who collaborating centre for regulatory ... - mida…€¦ · who collaborating centre for...
TRANSCRIPT

WHO Collaborating Centre
for Regulatory Control of Pharmaceuticals
Member of Pharmaceutical
Inspection Cooperation Scheme
MS ISO 9001:2008 Certified
Non-OECD Member
full adherence to the Mutual
Acceptance Data (MAD) System
MS ISO 17025:2005 Certified
NATIONAL PHARMACEUTICAL REGULATORY AGENCY
(NPRA)
(i) Guidelines on Registration of Orphan Drug (ii) Updates on the
Harmonization initiatives PPWG Protection of test data for
pharmaceuticals and biologics
SEMINAR ON CAPACITY BUILDING FOR PHARMACEUTICALS AND MEDICAL DEVICES INDUSTRIES 7 September 2016

Guidelines on Registration of Orphan Drug

ORPHAN DRUG IN MALAYSIA
• GUIDELINE & RELATED PROCESSES ARE IN DRAFTING STAGES
–Management of Orphan Drug by the Main Committee in DUNAs
3

TWG DESIGNATION ORHAN DRUGS • TWG Designation Orphan Drug has ben established
• Members
Government agencies
• NPRA - Chair
• Pharmaceutical Services Division, MOH
• Medical Practice Division & Development Division, MOH
• Genetics Department, Hospital Kuala Lumpur
Relevant stakeholders
• Malaysian Rare Disorders Society (MRDS)
• MOPI, PhAMA, MAPS
• Association of Private Hospitals of Malaysia (APHM)
• Others eg: Malaysia Lysosomal Disease Association, etc
4

TOR
• To discuss criteria
• To propose process flow for designation
5

criteria
• Agreed it is based on definition as per stated by Main Committee of Orphan Drug
Definition
• A medicine, vaccine or in vivo diagnostic agent that is intended to
treat, prevent or diagnose a rare disease
Rare Disease Definition
• A life-threatening or chronically debilitating rare condition with a
maximum prevalence of 1 in 12,000 persons per population.
6
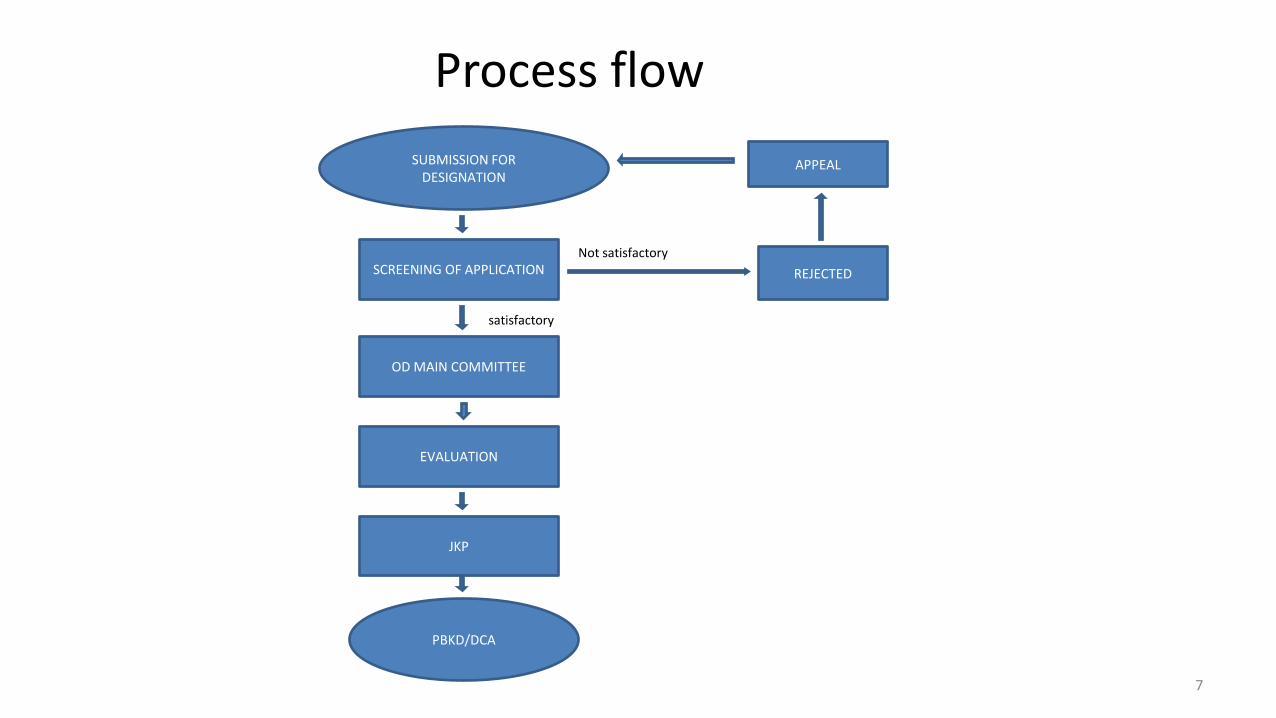
Process flow
SUBMISSION FOR DESIGNATION
PBKD/DCA
SCREENING OF APPLICATION
OD MAIN COMMITTEE
EVALUATION
JKP
APPEAL
REJECTED
Not satisfactory
satisfactory
7

Supporting documents
• Description of the condition – Details of condition, proposed indication
• Prevalence of the condition
– Local & international situation
• Other methods for diagnosis, prevention or treatment of the condition – Current available methods
• Description of product development
• Worldwide registration status
8

Registration requirement • Flexibility for some registration requirements
• Examples:-
NO REQUIREMENTS NCE BIOLOGICS GENERICS
1 Pre-clinical and clinical studies must be submitted to support the safety and efficacy of the product. The product must be registered in at least one reference country.
√ Case by case NA
2 If the product / active ingredient has been established, published literature is sufficient to support the safety and efficacy of the product and pre-clinical or new clinical studies are not required. The product must be registered in at least one country.
√ Case by case NA
3 Two year registration validity for products from non-PICS country, Will be subjected to GMP inspection by NPCB within that period of time.
√ Case by case √
4 Post-approval commitment for stability data and storage condition not at Zone IVB condition & should be complied during the renewal of product registration
√ Case by case √
9

POST-Registration requirement
• Surveillance procedures and requirements are as follows:
a) The product registration holder must report any adverse reactions involving orphan product to NPCB
b) Periodic Safety Updates Report (PSUR)/Periodic Benefit Risk Evaluation Report (PBRER) must be submitted to NPCB for orphan products in the category of new chemical entities/new drug products/ biologics within specified time frame agreed by the NPCB
c) Products will be sampled and tested to ensure that it complies to the established standards and specifications. Actions will be taken against products that do not comply to the established standards.
10

CONCLUSION
• Guideline/policy/process are still in development
• Further discussion on certain matters
• Will inform stakeholders once the Main Guideline & its relevant processes is finalized
11

• Update on the
Harmonization initiatives PPWG

The OBJECTIVE of ACCSQ-PPWG is to develop harmonization schemes of pharmaceutical regulations among the ASEAN member countries to complement and facilitate the objective of ASEAN Free Trade Area, particularly towards the elimination of technical barriers to trade posed by regulators, however without compromising on product quality, efficacy and safety.

PRODUCTS OF ASEAN HARMONIZED INITIATIVES – available on ASEAN website
ASEAN HARMONISATION INITIATIVES 14
1 •ASEAN Common Technical Dossier (ACTD)
2 •ASEAN Common Technical Requirement (ACTR) on ; - Quality, Safety and Efficacy - Administrative Data and Glossary
3 •Guideline on Analytical Validation - Thailand
4 •Guideline on Process Validation - Singapore
5 •Guideline on Stability Studies - Indonesia
6 •Guideline on BA/BE Studies - Malaysia
7 • Guideline on Variation - Malaysia

ASEAN HARMONISATION INITIATIVES 15
MRA on GMP Inspection signed 2009
ASEAN Sectoral MRA on GMP Inspection of Manufacturers of Medicinal Products –listed services
Malaysia
Singapore
Indonesia
Thailand
[Philippines and Vietnam have indicated interest to be listed – HoD meeting 18 Aug 2016]
Joint Sectoral Committee – to monitor implementation of GMP MRA
PIC/S members; Malaysia
Singapore
Indonesia
Thailand

PPWG Collaborations for GMP
PIC/S Korea PMDA

ASEAN MUTUAL RECOGNITION ARRANGEMENT FOR BIOEQUIVALENCE STUDY REPORTS OF GENERIC MEDICINAL PRODUCTS Effective - 5 years after enforcement of MRA ~ by 2021. Status as at 30 August 2016: MRA signed : Brunei, Indonesia, Lao and Malaysia Others by ad referendum - JSC to be formed - Workshop on Development of Procedures for JSC MRA BE report Feb 2017

No Follow up action Timeline Responsible Party
1 Seek input from Industry Associations (APC and APRIA) as
well as WHO about candidates products for JA
End of September 2016 All JACG members to seek information.
JACG Chair to consolidate.
2 Circulate to all JACG members and email discussions of
proposals to identify products
20 October 2016 Coordinated by JACG Chair
3 Issue notice to invite applications with time frame for
submission of 6 months from publication of notice
15 November 2016 All JACG members.
JACG Chair to coordinate.
4 Each JACG member to check model letters and adapt
language to their national context
15 November 2016 All JACG members.
5 Explore ways to establish secure communication
mechanism to exchange of confidential information among
ASEAN NRAs
15 November 2016 All JACG members with WHO support.
6 Application to be received 31 December 2016 All JACG Members
7 Application Review session End of March/Early April
2017
Reviewer from AMS, Lead Countries and
JACG Chair with WHO support
1STJOINT ASSESSMENT COORDINATING GROUP (JACG) MEETING 17 August 2016

• Update on the
Protection of test data for pharmaceuticals and biologics

Chapter Summary of chapter
CHAPTER 18 : Intellectual Property 3 . (i) Article 18.50 Protection of Undisclosed Test or Other Data
Protection of test data / Data Exclusivity (DE)for (i) small molecules In the TPPA, Malaysia will continue to provide test data protection for pharmaceuticals for five years. The DE period starts from the approval date in Malaysia.
The TPP will not affect Malaysia’s ‘access window’ (application for marketing approval must be submitted within 18 months from the date first approved in any other country), which is a condition before any data protection is granted This ‘access window’ is fundamental, and will ensure that new pharmaceuticals/biologics would enter into Malaysia early, thus enable Malaysians to have access to these life saving products.
Intellectual Property

Timeline for Patent Protection and DE
0 YEARS
20 YEARS
CLINICAL TRIALS On average takes 2-10 years
Patent is filed, patent protection begins
First marketing approval in any other country
Apply for marketing approval in Malaysia
Granted marketing approval in Malaysia
Data exclusivity for 5 years
Innovator test data can be used to approve a generic for marketing approval
PATENT PROTECTION TERM OF 20 YEARS
Acc
es s
Win
do
w
18
mo
nth
s
24
5 w
ork
ing
day
s
• Granting pharmaceutical test data protection within the patent term of the product will not delay access of generics and biosimilars to the public as the period of protection for the patent and test data is concurrent.
• Imposing the ‘access window’ condition will avoid overhang of a patent term.
Generics can be marketed at the end of patent term

Chapter Summary of chapter
CHAPTER 18 : Intellectual Property 3. (ii) Article 18.52 Biologics
Protection of test data / Data Exclusivity (DE)for (ii) biologics Agreement on biologics gives two options for TPP Parties: countries to offer 8 years market protection
for Biologics; or countries to offer 5 years of data protection,
and other measures such as regulatory measures and patent to deliver comparable market protection for biologics.
The biologics agreement also provides a review to be undertaken 10 years after entry into force of the TPPA.
Currently Malaysia does not provide DE for biologics. Malaysia is taking the second option of a minimum standard of 5-year exclusivity and other measures. Patient’s timely access to the latest biosimilars may be delayed, but this impact, however, may not be significant since the 18-month access window safeguard also applies.
Intellectual Property

Biosmilars takes more than 5 years to be developed Biosimilar (company)
-first approved Reference Product
(company) -first approved
Years gap before first biosimilars were
registered
SciTropin (Sandoz) EU 2006
Genotropin (Pfizer) Feb 1997
9 years
Binocrit (Sandoz) EU 2009
Eprex (J&J) Sept 1998
11 years
Zarzio (Sandoz) EU 2009
Neupogen (Roche) Aug 2002
6 years
Nivestim (Hospira) UK 2010
Neupogen (Roche) Aug 2002
8 years
Insugen (Biocon) Jan 2014
India 2006-not biosimilar pathway
Actrapid / Insulatard / Mixtard
(Novo Nordisk) Sept 1999
7 years
Remsima (Celtrion) Korea 2012
Remicade (J&J) Aug 1999
13 years
• DE protection for biologics is expected to have low potential in deterring new investments in biosimilars, as historical statistics suggest that new biosimilars were generally registered 6 to 13 years after the first marketing approval of their biologics counterparts were granted
• In contrast, the intellectual property protection to biologics could attract biologics manufacturers to expand operations in Malaysia

24
• Transition Periods 5 years for biologics • Within this transition period; legislative amendments
The Sale of Drugs Act 1952 – [expected to be tabled in Parliament 2017] The Control of Drugs and Cosmetics Regulations 1984
continue to provide capacity building and ensure enough personnel to process efficiently all applications for marketing approval for pharmaceuticals and biologics
enhance efficiency of on-line system to process application for medicines approval (QUEST)
