and to train
employees, and to purchase and install equipment needed to print and verify the UDI on labels.
In addition, other significant components of one-time costs include costs to redesign labels of
devices to incorporate the date format within 1 year and to allow space for the UDI barcode, and
the direct marking of certain devices.

7 CDRH200816
The largest anual cost components.include labor, operating, and maintenance associated
with equipment for printing operations, and labor related to software maintenance and training
needed to maintain the UDI information system.
2. Costs to Issuing Agencies
The estimated present value of costs over 10 years for two existing organizations,
curently performing functions similar to those of an issuing agency under the proposed rule, to
apply for FDA accreditation and comply with the proposed reporting requirements would be $0.9
milion at a 7 percent discount rate and $1.0 milion at 3 percent. The anualized costs over 10
years would be $0.1 milion at both 7 percent and 3 percent discount rates. In addition to these
two organizations, there may be other nonprofit organizations or State agencies that might apply
to FDA to become an issuing agency. In such cases, the estimated application preparation, legal,
and reporting costs would apply to other organizations.
3. Costs to FDA to Establish and Maintain the GUDID
The estimated present value over 10 years of the costs to FDA to establish and maintain
the GUDID would be $13.7 milion at a 7 percent discount rate and $16.1 milion at 3 percent.
The anualized costs over 10 years would be $1.8 milion at 7 percent and 3 percent.
4. Costs to Foreign Labelers
We lack suffcient information to quantify the potential impact of the proposed rule on
foreign establishments and thus exclude these establishments from our cost estimate. However,
we include a qualitative discussion of the potential impact ofthis rule on trade and the cost of
imported products, whose value is about one-fourh the value of domestic production. We
request comment from affected industries about their expected compliance costs and responses to
the proposed rule.

8 CDRH200816
5. Uncertainty
In this analysis, the lower and upper bounds of uncertainty surounding the central
estimate ofthe costs to domestic labelers are about 50 percent lower and 50 percent higher,
respectively. Applying a similar range of uncertainty to the total costs of the proposed rule to
domestic labelers, issuing agencies, and the FDA, over 10 years the total annualized domestic
costs would range from $34.9 millon to $101.8 millon at 7 percent and $34.1 milion to $99.7
millon at 3 percent.
6. Alternatives
The Agency analyzed a number of alternatives with varied requirements affecting the
coverage of devices, the content of the information required to be encoded in a UDI, and specific
provisions of the proposed rule. With respect to device coverage, we analyzed applying the UDI
requirements to class III devices only, and to class II and III devices only. The Agency also
analyzed costs for requiring the UDI to contain only the device identifier across all device
classes. Also included was an alternative that required a UDI labeling change without requiring
the submission of data to the GUDID.
Over 10 years at 7 percent, the anualized present value of the highest cost alternative is
about $95 milion. This alternative would apply the UDI requirements to class I, II, and III
devices, as well as unclassified devices, unless excepted by proposed 801.30(a)(3) through (12).
The lowest cost alternative would apply the UDI requirements to class III devices only. The
annualized present value of this alternative is about $11 milion.
B. Sumar of Regulatory Flexibility Analysis
FDA conducted a regulatory flexibility analysis of the impact of the proposed rule on
small entities. Ninety-six percent of the 4,693 affected labeler firms (i.e., 4,483 firms) are small

9 CDRH200816
according to Small Business Administration (SBA) size standards. Costs of compliance for
domestic labelers as a percentage of revenues exceed 1 percent for about 32 firms with fewer
than 19 employees that label devices subject to the direct marking requirements. Moreover, for
an estimated 8 firms with fewer than 5 employees, the burden of the proposed rule would
represent about 8 percent of their average revenues. If direct marking of devices were not
required, no firms would experience costs exceeding 1 percent of revenues.
C. Sumar of Benefits
The proposed rule would standardize how medical devices are identified and would
contribute to future potential public health benefits from initiatives associated with the increased
use of automated systems in healthcare. Most of these benefits, however, require complementary
developments and innovations in the private and public sectors, and investments by the
healthcare industry that are beyond the scope of this rule. Because such actions are uncertain, we
restrict our discussion of the potential public health benefits to those most likely to occur as
results of probable responses to the proposed rule in the private and public sectors.
The public health benefits from the UDI would be related to reductions in medical
device-related patient injuries and deaths. More accurate and prompt identification of problems
would enable more rapid action to reduce the incidence of the adverse events. Public health
safety alerts, for example, could be more accurate and timely. Recall actions could more
effectively target the problem device. The increased accuracy of adverse medical device
reporting and improved recalls should reduce the total number of adverse medical device events,
although we are unable to quantify that reduction.
Table 2. Economic Data: Costs and Benefits Accounting Statement (2010 dollars)
UnitsCategory Primary Low High Year
I Discount I PeriodNotes
Estimate Estimate Estimate Dollars Rate Covered

10 CDRH200816
BenefisAnnualized 7%Monetized 3%$milions/yearAnnualized 7%
Quantified 3%
Qualitative More accurate and promptidentification of device relatedadverse events would lead to morerapid action to reduce the incidenceof the adverse events and to moreeffectively target and managemedical device recalls.
CostsAnnualized $68.4 $34.9 $101. 2011 7% 10 years Costs toMonetized $66.9 $34.1 $99.7 2011 3% 10 years foreign$milions/year labelers are
not included.
Annualized 7%Quantified 3%
Qualitative
TransfersFederal 7%Annualized 3%Monetized$milions/yearFrom/To From: To:
Other 7%Annualized 3%Monetized$milions/yearFrom/To From: To:
EffectsState, Local or Tribal Governent: No effect
Small Business: The proposed rule may have a significant economic impact on a substantialnumber of small entities that label medical devices.
Wages: No effect
Growth: No effect
D. Need for Regulation and Sumar of the Proposed Rule
There curently is an imbalance between the entities that would incur the cost of
establishing a standardized system to uniquely identify medical devices and the entities that
might benefit from the use of such a system. Medical device labelers would incur the costs of

11 CDRH200816
placing a unique identifier on device labels and of providing medical device information to a
device database. Distributors, hospitals, GPOs, insurers and other groups could benefit from the
availability of a standardized device identifier and database. The medical device supply and use
chain is a disaggregated set of disparate industries. The transaction costs of bringing these
disparate parties together to create a standardized system are high. To date, the market has failed
to establish a standardized UDI system that meets the basic needs of medical device producers
and the users of medical devices. Governent can reduce transaction costs and increase net
social benefits by defining the basic requirements and structure of a UDI system and by
providing oversight to ensure that standards are followed. Once established, a standardized UDI
system may be used as a platform for investment in information technology enhancements that
can improve patient safety. Although the decisions to invest in health information systems that
would use a UDI would be made independently of the proposed rule, the availability ofa
standardized UDI system would advance the development of analytic tools and other information
technology dependent on device identifiers in health information systems, including database
querying and networking.
Section 226(a) of the FDAA (Public Law 101-85) created a new section of 519 (f) of
the FD&C Act (21 U.S.C. 360i(f)) stating that: "The Secretar shall promulgate regulations
establishing a unique device identification system for medical devices requiring the label of
devices to bear a unique identifier, unless the Secretary requires an alternative placement or
provides an exception for a paricular device or type of device. The unique identifier shall
adequately identify the device through distribution and use, and may include information on the
lot or serial number."

12 CDRH200816
The proposed rule would implement this provision of FDAA by requiring a UDI to
appear on the label and on the package of affected medical devices in an easily-readable plain-
text form and in a form using automatic identification and data capture (AID C) technology (note:
this analysis uses the term "barcode" as shorthand to refer to all forms of AIDC technology,
because that is the most commonly-used form of AIDC at present), by establishing a GUDID,
and by requiring device labelers to submit descriptive information about each version or model
of device labeled with a UDI to the GUDID. The agency has specified certain types of devices
that would be excepted from some or all of the UDI requirements.
This proposed rule would establish requirements for the UDI that must appear on each
label, and for procedures for using, changing, and discontinuing UDIs. A UDI would consist of a
fixed device identifier (a mandatory portion of a UD I that could be used to access data that
identifies the specific version or model of a device and the labeler of that device), and a variable
production identifier (a portion of the UDI that would be required to identify certain labeled
production information including: the lot or batch within which a device was manufactued, the
serial number, the expiration date, or the date of manufacture). The proposed rule identifies
general exceptions from the requirement for a label of a device to bear a UDI and describes the
process for other labelers to request an exception or alternative placement of the UD1. Class I .
devices would not be required to bear a production identifier. Moreover, those class I devices
that FDA has by regulation exempted from the good manufactuing practice (GMP) requirements
would not be required to bear a UD1. Certain devices for which the labeling requirement may
not be suffcient, for example, those that remain in use for an extended period of time and
devices that are likely to become separated from their labeling, would be directly marked with a
UD1. The proposed rule lists criteria for exceptions to the requirement for direct marking of

13 CDRH200816
devices. This proposed rule would also establish the accreditation requirements for agencies that
operate a system for the issuance ofUDIs and explains when FDA might act as an issuing
agency.
The proposed rule specifies the data for each version or model of a device that would
have to be submitted to the publicly available GUDID. Users of the GUDID would use the
device identifier portion of the UDI to query descriptive data about a specific device at any time
during the distribution and use of the medical device, including a generic descriptor (the
GMDN), the proprietar, trade, or brand name of the device, and other identifying information
and contact information.
E. Medical Device Manufacturing Industry Profie
The medical device industry is among the most competitive sectors in the United States.
It is characterized by a large number of innovátive firms that produce a wide aray of products.
As measured by the total value of shipments, medical device manufacturing industry production
was about $117.5 bilion in 2007 (see table 3 of this document), or about 2.2 percent ofthe total
value of shipments for all manufacturing industries in the United States. A large majority of
domestic medical device manufacturing establishments have fewer than 500 employees, and
roughly 75 percent of establishments have fewer than 50 employees, according to the U.S.
Census of Manufactues.
Medical device manufacturers are categorized within a number of distinct industries or
NAICS (North American Industry Classification System) codes by the Census of Manufacturers.
Medical devices var in size, complexity and packaging. The vast array of medical device types
covers implants (e.g., heart valves, stents, arificial knees), screening technologies (e.g., CT
scaners, MRI equipment), diagnostic tests, surgical instruents, hospital equipment and

14 CDRH200816
supplies, and devices sold at retail (glucose monitors and strips, bandages, canes). We include
this information for background, but note that the proposed rule includes an exception for over-
the-counter devices sold at retaiL. Medical devices might be packaged individually or in boxes of
hundreds. The useful life of a device ranges from a brief single use for some disposable items to
use over many years for capital equipment, implants, and other multiple-use devices. End users
of medical devices range from highly skiled specialists and medical practitioners working in a
number of different healthcare and emergency care settings, to consumers. Table 3 of this
document presents the major medical device manufactuing industries and the 2007 domestic
total value of shipments by the NAICS code as reported by the U.S. Census Bureau.
Table 3.--Medical Device Manufactuing Industr Tota Va ue of Shipments 2007 Do arsIndustr by NAICS Value of Shipments
($ bilion)NACIS 325413, In vitro diagnostic substances manufacturing $13.0NAICS 334510, Electromedical and electrotherapeutic apparatus mfg. $22.5NAICS 334517, Irradiation apparatus manufacturing $10.8NAICS 339112 Surgical and medical instrment manufactuing $29.6NAICS 339113 Surgical appliance and supplies manufacturing $31.5NAICS 339114, Dental equipment and supplies manufacturing $4.4NAICS 339115, Ophthalmic goods manufacturing $5.7Total $117.5
( ll)
Source: ERG Report, Table 3-2 (Ref. 1).Note: Numbers may not sum due to rounding.
1. Number of Labeler Establishments and Firms!
The diversity of products in the medical device industry prevents basing the cost analysis
on cost per product. Therefore, this analysis contains a simplifying assumption that costs would
be incured by labelers of affected medical devices on an establishment or firm basis, depending
on the type of cost. The proposed rule would affect initiallabelers of medical devices: that is,
those entities that manufacture, reprocess, or develop specifications for medical devices and that
cause a label to be applied to a medical device intended for interstate commerce. Repackagers
i An establishment is a business unit at a single location. A firm is comprised of all the establishments that
operate under the ownership or control of a single organization.

15 CDRH200816
and relabelers, or non-manufactuing labelers, would also be subject to requirements of the
proposed rule.
Based on the FDA registration and listing database, table 4 of this document shows the
estimated total number of labeler establishments by type of FDA registrant and whether the
establishment is located in the United States or in another country. Although not included in
table 4 of this document, these establishments are owned by about 12,484 firms: 6,569 domestic
and 5,915 foreign firms (ERG Report, Table 3-6, Ref. 1).
Table 4.--Number of Registered Establishments Considered Labelers under the Proposed RuleType of Registrant Domestic Foreign Total Registrants
Manufacturers 4,901 6,492 11,393
Reprocessors 21 3 24
Specification Developers 1,346 276 1,622
Relabelers and Repackagers 1,310 320 1,630
Total Labelers 7,578 7,091 14,669
Source. ERG Report, Appendix A (Ref. i).
To further characterize domestic labelers, Table 5 of this document presents a breakdown
of registered establishments by employment size. We use the same employment size categories
as the U.S. Census Bureau, whose size categories are more detailed for initiallabelers than for
non-manufactuing labelers.
a e .-- um er 0 omestic a e er sta is ents)v mp\ovment ize
Type of Labeler Emplo went SizeInitial Labelers 1-4 5-9 10-19 20-49 50-99 100- 250- 500- . 1,000 or Total
249 499 999 more
Manufacturers 1,630 794 695 698 419 369 185 68 42 4,901
Single-Use 0 11 0 2 2 2 4 0 0 21
DeviceReprocessorsSpecification 722 210 184 146 51 25 6 2 1 1,346
DevelopersNon- 1-4 5-9 10-49 50-99 100-249 250-499 500 or Total
Manufacturing more
LabelersRepackagers and 736 212 272 47 28 10 4 1,310
Relabelers
T bl 5 N b fD . L b i E bl hm b E S'
Source: ERG Report, Tables 3-5, 3-8, and 3-10 (Ref. 1).Numbers may not add due to rounding.
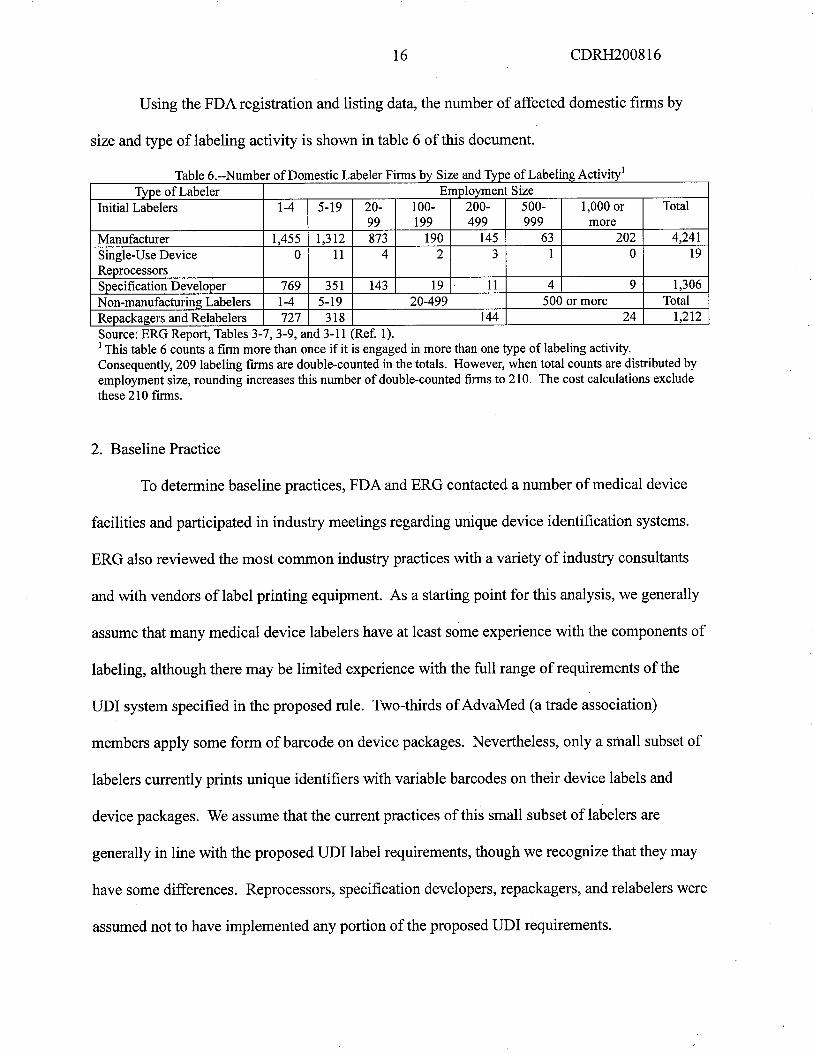
16 CDRH200816
Using the FDA registration and listing data, the number of affected domestic firms by
size and type of labeling activity is shown in table 6 of this document.
a e -- um ero omestic a e er irs)y ize an lype 0 a e ing. ctiVItyType of Labeler Employment Size
Initial Labelers 1-4 5-19 20- 100- 200- 500- 1,000 or Total99 199 499 999 more
Manufacturer 1,455 1,312 873 190 145 63 202 4,241
Single-Use Device 0 11 4 2 3 1 0 19
ReprocessorsSpecification Developer 769 351 143 19 11 4 9 1,306
Non-manufacturing Labelers 1-4 5-19 20-499 500 or more Total
Repackagers and Relabelers 727 318 144 24 1,212
T bl 6 N b fD . L b i F' b S' dT fL b l A" i
Source: ERG Report, Tables 3-7, 3-9, and 3-11 (Ref. 1).i This table 6 counts a firm more than once if it is engaged in more than one type of labeling activity.
Consequently, 209 labeling firs are double-counted in the totals. However, when total counts are distributed byemployment size, rounding increases this number of double-counted fis to 210. The cost calculations exclude
these 210 firs.
2. Baseline Practice
To determine baseline practices, FDA and ERG contacted a number of medical device
facilties and paricipated in industry meetings regarding unique device identification systems.
ERG also reviewed the most common industry practices with a variety of industry consultants
and with vendors of label printing equipment. As a staring point for this analysis, we generally
assume that many medical device labelers have at least some experience with the components of
labeling, although there may be limited experience with the full range of requirements of the
UDI system specified in the proposed rule. Two-thirds of AdvaMed (a trade association)
members apply some form of barcode on device packages. Nevertheless, only a small subset of
labelers currently prints unique identifiers with variable barcodes on their device labels and
device packages. We assume that the curent practices of this small subset of labelers are
generally in line with the proposed UDI label requirements, though we recognize that they may
have some differences. Reprocessors, specification developers, repackagers, and relabelers were
assumed not to have implemented any portion of the proposed UDI requirements.

17 CDRH200816
The detailed assumptions, calculations, and references supporting the cost analysis can
be found in the ERG report (Ref. 1). For more detail on baseline practices and costs, see section
4.2 and 4.3 of the ERG report.
For certain cost components, such as label printing equipment, a wide variety of possible
compliance strategies exist. To respond to the rule, firms would follow strategies that account
for their specific situation, including their production and packaging methods, the number of
lines and production speed, and the natue of existing labeling practices and systems.
F. Costs of the Proposed Rule
The proposed rule would require labelers of medical devices to place a UDI on the label
of a device, in an easily-readable plain-text form and in a form that uses AIDC technology. A
UDI would consist ofa fixed device identifier (a mandatory portion ofa UDI that could be used
to access data that identifies the specific version or model of a device and the labeler of that
device) and, for class II and class III devices, a variable production identifier (a portion of the
UDI that would be required when certain production information is displayed on the label
including: the lot or batch within which a device was manufactured, the serial number, the
expiration date, or the date of manufactue). The UDI would identify the device throughout its
distribution and use. Proposed section 801.30(a) lists general exceptions from the requirement
for the label ofa device to bear a unique device identifier. Proposed 801.30(a)(l) would except
over-the-counter devices sold at retaiL. The second exception is for class I devices that FDA has
exempted from the GMP regulations. The remaining exceptions list specific types of devices
that would be excepted. Under proposed 801.30(c) labelers of class I devices would not be
required to include the variable production identifier in their UDIs. Ifthe device is an
implantable device, is intended for more than a single use and must be sterilized before each use,

18 CDRH200816
or is stand-alone softare, the UDI would also have to appear on the device itself. The proposed
rule would establish a public database of information for devices labeled with a UDI and would
require the submission of information about each device to FDA.
To build the cost estimate for the proposed rule, we first estimate the costs of subjecting
most medical devices to the UDI requirements. These costs are shown in section F1 and F2 of
this document. For puroses of analysis, this does not include devices excepted from the
proposed rule by proposed 801.30(a)(3) through (12). Throughout this document, for simplicity,
we use "excepted devices" to refer to device types listed in proposed 801.30(a)(3) through (12).
In section F3 ofthis document, we adjust and redistribute the total number of affected
establishments. We separate counts of those establishments that would have lower burdens and
costs because of three additional exceptions. The first exception is that the label of class I device
would need to bear only the device identifier and not the production identifier in the UD1. The
second is that class I GMP exempt devices would be excepted from the proposed rule. Finally,
over-the-counter devices sold at retail, including such devices when delivered directly to
hospitals and other health care facilities, would not be required to bear a UD1. The total costs of
the proposed rule for all domestic labelers with immediate implementation are shown in section
F4. Section F5 shows the total costs of the proposed rule for alllabelers using the phased-in
implementation schedule.
Certain types of labelers would incur only some of the estimated regulatory costs.
Although included in the total number of affected labelers, the cost estimate generally excludes
any labe1er that exclusively handled excepted devices. In this analysis, we assumed that 70
percent of the smallest device manufactuers with fewer than 5 employees and 30 percent of
device manufacturers with 5 to 9 employees would handle only excepted devices. Therefore, of

19 CDRH200816
the 2,424 manufacturing establishments with fewer than 10 employees shown in table 5 of this
document (1,630 establishments with 1-4 employees + 794 establishments with 5-9 employees),
1,379 establishments (70 percent x 1,630 establishments + 30 percent x 794 establishments)
would incur only the minimal costs necessary to determine that all of their devices would be
excepted from the UDI requirements.
Furthermore, over-the-counter devices sold at retail are exempt from the requirements for
a UD1. However, these devices would stil be subject to other requirements of the proposed rule,
such as the date change. Although over-the-counter devices sold at retail are excepted, only
establishments that exclusively label such retail devices would avoid the UDI labeling costs.
Excluding establishments exclusively labeling excepted devices, this analysis assumes that
approximately 10 percent of establishments with fewer than 10 employees manufactue devices
exclusively for the retail market. Consequently, we estimate that only 104labelers ((2,424
establishments - 1,379 establishments) x 10 percent) market exclusively to retail sector.
The analysis also estimates that 3 percent of manufactuing establishments already
provide variable barcode information (lot number, serial number or dates) on their medical
device labels. This percentage represents roughly 108 manufacturing establishments that use
variable barcodes and have incorporated these barcodes into their electronic recordkeeping and
reporting systems. The 3 percent estimate is based on information from AdvaMed about the
curent barcoding practices of their members. As noted in the ERG report, AdvaMed members
primarily own establishments with 50 or more employees; 15 percent of member establishments
use variable barcodes and 85 percent of member establishments use only static barcodes (that
represent the fixed device identifier only). For this analysis, we assume that none ofthe
manufacturers with fewer than 50 employees use variable barcodes, but about 5 percent of these

20 CDRH200816
manufactuers use static barcodes. The manufacturers using variable barcodes may need to make
some modifications to their administrative systems, but we assume they are likely to have
previously absorbed most of the costs for complying with the proposed UDI regulations. For
more detail on our assumptions, see section 4.2 of the ERG report.
The costs of developing and installng a UDI capability would include:
Administration and Plan Development.--Develop a facility plan for implementing a UDI
system and prepare new or modified Standard Operating Procedures (SOPs) to meet FDA's
Quality System regulation.
Paricipate in a UDI System Operated by an Issuing Agency.-- The labeler wil choose
among systems offered by FDA-accredited issuing agencies. All issuing agencies wil provide
access to, and technical advice concerning, their systems for the assignment of device identifiers,
and wil charge a fee for their services.
Purchase Equipment.--Select and purchase equipment to print or place the UDI on
products or packages and verify the quality of the UDI marking. Printing labels may be
conducted in-house or by a contract printer.
Direct Marking.--Se1ect and purchase equipment to etch or otherwse permanently mark
the specific devices that would be subject to direct marking requirements, or apply for an
exception.
Label Redesign.--Redesign and print labels (or add a supplemental label) to add:
. A plain-text UDI
. A static barcode (or other AIDC technology) that represents the device
identifier (i.e., the version or model) when no production identifier appears on the label

21 CDRH200816
. A variable barcode that represents the device identifier and production
identifier when any production identifier appears on the label
. A symbol indicating the use of AIDC technology other than a barcode
. Correct date formats.
Software and Data Integration.--Integrate the UDI data into certain FDA-required device
records, which may require softare or other IT changes.
Recordkeeping and Reporting.--Provide initial information and ongoing updates to the
GUDID.
Table 7 of this document shows each major cost component and whether it accrues at the
level of the establishment or the firm. Curent practices and device types determine which cost
components would apply to a specific labeler. For example, a labeler applying variable barcodes
to device labels and packages, but not subject to direct marking requirements, would incur only
the costs for administration and plan development, label redesign, and submitting data to the
GUDID.
a e -- aior ost omponents 'Y rganizationa eve ncurring ostsCost Component
Organizational Admin. & Register Purchase Direct Label Softare & Record-Level Plan with an Equip- Marking Redesign Data keeping &
Develop- Issuing ment Integration Reportsment Agency
Establishment X X X X X
Fir X X
T bl 7 M' C C b 0 I L I I C
1. Costs for Initial Labelers
Administration and Plan Development.
All labelers of medical devices would need to read and understand the proposed rule to
determine how the rule wil affect them. These costs would be incured on an establishment
basis. Larger establishments with more complex operations involving many devices would need

22 CDRH200816
more time than smaller establishments with few devices. Once labelers understand the UDI
requirements of the proposed rule, they would evaluate their operations and devices and, if
subject to the UDI requirements, would develop a plan to implement these requirements and to
modify their SOPs. Some labelers of devices with identifiers that fully conform to the UDI
requirement would not need to develop an implementation plan to add the UDI to their device
labels and device packages. However, alllabelers with devices subject to the UDI requirements
would need to modify their SOPs to include the UDI in certain records and reports and to add
procedures to report device data to the GUDID. In addition to the UDI requirements, labelers
would need to review their device labels to determine whether they would need to modify the
date format on their device labels within 1 year.
The proposed rule includes effective dates for UDI requirements based on the class of a
device. Labelers with devices from more than one class would decide whether to develop an
implementation plan that follows the staggered effective dates or a plan to implement the UDI
requirements for all devices at one time, regardless of class. To minimize potential disruptions in
establishment operations, we assume that most labelers would likely opt for a plan that
implements the UDI requirements for all of their devices at the same time. As explained in more
detail in section 4.3 of the ERG report, we estimate that labelers would spend between 2.5 hours
and 720 hours to read and understand the proposed rule, to evaluate their devices and operations,
to develop an implementation plan and to modify SOPs, depending on the size of the
establishment and the level of effort required by the proposed rule. For example, establishments
that exclusively label excepted deyices need only read and understand the rule and would have
the smallest burden of 2.5 hours. Establishments with more than 500 employees that label

23 CDRH200816
devices that need to develop a full UDI implementation plan and to modify SOPs would have the
largest burden of 720 hours.
We anticipate that managers would perform these tasks. With an average hourly wage of
$75, including benefits, a very small establishment with fewer than 5 employees would spend
from $190 (2.5 hours x $75 per hour) to $2,250 (30 hours x $75 per hour), depending on the
types of devices labeled and the level of effort required. Similarly, a very large establishment
with more than 500 employees would spend from $2,250 (30 hours x $75 per hour) to $54,000
(720 hours x $75 per hour).
Costs for the estimated 1,379 establishments that exclusively label excepted devices
would equal about $0.3 milion ($190 per establishment x 1,379 establishments). Establishments
that curently print UDI-compliant identifiers on their device labels and packages would spend
about $0.1 milion ($190 per establishments x 105 establishments + $375 per establishment x 21
establishments + $750 per establishment x 37 establishments + $1,500 per establishment x 28
establishments + $2,250 per establishment x 22 establishments.) The remaining 4,677
establishments would incur costs equaling about $42.8 milion ($2,250 per establishment x 1,162
establishments + $4,500 per establishment x 721 establishments + $9,000 per establishment x
2,176 establishments + $18,000 per establishment x 359 establishments + $36,000 per
establishment x 167 establishments + $54,000 per establishment x 91 establishments.) As shown
in table 8 of this document, one-time costs for administration and plan development would total
$43.2 milion ($0.3 millon + $0.1 milion + $42.8 milion).
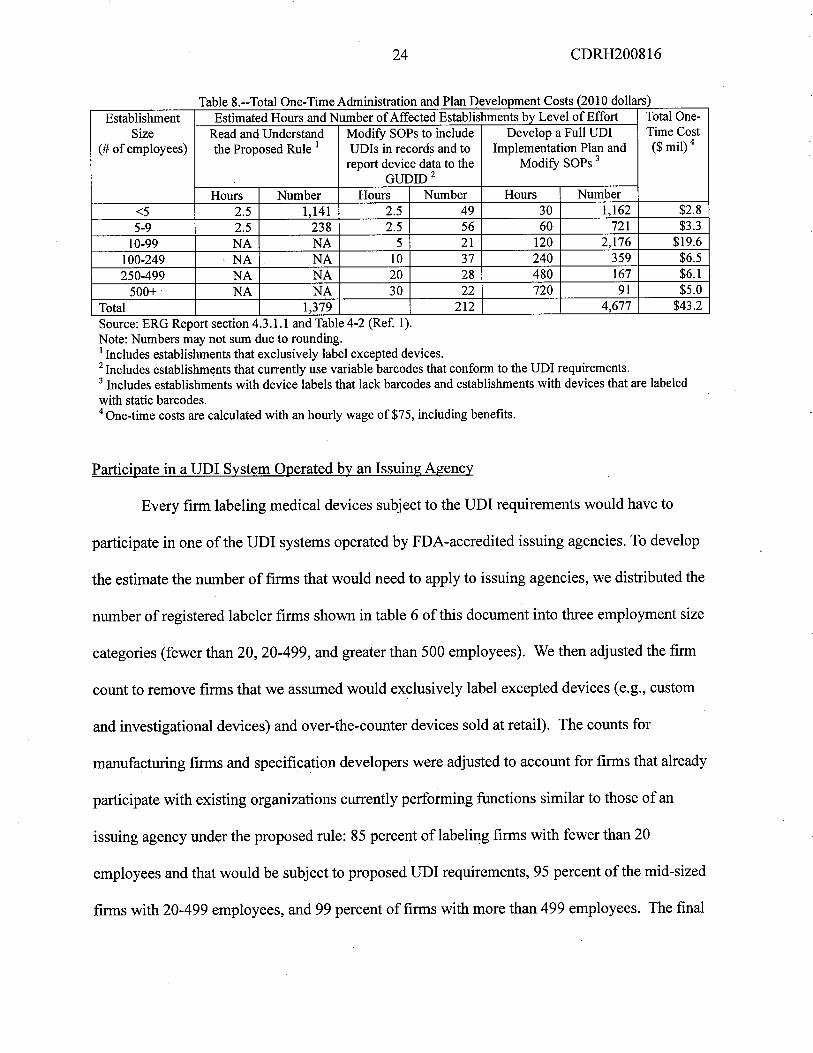
24 CDRH200816
Ta e 8.--Tota One-Time A inistration an P an Development Costs 20 i 0 0 ars
Establishment Estimated Hours and Number of Affected Establishments bv Level of Effort Total One-
Size Read and Understad Modify SOPs to include Develop a Full UDI Time Cost
(# of employees) the Proposed Rule i UDIs in records and to Implementation Plan and ($ mil) 4
report device data to the Modify SOPs 3
GUDID2Hours Number Hours Number Hours Number
..5 2.5 1,141 2.5 49 30 1,162 $2.85-9 2.5 238 2.5 56 60 721 $3.3
10-99 NA NA 5 21 120 2,176 $19.6100-249 NA NA 10 37 240 359 $6.5
250-499 NA NA 20 28 480 167 $6.1
500+ NA NA 30 22 720 91 $5.0Total 1,379 212 4,677 $43.2
bl dm' . d I ( d II )
Source: ERG Report section 4.3.i. and Table 4-2 (Ref. 1).Note: Numbers may not sum due to rounding.i Includes establishments that exclusively label excepted devices.2 Includes establishments that currently use variable barcodes that conform to ile UDI requirements.3 Includes establishments with device labels that lack barcodes and establishments with devices that are labeled
with static barcodes.4 One-time costs are calculated with an hourly wage of $75, including benefits.
Participate in a UDI System Operated by an Issuing Agency
Every firm labeling medical devices subject to the UDI requirements would have to
paricipate in one of the UDI systems operated by FDA-accredited issuing agencies. To develop
the estimate the number of firms that would need to apply to issuing agencies, we distributed the
number of registered labeler firms shown in table 6 of this document into three employment size
categories (fewer than 20, 20-499, and greater than 500 employees). We then adjusted the firm
count to remove firms that we assumed would exclusively label excepted devices (e.g., custom
and investigational devices) and over-the-counter devices sold at retail). The counts for
manufactuing firms and specification developers were adjusted to account for firms that already
paricipate with existing organizations curently performing functions similar to those of an
issuing agency under the proposed rule: 85 percent of labeling firms with fewer than 20
employees and that would be subject to proposed UDI requirements, 95 percent ofthe mid-sized
firms with 20-499 employees, and 99 percent of firms with more than 499 employees. The final

25 CDRH200816
count of firms not paricipating with existing organizations that would incur costs to apply to
issuing agencies equals 476.
To estimate paricipation fees, we relied on publicly available information from HIBCC
(Health Industry Business Communications Council@), an organization that provides services
similar to those that would be provided by an issuing agency under this proposed rule. HIBCC
charges one-time fees according to gross sales revenue and not by the number of products
requiring a unique identifier. HIBCC fees do not include recuring charges. The fee schedule for
GS-1, an organization that also provides similar services, is not publicly available. Using data
from the 2007 Census of U.S. Manufacturers, we estimate the average sales revenue for each size
category to estimate possible paricipation fees. The average sales revenue used to calculate the
three categories of firm fees are: less than $2 milion for the smallest size, under $30 milion for
the middle size, and over $500 milion for the largest size firms.
One-time costs to paricipate in an issuing agency's system would be $500 for the
smallest firms with 19 or fewer employees, $4,000 for firms with 20 to 499 employees, and
$20,000 for firms with 500 or more employees. Therefore, we estimate that the total one-time
costs for 476 firms to apply to issuing agencies would be approximately $0.6 milion. See table 9
of this document. The agency seeks comments on this estimate.
The proposed rule would require that issuing agencies be private nonprofit organizations
or State governent agencies. Moreover, FDA would be able to act as an issuing agency if a
significant number of small businesses would be substantially affected by the fees charged by all
accredited issuing agencies. Although we anticipate that these conditions would limit any
oligopoly power, we request comment from labelers on their experience with paricipation fees,
including recuring fees, charged by existing organizations.

26 CDRH200816
a e .-- osts to aricipate in an ccre ite I system oarsFir Employment Size Adjusted Number of Firs Cost per Fir To Aggregate Costs to
Paricipate Participate($ milion)
Fewer than 20 employees 397 $500 $0.2
20-499 employees 76 $4,000 $0.3
500 or more employees 4 $20,000 $0.1
Total 476 $0.6
T bl 9 C P .. A d dUD S (2010 d II )
Note: Numbers may not sum due to rounding.Source: ERG Report, Table 4-4 (Ref. 1).
Purchase Equipment
The costs to implement required label changes would var widely depending on printing
capabilities and equipment. We generally assume, however, that the methods used and
presentation of a machine readable UDI on labels are the same as those curently used for trade
puroses (e.g., standard linear or 2-D barcode). These barcodes allow for the representation of
the human readable form ofthe UDI above or below the barcode.
Baseline conditions for large establishments with complex automated production lines
would differ from baseline conditions for very small establishments with manual production
lines. The primary basis for the difference in manufacturer response is the prevalence of baseline
digital printing technology. This technology allows for on demand printing of new labels with
both plain text and barcoded UDIs that would incorporate the frequent changes needed to include
the variable component ofthe UDI (e.g., lot number, serial numbers, manufactuing date and
expiration date). We assume that about 3 percent of all manufactuers with automated lines have
installed equipment to print both static and variable barcodes on medical device labels and would
not need to invest in labeling equipment. Some labelers curently apply barcodes with only static
information (e.g., a device identifier that could be used to access information about the labeler
and the specific version or model of the device) that does not include a barcode equivalent of the
variable identifiers.

27 CDRH200816
Larger manufacturers, which are assumed to operate automated lines, indicated that the
two most commonly used labeling methods are (1) use of preprinted labels (including labels
produced by outside contractors) and (2) use of in-line printing systems, such as flexographic
printers (which use printing plates). A UDI requirement that includes variable information would
impose more frequent changes ofthe UDI on the label than would be required with a UDI that
only includes static information. More frequent changes would create a challenge for many
printing technologies, such as printing press technology, which is designed to produce large
numbers of labels or other printed material very cheaply, but is not designed to produce the
frequent label changes that would be necessary to produce barcoded production identifiers.
In response to the proposed rule, labelers may choose to do the following: (1) Continue
using outside contractors to print device labels that incorporate variable information; (2)
purchase and install equipment to print in-house a separate supplemental label with variable
information; (3) purchase and install equipment to move the entire label printing system in-
house; or (4) modify their curent in-house label printing system. The agency requests comments
on how industry expects to implement these provisions, as this may influence the cost estimates
at the final rule stage.
The equipment investment necessary to comply with the proposed rule would var
according to the type and number of production lines. Costs would be higher for establishments
with multiple production lines. The cost to add new equipment ranges from $450 (to add a
verifier in an establishment with 1 manual production line), to about $120,000 (to install a
complete printing system in an establishment with 6 or more automated production lines).
Adding supplemental label printing capabilities requires an investment of $450 in an

28 CDRH200816
establishment with 1 manual line, and from about $21,100 in an establishment with 1 automated
production line to about $31,700 in an establishment with 6 or more automated production lines.
Table 10 of this document presents a sumary of the costs for purchasing, installng and
maintaining equipment to add the UDI barcode to device labels. Takng into account curent
baseline practices, table 10 shows the probabilities and costs of possible compliance responses
for establishments with one manual production line and for establishments with one or more
automated production lines. The costs were calculated by first multiplying the percentage of
establishments in a size category anticipated to choose a paricular compliance response by the
percentage of all establishments in that category. For example, 35 percent (88 percent of 40
percent) of establishments curently using outside label printers would decide to add
supplemental label printing equipment. For this case, the costs for establishments with one
automated production line would equal about $16.2 millon (35 percent x 2,176 establishments
with 1 automated production line x $21,094 supplemental label printing equipment cost).
Similarly, the costs for establishments with 6 or more automated lines would equal about $1.2
millon (35 percent x 110 establishments with 6 or more lines x $31,719). As shown in table 10
ofthis document, the estimated total I-year investment in equipment would cost about $71.5
milion for all affected establishments.
In addition to the one-time investment, labelers would incur anual labor costs of$29.3
milion (cost per establishment to operate verifiers x the number of establishments for each type
of production line) and annual equipment operating costs of $7.2 milion (10 percent of the $71.5
millon investment cost). The estimated total anual costs to operate and maintain label printing
equipment would be $36.5 milion ($29.3 milion, + $7.2 milion). We request detailed

29 CDRH200816
comments from industry on these cost estimates, including the assumptions, many of which are
detailed in the ERG report.

30 CDRH200816
.. ...
...""
.. v
. .."
1....
1"...
..._.
.....
..~",
. _v.
."''''
'_...
.....
......
.... .._--i-&&_&&&_&&V~, - --
---
Per
Est
ablis
hmen
t Equ
ipm
ent a
nd L
abor
Cos
ts,
Man
ual
Aut
omat
edbv
Num
ber
of L
ines
iLi
nes
(%Li
nes
(%M
anua
lA
utom
ated
Est
ablis
h-E
stab
lish-
Est
ablis
hmen
ts, b
y B
asel
ine
Lab
el P
rint
ing
Syst
emm
ents
)ments) 2
1 line
1 lin
e 2-
3 lin
es4-
5 lin
es
6+ li
nes
Tot
al
Number of establishments, by assumed number of
prod
uctio
n lin
es1,
883
2,17
635
914
811
04,
677
Inst
all f
ull o
n-lin
e la
bel p
rint
ing
syst
em o
n au
tom
ated
line
NA
$43,
594
$46,
813
$93,
625
$119
,438
Inst
all s
uppl
emen
tal l
abel
sys
tem
on
auto
mat
ed li
neN
A$2
1,09
4$2
1,09
4$2
4,06
3$3
1,71
9
FTE
s to
ope
rate
ver
ifie
rs$0
0.15
0.30
0.60
1.00
Ann
ual
labo
r co
st to
ope
rate
ver
ifie
rs o
n au
tom
ated
line
3$0
$6,9
47$1
3,89
4$2
7,78
7$4
6,31
2
Eau
ipm
ent c
osts
to o
rint
labe
ls--
man
ualli
nes
$450
NA
NA
NA
NA
Est
ablis
hmen
ts u
sinl
I ou
tsid
e la
bel p
rint
ers
40%
40%
Equipment Costs ($ milion), by Number of
Line
s i
Switc
h to
new
out
side
labe
l pri
nter
, add
lot #
s (1
0% o
f 40
%)
4N
A4%
NA
NA
NA
NA
NA
NA
Mov
e en
tire
labe
l ope
ratio
n in
-hou
se (
2% o
f 40
%)
NA
1%N
A$0
.8$0
.1$0
.1$0
.1$1
.Add small supplemental
labe
l, ap
plie
d in
-hou
se (
88%
of
40%
)N
A35
%N
A$1
6.2
$2.7
$1.
$1.2
$21.
Man
. lin
e: s
witc
h to
new
out
side
labe
l pri
nter
, add
lot #
s (2
0% o
f 40
%)
8%N
AN
A(d
)N
AN
AN
AN
AN
A
Man
. lin
e: m
ove
entir
e la
bel o
pera
tion
in-h
ouse
(75
% o
f 40
%)
30%
NA
$0.3
NA
NA
NA
NA
$0.3
Man. line: add small supplemental
labe
l, ap
plie
ö in
-hou
se (
5% o
f 40%
)2%
NA
$0.0
NA
NA
NA
NA
$0.0
Est
ablis
hmen
ts p
rint
ing
labe
ls in
-hou
se w
ith p
rint
ing
syst
ems
that
do
not a
ccom
mod
ate
vari
able
info
rmat
ion
0%45
%
Mod
ifv
entir
e la
bel o
rint
ing
oper
atio
n (6
0% o
f 45
%)
0%27
%$0
$25.
6$4
.5$3
.7$3
.6$3
7.4
Add small supplemental
labe
l, ap
plie
d in
-hou
se (
40%
of
45%
)0%
18%
$0$8
.3$1
.4$0
.6$0
.6$1
0.9
Est
ablis
hmen
ts w
/labe
l ori
ntin
g sy
stem
s ac
com
mod
atin
g va
r. d
ata
60%
15%
Mod
ify
labe
l with
exi
stin
g pr
intin
g ea
uipm
ent (
100%
of
15%
)N
A15
%$0
NA
NA
NA
NA
NA
Man
. lin
e: m
odif
y la
bel w
/exi
stin
g eq
uipm
ent (
loo%
of
60%
)60
%N
A$0
.5N
AN
AN
AN
A$0
.5
Tot
al I
nves
tmen
t$7
1.5
Tot
al
labo
r $0
$15.
1$5
.0$4
.1$5
.1$2
9.3
Tot
al O
&M
(10
per
cent
of
equi
pmen
t cos
t) p
lus
Lab
or$3
6.5
Sou
rce:
ER
G R
epor
t, T
able
4-7
(R
ef. 1
)N
ote:
Num
bers
may
not
sum
due
to r
ound
ing.
i Fro
m E
RG
Tab
le 4
-6. N
umbe
rs o
f es
tabl
ishm
ents
are
fro
m E
RG
Tab
le 3
-8, a
djus
ted
for
the
3 pe
rcen
t of
man
ufac
ture
rs w
ho a
re a
ssum
ed to
be
prin
ting
vari
able
bar
code
s.
The
se c
ount
s ex
clud
e sm
all m
anuf
actu
rers
ass
umed
to b
e ex
clus
ivel
y m
anuf
actu
ring
exc
lude
d de
vice
s or
who
are
ass
umed
to b
e us
ing
UPC
s ex
clus
ivel
y.2
From
ER
G T
able
4-5
and
ER
G a
ssum
ptio
ns (
see
text
).3
Ass
umes
a w
age
rate
plu
s 29
per
cent
fri
nge
of $
22.2
7 pe
r ho
ur (
BL
S, 2
009)
for
insp
ecto
rs in
NA
ICS
339.
4 In
crem
enta
l cos
ts f
or o
utsi
de p
rint
er la
bels
ass
umed
pri
mar
ily c
osts
of
coor
dina
tion,
whi
ch is
pas
sed
thro
ugh
to la
bele
rs. S
ee E
RG
Tab
le 4
-12.

31 CDRH200816
Direct Marking.
The proposed rule would require manufacturers of implanted devices (devices intended
to be left in the body continuously for 30 days or more) and devices intended for multiple uses
(referred to as multiple-use devices) that require sterilization before each use to be permanently
marked with a UD1. The proposed rule would also require stand-alone software devices to be
directly marked.
Exceptions to Direct Marking of Devices.
The proposed regulation provides exceptions to the direct marking requirements.
Exception criteria for devices apply as follows: when marking would interfere with the safety or
effectiveness of the device; when a device canot be marked because it is not technologically
feasible to mark the device; when a device is intended to remain implanted continuously for a
period of less than 30 days, unless the Commissioner determines otherwise in order to protect
human health; when the device has been previously direct-marked; when the device is sold at
retail and bears a UPC; and softare that is not stand-alone software, but which is a component
of a medical device.
Exceptions from direct marking devices are expected because of the size of the device,
the diffculty in marking certain material, or to the lack of adequate surface space. We assume
that if no machine-readable mark can technologically be applied to the device, then no easily
readable plain-text UDI could be applied either. Thus, marking the device would not be
technologically feasible. We fuher assume that easily readable does not mean "with
magnification." With the diverse types of medical devices, we acknowledge that this conclusion
may overly simplify the challenges some labelers would face when deciding how to directly
mark devices. Because we lack detailed information about this issue, the agency requests

32 CDRH200816
comments on the technological feasibility of direct marking, the costs of technologically feasible
direct marking, and about the challenges direct marking would cause device labelers.
We estimate that 1,222 manufacturers and specification developers that listed either an
implant or multiple-use device that might need to be directly marked; 517 establishments with
multiple-use devices and 705 establishments with device implants. We assume that 75 percent of
labelers of multiple-use devices and 80 percent of implant labelers currently directly mark their
devices in some maner, and 20 percent of labelers that curently directly mark devices use
barcodes. Ofthose that do not mark devices, 5 percent of multiple-use device labelers and 15
percent of implant device labelers are assumed to manufactue devices that would be exempt
from direct marking (e.g., it is not feasible to mark the device, or direct marking would interfere
with the safety and effectiveness of the device).
The proposed rule would require labelers to document the basis for any exception in the
design history fie, and to notify FDA of the first two exceptions. We estimate the
documentation would require about 10 hours per exception. Using an average hourly wage cost
of $75, the average cost of an exception would be $750 per exception.
Based on discussions with vendors of direct marking equipment and manufacturers of
marked devices sumarized in the ERG report, we estimated that about 132 establishments
((517 multiple-use establishments x 0.05) + (705 implant establishments x 0.15)) would incur
costs to document exceptions to the direct marking requirement. Furhermore, these
establishments would incur anual costs to document exceptions for new products. The number
of initial exceptions per establishment is scaled up from one device for the smallest
establishments to 50 devices for the largest establishments. In subsequent years, establishments
might introduce an average of 0.3 new devices for the smallest establishments, up to an average

33 CDRH200816
of 13 products for the largest establishments. The estimated I-year costs for 132 establishments
to document exceptions to the direct marking requirements would total about $0.5 millon;
anual costs to document exceptions for new products would be approximately $0.1 milion.
Directly Marking Multiple-use and Implanted Devices:
Costs for Establishments that Currently Directly Mark Medical Devices.
Table 11 ofthis document presents the costs for software upgrades and redesign costs for
establishments curently marking implants and multiple-use devices. We estimate that
approximately 75 percent ofthe multiple-use establishments and 80 percent ofthe implant
establishments curently mark their products; 20 percent of these establishments curently use
barcodes. Thus, approximately 760 establishments that curently mark devices, but not with
barcodes, would incur software and redesign costs ((517 multiple-use establishments x .75
curently marking x .80 not using barcodes) + (705 implant establishments x .80 curently
marking x .80 not using barcodes)).
Establishments that curently mark their products, but not with barcodes, would incur
software costs of $600 to add barcode capabilities to existing marking systems. This estimate
assumes that space limitations would prevent directly marking with a plain text UDI and,
therefore, overstates costs. The total cost for software upgrades for direct marking would equal
about $0.5 milion. Affected establishments would also incur costs to redesign curent marks to
accommodate the UD1. The redesign is needed to add a UDI, either in plain text or 2-D barcode
format, to the existing mark. Redesign costs range from $1,250 per establishment for the
smallest establishments to $75,000 for the largest establishments. These redesign costs are
assumed the same as the costs to redesign the main packaging label, discussed in the Label
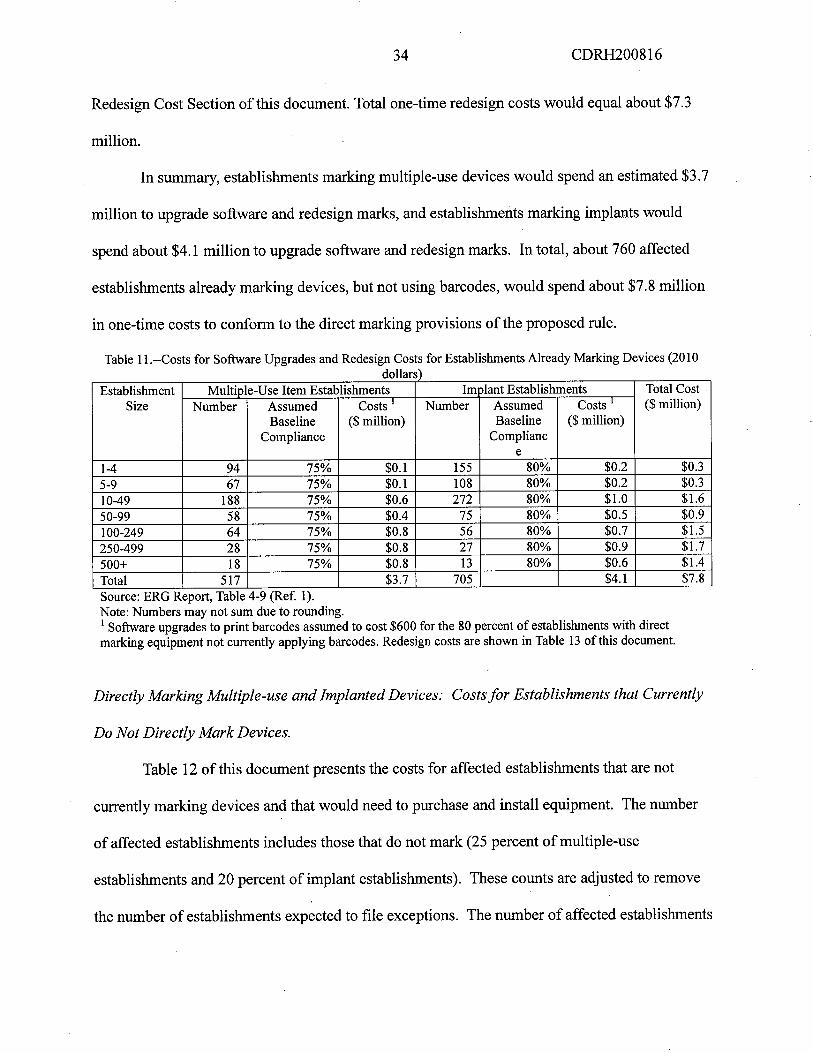
34 CDRH200816
Redesign Cost Section of this document. Total one-time redesign costs would equal about $7.3
milion.
In summary, establishments marking multiple-use devices would spend an estimated $3.7
milion to upgrade software and redesign marks, and establishments marking implants would
spend about $4.1 millon to upgrade softare and redesign marks. In total, about 760 affected
establishments already marking devices, but not using barcodes, would spend about $7.8 milion
in one-time costs to conform to the direct marking provisions ofthe proposed rule.
Table 11 .--Costs for Softare Upgrades and Redesign Costs for Establishments Already Marking Devices (2010dollars)
Establishment Multiple-Use Item Establishments 1m olant Establishments Total CostSize Number Assumed Costs i Number Assumed Costs 1 ($ milion)
Baseline ($ milion) Baseline ($ milion)Compliance Complianc
e
1-4 94 75% $0.1 155 80% $0.2 $0.3
5-9 67 75% $0.1 108 80% $0.2 $0.3
10-49 188 75% $0.6 272 80% $1.0 $1.6
50-99 58 75% $0.4 75 80% $0.5 $0.9100-249 64 75% $0.8 56 80% $0.7 $1.250-499 28 75% $0.8 27 80% $0.9 $1.7
500+ 18 75% $0.8 13 80% $0.6 $1.4
Total 517 $3.7 705 $4.1 $7.8
Source: ERG Report, Table 4-9 (Ref. 1).Note: Numbers may not sum due to rounding.i Softare upgrades to print barcodes assumed to cost $600 for the 80 percent of establishments with direct
marking equipment not currently applying barcodes. Redesign costs are shown in Table 13 of this document.
Directly Marking Multiple-use and Implanted Devices: Costs for Establishments that Currently
Do Not Directly Mark Devices.
Table 12 of this document presents the costs for affected establishments that are not
curently marking devices and that would need to purchase and install equipment. The number
of affected establishments includes those that do not mark (25 percent of multiple-use
establishments and 20 percent of implant establishments). These counts are adjusted to remove
the number of establishments expected to fie exceptions. The number of affected establishments

35 CDRH200816
that would incur costs to install and operate direct marking equipment equals 138 ((517 multiple-
use establishments x .25 curently not marking) - 26 multiple-use establishments filing
exceptions) + (705 implant establishments x .20 curently not marking) - 106 implant
establishments filing exceptions).
We anticipate that these establishments would likely choose C02 lasers or yttrium
aluminum garet (YAG) lasers to directly mark devices. Costs would be about $12,000 for a
C02 laser and $55,000 for a YAG laser, plus engineering costs, equaling an estimated 75 percent
of capital expenditures, for installation and costs for materials. Capital and installation costs for
smaller establishments with one production line that purchase C02 lasers would be about
$21,000 ($12,000 x 1.75 engineering and installation x 1 line). We assume that YAG lasers
would be used for all implanted products and in larger establishments. The capital cost for
smaller establishments with one production line to purchase and install a YAG laser would be
about $96,250 ($55,000 per unit x 1.75 engineering and installation x 1 line); costs are scaled up
for larger establishments with greater equipment needs. For example, the largest establishments
with 250 or more employees are judged to require 3-4 YAG lasers at $55,000 each, and 1-2 fully
automated lasers at $150,000 each plus engineering and installation.

36 CDRH200816
ble 12
II a
ndki
ng E
.bl
ishm
Cki
ng (
2010
dol
lars
)---- -
- --- ----
.. ~
..-~
..---
----
---
---0
--
- - - - -
. J
Est
ablis
hmen
tN
umbe
r of
Cap
ital C
osts
Plu
sPr
oces
sE
stab
lishm
ents
Nee
ding
One
Tim
e C
osts
5T
otal
O
ne
Tot
al
Size
Line
s i
Installation 2
Redesign 3
Equ
ipm
ent
4 ($
mili
on)
Tim
e C
osts
Ann
ual
CO
2Y
AG
s or
Mul
tiple
-Im
plan
tM
ultip
le-
Impl
ant
($ m
ilion
)Costs 6
Las
ers
High Speed
Use Item
Use
Item
($ m
ilion
)L
aser
s.
1-4
1$2
1,00
0$9
6,25
0$2
5,00
019
8$0
.5$1
.0$1
.4$0
.1
5-9
1$2
1,00
0$9
6,25
0$2
5,00
013
5$0
.3$0
.7$1
.0$0
.1
10-4
9i
$21,
000
$96,
250
$75,
000
3814
$0.9
$2.4
$3.3
$0.3
50-9
9i
$21,
000
$96,
250
$100
,000
124
$0.3
$0.7
$1.0
$0.1
100-
249
2N
A$1
92,5
00$1
50,0
0013
3$2
.6$1
.0$3
.5$0
.4
250-
499
4-6+
NA
$640
,938
$200
,000
61
$3.6
$1.2
$4.8
$0.5
500+
4-6+
NA
$820
,313
$250
,000
4i
$3.0
$0.7
$3.7
$0.4
Tot
al10
335
$11.
$7.6
$18.
7$1
.9
Sou
rce:
ER
G R
epor
t, T
able
4-
i 0 (
Ref
. 1).
Not
e: N
umbe
rs m
ay n
ot s
um d
ue to
rou
ndin
g.i A
ssum
ptio
ns a
bout
num
bers
of p
rodu
ctio
n lin
es fr
om E
RG
Rep
ort,
Tab
le 4
-7 (
Ref
. 1).
2 C
osts
for
the
C02
lase
r on
a p
er p
rodu
ctio
n lin
e ba
sis;
cos
ts f
or Y
AG
s an
d hi
gh s
peed
lase
rs a
re o
n a
per
esta
blis
hmen
t bas
is. C
apita
l cos
ts in
clud
e
engi
neer
ing
cost
s as
sum
ed a
t 75%
of
capi
tal c
ost.
Onl
y sm
alle
r op
erat
ions
pro
duci
ng m
ultip
le-u
se it
ems
are
assu
med
to u
se C
02 la
sers
due
to h
igh
cost
of
mat
eria
ls. C
osts
for
est
ablis
hmen
ts w
ith 2
50 o
r m
ore
empl
oyee
s in
clud
e co
sts
to in
stal
l 3-4
YA
G la
sers
at $
55,0
00 e
ach,
and
1-2
ful
ly a
utom
ated
lase
rs a
t$
1 50
,000
eac
h. I
mpl
ant e
stab
lishm
ents
wil
only
inst
all Y
AG
s or
hig
h sp
eed
lase
rs.
3 Pr
oces
s re
desi
gn c
osts
for
impl
ants
onl
y. C
osts
wil
var
wid
ely,
and
we
assu
me
that
cos
ts w
il in
crea
se w
ith e
stab
lishm
ent s
ize,
fro
m $
25,0
00 to
$25
0,00
0
per
esta
blis
hmen
t.4
Adj
ustin
g fo
r th
e nu
mbe
r of
est
ablis
hmen
ts a
pply
ing
for
exce
ptio
n an
d as
sum
ing
base
line
com
plia
nce
rate
s of
75
perc
ent f
or m
ultip
le-u
se d
evic
e
esta
blis
hmen
ts a
nd 8
0 pe
rcen
t for
impl
ant e
stab
lishm
ents
.5
One
tim
e co
sts
incl
ude
40 h
ours
per
line
to v
alid
ate
oper
atio
ns s
how
ing
no h
ealth
and
saf
ety
issu
es.
6 A
nnua
l cos
ts a
ssum
ed a
t 10
perc
ent o
f one
-tim
e co
sts.

37 CDRH200816
Process redesign costs for implant manufactuers range from $25,000 for small
establishments with one production line to $250,000 for the largest establishments with multiple
production lines. Furhermore, establishments that directly mark implants would incur one-time
costs of $3,000 per production line (40 hours at $75 per hour) for a manager to validate
operations and document that the safety of the devices has not been compromised. We estimated
validation costs based on collecting supporting documentation from literature searches of similar
devices or materials that have been previously marked without compromising safety. However,
because of the complexities of direct marking certain devices, the agency requests comment on
how industry would respond to validation requirements, including the cost of testing to
demonstrate that direct marking would not interfere with the safety or effectiveness of the device.
Moreover, we request comment from smalllabelers about the cost estimate and their expected
response to this requirement. One-time costs of direct marking would be about $11.1 milion for
multiple-use devices and about $7.6 milion for implanted devices, for a total of$18.7 milion.
Anual maintenance and operating costs would equal about 10 percent of the one-time
investment in direct marking equipment, or about $1.9 milion.
Costs for Stand-Alone Software Devices.
Stand-alone softare devices would be required to have a UDI present on the starup
page or in a menu, such as in the help menu under an "About * * *" selection. Because FDA has
provided, at a minimum, 3 years between promulgation and implementation, and because
softare revisions are made frequently, the work to add the UDI in these locations within the
softare would be integrated into regular revision and update cycles. Most of the time needed to
meet this requirement is for planing the implementation ofUDI in general, and this has been
accounted for in the Administration and Planing Cost Section of this document. Any additional .

38 CDRH200816
time needed to add the UDI to the softare itself (while the starp page is being edited to
contain a new version identifier or revision date) would be a negligible increment to the 30 to
720 hours allotted to the varous size establishments to plan for UDI implementation. Although
future software revisions would require new UDIs, these changes would be incorporated while
other revisions were being made to the softare. Moreover, because some softare might be
sold as a downloadable electronic file rather than as a packaged device, the traditional costs for
relabeling (e.g. printing and materials) are somewhat overstated. Therefore, the cost of including
a UDI in stand-alone software would be a negligible addition to costs already estimated for those
establishments.
Total Costs for Directly Marking Medical Devices.
The one-time total estimated costs to directly mark multiple-use and implanted devices
would be $27.0 milion, with anual costs for operation and maintenance of about $2.0 milion.
Incremental costs for direct marking stand-alone softare devices are assumed to be a negligible
addition to costs already estimated for affected establishments. Because of uncertainty about
curent compliance and labeler response to the direct marking requirements, we request detailed
comments from industry about the industry response and the one-time and annual incremental
costs for direct marking medical devices and filing exceptions.
Label Redesign.
The proposed rule would require that, within 1 year, labelers modify the format of dates
displayed on device labels. In addition, labelers of devices subject to the UDI requirements
would need to add the UDI to device labels according to the implementation schedule described
in the preamble of the proposed rule. Because the proposed rule would leave the remaining
content of device labels unchanged, labelers may choose to coordinate the label redesign at the

39 CDRH200816
establishment leveL. As the size of the establishment increases, the number of devices and
production lines increases and the required effort increases.
Labels can be permanently attached on the device itself or displayed on the packaging.
The cost estimates of the proposed requirement that the UDI be placed on the device label and
device package assume that new levels of packaging are not needed. For example, shelf packs of
class I devices containing identical multiples that are not individually packaged and labeled
would only need to add the UDI to the shelf pack itself. However, device packages that contain
other device packages would need different UDIs on the inner and outer device packages.
For the purose of this analysis, we assume that no device labels have dates presented in
the precise format that would be required by the proposed rule. Although labelers would have 1
year to redesign device labels, to avoid the cost of two label redesigns labelers would likely
redesign their device labels to add suffcient space for the UDI in human-readable and AIDC
format at the same time as they modify the date format. Labelers would print the UDI on the
redesigned label at the date specified in their implementation plan. Although the number of
labelers that include dates on their device labels is unkown, we conservatively anticipate that
most device labels have some type of date printed on the label (e.g., expiration date or date of
manufacture). Consequently, we estimate that labelers would incur one-time label redesign costs
in the first year.
Table 13 of this document presents the estimated one-time costs to redesign device
labels. As noted in the ERG report, these costs are estimated to range from $1,250 for
establishments with 1 to 4 employees to $75,000 for establishments with more than 500
employees. For about 4,900 labelers that would redesign device labels in the first year, the total
one-time costs would equal approximately $43.0 millon. However, we note that these estimates

40 CDRH200816
have a high degree of uncertainty (see the Uncertainty Section G). Consequently, we request
detailed comment from industry on this estimate. In addition, we request comment from industry
on whether the I-year effective date for complying with date formats is suffcient or whether this
requirement should coincide with the phase-in periods for the label to bear a UD1.
a e -- ne- ime ostto e esign an o i evice a e s 01 o ars
Employment Size Number of Costs Per Establishment Total One-Time CostsEstablishments ($ milion)
1-4 1,211 $1,250 $1.5-9 777 $2,500 $1.910-49 1,725 $5,000 $8.650-99 472 $10,000 $4.7100-249 396 $20,000 $7.9250-499 195 $50,000 $9.7500+ 113 $75,000 $8.5Total 4,889 $43.0
'L bl 13 0 T C Rd' d M dfy D . L b i i (2 0 d II )
Source: ERG Report, Table 4- 11 (Ref. 1).Note: Numbers may not sum due to rounding.i Because no labelers are assumed to present label information in the precise format that would be required by the
proposed rule, all labelers of non-excepted devices would need to redesign labels. We assumed that labelers ofexcepted devices would not incur costs to redesign labels. Labelers of devices sold at retail and labelers thatcurrently use variable barcodes that would only be changing date formats.
Supplemental labels, larger labels, and new label printing technologies would increase
the cost to produce device labels displaying the UD1. Using U.S. Census data on the value of
materials consumed, we estimate that materials for labels, such as paper and ink, represent about
0.2 percent of all material costs, or $58.1 milion anually. The increase in materials cost for
affected device labels is estimated at 10 percent, or about $5.8 milion anually.
Small establishments that choose to add manually a supplemental label would incur costs
to affx the supplemental labeL. An estimated 38 establishments with fewer than 10 employees (2
percent of the 1,883 establishments in this size category) would each spend about $2,625
anually to add the supplemental label to their devices (125 hours x $21 per hour). For all of
these labelers, anual incremental labor costs would be about $0.1 milion ($2,625 per
establishment x 38 establishments).

41 CDRH200816
Finally, labelers would incur costs for the increased amount of time needed to coordinate
print jobs with outside printing contractors. Both labelers and printing contractors would spend
additional time to ensure proper printing of the variable portion of the UD1. Similar to other
labeling costs, the time needed to coordinate label printing increases with the size of the
establishment. An estimated 271 establishments would continue to use outside contractors to
print labels with the UD1. We estimate that the total time needed to coordinate with an outside
printer would be 50 hours for establishments with 1 to 9 employees, 100 hours for establishments
with 10 to 49 employees, 200 hours for establishments with 50 to 99 employees, 800 hours for
establishments with 100 to 249 employees, 1,200 hours for establishments with 250 to 499
employees and 2,400 hours for establishments with 500 or more employees With a wage cost of
$75 per hour, the anual cost for 271 affected establishments to coordinate outside printing
would equal about $2.6 milion (50 hours x $75 per hour x 151 establishments + 100 hours x $75
per hour x 86 establishments + 200 hours x $75 per hour x 18 establishments + 800 hours x $75
per hour x 11 establishments + 1,200 hours x $75 per hour x 5 establishments + 2,400 hours x
$75 per hour x 0 establishments).
the total anual incremental costs to redesign and modify device labels to add the UDI
and change the date format would equal about $8.5 millon: $5.8 milion for additional materials,
$0.1 milion for additional time to apply supplemental labels, and $2.6 milion for additional
time to coordinate printing. Because of uncertainty about labeler response and possible curent
compliance, we request comment from industry about the one-time and anual incremental costs
for redesigned medical device labels.
Software and Data Integration.

42 CDRH200816
The proposed rule would require integration of the UDI into existing information systems
and installation of barcode printing software. Because information technology performs many
fuctions in an organization, medical device firms with multiple establishments would
coordinate decisions on information technology systems. at the firm level rather than at the
establishment leveL. Firms would need to add UDI barcodes to device labels, to incorporate the
UDI into device related records and correspondence with FDA, and to manage the device data
required for submission to the GUDID. The one-time investment in softare and related
measures include the costs to do the following: (1) Purchase the softare packages or softare
licenses needed to print barcodes; (2) add the UDI to existing information systems; (3) install,
test, and integrate the barco ding software with existing information technology systems; (4)
validate that softare meets FDA software validation requirements; and (5) train employees.
As of March 2010, there were 5,566 domestic initiallabeler firms in the FDA registration
and listing database (see Table 6 ofthis document). When these firms were distributed by type
as shown in Table 6, approximately 210 firms were double counted because they owned more
than one type of establishments (e.g., a firm could own a manufactuing establishment and a
specification development establishment). Adjusting for double counting, we estimated that
1,239 firms exclusively label excepted devices. In addition, an estimated 85 firms have
establishments that curently use variable barcodes and an estimated 95 firms have
establishments that exclusively label over-the-counter devices sold at retaiL. These firms have
already integrated identifiers and labeled device data into their information systems and have
softare systems in place that would comply with the proposed rule. Consequently, we
anticipate that any regulatory costs for changes to software that would be required by the
proposed rule would be negligible for these firms. In contrast, we anticipate that the remaining

43 CDRH200816
3,937 firms (5,566 firms - 210 double-counted firms- 1,239 firms with excepted devices - 180
firms that currently print UDI-compatible identifiers) that do not curently comply with the UDI
requirements of the proposed rule would incur one-time and anual costs for investment in
information technology and employee training.
An estimated 1,162 small firms operate one manual production line and could readily
adapt their information systems. In some cases, firms would manually track barcodes and device
information. However, to conform to the softare validation requirements of the quality system
regulations, we assume that all of these small firms would need to purchase a softare package
that includes FDA validation tools. Once installed and tested, these firms would require no
fuher validation of their information systems. The one-time costs for small firms with fewer
than 5 employees to purchase, install and test softare would total about $800, including $200
for the softare.
Larger firms with numerous medical devices might need to coordinate multiple
production lines and multiple establishments. As firm size increases, the complexity of
information management systems increases and firms would require more sophisticated barcode
softare packages and multiple softare licenses. As a result, the costs of softare and software
licenses increase as the size of the firm increases. The estimated costs of software range from
about $7,500 for firms with 5 to 19 employees to $130,000 for firms with 1,000 or more
employees.
Integrating device UDIs into existing management information systems requires a certain
level of effort to install new software, verify, test and validate that the new softare fuctions as
expected, and to make any changes to existing systems. Firms would also need to test and
validate any softare that would be used to submit device data to the GUDID. Similar to other

44 CDRH200816
costs, the level of effort to integrate UDIs increases as the size ofthe firm increases. Moreover,
adding barcode softare to complex management information systems would require additional
time to test and validate the softare. Some large firms have integrated enterprise resource
planing (ERP) systems; a complex computer system that links all ofthe firm's fuctions in a
standardized enterprise-wide environment to control the flow of information within the
organzation and control the flow of data with outside sources. Such systems handle asset
management, financial and human resources, production, design, sales and marketing. Large
firms that have fully integrated ERP systems would require extensive testing and validation to
ensure that modifying their systems to accommodate the UDI and the associated device data
would have no unforeseen effects on other aspects of the firm's information systems. Because
these types of systems are designed to control the flow of information, validation would be of
primary importance to the fuctioning of ERP systems. Consequently, there are considerable
cost differences for validation between firms with ERP systems and similar-sized firms without
ERP systems. As shown in Table 14 of this document, the one-time costs to purchase, install and
integrate, verify and test, and validate softare range from $9,500 for firms with 5 to 19
employees to $780,000 for firms with more than 1,000 employees.
Firms would also need to train employees to use the barco ding softare. Similar to the
investment in softare, the number of employees to train increases as the size of the firm
increases. For the initial employee training, firms would spend from $100 for the smallest firm
(fewer than 5 employees) to train 1 person, to $125,000 for the largest firms (more than 1,000
employees) to train 1,250 people. We consider initial employee training as a one-time cost of the
proposed rule.

45 CDRH200816
Including the initial training costs, the total one-time costs for software associated with
the UDI range from $900 for firms with fewer than 5 employees to $905,000 for firms with
1,000 or more employees. Table 14 of this document presents a detailed description of the
anticipated one-time software-related costs by size of the firm.
Once the softare has been installed and shown to fuction as expected, firms would stil
need to maintain and validate the software on an annual basis. Furhermore, some on-going
training of employees would be needed. These anual costs are shown in table 14 of this
document and range from $61 for firms with fewer than 5 employees to $94,650 for firms with
1,000 or more employees.
T bl 14 P F" S ft A d C t fì UDI C l b In't I L b I (2010 d II )a e .-- er ir 0 are ssociate os s or ompiiance y iia a e ers oarsCost Element Emplovment Size by Fir (Number of Emplovees)
1-4 i 5-19 20-99 i 100- 200-4994 500-999' 1000+ )
1993
One-Time Costs and Initial Employee TrainingSoftare $200 $7,500 $15,000 $30,000 $52,500 $75,000 $130,000
Installation, Integration, $600 $1,000 $5,000 $25,000 $45,000 $150,000 $250,000
Verification & TestingValidation $0 $1,000 $2,000 $3,500 $55,000 $250,000 $400,000
Training 6 $100 $1,000 $5,000 $17,500 $37,500 $75,000 $125,000
Per Firm One-Time Costs $900 $10,500 $27,000 $76,000 $190,000 $550,000 $905,000
Annual CostsTraining (25% of initial $25 $250 $1,250 $4,375 $9,375 $18,750 $31,250
training)Validation (10% of one-time $0 $100 $200 $350 $5,500 $25,000 $40,000
validation)Softare Maintenance $36 $1,350 $2,700 $5,400 $9,450 $13,500 $23,400
Contract (18% of one-timesoftare)
Per Fir Annual Costs $61 $1,700 $4,150 $10,125 $24,325 $57,250 $94,650
Source: ERG Report, Table 4-13 (Ref. i).Note: Numbers may not sum due to rounding.i Establishments have limited production; includes purchase of simple softare, simple testing and no validation.2 Includes one UDI server, one establishment and one production line.3 Requires more testing than firs with 20-99 employees; includes two softare licenses.
475 percent offirs purchase two softare licenses; 25 percent offirs have complex ERP systems requiring
more expensive softare and more time-consuming integration.5 Assumes more complex installation requirements associated with ERP systems with more establishments to
consider.6 Per employee cost of training equals $100.

46 CDRH200816
The industry totals for the one-time and anual costs for softare and related costs are
shown in table 15 ofthis document. An estimated 3,937 firms would spend about $174.0 milion
in one-time costs and about $21.1 milion in total annual costs. Because software is a major cost
of the proposed rule, we request detailed comment from industry on our estimate and about any
pertinent experiences they may have integrating new identifiers into their software systems.
Emplovment Size bv Firm (Number of Employees) Total
1-4 5-19 20-99 100-199 200-499 500-999 1000+
Number of Firms 1 1,162 1,403 980 172 96 36 89 3,937
One Time CostsPer Firm ($) $900 $10,500 $27,000 $76,000 $190,000 $550,000 $905,000
Industry Total ($ mil $1.0 $14.7 $26.5 $13.1 $18.2 $20.0 $80.5 $174.0
Annual CostsPer Firm ($) I $61 $1,700 $4,150 $10,125 $24,325 I $57,250 $94,650
Industry Total ($ mil I $0.1 $2.4 $4.1 $1.7 $2.3 I $2.1 $8.4 $21.
Table 15 --Softare Associated Costs for UDI Compliance by Initial Labelers (2010 dollars)
Source: ERG Report, Table 4-13 (Ref. 1) and Table 14 of this document.Note: Numbers may not sum due to rounding.i All firm counts are adjusted to account for: (a) 85 firms printing variable barcodes at this time, (b) 1,334 firms only labeling
excepted devices or only labeling over-the-counter devices sold at retail, and (c) 210 firms that were double counted whenbreaking out firms by establishment types owned (see Section 3 of the ERG Report).
Recordkeeping and Reporting.
In addition to proposing that the UDI be displayed on the labels of medical devices, the
proposed rule would require that labelers add the UDI to existing records and to include the UDI
in reports and submissions to FDA.2 For its par, FDA wil include the UDI in public health
communcations, such as public health notifications, recall alerts, cease distribution and
notification orders. One aspect of plan development includes the review of SOPs to ensure the
requirements of the proposed rule are met. During the review of SOPs, labelers would identify
and modify the procedures related to recordkeeping. Furhermore, we expect that integrating the
UDI into software systems would include adding theUDI to records. Consequently, the costs of
2 The UDI must also be included in reports of adverse events. We assume that the incremental time
needed to add the UDI to adverse event forms would be negligible.

47 CDRH200816
recordkeeping are largely captured in the administrative, direct marking, and softare cost
components. We assume that any additional effort would be minimaL.
Global Unique Device Identification Database (GUDID)
Labelers of devices required to display a UDI would also need to submit certain-data to
the GUDID. We anticipate that these costs would be incured on an establishment basis. Only
establishments that exclusively label excepted devices would not be required to submit data to.
the GUDID. All other labelers would incur some costs to submit required device data to the
GUDID.
Much of the required GUDID data is currently included on the device label and thus
would be readily accessible to labelers. Most device data would be submitted only once, when
the device labeled with the UDI enters commerce. Prior to data submission, however, labelers
would need to gather and prepare the data for submission. We anticipate that a manager would
perform this task. For small establishments with 1 to 9 employees, it would take a manager
about 3 hours and cost about $225 (3 hours x $75 per hour) to prepare the GUDID data. For
establishments with 10 to 49 employees, a manager would spend about 6 hours at a cost of about
$450 (6 hours x $75 per hour). Because larger establishments with 50 or more employees would
likely have incorporated all of the GUDID required data into their management information
systems when they integrated the UDI, we expected that the cost to gather UDI data for
submission to the GUDID would be negligible for these establishments.
The proposed rule would require that labelers electronically submit UDI data to the
GUDID. In most cases, labelers curently submit registration and listing data electronically to
the FDA Unified Registration and Listing System (FURLS). Therefore, we anticipate that
labelers would have little diffculty with the electronic submission of device data to the GUDID.

48 CDRH200816
Labelers would either enter and validate data submission via a web page, or convert data to the
SPL format for uploading and validate the uploaded data. We assume that smalllabelers wil
likely use a web-based form to submit data. To submit and validate data, it would cost about
$225 for an establishment with 1 to 9 employees (3 hours x $75 per hour) and $300 for an
establishments with 10 to 49 employees (4 hours x $75 per hour). Medium and large
establishments would incur a cost of about $100 to convert their data to SPL format and incur
labor costs of about $338 (4.5 hours x $75 per hour) to upload the SPL fie directly to the
GUDID and to validate the data.
The one-time costs to gather and submit GUDID data to FDA would equal $2.7 milion,
or about $0.9 milion ($450 per establishment x 1,988 establishments) for very small
establishments with 1 to 9 employees, about $1.3 milion ($750 per establishment x 1,725
establishments) for small establishments with 10 to 49 employees, and $0.5 milion ($438 per
establishment x 1,176 establishments) for medium and large establishments. Once submitted,
data for a paricular version or model would normally remain unchanged. Should changes be
necessary, however, both the web page and the SPL format would allow labelers to rapidly edit
and resubmit their data. To account for possible minor changes, we estimate that a manager in
each of the affected establishments would spend up to one hour anually to modify the GUDID
data. These total anual costs would equal about $0.4 milion ($75 per hour x 1 hour x 4,889
establishments). We request detailed comment from industry on these cost estimates.
2. Costs for Repackagers and Relabelers
Repackagers and relabelers would incur similar types.of compliance costs as initial
labelers, but have less complex systems and thus lower per firm and per establishment costs than
initiallabelers. For these labelers, we assume that the costs for direct marking of devices would

49 CDRH200816
be limited to costs of noting exceptions. Because we assume that no repackagers or relabelers
only handle excepted devices, for this analysis we use the establishment and firm counts from
tables 5 and 6 of this document (1,310 establishments and 1,212 firms.) Total one-time costs for
repackagers and relabelers would be $34.3 milion and anual costs would be $5.2 milion.
Similar to initiallabelers, the major one-time cost components for repackagers and relabelers
would be $13.1 milion for softare and training, and $11.3 milion for equipment. Other one-
time costs include $3.3 milion for administration and plan development, $1.6 milion to
paricipate in a UDI system operated by an issuing agency, $4.6 milion for label redesign, and
$0.4 milion for recordkeeping and reporting. Anual costs equal $3.1 milion for equipment,
$1.0 milion for incremental label materials, $1.1 milion for softare including training, and
$0.05 milion for recordkeeping and reporting. Over 10 years, the total annualized costs would
be $10.1 millon with a 7 percent discount rate and $9.2 milion with a 3 percent discount rate.
3. Efforts to Reduce the Scope and Regulatory Burdens for Certain Low Risk Devices
In this section, we adjust our establishment counts to incorporate the Agency's efforts to
reduce the burden for labelers of class I devices and labelers of over-the-counter devices sold at
retaiL. Specifically, labels of class I devices would not be required to bear the variable
production identifier portion of the UD1. In addition, labels of a class I device that FDA has by
regulation exempted from the GMP requirements and any over-the-counter device sold at retail,
including such devices when delivered directly to hospitals and other health care facilities, would
not be required to bear a UD1. However, the labels of class II and class III devices stil would be
required to include variable production information portion of the UD1. Direct marking
requirements would remain unchanged.

50 CDRH200816
The overall effect of these provisions is to apply the UDI requirements to fewer devices
and labelers. Our initial counts of domestic establishments from FDA's registration and listing
data are presented in table 5 of this document. For this section, we estimate the count of
establishments that would be subject to reduced compliance costs because they label only class I
or unclassified devices. Our estimate of the number of class I establishments includes those
establishments that handle unclassified devices. Table 16 presents the number of class II and
class III establishments, and the number of class I establishments. We separate the count of class
I establishments into establishments that exclusively handle class I devices exempt from GMP
requirements, and establishments that handle some non-GMP exempt devices.
Ta e - istri ution 0 sta is ents )y Device TypeType of Number of Number of Number of Total NumberLabeler Establishments Establishments Labeling Establishments Labeling of
Labeling Class II and Class I or Unclassified Class I or Unclassified EstablishmentClass II Devices i GMP Exempt Devices Non-GMP Exempt
Only 2
Devices 3
Manufacturer 3,088 399 1,414 4,901
Reprocessor 13 1 7 21
Specification 700 150 496 1,346
DeveloperTotal Initial 3,801 550 1,917 6,268LabelersRepackagers 481 129 700 1,310
and RelabelersAll Labelers 4,282 679 2,617 7,578
bl 16 D' 'b' fE bl hm b
Source: ERG Report, Table 6-23 (Ref. 1)i The UDI is required to include a device identifier and a production identifier.2 Devices from these establishments would be covered by general exception 80 i .30(a)(2); devices would not be
required to bear a UDI.3 Devices from these establishments would be covered by general exception 801 .30( c); the UDI is not required to
include a production identifier
We identified 2,467 initiallabeler establishments (550 + 1,917) labeling class I devices.
Similar to the adjustments described at the beginning of this Cost Section, establishments would
be removed from these counts if the device is subject to the general exceptions not specific to the

51 CDRH200816
class I exception. A final adjustment was made to remove the number of establishments that are
assumed to already include production information in the UDrJ.
These adjustments reduce the number of initiallabeler establishments labeling class I
devices from 2,467 to 1,8414, including 1,430 initiallabelers that exclusively handle non-GMP
exempt class I devices, and 410 initiallabelers that exclusively handle class I devices exempt
from the GMP regulations. These labelers would incur a subset of the costs discussed in sections
F1 of this document. For example, labelers of class I non-GMP-exempt devices would not incur
the costs to implement the production identifier portion ofthe UDI and labelers of class I GMP-
exempt devices would not incur the costs to implement the UDI, but would need to read the rule.
Labelers of class II and class III devices not covered by any ofthe general exceptions
would incur the costs to comply with the full UDI requirements. Compared to the counts shown
in table 8, only 2,836 initiallabeler establishments (4,677 - 1,841) would need to develop a full
UDI implementation plan under the proposed rule. Similarly, only 481 repackager or relabeler
establishments (1,310 - 829) would need to develop a full UDI implementation plan under the
proposed rule. See section 6.6 of the ERG report for more detaiL.
4. Cost ofthe Proposed Rule to Labelers with Simplifying Assumption
Table 17 sumarizes the total costs of the proposed rule for all domestic labelers under
the assumption of immediate implementation (i.e., assuming no phase-in). We use this
simplifying assumption to permit comparisons with the alternatives listed below. The total one-
time costs of the proposed rule would be $292.8 milion and anual costs would be $46.7
milion. The total anualized costs would be $88.4 milion per year at a 7 percent discount rate
over 10 years and $81.0 milion per year at 3 percent.
3 We did not adjust the counts ofrepackager and relabeler establishments in sections F2.4 Numbers are rounded and may not sum.

52 CDRH200816
T bl 1 7 ~ IF' Y; A dA l dC f h P diD All L b I i 2 (201 d II )a e .-- ota irst- ear, nnua an nnua ize osts 0 t e ropose Ru e or a e ers 0 0 arsCost Element First-Year Annual
($ milion) ($ milion)Labeling and Database Requirements
Administration and planning $37.1 NABarcode registration $2.0 NAEquipment and other investments $47.5 $22.6Incremental label materials and labor NA $7.6Label redesign $47.6 NASoftare (with training) $128.7 $14.2Recordkeeping and Reporting (GUDID) $2.9 $0.4
Total Labeling and Database Requirements $266.0 $44.7Direct Marking
Implants $12.0 $0.8Multiple-use devices $14.9 $1.
Total Direct Marking $27.0 $2.0
Total--All Cost Elements $292.8 $46.7Annualized Costs
. ($ milion)First-Year Costs, annualized at 7 percent over i 0 years $41.7Total Annualized Costs, with 7 % annualized 151- Year Costs $88.4First-Year Costs annualized at 3 percent over 10 years $34.3Total Annualized Costs, with 3% anualized lSI_Year Costs $81.0
Source: ERG Report, Table 6-41 (Ref. 1)Note: Numbers may not sum due to rounding.i The GMP-exempt exclusion cost savings is not fully reflected in administration and planning costs. The cost
savings shown reflect a cost savings for these establishments for reading and implementing a static barcodingrequirement (the administrative and planning time is assumed to be half of that for planning for a variable barcoderequirement). However, GMP-exempt establishments are expected to incur costs only to read and determine theyare not affected by the proposed rule. This task is less time intensive than a task that includes implementation ofstatic barco ding requirements.2 Includes the GUDID cost savings for establishments exclusively handling devices sold at retail that would be
excepted under section 801.0(a)(l).
5. Costs of the Proposed Rule to Labelers Under FDA's Proposed Implementation Schedule
The domestic industry costs presented in table 17 of this document treat all one-time
costs as occuring in the first year. However, the proposed effective dates would allow industry
up to 7 years to phase in requirements. This section presents costs in the year they would be
incured according to the proposed implementation schedule. Therefore, this section best
describes the total costs of the proposed rule for labelers.
The effective dates after publication of a final rule for medical devices to bear a UDI on
the label are:

53 CDRH200816
Class III devices, one year,
Class II devices, three years, and
Class I devices and devices not classified into class I, II, or III, 5 years.
The effective dates for devices that must be directly marked allow for two additional
years, depending on the regulatory class of each device.
By linking FDA's product code database, which provides the class of the devices for each
product code, with the registration and listing data, we created a count of domestic labelers by
highest class of device. This allows us to assign the one-time and recurring costs (shown in table
17) on the basis of the percentage of establishments with devices in each device class. For this
analysis, labelers are only counted once. For example, if a labeler handled class I and class III
devices, this labeler is added to the count of establishments with class III devices, but not added
to the count of establishments with class I devices.
Using this approach, we find that about 6 percent of affected establishments would come
into compliance in the first year--establishments that label class III devices but may also label
class II, class I and unclassified devices. Another 51 percent that label class II devices (and also
class I and unclassified devices, but not class III devices) would comply in year 3, and the
remaining 43 percent that label only class I and unclassified devices comply in year 5. Direct
marking costs are assumed to occur in year 3 for implant devices and in year 7 for multiple-use
devices. In addition, alllabelers would be affected by the I-year effective date to change date
format on device labels and incur the one-time labeling costs in the first year.
Table 18 of this document presents undiscounted regulatory costs for domestic labelers
and the present value of these costs over a 10-year time horizon with a 7 percent discount rate
and a 3 percent discount rate. As ilustrated, total present value of compliance costs to domestic
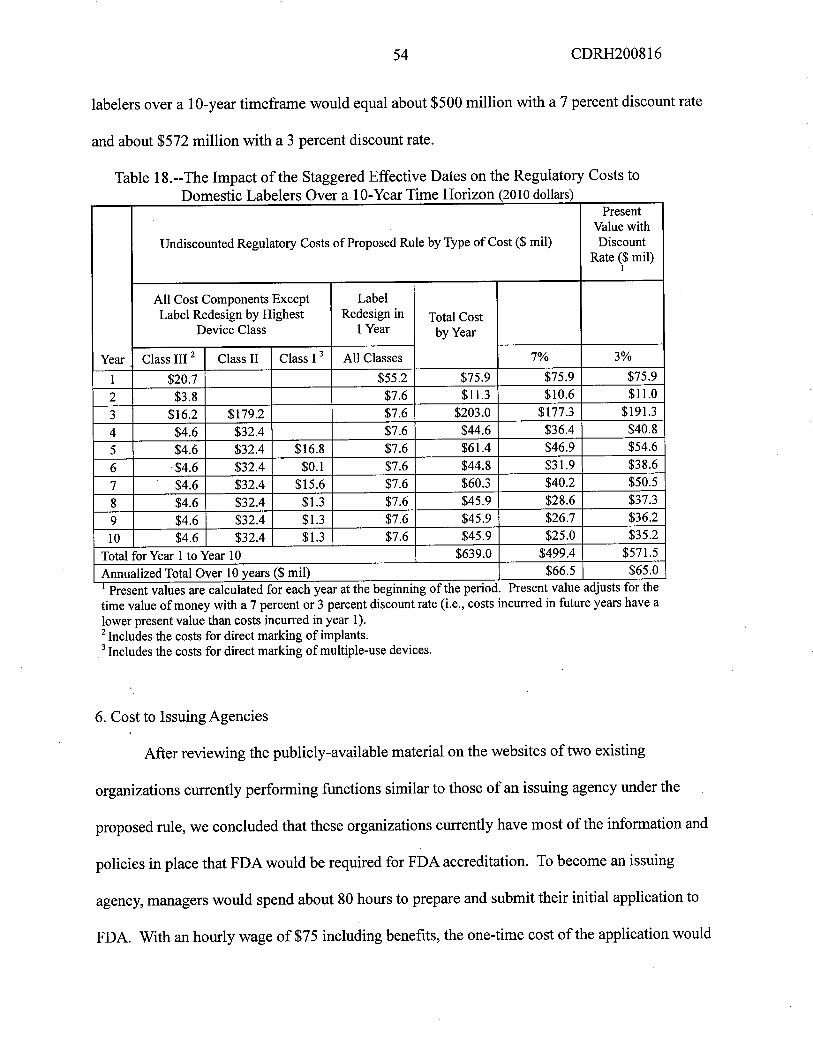
54 CDRH200816
labelers over a 10-year timeframe would equal about $500 milion with a 7 percent discount rate
and about $572 milion with a 3 percent discount rate.
Table 18.--The Impact of the Staggered Effective Dates on the Regulatory Costs toDomestic Labelers Over a.1 0- Year Time Horizon (2010 dollars)
PresentValue with
Undiscounted Regulatory Costs of Proposed Rule by Type of Cost ($ mil) DiscountRate ($ mil)
i
All Cost Components Except LabelLabel Redesign by Highest Redesign in Total Cost
Device Class 1 Year by Year
Year Class II 2 Class II Class I 3 All Classes 7% 3%
i $20.7 $55.2 $75.9 $75.9 $75.9
2 $3.8 $7.6 $11.3 $10.6 $11.0
3 $16.2 $179.2 $7.6 $203.0 $177.3 $191.4 $4.6 $32.4 $7.6 $44.6 $36.4 $40.8
5 $4.6 $32.4 $16.8 $7.6 $61.4 $46.9 $54.6
6 $4.6 $32.4 $0.1 $7.6 $44.8 $31.9 $38.6
7 $4.6 $32.4 $15.6 $7.6 $60.3 $40.2 $50.5
8 $4.6 $32.4 $1. $7.6 $45.9 $28.6 $37.3
9 $4.6 $32.4 $1. $7.6 $45.9 $26.7 $36.2
10 $4.6 $32.4 $1. $7.6 $45.9 $25.0 $35.2
Total for Year 1 to Year 10 $639.0 $499.4 $571.Annualized Total Over 10 years ($ mil) $66.5 $65.0i Present values are calculated for each year at the begining of the period. Present value adjusts for thetime value of money with a 7 percent or 3 percent discount rate (i.e., costs incurred in future years have alower present value than costs incurred in year i).2 Includes the costs for direct marking of implants.3 Includes the costs for direct marking of multiple-use devices.
6. Cost to Issuing Agencies
After reviewing the publicly-available material on the websites of two existing
organizations curently performing fuctions similar to those of an issuing agency under the
proposed rule, we concluded that these organizations curently have most of the information and
policies in place that FDA would be required for FDA accreditation. To become an issuing
agency, managers would spend about 80 hours to prepare and submit their initial application to
FDA. With an hourly wage of$75 including benefits, the one-time cost of the application would

55 CDRH200816
equal $6,000 for each organization. Assuming that these two existing organizations submit
applications, initial one-time application costs total $12,000 ($6,000 per organization x 2
organizations). Application renewals would require about 20 hours and cost $1,500 (20 hours x
$75 per hour) for each organization, for a total of $3,000 in recuring application renewal costs.
Because organizations accept significant legal responsibilities when they become issuing
agencies for FDA, we assumed that each organzation might spend up to $250,000 in the first
year for its executive and legal staffs to ensure that the organization and the interests of its
existing members would be suffciently protected. Furhermore, in subsequent years, each
issuing agency might incur about 10 percent of its initial costs for on-going executive and legal
reviews, or $25,000 anually. For the two existing organizations currently performing fuctions
similar to those of an issuing agency under the proposed rule, one-time review would therefore
cost $500,000 and anual review would cost $50,000.
Once an organization becomes an issuing agency for FDA, it would need to inform its
members about any requirements specific to FDA and whether and how their system might
change to conform to the requirements of the proposed rule. This would take an estimated 80
hours in the first year and cost $6,000 (80 hours x $75 per hour) for each organization, or a total
of$12,000.
In addition, to maintain accreditation, an issuing agency would have to submit a list of
their labelers directly to FDA. To accomplish this, an issuing agency would likely modify its
software system. We estimated that each organization would need about 20 hours for a softare
engineer to initially automate data collection of the required labeler information and about 12
hours anually for a manager to maintain the list. With an hourly wage rate of $125 including
benefits, it would cost about $2,500 (20 hours x $125 per hour) in the first year for a softare

56 CDRH200816
engineer to make changes. With an hourly wage rate of $75 including benefits, it would cost
about $900 (12 hours x $75 per hour) anually for a manager in each organization to maintain
the list of labelers.
The total initial cost for the two existing organizations curently performing fuctions
similar to those of an issuing agency under the proposed rule would equal about $0.5 milion
($12,000 to apply with FDA + $5,000 to modify the softare system + $12,000 to inform
existing members + $500,000 for executive and legal review). Anual costs would equal about
$0.1 millon ($3,000 to renew the application + $1,800 to maintain the list of labelers + $50,000
for executive and legal review).
In addition to these two organizations, there may be other nonprofit organizations or
State governent agencies that decide to apply to become an issuing agency. We assume that the
costs estimated for the two existing organizations would apply to any other organization that
applies to FDA to become an issuing agency.
7. Cost to FDA for the GUDID
We anticipate that contractors and FDA personnel would paricipate in the development
ofa separate database for UDI data. The GUDID would accept electronic submission ofUDI-
related device data, generate standard reports, and allow queries of publicly-available
information. As shown in table 19 of this document, FDA estimates that it would take about
15,100 hours of contractor and FDA personnel time to develop and launch the GUDID. With an
average hourly wage of$103, the one-time cost to develop and launch the GUDID would equal
about $1.6 milion (15,100 hours x $103 per hour). Anualized over 10 years, star-up costs for
the GUDID equal about $0.2 milion with either a 3 percent discount rate or a 7 percent discount
rate. Moreover, once the database is operational, FDA expects it wil take about 18,100 hours
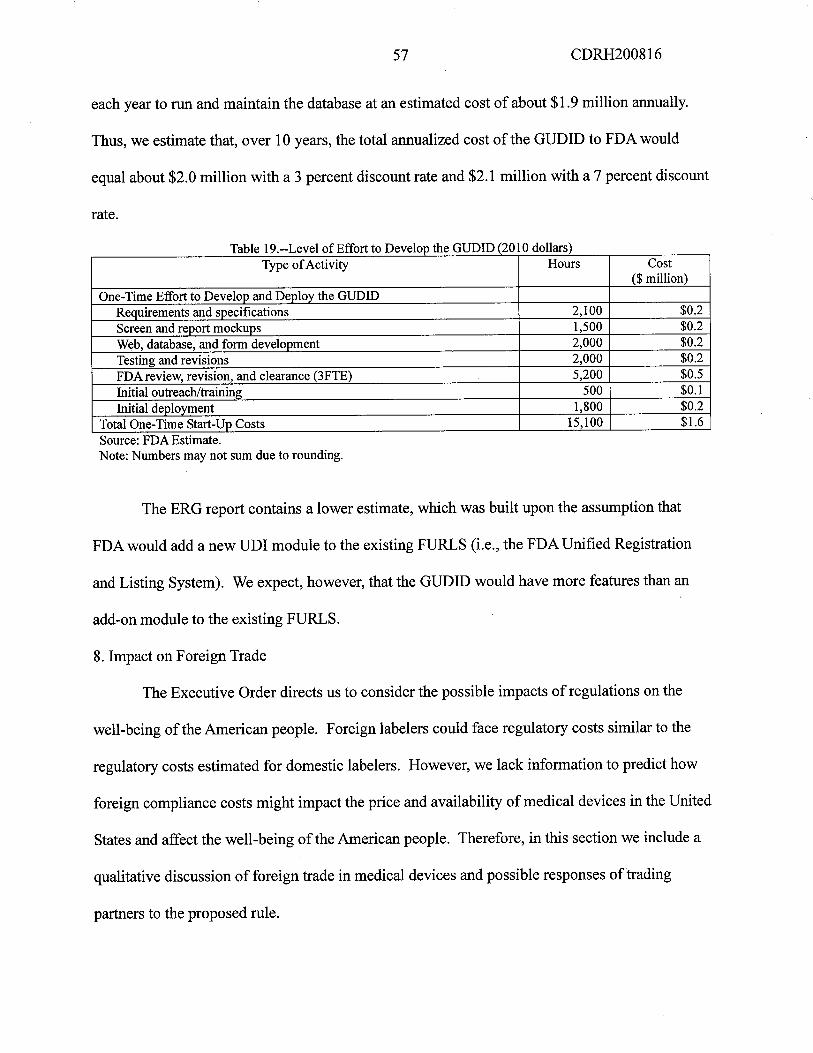
57 CDRH200816
each year to ru and maintain the database at an estimated cost of about $1.9 milion anually.
Thus, we estimate that, over 10 years, the total anualized cost of the GUDID to FDA would
equal about $2.0 milion with a 3 percent discount rate and $2.1 milion with a 7 percent discount
rate.
a e .-- eve 0 0 0 eve op t e o ars
Type of Activity Hours Cost($ milion)
One-Time Effort to Develop and Deploy the GUDIDRequirements and specifications 2,100 $0.2Screen and report mockups 1,500 $0.2
Web, database, and form development 2,000 $0.2Testing and revisions 2,000 $0.2FDA review, revision, and clearance (3FTE) 5,200 $0.5
Initial outreach/training 500 $0.1
Initial deplovment 1,800 $0.2Total One-Time Star-Up Costs 15,100 $1.6
T bl 19 L 1 fEffì rt t D h GUDID(2010d II )
Source: FDA Estimate.Note: Numbers may not sum due to rounding.
The ERG report contains a lower estimate, which was built upon the assumption that
FDA would add a new UDI module to the existing FURLS (i.e., the FDA Unified Registration
and Listing System). We expect, however, that the GUDID would have more features than an
add-on module to the existing FURLS.
8. Impact on Foreign Trade
The Executive Order directs us to consider the possible impacts of regulations on the
well-being of the American people. Foreign labelers could face regulatory costs similar to the
regulatory costs estimated for domestic labelers. However, we lack information to predict how
foreign compliance costs might impact the price and availability of medical devices in the United
States and affect the well-being of the American people. Therefore, in this section we include a
qualitative discussion of foreign trade in medical devices and possible responses oftrading
parners to the proposed rule.

58 CDRH200816
We used data on the value of imports to the United States and exports from the United
States of medical devices to assess the impact of the proposed rule on foreign trade. Anual
trade data is available for most of the medical device manufactung categories affected by the
proposed rule, including NAICS codes 339112 (surgical and medical instrment manufactuing),
339113 (surgical appliance and supplies manufacturing), 334510 (electromedical and
electrotherapeutic apparatus manufactuing), 334517 (Irradiation apparatus manufactuing),
339115 (ophthalmic goods manufactuing), and 339114 (dental equipment and supplies
manufactuing). Table 20 of this document shows that the anual value of trade (imports plus
exports) in these medical device manufacturing industries totals more than $60 bilion. The
export data includes some freight, insurance and other charges that are excluded from the import
data.
T bl 20 U' d S I dE f Sid M d I D' 2007 2009 ($ 1l ) ia e .-- nite tates mports an xports 0 e ecte e ica evices, - mi ionNAICS 2007 2008 2009
CODE Imports Exports Imports Exports Imports Exports
339112 $8,694.4 $8,816.7 $9,138.5 $9,979.4 $9,004.1 $10,038.7
339113 $7,072.9 $6,701.2 $9,007.5 $7,397.9 $8,568.4 $7,686.3
334510 $6,727.7 $7,206.9 $7,216.5 $8,070.4 $6,986.3 $8,094.5
334517 $3,574.2 $3,060.6 $3,721. $3,343.1 $3,097.2 $3,235.5
339115 $3,198.1 $1,340.1 $3,265.2 $1,396.0 $3,059.7 $1,291.339114 $1,187.9 $1,118.1 $1,278.7 $1,214.0 $1,249.2 $1,166.1
Total $30,455.3 $28,243.6 $33,627.6 $31,400.9 $31,964.9 $31,512.4
Source: United States International Trade Commission, Interactive Tariff and Trade Data Web;htt://dataweb.usitc.gov (Ref. 2).i In current dollars for the year reported.
A breakdown of the 2007-2009 trade data by country shows that almost every country in
the world ships medical devices to the United States, with a small number of countries
accounting for a large proportion of the value of medical device imports. Nevertheless, imports
from about 130 countries account for about 3 percent of the $32.1 bilion total average anual
value of imports from all countries in the world, or $1.1 bilion in import value.

59 CDRH200816
Table 21 of this document shows that the top ten countries for imports of medical devices
to the United States account for about 75 percent of the total average anual value of imports.
The top 4 countries account for over 50 percent of the value of imports. Mexico has the largest
share of imports, accounting for 15 percent of the total average anual imports. Ireland has a 14
percent share of total average anual imports and Germany has a 13 percent share of total
average anual imports. China accounts for 10 percent of the value of total average anual
imports.
Table 21.-- Top Ten Countries Shipping Medical Devices to the United States by Share of Total Average AnnualI Vd 12mport a ue'
Countr Value of Imports Average Annual Value of Share of Total Average
2007-2009 Imports Annual Imports
($ bilion) ($ bilion)World Total 96.2 32.1 100%
Mexico 14.1 4.7 15%
Ireland 13.1 4.4 14%
Germany 12.4 4.2 13%
China 9.8 3.3 10%
Japan 6.1 2.0 6%
Switzerland 4.5 1. 5%
United Kingdom 3.3 1. 3%
Italv 2.8 0.9 3%
Malaysia 2.7 0.9 3%
France 2.6 0.9 3%
Total Share of Imports for Top Ten Countries 75%i Source: U.S. Census Bureau, International Trade Statistics. Based on aggregate data forNAICS 339112,339113,339114,339115,334510.334517.htt://censtats.census.gov/naic3 6/naics3 6.shtml (Ref. 3).2 In current dollars for the year reported.
Trade data shows that medical devices from the United States are exported throughout
the world. Similar to imports of medical devices, a small number of countries receive the
majority of medical devices exports, based on the value of exports. Table 22 of this document
shows the top ten countries that purchase U.S. exports of medical devices. These countries
account for approximately two-thirds of the total average anual value of medical device exports.

60 CDRH200816
The top countries receiving medical devices from the United States are Japan, the Netherlands
and Canada each receiving at least 10 percent of the total average anual value of exports.
Table 22.-- Top Ten Countries Receiving Medical Devices from the United States by Share of Total AverageE V:l 12xport a ue '
Countr Value of Exports Average Annual Value Share of Total
2007-2009 ($ bilion) Average Annual
($ bilion) Exports
Japan 12.2 4.1 12%
Netherlands 11.0 3.7 10%
Canada 10.4 3.5 10%
Germany 9.0 3.0 9%
Belgium 6.2 2.1 6%
Mexico 5.7 1.9 5%
United Kingdom 4.8 1.6 5%
France 4.1 1.4 4%
Australia 3.7 1.2 3%
China 3.4 i. 3%
Total Share of Exports for Top Ten Countries 67%i Source. U.S. Census Bureau, International Trade Statistics. Based on aggregate data forNAICS 339112,339113,339114,339115,334510.334517.htt://censtats.census.gov/naic3 6/naics3 6.shtml (Ref. 4).2 In current dollars for the year reported.
As noted previously in this document, the total value of shipments for all device
manufactuing industries equaled about $117 billon in 2007 (table 3 of this document). The data
in table 23 of this document shows that imports and exports each represent about one-fourth of
the value of domestic production of these medical devices manufacturers. This percentage
demonstrates the importance of international trade to the medical device industry.
Foreign producers.
About one-half of the registered establishments that would be considered labelers and
affected by the proposed rule are located in countries other than the United States. Table 23 of
this document shows a distribution of these approximately 7,100 foreign labeler establishments
by the type of labeling activity. This list was generated using the same methodology used to
count the number of affected domestic labelers.

61 CDRH200816
bl b f bl h 'd dTa e 23.--Num er 0 Foreign Registered Esta is ments Consi ered Labelers un er the Proposed Rule
Type of Labeler Foreign Total Registrants Percentage of Total RegistrantsRegistrants
Manufacturers 6,492 11,393 57%Reprocessors 3 24 13%
Specification Developers 276 1,622 17%
Relabelers and Repackagers 320 1,630 20%Total 7,091 14,669 48%I.Source. ERG Report (Ref. 1).
We lack data on the structue of the foreign medical device industry, the size distribution
of foreign establishments and firms, the proportion of foreign output exported to the United
States, and the complexity of foreign medical device manufacturing facilities; data that would
allow us to predict likely responses of foreign labelers to the proposed rule and likely changes in
the cost of imported medical devices. However, the OECD publishes country data on relative
comparative advantage (RCA) by type of industry. Economic theory predicts that with
international trade, countries will employ resources in industries where they can effciently
produce goods. RCA gives us an indication of the degree of specialization of a paricular
industry in the global economy. Table 24 of this document presents a list of countries with
medical, precision, and optical instruent manufacturing that have RCA values exceeding one; a
value that indicates specialization in the industry. This suggests that the medical device industry
has developed as an important sector of the economy of these countries. Moreover, along with
the United States, table 24 ofthis document includes some countries such as Mexico, Germany,
Ireland, Japan, and the Netherlands that are among our top ten trading parners of medical
devices. Although uncertain, the cross trade in medical devices among countries with a
specialization in medical device manufactuing suggests that the foreign and domestic medical
device industries have developed similar standards and practices.

62 CDRH200816
Table 24.--0ECD Measure of Specialization of the Medical, Precision and Optical Instrment ManufacturingInd b Custr )v ountr
Countr Revealed Comparative Advantage For Manufacturers ofMedical, Precision and Optical Instrents (ISIC 33) i
Switzerland 4.549Korea 2.041
United States 1.843
Ireland 1.823
Japan 1.52
Denmark 1.74Germanv 1.277
United Kingdom 1.255
Netherlands 1.51France 1.073
Mexico 1.066
Source: OECD Micro Trade Indicators (by category of industr, ISle), data extracted on 28 Jul 2010 19:45 UTe
(GMT) from OECD.Stat; www.oecd.org/std/its/tradeindicators (Ref. 4).i The revealed comparative advantage (RCA) measures the intensity of trade specialization of a countr and is
calculated as the industr's share of exports from a countr divided by the industr's share of global exports. A
countr has not specialized in exports of the industr if the RCA is less than 1; a countr has specialized inexports of the industr if the RCA is greater than 1.
The number of foreign labelers expected to be affected by the proposed rule almost
equals the number of affected domestic labelers. This might suggest that under the proposed
rule, the incremental costs of foreign manufacturing would rise by about the same amount as the
incremental costs of U.S domestic manufacturing. Although we lack information on the types
and number of medical devices produced by these foreign firms, any disproportionate increase in
the cost of production of medical devices between foreign and domestic labelers could affect
international trade. Moreover, increases in the cost of production of medical devices in other
countries would be expected to increase the cost of imports of medical devices to American
consumers, as would be expected with the domestic labelers.
We estimated that the total anualized costs to domestic labelers would be about $66.5
millon with a 7 percent discount rate and about $65.0 milion with a 3 percent discount rate.
There is greater uncertainty in estimating the costs to foreign firms. As we have noted, although
much ofthe medical device trade with the U.S. is concentrated in a few countries, a large number

63 CDRH200816
of countries manufactue some types of medical devices. Because we lack suffcient information
to estimate the potential impact of this rule on foreign labelers or the impact on international
trade, we request comment from affected industries about their expected compliance costs and
responses to the proposed rule.
9. Sumar of Total Costs of the Proposed Rule
Table 25 of this document presents, for each affected sector, a sumary of the estimated
total present value and the anualized domestic costs of this proposed rule over 10 years using
discount rates of7 percent and 3 percent. Over 10 years, the total present value of the domestic
costs would be $514.0 millon using a 7 percent discount rate and $588.6 milion at 3 percent,
and the anualized costs would be $68.4 millon at a 7 percent discount rate and $66.9 milion at
3 percent.
Table 25.--Summary of the Estimated Regulatory Costs of the Proposed Rule 1,2 (2010 dollars)
Affected Sectors Total Present Value of Cost Total Anualized Costsover 10 years Over 10 Years
($ milion) ($ milion)3 Percent 7 Percent 3 Percent 7 Percent
Domestic Labelers $571.5 $499.4 $65.0 $66.5
Issuing Agencies $1.0 $0.9 $0.1 $0.1
FDA $16.1 $13.7 $1.8 $1.8
Imports Not Not Not Notquantified quantified quantified quantified
Total Domestic Cost of the Proposed$588.6 $514.0 $66.9 $68.4
Rulei Present value and annualized costs calculated at the begining of the penod.2 This summary table 25 is identical to table 1 of this document.
Costs to Domestic Labelers.
The majority of the costs of this proposed rule would be incured by labelers of medical
devices. Labelers include manufactuers, reprocessors, specification developers, repackagers,
and relabelers that cause a label to be applied to a medical device. Over 10 years the anualized

64 CDRH200816
costs to domestic labelers would be $66.5 milion at a 7 percent discount rate and $65.0 millon
at 3 percent. The largest components of one-time costs would include the costs to integrate the
UDI into existing information systems, to install, test and validate barcode printing software, and
to train employees, and to purchase and install equipment needed to print and verify the UDI on
labels. In addition, the redesign of all device labels to incorporate the date format within 1 year,
the redesign of the UDI barcode format, and the direct marking of certain devices are significant
components of one-time costs.
The largest anual cost components include labor, operating, and maintenance costs
associated with equipment for printing operations and labor related to software maintenance and
training needed to maintain the UDI data and UDI reporting systems.
Costs to Issuing Agencies.
The estimated present value of costs over 10 years for two existing organizations
curently performing fuctions similar to those of an issuing agency under the proposed rule, to
apply for FDA accreditation and comply with the proposed reporting requirements would be $0.9
milion at a 7 percent discount. rate and $1.0 milion at 3 percent. The anualized costs over 10
years would be $0.1 milion at a 7 percent and 3 percent discount rate. In addition to these two
organizations, there may be other nonprofit organizations or State or Federal Governent
agencies that might apply to FDA to become an issuing agency. In such cases, the estimated
application preparation, legal and reporting costs would apply to other organizations.
Costs to FDA to Establish and Maintain the GUDID.
The estimated present value over 10 years of the costs to FDA to establish and maintain
the GUDID would be $13.7 milion at a 7 percent discount rate and $16.1 milion at 3 percent.
The anualized costs over 10 years would be $1.8 milion at 7 percent and 3 percent.

65 CDRH200816
Costs to Foreign Labelers.
We lack suffcient information to quantify the potential impact of the proposed rule on
foreign establishments and thus exclude these establishments from our cost estimate. However,
we include a qualitative discussion of the potential impact of this rule on trade and the cost of
imported products. .
G. Analysis of the Uncertainty of Costs
The estimates of compliance cost presented in the Cost Section F of this document are
associated with uncertainty, with some cost categories more uncertain than others. This section
qualitatively discusses the uncertainty of the cost estimates for each of the major cost
components and presents an upper bound and lower bound estimate for each cost component, as
well as total cost. The domestic industry costs presented in this uncertainty analysis treat all one-
time costs as occuring in the first year and do not incorporate the proposed effective dates,
which would allow industry up to 7 years to phase in requirements. The agency welcomes
comments on assumptions and on estimates of cost used for this analysis.
The maximum number of domestic firms and establishments expected to be affected is
reasonably certain. All affected entities are already required to be registered with FDA. Any that
are not registered are out of compliance with FDA's registration and listing requirements. More
uncertain is the share of establishments involved in labeling devices for retail outlets only. These
uncertainties are handled within bounding estimates made for each cost category. These
bounding estimates depend on factors related to the uncertainty in each cost category.
Another uncertainty is how many establishments would only have devices that meet an
exception, and thus would be excepted from the UDI requirements. We estimated that 1,141
establishments in the 1-4 employee size group and 238 establishments in the 5-9 employee size

66 CDRH200816
group would meet one ofthe exceptions listed in 801.30(a)(3) - (12). However, if none of these
establishments met an exception, this would add $2.7 milion per year to the costs of the rule.
The first cost category, Planing and Administrative Costs, is our best estimate of the
time needed for companies to undertake basic compliance preparations, although some entities
might spend more or less time. The true overall average across most entities is unlikely to differ
too widely (i.e., an order of magnitude) from the estimate. However, the requirement to meet the
date format change in 1 year could have an effect on planing and administrative costs for
certain establishments. Establishments needing to make this change might need to change the
way they assign lot numbers (if their lot numbers are based on the date that appears on the label).
The number of establishments this requirement might affect is uncertain, but because of this
implementation period, we chose a relatively wide bounding assumption, setting costs between
50 percent lower and 50 percent higher than that estimated in the Cost Section F of this
document.
Costs to paricipate in an accredited UDI system are considered reasonably reliable. A
plus or minus 10 percent factor is used to bound the estimate for this cost category.
Somewhat less certain are the cost estimates for equipment. The costs for smaller
establishments are reasonably certain, but those for the largest establishments could vary widely
if certain types of device packages are being labeled. If establishments must create new levels of
packaging and labeling for certain devices, additional equipment for packaging and labeling
might need to be purchased than was estimated in the Cost Section F of this document. For
example, class II devices that are not labeled separately within another device package (a shelf
pack), combination products with a separable device that is not individually labeled, and certain
devices intended for more than one use that are currently placed unlabeled within kits could be

67 CDRH200816
affected. We do not have information about the prevalence of such devices or the number of
establishments to which this situation might apply.
Alternatively, establishments would be able to judge which of several options (e.g.,
switching from outside printing to in-house printing) are the least expensive for them in
complying with UDI requirements. We did not attempt to judge which options would be chosen
on the basis of cost, which could overstate the equipment costs. To account for these
uncertainties, we estimated costs using factors of plus or minus 50 percent for equipment costs.
It is possible that few establishments would need additional materials for labels. Because
the proposed rule would allow for a shelf pack to be labeled in lieu of requiring each individual
item therein to be labeled with a UDI, because 2D barcodes (which are very small) can be used
to represent UDI information, and because label redesign should solve many label size issues
without the need to expand label area, it is possible that the lower bound of the material costs
could be substantially smaller than our estimate.
The approximation of label materials costs (2 percent of all packaging materials costs)
and the potential cost increase associated with larger packaging and labeling areas (estimated at
10 percent) are also both uncertain, as is the cost implications of the need to change label designs
within 1 year of implementation. This requirement could lead to less cost-effective means of
complying, including the possibility for some classes of devices, the need to go through two
separate rounds of label redesign to accommodate, first, the date format change, and second the
UDI change. The proposed rule requests comments on the value of linking this requirement to
the effective date for the UDI requirement for each device class. However, we are not certain of
the number of such affected entities and may have overstated the costs under the timing
assumption that all affected establishments would redesign labels in the first year. Device

68 CDRH200816
labelers that are currently required to have dates on their labels have a previously established
date format and are not affected by the proposed rule requirement. The number of labelers who
choose to use a date on their labels is not known, but could be relatively smalL. All of these
uncertainties and assumptions could make estimated costs too low or too high. An uncertainty
factor of plus or minus 25 percent has been chosen for this cost category.
Label redesign costs are more speculative, given the range oftechnical, regulatory, and
marketing considerations at play. It is not known how many establishments might be able to
integrate UDI requirements into routine label redesign cycles, which could reduce the
incremental cost of label redesign. On the other hand, the long lead times offered by the
proposed implementation schedule implies that many establishments might be able to do this,
although the number who must meet an earlier deadline for date format changes is not known.
Alternatively, costs could be much higher at establishments with unusual packaging and labeling
issues, including any that are affected by the need to label and package at a new leveL. We used
a plus or minus 60 percent factor to create the upper and lower bound estimate for this cost item.
Softare costs are also considered highly speculative. Costs could be overstated because
we canot estimate how much of the integration costs would be performed as a result of
complying with the proposed rule and how much would be performed as a result of corporate
preferences for integration. The integration would, however, yield benefits in terms of
recordkeeping and reporting cost savings, so the lower bound factor reflects the judgment that
some integration might be performed to reduce incremental costs of recordkeeping and reporting.
We use uncertainty factors of plus or minus 50 percent for this cost item. GUDID costs are
considered reasonable estimates, so have been given factors of plus or minus 25 percent.

69 CDRH200816
Direct marking costs range in their certainty. Implant marking costs are considered the
most uncertain, due to the paucity of data about the extent to which implants are curently
directly marked. Although contacts have indicated that most implants that can be marked
(subject to size and material constraints) are directly marked and that health and safety issues
should not arise, we recognize that higher costs for marking implants could arise. On the other
hand, if all of the implants curently able to be marked are being marked, and those not currently
marked would meet the exceptions for direct marking, costs for marking implants could be
overstated. Additionally, if "technologically feasible" implies that a plain-text UDI would have
to be marked, even if it must be magnified to be read, this could substantially increase costs. If
any exceptions would need to be made on the basis of health and safety, which could be a much
lengthier process than the exception process considered in the Cost Section F of this document,
costs for exceptions would be higher. Also, if FDA were to deny a portion of the exceptions
curently estimated to be requested, substantially more establishments would need to install
equipment, increasing the equipment and operating costs for directly marking devices. We are
also not certain whether additional costs would be incured as a result of needing to mark devices
with plain text so small that it requires magnification to be read. Because of all of these
uncertainties, we estimate an uncertainty factor of plus or minus 80 percent.
For multiple-use devices, the uncertainty is significant, again due mainly to the paucity
of data on current marking practices and, to a lesser extent than that for implants, the issue of
technological feasibilty. Therefore, we selected a factor of plus or minus 50 percent to calculate
bounding estimates.
These factors produce the bounding estimates shown in table 26 of this document. As
table 26 shows, with uncertainty considered (and with no implementation schedule used), we

70 CDRH200816
estimated the anualized compliance cost of the proposed rule to U.S. labelers using a 7 percent
discount rate would be $44.5 milion per year at the low end and $131.8 milion per year at the
high end, with the central estimate equal to $88.4 milion per year. With a 3 percent discount
rate, the low end of anualized cost would be $41.5 millon and the high end would be $120.6
milion, with the central estimate of anualized costs equal to $81.0 milion.
Applying the bounding estimates to table 25 of this document with the phase-in
implementation, our best estimate of the total cost of the proposed rule for all domestic labelers,
issuing agencies and the FDA, the anualized present value of costs to initiallabelers of the
proposed rule over 10 years using a 7 percent discount rate would range from $34.9 milion to
$101.8 milion at 7 percent and $34.1 milion to $99.7 milion at 3 percent.

71 CDRH200816
Cos
t Ele
men
tFi
rst-
Yea
rL
owH
igh
Ann
ual
Low
Hig
h
($ m
ilion
)($
mili
on)
($ m
ilion
)R
ecur
ing
($ m
ilion
)($
mili
on)
($ m
ilion
)L
abel
ing
and
Dat
abas
e R
equi
rem
ents
Adm
inis
trat
ion
and
plan
ning
$37.
1$1
8.5
$55.
6N
AN
AN
A
Bar
code
Reg
istr
atio
n$2
.0$1
.8$2
.2N
AN
AN
AE
quip
men
t and
oth
er in
vest
men
ts$4
7.5
$23.
8$7
1.$2
2.6
$11.
3$3
3.8
Incr
emen
tal l
abel
mat
eria
lsan
d la
bor
NA
NA
NA
$7.6
$5.7
$9.5
Lab
el r
edes
ign
$47.
6$1
9.0
$76.
2N
AN
AN
AS
ofta
re (
with
trai
ning
)$1
28.7
$64.
4$1
93.1
$14.
2$7
.1$2
1.R
ecor
dkee
ping
& R
epor
ting
(GU
DID
)$2
.9$2
.2$3
.6$0
.4$0
.3$0
.5T
otal
Lab
elin
g an
d D
atab
ase
$265
.9$1
29.7
$402
.0$4
4.7
$24.
3$6
5.0
Req
uire
men
tsD
irect
Par
Mar
king
Impl
ants
$12.
0$2
.4$2
1.7
0.8
$0.2
$1.
Mul
tiple
-Use
Dev
ices
$14.
9$7
.5$2
2.4
$1.
$0.6
$1.7
Tot
al D
irect
Par
Mar
king
$27.
0$9
.9$4
4.0
$2.0
$0.7
$3.2
Total-All Cost Elements
$292
.8$1
39.6
$446
.0$4
6.7
$25.
1$6
8.3
Ann
ualiz
ed F
irst-
Yea
r C
osts
(7
perc
enti
$41.
7$1
9.9
$63.
5A
nnua
lized
Firs
t-Y
ear
Cos
ts (
3 pe
rcen
t)T
$34.
3$1
6.4
$52.
3T
otal
Anu
aliz
ed C
osts
($
mill
on)
Cen
tral
Low
Hig
hT
otal
Ann
ualiz
ed C
osts
(7
perc
ent)
2$8
8.4
$44.
9$1
3 1.
8
Tot
al A
nnua
lized
Cos
ts (
3 pe
rcen
t)T
$81.
0$4
1.4
$120
.6
Tab
le 2
6--B
ound
ing
Est
imat
es R
efle
ctin
g U
ncer
tain
ty in
the
Est
imat
esof
th
e T
otal
Dom
estic
Cos
t for
Ini
tial L
abel
ers
i (20
10 d
olla
rs)
Sou
rce:
ER
G R
epor
t, T
able
8-3
(R
ef. 1
).i C
ost e
stim
ates
ass
ume
imm
edia
te im
plem
enta
tion.
The
GM
P-ex
empt
exc
lusi
on c
ost s
avin
gs is
not
ful
ly r
efle
cted
in a
dmin
istr
atio
n an
d pl
anni
ng c
osts
. The
cost
sav
ings
sho
wn
refl
ect a
cos
t sav
ings
for
thes
e es
tabl
ishm
ents
for
rea
ding
and
impl
emen
ting
a st
atic
bar
codi
ng r
equi
rem
ent (
the
adm
inis
trat
ive
and
plaring time is assumed to be half of
that
for
pla
ring
for
a v
aria
ble
barc
ode
requ
irem
ent)
. How
ever
, GM
P-ex
empt
est
ablis
hmen
ts a
re e
xpec
ted
to in
cur
cost
s on
ly to
rea
d an
d de
term
ine
they
are
not
aff
ecte
d by
the
prop
osed
rul
e. T
his
task
is le
ss ti
me
inte
nsiv
e th
an a
task
that
incl
udes
impl
emen
tatio
n of
sta
ticba
rcod
ing
requ
irem
ents
.2
Firs
t-ye
ar c
osts
are
ann
ualiz
ed o
ver
10 y
ears
.

72
H. Benefits
The proposed rule would standardize how medical devices are identified and
contribute to futue potential public health benefits of initiatives aimed at optimizing the
use of automated systems in healthcare. We restrict our description to potential public
health benefits most likely to occur from the direct actions of the proposed rule. These
potential public health benefits would include:
. Improved reporting of postmarket adverse medical device events;
. Improved medical device recalls.
Both postmarket sureilance and recall actions are often hampered by poor
product identification. A UDI would not automatically solve these problems--human
reporting behavior and other data issues hamper surveilance efforts--but having a UDI
could reduce certain costs of product identification and thereby contribute to improved
detection of problem medical devices.
As discussed in section 1.B of the preamble to the proposed rule (Additional
Benefits), the development of a standardized UDI may contribute to the value of other
health information technology (HIT) initiatives. HIT is considered an important tool to
improve patient safety, and a range of HIT initiatives have begun. The adoption rates for
selected HIT initiatives in U.S. hospitals as of 2006 were estimated as follows: electronic
medical record = 37 percent; computerized physician order entry = 13.9 percent; bar-
coding at medication dispensing = 27.1 percent; and barco ding at medication
administration = 4.7 percent. Some researchers attribute financial and cultural bariers to
the initially observed slow rates of adoption. (Ref. 5) Although decisions to invest in HIT

73
would be made independently of the proposed rule, a UDI system may help to facilitate
the adoption and use of complementar HIT systems for improving patient safety.
A standardized UDI also could be used in national and National Institutes of
Health (NIH) device registries, in NIH studies, by the Center for Medicare and Medicaid
Services, and by private healthcare organizations. Such uses would require
complementary developments and innovations in the private and public sectors and
investment in technologies to use the UD1. Moreover, many of these actions would be
developed in futue years. Identifying and assessing these potential future benefits and
costs, however, is beyond the scope of this analysis. Nonetheless, the creation ofa
platform to link specific device information to research databases is likely to enhance the
value of such databases.
An additional benefit of standardizing UDI relates to the formatting of dates on
device labels. A standardized formatting of dates on medical device labels would
eliminate any possibility of confusion from date formats that might be interpreted in more
than one way. Examples of possible confusion due to inconsistent date formatting are
described at II.B.11 of the preamble to the proposed rule. The proposed date format may
contribute to more accurate identification of a device by making it possible to distinguish
between those devices that have passed an expiration or use-by date and those that have
not. More accurate identification would make it easier to both avoid the risks of using
"expired" devices and the costs of prematue disposal of devices that have not actually
reached an expiration or use-by date. We lack suffcient detail to estimate the individual
benefits associated with the date format. These benefits would be captued in the global
benefits presented below.

74
1. Improved Reporting of Adverse Medical Device Events
Baseline and Background.
The proposed rule would be expected to improve adverse medical device event
reporting by providing a reliable and unique identifier with which to report a problem
device. With more reliable identification of devices associated with an adverse medical
event, FDA would be able to improve postmarket sureilance of medical devices and to
detect problem devices more rapidly.
To describe how the UDI could improve postmarket sureilance of medical
devices, we begin with a characterization of the baseline level of adverse medical device
events that occur in normal medical practice with curent technology, and FDA
regulations and databases associated with adverse medical device event reporting. A few
studies have estimated the overall frequency of adverse medical device events. Although
these studies produce different estimates of the frequency of adverse medical device
events and embody much uncertainty, they suggest that a considerable number of medical
device-related adverse events occur each year. One study generated a national estimate
that in a one-year period, 455,000 visits to emergency deparments were for injuries
associated with medical devices. Of these, 58,000 patients were hospitalized, although
the cause of the injur could not be established (Ref. 6). Samore and others searched
patient records at a major tertiary teaching hospital to gather information on the number
of adverse device events (Ref. 7). Samore's team examined computer-flagged medical
records, telemetry problem checklists, clinical engineering work logs, patient surey
results, and other hospital data to identify possible adverse medical device events and to
determine whether such techniques could be used to identify events consistently. When

75
they combined the three detection methods, they estimated the incidence rate for adverse
medical device events was 83.7 per 1,000 hospital admissions. Because the study
collected data from only one hospital, we canot apply the rate to the 40 milion anual
hospital admissions but the results point to a high national incidence of adverse events.
Using a national sample of hospital discharge diagnoses for the years 1997-2003, Bright
and Shen found 820,000 to 1,100,000 diagnoses per year related to adverse medical
device events (Ref. 8).
FDA collects data on adverse medical device events as par of its regulatory
responsibilities under FDA's Medical Device Reporting (MDR) requirements. Medical
device manufacturers, importers and user facilities must report to FDA all deaths and
serious injuries that a medical device has or may have caused or contributed to. In
addition, manufacturers and importers must also report to FDA certain device
malfuctions. FDA provides a gateway for the electronic reporting of mandatory adverse
events (Electronic Medical Device Reporting (eMDR)). In addition, consumers and
others who are not required to report by the MDR rule can voluntarily report device
problems to MEDWATCH. Healthcare professionals and consumers are encouraged to
voluntarily report adverse events involved with medical devices to MEDWATCH.
All adverse medical device event reports received by FDA are entered into FDA's
Manufactuer and User Facilty Device Experience (MAUDE) database. FDA uses
MAUDE to help identify an increase in the number of reports associated with a device or
an increase in the severity of adverse events reported for a device. With this information,
FDA can further investigate newly identified problems related to medical devices and
determine appropriate regulatory responses.

76
The number of reports of serious outcomes submitted to FDA has increased
steadily since 2005. As stated in the preamble to the proposed rule, from 2005 through
2009 FDA received more than 17,700 reports involved a death, and more than 283,000
reports involved an injur. However, there can be more than one report submitted for an
adverse event. Table 27 of this document shows the number of adverse events from
2008-2010 by device classification. During this three year period, about 9,000 deaths and
150,000 serious injuries were associated with adverse medical device events. Because
we lack information about the total number of marketed devices by device class, we
canot determine the relative risks of a serious adverse event for the different device
classes. Nevertheless, this data suggests that adverse events associated with serious har
can occur with all classes of medical devices.
Table 27. -- Number and Share of Adverse Events by Device Class and Event Type(2008-2010)
Type of Adverse Event Share of
Other orTotal Events
Device ClassDeath
Serious Malfuction MissingNumber by
Injury of Events DeviceOutcome Class
Class III 1,810 30,501 41,036 2,482 75,829 20%Class II 3,326 55,987 80,367 4,620 144,300 38%
Class I 2,374 39,033 57,877 3,008 102,292 27%
Unclassified 1,408 22,500 30,091 1,642 55,641 15%
TOTAL 8,918 148,021 209,371 11,752 378,062 100%
Limitations of MAUDE Data.
Ideally, the MAUDE data should provide the FDA, the public, and researchers
with electronic search capabilities to track both general and specific measures of medical
device-related adverse events. However, analyzing medical device adverse event data and

77
adequately identifying the suspected medical devices can be hampered or delayed
because of inaccurate, incomplete, or inconsistent reports of an event.
For example, some medical device adverse event reports do not contain enough
information to identify the device involved with the adverse event, including
manufacturer name, model number, lot number, or date information. An informal review
of fatality reports (totaling 556 reports) in the MAUDE database that occured during a 2-
month period in 2006 revealed that 25 percent of the fatality reports were missing some
portion of manufacturer, model, and lot or date information. Moreover, at least 23 of
these fatality reports were associated with implanted devices and ventilators that lacked
model numbers, lot numbers or both. In some specific episodes where FDA staff was
initially unable to determine the device models or lots implicated in adverse events, the
medical devices were eventually recalled.
Other impediments to identifying a suspected device include: changes to model
numbers and brands made by distributors; interchangeable use of catalog numbers and
model numbers; and punctuation, abbreviation, and spellng of manufactuer names or
brand names.
Inaccurate and incomplete reporting of device identifiers causes FDA to devote
substantial resources finding and verifying the information necessary to identify these
devices before the adverse event data can be used. Moreover, without a uniform
identifier, the MAUDE database canot be effciently and effectively searched for reports
on specific devices. These shortcomings of the MAUDE data can hamper agency efforts
to assess subtle or complex patterns in the adverse event histories. Under these

78
conditions, FDA requires more time to identify patterns in device failure than needed if
devices could be readily and unambiguously identified.
How the UDI Requirements Can Improve the Curent Situation.
Near-Term Improvements.
The lack of an unambiguous device identifier limits the curent usefulness of the
MAUDE data. With the UDI, FDA would be able to immediately identify and validate
the device when an adverse event is reported. It would also make the device easily
searchable throughout the system, regardless of variants of manufactuer names, model,
or catalog numbers, or descriptors used to identify the device. A UDI could improve
FDA's ability to compile additional evidence on similar device types and reduce the time
needed to realize that a wider search for data on the device in question or enhanced
postmarketing sureilance would be waranted. Including data such as product codes or
GMDN in FDA's publicly available GUDID would provide an important data element
that could be used to allow searches to be performed quickly for similar devices
manufactued by multiple companies.
Future Improvements.
With the MAUDE data alone, the Agency is unable to compare failure
frequencies across similar devices or alternative treatments to assist in determining if the
problem is with the device itself (rather than just with a paricular lot or lots). Once
medical devices are identified with a UDI, there is the potential to increase the amount of
data available on medical devices and to improve postmarket sureilance. Linking
MAUDE data to other databases could increase the ability to use MAUDE to assess
causality. With widespread use, many diverse databases could be linked by the UD1.

79
Using linked data from many sources would allow for more robust data analysis of device
problems and outcomes and would allow FDA to perform independent postmarket
sureilance. This linking, however, would require that commercial and public databases
incorporate and use the UDI and make that data available for postmarket sureilance.
Commercial and public databases containing UDI-linked device use could be
used by FDA to estimate exposure. With exposure estimates, the agency would be able to
more accurately and quickly determine if adverse event reports indicated a public health
problem. Causality is always diffcult to determine in human activities, but the ability to
link product databases by the UDI could make more causal inferences possible. For
example, if a disproportionate number of adverse event reports come in for a paricular
device model relative to similar models of the same device, the agency could check the
gross numbers of adverse events against use to determine if the larger number reflects
simply greater exposure or if it reflects greater risk.
2. Improved Efficiency in Removing Recalled Devices from Use
Baseline and Background.
Recalls occur when a medical device is defective, when it could be a risk to
health, or when it is both defective and a risk to health. In most cases, companies
voluntarily recall medical devices when problems arise. FDA oversees recalls to ensure
that the actions the company takes are adequate to protect the public health. During a
medical device recall, FDA works with the recallng firm to obtain information about the
product, the problem, the recall strategy, and planed steps to prevent the problem from
happening again; conducts audits to make sure the recall efforts are appropriate and

80
effective; and makes sure the company takes necessary actions to prevent the problem
from happening again.
FDA classifies medical device recalls into three categories, representing the
potential risk to public health: class I, class II, and class II1. This classification process
usually takes place after the company has issued its recall.
. Class I recall: high risk
. Class II recall: less serious risk
. Class III recall: low risk
A class I recall is the most serious. In a class I recall, there is a reasonable chance
that the product wil cause serious health problems or death; the company whose product
is being recalled notifies its distributors or vendors and directs them to notify the intended
recipients of the device, including other vendors, hospitals, nursing homes, outpatient
treatment facilities, doctors, or individual patients. The notification usually contains the
name of the device being recalled, identifying lot or serial numbers, the reason for the
recall, and instructions about how to correct, avoid, or minimize the problem. FDA may
also issue a press release or public health notice during a class I recall.
A class II recall usually represents a less serious risk than a class I recall. In a
class II recall, there is either a possibility that the device will cause temporary or
reversible health problems, or there is a remote chance that the device wil cause serious
health problems; the company notifies its distributors or vendors and sometimes asks
them to notify the intended recipients of the device. FDA generally does not issue a press
release or expect the company to issue a press release for class II recalls, unless there is a
specific need to do so (for example, if the device could affect the health ofa large number
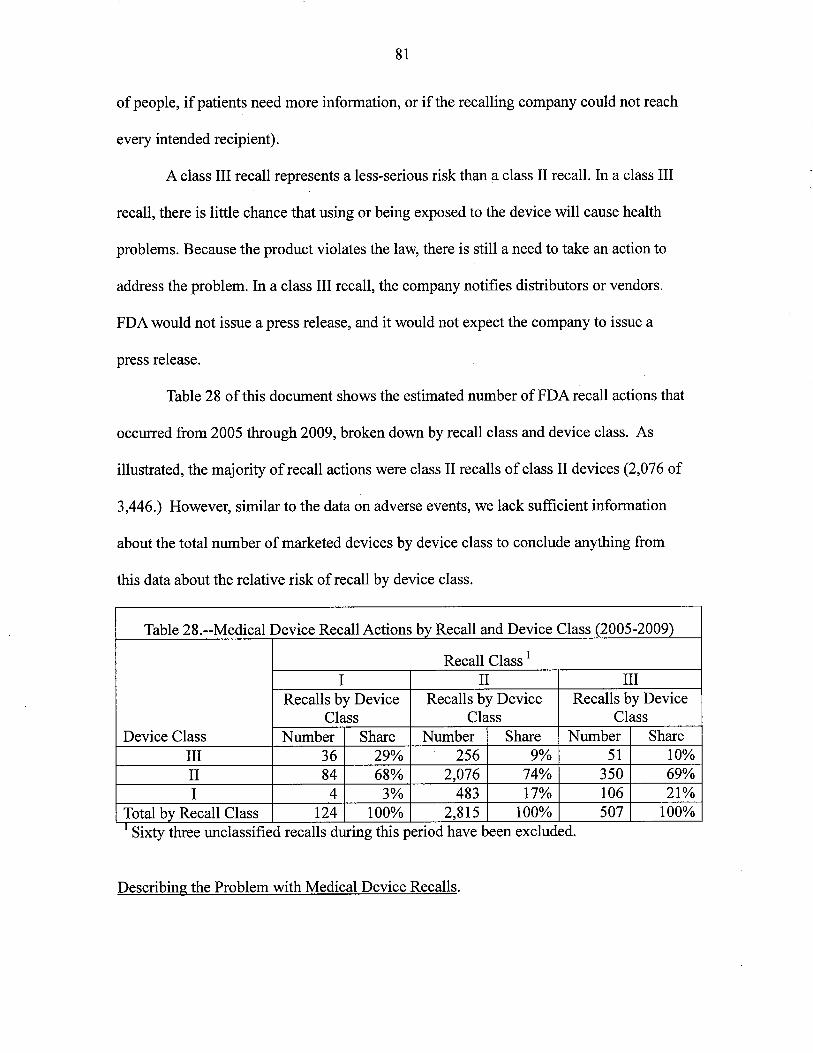
81
of people, if patients need more information, or if the recalling company could not reach
every intended recipient).
A class III recall represents a less-serious risk than a class II recall. In a class III
recall, there is little chance that using or being exposed to the device wil cause health
problems. Because the product violates the law, there is stil a need to take an action to
address the problem. In a class III recall, the company notifies distributors or vendors.
FDA would not issue a press release, and it would not expect the company to issue a
press release.
Table 28 of this document shows the estimated number of FDA recall actions that
occured from 2005 through 2009, broken down by recall class and device class. As
ilustrated, the majority of recall actions were class II recalls of class II devices (2,076 of
3,446.) However, similar to the data on adverse events, we lack suffcient information
about the total number of marketed devices by device class to conclude anything from
this data about the relative risk of recall by device class.
Table 28.--Medical Device Recall Actions by Recall and Device Class (2005-2009)
Recall Class 1
I II IIIRecalls by Device Recalls by Device Recalls by Device
Class Class ClassDevice Class Number Share Number Share Number Share
III 36 29% 256 9% 51 10%
II 84 68% 2,076 74% 350 69%I 4 3% 483 17% 106 21%
Total by Recall Class 124 100% 2,815 100% 507 100%i .Sixty three unclassified recalls during this penod have been excluded.
Describing the Problem with Medical Device Recalls.

82
Many of the problems described with respect to adverse medical device event
reporting also affect device recalls. With incomplete information or poor device
identification recalls are often incomplete or misdirected. Indeed, the same device may be
identified with several different descriptors. Identifying and locating all of the recalled
devices, while simultaneously not removing devices without problems, presents many
challenges, even when a single product is involved. When a recall action involves many
versions or types of a product, the problems of incomplete data are multiplied. With a
large number of products involved in a single recall action, product removal could be
slow and possibly incomplete, which suggests that potentially hazardous devices
occasionally remain in use beyond their recalL. Incompletely or slowly executed recalls of
potentially hazardous devices could lead to patient deaths or injuries: the longer a
defective or problem recalled device remains in use, the more likely it is to cause a
serious problem.
Although class I recalls generate the most thorough and careful recall efforts,
even these recalls can be hampered by incomplete product identification. For example, an
incomplete class I recall involved a brand of bronchoscope that was diffcult to sterilze
completely due to a design defect. Because of a failure in communication at one large
hospital, the recalled bronchoscopes continued to be used after the recall, resulting in a
pattern of infections among the affected patients.
How the UDI Requirements Can Improve the Situation.
Near- Term Improvements.
Increasing the speed and effectiveness of medical device recalls would reduce
adverse events associated with those recalled devices. Although the threat posed by

83
incomplete withdrawals of recalled devices exists, curent databases are inadequate to
estimate the numbers of patient injuries or deaths or injuries that might be averted with
more effective FDA management of device recalls. For example, in the case ofthe
incomplete class I recall of the bronchoscope, had a UDI system been incorporated in the
hospital's materials management system, while not suffcient to prevent the episode, it
might have helped the hospital to identify the recalled bronchoscopes more quickly and
completely remove them from service sooner, thereby reducing the numbers of patients
potentially exposed to infection.
Futue Improvements.
A device identifier, combined with a system that can capture the device identifiers
in patient records, would also have facilitated the search for at-risk patients (assuming
that electronic health systems were in place) by providing a computer searchable number
in the record, possibly preventing active infections or more quickly identifying infections
needing treatment. A comprehensively implemented UDI-based system would facilitate
more thorough and complete FDA dissemination of information about the specific
devices being recalled and FDA oversight of the recall action.
3. Reduced Device Related Medical Errors and UDI
Another potential benefit of the required UDI system would be reduced medical
errors from human and mechanical problems with medical devices. FDA's
MAUDEdatabase captues reports of device related medical errors. However, the
limitations of the MAUDE database described above prevent us from estimating the
frequency of reported medical errors associated with devices. Table 27 includes reports
of device related medical errors, but the frequency is not explicitly or easily enumerated.

84
Furhermore, medication errors, a subset of medical errors, may also be attributed
to medical devices. Although not nationally representative, an insight into the frequency
of device related medication errors comes from the 2007 IOM report on preventing
medication errors. The IOM report cites a study of medication errors by major cause
which attributes 4 percent of medication errors to devices (Ref. 9). Because we lack data
on the frequency of device related medical errors, the agency is requesting that
commenters provide specific data or anecdotal information on the natue and frequency
of device related medical errors including device-related medication errors.
Establishing a link between an FDA-mandated UDI system and a reduction in
medical errors is more complicated. The UDI and the GUDID would allow users to
electronically access specific product identification and information printed on the device
labeL. Although the final regulation would not require other entities to electronically
capture and use this information in automated systems, there is a clear intent for the UDI
to serve as a key that would link the GUDID with other complementary systems in use
now or that might be developed to reduce medical errors and improve patient safety.
Hospitals and other health-care facilities wil choose to make investments in the new
technology and methods if they expect it to be a cost-effective method to reduce errors
involving medical devices. To the extent that the FDA-requirement for a UDI increases
the perceived cost-effectiveness of scanner-based device use and thereby increases the
use of scaner-based treatment delivery, it could lead to reduced medical errors. The
identification technology, however, would not be the decisive consideration. Other
studies indicate that health-care facilities base the technology adoption decision on cost
and effectiveness.

85
As we point out in the discussion of adverse event reporting, medical errors may
be noticed sooner and preventive measures taken when adverse event reports are
collected in a central database (or linked databases under standardization). The adverse
medical device event that appears random to individual users can more readily be
identified as a design, performance, or user error when combined with like events and
analyzed using a unique identifier.
Putting a standardized unique device identifier on a device label is one step in
creating systems that could reduce device related medical errors. Changes in technology
and user practices are also required. The proposed rule would create a platform that
would enhance the value of the new electronic health technologies and thereby might
encourage their development. But the decision to invest and adopt the new technologies
would be made independently of the proposed rule.
4. The Public Health Implications of Better Device Analyses by FDA
The public health benefits from the UDI would come from related reductions in
medical device-related patient injuries and deaths. More accurate and prompt
identification of problems would enable more rapid action to reduce the incidence of the
adverse events. Public health safety alerts, for example, could be more accurate and
timely. FDA would be able to car out recall actions more effciently with more effective
targeting of the problem device.
The proposed rule would standardize how medical devices are identified. A
standardized UDI could serve as an electronic key to link device information among
existing and future databases related to device use and safety. Thus, a UDI for medical
devices could contribute to potential public health benefits of initiatives aimed at

86
optimizing the use of automated systems in healthcare, but we canot estimate those
futue benefits without knowing what those healthcare systems would be.
Because we have insuffcient information to quantify the public health gains from
this proposed rule, we carr out an ilustrative break-even analysis to determine the level
of effectiveness that would cover the total costs of the proposed rule. The total present
value ofthe costs of the proposed rule over 10 years would be about $515 milion using a
7 percent discount rate and about $590 milion using a 3 percent discount rate. The
average number of deaths associated with (although not necessarily caused by) reported
adverse medical device events was 2,973 per year from 2008 through 2010 (table 27 of
this document). We exclude all non-fatal adverse events from the calculations because
those events include a wide a variety of outcomes and are dominated by the value of
averted fatal events. The curent estimated value of a statistical life used in FDA analyses
is $7.9 millon (Ref. 10). Using 10-year averages and assuming that benefits begin in year
3, we find that less than a 0.5 percent decline in the average anual reported number of
deaths (about 14 averted deaths per year using a 7 percent discount rate and 13 averted
deaths per year using a 3 percent discount rate) would produce monetary benefits
approximately equal to the total present value of the costs of the proposed rule. Because
reported adverse device events represent a fraction of the number of actual adverse
device events, the percentage breakeven decline as a fraction of all adverse device events
would be smaller than 0.5 percent.
In sumary, the UDI should benefit FDA's adverse medical device event
reporting and surveilance efforts, and improve recall operations. Despite some
diffculties and incompleteness that are likely to remain in FDA's data, the enhancement

87
could lead to earlier, more definitive, or more frequent identification of problem devices.
The increased effectiveness of sureilance and the more effective management of recalls
should reduce the total number of adverse medical device events, although we are unable
to quantify that reduction.
1. Alternatives to the Proposed Regulation
The agency identified and assessed the costs for labelers of the following
alternatives to the proposed rule:
1. Full UDI requirements for unclassified and class I, II, and III devices.
2. A requirement for labeling only.
3 Apply UDI requirements only to class II and class III devices.
4. A UDI that includes only static information (variable information such as lot
or batch, serial number, and date, would not be required).
5. Apply UDI requirements only to class III devices.
The costs ofthese alternatives are summarized in table 37 of this document.
Consistent with analysis presented in the Cost Section F of this document, we assume for
all alternatives that labelers of excepted devices (devices covered by proposed
801.30(a)(3) - (12) general exceptions) would be excepted from the UDI requirements,
and some smalllabelers, assumed to exclusively distribute over-the-counter devices to
retail outlets. The proposed rule includes an implementation of seven years before all
requirements must be implemented. Because the implementation schedule could differ
across the alternatives considered, to simplify and to present a more robust comparison of
costs, in this section we assume immediate implementation. This means that all up front
costs and anual costs are assumed to occur beginning in the first year for all alternatives

88
and for the proposed rule. The best estimate of costs of the proposed rule to initial
labelers with the phased-in implementation schedule is shown in table 18.
The first alternative includes most of the requirements of the proposed rule, but
does not allow for certain reduced requirements for class 1 devices. The costs for the next
four alternatives allow the labeling and database requirements to vary and differences in
costs are compared to the highest cost alternative.
1. Full UDI Requirements for Unclassified and Class I, II, and III Devices
Under this alternative, all requirements of the proposed would rule apply to class
II and III devices. However, the label for class I devices would be required to also bear
the production identifier portion of its UDI and class I devices that FDA has exempted
from GMP regulations would not be included under a general exception. Direct marking
is unchanged.
The costs of this alternative are shown in table 29 of this document. Because
some class I labelers would be required to include variable information in the device
identifier portion of the UDI and the label of some GMP-exempt devices would be
required to bear a UDI under this alternative, one-time costs related to planing and
administration would be increased by $9.5 millon compared with the proposed rule
(table 17). In addition, the one-time costs for barcode registration and for recordkeeping
would be increased slightly, $0.1 milion for barcode registration and $0.3 milion for
recordkeeping. Anual costs associated with equipment and software would be increased
by $17.0 milion and $8.0 milion, with incremental label materials ($1.9 milion) and
recordkeeping and reporting to GUDID increased somewhat ($0.04 milion). Because all
device labelers are assumed subject to the date format requirements, we do not assume

89
changes to the cost due to label redesign. However, class I device labelers may spend
about $1.9 milion more anually in label material costs because the variable identifier
portion ofthe UDI may require more space than the fixed identifier only. Costs for
directly marking devices remain unchanged. The following paragraphs discuss more
fully the two largest categories of cost increases: equipment and software.
Under this alternative, all class I establishments not covered by the general
exceptions under 801.30(a)(3) - (12) would incur costs related to complying with the
variable barcode requirement. The costs would be dependent on the labeler's curent
printing capabilities and their compliance response which might include using outside
contractors to print labels that incorporate variable information, modifying curent in-
house label printing systems, or purchasing and installing equipment that would
incorporate the frequent changes needed to include the variable information. For the
approximately 1,840 class I initiallabelers, the one-time increase in costs would equal
$28.2 milion, and the anual cost increase would equal $14.4 milion. For the 830
repackagers and relabelers, the increase in costs would be $7.1 milion in one-time and
$2.7 anually.
Similarly, we assume that wider use of variable identifiers will require additional
softare and data integration costs. This may include costs related to purchasing and
installng software or modifying existing device tracking systems, and software
validation and training. To calculate the cost increase for softare, we assumed that each
class I firm operates only one establishment. This assumption may overestimate the
number of affected firms and thus overestimate the cost increase. The one-time increase
in costs would equal $52.4 milion, and the anual increase in cost would be $7.1 milion
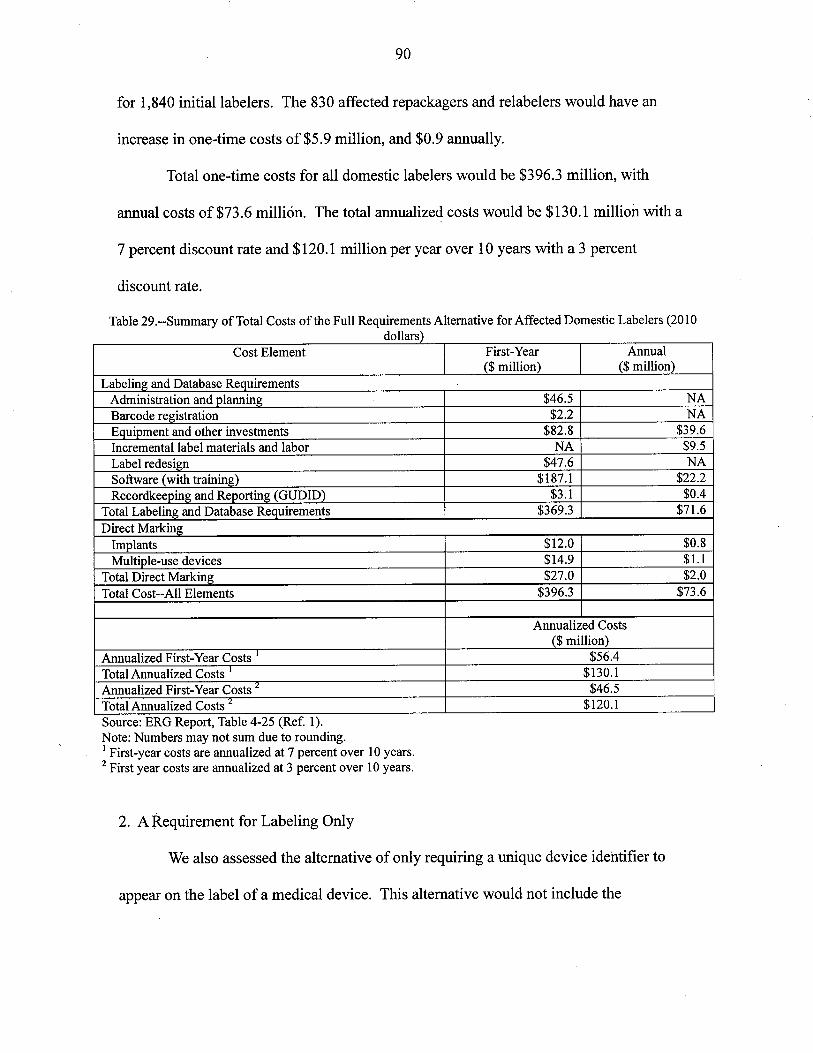
90
for 1,840 initiallabelers. The 830 affected repackagers and relabelers would have an
increase in one-time costs of$5.9 milion, and $0.9 anually.
Total one-time costs for all domestic labelers would be $396.3 milion, with
anual costs of$73.6 millón. The total anualized costs would be $130.1 millon with a
7 percent discount rate and $120.1 milion per year over 10 years with a 3 percent
discount rate.
Table 29.--Summary of Total Costs of the Full Requirements Alternative for Affected Domestic Labelers (2010dollars)
Cost Element First-Year Annual($ milion) ($ milion)
Labeling and Database RequirementsAdministration and planning $46.5 NABarcode registration $2.2 NA
Equipment and other investments $82.8 $39.6Incremental label materials and labor NA $9.5Label redesign $47.6 NASoftare (with training) $187.1 $22.2Recordkeeping and Reporting (GUDID) $3.1 $0.4
Total Labeling and Database Requirements $369.3 $71.6
Direct MarkingImplants $12.0 $0.8Multiple-use devices $14.9 $1.
Total Direct Marking $27.0 $2.0
Total Cost--All Elements $396.3 $73.6
Annualized Costs
($ milion)Annualized First-Year Costs i $56.4Total Annualized Costs i $130.1Annualized First-Year Costs 2 $46.5Total Annualized Costs ;¿ $120.1Source: ERG Report, Table 4-25 (Ref. 1).Note: Numbers may not sum due to rounding.i First-year costs are annualized at 7 percent over 10 years.2 First year costs are annualized at 3 percent over i 0 years.
2. A Requirement for Labeling Only
We also assessed the alternative of only requiring a unque device identifier to
appear on the label of a medical device. This alternative would not include the

91
requirements for direct marking of devices and for filing related exceptions, and would
not require device identifying information to be submitted to a GUDID.
The largest reduction in costs compared to the first alternative would be from not
requiring direct marking. One-time costs of $27.0 milion for implants and multiple-use
devices, and $2.0 milion in anual costs, would be avoided. Also, there would be
reduced costs of about $3.1 milion in one-time costs, and $0.4 milion anually related to
the GUDID. Although not included in the sumar cost comparisons of alternatives,
FDA would not incur one-time costs of about $1.6 milion and anual costs of$1.9
millon to set up and maintain the GUDID.
Total one-time industry costs of this alternative to only require a UDI on medical
device labels would be $366.2 milion and anual costs would be $71.2 milion. (See
table30 of this document.) The total anualized costs of this alternative would be $123.4
milion per year, using a discount rate of 7 percent over 10 years, and $114.2 milion per
year at 3 percent over 10 years. Under this scenario, all firms would have annual
compliance costs of less than 1 percent of revenues.

92
Table 30.--Total First-Year, Annual and Annualized Costs ofUDI Implementation for All Labelers Under theL b l 0 i Al . i (2010 d II )a e ing nlv temative o ars
Cost Element First-Year Annual($ milion) ($ milion)
Labeling and Database RequirementsAdministration and planning $46.5 NABarcode registration $2.2 NAEquipment and other investments $82.8 $39.6Incremental label materials and labor NA $9.5Label redesign $47.6 NASoftare (with training) $187.1 $22.2
Total--All Cost Elements $366.2 $71.2Annualized Costs
($ milion)Annualized First-Year Costs 1. $52.1Total Annualized Costs 2 $123.4Annualized First-Year Costs j $42.9Total Anualized Costs 3 $114.2Source: ERG Report, Table 6-2 (Ref. 1).Note: Numbers may not sum due to rounding.i Labelers include medical device manufacturers, reprocessors, specification developers, repackagers and
relabelers.2 First-year costs are annualized at 7 percent over 10 years.3 First year costs are annualized at 3 percent over 10 years.
3. Apply UDI Requirements Only to Class II and Class III Devices
Under this alternative, FDA would require labelers of class II and class III devices
to meet the UDI labeling and GUDID requirements, and all classes of devices would be
directly marked. Labelers of class I devices and unclassified devices would be exempt
from the general UDI labeling and database reporting provisions. If labelers of
unclassified devices were required to comply, this would slightly increase the estimate of
the total number of affected labelers and the costs of this alternative.
We used FDA's Registration and Listing database to match product codes to class
identifiers in FDA's product codes database. Class II and class III devices were
identified, and the counts of firms and establishments by type of labeling activity were
recalculated for this subset of labelers. Table 31 shows the revised count of domestic
class II and class III labeling establishments and firms by employment size. The total

93
number of affected labeler establishments under this alternative would be 4,282,
compared with 7,578 labeler establishments identified in tables 4 and 5 of this document.
Table 31 .--Number of Affected Domestic Establishments by Employment Size and Type of Labeling ActivityUnder a Class II and Class II UDI Alternative 1,2
Type of Labeler Emr: loyment Size
Initial Labelers 1-4 5-9 10- 20- 50- 100- 250- 500- 1000 or Total19 49 99 249 499 999 more
Manufacturers 1,027 500 438 440 264 233 117 43 26 3,088Single-Use Device 0 3 0 2 2 2 4 0 0 13
Reprocessors3Specification 376 109 96 76 26 13 3 1 i 700Developers
Em¡: loyment Size
Non-Manufacturing 1-4 5-9 10-49 50- 100- 250- 500 or more TotalLabelers 99 249 499Repackagers and 270 78 100 17 10 4 2 481
Relabelers1 Source. ERG Report, Tables 6-3,6-4, and 6-6 (Ref. i).2 Numbers may not add due to rounding.3 All counts ofreprocessors by size remain the same as those in table 5 of this document, except tlat we assumethat the reprocessors in the 5-9 employment size group are the likeliest to be currently reprocessing class Idevices. Therefore, all but three of these reprocessors are removed from the analysis to match the total number ofestablishments reprocessing class II and class II devices.
The number of affected domestic firms that manufactue class II and class III
devices by size and type oflabeling activity is shown in table 32 of this document. The
number oflabeling firms affected by this alternative is 3,673, compared with 6,778 firms
identified in table 6 of this document. We adjust these counts of affected establishments
and firms for exceptions and baseline compliance. We use the same methods for
calculating costs for class II and III establishments as described in the Cost Section F of
this document.

94
Table 32.--Number of Affected Domestic Firs by Size and Type of Labeling Activity Under a Class II and ClassII UDI Alternative 1,2
Type of Labeler Employment SizeInitial Labelers 1-4 5-19 20-99 100-199 200-499 500-999 1,000 or Total
moreManufacturer 877 791 526 114 88 38 122 2,556
Single-Use Device 0 3 3 2 2 i 0 11
ReprocessorsSpecification Developer 393 179 73 9 6 2 5 668
Employment SizeNon-manufacturing Labelers 1-4 5-19 20-499 500 or more Total
Repackagers and Relabelers 263 115 52 9 438i Source. ERG Report, Tables 6-5, and 6-7 (Ref. 1).2 Numbers may not add due to rounding.
The domestic costs of the alternative to apply the provisions of the proposed rule
to only class II. and class III devices and direct marking to all device classes are shown in
table 33 of this document. One-time costs to alllabelers to comply with only the labeling
and database requirements would be $212.0 milion and anual costs would be $44.4
milion. One-time and anual costs related to direct marking are unchanged at $27.0
milion and $2.0 milion. The total one-time costs ofthis alternative would be $238.9
milion and anual costs would be $46.4 milion.

95
Table 33.-- Total First-Year, Annual and Annualized Costs ofUDI Implementation for All Labelers Under theClass II and Class II Alternative i (2010 dollars)
Cost Element.
First-Year Annual($ milion) ($ milion)
Labeling and Database RequirementsAdministration and planing $27.8 NABarcode registration $0.9 NAEquipment and other investments $48.2 $23.7Incremental label materials and labor NA $7.9Label redesign $28.4 NASoftare (with training) $104.9 $12.6Recordkeeping and Reporting (GUDID) $1.8 $0.2
Total Labeling and Database Requirements $212.0 $44.4Direct Marking
Implants $12.0 $0.8Multiple-use devices $14.9 $1.
Total Direct Marking $27.0 $2.0Total--All Cost Elements $238.9 $46.4
Annualized Costs
($ milion)Annualized First-Year Costs L $34.0Total Anualized Costs Z $80.4Anualized First-Year Costs j $28.0Total Annualized Costs j $74.4
Source: ERG Report, Table 6-8 (Ref. 1)Note: Numbers may not sum due to rounding.i Labelers include medical device manufacturers, reprocessors, specification developers, repackagers and
relabelers of class II and class II devices.2 First-year costs are annualized at 7 percent over 10 years.3 First-year costs are annualized at 3 percent over 10 years.
The total anualized costs ofthis alternative would be $80.4 milion per year
using a 7 percent discount rate over 10 years, and $74.4 milion per year at 3 percent.
Impacts per firm would remain the same as for the proposed rule, because this alternative
would not affect per-establishment costs or the need for direct marking.
4. A Unique Device Identifier That Includes Only Static Information
Under this alternative, we modify the full UDI alternative such that labelers
would not be required to include variable information in the UDIs. The UDI would
include only the fixed portion that could be used to access data that identifies the specific
version or model of a device and the labeler of that device. Existing human-readable
variable information would continue to appear on medical device labels (e.g., the lot,

96
batch, serial number, expiration date or date of manufacture), consistent with most
curent practices. Under this alternative, more establishments would already comply with
the requirements because of existing use of static barcodes. We estimate that 2/3 of the
manufactuers with 50 or more employees use at least static barcoding. Those that do not
barcode static information are mostly small establishments. About 5 percent, of all
manufactuers with fewer than 50 employees are assumed to use a static barcode. No
reprocessors or specification developers are assumed to label with static barcodes.
Manufactuers would continue to use curent printing procedures and would not
need to purchase additional printing equipment. In addition, because variable
information would not be contained within the barcode, firms would be able to use their
curent systems of tracking lot, batch or serial numbers and no new software t~ integrate
variable information into existing systems or related training would be needed. Planing
and administrative costs would be reduced primarily because less time is needed to
develop plans for those labelers going from not printing any barcode to printing a static
barcode.
The one-time costs to register for barcodes, and one-time label redesign costs
would remain unchanged from the full requirement alternative. Certain anual costs for
supplemental labels and coordination with outside printers would be avoided. One-time
and anual costs for direct marking and GUDID would not change.
For repackagers and relabelers, this alternative would provide some reductions in
cost to repackagers and relabelers because the requirements to add a static barcode are
simpler to plan and cary out.

97
A sumar of the total costs of requiring static information in the UDI for all
labelers is presented in table 34 of this document. The one-time costs for labeling and
database requirements of a static barcode alternative would be $87.2 milion and anual
costs would be $3.4 milion. The most significant reductions in the costs compared with
the full requirements alternative would be about $82.8 millon in one-time costs and
$39.6 millon in anual costs for equipment and other investments, and $187.1 milion in
one-time costs and $22.2 milion anually for software and training. Costs for direct
marking would remain at $27.0 millon in one-time costs and $2.0 milion in anual
costs.
Table 34.-- Total First-Year, Annual and Annualized Costs of UDI Implementation for All Domestic LabelersUnder the Static Barcode Alternative i (2010 dollars)
Cost Element First-Year Annual
($ milion) ($ milion)Labeling and Database Requirements
Administration and planning $34.3 NABarcode registration $2.2 NAEquipment and other investments NA NAIncremental label materials and labor NA $3.0Label redesign $47.6 NA
Softare (with training) NA NA
Recordkeeping and Reporting (GUDID) $3.1 $0.4Total Labeling and Database Requirements $87.2 $ 3.4
Direct MarkingTotal Direct Marking $27.0 I $2.0
Total--All Cost Elements $114.2 $5.4Annualized Costs
($ millon)Annualized First-Year Costs 2 $16.3Total Annualized Costs Z $21.7Annualized First-Year Costs'- $13.4Total Annualized Costs 3 $18.8Source: ERG Report, Table 6-18 (Ref. 1).Note: Numbers may not sum due to rounding.i Labelers include medical device manufacturers, reprocessors, specification developers, repackagers and
relabelers.2 First-year costs are annualized at 7 percent over 10 years.3 First year costs are annualized at 3 percent over 10 years.

98
The total one-time costs ofthis alternative would be $114.2 milion and anual
costs would be $5.4 milion. The anualized costs would be $21. 7 milion per year at a 7
percent discount rate over 10 years, and $18.8 millon per year at 3 percent.
5. Apply UDI requirements only to Class III devices.
Under this alternative FDA would require only labelers of class III devices to
meet the UDI labeling and GUDID requirements, and only class III devices would be
directly marked. The general approach and the underlying assumptions used for
preparing this estimate are not comparable to the methods used to estimate alternatives 1
through 4 due to the very limited subset of firms and devices that would be covered by
the class III only alternative. Therefore, this estimate is intended only to provide a rough
estimate of the costs. The simplifying assumptions are discussed in more detail below.
The uncertainty surrounding this estimate is broader than the uncertainty discussed for
the other alternatives.
To analyze this alternative, we used the Registration & Listing database to match
product codes to class identifiers in FDA's product codes database. Those devices that
were identified as class III devices were captued in the analysis, and counts of
establishments by type (manufacturer, reprocessor, specification developer, and R/)
were recalculated for this subset of class III-only labelers. We assume for puroses of this
estimate that one firm operates one establishment. This assumption possibly overstates
firm-level costs estimated for the softare cost component because the assumption
overestimates the number of firms that would be affected. We then assumed that the
distribution by size and NAICS for both firms and facilities would be the same as used
for all affected entities. For this analysis, however, we assumed no class III devices are

99
over-the-counter devices sold at retail, so there was no adjustment made for such
exceptions. We also assumed that only 1 percent of multiple-use device establishments
label class III devices.
Table 35 presents the revised count of domestic class III labeling establishments
by employment size. The total estimated number of affected labeler establishments and
firms by employment size for this alternative based on the methodology described in the
preceding paragraph would be 444, compared with 7,578 labeler establishments and
6,778 labeler firms affected for all devices.
Table 35.--Number of Affected Domestic Establishments by Employment Size and Type of Labeling ActivityU d CL II UDI 0 i Al . i 2 (20 10 d II )n er a ass mv. temative ' oars
Type of Labeler Emplovment SizeInitial Labelers 1-4 5-9 10- 20- 50- 100- 250- 500- 1000 or Total
19 49 99 249 499 999 moreManufacturers 119 58 51 51 31 27 14 5 3 359Single-Use Device 0 0 0 0 0 0 0 0 0 0
ReprocessorsSpecification 34 10 9 7 2 1 0 0 0 64Developers
Employment SizeNon-Manufacturing 1-4 5-9 10-49 50- 100- 250- 500 or more TotalLabelers 99 249 499Repackagers and 12 3 4 1 0 0 0 21
Relabelersi Source. ERG Report, Tables 6-9,6-10 ,6-11 and 6-12 (Ref. i).2 Numbers may not add due to rounding. Although the total number of establishments and firs are assumed to
be equal, employment size categories for establishments vary somewhat from employment size categories forfirs; categories are more aggregated at the fir leveL.
We generally used the same methods for calculating costs as described in the Cost
Section F of this document. The analysis continues to assume the percentages of
establishments curently barcoding with variable barcodes remain the same under this
alternative.
For the direct marking cost estimate, we determined that very few multiple-use
devices would be classified as class III (many are surgical instruents that are class I

100
devices). We estimated that only 1 percent of multiple-use device establishments would
be affected by the direct marking requirement. To keep the number of affected direct
marking facilities from exceeding the number of total class III establishments, we also
assumed that 40 percent of implant manufactuers and 20 percent of specification writers
would label class III implants. The combination of these assumptions results in an
estimated 84 percent of all establishments handling class III devices needing to also mark
their devices.
The domestic costs of the alternative to apply the provisions of the proposed rule,
including direct marking, to only class III devices are shown in table 36 of this document.
One-time costs to labelers to comply only with the labeling and database requirements
would be $33.6 milion and anual costs would be $5.8 milion. One-time and anual
costs for direct marking would be $4.7 milion and $0.3 milion. The total anualized
cost estimated for this alternative would be $11.6 milion per year using a 7 percent
discount rate over 10 years and $10.6 milion using 3 percent. Although narowing the
scope of devices covered would reduce the compliance costs to industry substantially, a
unique device identifier would not be required for the majority of medical devices (class
II and I) that are associated with serious adverse events and with recalls. See tables 27
and 28.

101
Table 36.-- Total First-Year, Annual and Annualized Costs of UDI Implementation for All Domestic LabelersUnder the Class II Only Alternative i (2010 dollars)
Cost Element First-Year Annual($ milion) ($ milion)
Labeling and Database RequirementsAdministration and planing $3.1 NABarcode registration $0.07 NAEquipment and other investments $5.1 $2.6Incremental label materials and labor NA $0.6Label redesign $3.1 NASoftare (with training) $22.1 $2.6Recordkeeping and Reporting (GUDID) $0.2 $0.03
Total Labeling and Database Requirements $33.6 $5.8Direct Marking
Implants $4.5 $0.3Multiple-use devices $0.1 $0.01
Total Direct Marking $4.7 $0.3Total--All Cost Elements $38.3 $6.1
Annualized Costs
($ milion)First-Year Costs, annualized at 7 percent over 10 years $5.4Total Annualized Costs, with 7 % annualized i st. Year Costs $11.6First-Year Costs annualized at 3 percent over 10 years $4.5Total Annualized Costs, with 3% annualized 1 st_ Year Costs $10.6
Source: ERG Report, Table 6-14 (Ref. 1)Note: Numbers may not sum due to rounding.i Labelers include medical device manufacturers, reprocessors, specification developers, repackagers and
relabelers of class II devices.
6. Sumar of Alternatives
Table 37 of this document sumarizes the one-time and anual costs of the
proposed rule and of alternatives 1 through five, assuming immediate implementation.
U sing a 3 and 7 percent discount rate over 10 years, the total anualized cost of each
alternative and the difference in anualized costs compared with the previous alternative
are also presented.
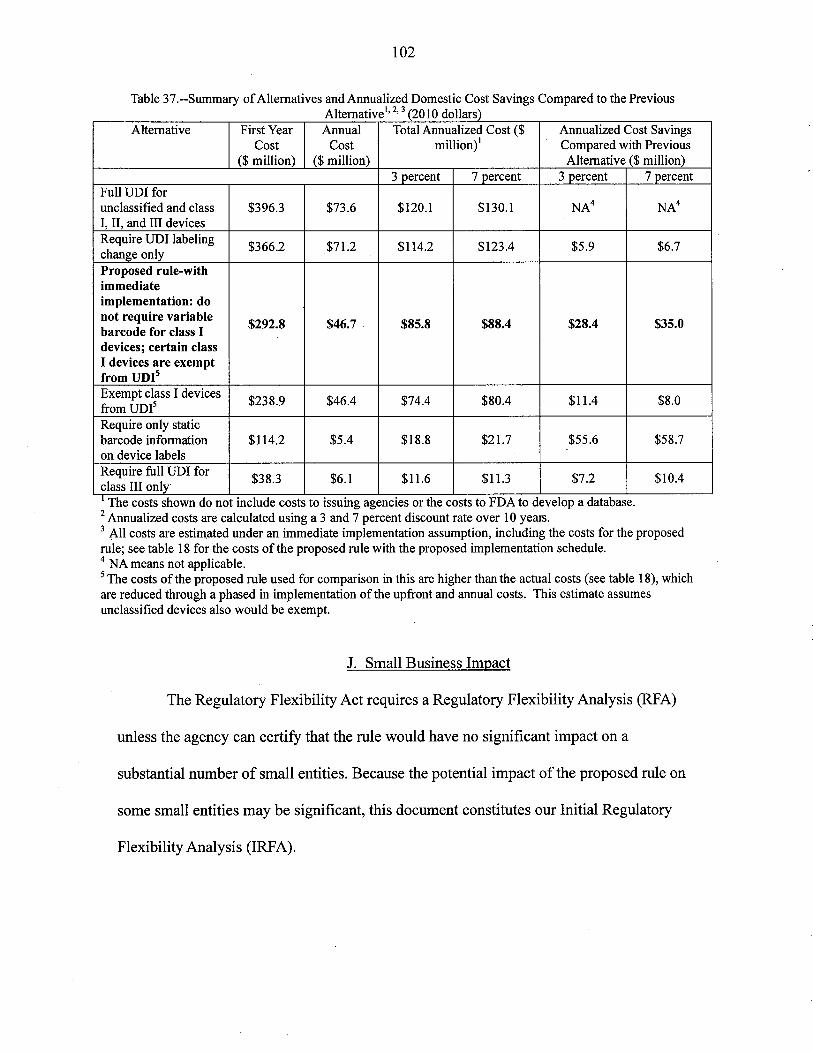
102
Table 37.--Summar of Alternatives and Annualized Domestic Cost Savings Compared to the PreviousAlternativel' 2, 3 (2010 dollars)
Alternative First Year Annual Total Annualized Cost ($ Annualized Cost SavingsCost Cost milion)
i Compared with Previous($ milion) ($ milion) Alternative ($ milion)
3 percent 7 percent 3 percent 7 percentFull UDI forunclassified and class $396.3 $73.6 $120.1 $130.1 NA4 NA4I, II, and II devicesRequire UDI labeling
$366.2 $71.2 $114.2 $123.4 $5.9 $6.7change only
Proposed rule-withimmediateimplementation: do
not require variable$292.8 $46.7 $85.8 $88.4 $28.4 $35.0barcode for class I
devices; certain class
I devices are exemptfrom UDi5Exempt class I devices
$238.9 $46.4 $74.4 $80.4 $11.4 $8.0from um5Require only staticbarcode information $114.2 $5.4 $18.8 $21.7 $55.6 $58.7on device labelsRequire full um for
$38.3 $6.1 $11.6 $11. $7.2 $10.4class II onlyi The costs shown do not include costs to issuing agencies or the costs to FDA to develop a database.2 Annualized costs are calculated using a 3 and 7 percent discount rate over 10 year.3 All costs are estimated under an immediate implementation assumption, including the costs for the proposed
rule; see table 18 for the costs ofthe proposed rule with the proposed implementation schedule.4 NA means not applicable.5 The costs of the proposed rule used for comparison in this are higher than the actual costs (see table 18), whichare reduced through a phased in implementation of the upfront and annual costs. This estimate assumesunclassified devices also would be exempt.
J. Small Business Impact
The Regulatory Flexibility Act requires a Regulatory Flexibility Analysis (RFA)
uness the agency can certify that the rule would have no significant impact on a
substantial number of small entities. Because the potential impact of the proposed rule on
some small entities may be significant, this document constitutes our Initial Regulatory
Flexibility Analysis (IRFA).

103
1. Need for the Rule and Objectives ofthe Rule
The proposed rule would fulfill the statutory requirement to establish a unique
device identification system for medical devices that would adequately identify a device
through distribution and use. Curently, medical devic,, manufactuers are not required to
use a standardized device identifier. The proposed rule would standardize how medical
devices are identified by requiring that medical devices be labeled with a UDI that is both
human and machine readable. In the near-term, we anticipate that UDI will help to
improve the effciency of recalls of medical devices and to improve medical device
adverse event reporting. In the future, standardized device identifiers would contribute to
the success of other initiatives aimed at optimizing the use of automated systems in
healthcare.
2. Number of Affected Small Entities
The proposed rule would affect labelers of medical devices. As discussed
previously in this document, 6,569 domestic firms would be considered labelers for the
puroses of this rule, including medical device manufactuers, medical device
reprocessors, specification developers, and firms that repackage or relabel medical
devices. Small firms that only handle devices covered by the general exceptions from the
proposed UDI requirements, including the GMP exempt class I labelers, would not be
affected by the UDI requirements ofthe proposed rule. We anticipate that the potential
impact of the rule on excepted firms would be minimal compared to the impact on small
firms that would be required to add the UDI to device labels. To avoid understating the
impact of the proposed rule on small entities, we concentrate our analysis on domestic
firms that would need to conform to the UDI requirements.

104
The SBA considers as small, medical device manufacturers with 500 or fewer
employees and medical device wholesalers with 100 or fewer employees. Device
manufactuers would be included in NAICS categories for manufacturing industries;
firms that repackage and relabel medical devices would be included in NAICS categories
for the merchant wholesale industry. Because no NAICS category exists for medical
device reprocessors, we use the size standard for NAICS 339112 to determine the number
of small reprocessors. Similarly, no NAICS category exists for medical device
specification developers. To determine the number of small specification developers, we
use the size standard for the medical device manufactuing industry (NAICS 3391).
Table 38 of this document shows the SBA size standards for the NAICS categories of
affected labelers.
Table 38.--Size Standards by Tvpe of Labeler and NAICSType of Labeler NAICS Description of Industr SBASize
Standard
(Number of
Employees)
Manufacturers 325413 In vitro diagnostic substances manufacturing 500
334510 Electromedical & electrotherapeutic apparatus 500manufacturing
334517 Irradiation apparatus manufactuing 500
339112 Surgical & medical instrment manufacturing 500
339113 Surgical appliance & supplies manufacturing 500
339114 Dental equipment & supplies manufacturing 500
339115 Ophthalmic goods manufacturing 500
Repackaging & 42345 Medical, Dental and Hospital Supplies Merchant 100
Relabeling Wholesalers Industr
42346 Ophthalmic Goods Merchant Wholesalers Industr 100
An estimated 1,873 small firms would meet the criteria for the general exceptions
(including the GMP exempt class I exception) from all UDI requirements ofthe proposed
rule. Table 39 ofthis document shows that of the estimated 4,693 domestic non-excepted
firms, 96 percent fall below the SBA size standard for small firms. For device
manufacturing, the percentage of small firms ranges from 88 percent for in vitro

105
diagnostic substances manufactuing to 98 percent for dental equipment and supplies
manufacturing. The percentage of small firms for the other types of labelers equals 96
percent for reprocessors, 95 percent for firms that repackage and relabel devices, and 99
percent for specification developers.
a e .--Num er an Percentage 0 ecte Sma InS ,y Type 0 La e erEmployment Number Percent of
Type of Labeler Size of Fins Small Fins1-4 5-19 20-499 Small Total
Initial Labeling Firms 1,060 1,339 1,051 3,451 3,612 96%Employment
Size1-4 5-19 20-99
Repackaging &Relabeling Fins 654 297 81 1,032 1,082 95%Total 1,714 1,636 1,132 4,483 4,693 96%
'L bl 39 b d fAffì d II F' b fbi
Source: ERG Report, Table 7-5 (Ref. 11).Numbers may not sum due to rounding.
3. Description of the Reporting and Recordkeeping Burdens and Personnel Skil Levels
Regardless of size, all firms subject to thè UDI requirements oftheproposed rule
would need to perform several actions, some of which include reporting and
recordkeeping. Because medical device labelers routinely prepare and submit reports to
FDA, none of these actions would require new skils. Moreover, alllabelers have
personnel who can prepare labels with the UDI and operate label printing or marking
equipment. Consequently, no new skils would be needed to conform to the requirements
of the proposed rule. Table 40 of this document describes the reporting and
recordkeeping burdens by major cost component.

106
a e .-- otentia eporting an ecor eeping ur ens on ma a e er irsCost Component Actions involving reporting or recordkeeping Percentage of Professional Skil
Small Firs LevelAdministration and Create new or modify existing SOPs--accounts for 100% lManagerialPlaring about 25 percent of cost component.Barcode Registration Complete registration form--a minor par of this 10% Managerial
componentEquipment Record outcome of the verification tests and necessary 100% Quality Control
emedial actions IispectorDirect Marking Document exceptions require 10 hours per exception 3% with Managerial
exceptionsVerify safety by preparing sumary of literature 3% verify safetyeviews
Softare Document testing, verification and validation 100% IIspector or qualityassurance; IT,
Except for smallest firs, automates UDI-related accounting orecordkeeping and report generation clerical staff for
eportsGUDID Primary reporting and recordkeeping requirement. 100% T, managerial,
Automated or web-based entr minimizes the time echnical or clericalneeded for these actions. Requires from 3 to 4.5 hours staff trained to
in first year and 1 hour annually in subsequent years. !upload data
T bl 40 P . lR dR dk B d S llLbl F"
4. Impact of the Rule on Small Entities
We use u.s. Census data on average industry receipts to estimate the impact of
the proposed rule on small entities. Table 41 of this document shows the average anual
receipts for small firms by NAICS and employment size. For this analysis, the average
anual receipts for NAICS 339112 serves as a proxy for average anual receipts of
reprocessing firms and the device manufacturing industry average anual receipts serves
as a proxy for average anual receipts for specification developers. The average anual
receipts of firms in NAICS 42345 and 42346 serves as a proxy for the average anual
receipts for firms that repackage and relabel medical devices.

107
bl b d S' f' ( II )Ta e 41.--Average nnua Receipts y Type an ize 0 Fir 2007 Do ars
Type of Labeler i Per Fir Average Annual Receipts ($1,000)
b, Employment Size0-4 5-19 20-499
Employees Employees EmployeesNACIS 325413 $890.4 $3,459.3 $28,350.9NAICS 334510 $520.4 $2,093.2 $21,094.8NAICS 334517 $594.1 $2,287.7 $18,572.2NAICS 339112 $443.0 $1,726.1 $15,901.6NAICS 339113 $365.8 $1,619.2 $13,649.7NAICS 339114 $330.7 $1,042.1 $16,218.1NAICS 339115 $1,643.6 $1,556.6 $8,124.2Reprocessors L $443.0 $1,726.1 $15,901.6Specification Developers j $568.2 $1,657.8 $15,742.1
0-4 5-19 20-99Employees Employees Employees
Repackaging and Relabeler Firs4 $807.5 $2,804.2 $14,287.5
A
Source: ERG Report, Tables 5-4 and 5-9 (Ref. 1), based on estimated receipts reported for 2007 (SBA, 2007).i NAICS codes for medical device manufacturing firs.
2 Estimated to equal annual receipts forNAICS 339112.3 Estimated to equal average receipts for the medical device manufacturing industr.4 Estimated to equal the average of annual receipts for NAICS 42345 and 42346.
To estimate the magnitude of the potential burden of the proposed rule on small
firms, we calculate the average anualized costs of the rule as a percentage of average
anual receipts. The detailed cost estimates discussed previously were adjusted from an
establishment basis to a firm basis and aggregated by firm size. Table 42 of this
document shows a breakdown by employment size for small firms of the total anualized
costs over 10 years with 3 percent and 7 percent discount rates. Firms that directly mark
devices would have higher anualized costs than similar-sized firms that would not need
to directly mark devices; firms directly marking implants would have the highest
anualized costs of all types of small firms. Furhermore, we only include the costs for
labelers required to include the variable information portion of the UD1. The average
anualized costs for labelers of class I devices excepted from including production
information would be substantially lower than the average anualized costs for labelers
required to include the production information. Consequently, our estimate of the

108
potential burden of the proposed rule for labelers with no devices requiring direct
marking significantly overestimates the burden for labelers of class I devices.
II )Table 42.--Annualized Domestic Costs of the Proposed Rule for Sma Firs by Type and Size (2010 dollarsType of Small Fir Anualized Per Fir Costs bv Emplovment Size
3 percent 7 percent
1-4 5-19 20-499 1-4 5-19 20-499
Initial Labelers with No Direct $1,199 $9,576 $31,369 $1,333 $10,345 $33,782Marking that Include VariableInformationInitial Labelers with Direct $28,190 $36,567 $101,659 $31,449 $40,461 $111,327Marking of ImplantsInitial Labelers with Direct $6,413 $14,790 $74,736 $7,150 $16,162 $81,147Marking of Multiple-Use
Devices1-4 5-19 20-99 1-4 5-19 20-99
Firs that Repackage and $969 $5,464 $23,997 i $1,072 $5,958 $25,8351Relabel DevicesSource: ERG Report, Tables 5-5 and 5-9 (Ref. 1).i Annualized costs for firs with 20- 1 99 employees.
Tables 43 and 44 of this document illustrate the burden ofthe proposed rule on
small firms that would not be expected to directly mark devices. We estimate the relative
burden of the proposed rule on different size firms as anualized costs as a percentage of
average anual receipts. As shown in tables 43 and 44 of this document, the burden for
firms not directly marking devices would not exceed 1 percent of average annual receipts
with 3 percent and 7 percent discount rates, with the burden estimated to be the largest for
small firms in NAICS 339114 that employ between 5 and 19 employees.

109
Table 43.--Relative Burden of the Proposed Rule by Industr Sectors Not Needing to Directly Mark DevicesIndustr Sector Annualized Costs as a Percentage of Average Anual Receipts
3 percent 7 percent1-4 5-19 20-499 1-4 5-19 . 20-499
NAICS 325413 0.1% 0.3% 0.1% 0.1% 0.3% 0.1%
NAICS 334510 0.2% 0.5% 0.1% 0.3% 0.5% 0.2%
NAICS 334517 0.2% 0.4% 0.2% 0.2% 0.5% 0.2%
NAICS 339114 0.4% 0.9% 0.2% 0.4% 1.0% 0.2%
NAICS 339115 0.1% 0.6% 0.4% 0.1% 0.7% 0.4%
Reprocessors NA 0.6% 0.2% NA 0.6% 0.2%
Specification Developers 0.2% 0.6% 0.2% 0.2% 0.6% 0.2%1-4 5-19 20-99 1-4 5-19 20-99
Repackage and Relabel 0.1% 0.2% 0.2%2 0.1% 0.2% 0.2%2
Source: Tables 41 and 42 of this document.i Excludes firs with devices that wil require direct marking, firs labeling excepted devices and class I devices
exempt from GMP regulations, and firs that currently use variable barcodes. As noted, costs are annualizedover 10 years with a 3 percent or 7 percent discount rate. Average per fir revenues from table 4 i of thisdocument.2 Based on per-fir costs for the 20-199 employment size; this likely overstates the impact of the proposed ruleon these small entities.
We anticipate that the proposed rule would create the greatest burden on small
firms required to directly mark devices. These firms would normally be included in
NAICS 339112 and NAICS 339113. Table 44 of this document shows that the burden on
firms in these industries that have no devices that require direct marking ranges from 0.2
to 0.6 percent of average anual receipts using both 3 percent and 7 percent discount
rates. By contrast, an estimated 32 small firms with 1 to 19 employees would incur
anualized costs to directly mark devices that exceed 1 percent of average annual
receipts. Firms required to directly mark implants would have a greater burden than
firms required to directly mark multiple-use devices. For the firms with 1 to 4 employees
that directly mark implants (e.g., 8 firms), anualized costs would be about 7.7 percent of
average anual receipts with a 3 percent discount rate and about 8.6 percent with a 7
percent discount rate, but for the firms that directly mark multiple-use devices (e.g., 19
firms) anualized costs would only be 1.4 percent of average anual receipts with a 3
percent discount rate and 1.6 percent with a 7 percent discount rate. The one-time cost of

110
equipment needed to directly mark implant devices represents 34 percent of the average
anual receipts and the one-time cost of equipment needed to directly mark multiple-use
devices represents 5 percent ofthe average anual receipts. For firms with 5 to 19
employees, anualized costs as a percentage of average anual receipts would total 2.3
percent with a 3 percent discount rate and 2.5 percent with a 7 percent discount rate for
firms with direct marking of implants (e.g., 5 firms), and only 0.9 percent with both 3
percent and 7 percent discount rates for firms with direct marking of multiple-use devices
(e.g., 13 firms). The one-time cost of equipment needed to directly mark implant devices
represents 8 percent of the average anual receipts and the one-time cost of equipment
needed to directly mark multiple-use devices represents 1 percent of the average anual
receipts. For more detail on the burden ofthe proposed rule on small firms, see section 5
and section 7 of the ERG report.
Average anualized costs exceed 1 percent of average anual receipts for about
0.7 percent of all affected smalllabelers. For about 7 percent of the firms with fewer than
20 employees that manufacture surgical and medical instruent (NAICS 339112),
average anualized costs as a percent of average anual receipts would exceed 1 percent.
The burden for about 2 percent of the firms with fewer than 20 employees that
manufactue surgical appliance and supplies (NAICS 339113) would exceed 2.3 percent.
Because of our uncertainty about the burden of the direct marking requirements on small
firms, we request detailed comment from small firms about our estimates of the potential
impact of the proposed rule and how they expect to respond to the direct marking
requirements. For more detail on the burden of the proposed rule on small firms, see
section 5 and section 7 of the ERG report.

111
Table 44.--Relative Burden of the Proposed Rule by Employment Size and Industr Sector on Small EntitiesRequired to Directly Mark Devices
Affected Industr by Type of Devices that Require Employment SizeDirect Marking 1-4 I 5-19 I 20-499
Number of Affected Firs iNAICS 339112 - Surgical and medical instrment manufactuing
Multiple-use items require direct marking 19 I 13 I 53
No devices reauire direct marking 57 I 201 I 226
NAICS 339113 - Surgical appliance and supplies manufacturingImplants reauire direct marking 8 I 5 I 17
No devices reauire direct marking 133 I 382 I 401
Anualized Per Fir Costs as Percent of Average Annual
Receipts2
NAICS 339112 - Surgical and medical instrment manufacturingMultiple-use items require direct marking 1.6% I 0.9% I 0.5%
No devices reauire direct marking 0.3% I 0.6% I 0.2%
NAICS 3391 13 - Surgical appliance and supplies manufacturingImplants reauire direct marking 8.6% I 2.5% I 0.8%
No devices require direct marking 0.4% I 0.6% I 0.2%Annualized Per Firm Costs as Percent of Average Annual
Receipts 3
NAICS 339112 - Surgical and medical instrment manufactuingMultiple-use items require direct marking 1.4% I 0.9% I 0.5%
No devices require direct marking 0.3% I 0.6% I 0.2%
NAICS 339113 - Surgical appliance and supplies manufacturingImplants require direct marking 7.7% I 2.3% I 0.7%
No devices reauire direct marking 0.3% I 0.6% I 0.2%
Source: Tables 41 and 42 of this document.i Firms are counted once (Le., firs with devices requiring direct marking are excluded from the count of firs
with no devices requiring direct marking).2 Costs annualized at 7 percent over 10 years.3 Costs annualized at 3 percent over i 0 years.
5. Alternatives Considered
We analyze the costs of several alternatives to the proposed rule in the
Alternatives Section I of this document. The costs and cost savings for the alternatives
are sumarzed in table 37. Because approximately 96 percent of the affected labelers
are small entities according to the SBA size standards, the impact on small firms would
be the essentially same as for the industry as a whole.
II. References
1. Eastern Research Group, Inc., "Unique Device Identification (UDI)
for Medical Devices," 2011.

112 .
2. U.S. International Trade Commission, Interactive Tariff and Trade
Data Web; http://dataweb.usitc.gov.
3. U.S. Census Bureau, U.S. International Trade Statistics, aggregate
data for NAICS 339112, 339113,339114,339115,334510, and 334517;
http://censtats.census.gov/naic3 6/naics3 6.shtml.
4. OECD Micro Trade Indicators (by category of industry, ISIC), data
extracted on 28 Ju12010 19:45 UTC (GMT) from OECD.Stat;
ww.oecd.org/std/its/tradeindicators.
5. Michael F. Furawa, T.S. Raghu, Trent J. Spaulding and Ajay
Vinze, "Adoption Of Health Information Technology For Medication Safety
In U.S. Hospitals, 2006" Health Affairs, 27, nO.3 (2008):865-875.
6. Heffin, B.J., T.P. Gross, and T.J. Schroeder, "Estimates of medical
device-associated events from emergency deparments," American Joural of
Preventive Medicine, 27 (3):246-253, 2004.
7. Samore, M.H., RS. Evans, A. Lassen, P. Gould, J. Lloyd, R.M.
Gardner, R Abouzelof, C. Taylor, D.A. Woodbur, M. Wily, and R.A.
Bright, "Sureilance of medical device-related hazards and adverse events in
hospitalized patients," Joural of the American Medical Association,
291(3):325-334,2004.
8. Bright, RA., "Sureilance of adverse medical device events," in
Brown, L.S., Bright, RA, and DR Travis, (Eds.), Medical Device
Epidemiology and Sureilance, pp. 43-61, New York: Wiley, 2007.

113
9. Leape, L. et aI, "Systems Analysis of Adverse Drug Events, Joural
oftheAmerican Medical Association, 274(1):35-43, 1995, as cited by
Institute of Medicine, "Preventing Medication Errors," National Academy
Press, 2007.
10. U.S. Environmental Protection Agency, National Center for
Environmental Economics, "Frequently Asked Questions on Mortality Risk
Valuation," 2010;
http://yosemite.epa. govl eel epa! eed.nsf/pages/MortalityRisk Valuation.html.
Updated to 2009 using U.S. Deparment of Commerce, Bureau of Economic
Analysis, National Economic Accounts: Current-dollar and "real" GDP,
2010a; http://ww. bea. gov Inational/.