utilization of large-insert libraries to genome analysis of … · genome analysis of genetically...
TRANSCRIPT

Utilization of Large-insert Libraries toGenome Analysis of Genetically
Uncharacterized OrganismsYuji Yasukochi
Insect Genome Research UnitNational Institute of Agrobiological Sciences, Japan

1 Introduction
Whole genome approaches are highly effective for characterizing model organisms; however, “clone byclone” approaches using large-insert genomic libraries are still useful for genetically uncharacterized or-ganisms lacking reference genome sequences. Although recent progress in next generation sequencing(NGS) technologies has been remarkable, the cost of de novo sequencing of higher eukaryote genomes istoo expensive for the small research community, especially for analyzing genetically heterogeneous species.In addition, in genetically uncharacterized species the need for high-quality sequence is limited to specificgenomic regions since widely conserved mechanisms tend to be analyzed using species with rich geneticresources which usually have precise and relatively complete genome sequences. Consequently, it is ex-pected that most determined sequences are not effectively utilized even if whole genome sequencing ofrelatively uncharacterized species is possible. Therefore, it is crucially important to minimize the genomicregion to be sequenced in-depth and to do it at minimum cost. Large-insert libraries are powerful tools forthis purpose. This section is a brief overview of BACs and fosmids, currently the most common large-insertvectors.
1.1 History of Large-insert Genomic Libraries
Until the 1980s, genomic libraries were tentatively constructed for screening with lambda phage vectors (upto 23kb). Most “negative” clones were then discarded after clones harboring the targeted sequences were,fortunately, isolated and stored. Construction and screening of genomic libraries were often limiting factorsfor the complete success of experiments. Furthermore, library screening and preparation of phage DNA arelaborious compared with plasmid vectors. It was evident that such an inefficient and poorly reproduciblesystem could not support analyses of higher eukaryotes with large and complicated genomes. Thus, manyefforts were made to improve insert size and stable maintenance of inserts. Later, the cosmid was developedby insertion of cos sequences required for in vitro packaging of lambda phage into plasmid vectors (Collins& Hohn, 1978). Once transected into Escherichia coli cells, cosmids can be treated in the same manneras plasmids. Approximately 40kb foreign fragments can be cloned into a cosmid. However, a cosmid isa multi-copy vector and inserts are sometimes unstably maintained, presumably because of recombinationvia repetitive sequences or the toxic effects of genes located on them.
The yeast artificial chromosome (YAC) was then developed (Burke et al., 1987) and widely used inearly genome projects. The remarkable ability of YACs to clone large foreign fragments (100kb – 1Mb)enabled coverage over the whole genome with a limited number of contiguous clones (contigs) (e.g., Caiet al., 1994). However, increased difficulty in library construction and low transformation efficiency madeit impossible to discard negative clones after screening, and the current strategy for usage of large-insertgenomic libraries was established. That is, all clones are stored without screening and repeated screening ofpermanently stocked libraries is performed by colony hybridization (Brownstein et al., 1989) or PCR-basedmethods (Green & Olson, 1990).
In spite of the great advantage in cloning huge genomic fragments, YACs have several disadvan-tages. One is a relatively high occurrence of chimerism, co-localization of non-contiguous fragments in asingle clone, which leads to incorrect genome assembly. In addition, YAC-DNA cannot be easily separatedfrom host genomic DNA since a YAC has a yeast centromere, telomere and an autonomously replicat-ing sequence and behaves just like the host chromosomes. This makes it necessary to recover DNA fromYAC-specific bands on agarose gels fractionated by pulsed-field gel electrophoresis for subcloning and se-

Figure 1: Schematic representation of improvements of large-insert vectors.
quencing of inserts. Furthermore, many researchers were not so familiar with genetic manipulation of yeastas E. coli.
In 1992, two vector systems, bacterial artificial chromosomes (BAC) and fosmids, were developed,which are commonly based on the E. coli F factor (Shizuya et al., 1992; Kim et al., 1992). The F factorincludes the parA and parB genes that stably maintain BACs and fosmids at one to two copies per E.coli cell, which reduces the potential risk of recombination and internal deletions of inserts. In addition,BACs and fosmids exist as supercoiled circular plasmids in E. coli, and can be isolated by a commonlyused alkaline lysis method at a low risk of host DNA contamination. An attempt to develop a similarlyeffective system was made by modification of the bacteriophage P1, resulting in the P1-derived artificialchromosome (PAC) which gained the ability to clone larger inserts but lost the ability of phage-mediatedintroduction into E. coli cells (Ioannou et al., 1994).
BACs immediately became widespread as an essential resource for construction of a minimum tilingpath for the “clone by clone” sequencing approach, although the average insert size of BAC libraries (100– 200kb) was generally shorter than that of YAC libraries. Fosmids have cos sequences used for in vitropackaging into lambda phage particles, which limits the maximum cloning capacity to approximately 40kb.On the other hand, the phage-mediated virulence transformation efficiency of fosmids is significantly higherthan BACs, which are introduced into E. coli cells by electroporation. Thus, the technical difficulty ofconstructing fosmid libraries is relatively lower than for BACs. Essential features of BACs and fosmids arecompared below (see section 1.2).
During the 1990s, a whole genome shotgun sequencing (WGS) approach was gradually adoptedfor analysis of organisms having a large genome size (Fleischmann et al., 1995; Adams et al., 2000), andpaired-end sequencing of NGS is now drastically reducing the time and cost required for the WGS strategy.However, large-insert libraries still play important roles in genome analysis (see section 1.3).

1.2 BAC or Fosmid?
BAC and fosmid libraries are the most commonly used long-insert libraries. The most important differencebetween them is that the maximum cloning capacity of fosmids is physically limited by the size of thelambda phage particle. In general, the average insert size of a BAC is 3 – 5 times as large as that of a fosmid,which greatly diminishes the number of stored or screened clones required for the same genome coverage.Reducing the number of stored clones saves the cost and labor for picking, replication and transportationof libraries, as well as valuable space in deep freezers. An increase in the number of clones also leads to anincrease in the cost of library screening. In addition, due to its longer insert size, a BAC contig can be easilyextended by chromosome walking compared with a fosmid. Therefore, BACs are particularly useful forsequence determination of the relatively long region necessary for map-based cloning or characterizationof complex duplicated or extremely long genes.
Nevertheless, BAC libraries are usually constructed by partial digestion of high molecular weightDNA with a restriction enzyme, and coverage at a specific chromosomal point is considerably influencedby the distribution of the restriction sites. This disadvantage can be reduced by construction of multiplelibraries using different restriction enzymes, although the cost required for library construction increases.In contrast, fosmid libraries are constructed by physical shearing of high molecular weight DNA whichare expected to cover a whole genome more evently. Thus, fosmids are valuable for filling gaps betweenBAC contigs, making it better to construct two or more BAC and fosmid libraries rather than multiple BAClibraries alone.
If already constructed libraries are available from genomic resource centers such as Children’s Hos-pital Oakland Research Institute (bacpac.chori.org/) and the Clemson University Genome Institute(www.genome.clemson.edu/services), the cost required for distribution is usually inexpensive. If not,custom library construction is needed. However, it might be troublesome to find a skillful constructor. Now,it is impractical for most researchers to master the technique of BAC library construction by themselves,since there are many commercial or non-profit organizations which undertake the task at a reasonable cost.From common starting materials, most constructors are able to construct excellent BAC libraries withoutfailure since they have already accumulated significant experience. In contrast, it is more challenging toconstruct BAC libraries from organisms for which the method of DNA preparation is not yet established.The quality of BAC libraries, typically represented by average insert size, critically depends on preparationof high molecular weight DNA, and shorter DNA cannot fully utilize the advantage of BACs for the max-imum cloning capacity. In these cases, construction of fosmid libraries should be considered as a betteroption.
The insert size of a fosmid is sufficient for analyzing genomic structure around genes of mediumor small size. For this purpose, fosmids are superior to BACs to ensure sufficient sequence depth andcoverage under the same sequencing conditions, which lessen additional labor necessary to fill gaps andcorrect uncertain reads. In addition, a significantly higher yield is achieved in the preparation of fosmid-DNA compared with BAC-DNA, especially for copy-control vectors whose copy-number per cell can beinduced up to 50. The higher yield of DNA improves the success rate of direct sequencing and reducesthe scale of DNA preparation, which saves additional cost and labor. Shorter inserts are also helpful tosubclone DNA fragments of interest into other vectors. Therefore, it is worthwhile to consider carefullywhether BACs are really the best option.
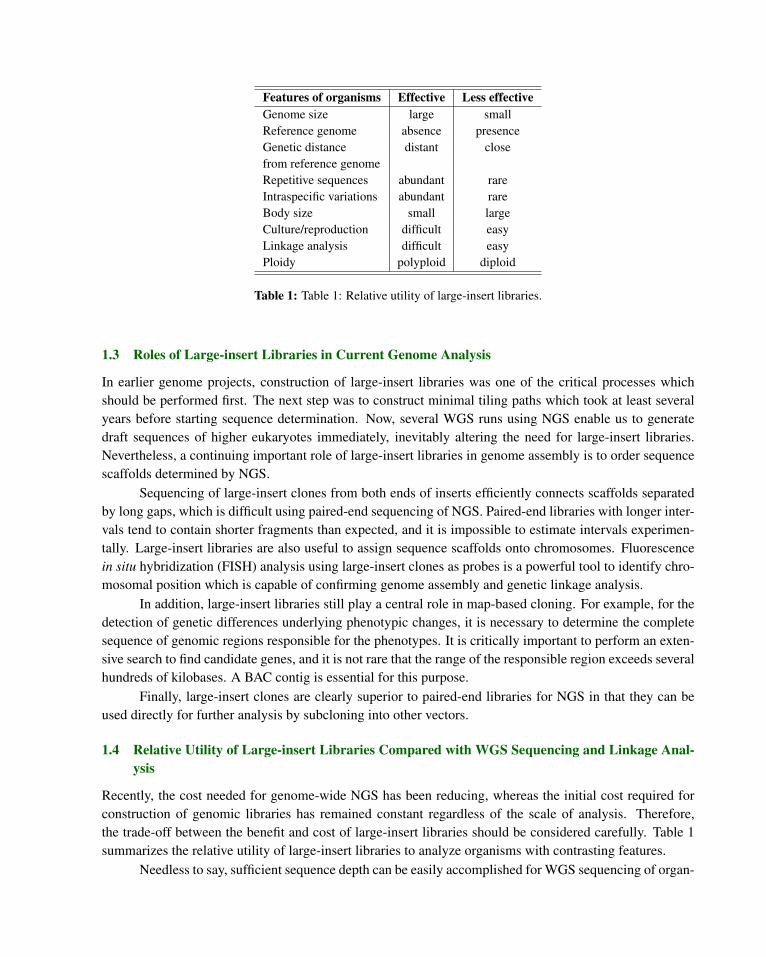
Features of organisms Effective Less effectiveGenome size large smallReference genome absence presenceGenetic distance distant closefrom reference genomeRepetitive sequences abundant rareIntraspecific variations abundant rareBody size small largeCulture/reproduction difficult easyLinkage analysis difficult easyPloidy polyploid diploid
Table 1: Table 1: Relative utility of large-insert libraries.
1.3 Roles of Large-insert Libraries in Current Genome Analysis
In earlier genome projects, construction of large-insert libraries was one of the critical processes whichshould be performed first. The next step was to construct minimal tiling paths which took at least severalyears before starting sequence determination. Now, several WGS runs using NGS enable us to generatedraft sequences of higher eukaryotes immediately, inevitably altering the need for large-insert libraries.Nevertheless, a continuing important role of large-insert libraries in genome assembly is to order sequencescaffolds determined by NGS.
Sequencing of large-insert clones from both ends of inserts efficiently connects scaffolds separatedby long gaps, which is difficult using paired-end sequencing of NGS. Paired-end libraries with longer inter-vals tend to contain shorter fragments than expected, and it is impossible to estimate intervals experimen-tally. Large-insert libraries are also useful to assign sequence scaffolds onto chromosomes. Fluorescencein situ hybridization (FISH) analysis using large-insert clones as probes is a powerful tool to identify chro-mosomal position which is capable of confirming genome assembly and genetic linkage analysis.
In addition, large-insert libraries still play a central role in map-based cloning. For example, for thedetection of genetic differences underlying phenotypic changes, it is necessary to determine the completesequence of genomic regions responsible for the phenotypes. It is critically important to perform an exten-sive search to find candidate genes, and it is not rare that the range of the responsible region exceeds severalhundreds of kilobases. A BAC contig is essential for this purpose.
Finally, large-insert clones are clearly superior to paired-end libraries for NGS in that they can beused directly for further analysis by subcloning into other vectors.
1.4 Relative Utility of Large-insert Libraries Compared with WGS Sequencing and Linkage Anal-ysis
Recently, the cost needed for genome-wide NGS has been reducing, whereas the initial cost required forconstruction of genomic libraries has remained constant regardless of the scale of analysis. Therefore,the trade-off between the benefit and cost of large-insert libraries should be considered carefully. Table 1summarizes the relative utility of large-insert libraries to analyze organisms with contrasting features.
Needless to say, sufficient sequence depth can be easily accomplished for WGS sequencing of organ-

isms with relatively small genomes at only the cost required for library construction and a whole-genomeapproach is an attractive option even if only a bit of the whole genome information is needed to assess thepotential utility of the rest of the determined sequences in the future. Similarly, a limited scale of sequenc-ing is informative if the genome sequence of a closely related species is precisely determined. Conversely,large-insert libraries are useful for organisms of considerably large genome size without any referencegenome sequence.
Repetitive sequences which are abundant in large eukaryote genomes often interfere with genomeassembly. Intra-specific variations also confound genome assembly, which is troublesome, especially forspecies lacking inbred lines selected for multiple generations. Very small body size makes this problemmore serious since template DNA used for NGS is derived from multiple genetically heterogeneous in-dividuals. In contrast, the sequence identity of DNA from a single large-insert clone is helpful to assureaccurate assembly of continuous and long sequences.
Metagenomics is another promising field for large-insert libraries. Metagenomic libraries are con-structed from DNA extracted from a mixture of multiple organisms collected from environmental samplessuch as soil, water or sediment (Rondon et al., 2000). Although NGS is quite effective for gene discov-ery of viable but non-culturable microorganisms, large-insert libraries have a distinct advantage to identifylarge-scale structural organization of such microorganisms, especially for those of low abundance in a pop-ulation.
The feasibility of linkage analysis is also an important factor, since the role of linkage analysis ingenome assembly overlaps considerably with that of physical mapping using large-insert libraries (e.g.,minimal tiling path, FISH analysis). Fine-scale linkage analysis is generally difficult for organisms of verysmall body size, few progeny per single mating, long generation time, or lack or excess of polymorphisms.The relationship between genetic and physical distances varies significantly depending on genomic regions.Physical mapping is more effective than linkage analysis to order sequence scaffolds located in cold spotswhere meiotic recombination rarely occurs.
Large-insert libraries are critical tools for genome analysis of polyploid organisms widely presentamong major crops. Similar sequences derived from different homoeologous chromosomes cause confu-sions in both genome assembly and linkage analysis. Longer and continuous sequences determined fromlarge-insert clones are a robust solution for the problem. Combined with flow cytometry technology, it ispossible to construct libraries specific for a portion of a particular chromosome (e.g., Sehgal et al., 2012).
For identification of novel phenotypes caused by a single gene mutation, a whole-genome approachis evidently inefficient to analyze slight genetic differences between newly generated and parental strains.Combined with linkage analysis, large-insert libraries are helpful to focus on specific genomic region re-sponsible for the phenotype and save time, cost and labor.
2 Utilization of Large-insert Libraries used in Lepidopteran Genetics andGenomics
Lepidoptera is an insect order consisting of more than 150,000 named species of butterflies and moths.Butterflies attract attention by their diverse and fascinating wing patterns, while herbivorous caterpillarshave aroused the hostility of farmers from ancient times. In addition, silk taken from moth cocoons has

been one of the most luxurious textile fibers. Based on both practical and intellectual interest, Lepidopteracomprise a major section of entomology and lepidopteran genetics began soon after the re-discovery ofMendelian inheritance, mainly based on the domesticated silkworm, Bombyx mori. However, it is only oneof several species which are now genetically characterized.
The body-size of evolved Lepidoptera is relatively large for an insect and several hundred progeny aregenerated from a single pair mating of many species, which facilitates detailed linkage analysis using PCR-based methods. Since B. mori has been domesticated for thousands of years and lost the capability of flightand escape, it is an ideal experimental animal by nature (For review, Banno et al., 2010). During the historyof more than a century, many spontaneous and artificially induced mutants have been isolated and storedas genetic resources (e.g.http://www.shigen.nig.ac.jp/silkwormbase/index.jsp). Genome se-quencing of B. mori was completed in 2008 (International silkworm genome consortium 2008), and morethan 30 genes causing mutant phenotypes have been identified by map-based cloning.
We have constructed several BAC and fosmid libraries from moths (Wu et al., 1999; Yoshido et al.,2011; Kamimura et al., 2012) and established a high-throughput PCR-based screening system for isolatingclones of interest from these and other libraries (Yasukochi et al., 2011a). Fluorescence in situ hybridization(FISH) probes are another application of large-insert clones which enables mapping of sequence scaffoldsonto chromosomes (Yoshido et al., 2005a). This technique is also useful for comparing genetically un-characterized species with model sequenced species, which facilitates genetic analysis in uncharacterizedspecies (Yasukochi et al., 2009; Yoshido et al., 2011). BAC libraries are also useful to analyze large geneclusters such as Hox and odorant receptors (Yasukochi et al., 2004; Yasukochi et al., 2011b). In this section,I describe utilization of lepidopteran large-insert libraries mainly based on our own work.
2.1 Genome Assembly
WGS of the B. mori genome was performed by Sanger sequencing before NGS was commonly utilized.Thus, coverage of sequence reads is not so high and end sequences of both BAC and fosmid clones playeda critical role for scaffolding of sequence contigs (International silkworm genome consortium 2008). Con-versely, end sequences were used for assignment of BAC and fosmid clones onto genomic sequences andchromosomes, which are available via a genome database, Kaikobase (Shimomura et al., 2009). How-ever, this approach is now too expensive and labor-intensive, and newer genome projects will stress morea combination of several NGS sequencing runs under different conditions (Zhan et al., 2011). Recently, adetailed genome sequence of a nymphalid butterfly, Heliconius melpomene, was reported, in which BACend sequences were used for assembly verification of sequence scaffolds determined by NGS (HeliconiusGenome Consortium, 2012).
As described below, gene order and chromosome organization are generally well conserved amongthe lepidopteran species examined so far, and precise genome assembly of a limited number of speciesis not necessarily a top priority. Considering the great diversity of Lepidoptera, it is more important tosequence as many species as possible for understanding molecular mechanisms underlying similar and dif-ferent phenotypes by direct and comparative analysis. In line with these aims, a community of entomologistrecently announced the launch of the “i5k” initiative, to sequence the genomes of 5000 species of insectsand other arthropods (Robinson et al., 2011). Easy access to genome sequences of a wide variety of specieswill greatly accelerate functional analysis in arthropods, and large-insert libraries will play an essential rolein narrowing down the genomic region to be analyzed.

2.2 Comparative Genomics
In general, differences in the gene order between two species increase in proportion to the time after theirdivergence by accumulation of chromosomal rearrangements and translocations of genes. However, theextent of conservation varies depending on the taxonomic group. We speculated that the gene order is well-conserved among lepidopteran species since most of their chromosome numbers are 28–31. In particular,the haploid karyotype of the common ancestor is likely to be n=31 since more than half of many independentlineages carry this chromosome number (Robinson, 1971). Therefore, we intended to construct a syntenymap covering a wide range of lepidopteran species.
First, an integrated map of B. mori was constructed, on which 523 BAC contigs including 342 genesand 85 expressed sequence tags (ESTs) were localized (Yasukochi et al., 2006). During the process, weconfirmed significant synteny and conserved gene order between B. mori and H. melpomene in four linkagegroups (Yasukochi et al., 2006). In the next step, it was necessary to add analyzed species. One optionwas to construct a detailed linkage map for each one. However, for this aim it is necessary to map mul-tiple (preferably 4 or more) highly conserved single-copy genes for each chromosome for comparison ofkaryotypes, since paralogs of multi-copy genes and small-scale chromosomal translocation prevent correctassignment of orthologous chromosomes. Although we established an effective method to generate poly-morphic genetic markers by partial sequencing of BAC clones harboring targeted genes (Yasukochi et al.,2006), it was still hard to detect polymorphisms from nearly one hundred conserved genes.
Therefore, we selected FISH analysis to compare genomes using BAC clones as probes (BAC-FISH)(Figure 2). BAC-FISH mapping does not require polymorphism in the genes examined, the use of numeroussibs from matings or multiple genetically homogeneous strains, which are essential for genetic linkageanalysis. Therefore, a relatively small number of heterogeneous insects collected from wild populationscan be analyzed directly (Yasukochi et al., 2009). Moreover, BAC-FISH is highly robust to intra- andinterspecific sequence variation and BAC probes may not necessarily be derived from the examined species(Yasukochi et al., 2009).
The availability of EST data and already constructed libraries influences the cost and time requiredfor BAC-FISH mapping. We selected four moths, the tobacco horn worm, Manduca sexta, the tobaccobudworm, Heliothis virescens, the European corn borer, Ostrinia nubilalis, and the diamondback moth,Plutella xylostella, as species to be analyzed immediately in this respect. We first constructed a BAC-FISH karyotype identifying all 28 chromosomes of M. sexta by mapping 124 loci using the correspondingBAC clones containing orthologous single-copy genes (Yasukochi et al., 2009). B. mori and M. sexta haveidentical haploid chromosome numbers of n = 28; nevertheless, one-to-one correspondence was observedonly for 25 chromosomes despite highly conserved gene order, indicating that at least three chromosomefusion events occurred independently in the the two lineages (Yasukochi et al., 2009).
We then isolated 108 – 184 BAC clones representing 101 – 182 conserved genes from the remainingthree moths, which are not closely related to each other, but share a putative ancestral haploid karyotype ofn = 31 (Yasukochi et al., 2011a). Isolated clones are used for FISH analysis in the same strategy (Saharaet al., manuscript in preparation). Recently, construction of a dense linkage map of P. xylostella based onrestriction-site associated DNA (RAD) sequencing was reported, and an orthologous relationship betweentwo P. xylostella and one B. mori chromosome was identified for B. mori chromosomes 11, 23 and 24(Baxter et al., 2011) (Figure 3). Our unpublished results for karyotypes of O. nubilalis and Helicoverpaarmigera probed with H. virescens BAC clones support these results, strongly suggesting that n = 31 is the

!Figure 2: Strategy of comparative FISH analysis. Known genes and ESTs of other moths wereused as queries against a B. mori genome database, Kaikobase (Shimomura et al., 2009). Genesand ESTs showing significant similarity to putative single-copy B. mori genome sequences wereselected and checked for localization of their B. mori orthologs. For location of confirmed genes,BAC or fosmid libraries were screened and isolated clones were used as BAC probes in FISHanalysis.
ancestral karyotype. In addition, RAD sequencing of H. melpomene with a haploid chromosome numberof 21 also showed that all the chromosomes are composed of 31 units (Heliconius Genome Consortium,2012) (Figure 3).
We also attempted to use fosmids in place of BACs as FISH probes to reduce the cost of library con-struction (Yoshido et al., 2011; Kamimura et al., 2012). A wild silkmoth, Samia cynthia, is unusual in thatgeographic subspecies have different chromosome numbers, ranging from 2n = 25−28, due to variable sexchromosome constitution (Yoshido et al., 2005b). FISH analysis was necessary to reveal the relationshipbetween highly different karyotypes of S. cynthia and B. mori; however, no BAC libraries of S. cynthia orrelated species were available. Thus, we constructed an S. cynthia fosmid library. Using 64 fosmid probesthat generated stable signals by adjusting experimental conditions, a low-number karyotype of S. cynthiawas revealed to be the result of chromosome fusions (Yoshido et al., 2011) (Figure 3). It is of great interestthat putative chromosome fusions occurred independently in each lineage and chromosome fission eventsrarely occurred in Lepidoptera, suggesting the utility of karyotyping to determine phylogenetic relation-ships among related species having variable chromosome numbers.
Large-insert libraries can also be utilized for sequence comparison of a relatively large region toanalyze micro-synteny (Papa et al., 2008; d’Alencon et al., 2010); however, the use of NGS is now more
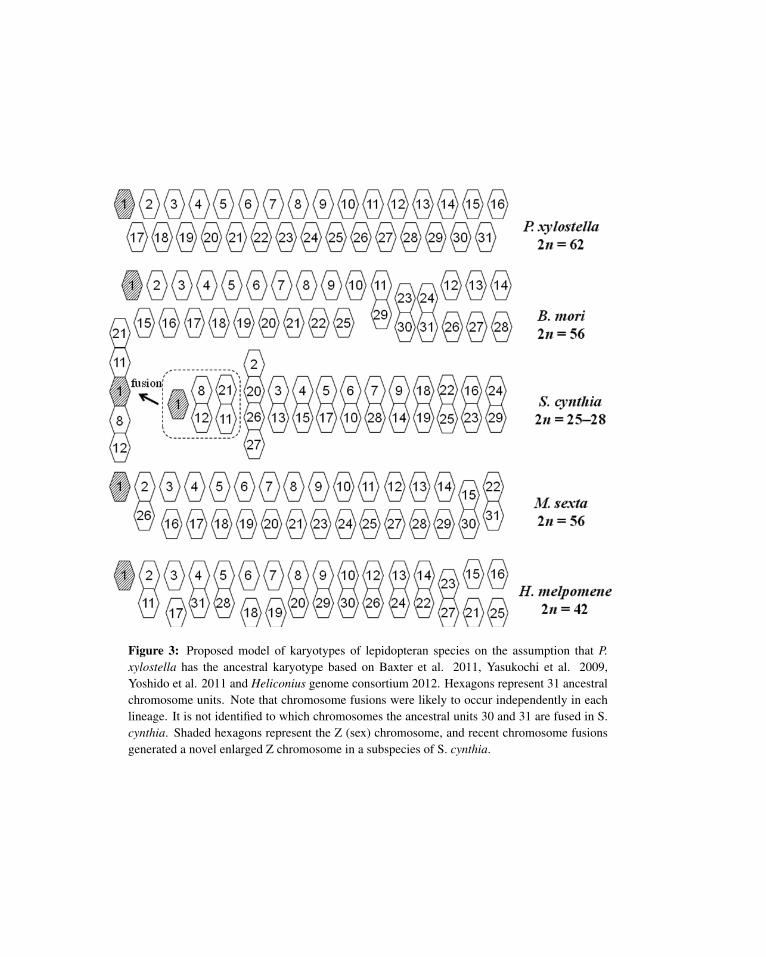
!Figure 3: Proposed model of karyotypes of lepidopteran species on the assumption that P.xylostella has the ancestral karyotype based on Baxter et al. 2011, Yasukochi et al. 2009,Yoshido et al. 2011 and Heliconius genome consortium 2012. Hexagons represent 31 ancestralchromosome units. Note that chromosome fusions were likely to occur independently in eachlineage. It is not identified to which chromosomes the ancestral units 30 and 31 are fused in S.cynthia. Shaded hexagons represent the Z (sex) chromosome, and recent chromosome fusionsgenerated a novel enlarged Z chromosome in a subspecies of S. cynthia.

advantageous for this purpose (Zhan et al., 2011; Heliconius Genome Consortium, 2012). Compared withrecent great progress in genome sequencing, genotyping is still a laborious and time-consuming effort. NGSis also applicable to genotyping (for a recent review, see Davey et al., 2011), and RAD sequencing reducesthe labor and cost per locus in large-scale analyses as reported in P. xylostella and H. melpomene (Baxter etal., 2011; Heliconius Genome Consortium, 2012). However, the cost required for these analyses is not solow and may be somewhat excessive for many purposes including the need for sufficient informatic support.In addition, it is difficult to focus on a specific chromosomal region using high throughput NGS methods.FISH analysis using large-insert libraries, though technically exacting and requiring access to sufficientmeiotic or mitotic tissue, is particularly suitable for comparative genomics covering a wide variety of lesscharacterized genomes.
2.3 Map-based Cloning
During the first one hundred years of silkworm genetics before the release of the genome sequence, onlyeight genes were identified to be associated with mutant phenotypes (Yasukochi et al., 2008). Now, map-based cloning of B. mori genes responsible for a mutant phenotype is so frequently reported that it is difficultto sum up exact number of identified genes. BAC and fosmid libraries play a critical role in these studies.However, most mutations so far examined are recessive and caused by loss of function.
Typically, map-based cloning in B. mori is first performed by detailed linkage analysis to narrow theresponsible region, followed by filling gaps in the reference genome sequence of the region using BAC andfosmid clones. Then, candidate genes are picked up from the sequences and expression and genomic struc-ture of the genes are analyzed in mutants and compared with wild-type. It is difficult to analyze a gain offunction mutation because large-insert libraries for genome sequencing are usually constructed from wild-type silkworms and additional work must be done to determine the genome structure of mutants. Therefore,construction of large-insert libraries from mutants is a crucial factor to characterize dominant mutations.Utilization of mutants harboring multiple dominant phenotypes for library construction promises to reducethe cost and labor per locus.
Map-based cloning in other lepidopteran species without a reference genome is still difficult (Baxteret al., 2010; Gahan et al., 2010); however, an increase in the number of sequenced genomes is anticipatedin the near future and will facilitate such projects.
2.4 Analysis of Clustered Genes
Analysis of clustered genes is an important application for large-insert libraries. Determination of precisesequence is important for understanding duplication and inactivation processes, as well as transcriptionalcontrol. We reported construction of BAC contigs covering the Hox genes of B. mori (Yasukochi et al.,2004). The Hox genes determine developmental fate along the anterior-posterior (A-P) axis of the embryoand are located along the chromosome in the same order as their functional domains along the A-P axis,which is well conserved among a wide range of animals. We first showed that the Hox gene cluster of B.mori is much longer than in other insects and the labial gene is located separately on the same chromosomeas that of other insects (Yasukochi et al., 2004). This structure was recently shown to be conserved in H.melpomene (Heliconius Genome Consortium, 2012), suggesting translocation of the labial gene occurredearly during the diversification of Lepidoptera.
Evolution of genes responsible for sex pheromone communication in moths is an attractive model for

investigating the relationship between the divergence of genes and mechanisms of speciation. A promisingstrategy is to compare the sequences of odorant receptor (OR) genes between closely related species that usedifferent female sex pheromone compounds. We isolated O. nubilalis BAC clones containing OR genes forFISH analysis and sequence determination, and found at least seven OR genes were in tandem arrays on theZ chromosome (Yasukochi et al., 2011b). In addition, a 181-bp direct repeat sequence encompassing exon 7and intron 7 was conserved among four of them, suggesting the possibility that gene duplication was causedby unequal crossovers via the repeat. The chromosomal region where the cluster was located, determinedby FISH analysis, was orthologous to BmOr1, a sex pheromone receptor gene of B. mori (Yasukochi et al.,2011b).
We have constructed fosmid libraries from O. furnacalis and O. latipennis, and sequence determina-tion of the genes is now underway (Yasukochi et al., unpublished results). In this case, we selected fosmidsinstead of BACs due to the reduced cost of library construction and ease of sequence determination. Thisstrategy is effective to concentrate on slight differences between mutant and wild-type or among closelyrelated species/subspecies when a BAC library is already constructed for a wild-type or model species.
2.5 PCR-based Screening
Genomic libraries without an efficient screening system are like a book lacking an index. End sequencingis the most stable but expensive way for anchoring clones to the reference genome sequence. However,a decreased tendency to perform end-sequencing for genome assembly makes library screening more im-portant. I previously described a method for PCR-based screening of BAC libraries (Yasukochi, 2002). Insitu hybridization using high-density replica (HDR) filters is another method to screen libraries. However,preparation of HDR filters is substantially impossible for ordinary laboratories without special equipmentand unsuitable for custom libraries. PCR-based screening can be carried out using standard thermal cyclerswithout any special skills, and stepwise changes in the scale of screening using a pooling strategy reducestime and labor. In addition, PCR-based screening can be easily performed for gene sequences downloadedfrom public databases, whereas DNA probes for in situ hybridization either have to be obtained from theoriginal investigators or prepared independently (Yasukochi et al., 2011a). Therefore, I strongly recom-mend PCR-based screening especially for fosmid libraries.
3 Conclusion
In spite of the current widespread use of NGS, large-insert libraries have irreplaceable roles in genomeanalysis as described above. It seems undeniable that utilization of large-insert libraries needs considerablecost, experience and patience to establish experimental systems, especially for uncharacterized organisms.However, it enables a tried-and-true approach for analyzing complex phenotypes without definitive cluesabout the underlying genes or mechanism.
Acknowledgement
I am grateful to M. R. Goldsmith for ciritical reading of the manuscript.

References
Adams, M. D., Celniker, S. E., Holt, R. A., Evans, C. A., Gocayne, J. D., et al. (2000). The genome sequence of Drosophilamelanogaster. Science, 287, 2185–2195.
Banno, Y., Shimada, T., Kajiura, Z., & Sezutsu, H. (2010). The silkworm-an attractive BioResource supplied by Japan. ExpAnim., 59, 139–146.
Baxter, S. W., Chen, M., Dawson, A., Zhao, J.-Z., Vogel, H., Shelton, A. M., Heckel, D. G., & Jiggins, C. D. (2010) Mis-spliced transcripts of nicotinic acetylcholine receptor a6 are associated with field evolved Spinosad resistance in Plutellaxylostella (L.). PLoS Genet, 6, e1000802.
Baxter, S. W., Davey,J. W., Johnston, J. S., Shelton, A. M., Heckel, D. G., Jiggins, C. D.,& Blaxter, M. L. (2011). Linkagemapping and comparative genomics using next-generation RAD sequencing of a non-model organism. PLoS ONE 6,e19315.
Brownstein, B. H., Silverman, G. A., Little, R. D., Burke, D. T., Korsmeyer, S. J., Schlessinger, D. & Olson, M. V. (1989).Isolation of single-copy human genes from a library of yeast artificial chromosome clones. Science, 244, 1348–1351.
Burke, D. T., Carle, G. F., & Olson, M. V. (1987). Cloning of large-segments of exogenous DNA into yeast by means ofartificial chromosome vectors. Science, 236, 806–812.
Cai, H., Kiefel, P., Yee, J., & Duncan, I. (1994). A yeast artificial chromosome clone map of the Drosophila genome. Genetics,136, 1385–1401.
Collins, J. & Hohn, B. (1978). Cosmids: A type of plasmid gene-cloning vector that is packageable in vitro in bacteriophageλ heads Proc. Nadl. Acad. Sci. USA, 75, 4242–4246.
d’Alencon, E., Sezutsu, H., Legeai, F., Permal, E., Bernard-Samain, S., Gimenez, S., Gagneur, Z., et al., (2010). Extensivesynteny conservation of holocentric chromosomes in Lepidoptera despite high rates of local genome rearrangements.Proc. Nadl. Acad. Sci. U.S.A., 107, 7680–7685.
Davey, J. W., Hohenlohe, P. A., Etter, P. D., Boone, J. Q., Catchen, J. M. & Blaxter, M. L. (2011). Genome-wide geneticmarker discovery and genotyping using next-generation sequencing. Nature Rev. Genet., 12, 499–510.
Gahan, L. J., Pauchet, Y., Vogel, H., & Heckel, D. G. (2010). An ABC transporter mutation is correlated with insect resistanceto Bacillus thuringiensis Cry1Ac toxin. PLoS Genet., 6, e1001248.
Green, E. D. & Olson, M. V. (1990). Systematic screening of yeast artificial-chromosome libraries by use of the polymerasechain reaction. Proc. Nadl. Acad. Sci. USA, 87, 1213–1217.
Heliconius Genome Consortium (2012). Butterfly genome reveals promiscuous exchange of mimicry adaptations amongspecies. Nature, 487, 94–98.
International Silkworm Genome Consortium. (2008). The genome of a lepidopteran model insect, the silkworm Bombyxmori. Insect Biochem. Mol. Biol. 38, 1036–1045.
Ioannou, P. A., Amemiya, C. T., Garnes, J., Kroisel, P. M., Shizuya, H., Chen, C, Batzer, M. A. & de Jong, P. J. (1994). Anew bacteriophage P1-derived vector for the propagation of large human DNA fragments. Nature genetics, 6, 84–89.
Kamimura, M., Tateishi, K., Tanaka-Okuyama, M., Okabe, T., Shibata, F., Sahara, K. & Yasukochi, Y. (2012). EST sequenc-ing and fosmid library construction in a non-model moth, Mamestra brassicae, for comparative mapping. Genome, 55, 775–781.
Kim, U-J., Shizuya, H., de-Jong, P. J., Birren, B., & Simon, M. I. (1992). Stable propagation of cosmid sized human DNAinserts in an F factor based vector. Nucl. Acids Res., 20, 1083–1085.

Papa, R., Morrison, C. M., Walters, J. R., Counterman, B. A., Chen. R., et al. (2008). Highly conserved gene order andnumerous novel repetitive elements in genomic regions linked to wing pattern variation in Heliconius butterflies. BMCGenomics, 9, 345.
Robinson, G. E., Hackett, K. J., Purcell-Miramontes, M., Brown, S. J., Evans, J. D., Goldsmith, M. R., Lawson, D., Okamuro,J., Robertson, H.M. & Schneider, D. J. (2011). Creating a buzz about insect genomes. Science, 331, 1386.
Robinson, R. (1971). “Lepidoptera Genetics,” Pergamon Press, Oxford
Rondon, M.R., August, P.R., Bettermann, A.D., Brady, S.F., Grossman, T.H., Liles, M.R., Loiacono, K.A., Lynch, B.A.,MacNeil, I.A., Minor, C., Tiong, C.L., Gilman, M., Osburne, M.S., Clardy, J., Handelsman, J., & Goodman, R.M.,(2000) Cloning the soil metagenome: a strategy for accessing the genetic and functional diversity of uncultured mi-croorganisms. Appl. Environ. Microbiol., 66, 2541–2547.
Sehgal, S. K., Li, W., Rabinowicz, P. D., Chan, A.,Simkova H., Dolezel, J., & Gill, B. S. (2012). Chromosome arm-specificBAC end sequences permit comparative analysis of homoeologous chromosomes and genomes of polyploid wheat.BMC Plant Biol., 12, 64.
Shimomura, M., Minami, H., Suetsugu, Y., Ohyanagi, H., Satoh, C., Antonio, B., Nagamura, Y., Kadono-Okuda, K., Ka-jiwara, H., Sezutsu, H., Nagaraju, J., Goldsmith, M.R., Xia, Q., Yamamoto, K. & Mita, K. (2009). KAIKObase: anintegrated silkworm genome database and data mining tool. BMC Genomics, 10, 486.
Shizuya, H., Birren, B., Kim, U. J., Mancino, V., Slepak, T., Tachiiri, Y., & Simon M (1992). Cloning and stable maintenanceof 300-kilobase-pair fragments of human DNA in Escherichia coli using an F-factor-based vector. Proc. Natl. Acad.Sci. USA 89, 8794–8797.
Yasukochi, Y. (2002). PCR-based screening for bacterial artificial chromosome libraries. Methods in Mol Biol 192, 401–420.
Yasukochi, Y., Ashakumary, L., Wu, C., Yoshido, A., Nohata, J., Mita, K., & Sahara, K. (2004). Organization of the Hoxgene cluster of the silkworm, Bombyx mori: A split of the Hox cluster in a non-Drosophila insect. Dev. Genes Evol,214, 606–614.
Yasukochi, Y., Fujii, H., & Goldsmith, M. R. (2008) Lepidoptera: Silkworm, Bombyx mori In Genome Mapping in AnimalsVolume: Insects, edited by Chitta Kole and Wayne Hunter, 43–57.
Yasukochi, Y., Tanaka-Okuyama, M., Shibata, F., Yoshido, A., Marec, F., Wu, C., Zhang, H., Goldsmith, M.R., & Sahara, K.(2009). Extensive conserved synteny of genes between the karyotypes of Manduca sexta and Bombyx mori revealed byBAC-FISH mapping. PLoS ONE 4, e7465.
Yasukochi, Y., Tanaka-Okuyama, M., Kamimura, M., Nakano, R., Naito, Y., Ishikawa, Y., & Sahara, K. (2011a). Isola-tion of BAC clones containing conserved genes from libraries of three distantly related moths: a useful resource forcomparative genomics of Lepidoptera. J Biomed. Biotechnol. 2011, 165894.
Yasukochi, Y., Miura, N., Nakano, R., Sahara, K., &. Ishikawa, Y. (2011b). Sex-linked pheromone receptor genes of theEuropean corn borer, Ostrinia nubilalis, are in tandem arrays. PLoS ONE 6, e18843.
Yoshido, A., Yasukochi, Y., & Sahara, K. (2005a). The Bombyx mori karyotype and the assignment of linkage groups.Genetics 170, 675–685.
Yoshido, A., Marec, F., Sahara, K., (2005b). Resolution of sex chromosome constitution by genomic in situ hybridizationand fluorescence in situ hybridization with (TTAGG) n telomeric probe in some species of Lepidoptera. Chromosoma114,193–202.
Yoshido, A., Yasukochi, Y., & Sahara, K. (2011). Samia cynthia versus Bombyx mori: comparative gene mapping between aspecies with a low-number karyotype and the model species of Lepidoptera. Insect Biochem. Mol. Biol. 41, 370–377.

Wu, C., Asakawa, S., Shimizu, N., Kawasaki, S., & Yasukochi, Y. (1999). Construction and characterization of bacterialartificial chromosome libraries from the silkworm, Bombyx mori. Mol. Gen. Genet. 261, 698–706.
Zhan, S., Merlin, C., Boore, J.L., & Reppert, S.M. (2011). The monarch butterfly genome yields insights into long-distancemigration. Cell 147, 1171–1185.