updated-early phase drug developmetn and population pk and its' value
TRANSCRIPT

Early Phase Development and Population PK and Its Value
Navigating the Transition from Animal to Man
CAPT E. Dennis Bashaw, Pharm.D. Dir. Division of Clinical Pharmacology-3
Office of Clinical PharmacologyOffice of Translational Sciences
US Food and Drug Administration

2
Disclaimer
• The presentation should not be considered, in whole or in part as beingstatements of policy or recommendation by the United States Government orthe US Food and Drug Administration.
• Throughout the presentation representative products or organizations may beused as examples to emphasize a point, no endorsement is either intended orimplied.

3
Outline
•Attrition in Drug Development–What is it really?
•Approaches For Dose Selection–General Approaches
•The High Risk Trial-General Procedures
•A Tale of Two Programs–TGN-1412
–BIA 10-2474
•Risk Recognition and Management

4
ATTRITION IN DRUG DEVELOPMENT
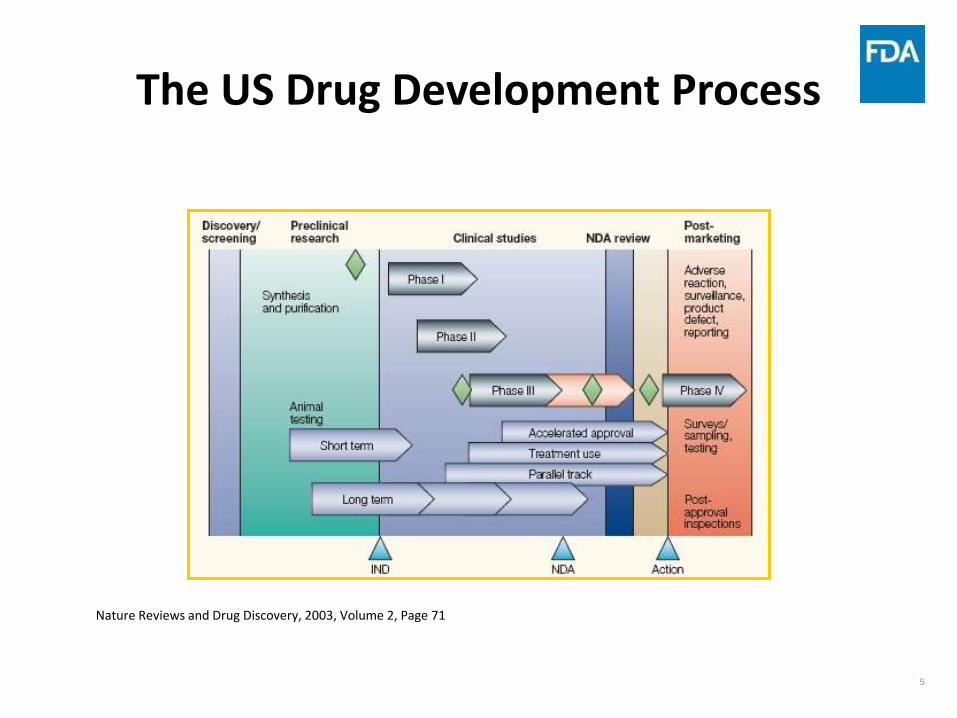
5
The US Drug Development Process
Nature Reviews and Drug Discovery, 2003, Volume 2, Page 71

6
Lengthy Process to Reach Market

7
Updated Drug Development Cost Figures
J Health Econ. 2016 May;47:20-33. doi: 10.1016/j.jhealeco.2016.01.012

8
The Cost of Research vs Ease of Conduct
High
Low
Single Center Randomized Trials
Low
Single Case
Reports
Cohort Studies
Case-Control Studies
Case Series
HighEase of conduct
Multi-Center Randomized Trials
Cost
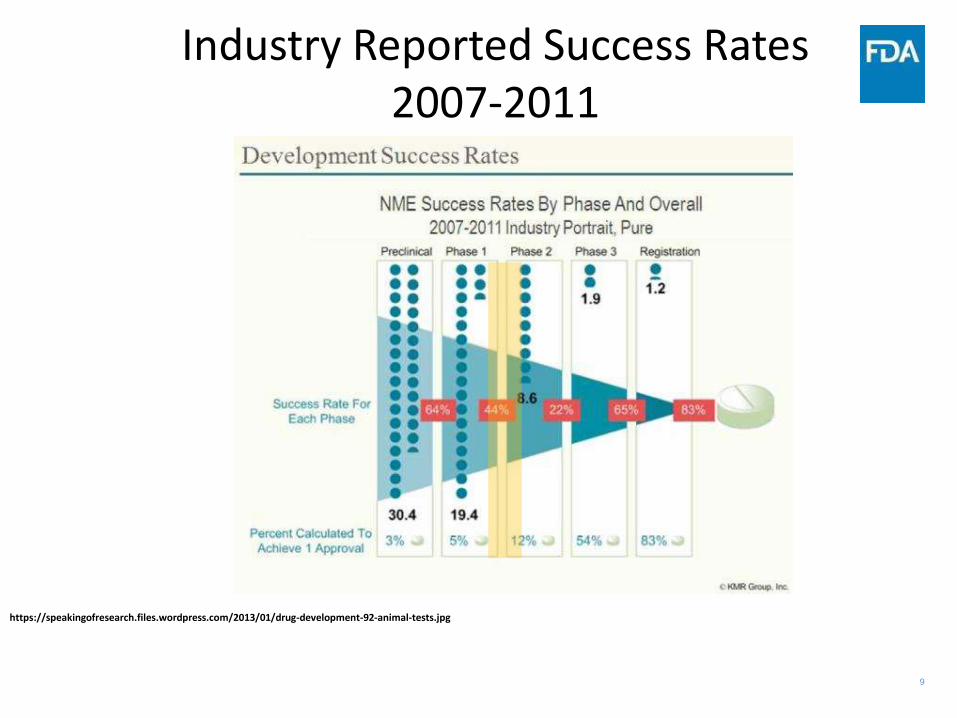
9
Industry Reported Success Rates 2007-2011
https://speakingofresearch.files.wordpress.com/2013/01/drug-development-92-animal-tests.jpg

10
Reasons for Lack of Success in Drug Discovery
Lack of fundamental knowledge regarding the causes of CNS disorders
Absence of biomarkers for diagnosing and monitoring these conditions
A paucity of animal models that are congruent with the human disease state
The likelihood that CNS conditions are multifactorial in their etiology
These factors are true for most therapeutic areas.
They are also factors that we can IMPACT.
Williams, Michael & Enna, S J “Prospects for neurodegenerative and psychiatric disorder drug discovery” Expert Opin. Drug Discov. (2011) 6(5):457-463

11
Work Streams for First-in-Human (FIH)Study Design
Risk Assessment
Preclinical Safety Evaluation
Biomarker Development
ResearchPharmacology
PK/PDModeling
First-in-Human
Study Design
Adapted from “Gibbs, JP-, “Prediction of Exposure–Response Relationships to Support First-in-Human Study Design”, The AAPS Journal, Vol. 12, No. 4, December 2010, pg 750-758

12
APPROACHES TO DOSE SELECTION

13
General Approaches to Starting Dose Selection
• Classic approach: NOAEL/safety factor (≥10)–Usually derived from doses rather than exposures (i.e., NOEL, PAD)
• the safety factor can be increased (>10) when there is a high level of concernfor the observed toxicity in the animal species (e.g., a steep dose-responsecurve for the appearance of severe toxicity or death in animals)
• the safety factor can be decreased (<10) when there is a low level of concernfor the identified toxicity in animals (e.g., mild adverse effects that can bemonitored and/or were shown to be reversible).
• Allometric Methods–Human equivalent dose (HED) based on animal NOAELs
• NOAEL-HED Approach–Combine NOAELs with HED and a safety Factor.
Guidance for industry and reviewers: Estimating the safe starting dose in clinical trials for therapeutics in adult healthy volunteers, July 2005, http://www.fda.gov/CDER/guidance/5541fnl.pdf

14
Definitions and their Relationship
• NOAEL – no observed adverse effect level,
• PAD – pharmacologically active dose,
• MABEL – minimum anticipated/acceptable biological effect level,
• HED – human equivalent dose, AUC – area under the concentration-time curve,
• Cmax – maximum concentration,
• MRSD – maximum recommended starting dose.

15
ICH Guidance on Approaches to Selection
• The selection of the starting dose for healthyvolunteers is often based on the NOAELdetermined in pivotal toxicology studies.–ICH Guidance M3(R2), “Nonclinical Safety Studies for the Conduct of
Human Clinical Trials and Marketing Authorization forPharmaceuticals
• Normally, two species prior to and during the clinical testing ofinvestigational products.
• In some cases, toxicity testing in a single species can be justified, especiallyfor biotechnology-derived products (ICH Guidance S6(R1), Preclinical SafetyEvaluation of Biotechnology-Derived Pharmaceuticals).

16
FDA Guidance
• The FDA guidance on safe starting dose selection inhealthy volunteers provides an algorithm fordetermining the MRSD (maximum recommendedstarting dose). This algorithm includes the selection of amost appropriate species for determining the MRSD.
http://www.fda.gov/downloads/Drugs/Guidances/UCM078932.pdf

17
Guidance Algorithm
The provided algorithm is a “GUIDANCE”, clinical judgement must be used at all times in selecting starting doses for patient safety

18
THE HIGH RISK TRIAL-GENERAL PROCEDURES

19
In general, the higher the potential risk associated with an investigational medicinalproduct (IMP) and its pharmacological target, the greater the precautionary measures thatshould be exercised in the design of the first-in-human study. The protocol should describethe strategy for managing risk including a specific plan to monitor for and manage likelyadverse events or adverse reactions as well as the procedures and responsibilities formodifying or stopping the trial if necessary. The sponsor should arrange for peer review ofthe protocol and the associated risk factors and to assure that they have been properlyconsidered and planned for.
http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002988.pdf

20
MABEL Approach
• Recommended for high-risk, first-in-man trials
–Usually done in patients where risk tolerance is different and in whom the prospect of therapeutic benefit must be considered
• Minimal Anticipated Biological Effect Level (MABEL)
–Based on all RELEVANT in-vitro and in-vivo PK/PD data including•Receptor binding studies
•Concentration/response data
• Exposure at pharmacologic doses in a relevant animal model/species
–Starting Dose=MABEL dose/Safety Factor• If NOAEL-HED dose is lower, use this dose instead
•Patient Safety is always a concern

21
Characteristics of a Starting Dose
• A Safe Starting Dose
–Does not cause any clinical measurable effects
•no pharmacodynamic effects
•nor toxic effects
•dose prior MED / PAD (minimal effective dose, pharmacologically active dose)
–The next higher dose causes first pharmacological effects (if detectable in healthy volunteers) without toxic effects
•MED
Log Dose
Effe
ct

22
General Procedures
• Cohort Size
– Larger cohorts allow for better estimates and earlier refinement of escalation strategy
– Larger cohorts put more subjects at risk and increase the costs of clinical development programmes
–Common standard is an A + P design
• A = 6 to 10 subjects on active therapy
• P = 2 to 4 subjects receiving placebo

23
General Procedures
• Dose Escalation-Relevant factors
–Steepness of the slope of dose/effect and dose/toxicity relationship
•Greatly affected by cohort size
–Therapeutic range in non-clinical models
–Predictability of the effects of the next dose step
–Potential pharmacodynamic effects (if any)
–Potential toxic effects

24
Dosing in High-Risk Trials
• Initial sequential dose administration design within each cohort
• Adequate period of observation between the administration of each subject depending on estimated PK and PD data
• Before administration of the next cohort all results from all subjects of the subsequent cohort(s) must be reviewed
• PK and PD data from the previous cohorts should be compared to known non-clinical PK, PD and safety information
• Patient Safety is always a concern
• Stopping rules must be clear and unambiguous

25
A TALE OF TWO PROGRAMS

26
TGN1412

27
TGN1412
TGN1412 was developed as a therapeutic agent for variousdiseases in which T cells are involved in the pathogenesis ofchronic inflammation or hematological malignancies such asleukemia.
TGN1412 was genetically engineered by transfer of thecomplementarity determining regions (CDRs) from a monoclonalmouse anti-human CD28 antibody into human heavy and lightchain variable region frameworks. These variable regions weresubsequently recombined with a human gene coding for the IgG4chain.
–The human constant domain and variable domain framework structures wereexpected to confer decreased immunogenicity and an optimum of antibodyeffector functioning within the human immune system.

28
Clinical Trial Chronology
• TeGenero “first in human” (FIH) study of TGN1412–Direct immune stimulation
• Trial initiated March 13, 2006• Four single doses of 0.1, 0.5, 2.0 and 5.0 mg/kg planned
in 4 groups of 8 subjects • 1st cohort: –0.1 mg/kg IV at 2 mg/min to 6 subjects in the course of one
hour (i.e., one subject dosed every 10 minutes)–Placebo: 2 subjects
G. Suntharalingam, et al., “Cytokine Storm in a Phase 1 Trial of the Anti-CD28 Monoclonal Antibody TGN1412” NEJM, 355;10, pg 1018-1028

29
Clinical Course
• Within 90 min dosing–Severe headache, lumbar myalgia, pyrexia, rigors–Nausea, vomiting, diarrhea–Amnestic episodes, restlessness–Erythema, desquamation–Peripheral vasodilation, hypotension, tachycardia
• Subjects/patients admitted to ICU 12-16 hours after dosing–Multisystem failure–Metabolic acidosis, Disseminated Intravascular Coagulation–Respiratory failure, bilateral infiltrates
• Cytokine storm–Lymphopenia, monocytopenia–↑ ↑ TNF α–↑ IL-2, IL-6, IL-10–↑ IFN-γ
G. Suntharalingam, et al., “Cytokine Storm in a Phase 1 Trial of the Anti-CD28 Monoclonal Antibody TGN1412” NEJM, 355;10, pg 1018-1028

30
Therapy
• Aggressive supportive management–Steroids
–Daclizumab (anti-IL2 receptor antagonist)
–Pulmonary support, dialysis, fresh frozen plasma
–Irradiated cells to decrease GVHD
EXPERT SCIENTIFIC GROUP ON PHASE ONE CLINICAL TRIALS, www.tsoshop.co.uk
Time Course of Immunologic Effects

31
Follow-up at 30 days
• 4 patients better after 48 hours
• 1 has gangrenous digits
• 5 with late myalgia, HA, difficulty concentrating, short-term word-finding problems
• 3 with delayed hyperalgesia
• 2 with peripheral numbness
G. Suntharalingam, et al., “Cytokine Storm in a Phase 1 Trial of the Anti-CD28 Monoclonal Antibody TGN1412” NEJM, 355;10, pg 1018-1028

32
TGN1412-What Went Wrong?Animal-Human Pharmacology
• Potential differences between humans andmonkeys CD28 structure difference in three
transmembrane residues CD28SA binding kinetics and calcium
response (sustained in humans) Immunological Synapse (IS) formation
involving CD28 crosslinking Greater T-cell adhesion to endothelial
cells in humans Greater immunoregulation in animals
Hansel, T., et al, “The Safety and Side Effects of Monoclonal Antibodies”, Nature Reviews Drug Discovery, 2010 Apr; 9(4): 325-38

33
TGN1412-What Went Wrong?Animal-Human Pharmacology
Summary of in vitro activation and proliferation responses of human and Cynomolgus macaque lymphocytes to immobilized TGN1412
EXPERT SCIENTIFIC GROUP ON PHASE ONE CLINICAL TRIALS, www.tsoshop.co.uk

34
TGN1412-What Went Wrong?Receptor Dynamics & Occupancy
TGN1412Anti-CD28 mAb
CD28T-Cell membrane
TGN1412:CD28Complex
Kd = 1.88 nM
0.1 mg/kg IV at 2 mg/minMW= 150,000 DaltonsPlasma Volume= 2.5L
18.7 nM post-dosing
Using Standard Blood Values
T-cells 1.9x106 mL-1
CD28 receptors per cell ~150,000
CD28 ~ 0.95nM
0.86 nM at Equilibrium
0.86/0.95 = 90.5% Occupancy!
EXPERT SCIENTIFIC GROUP ON PHASE ONE CLINICAL TRIALS, www.tsoshop.co.uk

35
General Information Flow for Determining a FIH Dose: TGN1412
Tibbits J., et al, “Practical approaches to dose selection for first-in-human clinical trials with novel biopharmaceuticals” Regulatory Toxicology and Pharmacology, Volume 58, Issue 2, 2010, 243 - 251

36
BIA 10-2474

37
BIA 10-2474
*By Roadster29 - Own work, CC BY-SA 4.0, https://commons.wikimedia.org/w/index.php?curid=46531418
BIA 10-2474 is an experimental fatty acid amide hydrolase inhibitor developed by the Portuguese pharmaceutical company Bial-Portela & Ca. SA. It interacts with the human endocannabinoid system. A Phase I trial incorporating single dose, multiple dose, and a food effect study was underway in Rennes, France, in January 2016, in which serious adverse events occurred affecting five participants, including the death of one man.

38
Trial Design
J Pharmacol Pharmacother. 2016 Jul-Sep; 7(3): 120–126. doi: 10.4103/0976-500X.189661

39
What Happened?Minutes of the Temporary Specialist Scientific Committee (TSSC)
meeting on "FAAH(Fatty AcidAmideHydrolase) Inhibitors”15 February 2016.
FULL REPORT (28 pages)http://ansm.sante.fr/var/ansm_site/storage/original/application/744c7c6daf96b141bc9509e2f85c227e.pdf
MINUTES/SUMMARY (14 pages)http://ansm.sante.fr/content/download/86439/1089765/version/1/file/CR_CSST- FAAH_15-02-2016_Version-Anglaise.pdf

40
RISK RECOGNITION AND MANAGEMENT

41
EMA First-in-Human Study GuidelineRevised July 25, 2017
http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2017/07/WC500232186.pdf

42
EMA First-in-Human Study GuidelineRevised July 25, 2017

43
Risk
• Drug Development is an inherently riskybusiness
• Attrition at each stage of the program isdue to a combination of factors
• EARLY attrition is preferred over lateattrition due to the associated financialand “opportunity” cost

44
Risk Tolerance Evolves
Lack of Appropriate Head Gear
Lack of Protective Clothing No Safety
Rigging
Hard HatsSafety Rigging
High VisibilityClothing
RCA Building-1932 Heron Tower in central London-2011
Bottle of Whisky
Milk Tea?

45
Patrick Muller and Mark Milton, “The Determination and Interpretation of the Therapeutic Index in Drug Development” Nature Reviews Drug Discovery,2012, vol. 11, pg 751-751
TGN 1412
Modified from:
Idealized Efficacy and Safety vs. TGN1412 and BIA 10-2474
BIA 10-2474

46
Post-TGN1412And BIA 10-2474
• The events surrounding both drugs has been a wake-up call tothe drug development industry to the recognition that weneeded to re-examine both our processes and our tools.
• We need to pay more attention to the use of in vitro bindingand inhibition assays
• We need to take the “world view” of information and not viewdisparate data sets separately
• As we use more and more targeted therapies, we must neverrelax our dedication to safety, but by the same token neverallow us to have “paralysis of will” in determining reasonablerisk.

47
Confluence of Decision-making in FIH
Understanding
of Disease
How well do we truly understand the underlying biological
system?
Risk Tolerance
Given the potential benefit what level of risk are we willing to
consider
Animal Data
MABEL, HED, NOAEL,PAD,
FIH Dose
SelectionBest Proposed Dose
Population PKModeling

48
Development of Safe and Effective Drugs Requires a Team Effort
Academia
IndustryInternational
Collaboration
Patient
AdvocacyRegulators
Benefits
To All

49
As drugs become more potent wemust all work to properly• Assess the risk to the patient• Monitor the patient appropriately• Review our tools to make sure
they are up to date and “best inclass”
• Identify potential weaknesses inour programs
• Control the flow of the studyaccording to the protocol
• Mitigate in a real-time activemanner any unforeseen adverseevents
Elements of a Risk Mitigation Planning Process

50
Closing ThoughtRisk Mitigation via Sherlock Holmes
“…If you should find yourself in doubt or in danger --"
"Danger! What danger do you foresee?"
Holmes shook his head gravely. "It would cease to be a danger if we could define it," said he.
The Adventure of the Copper Beeches

51
Contact Information
CAPT Edward D. Bashaw, PharmD.Director, Div. of Clinical Pharmacology -3US FDA10903 New Hampshire AveBuilding 51, Rm [email protected]

52
Acknowledgements• The Staff of the Division of Clinical Pharmacology-3
• The Office of Clinical Pharmacology
• The Office of Translational Sciences
• Hazem E. Hassan, PHD, MS, RPH, RCDS and the other organizers of this meeting