vaccines and immunotherapies for the prevention of ... and immunotherapies for the... · continuing...
TRANSCRIPT

CONTINUING MEDICAL EDUCATION
Vaccines and immunotherapies for the prevention ofinfectious diseases having cutaneous manifestations
Jashin J. Wu, MD,a David B. Huang, MD, MPH,b Katie R. Pang, MD,a andStephen K. Tyring, MD, PhD, MBAc
Houston, Texas
Although the development of antimicrobial drugs has advanced rapidly in the past several years, suchagents act against only certain groups of microbes and are associated with increasing rates of resistance.These limitations of treatment force physicians to continue to rely on prevention, which is more effectiveand cost-effective than therapy. From the use of the smallpox vaccine by Jenner in the 1700s to the currentconcerns about biologic warfare, the technology for vaccine development has seen numerous advances.The currently available vaccines for viral illnesses include Dryvax for smallpox; the combination measles,mumps, and rubella vaccine; inactivated vaccine for hepatitis A; plasma-derived vaccine for hepatitis B; andthe live attenuated Oka strain vaccine for varicella zoster. Vaccines available against bacterial illnessesinclude those for anthrax, Haemophilus influenzae, and Neisseria meningitidis. Currently in developmentfor both prophylactic and therapeutic purposes are vaccines for HIV, herpes simplex virus, and humanpapillomavirus. Other vaccines being investigated for prevention are those for cytomegalovirus, respiratorysyncytial virus, parainfluenza virus, hepatitis C, and dengue fever, among many others. Fungal andprotozoan diseases are also subjects of vaccine research. Among immunoglobulins approved for prophy-lactic and therapeutic use are those against cytomegalovirus, hepatitis A and B, measles, rabies, and tetanus.With this progress, it is hoped that effective vaccines soon will be developed for many more infectiousdiseases with cutaneous manifestations. (J Am Acad Dermatol 2004;50:495-528.)
Learning objective: At the completion of this learning activity, participants should be familiar with thecurrent and experimental vaccines and immunotherapies for infectious diseases with cutaneous manifes-
tations.From the Center for Clinical Studiesa; Department of InternalMedicine, Division of Infectious Diseases, Baylor College ofMedicineb; and Departments of Dermatology, Microbiology-Immunology, and Internal Medicine, University of TexasMedical Branch.c
Funding sources: None.Disclosure: Dr Tyring has conducted research on vaccines for vesic-
ular stomatitis virus, herpes simplex virus, human papillomavi-ruses, and HIV with grants from Merck, Chiron, GlaxoSmithKline,MedImmune, and VaxGen to the University of Texas MedicalBranch and the Center for Clinical Studies.
Presented in part at the 61st Annual Meeting of the American Acad-emy of Dermatology, San Francisco, California, March 21, 2003.Also presented in part at the 62nd Annual Meeting of the Amer-ican Academy of Dermatology, Washington, DC, February 6-11,2004.
Reprint requests: Stephen K. Tyring, MD, PhD, MBA, Professor ofDermatology, University of Texas Health Science Center,Houston, 2060 Space Park Dr, Ste 200, Houston, TX 77058. E-mail:[email protected].
0190-9622/$30.00© 2004 by the American Academy of Dermatology, Inc.doi:10.1016/j.jaad.2003.12.003
T he medical approach to infectious diseases isfraught with limitations. Antimicrobial drugsare available for only a few groups of mi-
crobes. These drugs provide no cure for infectionssuch as HIV or herpes simplex virus (HSV) butmerely alter the clinical course of disease while themedication is being taken. Because of the void ofadequate antimicrobial treatment, the prevention ofthese infections is all the more crucial.
Vaccination, with public health measures, is thebest option for controlling the spread of infectiousdiseases. Vaccination is ranked as the greatestpublic health achievement in the last century, con-tributing the most to decreased global morbidityand mortality.1,2 Since 1988 more than 2 billionchildren have been immunized, saving more than3 million pediatric lives per year.3 The significantimpact of immunization is well illustrated by thecomplete eradication of smallpox in 1977. Globaleradication of poliomyelitis is currently underway, and although the goal of total elimination ofthis disease by the end of 2000 unfortunately was
495

496 Wu et al J AM ACAD DERMATOL
APRIL 2004
not met,4 the eradication is estimated to be com-plete by 2005.5 A number of other vaccines haveled to notable decreases in infections and compli-cations. For instance, after years of widespreadmeasles vaccination, a record low of 37 measlescases was reported in the United States in 2002.6
Vaccines for Haemophilus influenzae and Neisse-ria meningitidis have greatly decreased the inci-dence of these diseases. These currently availablevaccines also provide a basic framework ofknowledge and experience with which other vac-cines can be developed.
Unfortunately, the progress in finding new vac-cines cannot be rapid enough. The incidence ofmany viral infections, such as HIV, HSV, and hu-man papillomaviruses (HPVs), is increasing de-spite the currently available antiviral agents. Thedevelopment of prospective vaccines is generallymeasured in decades (Table I), marked by smallincrements of advancement before the final prod-uct becomes available.7 Numerous different vac-cine candidates often are developed and evalu-ated over many years before a single effectivevaccine is licensed.
Abbreviations used:
AVA: anthrax vaccine adsorbedCDC: Centers for Disease Control and
PreventionCRM197: cross-reacting, mutant, nontoxic
diphtheria toxinDTaP: diphtheria and tetanus toxoids and
acellular pertussis vaccineDTP: diphtheria and tetanus toxoids and
whole-cell pertussis vaccineHbOC: Haemophilus influenzae type b
(polyribosyl ribitol phosphate)oligosaccharides conjugated to CRM197
HBsAg: hepatitis B surface antigenHib: Haemophilus influenzae type bHPV: human papillomavirusHSV: herpes simplex virusIVIG: intravenous immunoglobulinLACK: Leishmania homologue of receptors for
activated C kinaseMMR: measles-mumps-rubellaOspA: 31-kd outer-surface proteinOspC: 23-kd outer-surface proteinPA: protective antigenPGA: poly-�-linked D-glutamic acidPRP: polyribosyl ribitol phosphatePRP-D: polyribosyl ribitol phosphate conjugated
to diphtheria toxoidPRP-OMP: polyribosyl ribitol phosphate conjugated
to an outer-membrane protein complexrOspA: recombinant 31-kd outer-surface proteinSSPE: subacute sclerosing panencephalitisVIG: vaccinia immunoglobulinVLP: virus-like particles
CURRENTLY LICENSED VACCINESViruses
Smallpox. In 1796 Edward Jenner first demon-strated that inoculation of cowpox virus into humanskin could lead to protection from subsequent small-pox infection.8 He named the inoculation substancevaccine, based on the Latin word vacca, meaningcow. The most recent vaccines used for smallpoxvaccination were derived from a species similar tocowpox, called vaccinia virus. Vaccinia virus is amember of the orthopox virus family, which in-cludes variola virus, the virus that causes smallpox,and other viruses such as cowpox and monkeypox.Several strains of the live attenuated virus vaccinewere employed in the eradication of disease. Thesmallpox vaccine has been the prototype of successof a viral vaccine. Prior to immunization, smallpoxinfection relentlessly killed hundreds of millions ofpersons. The complete eradication of this diseasecould easily be considered the greatest success storyin medical history.
Vaccine production ended 2 decades ago afterthe eradication of smallpox,9 and approximately 60million vaccine doses remain worldwide.10 Becauseof recent concerns over potential biologic warfare inthe future, the threat of smallpox may not have been
Table I. Timeline of vaccine development*
1796—Smallpox1800 1881—Anthrax
1885—Rabies1900 1945—Influenza
1953—Yellow fever1963—IPV, OPV (Poliomyelitis)1963—Measles1967—Mumps1969—Rubella1975—Neisseria meningitidis1980—Adenovirus1980—Rabies (IMOVAX: first FDA-approved
rabies vaccine)1981—Hepatitis B1987—Haemophilus influenzae1992—Japanese encephalitis1992—DTaP (diphtheria-tetanus-acellular
pertussis)1995—Hepatitis A, Varicella1998—Lyme disease†
1998—Rotavirus†
2000 2000—Streptococcus pneumoniae2000—Dryvax (smallpox)2002—Pediarix (DTaP-HepB-IPV)
*Dates for smallpox, anthrax, and rabies vaccines are of the first pub-lished results of vaccine usage. Remaining dates are those of ap-proval of vaccines by the Food and Drug Administration (FDA).†No longer available.

Wu et al 497J AM ACAD DERMATOL
VOLUME 50, NUMBER 4
completely eliminated with eradication of the dis-ease. Therefore, renewed interest in the productionof smallpox vaccines has developed.
The destruction of the 2 remaining smallpox virusreserves, in Atlanta and Moscow, has been a sourceof ongoing debate. Opponents of destruction con-tend that the virus stocks would be helpful for futureresearch, such as smallpox pathogenesis and theproduction of new antiviral agents.11,12 Proponentsargue that the virus genome has already been clonedand sequenced and is unnecessary for such re-search.13 The World Health Assembly in May 1999resolved to postpone the previously arranged small-pox destruction scheduled for June 30 of that year.14
All US smallpox vaccine formulations contain theNew York City Board of Health vaccinia strain,which is less reactogenic than other strains.15 TheU.S. National Pharmaceutical Stockpile contains 4formulations of smallpox vaccine: 2 previously man-ufactured calf-lymph-derived vaccines, Dryvax(Wyeth Laboratories, Marietta, Pa) and Aventis Pas-teur vaccine (Swiftwater, Pa); and two newly devel-oped vaccines from Acambis/Baxter Pharmaceuti-cals (Cambridge, Mass), ACAM1000 and ACAM2000(Centers for Disease Control [CDC] Drug Services,unpublished data, 2002). ACAM1000 is grown inhuman embryonic lung cell culture (MRC-5), andACAM2000 is grown in African green monkey kid-ney cells (Vero cells).
Dryvax is licensed and is currently being used forimmunization against smallpox in public health andhealth-care response teams and laboratory workerswho are involved with research activities that in-volve vaccinia virus. This vaccine will be used tofulfill the recommendations of the national AdvisoryCommittee on Immunization Practices. An emer-gency vaccination strategy has been developed inthe event of confirmed cases of smallpox. Prioritywill be given to those with early diagnosis, all whohad been in contact with the patient since onset offever, all household members of the contacts, andhealth-care workers, public health personnel, firstresponders, and other personnel who will be assist-ing with outbreak control measures and emergencyresponse activities.16 Dryvax is freeze-dried and re-constituted before use with a diluent that contains50% glycerin and 0.25% phenol. Contraindications touse of this vaccine in the absence of smallpox in-clude allergies to polymyxin B sulfate, streptomycinsulfate, chlortetracycline hydrochloride, and neomy-cin sulfate. In the case of contact with persons withsmallpox or in the presence of smallpox, the vaccineshould be administered along with antihistamines orglucocorticoids.
The CDC is holding the other 3 vaccines in re-serve.17 Studies are currently under way to deter-mine the reactogenicity of the 2 newer cell culturevaccines. The tissue culture cell vaccines are beingdeveloped in hopes of supplanting the calf-lymphvaccine if a more extensive vaccination program isneeded.18
All 4 vaccines are administered by means of in-oculation into the superficial layers of the skin,which allow the virus to grow and induce an immu-nologic response that protects the host againstsmallpox. The vaccines elicit antibody and cell-me-diated immunity. A successful vaccination, definedas an antibody titer of 1:10 or greater, occurs in morethan 95% of persons within 1-2 weeks of immuniza-tion.19 The protective duration of the vaccine occursfor 3-5 years and begins fading after 5 years, withminimal protection after 20 years. In persons whohave undergone revaccination with smallpox, resid-ual immunity may last 30 years or more. Individualsundergoing postexposure vaccination should re-ceive the smallpox vaccination within 3 days ofexposure to prevent the natural history of small-pox.20 Some epidemiologic evidence suggests thatvaccination up to 4-7 days after exposure also mayalter the severity of the disease and provide protec-tion from death.19
Adverse reactions of the smallpox vaccine aresimilar to those caused by other vaccines and in-clude local skin reactions, fever, erythema multi-forme, and anaphylaxis. There recently have been30 cases of cardiac-related events, such as myocar-ditis, pericarditis, myocardial infarction, and anginaduring the civilian smallpox vaccination program.21
Other adverse effects specific to smallpox vacci-nation include eczema vaccinatum, fetal vaccinia,generalized vaccinia, inadvertent inoculation, ocularvaccinia (Fig 1), progressive vaccinia, and postvac-cinial encephalopathy and encephalomyelitis.22
These specific complications of smallpox vaccina-
Fig 1. Ocular vaccinia secondary to digital spread fromthe inoculation site.

498 Wu et al J AM ACAD DERMATOL
APRIL 2004
tion can be treated with vaccinia immunoglobulin(VIG), cidofovir, and ophthalmic antiviral agents,although none of these therapies have undergoneclinical trials.22 VIG is considered first-line treatmentbecause of worldwide historical experience with itin the treatment of vaccinia-related adverse events.However, supplies of VIG are limited. Although ci-dofovir has never been used to treat vaccinia infec-tions in human beings, the first use of cidofovir in 3HIV-positive men with recurrent molluscum conta-giosum virus cleared the lesions.23 These were thefirst documented cases in which cidofovir was usedto treat poxvirus infections in human beings.
Smallpox vaccination is contraindicated for per-sons with any of the following conditions or whohave had close contact with any of the followingconditions: atopic dermatitis or active acute orchronic exfoliative skin conditions that disrupt theepidermis, Darier disease, pregnancy or a desire tobecome pregnant within the 28 days after vaccina-tion, and compromised immune systems.22 Othercontraindications that apply only to potential vac-cine candidates but do not include their close con-tacts are moderate to severe intercurrent illness,smallpox vaccine-component allergies, age �18years, breastfeeding, and the use of topical ocularsteroid medications.22
Measles, mumps, and rubella. Vaccination forthese 3 classic childhood diseases typically is admin-istered as a combination measles-mumps-rubella(MMR) vaccine (Tables II and III). Live virus vac-cines for all 3 viruses were introduced in the 1960s,and after widespread implementation in the UnitedStates, the number of cases of these infections re-ported annually has declined by more than 98%.24
This result is largely due to the recommendation thatall states require a 2-dose MMR vaccine in childrenbefore they enter school. The use of 2-dose MMRvaccine is to induce immunity in that small percent-age of persons with failure to develop an immuno-logic response to one or more components of thefirst dose. The most recent recommendations by theCDC are that vaccination with the first MMR dose beadministered at 12-15 months and the second doseat 4-6 years of age.25 Each MMR vaccine contains 0.3mg of human albumin, 25 �g of neomycin, 14.5 mgof sorbitol, and 14.5 mg of hydrolyzed gelatin(Merck, manufacturer’s package insert). Becausethese vaccines consist of live attenuated viruses,they should not be administered to pregnant womenor those planning to become pregnant within thefollowing 3 months. The theoretical risk of congen-ital rubella syndrome after immunization has beenthe primary concern. However, a study of 321women who had received the rubella vaccine 3
months before or after conception revealed no con-genital malformations compatible with congenitalrubella infection.26 Immunization is also contraindi-cated in immunosuppressed patients, although itcan be administered to individuals with asymptom-atic HIV infection as well as to persons with mildimmunosuppression. In healthy individuals, minorillnesses with or without fever are not a contraindi-cation to vaccination. Patients with a history of ana-phylactic hypersensitivity to neomycin should notreceive the MMR vaccine. The vaccine can be ad-ministered to patients with an allergy to eggs, sincethe risk of severe anaphylactic reactions is exceed-ingly low.25 It is recommended that these patients beobserved for 90 minutes after immunization.27
Measles. Measles virus has been noted to bethe most infectious disease of humankind in terms ofthe minimal number of virions necessary to produceinfection.28 An estimated 75% of susceptible familycontacts who are exposed to a case of measlesdevelop the disease.29 Because human beings arethe only reservoir for measles virus, global eradica-tion is technically feasible. The World Health Orga-nization, the CDC, and the Pan American HealthOrganization convened in 1996 and adopted thegoal of global eradication by 2005-2010.30 Owing touniversal childhood immunization in the UnitedStates, measles is no longer considered an indige-nous disease in this country.31 The reported inci-dence of measles has decreased by �99% since themeasles vaccine became available.
In 2000 there were 86 reported measles cases, ofwhich 26 were internationally imported. Of the re-maining 60 indigenous cases, 9 were definitely dueto imported virus, 18 were linked to imported virus,and 33 were of unknown source.32 In 2001 therewere 116 reported cases of measles, 33 but in 2002only 37 measles cases were reported in the UnitedStates.33 The increase of 35% from 2000 to 2001should remind us that we need to guard againstcomplacency. Lack of compliance with routine MMRvaccination in the past led to a resurgence of mea-sles infection in the United States between 1989 and1991, with some deaths reported.30 Moreover, morethan 1 million children die of measles each year inthird-world countries.34
In 1963 the initial measles vaccine, which was alive attenuated vaccine, was licensed. In 1968 thecurrent measles vaccine was licensed. It uses theEnders-Edmonston virus strain, a further attenuatedversion of the live preparations previously available,resulting in fewer adverse reactions in recipients. Itis produced by culturing the Moraten virus strain inchick embryo cells. Measles vaccination produces amild or unapparent infection that is noncommuni-

Wu et al 499J AM ACAD DERMATOL
VOLUME 50, NUMBER 4
cable. Both humoral and cellular immune responsesdevelop as a result.35 After receiving 2 doses ofvaccine, 95% to 99% of recipients develop serologicevidence of immunity to measles.36,37 Immunity isthought to be lifelong, similar to that acquired afterinfection with the wild-type virus.38 However rare,measles infection has been reported in patientswith previously documented postimmunizationseroconversion.39,40
Adverse effects after measles vaccination typi-cally are mild. Five percent to 15% of recipientsdevelop a fever of at least 103°F for 1 to 2 days,generally 5 to 12 days after immunization.41 Thesepersons are largely asymptomatic, but some may
Table II. Vaccines: recommendations for immunizatio
Vaccine Target population
Adenovirus Military personnelAnthrax Military personnel, and
those involved inproducing quantities orconcentrations of Bacillusanthracis cultures, orthose with high potentialfor aerosol exposure
DT (diphtheria-tetanus) All adults
Hepatitis A* Persons with behavioral,medical, or occupationalindications
Hepatitis B* Persons with behavioral,medical, or occupationalindications
Influenza Persons over 50 y; thosewith medical oroccupational indications,and home contacts ofpersons with indications
HDCV, Human diploid cell vaccine; ID, intradermal; IM, intramuscula*A combination vaccine (Twinrix), now available, contains Havrixvaccinations from 5 to 3.
develop a transient viral exanthem.24 An associ-ated encephalitis or encephalopathy rarely hasbeen reported after immunization and occurs infewer than 1 in 1 million vaccinees.42 There havebeen early concerns about the association of mea-sles vaccination and subacute sclerosing panen-cephalitis (SSPE), since this complication may oc-cur with natural infection. A small number ofreports have described the occurrence of SSPE inpersons with a history of vaccination but noknown history of infection.43-45 More recent evi-dence indicates that at least some of those caseshad unrecognized natural measles infection priorto vaccination, and the SSPE was directly related
adults
Dosage Comments
1 dose3 doses: 0, 2, and 4 wk;
boosters at 6, 12, and18 mo
Current recommendationis for annual boosters.
1 dose booster every10 y; for adults, theprimary series is 3doses; the first 2 dosesgiven at least 4 wkapart and the thirddose, 6-12 mo after thesecond
Give 1 dose if patient hadreceived the primaryseries and the lastvaccination was �10 yago.
2 doses: 0 and 6-12 mo Other indications aretravel to or work incountries that havehigh incidence ofhepatitis A.
3 doses: 0, 1-2, and 4-6mo
Other target populationsare inmates ofcorrectional facilities,home contacts and sexpartners of persons withchronic hepatitis B viral(HBV) infection, andtravelers to countrieswith high prevalence ofchronic HBV infection.
1 dose annually October or November isthe optimal time, butDecember to March isacceptable.
Cont’d on page 500
purified chick embryo cell culture vaccine; SC, subcutaneous.itis A) and Engerix-B (Hepatitis B). It thus reduces the number of
ns in
Route
OralSC
IM
IM
IM
IM
r; PCEC,(Hepat
500 Wu et al J AM ACAD DERMATOL
APRIL 2004
to the infection.25 Widespread measles immuniza-tion has nearly eliminated SSPE in the UnitedStates, and the live measles vaccine does not in-crease the risk for this complication.25
Mumps. Since the introduction of live mumpsvaccine in 1967 and its recommended use in 1977,the incidence of mumps has decreased steadily bymore than 99%. The live attenuated mumps vaccine(Jeryl-Lynn strain) is prepared in chick embryo cellculture. Immunization produces a mild subclinicalinfection that is noncommunicable. Early clinicalstudies have shown that 97% of children and 93% ofadults develop serologic evidence of immunity aftervaccination.46-48 Outbreak-based studies, however,
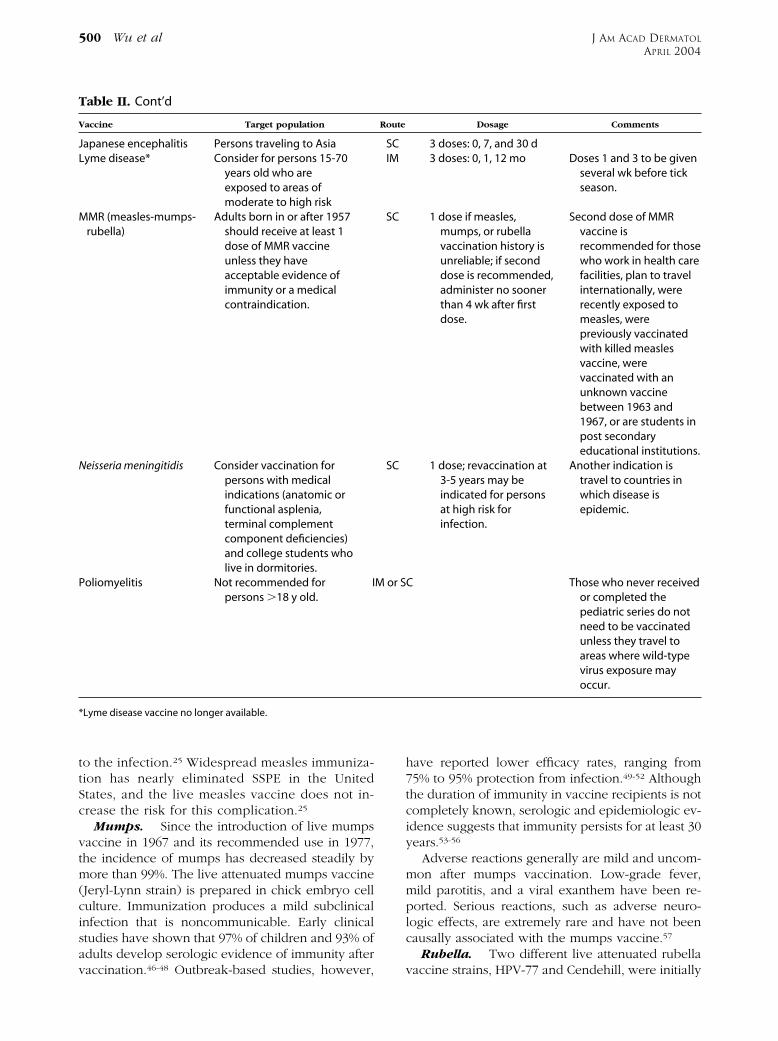
Table II. Cont’d
Vaccine Target population
Japanese encephalitis Persons traveling to AsiaLyme disease* Consider for persons 15-70
years old who areexposed to areas ofmoderate to high risk
MMR (measles-mumps-rubella)
Adults born in or after 1957should receive at least 1dose of MMR vaccineunless they haveacceptable evidence ofimmunity or a medicalcontraindication.
Neisseria meningitidis Consider vaccination forpersons with medicalindications (anatomic orfunctional asplenia,terminal complementcomponent deficiencies)and college students wholive in dormitories.
Poliomyelitis Not recommended forpersons �18 y old.
I
*Lyme disease vaccine no longer available.
have reported lower efficacy rates, ranging from75% to 95% protection from infection.49-52 Althoughthe duration of immunity in vaccine recipients is notcompletely known, serologic and epidemiologic ev-idence suggests that immunity persists for at least 30years.53-56
Adverse reactions generally are mild and uncom-mon after mumps vaccination. Low-grade fever,mild parotitis, and a viral exanthem have been re-ported. Serious reactions, such as adverse neuro-logic effects, are extremely rare and have not beencausally associated with the mumps vaccine.57
Rubella. Two different live attenuated rubellavaccine strains, HPV-77 and Cendehill, were initially
Dosage Comments
3 doses: 0, 7, and 30 d3 doses: 0, 1, 12 mo Doses 1 and 3 to be given
several wk before tickseason.
1 dose if measles,mumps, or rubellavaccination history isunreliable; if seconddose is recommended,administer no soonerthan 4 wk after firstdose.
Second dose of MMRvaccine isrecommended for thosewho work in health carefacilities, plan to travelinternationally, wererecently exposed tomeasles, werepreviously vaccinatedwith killed measlesvaccine, werevaccinated with anunknown vaccinebetween 1963 and1967, or are students inpost secondaryeducational institutions.
1 dose; revaccination at3-5 years may beindicated for personsat high risk forinfection.
Another indication istravel to countries inwhich disease isepidemic.
C Those who never receivedor completed thepediatric series do notneed to be vaccinatedunless they travel toareas where wild-typevirus exposure mayoccur.
Route
SCIM
SC
SC
M or S

Wu et al 501J AM ACAD DERMATOL
VOLUME 50, NUMBER 4
developed and licensed in the United States in 1969.They were replaced in 1979 by the RA 27/3 (rubellaabortus 27, explant 3) vaccine, which is grown inhuman diploid fibroblast cell culture. This vaccineproduces nasal antibodies, as well as higher andmore persistent antibody titers, that better mimic theimmune protection developed after natural infec-tion.58,59 Vaccination induces an antibody responsein more than 97% of recipients.47,60 Immunity invaccine recipients is thought to be lifelong and hasbeen shown to persist for at least 16 years.61,62 Inaddition, this vaccine was associated with fewer
Table II. Cont’d
Vaccine Target population
Rabies Persons at risk of rabiesexposure or thoserecently exposed.
Smallpox Those with early diagnosis,all who had been incontact with the patientsince onset of fever, allhousehold members ofthe contacts, smallpoxpublic health and health-care response teams, andlaboratory workers whoare involved with vacciniavirus
Streptococcuspneumoniae
Persons � 65 years old andthose with chroniccardiovascular, hepatic,pulmonary, or renaldiseases, diabetesmellitus, anatomic orfunctional asplenia, orimmunosuppression
I
Varicella zoster Susceptible adults;recommended for thosewho do not have reliableclinical history of varicellainfection or serologicevidence of varicellazoster virus infection
Yellow fever Persons traveling toendemic countries (partsof Africa, South America,and southeast Asia)
adverse events compared with the previous rubellavaccines.
Adverse effects after rubella vaccination typicallyare mild. Five percent to 15% of vaccinated childrendevelop fever, lymphadenopathy, or a viral exan-them, usually 5 to 12 days after vaccination.57,63
Arthralgias and arthritis are frequent complicationsin adult vaccinees, particularly women, and maydevelop in 25% to 40% of this population,64-66 butoccurrences in children are rare (0.5%).64 These jointsymptoms typically begin within the first 3 weeksafter vaccination and remit within 11 days.26 The
Dosage Comments
Preexposure: 3 doses at0, 7, and 21 or 28 d
HDCD: ImovaxPCEC: PabAvertRabies Vaccine AbsorbedHDCD: Imovax Rabies ID
Postexposure: 5 dosesat 0, 3, 7, 14, and 28 d
HDCVRabies Vaccine AbsorbedPCEC
Previously vaccinatedpersons require only 2doses after rabiesexposure at 0 and 3 d.
1 dose; if there is noevidence of a localvaccine reaction, canrepeat vaccinationafter 7 d.
Postexposure vaccination;should receive thesmallpox vaccinationwithin 3 d.
C 1 dose Other populations areAlaskan natives, certainAmerican Indianpopulations, andresidents of nursinghomes and other long-term-care facilities.
2 doses: 0, and 4-8 wk
1 dose Booster is given every 10years for recertificationfor travel into endemiccountries.
Route
IMIMIMID
IMIMIMSC
M or S
SC
SC

502 Wu et al J AM ACAD DERMATOL
APRIL 2004
knees and the fingers are most frequently involved,but any joint may be affected.63
Hepatitis A virus. Both inactivated and attenu-ated forms of hepatitis A vaccines have been devel-oped and studied. However, the inactivated vaccineis the only type licensed and available in the UnitedStates (Havrix and Vaqta). These vaccines are prop-
Table III. Vaccines: recommendations for immunizat
Vaccine Target population
DTaP (diphtheria,tetanus, acellularpertussis)
Infants and toddlers
DT (diphtheria-tetanus)
Children aged 11-12 y
Hepatitis A* Children
Hepatitis B* Children
Hib (Haemophilusinfluenzae type b)
Infant and toddlers
Influenza Children older than 6 mo whohave chronic cardiovascular,pulmonary, or renal diseases,chronic metabolic diseases,hemoglobinopathies,immunosuppression, andthose receiving long-termaspirin therapy (risk ofReye syndrome)
MMR (measles,mumps, rubella)
Children
IM, Intramuscular; HBsAg, hepatitis B surface antigen; IPV, inactivate*A combination vaccine (Twinrix), now available, contains Havrixvaccinations from 5 to 3.
agated in human diploid fibroblast culture and inac-tivated by formalin. Immunization generally in-volves 2 doses given 6-12 months apart in adults andchildren 2 years and older. Studies of both availableinactivated vaccines show excellent as well as com-parable immunogenicity and efficacy rates. Overall,97% to 99% of recipients develop protective levels of
children
te Dosage Comments
4 doses: 2, 4, 6, and 15-18mo
The fourth dose of DTaPmay be given as earlyas age 12 mo if 6 mohave elapsed since thethird dose, and thechild is unlikely toreturn at age 15-18 mo.
Recommended if at least5 y have elapsed sincethe last dose ofdiphtheria and tetanustoxoid-containingvaccine.
Recommended routineDT boosters every 10 y
2 doses: Havrix, �2 y: 0and 6-12 mo
Vaqta, 2-17 y: 0 and 6-18mo; �17 y: 0 and 6 mo
Minimum intervalbetween doses is 6 mo.
3 doses: infants, birth to2 mo, 1-4 mo, and 6-18mo; children andadolescents; 0, 2, and4 months
Infants with HBsAg-positive mothersreceive first dose within12 h of birth, seconddose at 1 mo, and thirddose at 6 mo.
4 doses: 2, 4, 6, and12-15 mo
If PRP-OMP (Pedvax HIBor ComVax [Merck]) isgiven at ages 2 and 4mo, the 6-mo dose isnot required.
6 mo-8 y, 1 or 2 doses ofsplit virus only, at least1 mo apart; 9-12 y,1 dose of split virusonly; �12 yrs, 1 doseof whole or splitvirus
October and Novemberfor those moresusceptible to flucomplications, andNovember andDecember for all others
2 doses: 12-15 mo and4-6 y
Second dose may beadministered duringany visit, provided atleast 4 wk have elapsedsince the first dose;second dose should begiven by 11 to 12 y
vaccine; OPV, oral polio vaccine; SC, subcutaneous.itis A) and Engerix-B (Hepatitis B). It thus reduces the number of
ions in
Rou
IM
IM
IM
IM
IM
IM
SC
d polio(Hepat

Wu et al 503J AM ACAD DERMATOL
VOLUME 50, NUMBER 4
antibodies 1 month after the first dose, and 99% to100% of recipients are protected 1 month after thesecond dose.67-72 When studied in placebo-con-trolled clinical trials in Thailand (which has highrates of hepatitis A), 2 doses of the inactivated vac-cine were 94% effective in protecting against hepa-titis A infection.73 A similar study in New York chil-dren showed 100% efficacy after a single dose ofvaccine.74
Because of limited long-term data about this vac-cine, the duration of immunity has yet to be deter-mined. In one study of a 3-dose series in adults,detectable antibodies were documented in all sub-jects 4 years after immunization.75 According to ki-netic models of antibody concentration decline, pro-tective levels of hepatitis A antibodies can beexpected to persist for 20 years76 and perhaps up to30 years.77 A separate mathematical evaluation oflong-term immunity after a primary dose andbooster dose for hepatitis A has calculated that pro-tective antibody levels should persist for 24 to 47years.78 It is unknown whether vaccine-induced im-munity will persist beyond the loss of detectableantibody levels, as occurs with hepatitis B immuni-zation, but this phenomenon has been suggested tooccur.78
The hepatitis A vaccine is recommended for per-sons at least 2 years of age living in or traveling to
Table III. Cont’d
Vaccine Target population
Poliomyelitis Children
Rotavirus Infants
Streptococcuspneumoniae
All children aged 2-23 mo, andrecommended for certainchildren aged 24-59 mo
Varicella zoster Children
IM, Intramuscular; HBsAg, hepatitis B surface antigen; IPV, inactivate*A combination vaccine (Twinrix), now available, contains Havrixvaccinations from 5 to 3.
areas of high endemicity for hepatitis A. It is alsorecommended for persons with chronic liver dis-eases due to causes other than hepatitis A, personsengaging in high-risk sexual activity, residents of acommunity experiencing an outbreak of hepatitis A,and users of illicit injectable drugs. In addition, thehepatitis A vaccine currently is recommended forroutine pediatric use in some states and regions.
Adverse effects with hepatitis A vaccination gen-erally are mild, and no serious side effects have beenattributed to the vaccine in clinical trials.73 Otherthan soreness at the injection site, the most commonadverse effects include headache (14%) and malaise(7%) in adults and feeding problems (8%) and head-ache (4%) in children.
Hepatitis B virus. Prior to the development ofthe hepatitis B vaccine, an estimated 200,000-300,000 persons worldwide were being infectedwith the hepatitis B virus (HBV) annually.79 Immu-nization for hepatitis B became a reality in 1981when the plasma-derived vaccine was licensed inthe United States. This vaccine was highly effectivein inducing immunity but was associated with sev-eral drawbacks. The supply of suitable carrierplasma necessary to make the vaccine was not suf-ficient for large-scale production. Also, despite thechemical treatment of plasma products for safety,there was some concern about the risk, albeit small,
te Dosage Comments
l
Sequential series, 4 doses:2 mo, IPV; 4 mo, IPV.
6-18 mo, OPV; 4-6 y,OPV
Regimens with all IPV orall OPV are given in thesame time frame; allIPV doses are indicatedfor immunosuppressedpatients or contacts; allOPV dosing is acceptedin certaincircumstances only.
l 3 doses: 2, 4, and 6 mo Vaccine is currentlysuspended forinvestigation regardingthe association withintussuception.
IM 4 doses: 2, 4, 6, and 12-15mo
1 dose at ages 12 mo to13 y; 2 doses (4-8 wkapart) in susceptiblepersons �13 y
Do not give to childrenyounger than 12 mo.
vaccine; OPV, oral polio vaccine; SC, subcutaneous.itis A) and Engerix-B (Hepatitis B). It thus reduces the number of
Rou
SC
Ora
Ora
SC or
SC
d polio(Hepat

504 Wu et al J AM ACAD DERMATOL
APRIL 2004
of HIV transmission.80 Both of these issues wereaddressed in 1986, when the yeast recombinant hep-atitis B vaccine was licensed. This particular vaccinehas been a major breakthrough for the field of med-icine. It was the first licensed recombinant viral vac-cine prototype, as well as the first effective viralvaccine for a sexually transmitted disease. This vac-cine is produced by means of recombinant DNAtechnology, which involves insertion of the gene forhepatitis B surface antigen (HBsAg) into the yeastSaccharomyces cerevisiae (baker’s yeast). Clinicalstudies in high-risk homosexual men demonstratedthree-dose vaccine efficacy of 82% to 93% in pre-venting acute hepatitis B.81,82 Overall, approximately5% of immunocompetent adults fail to develop sig-nificant antibody titers after hepatitis B vaccination.Nearly 99% of children respond to vaccination,83
while only 50% to 70% of those over age 60 acquireimmunity.84,85 Variables associated with a lower like-lihood of seroconversion include immunosuppres-sion, renal failure, prematurity with low birthweight, age older than 40 years, obesity, and smok-ing.86-88 Because of the decreased rates of serocon-version in specific populations, additional researchis focused on methods of increasing immunogenic-ity to hepatitis B vaccines. Alternative delivery sys-tems, such as adenoviruses and vaccinia vectors, areunder evaluation. Clinical trials are currently inves-tigating the addition of adjuvants to the current re-combinant vaccine in order to increase the hostimmune response.89,90 Several different types of vac-cines also in development include DNA vaccines91
and Pre-S vaccines, which incorporate the surfaceproteins of the hepatitis B virus.92-94
The duration of immunity afforded by vaccinationmerits further long-term studies, but according topresent data, long-term efficacy is expected.88 Anti-body levels decline rapidly in the first year aftervaccination and then level off to a slow pace ofdecline.95 The loss of detectable antibodies to hep-atitis B years after vaccination does not necessarilyindicate a lack of immunity. The majority of personsare protected by immunologic memory in B lym-phocytes, which mount an anamnestic response tonatural infection.96 Rare cases of hepatitis B infectionin previously vaccinated patients have been de-scribed.97,98 These patients generally have subclini-cal disease, and none has developed chronic infec-tion or serious complications.88
The regimen for immunization includes 3 doses,given at months 0, 1, and 6. Hepatitis B vaccinationis recommended for adults at risk (ie, persons livingin or traveling to areas of high endemicity of hepa-titis B, health care personnel, morticians, personsengaging in high-risk sexual activity, persons with
chronic liver disease due to causes other than hep-atitis B, prisoners, users of illicit injectable drugs,and police and fire department personnel who ren-der first aid) and all children aged 0-18 years. Be-cause of the current widespread use in children, athimerosal-free vaccine was recently approved bythe FDA. Thimerosal is a preservative that containsmercury, which has prompted the limitation of itsuse in children.99 In persons in whom vaccine-in-duced protection is less complete, such as in hemo-dialysis patients, the need for a booster dose shouldbe assessed by means of annual antibody testing.
Adverse effects after hepatitis B vaccination aregenerally mild and well tolerated. Most commonlyreported is fatigue (15%), followed by headache(9%) and fever (1%-9%).100,101 A postmarketing clin-ical surveillance of 4.5 million doses of hepatitis Bvaccine over 5 years revealed no serious or severereactions attributable to the vaccine.102 Several iso-lated reports since that time have described the rareoccurrence of adverse effects such as thrombocyto-penic purpura,103-105 vasculitis,106,107 rheumatoid ar-thritis,108 lichen planus,109 and lichenoid reaction.110
However, it appears that these conditions do notoccur at a higher rate than in the unvaccinated pop-ulation. Large-scale hepatitis B vaccination programshave been unable to establish any association be-tween the vaccine and severe adverse effects otherthan rare episodes of anaphylaxis.101,111
On May 11, 2001, the FDA licensed a new com-bination vaccine that protects people at least 18years of age against hepatitis A virus and hepatitis Bvirus. This vaccine, Twinrix, combines 2 alreadyapproved vaccines, Havrix and Engerix-B, so thatpersons at high risk for exposure to both viruses canbe immunized against both at the same time. Thecombination reduces the number of injections from5 to 3. This vaccine is given at the same schedulethat is used for the hepatitis B vaccine, administeredat months 0, 1, and 6. The adverse effects of thisvaccine are similar in type and frequency to those ofthe monovalent hepatitis A and hepatitis B vaccines.The data from 11 clinical trials indicate that 99.9% ofvaccinees developed seroconversion for antibodiesagainst hepatitis A virus and 98.5% antibodiesagainst HBsAg, with persistence up to 4 years(GlaxoSmithKline Biologicals, unpublished data,2001).
In 2002, the FDA approved DTaP-HepB-IPV (Pe-diarix, SmithKline Beecham Biologicals, Rixensart,Belgium), a combined diphtheria and tetanus tox-oids and acellular pertussis adsorbed (DTaP),hepatitis B (HepB) (recombinant), and inactivatedpoliovirus vaccine (IPV). After 3 doses of DTaP-HepB-IPV, the immunologic responses were similar

Wu et al 505J AM ACAD DERMATOL
VOLUME 50, NUMBER 4
to those following 3 doses of separately adminis-tered vaccines for diphtheria, tetanus, 3 pertussisantigens, hepatitis B, and poliovirus types 1, 2, and3.112,113 However, there are no immunologic dataregarding simultaneous administration of DTaP-HepB-IPV and both Haemophilus influenzae type b(Hib) conjugate vaccine and pneumococcal conju-gate vaccine.114 The rates of most solicited local andsystemic adverse events, with the exception of fever,after administration of DTaP-HepB-IPV were similarto rates that followed separate administration of US-licensed vaccines.113,115 Furthermore, administrationof DTaP-HepB-IPV together with Hib vaccine wasassociated with higher rates of fever in comparisonwith separately administered vaccines.112,116
The FDA has approved Pediarix for use in infantsaged 2, 4, and 6 months. The Advisory Committeeon Immunization Practices recommends that for in-fants who are born to women who are HBsAg-positive or whose HBsAg status is unknown, a birthdose of single-antigen vaccine is preferred, but thebirth dose can then be followed by 3 doses of Pe-diarix at ages 2, 4, and 6 months.117 The recom-mended period between doses is 6-8 weeks, prefer-ably 8 weeks,114 and the third dose of Pediarixshould be given at least 16 weeks after the first doseand at least 8 weeks after the second dose but notbefore age 6 months. It should not be given to anyinfant aged �6 weeks or any person aged �7years.117
Varicella-zoster virus (Fig 2). Prior to thewidespread availability of varicella vaccine, yearlyUS figures for varicella disease included approxi-mately 4 million cases, 11,000 hospitalizations, and100 deaths.118 The currently available varicella vac-cine in the United States is a live attenuated Okastrain vaccine approved in 1995. The vaccine is verysafe and effective.119-121 Clinical trials began morethan 20 years earlier in Japan, after the vaccine wasdeveloped by means of attenuation of virus isolatedfrom the vesicular fluid of a healthy boy (with thesurname Oka) who had natural varicella infection.122
These initial studies showed a 90% seroconversionrate 4 weeks after vaccination with few clinical re-actions.123 Follow-up studies showed that the vac-cine protected against chickenpox for at least 17 to19 years, and all of the subjects had persistent anti-bodies and delayed-type skin reactions to the vari-cella-zoster antigen.124 In the United States, a dou-ble-blind, placebo-controlled study of the Okavaccine in 914 children revealed an efficacy of 100%at 9 months.125 After a 7-year follow-up, 95% of thevaccinated subjects remained free of clinical diseasewith chickenpox.126 In comparison with the diseaserates of unvaccinated children in the United States, it
appears that the Oka vaccine reduces the rate ofvaricella in children participating in the clinical trialsby 65% to 90%.122 Additional studies in the UnitedStates have shown that the Oka vaccine induceshumoral and cell-mediated immunity in healthy chil-dren,127-129 both of which have been shown to per-sist for at least 8 years.130 Delayed-type hypersensi-tivity skin reactions to varicella-zoster virus antigenshave also been shown to occur for at least 10 yearsafter vaccination.131 Case series have also shownthat vaccinated persons have less severe varicella(�50 lesions, no fever, and shorter duration of ill-ness) than those unvaccinated.132,133 Recent data in-dicate that the varicella vaccine effectiveness was�95% for preventing disease and 100% for prevent-ing moderate or severe disease in susceptible con-tacts when given within 36 hours of exposure.134
Studies of adolescents and adults have shownthat 2 doses administered 4-8 weeks apart werenecessary to produce seroconversion rates and an-tibody responses similar to those obtained in healthychildren.135 Vaccination is recommended for suscep-tible adults, particularly those in high-risk situations(eg, health-care personnel). The vaccine is recom-mended for all children who have no history ofchickenpox and is required for all who attend schoolin most states. Clinical studies of the use of vaccina-tion in immunosuppressed children and adoles-cents, particularly in those with acute lymphocyticleukemia, have been performed.136-138 Results indi-cated that vaccination is safe for those who are atleast 1 year away from induction chemotherapy ifthe current chemotherapy is halted around the timeof vaccination and the patient’s lymphocyte countsare �700/mm3. The immune response in these per-sons is lower than that of healthy recipients, thusrequiring the administration of 2 doses separated by3 months. Transmission of varicella from these vac-cinees may occur if a vaccine-associated rash devel-ops, although the risk of transmission is about onefourth that of natural varicella (20%-25% vs 87%).Another study of hematopoietic-cell transplant re-cipients, without a history of zoster, showed thatzoster developed in only 13% of those who had
Fig 2. Herpes-zoster in the distribution of dermatomeL2.

506 Wu et al J AM ACAD DERMATOL
APRIL 2004
received the inactivated varicella vaccine but in 30%of those who were not vaccinated.140 These datasuggest that the varicella vaccine may boost immu-nity and prevent zoster.
Waning cellular immunity is strongly correlatedwith the development of herpes zoster.141-143 Levinet al144 studied the immune response of elderly per-sons who received the live attenuated vaccine andfound that approximately 10% to 15% of the vacci-nees failed to develop increased immunity. Overall,however, the calculated half-life of the enhancedimmunity in this study was 54 months. The long-term duration of the booster effect had a positivecorrelation with the dose of the administered vac-cine. In a follow-up study 6 years after vaccination,Levin et al145 found that the varicella-zoster virus-responding T-cell frequency was still significantlyimproved over initial baseline measurements, aswell as expected measurements for this age cohort.In this vaccinated population, the frequency of her-pes zoster was within the range of expected inci-dents for this age cohort. However, in all of the casesof herpes zoster in the study, the number of lesionswas small, the associated pain was minimal, andpostherpetic neuralgia did not occur. These findingssuggest that vaccination in the elderly may be able toattenuate the course of herpes zoster.146
Multiple studies have reported a modified varicel-la-like syndrome in some vaccinated children afterexposure to the natural wild-type varicella vi-rus.126,132,147 The average yearly rate of modifiedvaricella-like syndrome varies from 0 to 2.72%among children who have been vaccinated with theU.S.-licensed Oka strain vaccine. These children typ-ically develop a milder form of disease with fewerthan 50 lesions. Most children do not have associ-ated fever, and only 50% of them develop vesicularlesions.148 None of the cases has been associatedwith systemic or serious disease. It has been notedthat more complete and long-lasting protection fromvaricella is associated with a stronger antibody re-sponse to vaccination.132
It is known that immunization with the Oka strainvaccine can lead to latent infection. In children, theincidence of primary varicella is between 18 and 77per 1 million person years of follow-up.149 Herpeszoster can develop later, either from this vaccine-type virus or from natural wild-type varicella-zostervirus.150,151 There have been several reports of mildherpes zoster in previously healthy children whohad received the varicella vaccine. The incidence isless than that seen in children with prior chicken-pox,152 such that vaccinated children may perhapshave a decreased risk for herpes zoster.
The Oka vaccine should be given as a single doseto children 12 months to 12 years of age. Personsover the age of 13 should receive 2 doses, 4-8 weeksapart. The duration of protection is unknown at thistime, and the need for a booster immunization isuncertain. It has been observed that vaccinees whoare exposed to natural varicella have a boost inantibody levels. However, it is postulated that in ahighly vaccinated population, a lack of exposure tonatural varicella may result in waning immunity forsome.
The Oka vaccine is generally well tolerated. Inboth adults and children, the most common sideeffect is mild tenderness, erythema, or induration atthe injection site (19.3% to 24.4%). Fever occurred in10.2% to 14.7% of clinical trial subjects, and a gen-eralized varicella-like rash developed in 3.8% to5.5%. A localized varicella-like rash at the injectionsite may also occur. The likelihood of transmissionof the vaccine virus from a healthy vaccinee is lowbut may be more likely if a rash develops aftervaccination, especially in those who are immuno-compromised hosts. One case of transmission froma vaccinated child to a susceptible mother has beenreported in the United States, but it is suspected thatthe child may have been concurrently infected withnatural wild-type varicella. Persons who undergovaccination should avoid close association with sus-ceptible high-risk individuals for up to 6 weeks, ifpossible. This vaccination is also contraindicated inpregnant women or in any woman planning to be-come pregnant within 3 months, since vaccinationinvolves use of a live attenuated virus and naturalvaricella is known to cause fetal harm.
BacteriaAnthrax. Interest in Bacillus anthracis was
raised after the deliberate contamination of mailwith spores after the September 11, 2001, attacks onthe United States. This aerobic or facultative anaer-obic gram-positive rod forms highly resistant endo-spores under extreme conditions. The spores causehuman disease by means of direct contact, ingestion,or inhalation. Cutaneous anthrax occurs when abreak in the skin allows spores to enter. The incu-bation time is usually 2-3 days but can occur as soonas 12 hours or as late as 2 weeks after exposure.153
Initially, a papule forms, and within 24 hours, a ringof vesicles follows. These lesions ulcerate and be-come black, necrotic, and edematous. Mortality isabout 20% in untreated cases.
The toxins of B anthracis come from 2 genes,pX01 and pX02.154,155 The gene pX01 encodes pro-tective antigen (PA), lethal factor, and edema factor,which make up the anthrax toxin. The latter 2 com-

Wu et al 507J AM ACAD DERMATOL
VOLUME 50, NUMBER 4
bine with the PA to form lethal toxin and edematoxin. Edema toxin causes unregulated adenylatecyclase activity, which leads to production of un-physiologically high concentrations of cyclic aden-osine monophosphate.156 This event causes massiveedema and organ failure. Plasmid pX02 encodes thepoly-�-linked D-glutamic acid (PGA) capsule, whichprevents the antiphagocytic activity of macro-phages.157 Strains of B anthracis that lack pX02 areavirulent.
As early as 1881, Pasteur demonstrated the pro-tective efficacy of the first anthrax vaccine when heinjected sheep with heat-attenuated B anthracis. Inthe 1930s, widespread vaccination with attenuatedstrains such as the Sterne strain significantly de-creased the incidence of anthrax in domesticatedanimals in industrialized countries. In the UnitedStates, the licensed human vaccine (anthrax vaccineadsorbed [AVA], newly renamed BioThrax) is a cul-ture supernatant of a toxigenic, nonencapsulated Banthracis strain, V770-NP1-R, derived from theSterne strain.158 A similar culture supernatant–derived human vaccine (PL 1511/0058) is producedin the United Kingdom.159
A 1950s study of wool sorters immunized with avaccine similar to AVA, coupled with long experi-ence with AVA and the United Kingdom vaccine,have shown that a critical level of serum antibodiesto the PA confers immunity to anthrax.160,161 Afterregular immunizations the previous year, staff of theGovernment Wool Disinfection Station in Liverpoolwere free of the disease with a total protective effi-cacy of 92.5%.162 Laboratory animals and cattle alsoare protected by AVA from both cutaneous andinhalational B anthracis challenges.163,164
The CDC recommends anthrax vaccinations forpersons engaged in the production of quantities orconcentrations of B anthracis cultures, those en-gaged in activities with a high potential for aerosolexposure, persons in the military, and other selectpopulations for which a calculable risk can be as-sessed.165 Preexposure vaccinations currently arenot recommended for emergency first responders,medical practitioners, and private citizens.
Although efficacious and safe,166 AVA has multi-ple limitations. First, standardization of AVA is basedon the protection of guinea pigs challenged intracu-taneously with B anthracis spores.164 PA is not mea-sured in the vaccine, and there is no standardizedassay of PA antibodies in animals or humans vacci-nated with AVA. Second, the schedule of AVA ad-ministration (subcutaneous injections at 0, 2, and 4weeks and boosters at 6, 12, and 18 months withrecommended annual boosters to maintain immu-nity) is likely suboptimal. This schedule was de-
signed for rapid induction of immunity,167 but arecent study showed that increasing the interval be-tween the first 2 injections enhances the level ofAVA-induced antibodies to PA.168 Furthermore, aschedule of injections at 6, 12, and 18 months is notwell defined. The long-term protective efficacy inhuman beings is unknown. In animal studies, theprotective efficacy may last up to 2 years after 2inoculations.169,170 Finally, AVA contains other cellu-lar elements that contribute to the relatively high rateof local and systemic adverse reactions.166 The mostcommon adverse effects included injection-site hy-persensitivity, edema, pain, headache, arthralgia, as-thenia, and pruritus. In 20% of recipients, mild cu-taneous reactions included erythema, edema, andinduration. In �1% of recipients, systemic reactionsincluded fever, chills, nausea, and body aches.160
Spores and PGA have also been studied as pos-sible targets for immunization. PA antibodies havebeen found to have unexpected sporicidal proper-ties in vivo. PA antibodies both enhance phagocy-tosis of spores by macrophages and suppress ger-mination.171 Active immunization of guinea pigs andmice with formalin-inactivated spores also confersimmunity to infection.172,173
Encapsulated B anthracis strains are virulent inmice, regardless of whether they produce toxin.174
PGA is the major virulence factor in mice, and vac-cines based on PA show reduced efficacy in thoseanimals.172 It may be easier to demonstrate a role foranti-PGA antibodies in protective immunity with theuse of mice. However, there is no definitive test ofwhether anticapsular antibodies contribute to hu-man immunity to B anthracis. Furthermore, it willbe difficult to extrapolate conclusions about capsu-lar antibodies from mouse anthrax infection to hu-man anthrax infection, given that the contributionsof PGA and PA to pathogenesis differ substantiallybetween mice and humans.
Live vaccines are potential candidates for admin-istration as an oral, single dose. They consist oforganisms in which the gene encoding the vaccinehas been introduced. After delivery, the microorgan-ism either colonizes the mucosal surface or causes alimited infection during which the vaccine candidateis expressed and presented to the immune system.Two live vector systems have been described forPA.173,175 The main disadvantage of live vaccines isthe need to culture and store the organism prior touse.
By contrast, DNA vaccines are extremely simpleand cost-effective. They can be defined broadly asplasmid DNA expression vectors that result in ex-pression of an antigen in situ leading to the induc-tion of antigen-specific immunity.176 DNA-based

508 Wu et al J AM ACAD DERMATOL
APRIL 2004
vaccines have multiple advantages over conven-tional vaccines. Unlike subunit vaccines, which re-quire extensive purification, DNA is relatively cheapto produce. The vaccines are heat-stable and areamenable to genetic manipulation. DNA vaccinesbased on PA have been constructed and have beenshown to be capable of inducing PA immune re-sponses177 and of protecting mice against lethaltoxin.178
In the short term, improved anthrax vaccines willfocus on PA. The National Institute of Allergy andInfectious Diseases has an accelerated program forvaccine development to make 25 million doses of arecombinant PA vaccine available within 2 years.179
Lyme disease (Fig 3). Lyme disease is the mostcommonly diagnosed vector-borne disease in theUnited States. Since 1982 the annual incidence ofLyme disease has increased more than 25-fold.180
Between 1993 and 1997, an average of 12,451 newcases were reported each year to the CDC. Lymedisease is a tick-borne zoonosis caused by an infec-tion with the spirochete Borrelia burgdorferi.
B burgdorferi has several outer surface proteins.In naturally acquired infections, animals and hu-mans mount an immune response against the spiro-chete’s flagellin (41 kd) and the 23-kd outer-surfaceprotein (OspC).181 However, in culture, the domi-nant outer-surface protein expressed is OspA, a31-kd lipoprotein.182,183 Experiments have shownthat OspA is expressed abundantly by B burgdorferiin the midgut of infected ticks.183 When the tickattaches itself to a host, the spirochetes undergo asubstantial antigenic change during their migrationto the tick’s salivary gland, where OspA expressionis decreased and OspC expression is increased.184,185
This temperature-sensitive ability is likely an ac-quired protective mechanism that enables B burg-dorferi to become established in warm-blooded an-imals. OspA is uniform across different isolates inthe United States, but OspC is highly variable amongdifferent regions. An immune response directed
Fig 3. Erythema migrans of the inguinal region, a symp-tom of Lyme disease.
against OspC after Lyme disease is unlikely to pro-tect against a heterologous infection. However,cross-strain protection would be expected with animmune response directed against the spirochete’sOspA in the tick gut.186
In 1990, a mouse model of Lyme disease wasdeveloped. Shortly afterward, a recombinant OspA(rOspA) vaccine was shown to be protective againstsyringe-inoculated infection.187,188 Both passive im-munization and active immunization were shown tobe protective. The rOspA vaccine was then shown tobe protective against naturally occurring tick-trans-mitted infection.189-191 The effectiveness of rOspAvaccine initially was surprising because organismsisolated from infected animals do not express OspAuntil naturally occurring late infections. It was dis-covered that the vaccine acts primarily in the tickmidgut rather than in the vaccinated host.192 As afeeding tick ingests a blood meal, the ingested bloodhas circulating anti-OspA antibodies that destroy Bburgdorferi in the midgut of the tick. This finding issignificant because it suggests that a high level ofcirculating antibodies has to be maintained in thehost to protect against infection at the time of a biteby an infected tick.
The rOspA vaccine was also shown to be effec-tive in protecting dogs and rhesus monkeys againstLyme disease.193,194 On the basis of these promisingresults, human immunogenicity, efficacy, and safetytrials were performed to evaluate recombinant, lipi-dated OspA vaccine (LYMErix) of B burgdorferisensu stricto. The rOspA protein is expressed inEscherichia coli and purified. LYMErix was found tobe immunogenic and safe in phases I and II clinicaltrials.195,196 In a randomized, controlled clinicalphase III trial,197 10,936 people were recruited from31 sites in Lyme-endemic areas. Each subject re-ceived 3 doses of vaccine (30 �g) or placebo on a 0-,1-, and 12-month schedule and timed so that eachsubject received the initial vaccination several weeksbefore B burgdorferi transmission season. Thesesubjects were followed for clinical manifestations ofLyme disease or adverse events for 20 months.
Vaccine efficacy in protecting against sympto-matic infection (ie, the presence of erythema mi-grans or objective neurologic, musculoskeletal, orcardiovascular manifestations) was 49% after 2 dosesand 76% after 3 doses. The efficacy in protectingagainst asymptomatic infection (no recognizedsymptoms) was 83% after 2 doses and 100% after 3doses. All subjects had laboratory-confirmed infec-tion by means of cultural isolation, polymerasechain reaction positivity, or seroconversion. A sub-set of subjects had serologic testing for immunoge-nicity to correlate to protection. Protective immunity

Wu et al 509J AM ACAD DERMATOL
VOLUME 50, NUMBER 4
was achieved after 2 doses, but antibody levels de-clined rapidly, resulting in protective levels for lessthan a full year. With 3 doses, a significant anam-nestic response developed with maintenance of pro-tective antibody levels throughout the following ex-posure season.
The most common adverse event was soreness atthe injection site (24.1% of vaccine recipients versus7.6% of placebo recipients). Other side effects in-cluded arthralgia, myalgias, flulike symptoms, andfever and chills, but all were uncommon (less than3.2%). The vaccine was found to be safe in subjectswith a history of Lyme disease. However, there wasa greater incidence of musculoskeletal symptomswithin the first 30 days after vaccination in subjectspreviously afflicted with Lyme disease than in sub-jects without Lyme disease (20% versus 13%).198 Af-ter the first 30 days since vaccination, there was nosignificant difference between vaccine and placeborecipients with a history of Lyme disease (33% ver-sus 35%).197
The Pasteur Merieux Connaught vaccine, Imu-Lyme, was studied in a similar phase III trial involv-ing 10,305 adult subjects.199 The immunogen is alsorecombinant OspA lipoprotein, but unlike the LY-MErix vaccine, it does not have an aluminum adju-vant. Subjects received 3 doses of 30 �g of vaccineor placebo on a 0-, 1-, and 12-month schedule. At 12months, 7,515 subjects received booster doses. Theprimary end point was the total number of newclinical cases of Lyme disease, confirmed by meansof serology. The efficacy of vaccine was 68% after 2doses of vaccine in the first year of study and 92% inthose who received the third dose of vaccine in thesecond year. Local injection site reactions were morecommon in the vaccine group during the first week;otherwise, there was no difference between the vac-cine and placebo groups in the occurrence of lateadverse reactions.
An analysis of the cost per quality-adjusted life-year gains with vaccination showed use of the Lymevaccination to be economically attractive only inpersons whose seasonal risk for Lyme disease ex-ceeds 1%.200 In areas with a seasonal attack rate of0.5%, the cost per quality-adjusted life-year gainedwas $145,200. Areas with annual attack rates of 2.5%to 10% have been reported,201 but large endemicareas, such as those in which the 2 large Lymedisease vaccine trials were done, had annual attackrates of about 1%. In 1998, the LYMErix vaccine wasapproved by the FDA for use in persons aged 15 to70 years on a 0-, 1-, and 12-month schedule. TheCDC recommends use of the Lyme disease vaccinein persons aged 15-70 years who reside, work, or
play in areas of high or moderate risk and in travel-ers to areas of high or moderate risk with prolongedand frequent exposures to infected ticks.
Several specific issues surround the safety ofLyme vaccines.202 The first is a theoretical concernthat OspA-based vaccine could induce inflammatoryarthritis in genetically susceptible persons throughan autoimmune mechanism. The second issue is thequestion of cross-strain protection; will the currentvaccine protect against all strains of B burgdorferi inthe United States? The third is concern that boosterdoses will be required to maintain adequate circu-lating antibody levels, but the need for and safety ofbooster doses is not known. The fourth issue iswhether the vaccine might alter the clinical course ofinfection if antibody levels fall below the levelneeded for complete protection. Another concern iswhether alternative vaccine schedules might allowthe development of protective immunity in less than1 year. The final concern is the lack of informationabout the safety and immunogenicity of the vaccinein children.
Although the vaccine appeared safe and effective,it was taken off the market in early 2003 because ofa lack of general use in the medical community.
Haemophilus influenzae. The early 1980s sawthe introduction of the first vaccine for Hib, whichconsisted of polyribosyl ribitol phosphate (PRP), thecapsular polysaccharide of Hib. A large field trialperformed in Finland showed that antibody re-sponse to plain PRP vaccine correlated with preven-tion of invasive Hib disease.203 One month aftervaccination, an antibody concentration of 1 g/mL inserum was associated with long-term protectionfrom disease.204
In December 1987, the first Hib conjugate vac-cine, PRP-D (PRP conjugated to diphtheria toxoid;ProHIBIT, Aventis Pasteur) was licensed in theUnited States. The PRP-D vaccine was found to pre-vent Hib disease in Finnish infants immunized dur-ing the first 6 months of life.205,206 However, a studyof native Alaskan infants immunized at 2, 4, and 6months showed no significant protection,207 and theFDA rejected the application to license PRP-D foruse in infants.208 In 1987 the FDA approved PRP-Dfor use only in children older than 18 months.209
Two subsequent Hib conjugate vaccines werelicensed in the United States for use with infantsbeginning at 2 months of age: HbOC (Hib [PRP]oligosaccharides conjugated to CRM197, a cross-re-acting, mutant, nontoxic diphtheria toxin; HibTITER,Wyeth Laboratories) and PRP-OMP (PRP conjugatedto an outer-membrane protein complex; PedvaxHib,Merck Sharp and Dohme).210,211 Three randomized,

510 Wu et al J AM ACAD DERMATOL
APRIL 2004
placebo-controlled trials conducted in infants immu-nized beginning at 2 months of age showed theefficacy of these vaccines.212-214 Further, these 2 vac-cines were found to be significantly more immuno-genic in infants than PRP-D.211,215,216
In 1993, the FDA approved 2 more Hib conjugatevaccines, PRP-T (PRP conjugated to tetanus toxoid)vaccine (ActHIB, Aventis Pasteur) and a combineddiphtheria and tetanus toxoids and whole-cell per-tussis vaccine (DTP) and Haemophilus b conjugatevaccine (Tetramune, Lederle-Praxis Biologicals).217
Two studies showed that PRP-T has an immunoge-nicity profile similar to that of HbOC vaccine ininfants immunized at 2, 4, and 6 months of age.218,219
The DTP-HbOC vaccine was also found to be safeand more immunogenic than separate vaccinationwith DTP and HbOC.220,221
Before Hib conjugate vaccines became available,Hib was the principal cause of bacterial meningitisand a major cause of other serious invasive diseasesamong children aged �5 years in the UnitedStates.222,223 In the prevaccine era, the incidence ofHib invasive disease was 100 per 100,000 amongchildren aged �5 years. In 1991 all infants at age 2months were recommended to receive Hib conju-gate vaccines, and by 1996 the incidence of Hibinvasive disease among children aged �5 years haddeclined by more than 99%.222,224 During 1998-2000,a total of 824 Hib invasive disease cases was re-ported among children younger than 5 years, andrates were 1.4 per 100,000 children in 1998 and 1999and 1.6 in 2000.225 The Hib conjugate vaccines havebeen equally efficacious in the rest of the world, aswell.226,227
Neisseria meningitidis (Fig 4). The importantdeterminant of virulence of Neisseria meningitidis isthe polysaccharide capsule. Mutants without capsu-lar expression are nonpathogenic, since they arekilled by complement. There are licensed polysac-charide vaccines against serogroups A, C, Y, and
Fig 4. Disseminated meningococcal disease with second-ary disseminated intravascular coagulation resulting in ec-chymoses and purpura fulminans.
W135, but there currently is none against serogroupB.
Thirty years ago, the first successful capsularpolysaccharide vaccines against groups A and Cwere developed for epidemics of meningitis amongU.S. military recruits.228-230 Studies performed in theUnited States, Africa, and Europe showed that thesevaccines were safe and induced bactericidal anti-bodies in older children and adults.231-234 However,most polysaccharide vaccines are poor immunogensin infants and do not elicit immunologic memory inpeople of any age. Group A polysaccharide is im-munogenic in children �2 years of age, but protec-tive immunity after 1 dose lasts a short duration, andeven after repeated injections, booster responses arelow.235-238 Furthermore, repeated vaccination withgroup C polysaccharide vaccine causes immune hy-poresponsiveness, which may cause difficulties forpatients who need long-term protection.239-241
As with Hib vaccines, meningococcal conjugatevaccines were developed and found to induce long-term protection and to be safe and immunogenic inyoung infants.242 In November 1999, meningococcalgroup C conjugate vaccine entered routine immuni-zation in the United Kingdom. Infants received 3doses of vaccine at 2, 3, and 4 months of age as theirroutine primary immunizations. Also, a catch-upcampaign between November 1999 and December2000 was targeted at all children of �4 months and�18 years of age. Prospective nationwide surveil-lance showed that confirmed cases of invasivegroup C disease fell overall by 85%, in both theprimary immunized and the catch-up popula-tions.243 However, a concern about mass immuniza-tion with meningococcal group C conjugate vaccineis capsular group replacement by strains not con-tained in the vaccine.244,245 Capsule switching be-tween B and C strains has been documented.244,246
However, careful and extensive surveillance in themeningococcal group C conjugate vaccine study inthe United Kingdom showed no evidence of capsu-lar group replacement.
Multiple clinical trials have shown that bivalent Aplus C polysaccharide conjugate vaccines are welltolerated and immunogenic in infants, toddlers, andadults.238,247-249 The currently available formulationin the United States is the tetravalent A, C, Y, W-135vaccine (Menomune-A, C, Y, W-135; Aventis Pas-teur). It is safe and has an efficacy of 85%,250 and itcould prevent more than 60% of cases in collegestudents.251 The current recommendation for the tet-ravalent vaccine is selective immunization based onrisk factors for meningococcal disease, instead ofuniversal immunization in the pediatricpopulation.252,253

Wu et al 511J AM ACAD DERMATOL
VOLUME 50, NUMBER 4
INVESTIGATIONAL VACCINESMany of the vaccines currently under investiga-
tion more than likely will expand the focus of im-munization. All the vaccines available up to the endof the 20th century have been used solely to preventdisease. However, several new candidate vaccinesare being developed and evaluated in the treatmentof already acquired infections. Table IV lists thepredominant diseases for which candidate vaccinesare presently under investigation.
VirusesHIV. As the AIDS epidemic persists and spreads
unabated in much of the world, the search for aneffective HIV vaccine is becoming critical. In 1997President Clinton challenged scientists to develop aneffective HIV vaccine by the year 2007. Since clinicaltrials first began in 1987, at least 34 different HIVcandidate vaccines have begun phase I trials, and ahandful of these have progressed to phase II tri-als.254 Seventy-four additional HIV vaccine candi-dates are reported to be in research and develop-ment or preclinical testing in animals, and thisnumber probably has increased.254 The predomi-nant types of HIV vaccines under investigation arelisted in Table V. Thus far, only one AIDS vaccinehad advanced to phase III trials, the VaxGen AIDSvaccine AIDSVax, which was evaluated in theUnited States and in Thailand. Earlier research sug-gests that this recombinant gp120 vaccine stimulatesantibody production but may not induce cellularimmunity. HIV research has shown that the induc-tion of cytotoxic T lymphocytes may be an impor-tant correlate for protective efficacy of HIV vac-cines.255 However, the recombinant gp120 HIVvaccine failed. In the 3-year study, of 5,009 volun-teers at 59 trial sites in the United States, Canada, theNetherlands, and Puerto Rico, 5.7% of those whowere vaccinated developed HIV compared with5.8% of those given placebo.256
Recombinant subunit HIV vaccines are geneti-cally engineered from HIV surface envelope pro-teins, such as gp120 or gp160. Because they do notcontain live virus or DNA, there is no risk of causinginfection. A therapeutic trial was carried out withgp160 subunit immunization every 3 months for 3years in HIV-positive persons, in addition to antiret-roviral therapy.257 Results demonstrated a modesteffect on CD4 counts but no clinical benefit. Theseresults were consistent with similar, earlierstudies.258,259
Recombinant live-virus vector vaccines involvethe use of virus carriers that are genetically engi-neered to express particular HIV genes. The firstcandidate to be tested was a vaccinia vector with the
insertion of HIV gp160 gene. The vaccine aloneinduced little antibody.254 However, when it wasused as a primer and boosted with the recombinantgp160 vaccine, results showed strong induction ofcellular immunity and antibody responses.260 PhaseI trials are under way for a recombinant vaccinia HIVprimer followed by boosting with a recombinantgp120 vaccine.254
Because of concerns over shedding of the vac-cinia virus and possible disseminated disease in im-munosuppressed persons, more attention has beenfocused on canarypox and adenovirus vectors,which can infect humans but cannot replicate. Rep-lication in humans continues long enough to pro-duce the necessary HIV proteins before abortion ofthe cycle.261 Early results have shown that recombi-nant canarypox vector vaccines can induce humoraland cellular immune responses, including cytotoxiclymphocytes.262 The greatest interest for these vac-cine candidates lies in the prime and boost ap-proach. The canarypox vaccine primer induces a
Table IV. Infectious diseases with cutaneousmanifestations for which vaccines are underdevelopment
Prophylactic Therapeutic
HIV HIVHSV HSVHPV HPVCytomegalovirus Varicella-zoster virusRespiratory syncytial virusParainfluenza virusHepatitis CDengue feverEpstein-Barr virusAnthraxLyme diseaseNeisseria meningitidis serotype BNontypeable Haemophilus
influenzaeRocky Mountain spotted feverStaphylococcus aureusStaphylococcus epidermidisStreptococcus pyogenesPseudomonas aeruginosaFrancisella tularensisBrucella melitensisLeptospira interrogansCandida speciesAspergillus speciesCryptococcus neoformansCoccidioides immitisBlastomyces dermatitidisHistoplasma capsulatumLeishmania

512 Wu et al J AM ACAD DERMATOL
APRIL 2004
strong cellular immunity, followed by a recombinantsubunit vaccine that boosts the antibody re-sponse.261 The combination of both vaccines in-duces a stronger immune response than either onealone.255,263 Recent results from a phase II trialshowed that 93% of subjects who received the com-bination of vaccines developed neutralizing anti-bodies.264 Also, almost one-third of the recipientsdeveloped a cytotoxic lymphocyte response. Addi-tional studies have investigated canarypox vectorsexpressing gp160 or gp120/gag/pol HIV-1 antigensgiven along with recombinant gp160 or gp120 sub-unit vaccines.265 In a comparison of data from dif-ferent trials of several candidate canarypox vectorHIV vaccines, more than half of the recipients de-veloped durable, HIV-specific cytotoxic T-lympho-cyte responses.266 Researchers suggest that abroader recombinant vector vaccine would proba-bly increase the percentage of responders.266
A trial in Uganda of the effect of a canarypoxvector vaccine alone commenced in February1999.267 The vaccine, called Alvac vCP205, contains3 HIV genes in a weakened version of canarypoxvirus. The particular genes come from clade B vi-ruses, which are the predominant subtype of HIVfound in the United States and Europe. However,the majority of HIV infections that occur in Ugandaare due to clades A and D. This study will firstevaluate the cross-reactivity among these viral sub-
Table V. Predominant types of HIV vaccines underinvestigation
DNA plasmid: gp120, gp160, GagPol, Pol, techniques suchas multiplasmids, interleukin-2 adjuvants, geneticadjuvants, conventional adjuvants, DNA priming,microspheres, and in vivo electroporation
Epitopes: CTL or HTLLive attenuated virus: HIV, simian-human
immunodeficiency virusPeptides: synthetic V3-peptides, Tat, lipopeptidesRecombinant live vector: adeno-associated viruses,
replication-defective adenovirus serotype 5 vector,alphaviruses, canarypox and other avian and mammalianpoxviruses, herpes simplex virus type-1, picornaviruses,rhabdoviruses, vaccinia, modified vaccinia virus Ankaravector, vesicular stomatitis virus, bacterial vectors
Recombinant subunit: rgp120, rgp160, EnvRetroviral vectors: recombinant murine retrovirusRNA optimized constructs: pGag, pEnvSelf-replicating RNA: replicon particles based on
Venezuelan equine encephalitis virusVirus-like particles (VLP): p170/p24:Ty VLPWhole inactivated virus: envelope depleted whole
inactivated HIV-1
CTL, Cytotoxic T lymphocyte; HTL, helper T lymphocyte.
units and compare the immune responses inrecipients.
Use of the vesicular stomatitis virus as a vector toexpress HIV protein has also shown promise. Onestudy involved vaccination of 7 rhesus monkeyswith live attenuated vesicular stomatitis virus ex-pressing Env and Gag, and the animals were chal-lenged with simian-human immunodeficiency vi-rus.268 Two years after the challenge, none of theanimals has developed AIDS and 6 have no detect-able virus.269 Another study involved evaluation ofimmune responses of animals vaccinated either in-tranasally or intramuscularly with the vesicular sto-matitis virus vector expressing Env and Gag.269 Aftera challenge with simian and human immunodefi-ciency virus, the animals vaccinated intranasally hada slight loss of CD4 T lymphocytes. However, theanimals vaccinated intramuscularly had a significantdecrease in CD4 T lymphocytes, which returned to70% of baseline. The virus load was undetectable inboth groups.
DNA vaccines are another promising prospect forHIV immunization. With this approach, purifiedDNA that encodes for particular immunogenic anti-gens is injected. These antigens are presented to thehost immune system in their native form and areprocessed in a way similar to that for a natural viralinfection.262,270 Therapeutic immunization with aplasmid/gp160 and gag� pol DNA vaccine in HIV-positive chimpanzees revealed a significant de-crease in viral load and a boost in the immuneresponse.270 Studies in seronegative primates dem-onstrated the induction of neutralizing antibodiesand cytotoxic T-lymphocyte responses, but the vac-cine did not protect against infection.254 A phase Iclinical trial of 2 DNA vaccine candidates is currentlyin progress.254
Several other approaches to HIV vaccine devel-opment are under investigation. Live attenuated vi-rus vaccines are known to generate a broad anddurable immune response, but they have not beentested in humans owing to potential safety concernswith live HIV virus.254 Whole-inactivated vaccinesare generally thought to be safer than live attenuatedones. However, inactivation of the virus often leadsto a vaccine that is less potent or immunogenic.Studies of whole-killed virus vaccines in chimpan-zees thus far have not been able to demonstrateprotection from HIV infection.254 In addition, there isconcern that inadvertently incomplete inactivationcould lead to HIV infection of vaccine recipients.Virus-like particles (VLP) are a safer option, sincethey consist of a noninfectious HIV “look-alike” thatdoes not contain the HIV genome. One such candi-date, known as p17/p24:TY, has reached the stage

Wu et al 513J AM ACAD DERMATOL
VOLUME 50, NUMBER 4
of clinical trials. Early results have shown that thisvaccine leads to low levels of HIV binding antibod-ies and T-cell memory responses but induces verylittle cytotoxic T-lymphocyte activity.254 Other VLPcandidates are under development. Among themany important controversies that exist in HIV vac-cine development is whether neutralizing antibodiesas typically measured are relevant to clinicalprotection.
HSV (Fig 5). The search for a vaccine for herpessimplex virus (HSV 1 and 2) spans 9 decades. In the1920s, untreated vesicular fluid from herpes lesionswas injected into patients in an attempt to induceimmunity.254 This method, to say the least, did notwithstand the test of time. Inactivated whole virusvaccines were developed in the 1930s and weremade from HSV-infected animal tissue, such as rab-bit brain.271 Despite the many advances made withinactivated virus vaccines through the years, none ofthe candidates proved to be sufficiently immuno-genic. With increasing technology, several differentapproaches for HSV vaccines are currently in devel-opment and under evaluation.
Two separate recombinant subunit vaccines havebeen investigated in phase III trials. One such can-didate developed by Chiron contained HSV-2 sur-face glycoproteins B and D and the adjuvant MF59.The development of this vaccine was halted prema-turely because results demonstrated overall lack ofefficacy for both preventive and therapeuticuse.272,273 A second recombinant vaccine was devel-oped by SmithKline Beecham and contains the gly-coprotein D and the adjuvant monophosphoryl lipidA immunostimulant.274 Results of clinical trials withthis candidate indicate that it has a clinical efficacy of73% in protecting women who are serologically neg-ative for both HSV-1 and HSV-2 from acquiringHSV-2 disease.275 Another approach combines thesafety profile of a killed vaccine with the immuno-genic potential of a live virus vaccine.276 The dis-abled infectious single-cycle vaccine lacks the gly-coprotein H gene necessary for virus entry into cells.After a single replication cycle, the virus is unable tospread to surrounding cells and thus remains non-infectious. Studies in guinea pigs showed encourag-ing results for both preventive and therapeutic treat-ment.276,277 Although phase I studies showed thecandidate to be safe and well tolerated, phase IItrials showed that the vaccine failed as therapy.Trials are also planned to evaluate the efficacy inpreventing infection in seronegative partners in dis-cordant couples.254
DNA vaccines are also in development for HSVimmunization. Animal studies that involve inocula-tions of plasmid DNA carrying the desired viral
genes have shown promising results for the preven-tion of infection.278,279 These vaccines are able toexpress only 1 or 2 viral antigens but can inducecell-mediated immunity without the need for potentadjuvants. One such candidate that encodes glyco-protein D2 is currently in phase I clinical trials, andseveral others are in preclinical development.
Live attenuated HSV vaccines have been ratherdifficult to develop, as viruses that are the safest andmost attenuated tend to lack immunogenicity. Re-search in the past has shown that stable attenuationof HSV was not achieved after passage in cell cul-ture. After immunization, the vaccine strain wouldthen have the potential to revert to its virulent stateand cause disease. A genetically engineered HSVmutant vaccine was found to be safe and effective inanimal studies280 but in humans was overly attenu-ated and lacked sufficient immunogenicity.281 Newgenetically engineered strains are currently underdevelopment.
HPV (Fig 6). Certain types of HPV are associatedwith the development of cervical cancer. Thus thesearch for a prophylactic or therapeutic HPV vaccinehas been an important endeavor. Although morethan 30 types of HPV are known to be sexuallytransmissible, the major types associated with ma-lignancy (16, 18, 31, 33, 45, 52, 58) and condylomata(6 and 11) are relatively few in number, allowing formore focused strategies for immunization againstthese specific types. Vaccine development has been
Fig 5. Herpes simplex type 2 infection of the buttocks.

514 Wu et al J AM ACAD DERMATOL
APRIL 2004
hampered in the past because of the inability toculture HPV. However, an in vitro culture system forHPV has recently been developed, furthering theprospect for advances in this field.254
VLP are produced by means of recombinant DNAtechnology and are designed to self-assemble intoconformations that resemble natural HPV. Thesevaccines contain no viral DNA and thus carry no riskof infection or oncogenic exposure. VLP have beendesigned for all of the major HPV subtypes, andclinical trials are currently under way for HPV-11 L1VLP,282 HPV-6 L1 VLP,283 and HPV-16 L1 VLP.284 Arecent double-blind, placebo-controlled studyshowed that persons who were HPV-16 negativeand received the HPV-16 vaccine had a significantlyreduced incidence of both HPV-16 infections andrelated cervical intraepithelial neoplasia at a medianfollow-up of 17 months.285
Fusion protein vaccines are currently under eval-uation for the immunotherapy of cervical cancer andgenital warts. TA-HPV is a live recombinant vacciniavirus that has been engineered to express the E6 andE7 protein genes for HPV 16 and 18 as a treatmentfor cervical cancer.254 This method also involvesutilization of the viral vector approach, with vacciniaas a vehicle. Viral vector vaccines can be polyvalentand have the potential to produce immunity similarto that induced by live attenuated vaccines. A phaseI-II clinical trial of TA-HPV286 has shown encourag-ing results, and further studies are under way.TA-GW is a recombinant fusion protein vaccine con-sisting of HPV-6 L2 and E7 proteins and is underinvestigation for the treatment of genital warts. Aphase IIa clinical trial showed the vaccine to beimmunogenic, with encouraging clinical re-sponses.287 A third protein vaccine, TA-CIN, is inpreclinical development for the treatment of cervicaldysplasia.254
Peptide-based vaccines have been shown to beprotective against HPV-induced tumors in mice, al-though the T-cell repertoires in mice and humansdiffer. Two early-stage human clinical trials are un-
Fig 6. High-grade cervical dysplasia secondary to HPV-16.
der way, one involving HLA-A*0201 bindingHPV16-E7 peptides, to assess the possible therapeu-tic implications these vaccines may offer.157 Otherinvestigational approaches to HPV immunization in-clude DNA vaccines,288 bacterial vectors,289-291 anddendritic cells pulsed with HPV epitopes.292
In 1 study, the cost-effectiveness of vaccinatingadolescent girls who are at high risk of HPV infec-tion was evaluated.293 Relative to current practice, avaccine with a 75% probability of immunity againstHPV infection resulted in a life-expectancy gain of2.8 days or 4.0 quality-adjusted days at a cost of $246(incremental cost-effectiveness of $22,755 per qual-ity-adjusted life year). If all 12-year-old girls in theUnited States (approximated at 1,988,600) were vac-cinated, more than 224,255 cases of HPV, 3,317cases of cervical cancer, and 1,340 deaths from cer-vical cancer would be prevented.
FungiThe idea of an antifungal vaccine has been inves-
tigated for more than 40 years.294 Development of aneffective fungal vaccine is difficult because fungi,like other eukaryotic organisms, are orders of mag-nitude more complex than bacteria or viruses.295
The main obstacle of vaccine development is theisolation of the smallest possible immunogenic sub-stance from multiple proteins and carbohydratesthat safely confers lasting immunity against infec-tion. A second major challenge is the lack of scien-tific knowledge about natural fungal immunity andthe relative contributions of the innate, acquiredhumoral and cellular systems.296,297 Currently, thereis no FDA-approved fungal vaccine for clinical use.
Attenuated low-virulence strains of Candida spphave been used in experimental animal models withsome success.298-300 Other preparations, such as in-activated whole cells of the fungus, secretory andcell surface–located molecules, and major cytoplas-mic and cell wall enzymes, have been used withvarying levels of success.301-304
The challenge in developing a candida vaccine isto purify antigens associated with invasive forms,not colonized forms. In one report, heat-killed wild-type Candida albicans was combined with a noveladjuvant from a Vibrio cholerae enterotoxin to aug-ment the immune response.305 The vaccine wasgiven intranasally to mice, and there was a responseof induced cellular and humoral immunity by anti-body titers and a footpad delayed hypersensitivity inresponse to cutaneous antigen inoculation. Therewas a 90% survival rate among vaccinated mice,compared with 0 among the control animals, whenthey were challenged with a moderate inoculum oflive, virulent organisms.

Wu et al 515J AM ACAD DERMATOL
VOLUME 50, NUMBER 4
In another trial, researchers examined the immu-nogenicity of a specific candida antigen, mannan,which is a carbohydrate associated with the cell wallduring adhesion to host macrophages. Poorly immu-nogenic by itself, when encapsulated within a lipo-some, it is effective. It has a short shelf life andrequires multiple dosing.306 The same researchersconjugated mannan to bovine albumin and with 2doses of vaccine, and survival improved from 0 at 20days in controls to 80% at 65 days in vaccinatedanimals. The vaccine also showed transferable pro-tective immunity to naıve animals.307
A conjugate vaccine of cryptococcal capsular glu-curonoxylomannan covalently linked to tetanus tox-oid confers 70%-80% protection in mice by stimulat-ing both cellular and humoral immunity.308 Passiveimmunization with monoclonal antibodies againstCryptococcus neoformans has been successfully em-ployed in a murine model of disseminated dis-ease.309
A new strategy is the use of vaccines to neutralizefungal virulence factors. Fungal phospholipaseshave been found to be important mediators of fun-gal infection.310 �-Blockers, which have antiphos-pholipase activity, have been used successfully incombination with fluconazole to increase survival inmice with disseminated candidiasis.311
Table VI. Immunoglobulins: indications for administr
Infectious Disease
Cytomegalovirus Cyr
Hepatitis A WiHepatitis B He
pMeasles WiRabies RaRespiratory syncytial virus Pro
dTetanus Sin
tVaccinia FoVaricella Wi
vi
Other uses for intravenous immunoglobulin PriasspBhia
ProtozoaLeishmaniasis (Fig 7). The World Health Orga-
nization considers leishmaniasis to be one of themost significant parasitic diseases, with about 350million people at risk of contracting it. The bestoption in controlling leishmaniasis is considered tobe vaccination.312,313 Numerous antigens have beentested with variable success against cutaneous leish-maniasis in in vitro and mouse models,314 but therecurrently are no commercially available effectivevaccines. The general opinion is that a vaccineagainst leishmaniasis is feasible.314 Vaccines havethe additional advantage of avoiding the problemsof drug resistance, which has been an increasingproblem in the control of leishmaniasis.
Since cured individuals are protected from furtherdisease, vaccine development mainly has been fo-cused on cutaneous leishmaniasis. The develop-ment of a Leishmania vaccine has been hamperedby several factors, including the antigenic diversity,the complex life cycle, the use of experimental an-imal models not representative of human disease,and the lack of sufficient financial support to de-velop a vaccine for the population of third-worldnations.
Recently, the UNDP/World Bank/WHO SpecialProgramme for Research and Training in Tropical
Approved Indication
alovirus prophylaxis in seronegative renal transplantnts of a kidney from a cytomegalovirus-positive donorweeks of exposure in susceptible personsB exposure in susceptible persons, first dose as soon ase and the second within a monthdays of exposure in susceptible personsposure in previously unvaccinated personsxis in high-risk infants (e.g., those with bronchopulmonaryia, prematurity, or chronic lung disease)se after exposure; some recommend an injection aroundof infectionlications of vaccinia vaccinationhours of exposure for susceptible persons exposed to
a who have a high risk for complications (e.g.,ocompromised patients and neonates)
mmunodeficiencies (ataxia telangiectasia,aglobulinemia, common variable immunodeficiency,
combined immunodeficiency, etc.), Guillain-Barreme with ascending paralysis, immune thrombocytopenica, bone marrow transplantation, Kawasaki disease, chronicmphocytic leukemia, and in children with AIDS who aremmaglobulinemic or have had 2 or more serious bacterialns, parvovirus B19 infection, failure to make specificies after immunization, or enterovirus infection
ation
tomegecipiethin 2patitisossibl
thin 6bies exphylaysplasgle dohe siter compthin 96aricell
mmunmary igammevereyndrourpur-cell lyypoga
nfectiontibod

516 Wu et al J AM ACAD DERMATOL
APRIL 2004
Diseases has organized a comparative study of sev-eral leading recombinant antigens to test candidateantigens as potential vaccine candidates.315 Thestudy involved use of recombinant antigens fromLeishmania major, L braziliensis, and L mexicanaexpressed in Escherichia coli, either as a single re-combinant protein or as a mixture. The trial con-sisted of testing the antigens for human peripheralblood mononuclear cell proliferation and conduct-ing challenge experiments in immunized mice.
Experiments of peripheral blood mononuclearcell stimulation showed that most antigens inducedsome interferon-� production and proliferative re-sponses. This finding suggests that the selected an-tigens have some degree of immunogenicity in hu-mans. However, none of the antigens inducedsignificant protection in the mouse challenge exper-iments. Because of these conflicting results, it isdifficult to draw any definitive conclusions. The use-fulness of these antigens as a human vaccine stillremains unknown.
DNA vaccination may be a new approach whereconventional vaccines have failed.316,317 In DNA vac-cination, plasmid DNA containing the gene that en-codes the vaccine candidate is introduced into thetissue through intramuscular injection or particlebombardment. The DNA is taken up by cells andtranslocated to the nucleus, where it is transcribedinto RNA and then translated in the cytoplasm. Al-though the uptake and expression in DNA are ex-tremely low, the antigen is expressed in enoughquantity to induce a specific and potent immuneresponse and to confer future protection.318
The effectiveness of DNA vaccines against infec-tious diseases has been demonstrated in several an-imal models, including a murine model for leish-maniasis in which a GP63-based DNA vaccine wasstudied.319 Vaccination with DNA encoding Leish-mania homologue of receptors for activated C ki-
Fig 7. Cutaneous leishmaniasis in a Brazilian patient.
nase (LACK) conferred protection to mice againstinfection with L major.320 Although the amino acidsequence of the LACK antigen was highly con-served, the efficacy of this vaccine antigen againststrains other than L major could not be shown. DNAvaccination with a L donovani LACK DNA vaccineinduced a strong parasite-specific TH1 immune re-sponse (interferon-� but not interleukin-4 produc-tion) and primed for an in vivo T-cell response toinoculated parasites in one study. However, it didnot induce protection against cutaneous or systemicL donovani challenge in mice.321
DNA vaccination holds enormous promise for thefuture of vaccine development against infectiousagents, especially in developing countries. Such vac-cines are attractive because they ensure correct fold-ing of the antigen, generate appropriate immuneresponses, are simple to produce, and do not re-quire adjuvants, all of which may make them stableand affordable.314 DNA vaccines are also most likelyto confer long-term cellular immunity.322 BeforeDNA vaccines can be studied in clinical trials, it isimportant that safety issues, such as the induction ofautoimmune disease and the integration of the vac-cine DNA in the human genome, be appropriatelyaddressed.323,324
Traditional molecular biologic methods work onone gene at a time. Because of the limited output ofsuch methods, the whole picture of gene function isdifficult to obtain. With the recently developed mi-croarray technology, DNA molecules representingmany genes are placed in thousands of discretespots on a microscope slide. This technique allowsscientists to look at many genes at once and todetermine which ones are expressed in a particularcell type.
In 1994 the Leishmania Genome Network waslaunched in Rio de Janeiro, Brazil, and was aimed atsequencing the genome of the reference strain Leish-mania major Friedlin.325 It is expected that microar-ray technology, together with the Leishmania ge-nome project, will allow the identification of manynew drug and vaccine targets. From initial studies,more than 100 unknown genes were identified andare now being tested as new vaccine candidates.315
IMMUNOGLOBULINSTable VI lists the current immunoglobulins that
are used for infectious diseases. Immunoglobulinsare proteins made by B lymphocytes and plasmacells as part of the humoral portion of the immunesystem. Commercial sterile preparations are madefrom pooled human plasma of several thousanddonors and consist of purified immunoglobulin Galong with small amounts of other globulins. These

Wu et al 517J AM ACAD DERMATOL
VOLUME 50, NUMBER 4
preparations were first used to treat immune defi-ciency diseases in 1952, with the discovery of Bru-ton’s agammaglobulinemia.326 Early preparationswere associated with frequent side effects whenthey were given intravenously and thus requiredfrequent and painful intramuscular administration.In 1981 an improved preparation of immunoglobu-lin was licensed for intravenous use (IVIG). FDA-approved indications include the following 6 condi-tions: primary immunodeficiencies, immune-mediated thrombocytopenia, Kawasaki disease,recent bone marrow transplantation (in persons atleast 20 years of age), chronic B-cell lymphocyticleukemia, and pediatric HIV-1 infection.327 IVIG isalso used in clinical practice for numerous otherconditions, such as multiple sclerosis. Intravenouspreparations are manufactured by several differentcompanies, and owing to differences in the produc-tion process and donor populations, the productsavailable may vary considerably.328
The intramuscular form of immunoglobulin is stillavailable and is approved for the prophylaxis ofhepatitis A and measles. Several immunoglobulinpreparations with high titers to individual viruses arealso available for the prophylaxis of specific viralinfections. These hyperimmune globulins are avail-able for rabies, varicella, cytomegalovirus, respira-tory syncytial virus, and hepatitis B.
IVIG has also been shown to be of potential benefitfor HIV-infected children. This population frequentlysuffers from bacterial infections with common encap-sulated bacteria, whereas infected adults more fre-quently develop opportunistic infections.329 Severalsmall studies with IVIG have shown decreased bacte-rial infections and sepsis, as well as improved survivalin HIV-positive children.330-332 Another study of HIV-infected children who were undergoing treatment withzidovudine showed a benefit with the use of IVIG, butonly in those subjects not receiving trimethoprim-sul-famethoxazole as antibiotic prophylaxis.333 The exactrole of IVIG in this situation remains unclear.
Intramuscular immunoglobulin can be given forthe prevention of measles in susceptible individuals(those with no previous infection or immunization).Its use is indicated for exposed persons with anincreased risk of complications from disease, such asimmunocompromised patients or children less than1 year old. This treatment should be given within 6days of exposure. The second indication for intra-muscular immunoglobulin is the prophylaxis of hep-atitis A. The protective effect is of most value if givenprior to or immediately after exposure, and at leastwithin 2 weeks of exposure. If given within severaldays of exposure, the immunoglobulin prevents in-fection in 80% to 95% of patients.334,335 This treat-
ment was also indicated for travelers who plan tostay in areas with poor sanitation, but the hepatitis Avaccine is now the preferred method for hepatitis Aprevention in travelers.336
Hepatitis B immune globulin is given with hepa-titis B vaccine in certain situations as part of therecommended postexposure prophylaxis regimen.The following susceptible persons who were ex-posed to hepatitis B virus should receive hepatitis Bhyperimmune globulin in addition to immunization:persons with acute exposure to HbsAg-positiveblood, infants with perinatal exposure to HBsAg-positive mothers, persons with sexual exposure toHBsAg-positive partners, and infants with anHBsAg-positive primary caregiver. Hepatitis hyper-immune globulin has been evaluated for the pro-phylactic treatment of HBsAg-positive liver trans-plant recipients. High-dose, long-term treatment hasled to increased survival and decreased serologicrecurrence in a number of studies.337,338 However,maintenance treatment is required for the preven-tion of recurrence, and long-term treatment is ex-pensive.339
Varicella-zoster immune globulin should be ad-ministered to susceptible persons exposed to vari-cella virus who have an increased risk for complica-tions (i.e., immunocompromised persons). Thepostexposure prophylaxis should be administeredas soon as possible and no later than 96 hours afterexposure.340 The protection lasts at least 3 weeksafter the injection.
Adverse reactions to any of the immunoglobulinpreparations are rare. The incidence of systemic sideeffects is generally less than 5%, and these reactionsare typically mild and self-limited. Fever, chills,headache, backache, nausea, vomiting, chest tight-ness, myalgias, and dyspnea have all been reported.With intravenous preparations, slowing of the infu-sion rate can be of benefit in alleviating the sideeffects. Hydrocortisone and antihistamines are alsouseful. Anaphylactic reactions may also occur inimmunoglobulin A–deficient patients receivingIVIG, but this complication is rare.341 From 1985 to1998, acute renal failure has been described in 120IVIG recipients worldwide.327 Acute renal failure ap-pears most closely associated with IVIG prepara-tions with high sucrose content. The majority ofaffected patients developed renal failure within 7days of IVIG administration, and 40% required dial-ysis owing to the degree of failure. Although thiscomplication remains infrequent, it is associatedwith significant morbidity and mortality. Other un-common adverse effects include encephalopathyand thromboembolism.

518 Wu et al J AM ACAD DERMATOL
APRIL 2004
Because these biologic products are derived fromhuman plasma, viral contamination poses a poten-tial, though small, risk. In 1994 the association of 2different IVIG preparations with hepatitis C contam-ination led to numerous cases of acute hepatitis Cinfection (more than 100 cases in the United States).The manufacturers added a solvent-detergent treat-ment for viral inactivation, and users of the productsno longer are considered to be at risk for hepatitisC.342 All current IVIG preparations come fromdonors who are screened for hepatitis B and C,HIV, and elevated levels of liver enzymes. Also,the majority of manufacturers include a viral inac-tivation step in the production process. No casesof HIV transmission have been reported withIVIG.
CONCLUSIONIt is clear that vaccination is one of the greatest
achievements in the history of mankind. Immuniza-tion should be used in conjunction with publichealth measures such as sanitation, blood productscreening, safe sex techniques, vector control, di-etary education, and exercise promotion. Althoughimmunization is considered widely as a major suc-cess, there are the public health challenges of im-plementing vaccine programs and allaying un-founded fears of the public about immunization sideeffects. Vaccine-preventable infectious diseases stillremain major causes of morbidity and mortality inthe world. Many children in the world still die frommeasles, tetanus, and pertussis,343 and influenza-related pneumonia and sepsis are among the top 10causes of death in the United States and in theworld.344,345 Despite the data that show efficacy ofvaccinations, there is a small movement of antivac-cination activists who may be partly contributing tothe lower rates of adult immunization.346,347
The implementation of routine immunizationsnot only has a significant impact on the overallincidence of disease but also markedly decreases thedirect and indirect costs associated with health care.For instance, a 1994 study of the cost-effectivenessof a varicella vaccination program in the UnitedStates showed savings of $384 million dollars peryear.348 The cost reduction with varicella is mostlydue to a decrease in time lost from work by care-givers, although this savings is significant. Vaccinesfor more serious diseases that often require hospi-talization, such as respiratory syncytial virus in in-fants, will likely result in a more beneficial cost-effective profile. Historically, the cost savings of theeradication of smallpox, a disease that killed mil-lions of people, approaches infinity when the mil-lions more that would have been affected are con-
sidered. With recent current events, the use ofweapons of mass destruction, specifically smallpoxand anthrax, is now a realistic and potential biologicthreat. A smallpox vaccination program has nowbeen initiated by the United States in preparation forthe possibility of terrorist attacks and in protectingpersons in the front lines of a biologic attack. Theseindividuals include military personnel, medical pro-fessionals, and emergency personnel and responseteams who would be first on the scene in a smallpoxemergency. A similar situation exists for poliomyeli-tis, which is expected to be eradicated worldwide inthe near future. The cost savings for an HIV vaccinewould also be phenomenal, considering the long-term treatment and numerous complications in-volved with this chronic infection.
Immunization has successfully led to the reduc-tion in incidence of numerous diseases. Careful de-velopment and clinical evaluation have providedsafe and effective vaccines with few adverse effects.Many reported adverse reactions after vaccinationmay be coincidental and have no proven directrelationship with the vaccine in question. Althoughserious side effects may rarely occur from vaccines,a much greater risk for morbidity and mortality re-sults from the failure to become immunized. Onevaccine, however, was recently removed from themarket owing to safety issues. Rotashield was a live,oral, tetravalent rotavirus vaccine that was associ-ated with several cases of intussusception and isconsidered to be causal.349 Most associations be-tween vaccines and adverse events are not, how-ever, shown to be causal. For example, the MMRvaccine was reported recently not to have a causalrelationship to autism.350-352 Likewise, a causal rela-tionship between the hepatitis B vaccine and a va-riety of autoimmune diseases has been disproved.This vaccine does not increase the risk of multiplesclerosis353 nor does it cause a relapse of preexistingmultiple sclerosis.354 A claim that the rising rates ofdiabetes mellitus in children may be due to in-creased exposure to vaccination has been rigor-ously refuted.355,356 Nevertheless, suspected rela-tionships between vaccines and adverse eventsneed to be reported to the Vaccine Adverse EventReporting System (800-822-7967) so that the ex-cellent safety record of vaccines can be main-tained.
The technology of vaccine development has pro-gressed dramatically in the last decade. While moreconventional methods have consisted of whole-killed or live attenuated viruses, more recent ad-vances include genetically engineered vectors andVLP, among many others. Anticipated vaccine de-velopments in the future show exciting promise in

Wu et al 519J AM ACAD DERMATOL
VOLUME 50, NUMBER 4
several areas, such as immunization with plants.Potatoes, tomatoes, and bananas are currently un-dergoing genetic engineering to express immuniz-ing antigens against infections such as hepatitis Bvirus and Norwalk virus.357,358 This form of vaccina-tion would offer a convenient, painless, and inex-pensive approach to widespread control of diseaseand thus would be accessible to developing coun-tries.
It is anticipated that the future will bring safe andeffective vaccines for a variety of infectious diseases.Although few vaccines are available for the therapyof infectious diseases, our concept of vaccines isnow being expanded with ongoing clinical trials oftherapeutic vaccines.
REFERENCES1. CDC. Ten great public health achievements: United States,
1900-1999. MMWR Morb Mortal Wkly Rep 1999;48:241-3.2. CDC. Achievements in public health, 1900-1999: impact of vac-
cines universally recommended for children—United States,1990-1998. MMWR Morb Mortal Wkly Rep 1999;48:243-8.
3. Bonanni P. Demographic impact of vaccination: a review. Vac-cine 1999;17(Suppl 3):S120-5.
4. Wright PF, Kim-Farley RJ, de Quadros CA, Robertson SE. Strat-egies for the global eradication of poliomyelitis by the year2000. N Engl J Med 1991;325:1774-9.
5. Brundtland GH. State of the world’s vaccines and immuniza-tions. JAMA 2002;288:2532.
6. CDC. Measles update: measles activity through week 52 (end-ing Dec 28, 2002). National Immunization Program 2003;3:1-4.
7. Heyward W, MacQueen K, Goldenthal K. HIV vaccine develop-ment and evaluation: realistic expectations. AIDS Res Hum Ret-roviruses 1998;14(Suppl 3):S205-10.
8. Henderson DA, Moss B. Smallpox and vaccinia. In: Plotkin SA,Orenstein WA, editors. Vaccines. 3rd ed. Philadelphia: WBSaunders; 1999. p. 74-97.
9. Is smallpox history? Lancet 1999;353:1539.10. LeDuc JW, Becher J. Current status of smallpox vaccine. Emerg
Infect Dis 1999;5:593-4.11. Roizman B, Joklik W, Fields B, Moss B. The destruction of small-
pox virus stocks in national repositories: a grave mistake and abad precedent. Infect Agents Dis 1994;3:215-7.
12. Joklik W. The remaining smallpox virus stocks are too valuableto be destroyed. Scientist 1996;10:11.
13. Breman JG, Henderson DA. Poxvirus dilemmas: monkeypox,smallpox, and biologic terrorism. N Engl J Med 1998;339:556-9.
14. Smallpox eradication: destruction of variola virus stocks. Paperpresented at: World Health Assembly, May 24, 1999.
15. Fenner F, Henderson DA, Arita I, Jezek Z, Ladnyi ID. Smallpoxand its eradication. Geneva (Switzerland): World Health Orga-nization; 1988.
16. Lane JM, Goldstein J. Evaluation of 21st-century risks of small-pox vaccination and policy options. Ann Intern Med 2003;138:488-93.
17. CDC. Smallpox response plan and guidelines (version 3.0). At-lanta (GA): U.S. Department of Health and Human Services;2002.
18. Bicknell W, James K. The new cell culture smallpox vaccineshould be offered to the general population. Rev Med Virol2003;13:5-15.
19. Arita I. Duration of immunity after smallpox vaccination: astudy on vaccination policy against smallpox bioterrorism inJapan. Jpn J Infect Dis 2002;55:112-6.
20. Breman J, Henderson D. Diagnosis and management of small-pox. N Engl J Med 2002;346:1300-8.
21. CDC. Update: cardiac-related events during the civilian small-pox vaccination program—United States, 2003. MMWR MorbMortal Wkly Rep 2003;52:492-6.
22. Cono J, Casey CG, Bell DM, CDC. Smallpox vaccination and ad-verse reactions. Guidance for clinicians. MMWR Recomm Rep2003;52:1-28.
23. Meadows KP, Tyring SK, Pavia AT, Rallis TM. Resolution of recal-citrant molluscum contagiosum virus lesions in human immu-nodeficiency virus–infected patients treated with cidofovir.Arch Dermatol 1997;133:987-90.
24. Wharton M, Cochi S, Williams W. Measles, mumps, and rubellavaccines. Infect Dis Clin North Am 1990;4:47-73.
25. CDC. Measles, mumps, and rubella: vaccine use and strategiesfor elimination of measles, rubella, and congenital rubella syn-drome and control of mumps—recommendations of the Ad-visory Committee on Immunization Practices (ACIP). MMWRMorb Mortal Wkly Rep 1998;47:1-57.
26. Preblud SR. Some issues relating to rubella vaccine. JAMA1985;254:253-6.
27. James JM, Burks AW, Roberson PK, Sampson HA. Safe adminis-tration of the measles vaccine to children allergic to eggs.N Engl J Med 1995;332:1262-6.
28. Gellin BG, Katz SL. Putting a stop to a serial killer: measles. J In-fect Dis 1994;170(Suppl 1):S1-2.
29. Hope-Simpson RE. Infectiousness of communicable diseasesin the household (measles, chicken pox, and mumps). Lancet1952;2:549-54.
30. CDC. Measles eradication: recommendations from a meetingcosponsored by the World Health Organization, the Pan Amer-ican Health Organization, and CDC. MMWR Morb Mortal WklyRep 1997;46:1-20.
31. CDC. Measles: United States, 1999. MMWR Morb Mortal WklyRep 2000;49:557-60.
32. CDC. Measles: United States, 2000. MMWR Morb Mortal WklyRep 2002;51:120-3.
33. CDC. Measles: United States, 2002. MMWR Morb Mortal WklyRep 2003;50:i-xxiv, 1-136.
34. Murray C, Lopez A. Mortality by cause for eight regions of theworld: global burden of disease study. Lancet 1997;349:1269-76.
35. Krause PJ, Cherry JD, Carney JM, Nalditch MJ, O’Connor K. Mea-sles-specific lymphocyte reactivity and serum antibody in sub-jects with different measles histories. Am J Dis Child 1980;134:567-71.
36. Watson JC, Pearson JA, Markowitz LE, Baughman AL, ErdmanDD, Bellini WJ, et al. An evaluation of measles revaccinationamong school-entry-aged children. Pediatrics 1996;97:613-8.
37. Davis RM, Whitman ED, Orenstein WA, Preblud SR, MarkowitzLE, Hinman AR. A persistent outbreak of measles despite ap-propriate control measures. Am J Epidemiol 1987;126:438-49.
38. Krugman S. Further-attenuated measles vaccine: characteris-tics and use. Rev Infect Dis 1983;5:477-81.
39. Mathias RG, Meekison WG, Arcand TA, Schechter MT. The roleof secondary vaccine failures in measles outbreaks. Am J PublicHealth 1989;79:475-8.
40. Reyes MA, de Borrero MF, Roa J, Bergonzoli G, Saravia NG. Mea-sles vaccine failure after documented seroconversion. PediatrInfect Dis J 1987;6:848-51.
41. Pelota H, Heinonen OP. Frequency of true adverse reactions tomeasles-mumps-rubella vaccine. A double-blind placebo-con-trolled trial in twins. Lancet 1986;1:939-42.

520 Wu et al J AM ACAD DERMATOL
APRIL 2004
42. CDC. Measles: United States, 1988. MMWR Morb Mortal WklyRep 1989;38:601-5.
43. Modlin JF, Jabbour JT, Witte JJ, Halsey NA. Epidemiologic stud-ies of measles, measles vaccine, and subacute sclerosing pan-encephalitis. Pediatrics 1977;59:505-12.
44. CDC. Subacute sclerosing panencephalitis surveillance: UnitedStates. MMWR Morb Mortal Wkly Rep 1982;31:585-8.
45. Dyken PR. Subacute sclerosing panencephalitis, current status.Neurol Clin 1985;3:179-96.
46. Hilleman MR, Buynak EB, Weibel RE, Stokes J. Live, attenuatedmumps vaccine. N Engl J Med 1968;278:227-32.
47. Weibel RE, Stokes J Jr, Buynak EB, Whitman JEJ, Hilleman MR.Live attenuated mumps: virus vaccine. 3. Clinical and serologicaspects in a field evaluation. N Engl J Med 1967;276:245-51.
48. Sugg WC, Finger JA, Levine RH, Pagano JS. Field evaluation oflive virus mumps vaccine. J Pediatr 1968;72:461-6.
49. Chaiken BP, Williams NM, Preblud SR, Parkin W, Altman R. Theeffect of a school entry law on mumps activity in a school dis-trict. JAMA 1987;257:2455-8.
50. Kim-Farley R, Bart S, Stetler H, Orenstein WA, Bart K, SullivanKM, et al. Clinical mumps vaccine efficacy. Am J Epidemiol1985;121:593-7.
51. Sullivan KM, Halpin TJ, Marks JS, Kim-Farley R. Effectiveness ofmumps vaccine in a school outbreak. Am J Dis Child 1985;139:909-12.
52. Wharton M, Cochi SL, Hutcheson RH, Bitowish JM, Schaffner W.A large outbreak of mumps in the post vaccine era. J Infect Dis1988;158:1253-60.
53. Hersh BS, Fine PE, Kent WK, Cochi SL, Kahn LH, Zell ER, et al.Mumps outbreak in a highly vaccinated population. J Pediatr1991;119:187-93.
54. CDC. Mumps surveillance: United States, 1988-1993. MMWRMorb Mortal Wkly Rep 1995;44(S-3):1-14.
55. Weibel RE, Buynak EB, McLean AA, Hilleman MR. Persistence ofantibody after administration of monovalent and combinedlive attenuated measles, mumps, and rubella virus vaccines.Pediatrics 1978;61:5-11.
56. Chang TW, DesRosiers S, Weinstein L. Clinical and serologicstudies of an outbreak of rubella in a vaccinated population.N Engl J Med 1970;283:246-8.
57. Bakshi SS, Cooper LZ. Rubella and mumps vaccines. PediatrClin North Am 1990;37:651-68.
58. Plotkin SA, Farquhar JD, Ogra PL. Immunologic properties ofRA 27-3 rubella virus vaccine. A comparison with strains pres-ently licensed in the United States. JAMA 1973;225:585-90.
59. Gershon AA, Frey HM, Borkowsky W, Steinberg S. Live attenu-ated rubella virus vaccine: comparison of responses to HPV-77-DE5 and RA 27/3 strains. Am J Med Sci 1980;279:95-7.
60. Hilleman M, Weibel R, Buynak E, Stokes JJ, Whitman JJ. Liveattenuated mumps-virus vaccine: 4. Protective efficacy asmeasured in a field evaluation. N Engl J Med 1967;276:252-8.
61. Chu SY, Bernier RH, Stewart JA, Herrmann KL, Greenspan JR,Henderson AK, et al. Rubella antibody persistence after immu-nization: sixteen-year follow-up in the Hawaiian islands. JAMA1988;259:3133-6.
62. O’Shea S, Best JM, Banatvala JE, Marshall WC, Dudgeon JA. Ru-bella vaccination: persistence of antibodies for up to 16 years.Br Med J (Clin Res Ed) 1982;285:253-5.
63. Kimberlin DW. Rubella immunization. Pediatr Ann1997;26:366-70.
64. Freestone DS, Prydie J, Smith SG, Laurence G. Vaccination ofadults with Wistar RA 27/3 rubella vaccine. J Hyg (Lond) 1971;69:471-7.
65. Polk BF, Modlin JF, White JA, DeGirolami PC. A controlled com-parison of joint reactions among women receiving one of tworubella vaccines. Am J Epidemiol 1982;115:19-25.
66. Preblud S, Serdula M, Frank JJ, Brandling-Bennet A, Hinman A.Rubella vaccination in the United States: a ten-year review. Epi-demiol Rev 1980;2:171-94.
67. Balcarek KB, Bagley MR, Pass RF, Schiff ER, Krause DS. Safetyand immunogenicity of an inactivated hepatitis A vaccine inpreschool children. J Infect Dis 1995;171(Suppl 1):S70-2.
68. Clemens R, Safary A, Hepburn A, Roche C, Stanbury WJ, AndreFE. Clinical experience with an inactivated hepatitis A vaccine.J Infect Dis 1995;171(Suppl 1):S44-9.
69. Horng YC, Chang MH, Lee CY, Safary A, Andre FE, Chen DS.Safety and immunogenicity of hepatitis A vaccine in healthychildren. Pediatr Infect Dis J 1993;12:359-62.
70. Westblom TU, Gudipati S, DeRousse C, Midkiff BR, Belshe RB.Safety and immunogenicity of an inactivated hepatitis A vac-cine: effect of dose and vaccination schedule. J Infect Dis 1994;169:996-1001.
71. Block SL, Hedrick JA, Tyler RD, Smith RA, Calandra G, PattersonC, et al. Safety, tolerability and immunogenicity of a formalin-inactivated hepatitis A vaccine (VAQTA) in rural Kentucky chil-dren. Pediatr Infect Dis J 1993;12:976-80.
72. Werzberger A, Mensch B, Kuter B, Brown L, Lewis J, Sitrin R, et al.A controlled trial of a formalin-inactivated hepatitis A vaccinein healthy children. N Engl J Med 1992;327:453-7.
73. Innis BL, Snitbhan R, Kunasol P, Laorakpongse T, Poopatanak-ool W, Kozik CA, et al. Protection against hepatitis A by an in-activated vaccine. JAMA 1994;271:1328-34.
74. CDC. Prevention of hepatitis A through active or passive immu-nization: recommendations of the Advisory Committee on Im-munization Practices (ACIP). MMWR Morb Mortal Wkly Rep1996;45:1-30.
75. Wiens BL, Bohidar NR, Pigeon JG, Egan J, Hurni W, Brown L, etal. Duration of protection from clinical hepatitis A disease aftervaccination with VAQTA. J Med Virol 1996;49:235-41.
76. Damme PV, Thoelen S, Cramm M, Groote KD, Safary A, MeheusA. Inactivated hepatitis A vaccine: reactogenicity, immunoge-nicity, and long-term antibody persistence. J Med Virol 1994;44:446-51.
77. Wiedermann G, Kundi M, Ambrosch F, Safary A, D’Hondt E,Delem A. Inactivated hepatitis A vaccine: long term antibodypersistence. Vaccine 1997;15:612-5.
78. Nalin DR, Kuter BJ, Brown L, Patterson C, Calandra GB, Werz-berger A, et al. Worldwide experience with the CR326F-derivedinactivated hepatitis A virus vaccine in pediatric and adult pop-ulations: an overview. J Hepatol 1993;18(Suppl 2):S51-5.
79. Armstrong GL, Mast EE, Wojczynski M, Margolis HS. Childhoodhepatitis B virus infections in the United States before hepatitisB immunization. Pediatrics 2001;108:1123-8.
80. Douglas RG. The heritage of hepatitis B vaccine. JAMA 1996;276:1796-8.
81. Francis DP, Hadler SC, Thompson SE, Maynard JE, Ostrow DG,Altman N, et al. The prevention of hepatitis B with vaccine.Report of the Centers for Disease Control multicenter efficacytrial among homosexual men. Ann Intern Med 1982;97:362-6.
82. Szmuness W, Stevens CE, Harley EJ, Zang EA, Oleszko WR, Wil-liam DC, et al. Hepatitis B vaccine: demonstration of efficacy ina controlled trial in a high-risk population in the United States.N Engl J Med 1980;303:833-41.
83. Katkov WN, Dienstag JL. Hepatitis vaccines. Gastroenterol ClinNorth Am 1995;24:147-59.
84. Denis F, Mounier M, Hessel L, Michel JP, Gualde N, Dubois F, etal. Hepatitis B vaccination in the elderly. J Infect Dis 1984;149:1019.
85. Heyward WL, Bender TR, McMahon BJ, Hall DB, Francis DP,Lanier AP, et al. The control of hepatitis B virus infection withvaccine in Yupik Eskimos. Demonstration of safety, immuno-

Wu et al 521J AM ACAD DERMATOL
VOLUME 50, NUMBER 4
genicity, and efficacy under field conditions. Am J Epidemiol1985;121:914-23.
86. Roome AJ, Walsh SJ, Cartter ML, Hadler JL. Hepatitis B vaccineresponsiveness in Connecticut public safety personnel. JAMA1993;270:2931-4.
87. Wood RC, MacDonald KL, White KE, Hedberg CW, Hanson M,Osterholm MT. Risk factors for lack of detectable antibody fol-lowing hepatitis B vaccination of Minnesota health care work-ers. JAMA 1993;270:2935-9.
88. Zimmerman RK, Ruben FL, Ahwesh ER. Hepatitis B virus infec-tion, hepatitis B vaccine, and hepatitis B immune globulin. JFam Pract 1997;45:295-315.
89. Thoelen S, Damme PV, Mathei C, Leroux-Roels G, Desombere I,Safary A, et al. Safety and immunogenicity of a hepatitis B vac-cine formulated with a novel adjuvant system. Vaccine 1998;16:708-14.
90. Heineman TC, Clements-Mann NL, Poland GA, Jacobson RM,Izu AE, Sakamoto D, et al. A randomized, controlled study inadults of the immunogenicity of a novel hepatitis B vaccinecontaining MF59 adjuvant. Vaccine 1999;17:2769-78.
91. Jilg W. Novel hepatitis B vaccines. Vaccine 1998;16(suppl):S65-8.
92. Hourvitz A, Mosseri R, Solomon A, Yehezkelli Y, Atsmon J,Danon YL, et al. Reactogenicity and immunogenicity of a newrecombinant hepatitis B vaccine containing Pre S antigens: apreliminary report. J Viral Hepat 1996;3:37-42.
93. Yap I, Guan R, Chan SH. Recombinant DNA hepatitis B vaccinecontaining Pre-S components of the HBV coat protein: a pre-liminary study on immunogenicity. Vaccine 1992;10:439-42.
94. Bertino JS, Tirrell P, Greenberg RN, Keyserling HL, Poland GA,Gump D, et al. A comparative trial of standard or high-dose Ssubunit recombinant hepatitis B vaccine versus a vaccine con-taining S subunit, Pre-S1, and Pre-S2 particles for revaccinationof healthy adult nonresponders. J Infect Dis 1997;175:678-81.
95. Jilg W, Schmidt M, Deinhardt F. Persistence of specific antibod-ies after hepatitis B vaccination. J Hepatol 1988;6:201-7.
96. Stevens CE, Taylor FE, Tong MJ, et al. Hepatitis B vaccine: anoverview. In: Vyas GN, Dienstag JL, Hoofnagle JH, editors. Viralhepatitis and liver disease. Orlando: Grune & Stratton; 1984. p.275-91.
97. Hadler S, Francis D, Maynard J, Thompson S, Judson F, Echen-berg D, et al. Long-term immunogenicity and efficacy of hep-atitis B vaccine in homosexual men. N Engl J Med 1986;315:209-14.
98. Stevens C, Toy P, Taylor P, Lee T, Yip H. Prospects for control ofhepatitis B virus infection: implications of childhood vaccina-tion and long-term protection. Pediatrics 1992;90:170-3.
99. CDC. Notice to readers: availability of hepatitis B vaccine thatdoes not contain thimerosal as a preservative. MMWR MorbMortal Wkly Rep 1999;48:780-2.
100. Andre F, Safary A. Summary of clinical findings on Engerix-B, agenetically engineered yeast derived hepatitis B vaccine. Post-grad Med J 1987;63(Suppl 2):169-78.
101. CDC. Update: vaccine side effects, adverse reactions, contrain-dications and precautions—recommendations of the Advi-sory Committee on Immunization Practices (ACIP). MMWRMorb Mortal Wkly Rep 1996;45:1-35.
102. Andre FE. Overview of 5-year clinical experience with a yeast-derived hepatitis B vaccine. Vaccine 1990;8(suppl):74-8.
103. Neau D, Bonnet F, Michaud M, Perel Y, Longy-Boursier M, Rag-naud JM, et al. Immune thrombocytopenic purpura after re-combinant hepatitis B vaccine: retrospective study of sevencases. Scand J Infect Dis 1998;30:115-8.
104. Ronchi F, Cecchi P, Falcioni F, Marsciani A, Minak G, Muratori G,et al. Thrombocytopenic purpura as adverse reaction to re-combinant hepatitis B vaccine. Arch Dis Child 1998;78:273-4.
105. Poullin P, Gabriel B. Thrombocytopenic purpura after recombi-nant hepatitis B vaccine. Lancet 1994;334:1293.
106. Hello CL, Cohen P, Bousser MG, Letellier P, Guillevin L. Sus-pected hepatitis B vaccination related vasculitis. J Rheumatol1999;26:191-4.
107. Allen MB, Cockwell P, Page RL. Pulmonary and cutaneous vas-culitis following hepatitis B vaccination. Thorax 1993;48:580-1.
108. Pope JE, Stevens A, Howson W, Bell DA. The development ofrheumatoid arthritis after recombinant hepatitis B vaccination.J Rheumatol 1998;25:1687-93.
109. Rebora A, Rongioletti F, Drago F, Parodi A. Lichen planus as aside effect of HBV vaccination. Dermatology 1999;198:1-2.
110. Saywell CA, Wittal RA, Kossard S. Lichenoid reaction to hepati-tis B vaccination. Australas J Dermatol 1997;38:152-4.
111. Chen DS. Control of hepatitis B in Asia: mass immunizationprogram in Taiwan. In: Hollinger FB, Lemon SM, Margolis HS,editors. Viral hepatitis and liver disease. Baltimore: Williams &Wilkins; 1991. p. 716-9.
112. Yeh SH, Ward JI, Partridge S, Marcy SM, Lee H, Jing J, et al. Safetyimmunogenicity of a pentavalent diphtheria, tetanus, pertus-sis, hepatitis B and polio combination vaccine in infants. Pedi-atr Infect Dis J 2001;20:973-80.
113. Partridge S, Yeh SH. Clinical evaluation of a DTaP-HepB-IPVcombined vaccine. Am J Manag Care 2003;9:S13-22.
114. Pediarix [Diphtheria and Tetanus Toxoids and Acellular Pertus-sis Adsorbed, Hepatitis B (Recombinant) and Inactivates Polio-virus Vaccine Combined] prescribing information. Rixensart[Belgium]: SmithKline Beecham Biologicals; 2002.
115. CDC. FDA licensure of diphtheria and tetanus toxoids and acel-lular pertussis adsorbed, hepatitis B (recombinant), and polio-virus vaccine combined (Pediarix) for use in infants. MMWRMorb Mortal Wkly Rep 2003;52:203-4.
116. Zepp F, Schuind A, Meyer C, Sanger R, Kaufhold A, Willems P.Safety and reactogenicity of a novel DTPa-HBV-IPV combinedvaccine given along with commercial Hib vaccines in compar-ison with separate concomitant administration of DTPa, Hib,and OPV vaccines in infants. Pediatrics 2002;109:e58.
117. CDC. FDA licensure of diphtheria and tetanus toxoids and acel-lular pertussis adsorbed, hepatitis B (recombinant), and polio-virus vaccine combined (Pediarix) for use in infants. Erratum in:MMWR Morb Mortal Wkly Rep 2003;52:379.
118. CDC. Prevention of varicella: updated recommendations of theAdvisory Committee on Immunization Practices (ACIP). MMWRMorb Mortal Wkly Rep 1999;48:1-5.
119. Arvin AM. Varicella vaccine: the first six years. N Engl J Med2001;344:1007-9.
120. Vazquez M, LaRussa PS, Gershon AA, Steinberg SO, Freudig-man K, Shapiro ED. The effectiveness of the varicella vaccine inclinical practice. N Engl J Med 2001;344:955-60.
121. Wise RP, Salive ME, Braun MM, Mootrey GT, Seward JF, Rider LG,et al. Postlicensure safety surveillance for varicella vaccine.JAMA 2000;284:1271-9.
122. White C. Varicella-zoster virus vaccine. Clin Infect Dis 1997;24:753-63.
123. Takahashi M. Clinical overview of varicella vaccine: develop-ment and early studies. Pediatrics 1986;78:736-41.
124. Asano Y, Suga S, Yoshikawa T, Kobayashi I, Yazaki T, Shibata M,et al. Experience and reason: twenty-year follow-up of protec-tive immunity of the Oka strain live varicella vaccine. Pediatrics1994;94:524-6.
125. Weibel R, Neff B, Kuter B, Guess H, Rothenberger C, FitzgeraldA, et al. Live attenuated varicella virus vaccine. Efficacy trial inhealthy children. N Engl J Med 1984;310:1409-15.
126. Kuter B, Weibel R, Guess H, Matthews H, Morton D, Neff B, et al.

522 Wu et al J AM ACAD DERMATOL
APRIL 2004
Oka/Merck varicella vaccine in healthy children: final report ofa 2-year efficacy study and 7-year follow-up studies. Vaccine1991;9:643-7.
127. Watson BM, Keller PM, Ellis RW, Starr SE. Cell-mediated im-mune responses after immunization of healthy seronegativechildren with varicella vaccine: kinetics and specificity. J InfectDis 1990;162:794-9.
128. White C, Kuter B, Hildebrand C, Isganitis K, Matthews H, MillerW, et al. Varicella vaccine (Varivax) in healthy children and ad-olescents: results from clinical trials, 1987 to 1989. Pediatrics1991;87:604-10.
129. Watson B, Boardman C, Laufer D, Piercy S, Tustin N, Olaleye D,et al. Humoral and cell-mediated immune responses in healthychildren after one or two doses of varicella vaccine. Clin InfectDis 1995;20:316-9.
130. Watson B, Gupta R, Randall T, Starr S. Persistence of cell-medi-ated and humoral immune responses in healthy children im-munized with live attenuated varicella vaccine. J Infect Dis1994;169:197-9.
131. Asano Y, Nagai T, Miyata T, Yazaki T, Ito S, Yamanishi K, et al.Long-term protective immunity of recipients of the OKA strainof live varicella vaccine. Pediatrics 1985;75:667-71.
132. White C, Kuter B, Ngai A, Hildebrand C, Isganitis K, Patterson C,et al. Modified cases of chickenpox after varicella vaccination:correlation of protection with antibody response. Pediatr In-fect Dis J 1992;11:19-23.
133. Watson B, Piercy S, Plotkin S, Starr S. Modified chickenpox inchildren immunized with the Oka/Merck varicella vaccine. Pe-diatrics 1993;91:17-22.
134. Watson B, Seward J, Yang A, Witte P, Lutz J, Chan C, et al. Post-exposure effectiveness of varicella vaccine. Pediatrics2000;105:84-8.
135. Kuter BJ, Ngai A, Patterson CM, Staehle BO, Cho I, Matthews H,et al. Safety, tolerability, and immunogenicity of two regimensof Oka/Merck varicella vaccine (Varivax) in healthy adolescentsand adults: Oka/Merck Varicella Vaccine Study Group. Vaccine1995;13:967-72.
136. Hardy I, Gershon AA, Steinberg SP, LaRussa P. The incidenceof zoster after immunization with live attenuated varicellavaccine. A study in children with leukemia. The VaricellaVaccine Collaborative Study Group. N Engl J Med 1991;325:1545-50.
137. Heller L, Berglund G, Ahstrom L, Hellstrand K, Wahren B. Earlyresults of a trial of the Oka-strain varicella vaccine in childrenwith leukaemia or other malignancies in Sweden. PostgradMed J 1985;61(Suppl 4):79-83.
138. Gershon A, Steinberg S, Gelb L, Galasso G, Borkowsky W, La-Russa P, et al. Live attenuated varicella vaccine: efficacy for chil-dren with leukemia in remission. JAMA 1984;252:355-62.
139. Tsolia M, Gershon AA, Steinberg SP, Gelb L. Live attenuatedvaricella vaccine: evidence that the virus is attenuated and theimportance of skin lesions in transmission of varicella-zostervirus. National Institute of Allergy and Infectious Diseases Vari-cella Vaccine Collaborative Study Group. J Pediatr 1990;116:184-9.
140. Hata A, Asanuma H, Rinki M, Sharp M, Wong RM, K KB, et al. Useof an inactivated varicella vaccine in recipients of hematopoi-etic-cell transplants. N Engl J Med 2002;347:26-34.
141. Miller A. Selective decline in cellular immune response to vari-cella-zoster in the elderly. Neurology 1980;30:582-7.
142. Burke BL, Steele RW, Beard OW. Immune responses to varicella-zoster in the aged. Arch Intern Med 1982;142:291-3.
143. Hayward A, Levin M, Wolf W, Angelova G. Varicella-zoster virus-specific immunity after herpes zoster. J Infect Dis 1991;163:873-5.
144. Levin M, Murray M, Zerbe G, White C, Hayward A. Immune re-sponses of elderly persons 4 years after receiving a live atten-uated varicella vaccine. J Infect Dis 1994;170:522-6.
145. Levin MJ, Barber D, Goldblatt E, Jones M, LaFleur B, Chan C, etal. Use of a live attenuated varicella vaccine to boost varicella-specific immune responses in seropositive people 55 years ofage and older: duration of booster effect. J Infect Dis 1998;178(Suppl 1):S109-12.
146. Levin JM. Use of varicella vaccines to prevent herpes zoster inolder individuals. Arch Virol Suppl 2001;39:151-60.
147. Clements DA, Armstrong CB, Ursano AM, Moggio MM, WalterEB, Wilfert CM. Over five-year follow-up of Oka/Merck varicellavaccine recipients in 465 infants and adolescents. Pediatr In-fect Dis J 1995;14:874-9.
148. Bernstein HH, Rothstein EP, Watson BM, Reisinger KS, BlatterMM, Wellman CO, et al. Clinical survey of natural varicella com-pared with breakthrough varicella after immunization with liveattenuated Oka/Merck varicella vaccine. Pediatrics1993;92:833-7.
149. Guess HA, Broughton DD, Melton LJ, Kurland LT. Epidemiologyof herpes zoster in children and adolescents: a population-based study. Pediatrics 1985;76:512-7.
150. Hammerschlag MR, Gershon A, Steinberg S, Clarke L, Gelb L.Herpes zoster in an adult recipient of live attenuated varicellavaccine. J Infect Dis 1989;160:535-7.
151. Gelb LD, Dohner DE, Gershon AA, Steinberg SP, Waner JL, Ta-kahashi M, et al. Molecular epidemiology of live, attenuatedvaricella virus vaccine in children and in normal adults. J InfectDis 1987;155:633-40.
152. White CJ. Clinical trials of varicella vaccine in healthy children.Infect Dis Clin North Am 1996;10:595-608.
153. Leppla S, Robbins J, Schneerson R, Shiloach J. Developmentof an improved vaccine for anthrax. J Clin Invest 2002;110:141-4.
154. Green BD, Battisti L, Koehler TM, Thorne CB, Ivins BE. Demon-stration of a capsule plasmid in Bacillus anthracis. Infect Immun1985;49:291-7.
155. Uchida I, Hashimoto K, Terakado N. Virulence and immunoge-nicity in experimental animals of Bacillus anthracis strains har-bouring or lacking 110 MDa and 60 MDa plasmids. J Gen Mi-crobiol 1986;132:557-9.
156. Leppla SH. The bifactorial Bacillus anthracis lethal and oedematoxins. In: Alouf JE, Freer JH, editors. Comprehensive source-book of bacterial protein toxins. London: Academic Press,1999. p. 243-63.
157. Makino S, Uchida I, Terakado N, Sasakawa C, Yoshikawa M. Mo-lecular characterization and protein analysis of the cap region,which is essential for encapsulation in Bacillus anthracis. J Bac-teriol 1989;171:722-30.
158. Puziss M. Large-scale production of protective antigen of Ba-cillus anthracis in anaerobic cultures. Appl Microbiol 1963;11:330-4.
159. Hambleton P, Carman JA, Melling J. Anthrax: the disease inrelation to vaccines. Vaccine 1984;2:125-32.
160. Brachman PS. Field evaluation of a human anthrax vaccine.Am J Public Health 1962;52:632-45.
161. FDA. Biological products: bacterial vaccines and toxoids; im-plementation of efficacy review. Proposed rule. Federal Regis-ter 50:1002-117.
162. Ministry of Labor (United Kingdom). Report of the Committeeof Inquiry on Anthrax. London: Her Majesty’s Stationery Office,1959. p. 83.
163. Wright GG, Green TW, Kanode RG. Studies on immunity in an-thrax. Immunizing activity of alum-precipitated protective an-tigen. J Immunol 1954;73:387-91.

Wu et al 523J AM ACAD DERMATOL
VOLUME 50, NUMBER 4
164. Fellows PF, Linscott MK, Ivins BE, Pitt ML, Rossi CA, Gibbs PH, etal. Efficacy of a human anthrax vaccine in guinea pigs, rabbits,and rhesus macaques against challenge by Bacillus anthracisisolates of diverse geographical origin. Vaccine2001;19:3241-7.
165. CDC. Biosafety in microbiological and biomedical laboratories.4th ed. Washington, DC: US Department of Health and HumanServices; 2000. p. 88-9.
166. Is it safe? Does it work? In: Joellenbeck LM, Zwanziger LL, DurchJS, Strom BL, editors. The anthrax vaccine. Washington, DC:National Academy Press; 2002. p. 265.
167. Puziss M, Wright GG. Studies on immunity in anthrax. Gel-ad-sorbed protective antigen for immunization of man. J Bacteriol1963;85:230-6.
168. Pittman PR, Mangiafico JA, Rossi CA, Cannon TL, Gibbs PH,Parker GW, et al. Anthrax vaccine: increasing intervals betweenthe first two doses enhances antibody response in humans.Vaccine 2000;19:213-6.
169. Turnbull PC, Broster MG, Carman J, Manchee RJ, Melling J. De-velopment of antibodies to protective antigen and lethal fac-tor components of anthrax toxin in humans and guinea pigsand their relevance to protective immunity. Infect Immun1986;52:356-63.
170. Ivins BE, Pitt ML, Fellows PF, Farchaus JW, Benner GE, Waag DM,et al. Comparative efficacy of experimental anthrax vaccinecandidates against inhalation anthrax in rhesus macaques.Vaccine 1998;16:1141-8.
171. Welkos S, Little S, Friedlander A, Fritz D, Fellows P. The role ofantibodies to Bacillus anthracis and anthrax toxin componentsin inhibiting the early stages of infection by anthrax spores.Microbiology 2001;147:1677-85.
172. Brossier F, Levy M, Mock M. Anthrax spores make an essen-tial contribution to vaccine efficacy. Infect Immun 2002;70:661-4.
173. Cohen S, Mendelson I, Altboum Z, Kobiler D, Elhanany E, Bino T,et al. Attenuated nontoxinogenic and nonencapsulated re-combinant Bacillus anthracis spore vaccines protect againstanthrax. Infect Immun 2000;68:4549-58.
174. Welkos SL, Vietri NJ, Gibbs PH. Non-toxigenic derivatives of theAmes strain of Bacillus anthracis are fully virulent for mice: roleof plasmid pX02 and chromosome in strain-dependent viru-lence. Microb Pathog 1993;14:381-8.
175. Barnard JP, Friedlander AM. Vaccination against anthrax withattenuated recombinant strains of Bacillus anthracis that pro-duced protective antigen. Infect Immun 1999;67:562-7.
176. Hassett DE, Whitton JL. DNA immunization. Trends Microbiol1996;4:307-12.
177. Williamson ED, Beedham RJ, Bennett AM, Miller J, Baillie LW.Presentation of protective antigen to the mouse immune sys-tem: immune sequelae. J Appl Microbiol 1999;87:315-7.
178. Gu ML, Leppla SH, Klinman DM. Protection against anthrax byvaccination with a DNA plasmid encoding anthrax protectiveantigen. Vaccine 1999;17:340-4.
179. Development and testing of vaccines against anthrax. Requestfor proposal. Bethesda (MD): National Institute of Allergy andInfectious Diseases; 2002.
180. Advisory Committee on Immunization Practices (CDC). Rec-ommendations for the use of Lyme disease vaccine. MMWRMorb Mortal Wkly Rep 1999;48:1-25.
181. Montgomery RR, Malawista SE, Feen KJ, Bockenstedt LK. Directdemonstration of antigenic substitution of Borrelia burgdorferiex vivo: exploration of the paradox of the early immune re-sponse to outer surface proteins A and C in Lyme disease. J ExpMed 1996;183:261-9.
182. Fikrig E, Feng W, Aversa J, Schoen RT, Flavell RA. Differentialexpression of Borrelia burgdorferi genes during erythema mi-grans and Lyme arthritis. J Infect Dis 1998;178:1198-201.
183. Schwan TG, Piesman J, Golde WT, Dolan MC, Rosa PA. Induc-tion of an outer surface protein on Borrelia burgdorferi duringtick feeding. Proc Natl Acad Sci USA 1995;92:2909-13.
184. da Silva AM, Telford SR, Brunet L, Barthold S, Fikrig E. Borreliaburgdorferi OspA is an arthropod-specific transmission-block-ing Lyme disease vaccine. J Exp Med 1996;183:271-5.
185. da Silva AM, Zeidner N, Zhang Y, Dolan M, Piesman J, Fikrig E.Influence of outer surface protein A antibody on Borrelia burg-dorferi within feeding ticks. Infect Immun 1999;67:30-5.
186. Probert WS, Crawford M, Cadiz RB, LeFebvre RB. Immunizationwith outer surface protein (Osp) A, but not OspC, providescross-protection of mice challenged with North American iso-lates of Borrelia burgdorferi. J Infect Dis 1997;175:400-5.
187. Fikrig E, Barthold S, Kantor F, Flavell R. Long-term protection ofmice from Lyme disease by vaccination with OspA. Infect Im-mun 1992;60:773-7.
188. Fikrig E, Barthold SW, Kantor FS, Flavell RA. Protection of miceagainst the Lyme disease agent by immunizing with recombi-nant OspA. Science 1990;250:553-6.
189. Gern L, Hu CM, Voet P, Hauser P, Lobet Y. Immunization with apolyvalent OspA vaccine protects mice against Ixodes ricinustick bites infected by Borrelia burgdorferi ss, Borrelia garinii andBorrelia afzelii. Vaccine 1997;15:1551-7.
190. Golde WT, Burkot TR, Piesman J, Dolan MC, Capiau C, Hauser P,et al. The Lyme disease vaccine candidate outer surface pro-tein A (OspA) in a formulation compatible with human useprotects mice against natural tick transmission of B. burgdor-feri. Vaccine 1995;13:435-41.
191. Telford SR, Kantor FS, Lobet Y, Barthold SW, Spielman A, FlavellRA, et al. Efficacy of human Lyme disease vaccine formulationsin a mouse model. J Infect Dis 1995;171:1368-70.
192. Fikrig E, Telford SR, Barthold SW, Kantor FS, Spielman A, FlavellRA. Elimination of Borrelia burgdorferi from vector ticks feedingon OspA-immunized mice. Proc Natl Acad Sci USA 1992;89:5418-21.
193. Chang YF, Appel MJ, Jacobson RH, Shin SJ, Harpending P,Straubinger R, et al. Recombinant OspA protects dogs againstinfection and disease caused by Borrelia burgdorferi. Infect Im-mun 1995;63:3543-49.
194. Van Hoecke C, Comberbach M, De Grave D, Desmons P, Fu D,Hauser P, et al. The outer surface protein A (OspA) vaccineagainst Lyme disease: efficacy in the rhesus monkey. Vaccine1997;15:1872-87.
195. Van Hoecke CV, Comberbach M, De Grave D, Desmons P, Fu D,Hauser P, et al. Evaluation of the safety, reactogenicity and im-munologenicity of three recombinant outer surface protein(OspA) lyme vaccines in healthy adults. Vaccine1996;14:1620-6.
196. Hoecke CV, Lebacq E, Beran J, Parenti D. Alternative vaccina-tion schedules (0, 1, and 6 months versus 0, 1, and 12 months)for a recombinant OspA Lyme disease vaccine. Clin Infect Dis1994;28:1260-4.
197. Steere AC, Sikand VK, Meurice F, Parenti DL, Fikrig E, Schoen RT,et al. Vaccination against Lyme disease with recombinant Bor-relia burgdorferi outer-surface lipoprotein A with adjuvant.N Engl J Med 1998;339:209-15.
198. Schoen RT, Meurice F, Brunet C, Cretella S, Krause DS, Craft JE,et al. Safety and immunogenicity of an outer surface protein Avaccine in subjects with previous Lyme disease. J Infect Dis1995;172:1324-9.
199. Sigal LH, Zahradnik JM, Lavin P, Patella SJ, Bryant G, Haselby R,et al. A vaccine consisting of recombinant Borrelia burgdorferi

524 Wu et al J AM ACAD DERMATOL
APRIL 2004
outer-surface protein A to prevent Lyme disease. N Engl J Med1998;339:216-22.
200. Shadick NA, Liang MH, Phillips CB, Fossel K, Kuntz KM. The cost-effectiveness of vaccination against Lyme disease. Arch InternMed 2001;161:554-61.
201. Lastavica CC, Wilson ML, Berardi VP, Spielman A, Deblinger RD.Rapid emergence of a focal epidemic of Lyme disease incoastal Massachusetts. N Engl J Med 1989;320:133-7.
202. Rahn DW. Lyme vaccine: issues and controversies. Infect DisClin North Am 2001;15:171-87.
203. Peltola H, Kayhty H, Sivonen A, Makela PH. Haemophilus influ-enzae type b capsular polysaccharide vaccine in children: adouble-blind field study of 100,000 vaccinees 3 months to 5years of age in Finland. Pediatrics 1977;60:730-7.
204. Kayhty H, Peltola H, Karanko V, Makela PH. The protective levelof serum antibodies to the capsular polysaccharide of Hae-mophilus influenzae type b. J Infect Dis 1983;147:1100.
205. Eskola J, Peltola H, Takala AK, Kayhty H, Hakulinen M, KarankoV, et al. Efficacy of Haemophilus influenzae type b polysaccha-ride diphtheria toxoid conjugate vaccine in infancy. N EnglJ Med 1987;317:717-22.
206. Eskola J, Peltola H, Kayhty H, Takala AK, Makela PH. Finnishefficacy trials with Haemophilus influenzae type b vaccines. J In-fect Dis 1992;165(Suppl 1):S137-8.
207. Ward J, Brenneman G, Letson GW, Heyward WL. Limited effi-cacy of a Haemophilus influenzae type b conjugate vaccine inAlaska Native infants. The Alaska H. influenzae Vaccine StudyGroup. N Engl J Med 1990;323:1393-401.
208. Granoff DM. Assessing efficacy of Haemophilus influenzae typeb combination vaccines. Clin Infect Dis 2001;33(Suppl 4):S278-87.
209. CDC. Haemophilus influenzae type b vaccine. MMWR MorbMortal Wkly Rep 1988;36:832.
210. CDC. Food and Drug Administration approval of use of a Hae-mophilus b conjugate vaccine for infants. MMWR Morb MortalWkly Rep 1990;39:925-6.
211. CDC. Haemophilus b conjugate vaccines for prevention of Hae-mophilus influenzae type b disease among infants and childrentwo months of age and older: recommendations of the Immu-nization Practices Advisory Committee (ACIP). MMWR MorbMortal Wkly Rep 1991;40:1-7.
212. Black SB, Shinefield HR, Fireman B, Hiatt R. Safety, immunoge-nicity, and efficacy in infancy of oligosaccharide conjugateHaemophilus influenzae type b vaccine in a United States pop-ulation: possible implications for optimal use. J Infect Dis 1992;165(Suppl 1):S139-43.
213. Black S, Shinefield H, Fireman B, Hiatt R, Polen M, Vittinghoff E.Efficacy in infancy of oligosaccharide conjugate Haemophilusinfluenzae type b vaccine in a United States population of61,080 children. Pediatr Infect Dis J 1991;10:97-104.
214. Santosham M, Wolff M, Reid R, Hohenboken M, Bateman M,Goepp J, et al. The efficacy in Navajo infants of a conjugatevaccine consisting of Haemophilus influenzae type b polysac-charide and Neisseria meningitidis outer-membrane proteincomplex. N Engl J Med 1991;324:1767-72.
215. Daum RS, Granoff DM. Lessons from the evaluation of immu-nogenicity. In: Ellis RW, Granoff DM, editors. Development andclinical uses of Haemophilus b conjugate vaccine. New York:Marcel Dekker; 1994. p. 291-312.
216. Decker MD, Edwards KM, Bradley R, Palmer P. Comparative trialin infants of four conjugate Haemophilus influenzae type b vac-cines. J Pediatr 1992;120:184-9.
217. CDC. FDA approval of use of a new Haemophilus b conjugatevaccine and a combined diphtheria-tetanus-pertussis andHaemophilus b conjugate vaccine for infants and children.MMWR Morb Mortal Wkly Rep 1993;42:296-8.
218. Holmes SJ, Fritzell B, Guito KP, Esbenshade JF, Blatter MM, Re-isinger KS, et al. Immunogenicity of Haemophilus influenzaetype b polysaccharide-tetanus toxoid conjugate vaccine in in-fants. Am J Dis Child 1993;147:832-6.
219. Fritzell B, Plotkin S. Efficacy and safety of a Haemophilus influ-enzae type b capsular polysaccharide-tetanus protein conju-gate vaccine. J Pediatr 1992;121:355-62.
220. Paradiso PR, Hogerman DA, Madore DV, Keyserling H, King J,Reisinger KS, et al. Safety and immunogenicity of a com-bined diphtheria, tetanus, pertussis and Haemophilus influ-enzae type b vaccine in young infants. Pediatrics 1993;92:827-32.
221. Huang LM, Chang PF, Lee PI, Chiu HH, Tsai SY, Lee CY. Immu-nogenicity and safety of Haemophilus influenzae type b con-jugate vaccine (HibTiter) and a combination vaccine ofdiphtheria, tetanus, pertussis and HibTITER (Tetramune) intwo-month-old infants. J Microbiol Immunol Infect 1998;31:180-6.
222. Adams WG, Deaver KA, Cochi SL, Plikaytis BD, Zell ER,Broome CV, et al. Decline of childhood Haemophilus influen-zae type b (Hib) disease in the Hib vaccine era. JAMA 1993;269:221-6.
223. Ward JI, Zangwill KM. Haemophilus influenzae vaccines. In: Plot-kin SA, Orenstein WA, editors. Vaccines. 3rd ed. Philadelphia:WB Saunders; 1999. p. 183-221.
224. CDC. Progress toward elimination of Haemophilus influen-zae type b disease among infants and children: UnitedStates, 1987-1997. MMWR Morb Mortal Wkly Rep 1998;47:993-8.
225. CDC. Progress toward elimination of Haemophilus influenzaetype b invasive disease among infants and children: UnitedStates, 1998-2000. MMWR Morb Mortal Wkly Rep2002;51:234-7.
226. Booy R, Heath PT, Slack MP, Begg N, Moxon ER. Vaccine failuresafter primary immunisation with Haemophilus influenzaetype-b conjugate vaccine without booster. Lancet 1997;349:1197-202.
227. Dagan R, Fraser D, Greif Z, Keller N, Kaufstein M, Shazberg G, etal. A nationwide prospective surveillance study in Israel to doc-ument pediatric invasive infections, with an emphasis on Hae-mophilus influenzae type b infections. Israeli Pediatric Bactere-mia and Meningitis Group. Pediatr Infect Dis J 1998;17(Suppl9):S198-203.
228. Gotschlich EC, Goldschneider I, Artenstein MS. Human immu-nity to the meningococcus IV. Immunogenicity of group A andgroup C meningococcal polysaccharides. J Exp Med 1969;129:1367-84.
229. Artenstein MS, Gold R, Zimmerly JG, Wyle FA, Schneider H, Har-kins C. Prevention of meningococcal disease by group C poly-saccharide vaccine. N Engl J Med 1970;282:417-20.
230. Liu TY, Gotschlich EC, Jonssen EK, Wysocki JR. Studies on themeningococcal polysaccharides. I. Composition and chemicalproperties of the group A polysaccharide. J Biol Chem 1971;246:2849-58.
231. Makela PH, Peltola H, Kayhty H, Jousimies H, Pettay O, RuoslahtiE, et al. Polysaccharide vaccines of group A Neisseria meningi-tidis and Haemophilus influenzae type b: a field trial in Finland.J Infect Dis 1977;136(suppl):S43-50.
232. Wahdan MH, Rizk F, el-Akkad AM, el-Ghoroury AA, Hablas R,Girgis NI, et al. A controlled field trial of a serogroup A menin-gococcal polysaccharide vaccine. Bull World Health Organ1973;48:667-73.
233. Wahdan M, Sallam S, Hassan M, Gawad AA, Rakha A, Sippel J, etal. A second controlled field trial of a serogroup A meningococ-cal polysaccharide vaccine in Alexandria. Bull World Health Or-gan 1977;55:645-51.

Wu et al 525J AM ACAD DERMATOL
VOLUME 50, NUMBER 4
234. Sanborn WR, Bencic Z, Cvjetanovic B, Gotschlich EC, PollockTM, Sippel JE. Trial of a serogroup A meningococcus polysac-charide vaccine in Nigeria. Prog Immunobiol Stand1972;5:497-505.
235. Gold R, Lepow ML, Goldschneider I, Draper TL, Gotschlich EC.Clinical evaluation of group A and group C meningococcalpolysaccharide vaccines in infants. J Clin Invest 1975;56:1536-47.
236. Gold R, Lepow M, Goldschneider I, Draper T, Gotschlich E.Antibody responses of human infants to three doses ofgroup A Neisseria meningitidis polysaccharide vaccine ad-ministered at two, four, and six months of age. J Infect Dis1978;138:731-5.
237. Reingold AL, Broome CV, Hightower AW, Ajello GW, Bolan GA,Adamsbaum C, et al. Age-specific differences in duration ofclinical protection after vaccination with meningococcal poly-saccharide A vaccine. Lancet 1985;2:114-8.
238. Campagne G, Garba A, Fabre P, Schuchat A, Ryall R, BoulangerD, et al. Safety and immunogenicity of three doses of a Neisse-ria meningitidis A � C diphtheria conjugate vaccine in infantsfrom Niger. Pediatr Infect Dis J 2000;19:144-50.
239. Lepow ML, Goldschneider I, Gold R, Randolph M, GotschlichEC. Persistence of antibody following immunization of chil-dren with groups A and C meningococcal polysaccharide vac-cines. Pediatrics 1977;60:673-80.
240. Granoff D, Gupta R, Belshe R, Anderson E. Induction of immu-nologic refractoriness in adults by meningococcal C polysac-charide vaccination. J Infect Dis 1998;178:870-4.
241. MacDonald NE, Halperin SA, Law BJ, Forrest B, Danzig LE,Granoff DM. Induction of immunologic memory by conju-gated vs plain meningococcal C polysaccharide vaccine intoddlers: a randomized controlled trial. JAMA1998;280:1685-9.
242. Jodar L, Feavers IM, Salisbury D, Granoff DM. Development ofvaccines against meningococcal disease. Lancet2002;359:1499-508.
243. Ramsay ME, Andrews N, Kaczmarski EB, Miller E. Efficacy of me-ningococcal serogroup C conjugate vaccine in teenagers andtoddlers in England. Lancet 2001;357:195-6.
244. Swartley JS, Marfin AA, Edupuganti S, Liu LJ, Cieslak P, PerkinsB, et al. Capsule switching of Neisseria meningitidis. Proc NatlAcad Sci USA 1997;94:271-6.
245. Maiden MC, Spratt BG. Meningococcal conjugate vaccines:new opportunities and new challenges. Lancet1999;354:615-6.
246. Vogel U, Claus H, Frosch M. Rapid serogroup switching in Neis-seria meningitidis. N Engl J Med 2000;342:219-20.
247. Anderson EL, Bowers T, Mink CM, Kennedy DJ, Belshe RB, Hara-keh H, et al. Safety and immunogenicity of meningococcal Aand C polysaccharide conjugate vaccine in adults. Infect Im-mun 1994;62:3391-5.
248. Lieberman JM, Chiu SS, Wong VK, Partidge S, Chang SJ, ChiuCY, et al. Safety and immunogenicity of a serogroup A/C Neis-seria meningitidis oligosaccharide-protein conjugate vaccinein young children. JAMA 1996;275:1499-503.
249. Fairley CK, Begg N, Borrow R, Fox AJ, Jones DM, Cartwright K.Conjugate meningococcal serogroup A and C vaccine: reacto-genicity and immunogenicity in United Kingdom infants. J In-fect Dis 1996;174:1360-3.
250. Rosenstein N, Levine O, Taylor JP. Efficacy of meningococcalvaccine and barriers to vaccination. JAMA 1998;279:435-9.
251. Meningococcal disease prevention and control strategies forpractice-based physicians (addendum: recommendations forcollege students). Committee on Infectious Diseases. Pediat-rics 2000;106:1500-4.
252. CDC. Prevention and control of meningococcal disease and
meningococcal diseases and college students: recommenda-tions of the Advisory Committee on Immunization Practices(ACIP). MMWR Morb Mortal Wkly Rep 2000;49:13-20.
253. Pediatrics AAo. Meningococcal infections. In: Pickering L, edi-tor. Red book. Report of the Committee on Infectious Diseases.25th ed. Elk Grove Village (IL): American Academy of Pediatrics;2000.
254. The Jordan report: accelerated development of vaccines. Be-thesda (MD): National Institute of Allergy and Infectious Dis-eases, 1998.
255. Evans TG, Keefer MC, Weinhold KJ, Wolff M, Montefiori D, GorseGJ, et al. A canarypox vaccine expressing multiple human im-munodeficiency virus type 1 genes given alone or with Rgp120elicits broad and durable CD8� cytotoxic T lymphocyte re-sponses in seronegative volunteers. J Infect Dis1999;180:290-8.
256. McCarthy M. HIV vaccine fails in phase 3 trial. Lancet 2003;361:755-6.
257. Sandstom E, Wahren B. Therapeutic immunisation with recom-binant gp160 in HIV-1 infection: a randomised double-blindplacebo-controlled trial. Nordic VAC-04 Study Group. Lancet1999;353:1735-42.
258. Birx DL, Davis C, Ruiz N, et al. Results of a phase II double-blindmulticenter, placebo controlled HIV therapeutic vaccine trial[abstract]. International Conference on AIDS. Vancouver, July1996. Abstract 175.
259. Tsoukas CM, Raboud J, Bernard NF, et al. Immunization of pa-tients with HIV infection: a study of the effect of VaxSyn, a re-combinant HIV envelope subunit vaccine, on progression ofimmune deficiency. AIDS Res Hum Retroviruses 1998;14:483-90.
260. Graham BS, Matthews TJ, Belshe RB, Clements ML, Dolin R,Wright PF, et al. Augmentation of human immunodeficiencyvirus type 1 neutralizing antibody by priming with gp160 re-combinant vaccinia and boosting with rgp160 in vaccinia-na-ıve adults. J Infect Dis 1993;167:533-7.
261. Frey SE. HIV vaccines. Infect Dis Clin North Am 1999;13:95-112.
262. Oxford JS, Addawe M, Lambkin R. AIDS vaccine develop-ment: let a thousand flowers bloom. J Clin Pathol 1998;51:725-30.
263. Clements-Mann ML, Weinhold K, Matthews TJ, Graham BS,Gorse GJ, Keefer MC, et al. Immune responses to human immu-nodeficiency virus (HIV) type 1 induced by canarypox express-ing HIV-1MN gp120, HIV-1SF2 recombinant gp120, or bothvaccines in seronegative adults. NIAID AIDS Vaccine EvaluationGroup. J Infect Dis 1998;177:1230-46.
264. Belshe RB, Stevens C, Gorse G, et al. Phase II evaluation of a liverecombinant canarypox (AVLAC) vector HIV-1 vaccine with orwithout gp120 subunit HIV-1 vaccine [abstract]. InternationalSociety for Sexually Transmitted Diseases Research; July 11-14,1999; Denver, CO. Abstract 227.
265. Ferrari G, Humphrey W, McElrath MJ, Excler JL, Duliege AM,Clements ML, et al. Clad B-based HIV-1 vaccines elicit cross-clade cytotoxic T lymphocyte reactivities in uninfected volun-teers. Proc Natl Acad Sci USA 1997;94:1396-401.
266. Evans T, Corey L, Clements-Mann ML, et al. CD8�CTL inducedin AIDS Vaccine Evaluation Group phase I trials using canary-pox vectors (ALVAC) encoding multiple HIV gene products(vCP125, vCP205, vCP300) given with or without subunitboost. Paper presented at: World AIDS Conference; June 28-July 3, 1998; Geneva, Switzerland.
267. Doepel LK. NIAID opens first AIDS vaccine trial in Africa. NIAIDNews. National Institute of Allergy and Infectious Diseases,February 8, 1999.

526 Wu et al J AM ACAD DERMATOL
APRIL 2004
268. Rose NF, Marx PA, Luckay A, Nixon DF, Moretto WJ, DonahoeSM, et al. An effective AIDS vaccine based on live attenuatedvesicular stomatitis virus recombinants. Cell 2001;106:539-49.
269. Stephenson J. In HIV vaccine efforts, new strategies and pa-tience are needed in equal measure. JAMA 2002;288:2671-2.
270. Boyer JD, Ugen KE, Wang B, Agadjanyan M, Gilbert L, BagarazziML, et al. Protection of chimpanzees from high-dose heterolo-gous HIV-1 challenge by DNA vaccination. Nature Med 1997;3:526-32.
271. Frank SB. Formulized herpes virus therapy and neutralizingsubstance in herpes simplex. J Invest Dermatol 1938;1:267-82.
272. Straus S, Wald A, Kost R, McKenzie R, Langenberg A, Hohman P,et al. Immunotherapy of recurrent genital herpes with recom-binant herpes simplex virus type 2 glycoproteins D and B: re-sults of a placebo-controlled vaccine trial. J Infect Dis 1997;176:1129-134.
273. Corey L, Langenberg AG, Ashley R, Sekulovich RE, Izu AE, Doug-las JM, et al. Recombinant glycoprotein vaccine for the preven-tion of genital HSV-2 infection: two randomized controlled tri-als. JAMA 1999;282:331-40.
274. Leroux-Roels G, Moreau E, Desombere I, et al. Safety, hu-moral and cellular immune responses of three differentdoses of glycoprotein D(gD2t) in herpes simplex vaccinewith aluminum and MPL. Paper presented at: InterscienceConference on Antimicrobial Agents and Chemotherapy;1997; New Orleans.
275. Stanberry LR, Spruance SL, Cunningham AL, Bernstein DI, Min-del A, Sacks S, et al. GlaxoSmithKline Herpes Vaccine EfficacyStudy Group. Glycoprotein-D-adjuvant vaccine to preventgenital herpes. N Engl J Med 2002;347:1652-61.
276. Boursnell ME, Entwisle C, Blakely D, Roberts C, Duncan IA,Chisholm SE, et al. A genetically inactivated herpes simplexvirus type 2 (HSV-2) vaccine provides effective protectionagainst primary and recurrent HSV-2 disease. J Infect Dis 1997;175:16-25.
277. McLean CS, Challanin DN, Duncan I, Boursnell ME, Jennings R,Inglis SC. Induction of a protective immune response by mu-cosal vaccination with a DISC HSV-1 vaccine. Vaccine 1996;14:987-92.
278. McClements WL, Armstrong ME, Keys RD, Liu MA. Immuniza-tion with DNA vaccines encoding glycoprotein D or glycopro-tein B, alone or in combination, induces protective immunity inanimal models of herpes simplex virus-2 disease. Proc NatlAcad Sci USA 1996;93:11414-20.
279. Rouse BT, Nair S, Rouse RJ, Yu Z, Kuklin N, Karem K, et al. DNAvaccines and immunity to herpes simplex virus. Curr Top Mi-crobiol Immunol 1998;226:69-78.
280. Meignier B, Longnecker R, Roizman R. In vivo behavior of ge-netically engineered herpes simplex viruses R7017 and R 7020:construction and evaluation in rodents. J Infect Dis 1988;58:602-14.
281. Cadoz M, Micoud M, Seigneurin JM. Phase I trial of R7020: alive-attenuated recombinant herpes simplex virus (HSV) can-didate vaccine. Paper presented at: Interscience Conferenceon Antimicrobial Agents and Chemotherapy; 1992; Anaheim[CA].
282. Reichman R, Balsley J, Carlin D, et al. Evaluation of the safetyand immunogenicity of a recombinant HPV-11 L1 virus likeparticle vaccine in healthy adult volunteers. Paper presentedat: International Papillomavirus Conference; January 9-15,1999; Charleston [SC].
283. Zhang LF, Zhou J, Shao C, et al. A phase 1 trial of HPV 6 B viruslike particles as immunotherapy for genital warts. Paper pre-
sented at: International Papillomavirus Conference; January9-15, 1999; Charleston [SC].
284. Silva DD, Nieland J, Greenstone H, Schiller J, Kast W. Chimericpapillomavirus virus-like particles induce antigen-specifictherapeutic immunity against tumours expressing the HPV-16E7 protein. Paper presented at: International PapillomavirusConference; January 9-15, 1999; Charleston [SC].
285. Koutsky LA, Ault KA, Wheeler CM, Brown DR, Barr E, Alvarez FB,et al. A controlled trial of a human papillomavirus type 16 vac-cine. N Engl J Med 2002;347:1645-51.
286. Borysiewicz LK, Fiander A, Nimako M, Man S, Wilkinson GW,Westmoreland D, et al. A recombinant vaccinia virus encod-ing human papillomavirus types 16 and 18, E6 and E7 pro-teins as immunotherapy for cervical cancer. Lancet 1996;347:1523-7.
287. Lacey CJ, Monteiro EF, Thompson HS, et al. A phase IIa study ofa therapeutic vaccine for genital warts. Paper presented at:Medical Society for the Study of Venereal Disease; July 1997;Oxford, England.
288. Duggan-Keen MF, Brown MD, Stacey SN, Stern PL. Papilloma-virus vaccines. Front Biosci 1998;3:1192-208.
289. Nardelli-Haefliger D, Roden RB, Benyacoub J, Sahli R, Kraehen-buhl JP, Schiller JT, et al. Human papillomavirus type 16 virus-like particles expressed in attenuated Salmonella typhimuriumelicit mucosal and systemic neutralizing antibodies in mice.Infect Immun 1997;65:3328-36.
290. Jensen ER, Selvakumar R, Shen H, Ahmed R, Wettstein FO,Miller JF. Recombinant Listeria monocytogenes vaccinationeliminates papillomavirus-induced tumors and preventspapilloma formation from viral DNA. J Virol 1997;71:8467-74.
291. Krul M, Tijhaar E, Kleijne J, Loon AV, Nievers M, Schipper H.Induction of an antibody response in mice against human pap-illomavirus (HPV) type 16 after immunization with HPV recom-binant Salmonella strains. Cancer Immunol Immunother 1996;43:44-8.
292. Ossevoort MA, Feltkemp MC, VanVeen KJ, Melief CJ, KastWM. Dendritic cells as carriers for a cytotoxic T-lymphocyteepitope-based peptide vaccine in protection against a hu-man papillomavirus type 16-induced tumor. J Immunother1995;18:86-94.
293. Sanders GD, Taira AV. Cost effectiveness of a potential vac-cine for human papillomavirus. Emerg Infect Dis 2003;9:37-48.
294. Kong YC, Levine HB. Experimentally induced immunity in themycoses. Bacteriol Rev 1967;31:35-53.
295. Neely MN, Ghannoum MA. The exciting new future of anti-fungal therapy. Eur J Clin Microbiol Infect Dis 2000;19:897-914.
296. Casadevall A, Cassone A, Bistoni F, Cutler JE, Magliani W,Murphy JW, et al. Antibody and/or cell-mediated immunity,protective mechanisms in fungal disease: an ongoing di-lemma or an unnecessary dispute? Med Mycol 1998;36(Suppl1):95-105.
297. Deepe GS. Prospects for the development of fungal vaccines.Clin Microbiol Rev 1997;10:585-96.
298. Bistoni F, Vecchiarelli A, Cenci E, Puccetti P, Marconi P, Cas-sone A. Evidence for macrophage-mediated protectionagainst lethal Candida albicans infection. Infect Immun 1986;51:668-74.
299. Cassone A, Boccanera M, Adriani D, Santoni G, Bernardis FD.Rats clearing a vaginal infection by Candida albicans acquirespecific, antibody-mediated resistance to vaginal reinfection.Infect Immun 1995;63:2619-24.
300. Fidel PL, Sobel JD. Protective immunity in experimental Can-dida vaginitis. Res Immunol 1998;149:361-73.

Wu et al 527J AM ACAD DERMATOL
VOLUME 50, NUMBER 4
301. Bernardis FD, Boccanera M, Adriani DA, Spreghini E, SantoniG, Cassone A. Protective role of antimannan and anti-aspar-tyl proteinase antibodies in an experimental model of Can-dida albicans vaginitis in rats. Infect Immun 1997;65:3399-409.
302. Han Y, Morrison RP, Cutler JE. A vaccine and monoclonal anti-bodies that enhance mouse resistance to Candida albicansvaginal infection. Infect Immun 1998;66:5771-76.
303. Mencacci A, Torosantucci A, Spaccapelo R, Romani L, Bistoni F,Cassone A. A mannoprotein constituent of Candida albicansthat elicits different levels of delayed-type hypersensitivity, cy-tokine production, and anticandidal protection in mice. InfectImmun 1994;62:5353-60.
304. Mizutani S, Endo M, Ino-ve T, Kurosawa M, Uno Y, Saito H, et al.CD4-T-cell-mediated resistance to systemic candidiasis in-duced by a membrane fraction of Candida albicans. Antimi-crob Ag Chemother 2000;44:2653-8.
305. Cardenas-Freytag L, Cheng E, Mayeux P, Domer JE, ClementsJD. Effectiveness of a vaccine composed of heat-killed Candidaalbicans and a novel mucosal adjuvant, LT(R192G), against sys-temic candidiasis. Infect Immun 1999;67:826-33.
306. Han Y, Cutler JE. Antibody response that protects against dis-seminated candidiasis. Infect Immun 1995;63:2714-9.
307. Han Y, Ulrich MA, Cutler JE. Candida albicans mannan extract-protein conjugates induce a protective immune responseagainst experimental candidiasis. J Infect Dis 1999;179:1477-84.
308. Devi SJ. Preclinical efficacy of a glucuronoxylomannan-tetanustoxoid conjugate vaccine of Cryptococcus neoformans in a mu-rine model. Vaccine 1996;14:841-4.
309. Mukherjee J, Zuckier LS, Scharff MD, Casadevall A. Therapeuticefficacy of monoclonal antibodies to Cryptococcus neoformansglucuronoxylomannan alone and in combination with am-photericin B. Antimicrob Agents Chemother 1994;38:580-7.
310. Ghannoum MA. Potential role of phospholipases in virulenceand fungal pathogenesis. Clin Microbiol Rev 2000;13:122-43.
311. Hanel H, Kirsch R, Schmidts HL, Kottmann H. New systemati-cally active antimycotics from the beta-blocker category. My-coses 1995;38:251-64.
312. Tesh RB. Control of zoonotic visceral leishmaniasis: is it time tochange strategies? Am J Trop Med Hyg 1995;52:287-92.
313. Dye C. The logic of visceral leishmaniasis control. Am J TropMed Hyg 1996;55:125-30.
314. Handman E. Leishmaniasis: current status of vaccine develop-ment. Clin Microbiol Rev 2001;14:229-43.
315. Schallig HD, Oskam L. Molecular biological applications in thediagnosis and control of leishmaniasis and parasite identifica-tion. Trop Med Int Health 2002;7:641-51.
316. Ulmer JB, Sadoff JC, Liu MA. DNA vaccines. Curr Opin Immunol1996;8:531-6.
317. Wahren B. Gene vaccines. Immunotechnology 1996;2:77-83.318. Hassan UA, Abai AM, Harper DR, Wren BW, Morrow WJ. Nucleic
acid immunization: concepts and techniques associated withthird generation vaccines. J Immunol Methods 1999;229:1-22.
319. Walker PS, Scharton-Kersten T, Rowton ED, Hengge U, BoulocA, Udey MC, et al. Genetic immunization with glycoprotein 63cDNA results in a T helper cell type 1 response and protectionin a murine model of leishmaniasis. Hum Gene Ther 1998;9:1899-907.
320. Gurunathan S, Sacks DL, Brown DR, Reiner SL, Charest H,Glaichenhaus N, et al. Vaccination with DNA encoding the im-munodominant LACK parasite antigen confers protective im-munity to mice infected with Leishmania major. J Exp Med1997;186:1137-47.
321. Melby PC, Yang J, Zhao W, Perez LE, Cheng J. Leishmania dono-vani p36 (LACK) DNA vaccine is highly immunogenic but not
protective against experimental visceral leishmaniasis. InfectImmun 2001;69:4719-25.
322. Gurunathan S, Wu C, Freidag BL, Seder RA. DNA vaccines: a keyfor inducing long-term cellular immunity. Curr Opin Immunol2000;12:442-7.
323. Klinman DM, Takeshita F, Kamstrup S, Takeshita S, Ishii K, IchinoM, et al. DNA vaccines: capacity to induce auto-immunity andtolerance. Dev Biol 2000;104:45-51.
324. Mor G, Eliza M. Plasmid DNA vaccines. Immunology, toleranceand autoimmunity. Mol Biotechnol 2001;19:245-50.
325. Ivens AC, Blackwell JM. The Leishmania genome comes of age.Parasitol Today 1999;15:225-31.
326. Dickler HB, Gelfand EW. Current perspectives on the use ofintravenous immunoglobulin. Adv Intern Med 1996;41:641-80.
327. CDC. Renal insufficiency and failure associated with immuneglobulin intravenous therapy: United States, 1985-1998.MMWR Morb Mortal Wkly Rep 1999;48:518-21.
328. Bean B. Antiviral therapy: current concepts and practices. ClinMicrobiol Rev 1992;5:146-82.
329. Bernstein LJ, Krieger BZ, Novick B, Sicklick MJ, Rubinstein A.Bacterial infections in the acquired immunodeficiency syn-drome of children. Pediatr Infect Dis J 1985;4:472-5.
330. Calvelli TA, Rubinstein A. Intravenous gamma-globulin in in-fant acquired immunodeficiency syndrome. Pediatr Infect Dis J1986;5(Suppl 3):S207-10.
331. Gupta A, Novick BE, Rubinstein A. Restoration of suppressorT-cell functions in children with AIDS following intravenousgamma globulin treatment. Am J Dis Child 1986;140:143-6.
332. Oleski JM, Connor EM, Bohila R, et al. Treatment of immunode-ficiency virus antibody positive children with intravenous im-munoglobulin. Vox Sang 1987;52:162-75.
333. Spector SA, Gelber RD, McGrath N, Wara D, Barzilai A, Abrams E,et al. Controlled trial of intravenous immune globulin for theprevention of serious bacterial infections in children receivingzidovudine for advanced human immunodeficiency virus in-fection. N Engl J Med 1994;331:1181-7.
334. Hadler SC, Erben JJ, Matthews D, Starko K, Francis DP, MaynardJE. Effect of immunoglobulin on hepatitis A in day-care cen-ters. JAMA 1983;249:48-53.
335. Krugman S, Ward R, Giles JP, Jacobs AM. Infectious hepatitis.Studies on the effect of gamma globulin and on the incidenceof inapparent infection. JAMA 1960;174:883-90.
336. Woodson RD, Clinton JJ. Hepatitis prophylaxis abroad. JAMA1976;209:1053-9.
337. Lauchart W, Muller R, Pichlmayr R. Immunoprophylaxis of hep-atitis B viral reinfection in recipients of human liver allografts.Transplant Proc 1987;19:2387-9.
338. Samuel D, Bismuth A, Mathieu D, Arulnaden JL, Reynes M,Benhamou JP, et al. Passive immunoprophylaxis after livertransplantation in HBsAg-positive patients. Lancet 1991;337:813-5.
339. Grellier L, Dusheiko GM. Hepatitis B virus and liver transplanta-tion: concepts in antiviral prophylaxis. J Viral Hepat 1997;4(Suppl 1):111-6.
340. CDC. Prevention of varicella: recommendations of the Advi-sory Committee on Immunization Practices (ACIP). MMWRMorb Mortal Wkly Rep 1996;45:1-36.
341. Burks AW, Sampson HA, Buckley RH. Anaphylactic reactionsafter gamma globulin administration in patients with hypo-gammaglobulinemia. Detection of IgE antibodies to IgA.N Engl J Med 1986;314:560-3.
342. IVIG transmission of hepatitis C. FDA Med Bull 1994;24:3-4.343. Maldonado YA. Current controversies in vaccination: vaccine
safety. JAMA 2002;288:3155-8.344. Murray CJ, Lopez AD. Global mortality, disability, and the con-

528 Wu et al J AM ACAD DERMATOL
APRIL 2004
tribution of risk factors: Global Burden of Disease Study. Lancet1997;349:1436-42.
345. National Center for Health Statistics. National vital statisticssystem for numbers of death. Washington, DC: US Bureau ofCensus; 2001.
346. Davies P, Chapman S, Leask J. Antivaccination activists on theworld wide web. Arch Dis Child 2002;87:22-5.
347. Wolfe RM, Sharp LK, Lipsky MS. Content and design attributesof antivaccination web sites. JAMA 2002;287:3245-8.
348. Lieu TA, Cochi SL, Black SB, Halloran ME, Shinefield HR, HolmesSJ, et al. Cost-effectiveness of a routine varicella vaccinationprogram for US children. JAMA 1994;271:375-81.
349. Murphy TV, Gargiullo PM, Massoudi MS, Nelson DB, JumaanAO, Okoro CA, et al. Intussusception among infants given anoral rotavirus vaccine. N Engl J Med 2001;344:564-72.
350. Dales L, Hammer SJ, Smith NJ. Time trends in autism and inMMR immunization coverage in California. JAMA 2001;285:1183-5.
351. Kaye JA, Melero-Montes MdM, Jick H. Mumps, measles, andrubella vaccine and the incidence of autism recorded bygeneral practitioners: a time trend analysis. BMJ 2001;322:
460-3.352. Madsen KM, Hviid A, Vestergaard M, Schendel D, Wohlfahrt J,Thorsen P, et al. A population-based study of measles, mumps,and rubella vaccination and autism. N Engl J Med 2002;347:1477-82.
353. Ascherio A, Zhang SM, Hernan MA, Olek MJ, Coplan PM,Brodovicz K, et al. Hepatitis B vaccination and the risk of multi-ple sclerosis. N Engl J Med 2001;344:327-32.
354. Confavreux C, Suissa S, Saddier P, Bourdes V, Vukusic S. Vacci-nations and the risk of relapse in multiple sclerosis. Vaccines inMultiple Sclerosis Study Group. N Engl J Med 2001;344:319-26.
355. Karvonen M, Cepaitis Z, Tuomilehto J. Association betweentype 1 diabetes and Haemophilus influenzae type b vaccina-tion: birth cohort study. BMJ 1999;318:1169-72.
356. Graves PM, Barriga KJ, Norris JM, Hoffman MR, Yu L, Eisenbarth GS,et al. Lack of association between early childhood immunizationsand beta-cell autoimmunity. Diabetes Care 1999;22:1694-7.
357. Tacket CO, Mason HS, Losonsky G, Clements JD, Levine MM, Arn-tzen CJ. Immunogenicity in humans of a recombinant bacterialantigen delivered in a transgenic potato. Nat Med 1998;4:607-9.
358. Arakawa T, Langridge WH. Plants are not just passive creatures!
Nat Med 1998;4:550-1.