yap is essential for treg mediated suppression of …...2018/06/15 · tumor immunity and the...
TRANSCRIPT

1
YAP is essential for Treg mediated suppression of anti-tumor immunity
Xuhao Ni1,2**, Jinhui Tao1,3,6**, Joseph Barbi1,4**, Qian Chen3,7, Benjamin V. Park1,
Zhiguang Li1,8, Nailing Zhang3, Andriana Lebid1, Anjali Ramaswamy1, Ping Wei1, Ying
Zheng1, Xuehong Zhang1,8, Xingmei Wu1,5♯, Paolo Vignali1♮, Cui-Ping Yang1,9, Huabin Li5,
Drew Pardoll1, Ling Lu2*, Duojia Pan3 *♦, Fan Pan1*
1 Immunology and Hematopoiesis Division, Department of Oncology, Bloomberg-Kimmel Institute, Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins University School of Medicine, Baltimore, MD 21287, USA 2 Translational Medicine Research Center, Affiliated Jiangning Hospital, and Liver Transplantation Center, First Affiliated Hospital, Nanjing Medical University, Nanjing, China. 3 Department of Molecular Biology and Genetics, Howard Hughes Medical Institute, Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA 4 Department of Immunology, Roswell Park Comprehensive Cancer Center, Buffalo, NY, 14263, USA 5 Department of Otolaryngology, Head and Neck Surgery, Affiliated Eye, Ear, Nose and Throat Hospital, Fudan University, Shanghai, 200031, China. 6 Department of Rheumatology & Immunology, The First Affiliated Hospital of University of Science and Technology of China, No. 17 LuJiang Road, Hefei 230001, Anhui, China. 7 Thorgene Co., Ltd. 88 Kechuang 6th Street, Yizhuang Biomedical Park, Beijing, China. 8 Center of Genome and Personalized Medicine, Institute of Cancer Stem Cell, Dalian Medical University, Dalian, Liaoning 116044, China 9 Department of Gastroenterology, Rujin Hospital North, Shanghai Jiaotong University School of Medicine, Shanghai 20181, China * To whom correspondence should be addressed: Fan Pan Department of Oncology 1650 Orleans Street, CRB1 Rm452 Baltimore, MD 21287 [email protected] (F.P.) Duojia Pan Department of Physiology Howard Hughes Medical Institute UT Southwestern Medical Center 5323 Harry Hines Blvd. Dallas, TX 75390-9040 [email protected] (D.P.)
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

2
Ling Lu 300 Guangzhou St, Surgery Bldg, 21 Floor Nanjing, Jiangsu, China [email protected] (L.L.)
** Equal contribution to this work, ♯Current Address: First Affiliated Hospital of Sun Yat-
sen University, ♮Current Address: University of Pittsburgh School of Medicine, ♦Current
Address: UTSouthwestern Medical Centre.
Running title: YAP modulates regulatory T cells activity
Disclosure of potential Conflicts of Interest: No potential conflicts of interest were
disclosed by all authors.
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

3
Abstract
Regulatory T cells (Tregs) are critical for maintaining self-tolerance and immune
homeostasis, but their suppressive function can impede effective anti-tumor immune
responses. Foxp3 is a transcription factor expressed in Tregs that is required for their
function. However, the pathways and microenvironmental cues governing Foxp3
expression and Treg function are not completely understood. Herein, we report that Yes-
associated protein (YAP), a co-activator of the Hippo pathway, is highly expressed in
Tregs and bolsters Foxp3 expression and Treg function in vitro and in vivo. This
potentiation stemmed from YAP-dependent upregulation of Activin signaling which
amplifies TGFβ/SMAD activation in Tregs. YAP-deficiency resulted in dysfunctional
Tregs unable to suppress anti-tumor immunity or promote tumor growth in mice.
Chemical YAP antagonism and knockout or blockade of the YAP-regulated Activin
Receptor similarly improved anti-tumor immunity. Thus we identify YAP as an
unexpected amplifier of a Treg-reinforcing pathway with significant potential as an anti-
cancer immunotherapeutic target.
Significance
Tregs suppress anti-tumor immunity, and pathways supporting their function can be
novel immunotherapy targets. Here, the selective expression of YAP by Tregs, its
importance for their function, and its unexpected enhancement of pro-Treg
Activin/SMAD signaling are reported as are validations of potential cancer-fighting
antagonists of YAP and its regulatory targets.
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

4
INTRODUCTION
Regulatory T cells (Tregs) play critical roles in promoting immunological self-
tolerance and immune homeostasis by suppressing aberrant or excessive immune
responses that could give rise to autoimmune diseases (1). However, their ability to
dampen the activation of other leukocytes also can pose a major barrier to effective anti-
tumor immunity and the sterile cure of chronic infections (2). The signature forkhead
family transcription factor Foxp3 anchors the gene expression profile that is responsible
for the characteristic suppressive function of Tregs. Clearly demonstrating the
importance of this factor, mutations to the gene encoding Foxp3 can lead to fatal
autoimmune disorders in Scurfy mice and in human IPEX patients alike (3,4). Despite
the undeniable importance of Foxp3 for Treg function and immune control, our grasp of
the factors and mechanisms governing its expression remains incomplete.
The signaling pathways triggered in response to certain cytokines (e.g. IL-2 and
TGFβ) can be critical for induction and maintenance of Foxp3 expression in Tregs (5).
TGFβ is a potent inducer of Foxp3 expression in vitro and in vivo, and through activation
of SMAD signaling molecules, which serve as critical facilitators and regulators of TGFβ-
initiated signaling events and downstream gene activation (6,7). TGF- signaling has
also been reported to be critical for maintaining Foxp3 expression and Treg function
(8,9). Likewise SMAD2 and SMAD3 are also apparently needed for the optimal
phenotypic stability of Tregs (10). Importantly, mechanisms for the augmentation or
amplification of TGF/SMAD signaling in Tregs can stabilize or enhance the suppressive
function of these cells (11), and may be crucial determinants of Treg performance in a
variety of microenvironmental niches.
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

5
YAP (Yes-associated protein) is a transcriptional coactivator that
developmentally regulates organ size (12,13). YAP is frequently elevated in a number of
cancer types such as lung, colorectal, ovarian, liver and prostate cancers, where it acts
as a powerful tumor promoter, and its activation is a frequent event in the tumor
progression (14). The Hippo pathway is believed to be the major regulator of YAP
nuclear localization, activity, and tumorigenic potential (15-17). However, the
physiological role of YAP in immune system is unknown.
Unexpectedly, we found YAP to be highly expressed by Tregs. In this report, we
characterize the role of YAP in these important cellular mediators of immune control. Our
studies revealed that in the absence of YAP, Tregs failed to suppress immune activation
in vitro as well as in vivo. We also found that YAP potentiates the signaling events
triggered by dimeric members of the TGFβ cytokine superfamily known as Activins by
activating expression of a key signaling component of the Activin Receptor complex.
Interestingly, we found that not only is this signaling axis active in Tregs, it also could
effectively amplify TGFβ/SMAD signaling and the promotion of Treg differentiation and
function. Moreover, disrupting this YAP/Activin/SMAD axis dramatically slowed the
growth of tumors in mice including a highly aggressive melanoma model. This
experimental treatment also enhanced the anti-tumor efficacy of an anti-tumor vaccine
suggesting that the targeting of this YAP/Activin/SMAD axis can be used to improve anti-
cancer immunotherapy efficacy
RESULTS
YAP expression is induced by TCR signaling and highly expressed by Tregs and
supports their function
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

6
YAP is a transcriptional co-activator known for its role in the Hippo signaling
pathway (13). As such, its importance in tumorigenesis and organ size determination is
well recognized (14). However, little is known about the role of the Hippo pathway and
YAP in immune cells. Reports of crosstalk between the Hippo and TGFβ signaling
pathways (18,19) led us to speculate that elements of the former may have a role in the
mechanisms governing immune activation and tolerance.
We therefore screened YAP expression across different subsets of murine CD4+
T cells in order to assess the likelihood that Hippo signaling plays a role in these
functionally distinct T cell lineages. Little-to-no YAP mRNA was detected in naïve CD4+
T cells, but notably, YAP expression was uniquely induced during the early stages of
iTreg differentiation. Meanwhile, other T effector subsets (Th0, Th1, Th2, and Th17 cells)
failed to markedly up-regulate YAP mRNA (Fig. 1A). Interestingly, transient YAP
message accumulation was noted during Th17 skewing. However, after 12 hours post-
stimulation, YAP transcript levels returned to baseline in these T cells (Fig. 1A,
Supplementary Fig. S1A). Importantly, considerable levels of YAP protein were found
in cells of the iTreg subset and not other Thelper lineages (Fig. 1B). Human Tregs
isolated from the peripheral blood of healthy donors also display higher levels of YAP
mRNA than their conventional CD4+ (non-Treg) counterparts (Fig. 1C). These results
implicate YAP as a transcription factor preferentially expressed by developing and
established Tregs of mice and humans.
Since YAP is a major component of the Hippo pathway, we assessed levels of
several Hippo signaling factors known to be upstream of the transcription factor across T
cell subsets to determine if these also are expressed preferentially by Tregs.
Interestingly, we found that LATS1/2 and MST1/2, unlike YAP, were not up-regulated by
iTreg skewing conditions (Supplementary Fig. S1B). These findings suggest that unlike
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

7
other Hippo pathway factors, YAP is uniquely up-regulated in developing Tregs, and
they imply a role for YAP in the biology of these cells outside of its traditional role.
In order to dissect the potential role of YAP in the biology of CD4+ T cells, including
Tregs, we crossed YAPfl/fl mice to CD4-cre transgenic mice to generate animals with a
T cell-specific deletion of YAP. These conditional knockout mice (YAP cKO) developed
normally without apparent defects in T cell development or peripheral immune cell
populations (Supplementary Fig. S2). Additionally, no obvious spontaneous immune
pathologies were noted in these mice. Likewise the lung, kidney, liver, small intestine
and stomach of mice with Treg-specific YAP deficiency (generated crossing YAPfl/fl
mice to Foxp3Cre+ transgenic mice) appeared comparable to wild type (WT) littermates
(Supplementary Fig. S3). We used both strains to assess the impact of YAP-deficiency
on Thelper cytokine production and lineage commitment. To this end, we isolated naïve
CD4+ T cells from YAP cKO and WT mice for activation under different helper CD4+ T
cell (Th) polarizing conditions for 72 hours. YAP cKO CD4+ T cells express moderately
higher levels of IL-2 and IFN-γ upon unbiased activation (Th0 conditions) (Fig. 2A). YAP
cKO CD4+ T cells also express a greater amount of IL-17A than WT CD4+ T cells under
Th17 polarizing conditions (Fig. 2B), and, consistently, YAP cKO CD4+ T cells
expressed higher levels of il17a mRNA than WT cells (Fig. 2C). A modest decrease in
Foxp3+ cells was also seen in YAP cKO derived T cells cultured under Th17 conditions
(Fig. 2B). These observations, coupled with our earlier discovery that YAP is up-
regulated in iTregs, led us to suspect that YAP positively affects the generation of iTregs
in vitro over other CD4+ T cell fates. In line with this, the percentages of Foxp3+ cells
induced from naïve YAP cKO T cells activated under iTreg skewing conditions were
modestly, yet significantly lower than those seen in polarized WT CD4+ T
(Supplementary Fig. S4). Naïve CD4+ T cells isolated from YAPfl/fl Foxp3Cre+ mice
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

8
were also consistently less able to up-regulate Foxp3 than WT controls in response to
activation and various concentrations of the Treg-promoting cytokine TGF-β. Here, YAP-
deficiency specifically in T cells having already “turned on” Foxp3 expression reduced
the intensity of signal for the Treg transcription factor (Fig. 2D). Taken together, these
findings suggest that YAP likely plays an important role in the initiation or maintenance
of Treg differentiation.
In addition to Foxp3 induction, we also hypothesized that YAP might contribute to
the suppressive function of Tregs as well. Indeed, an in vitro suppression assay showed
that while WT Tregs readily dampened the proliferation of naïve T cells, YAP cKO Treg
were much less effective suppressors (Fig. 2E). In all, these findings implicate YAP as a
Treg-associated factor with a role in both the generation and function of these cells.
YAP-deficiency enhances anti-melanoma immunity
While Tregs are necessary to maintain immune homeostasis, they pose an obstacle in
mounting effective anti-tumor immune responses, and their suppressive function
dampens the efficacy of anti-cancer immunotherapies (20). For these reasons, therapies
aimed at inhibiting Treg activity are promising additions to the cancer immunotherapy
arsenal (21). We hypothesized that the apparent loss of Treg suppressive function seen
in the absence of YAP could enhance anti-tumor immune responses. To test this, WT
and YAP cKO mice were challenged with B16-melanoma, an aggressive “non-
immunogenic” cancer model. Tumor growth was measured in these mice over time, and,
strikingly, we found that YAP cKO mice controlled the subcutaneous growth of the
implanted melanoma cells while tumors grow robustly in WT mice (Fig. 3A, B). In line
with our in vitro findings, the activation of CD4+ and CD8+ tumor infiltrating lymphocytes
(TILs) from YAP cKO mice were apparently much less restrained than that of their WT
counterparts. Intracellular cytokine staining revealed these cells produced significantly
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

9
higher levels of IFNγ and TNFα (Fig. 3C) compared to those from WT tumors. These
results suggest that in the absence of YAP in T cells, a more robust anti-tumor immune
response is mounted.
Tumor challenge of mice with Treg-restricted YAP deficiency yielded similar
results. While WT controls expectedly permitted rapid tumor development, YAPfl/fl
Foxp3Cre+ mice maintained small tumors infiltrated by elevated populations of
inflammatory cytokine producing leukocytes. Specifically, producers of the tumoricidal
Th1 cytokine, IFNγ were found at higher frequencies and in greater numbers in the
tumors of YAPfl/fl Foxp3Cre+ mice than those of WT controls (Fig. 3D, E). Analysis of
Foxp3 expression by CD4+ TILs revealed that deletion of YAP in Tregs reduces the
frequency of suppressive Foxp3+ Tregs in the tumor microenvironment (Fig. 3F, left
and middle). The relative balance (i.e the ratio) of Tregs and potential effector CD8+ T
cells was similarly shifted in the tumors of mice with Treg-specific YAP deficiency
compared to those of WT controls (Fig. 3F, right). Treg-specific YAP-deficiency also
slowed the growth of tumors caused by implanted MC38 adenocarcinoma cells
(Supplementary Fig. S5A, B). Not only were MC38 tumors much smaller in YAPfl/fl
Foxp3Cre+ mice 21 days after injection, the relative proportions of Foxp3+ Tregs among
tumor-infiltrating T cells were reduced compared to WT tumors. In contrast, the
frequencies of intratumoral producers of IFNγ and TNFα were elevated in the absence of
Treg-specific YAP expression (Supplementary Fig. S5C-D). Corroborating results were
seen in the injectable EL4 thymoma model in which Treg-restricted YAP knockout
resulted in dramatically stunted tumor growth relative to WT mice. As with other tumor
models, this derailed tumor progression was concurrent with reduced Treg proportions
and an elevated presence of pro-inflammatory cytokine producing T cells in the tumor
microenvironment (Supplementary Fig. S6A-D). These experiments make a strong
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

10
case for YAP’s role as both a facilitator of Treg presence in the tumor niche and a potent
and broadly active driver of Treg-enforced inhibition of endogenous anti-tumor immunity.
Some of the most promising immunotherapeutic agents (i.e. PD-1 and CTLA-4
antagonist antibodies) show even greater anti-tumor effect when administered in concert
(22-24) or alongside tumor vaccine strategies (25-28). We therefore tested the
therapeutic potential of YAP targeting as an immunotherapeutic approach to combat
cancer. Administration of a known YAP inhibitor, Verteporfin (VP) (29), to melanoma
bearing mice resulted in modest reduction in tumor size (Fig. 3G). Treatment of
melanoma-bearing YAPfl/fl Foxp3Cre+ mice with VP, on the other hand, failed to alter
the already stunted progression of tumors in these mice (Supplementary Fig. S7A),
suggesting that potential off-target effects of this drug, or any direct effects on tumor
cells are not likely contributing to these in vivo observations. We also tested the effects
of combining VP with the proven immunotherapeutic agents anti-PD1 antibody and GM-
Vac (irradiated GMCSF-producing B16 cells). Both anti-PD1 and GM-Vac treatments
were able to slow tumor growth somewhat as monotherapies. Notably, combinatorial
treatment with VP and anti-PD1 neutralizing antibody suppressed tumor progression to a
greater extent than any monotherapy tested. Even more dramatic was the synergistic
effects of VP and GM-Vac, which prevented the development of tumors beyond a barely
detectable size (Fig. 3G). The decidedly improved anti-tumor efficacy seen upon
combination of either anti-PD1 or GM-Vac treatment with VP was associated with
enhanced proportions of IFNγ-producing CD8+ and CD4+ T cells and a compromised
Treg presence in the tumor microenvironment (Fig. 3H and Supplementary Fig. S7B-
D). These findings strongly suggest a major role for Treg-derived YAP in crafting the
immunosuppressive nature of the tumor microenvironment. They also suggest the
potential of immunotherapeutic approaches that include YAP targeting agents.
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

11
YAP potentiates expression of genes involved in TGF/SMAD and Activin
signaling
To gain insight into the mechanism by which YAP contributes to Tregs and their
enforcement of immune suppression, we isolated Tregs from mice lacking YAP
specifically in these cells (YAP cKO) and subject them to RNASeq analysis along with
WT Tregs (YAPwt/wt Foxp3cre+) and naïve CD4+ T cells from both mice. The results of
this analysis revealed that YAP-deficient Tregs display reduced expression of several
genes known to be important in the signaling pathway triggered by the anti-inflammatory
cytokine TGF. Interestingly, one of the genes most down-regulated in the absence of
YAP was that encoding the signaling component of the Activin Receptor complex known
as Acvr1c (Fig. 4A and Supplementary Fig. S8A). Confirming a role for YAP in
potentiating Acvr1c expression, we found that WT CD4+ T cells display considerable up-
regulation of the transcript for this receptor subunit during in vitro Treg differentiation
while their YAP-deficient counterparts did not. Interestingly, freshly isolated nTregs
expressed modest levels of Acvr1c. However, upon activation, these Tregs dramatically
activated activin receptor expression in a YAP-dependent manner (Supplementary Fig.
S8B). Since neither nTreg nor differentiating iTregs from YAP fl/fl Foxp3Cre+ mice
expressed considerable ACVR1C mRNA levels, Activin A expression and
responsiveness may have considerable influence over the biology of multiple Treg
populations.
It has been suggested that Activin can promote TGF signaling. Pathway
Analysis of our RNASeq results showed that the gene expression patterns most
impacted by YAP-deficiency in Tregs were highly relevant to immune control and the
diverse autoimmune pathologies resulting from the breakdown of such control. Among
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

12
these, the genes associated with the TGF signaling cascade were markedly altered
(Supplementary Fig. S8C). Furthermore RT-PCR analysis also showed reduced
transcript levels for several known TGF-responsive genes in Tregs from YAPfl/fl
Foxp3Cre+ mice (Supplementary Fig. S8D). In light of these findings, we suspected
that YAP contributes to Treg-mediated immune control at least in part by bolstering
TGF/SMAD signaling through the Activin/AcVR1C axis in these suppressor cells.
While Activin mRNA levels were low in naïve CD4+ T cells, in vitro differentiating
Tregs (naïve CD4+ T cells activated with anti-CD3/CD28 in the presence of IL-2 and
TGF) up-regulated Activin expression over time (Supplementary Fig. S9A). The
kinetics of this up-regulation paralleled the appearance of Foxp3 expression in these
cells (Supplementary Fig. S9B). qRT-PCR analysis also showed that expression of the
Activin Receptor (AcVR1C) was similarly low in naïve CD4+ T cells, but was robustly up-
regulated under in vitro culture conditions that generate iTreg (Supplementary Fig.
S8B, S9C). We went on to dissect which Treg-inducing stimuli was chiefly responsible
for inducing expression of YAP and elements of Activin/ACVR1C signaling. To this end,
naïve CD4+ T cells were activated in vitro with anti-CD3/CD28 antibodies, either alone
or in the presence of IL-2, TGF, or IL-2 and TGF. As expected, activation alone failed
to induce up-regulation of these genes or the canonical Treg transcription factor, Foxp3.
The cytokine TGF did trigger significant expression of Foxp3, as expected, but YAP as
well. Exposure to IL-2 along with TGF (but not IL-2 alone) greatly augmented
expression of YAP and Foxp3. Of the conditions tested, those up-regulating robust YAP
also brought about expression of Activin and ACVR1C (Supplementary Fig. S9D-G).
These findings further align the up-regulation of YAP expression and Activin signaling
with the Treg lineage, and shed some light on the largely unknown cast of molecular
characters regulating these processes in T cells.
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

13
To gain further insight into the mechanism of YAP-mediated ACVR1C up-
regulation, we explored the potential involvement of a known YAP-collaborating factor.
Mature YAP protein is known to contain a TEAD-binding domain, and prior studies
(largely conducted in non-T cells) have identified numerous target genes controlled by
the cooperation of these factors. Suggesting that transcription at the AcVR1c locus is
activated through YAP-TEAD interaction, the promoter sequence of this gene was found
to contain two TEAD consensus binding sites (Fig. 5A). To test the importance of TEAD
binding for YAP-dependent AcVR1c expression, we prepared luciferase-based reporter
constructs under the control of wild type murine AcVR1c promoter sequence. Mutant
constructs having either or both of the TEAD sites ablated were also designed (Fig. 5B).
Each AcVR1c-luciferase reporter construct was delivered into Jurkat T cells along with
an expression vector encoding YAP (“YAP1wt”) or a mutant version of this transcription
factor unable to interact with TEAD (“YAP1mut”) owing to an S-to-A mutation at residue
94 (“S94A”). In this system, expression of TEAD1 or YAP1wt alone induced only modest
activation of AcVR1c expression. In contrast, robust luciferase signal was detected when
wild type YAP and TEAD were co-expressed. Mutation of YAP’s TEAD-interaction site,
however, resulted in far less reporter activity (Fig. 5C) supporting the notion that YAP-
TEAD cooperation is necessary for optimal AcVR1c expression. Similarly, loss of a
single TEAD binding site in the promoter sequence reduced YAP-induced transcription
while mutation of both sites resulted in a significant and near complete loss of reporter
signal (Fig. 5D). These results clearly implicate a molecular partnership between YAP
and TEAD in the potentiation of Activin signaling through AcVR1c expression. This point
was further supported by ChIP assays showing both YAP1 and TEAD1 are enriched at
the AcVR1c locus in WT iTreg cells (Fig. 5E). Notably, in YAP cKO derived iTregs,
TEAD1 was still found interacting with the AcVR1c locus despite the absence of YAP
(Fig. 5E). These findings illuminate the mechanism behind YAP’s activation of activin
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

14
signaling.
Activin enhances SMAD/TGF signaling and Treg differentiation
Since Activin has been reported to promote SMAD signaling in non-T cells (30), we
tested whether Activin signaling in T cells could have a similar effect. SMAD activity was
assessed by western blot analysis of SMAD phosphorylation. Indeed, we found that
while untreated CD4+ T cells did not contain discernable levels of active
(phosphorylated) SMAD molecules, treatment with 5 or 10 ng/ml of Activin A resulted in
elevation of phospho-SMAD levels. As expected, TGF- treatment (0.5 or 2 ng/ml) also
induced SMAD phosphorylation. Importantly, combined Activin and TGF treatment
resulted in even further activation of the SMAD signaling pathway (Fig. 6A). These
findings suggest that Activin signaling can augment signaling along the TGF/SMAD
axis – a signaling pathway crucial for multiple aspects of Treg biology and immune
tolerance (7).
TGF/SMAD mediated events are important during the up-regulation of Foxp3 and the
generation of Tregs from naïve CD4+ T cell precursors. We next investigated whether
YAP-dependent Activin signaling can participate in the driving of this process. Having
shown that YAP plays an important role in promoting or maintaining Foxp3 expression
induced in the presence of various TGF concentrations (Fig. 2D), and having
implicated the transcription factor in the regulation of TGF-sensitive genes (Fig. 4,
Supplementary Fig. S8), we therefore postulated that YAP-mediated up-regulation of
AcVR1C and SMAD signaling might provide a crucial amplification of this important
Treg-supporting signaling pathway that allows for more robust or sustained Foxp3
expression.
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

15
To explore the involvement of Activin/AcVR1C signaling in the enhancement of Treg
differentiation by YAP, the effects of supplemental Activin A on in vitro Treg commitment
was also investigated. As expected, activation of naïve CD4+ T cells without TGF
yielded little-to-no Foxp3 induction regardless of YAP expression. Strikingly, activation of
WT cells with exogenous Activin A, even in the absence of TGF, generated a
population of Foxp3+ cells. While a suboptimal concentration of TGF resulted in
modest up-regulation of Foxp3 (mirroring the effects on SMAD activation), combined
treatment of WT naïve CD4+ T cells with low doses of TGFβ plus Activin A resulted in
synergistic promotion of Foxp3+ T cell induction. This induction of Tregs by Activin A
treatment alone was largely not seen upon Foxp3-driven knockout of YAP, and while
dual treatment of differentiating YAPfl/fl Foxp3Cre+ iTregs did enhance the generation of
Foxp3+ cells, it was to an extent far less than that seen in their WT counterparts (Fig.
6B). These results suggest that Activin signaling via YAP-dependent AcVR1C
expression on Treg not only augments TGF signaling but also can drive the process of
Foxp3 up-regulation. Supporting this notion, Naïve T cells lacking AcVR1c were found to
be less sensitive to TGFβ-induced iTreg differentiation their WT counterparts, particularly
when TGFβ concentrations were low (Supplementary Fig. S10A, B). Interestingly,
exogenous activin supplementation could do little to rescue the deficient Foxp3 induction
seen in naïve CD4+ T cells lacking either SMAD2 or SMAD3, or, for that matter, the
pronounced defect in iTreg generation seen in T cells genetically lacking both SMAD
molecules (Supplementary Fig. S11). This observation confirms that functional SMAD
signaling is required for activin-mediated enhancement of Treg generation, in agreement
with prior studies (31). In all, these findings are very much in line with a role for YAP-
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

16
driven Activin signaling in the augmentation of signaling down the SMAD/TGFβ axis in T
cells.
Activin-mediated support of Treg function is YAP/AcVR1C-dependent
YAP deficiency leads to improved anti-tumor immunity and a Treg pool that is insensitive
to an activator of the TGF/SMAD signaling pathway (i.e. Activin). We therefore
hypothesized that YAP facilitates robust Treg function in vivo through the induction of
AcVR1C, which in turn amplifies the pro-Treg signaling cascades. In order to determine
if the Treg-promoting effects of YAP were due to the up-regulation of AcVR1C, we set
out to test whether the defective Treg function seen in YAP knockouts could be restored
by ectopic expression of AcVR1C. In an in vitro suppression assay, as expected,
YAPfl/flFoxp3Cre+ derived Tregs transduced with an empty vector control expressed
reduced levels of ACVR1C protein and were much less efficient suppressors of naïve
CD4+ T cell proliferation than their WT counterparts. However, lentiviral-based delivery
of an ACVR1C-encoding expression construct into YAPfl/fl Foxp3Cre+ derived Tregs
more than rescued receptor expression, which greatly enhanced their suppressive
potency beyond even that of WT Tregs (Fig. 6C). These results support the conclusion
that Activin signaling through AcVR1C (up-regulated by YAP) can amplify the
suppressive potency of established Tregs as well as the TGF-driven differentiation of
iTregs and potentially other facets of this cytokine’s broadly immunosuppressive action.
Importantly, they also suggest that targeting either YAP or Activin signaling is likely to
undermine the tolerance promoting attributes of TGF and both subsets of Foxp3+ Treg
cells in the cancer setting. These approaches may provide avenues to enhanced anti-
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

17
tumor immunity either as novel treatments on their own or as potent enhancers of other
promising immunotherapeutic agents.
Activin blockade or AcVR1c knockout inhibits tumor growth
As an instigator of an apparent feed-forward loop capable of amplifying TGFβ/SMAD
activity, YAP presents a tempting target for those aiming to break tolerance in the cancer
setting. However, the targeting of YAP in cancer patients may prove problematic owing
to the molecule’s intracellular location and the chemical drawbacks of known inhibitors
(e.g. VP has noted solubility issues (29)). Therefore, the Activin/AcVR1C interaction is
likely to serve as a desirable alternative strategy. Having demonstrated the positive
effects of Activin signaling on the TGF/SMAD signaling pathway and the processes of
Treg generation and function, which can oppose immune-mediated tumor cell killing, we
suspected that disrupting Activin function should enhance anti-tumor immunity. We
therefore tested the potential of Activin targeting as an immunotherapeutic approach to
combat cancer. Administration of anti-activin monoclonal antibody to mice injected
subcutaneously with B16 melanoma markedly stunted the development of tumors
relative to an inert isotype control (Fig. 7A). We also tested the value of combining anti-
activin blocking antibody treatment with the anti-cancer vaccine GM-Vac. Treatment with
GM-Vac alone was able to partially slow the growth of tumors to an extent similar to anti-
activin monotherapy. However, combining anti-activin treatment with GM-Vac was able
to arrest tumor growth at a barely detectable size (Fig. 7A). Anti-activin treatment also
successfully reduced the frequency of Foxp3+ Tregs among TILs, and while GM-Vac -
receiving mice displayed some reduced Treg presence in their tumors, combined GM-
Vac and activin blockade resulted in dramatic loss of these suppressor T cells from the
tumor microenvironment (Fig. 7B). The effect of blocking activin on Tregs coincided with
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

18
increased frequencies of IFNγ-producing CD8+ and CD4+ T cells, an observation even
more prominent upon combination of GM-Vac and anti-activin treatments (Fig. 7C).
These results demonstrate the susceptibility of the YAP/AcVR1c/Activin axis to
therapeutic targeting at multiple points.
Along this line, B16 tumor growth was also markedly slower in AcVR1c knockout
mice compared to WT controls (Fig. 7D). Correspondingly, the TILs from AcVR1c-
deficient mice contained fewer Foxp3+ Tregs than their WT counterparts and displayed
a selective elevation of IFNγ-producing T cells (Fig. 7E, F). As with chemical YAP
inhibition and antibody-mediated activin blockade, administering GM-Vac to AcVR1c
knockout mice enhanced the already considerable anti-tumor effect of genetic AcVR1c
ablation (Supplementary Fig. S12). From these results it is clear that disrupting any of
the several elements of the YAP/Activin/SMAD axis can undermine immune suppression
and oppose tumor progression in mice.
In all, our findings support the conclusion that signaling along the YAP-regulated
Activin/ACVR1C axis can support Treg generation and function and potentially other
broadly immune-suppressing effects of the TGF/SMAD pathway. Importantly, they also
suggest that targeting this axis is likely to undermine the immune suppressive attributes
of TGF and Foxp3+ Treg cells in the cancer setting - either alone, or in combination
with other promising immunotherapeutic agents (e.g. immune checkpoint blocking
antibodies, anti-cancer vaccines).
DISCUSSION
Tregs are indispensable for restraining potentially lethal self-directed (autoimmune)
responses or over-exuberant ones mounted against normally harmless commensal
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

19
microbes (IBD) (1). However, in cancer patients, Tregs can be greatly enriched within
tumors and sometimes systemically (32). The suppressive function of these cells in this
setting dampens the effectiveness of tumor-directed immunity and is a major obstacle for
developing effective anti-cancer immunotherapies (21).
As part of an ongoing effort to identify precise mechanisms of Treg generation,
maintenance and function in the context of cancer, we have made the surprising
discovery that YAP, a transcription factor critical in developmental regulation of organ
size, is in fact an important factor in the generation and function of Tregs. Deletion of
Yap in T cells somewhat enhances both Th1 and Th17 development but most
impressively diminishes generation of iTreg under conditions of limiting TGFβ. YAP-
deficiency also negatively impacts the suppressive function of Tregs. The inability of
Tregs to suppress immunity in vivo in the absence of YAP was dramatically illustrated by
our B16 melanoma tumor model experiments (Fig. 3). The poorly immunogenic tumor
failed to grow in mice with Treg-specific Yap deletion, which displayed markedly
enhanced indicators of proinflammatory anti-tumor immunity compared to wild type
controls. This improved deployment of anti-tumor immunity was seen alongside a
markedly diminished Treg presence in the tumor microenvironment (Fig. 3E, F) –
observations also seen upon Treg-specific YAP deficiency across other, distinct tumor
models as well. These findings strongly suggest that YAP is important for the
accumulation and suppressive function of Tregs in the tumor microenvironment.
Furthermore, they imply that targeting YAP should be a potent means of overcoming
immune suppression in the cancer setting and improving the efficacy of endogenous and
therapeutically induced tumor killing by leukocyte. Further characterization of YAP
expression by Treg subsets found in different healthy and diseased tissues (including
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

20
tumors) should more clearly define this factor’s role in immune control in specific
physiological contexts.
Here we present a body of data strongly suggesting a Treg-specific role for YAP
in promoting the immune suppression capable of allowing the persistence and
progression of tumors in the cancer setting. Indeed, YAP-expression patterns and the
dramatically stunted tumor growth seen in YAPfl/fl Foxp3Cre+ mice support this.
However, comparing the degree of anti-tumor effect resulting from T cell- and Treg-
driven YAP deficiency, it appears that a slightly more dramatic effect is seen in the
former case. While the bulk of the effect seen in Fig. 3A is phenocopied by the more
restrictive deletion of YAP in only Foxp3+ cells (Fig. 3D), it is possible that YAP may
play a tumor-abetting role in some other T cell population capable of inducing the factor
in the cancer setting. While such YAP expression appears to have relatively minor
consequences next to Treg-derived YAP at least in the tumor models used in our study,
future work may bring to light additional layers of YAP’s pro-tumor effects involving cells
beyond Foxp3+ Tregs (such as anergized or exhausted T cells, non-Foxp3-expressing
TR1 Treg cells, etc.). These too may be susceptible to YAP-targeting strategies, which,
based on our results, clearly should have potent anti-tumor effects.
Indeed, using a known YAP antagonist with modest inhibitory activity (29) we confirmed
the potential of YAP as a target for Treg-undermining immunotherapies. While inhibiting
YAP alone slightly decreased tumor growth, we observed strong synergy in anti-tumor
activity and immunity boosting effects when the drug was combined with a tumor vaccine
and checkpoint inhibitor treatment that alone possess much less potent effects. These
findings suggest that YAP-targeting approaches should increase the efficacy of current
immunotherapies, potentially by enhancing the presence of activated effector leukocytes
in the tumor microenvironment.
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

21
Analysis of the downstream targets of YAP activity in Treg identified ACVR1C led to the
finding that the Activin-Activin Receptor signaling axis plays a major role in the
augmentation of TGF-β/SMAD signaling and Treg generation and function (summarized
in Supplementary Fig. S13). This pathway is highly important for the induction of
extrathymic Foxp3+ T cells from naïve CD4+ precursors as SMADS bind critical
enhancer regions for the Foxp3 gene (6,33). It is also important for sustaining Foxp3
expression and suppressive function in Tregs (7), and TGF- has been implicated as a
promoter of survival and phenotypic stabilization of thymic Tregs (34,35). With such
reliance on TGF and SMAD signaling, it stands to reason that Tregs employ
mechanisms to optimize or amplify the downstream signaling events and resultant gene
regulation triggered by this pathway. Such amplification mechanisms can be important
for maintaining the gene expression and phenotype traits underlying the suppressive
function of Tregs. Documented examples include the enzymatic conversion of latent
TGF to its active form (36) and the triggering of SMAD activation by galectin and CD44
(11). The up-regulation of YAP and subsequently ACVR1C – the receptor for a known
enhancer of SMAD signaling (i.e. Activin) in T cells exposed to TGFβ suggests the
existence of a positive feedback loop for this decidedly pro-Treg cytokine where
Activin/ACVR1C signaling can enhance the downstream signaling events triggered by
TGFβ. Reports of Activin expression in several tumor types (37,38) support the notion
that tumor-accumulating Tregs benefit particularly from Activin/ACVR1C signaling
facilitated by YAP induction.
Our proof-of-concept experiments demonstrate that this pro-Treg amplification
mechanism is susceptible to therapeutic disruption. Particularly, our findings suggest
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

22
that antibody-mediated Activin blockade may prove a most effective means for the
disruption of Treg- and tumor-abetting TGFβ activation in cancer patients. Additionally,
the development and vetting of therapeutic antibodies capable of neutralizing activin,
AcVR1C or blocking its association with AcVR1C in cancer patients may lead to new
and potent immunotherapeutic regimens that prevent anti-tumor immunity from stifling
Treg-enforced tolerance. On the other hand, our findings suggest that supplementation
of activin or other therapeutic enhancements of the Activin/ACVR1C axis could have
considerable potential as a strategy to correct inadequate immune regulation in settings
of autoimmune (e.g. Multiple Sclerosis) or inflammatory disease (e.g. Inflammatory
Bowel Disease). Future application of YAP inhibitors or Activin/ACVR1C ablation in
mouse models relevant to these and other pathologies of immune dysregulation will
shed light on whether this pro-Treg loop is generally important for immune control or if it
is principally operative in the tumor setting.
Our findings are, to our knowledge, the first to implicate YAP as a transcriptional
facilitator of Treg differentiation and function. While this molecule has been previously
studied for its regulation of development, organ size, regeneration, and tumorigenesis
(39), and its role as a transcriptional effector of gene expression downstream of the
Hippo pathway is well established. The importance of the Hippo pathway and its
associated cofactors in Tregs and immune control is only beginning to be understood. A
recent study showed that the Hippo pathway kinase known as Mst1 plays an important
role in stabilizing Foxp3 protein levels and supporting Treg function (40). Our present
findings reveal that YAP potentiates Treg-supporting SMAD activity in T cells through
Activin signaling. Notably though, this unexpected role appears to be independent of
other Hippo factors (i.e. Mst1/2, Lats1/2) as these, unlike YAP, were not highly up-
regulated in developing Tregs. Interestingly, another Hippo effector known as TAZ
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

23
(regarded to be a YAP paralog) was recently identified as a promoter of Th17
differentiation in naïve CD4+ T cells and an negative regulatory of Foxp3 function and
expression in these cells (41). This role for TAZ in the generation of proinflammatory T
cells was also apparently beyond its traditional Hippo-dependent role. Taken together,
these newly uncovered immunological roles played by YAP and TAZ suggest that
different molecular players in the Hippo pathway can have functionally opposite and
mechanistically distinct roles in determining the balance between inflammation and
tolerance. Further dissection of this pathway in T cells should add considerably to our
understanding of this balance and, based on our current study’s findings, may lead to
potent new immunotherapy approaches.
METHODS
Mice. C57/BL6 YAP fl/fl mice were generous gifts of Dr. Duojia Pan. C57/BL6 AcVR1c
knockout mice were gifts from Dr. Ning Lu. C57/BL6 CD4-cre and Foxp3-YFP-Cre
transgenic mice were purchased from the Jackson Laboratory. SMAD2-/-, SMAD3-/- and
SMAD2/3 double knockout mice on a C57BL/6 background were originally obtained from
Dr. Se-Jin Lee’s laboratory and where previously described (42). All animal experiments
performed where approved by the Johns Hopkins University Institutional Animal Care
and Use Committee (IACUC).
In vitro T-cell differentiation. Naïve CD4+ T cells (CD4+ CD25- CD62LHIGH) were
sorted on a FACS Aria II high-speed sorter. The sorted cells were activated with plate-
bound anti-CD3 (1µg/ml) and soluble anti-CD28 (2 or 4 µg/ml) in a 24-well plate with the
following polarizing conditions: Th1 (IL-12 (10ng/ml), anti-IL-4 (10µg/ml), Th2 (IL-4
(10ng/ml), anti-IFNγ (10µg/ml), anti-IL-12 (10µg/ml)), Th17 (IL-6 (10ng/ml), TGFβ1
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

24
(1.25ng/ml), IL-23 (10ng/ml), IL-1β (10ng/ml), anti-IFNγ (10µg/ml), anti-IL-4 (10µg/ml),
Treg (TGFβ1 (5ng/ml, or as indicated), IL-2 (100IU/ml) typically for 4 days, unless
otherwise indicated.
Human T cell isolation from Peripheral Blood. De-identified human peripheral blood
was obtained from blood bank in strict accordance with Johns Hopkins University School
of Medicine’s Institutional Review Board guidelines. Samples from a total of ten healthy,
adult volunteers (age range, 30 to 46 years). Peripheral blood mononuclear cells were
extracted from whole blood through a gradient of Ficoll-Paque PLUS (GE Healthcare).
CD4+ T cells were enriched using a Dynabeads Untouched CD4 T-cell isolation kit
(Invitrogen). Regulatory T-cells (Tregs) were identified and flow sorted via the following
staining profile: CD3+/CD4+/CD8–/CD25HIGH/CD127low/CD39+. Non-Treg CD4+ T cells
were sorted as previously described (43).
In vitro suppression assay. 0.1X106 WT naïve CD4+ T cells were labelled with CFSE
and cultured in a 96-well bottom plate with anti-CD3/CD28-conjugated beads at a cell to
bead ratio of 1:1. Serially diluted Treg cells (CD4+ CD25HIGH) were co-cultured for 72hrs
and cellular proliferation by CFSE was measured by flow cytometry.
Lentivirus production and transduction. HEK293T cells were purchased from the
ATCC in 2015 and were kept as a frozen stock. This cell line has not been authenticated
by the laboratory. Recombinant lentiviruses were generated using a three-plasmid
system as described previously (44). The AcVR1c cDNA was cloned into the modified
pLV lentiviral vector carrying CMV driven Thy1.1 as a transduction efficiency marker.
Virus was harvested at 48 and 72 h after transfection and titer was determined based on
percentages of Thy1.1-positive Jurkat T cells after transduction with serially diluted viral
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

25
supernatant. The titer, calculated as transducing units (TU)/ml of supernatant, was from
2x10^6 to 8x106 TU/ml. The virus-containing supernatant was concentrated using an
Amicon Ultra Concentrator (Millipore) and stored at -80°C. Gene transduction into
CD4+CD25- conventional T cells and CD4+CD25+ Tregs was performed by stimulating
cells with plate-bound anti-CD3 (10μg/ml) and soluble anti-CD28 (1μg/ml) with 60U/ml
human recombinant IL-2 for 16 h. Activated T cells were transduced with viral
supernatants supplemented with 60U/ml IL-2 and 8 μg ml-1 polybrene, followed by
centrifugation for 1h at 2,500 rpm. After transduction, 20U/ml human recombinant IL-2
(eBioscience) was added to the culture. At 40 h after transduction, Thy1.1+ Treg cells
were sorted for western blot, and/or suppression assay as indicated.
RNASeq Analysis. Spleen and peripheral lymph nodes were harvested from YAPwt/wt;
CD4-Cre- Wild-type (WT) and YAP flox/flox (fl/fl); CD4-Cre+ mice (n=5/group). CD4+ T
cells were magnetically enriched, and naïve (CD4+ CD62L+ CD25-) T cells and natural
Tregs (nTregs, CD4+ CD62L+/- CD25HIGH) cells were flow sorted from each group. For
activation condition, sorted nTreg cells were further activated with 2µg/ml of plate-coated
αCD3 and 2µg/ml of soluble αCD28 with TGF-β1 (5ng/ml) and IL-2 (100U/ml) for 24hrs.
2×106 nTreg (no stimulation or stimulation) from WT and YAP cKO mice were harvested
and washed with 1X PBS twice and immediately snap-frozen until further RNA-seq
analysis.
Construction of RNA-seq libraries. Total RNA was isolated by TRIZOL from naive
CD4+ T cells, or natural Treg cells with or without the stimulation anti-CD3/CD28 for
48hr from wild type or YAP cKO mice. RNA quality was monitored on Bioanalyzer.
Strand-specific RNA-seq libraries were prepared using TruSeq Stranded Total RNA LT
Sample Prep Kit (with Ribo-Zero Gold, RS-122-2301, Illumina) from 322 ng of total RNA
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

26
by following manufacturer protocols. Briefly, ribosomal RNAs (rRNAs) were depleted
using biotinylated, target-specific oligos combined with Ribo-Zero rRNA removal beads.
After purification, RNA was fragmented using divalent cations under elevated
temperature, and transcribed into first strand cDNA using reverse transcriptase and
random primers, followed by second strand cDNA synthesis using DNA Polymerase I
and RNase H. A single 'A' base was added to these cDNA fragments that were
subsequently ligated with the adapter. The products were enriched with 12-cycle PCR.
The concentration of final cDNA libraries in 30 ul ddH2O reached 24-27 ng/ul as
determined on Qubit 2.0.
Analysis of RNA-seq data. Sequencing was performed on Illumina Hiseq2000 at
Beijing Genomics Institute with the type of paired-end, 100bp. Data quality was
assessed by FastQC software
(http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Mapping to a mouse
reference genome (mm10) was conducted by TopHat. Differentially-Expressed genes
were called by Cuffdiff (45). The genes with p value < 0.05 and absolute values of log2-
transformed fold changes larger than 1.5 between WT and YAP cKO T cells were
considered differentially expressed. A heat map was generated in R statistical software
using the geom_tile function under ggplot2 package. Clustering was done with the
complete linkage and euclidean distance using hclust function in R statistical software.
Pathway Analysis (Ingenuity) was carried out as described previously (46).
Flow cytometry. For extracellular staining, harvested cells were washed and incubated
in PBS containing 1% FBS containing the below fluorochrome-conjugated antibodies in
a U-bottom 96-well plate. For intracellular cytokine staining, harvested cells were re-
stimulated in PMA and Ionomycin in the presence of Golgi-Plug (BD Biosciences). After
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

27
5 hour incubation, the cells were fixed/permeabilized (eBioscience) and incubated with
antibodies (see Supplementary Table1A for a comprehensive list). For cellular
proliferation, cell Trace CFSE cell proliferation kit (Invitrogen) was used per
manufacture’s manual.
Quantitative Real-Time PCR. RNA was extracted using Trizol (Invitrogen) followed by
cDNA synthesis reaction using SuperScript III (Invitrogen) in a 20ul reaction/well. The
same amount of RNA was used in each cDNA synthesis reaction measured by
NanoDrop Spectrophotometer (ThermoScientific). The same volume of cDNA per
sample was prepared for real-time PCR analysis using SYBR Green (Pierce) and the
indicated primers to assess transcript levels of each gene.
Tumor growth experiments. Murine B16-melanoma cells, MC38-colon cancer, and
EL4 thymoma cell lines were purchased from the ATCC and kept as frozen stock in
2015. These cell lines have not been authenticated by the laboratory. Cells were
cultured in vitro in DMEM plus 10% heat inactivated Fetal Bovine Serum and where
detached by trypsinization and washed prior to subcutaneously (s.c.) injection into the
shaved side flank of the indicated strains of female mice between the ages of 6-8 weeks
on a C57Bl/6 background (1x105 cells). In some experiments, 1-5x104 B16 melanoma
cells were injected each mouse in the footpad. Where indicated, once tumors were
palpable (7-10 days post-injection), 100 ml of 1x106 lethally irradiated (150Gy) B16 GM-
vaccine cells (GM-VAX) were injected s.c. into the contralateral limb. A hybridoma cell
line expressing a blocking anti-PD-1 antibody (clone G4) was obtained from Dr. Charles
Drake (JHH). 100 μg/mouse/injection of anti-PD-1 (G4) was injected intraperitoneally
twice a week once tumors were palpable (7-10 days) in conjunction with vaccine and
Verteporfin (USP, USP-1711461) treatments. Verteporfin was dosed at 2mg/mouse
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

28
diluted to 200µl with PBS and injected intraperitoneally every two days. Activin
neutralization antibodies and isotype control IgG were purchased from R&D.
100μg/mouse/injection of activin neutralizing antibodies was given intraperitoneally twice
a week. For all these experiments, 5-10 mice were used per group. Tumor progression
was assessed by measuring changes in tumor length (L) and width (W) and tumor
volume (V) over time. Tumor volume was calculated using the formula (L*W2)/2.
Molecular Cloning and Site-Directed Mutagenesis: Mouse AcVR1c promoter (1.2 kb)
was cloned from the genomic DNA of isolated CD4+ T cells and the sequence was
confirmed. The amplified clones were ligated to SacI/XhoI-digested pGL4.1-Basic Vector
(Promega) using the In-Fusion Cloning Kit (Clontech). Site-directed mutagenesis was
carried out using QuikChange Lightning Kit (Agilent Technologies).
Transient Transfection and Luciferase Assay: Jurkat T cells (clone E6-1) were
purchased from the ATCC in 2016 and were kept as a frozen stock. This cell line has not
been authenticated by the laboratory. Jurkat T cells (1.5 × 107) were transfected with 5
μg pGL4.1-AcVR1c, 1 μg of pRL-TK Vector (Promega) and other indicated plasmids by
electroporation using Nucleofector II (Amaxa/Lonza). The cells were rested for overnight
and stimulated with mock or PMA/Ionomycin for 8 hr before harvested and lysed
followed by luminescence measurement using a Dual-Luciferase Assay (Promega) as
per manufacturer’s instructions.
ChIP Assay: ChIP assay was performed according to the manufacturer's guidance
(Invitrogen MAGnify ChIP system). Briefly, sorted CD4+ iTreg cells were activated with
αCD3/αCD28–conjugated beads for overnight and fixed with 2% formaldehyde.
Sonicated DNA was immunoprecipitated with anti-YAP1 (Cell Signaling Technology),
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

29
and anti-TEAD1 (Santa Cruz Biotechnology). The immunoprecipitated chromatin was
analyzed on Roche LightCycler 480 by SYBR Green using the following primers for
AcVR1c promoter.
Statistical Analyses: Values are presented as means ± SEM where appropriate.
Statistical differences among multiple groups were determined using a two way analysis
of variance (ANOVA) with NewmaneKeuls Multiple Comparison Test, unless otherwise
indicated. Unpaired, two-tailed student’s t-tests were used for single-comparisons. In
general, P values <0.05 were considered significant and are indicated as follows:
*P<0.05, **P<0.01, ***P<0.002, ****P<0.001, ns: not significant. GraphPad Prism 7 was
used to calculate P values.
Data Availability: RNAseq dataset has been uploaded to an appropriate online
repository. The GEO accession number is GSE112593.
ACKNOWLEDGEMENTS
F. Pan’s research is supported by the Bloomberg-Kimmel Institute (Immunometabolism
Program & Immune Modulation Program), the Melanoma Research Alliance, the NIH
(RO1AI099300, RO1AI089830 and R01AI137046), The DoD (PC130767); J. Barbi’s
research is supported by the Melanoma Research Foundation, Phi Beta Psi, the Roswell
Park Alliance Foundation and NCI grant P30CA016056. The Li Lab was supported by
the National Natural Science Committee of China (No. 81725004) and Shanghai
Science and Technology Committee (No.16410723600). L. Lu’s research is supported
by the National Natural Science Fund of China (grants 81571564, grant 81521004 and
81522020) and the Foundation of Jiangsu Collaborative Innovation Center of Biomedical
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

30
Functional Materials (LL). D. Pan is an investigator of the Howard Hughes Medical
Institute.
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

31
REFERENCES
1. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell 2008;133(5):775-87 doi 10.1016/j.cell.2008.05.009.
2. Whiteside TL. What are regulatory T cells (Treg) regulating in cancer and why? Semin Cancer Biol 2012;22(4):327-34 doi 10.1016/j.semcancer.2012.03.004.
3. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet 2001;27(1):20-1 doi 10.1038/83713.
4. Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet 2001;27(1):68-73 doi 10.1038/83784.
5. Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol 2012;30:531-64 doi 10.1146/annurev.immunol.25.022106.141623.
6. Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, Rudensky AY. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature 2010;463(7282):808-12 doi 10.1038/nature08750.
7. Tran DQ. TGF-beta: the sword, the wand, and the shield of FOXP3(+) regulatory T cells. J Mol Cell Biol 2012;4(1):29-37 doi 10.1093/jmcb/mjr033.
8. Marie JC, Letterio JJ, Gavin M, Rudensky AY. TGF-beta1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J Exp Med 2005;201(7):1061-7 doi 10.1084/jem.20042276.
9. Liu Y, Zhang P, Li J, Kulkarni AB, Perruche S, Chen W. A critical function for TGF-beta signaling in the development of natural CD4+CD25+Foxp3+ regulatory T cells. Nat Immunol 2008;9(6):632-40 doi 10.1038/ni.1607.
10. Takimoto T, Wakabayashi Y, Sekiya T, Inoue N, Morita R, Ichiyama K, et al. Smad2 and Smad3 are redundantly essential for the TGF-beta-mediated regulation of regulatory T plasticity and Th1 development. J Immunol 2010;185(2):842-55 doi 10.4049/jimmunol.0904100.
11. Wu C, Thalhamer T, Franca RF, Xiao S, Wang C, Hotta C, et al. Galectin-9-CD44 interaction enhances stability and function of adaptive regulatory T cells. Immunity 2014;41(2):270-82 doi 10.1016/j.immuni.2014.06.011.
12. Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, et al. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 2007;130(6):1120-33 doi 10.1016/j.cell.2007.07.019.
13. Yu FX, Zhao B, Guan KL. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015;163(4):811-28 doi 10.1016/j.cell.2015.10.044.
14. Pan D. The hippo signaling pathway in development and cancer. Dev Cell 2010;19(4):491-505 doi 10.1016/j.devcel.2010.09.011.
15. Kapoor A, Yao W, Ying H, Hua S, Liewen A, Wang Q, et al. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell 2014;158(1):185-97 doi 10.1016/j.cell.2014.06.003.
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

32
16. Shao DD, Xue W, Krall EB, Bhutkar A, Piccioni F, Wang X, et al. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell 2014;158(1):171-84 doi 10.1016/j.cell.2014.06.004.
17. Ganem NJ, Cornils H, Chiu SY, O'Rourke KP, Arnaud J, Yimlamai D, et al. Cytokinesis failure triggers hippo tumor suppressor pathway activation. Cell 2014;158(4):833-48 doi 10.1016/j.cell.2014.06.029.
18. Varelas X, Samavarchi-Tehrani P, Narimatsu M, Weiss A, Cockburn K, Larsen BG, et al. The Crumbs complex couples cell density sensing to Hippo-dependent control of the TGF-beta-SMAD pathway. Dev Cell 2010;19(6):831-44 doi 10.1016/j.devcel.2010.11.012.
19. Fujii M, Toyoda T, Nakanishi H, Yatabe Y, Sato A, Matsudaira Y, et al. TGF-beta synergizes with defects in the Hippo pathway to stimulate human malignant mesothelioma growth. J Exp Med 2012;209(3):479-94 doi 10.1084/jem.20111653.
20. Nishikawa H, Sakaguchi S. Regulatory T cells in tumor immunity. Int J Cancer 2010;127(4):759-67 doi 10.1002/ijc.25429.
21. Klages K, Mayer CT, Lahl K, Loddenkemper C, Teng MW, Ngiow SF, et al. Selective depletion of Foxp3+ regulatory T cells improves effective therapeutic vaccination against established melanoma. Cancer Res 2010;70(20):7788-99 doi 10.1158/0008-5472.CAN-10-1736.
22. Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A 2010;107(9):4275-80 doi 10.1073/pnas.0915174107.
23. Snyder A, Wolchok JD, Chan TA. Genetic basis for clinical response to CTLA-4 blockade. N Engl J Med 2015;372(8):783 doi 10.1056/NEJMc1415938.
24. Callahan MK, Postow MA, Wolchok JD. CTLA-4 and PD-1 Pathway Blockade: Combinations in the Clinic. Front Oncol 2014;4:385 doi 10.3389/fonc.2014.00385.
25. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 2015;27(4):450-61 doi 10.1016/j.ccell.2015.03.001.
26. Duraiswamy J, Kaluza KM, Freeman GJ, Coukos G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res 2013;73(12):3591-603 doi 10.1158/0008-5472.CAN-12-4100.
27. Sommermeyer D, Hudecek M, Kosasih PL, Gogishvili T, Maloney DG, Turtle CJ, et al. Chimeric antigen receptor-modified T cells derived from defined CD8 and CD4 subsets confer superior antitumor reactivity in vivo. Leukemia 2015 doi 10.1038/leu.2015.247.
28. Jensen MC, Riddell SR. Design and implementation of adoptive therapy with chimeric antigen receptor-modified T cells. Immunol Rev 2014;257(1):127-44 doi 10.1111/imr.12139.
29. Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA, et al. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

33
oncogenic activity of YAP. Genes Dev 2012;26(12):1300-5 doi 10.1101/gad.192856.112.
30. Schmierer B, Schuster MK, Shkumatava A, Kuchler K. Activin a signaling induces Smad2, but not Smad3, requiring protein kinase a activity in granulosa cells from the avian ovary. J Biol Chem 2003;278(23):21197-203 doi 10.1074/jbc.M212425200.
31. Huber S, Stahl FR, Schrader J, Luth S, Presser K, Carambia A, et al. Activin a promotes the TGF-beta-induced conversion of CD4+CD25- T cells into Foxp3+ induced regulatory T cells. J Immunol 2009;182(8):4633-40 doi 10.4049/jimmunol.0803143.
32. Miller AM, Lundberg K, Ozenci V, Banham AH, Hellstrom M, Egevad L, et al. CD4+CD25high T cells are enriched in the tumor and peripheral blood of prostate cancer patients. J Immunol 2006;177(10):7398-405.
33. Josefowicz SZ, Niec RE, Kim HY, Treuting P, Chinen T, Zheng Y, et al. Extrathymically generated regulatory T cells control mucosal TH2 inflammation. Nature 2012;482(7385):395-9 doi 10.1038/nature10772.
34. Zheng SG, Wang J, Horwitz DA. Cutting edge: Foxp3+CD4+CD25+ regulatory T cells induced by IL-2 and TGF-beta are resistant to Th17 conversion by IL-6. J Immunol 2008;180(11):7112-6.
35. Ouyang W, Beckett O, Ma Q, Li MO. Transforming growth factor-beta signaling curbs thymic negative selection promoting regulatory T cell development. Immunity 2010;32(5):642-53 doi 10.1016/j.immuni.2010.04.012.
36. Worthington JJ, Kelly A, Smedley C, Bauche D, Campbell S, Marie JC, et al. Integrin alphavbeta8-Mediated TGF-beta Activation by Effector Regulatory T Cells Is Essential for Suppression of T-Cell-Mediated Inflammation. Immunity 2015;42(5):903-15 doi 10.1016/j.immuni.2015.04.012.
37. Loomans HA, Andl CD. Intertwining of Activin A and TGFbeta Signaling: Dual Roles in Cancer Progression and Cancer Cell Invasion. Cancers (Basel) 2014;7(1):70-91 doi 10.3390/cancers7010070.
38. Lonardo E, Hermann PC, Mueller MT, Huber S, Balic A, Miranda-Lorenzo I, et al. Nodal/Activin signaling drives self-renewal and tumorigenicity of pancreatic cancer stem cells and provides a target for combined drug therapy. Cell Stem Cell 2011;9(5):433-46 doi 10.1016/j.stem.2011.10.001.
39. Moroishi T, Hansen CG, Guan KL. The emerging roles of YAP and TAZ in cancer. Nat Rev Cancer 2015;15(2):73-9 doi 10.1038/nrc3876.
40. Li J, Du X, Shi H, Deng K, Chi H, Tao W. Mammalian Sterile 20-like Kinase 1 (Mst1) Enhances the Stability of Forkhead Box P3 (Foxp3) and the Function of Regulatory T Cells by Modulating Foxp3 Acetylation. J Biol Chem 2015;290(52):30762-70 doi 10.1074/jbc.M115.668442.
41. Geng J, Yu S, Zhao H, Sun X, Li X, Wang P, et al. The transcriptional coactivator TAZ regulates reciprocal differentiation of TH17 cells and Treg cells. Nat Immunol 2017;18(7):800-12 doi 10.1038/ni.3748.
42. Park BV, Freeman ZT, Ghasemzadeh A, Chattergoon MA, Rutebemberwa A, Steigner J, et al. TGFbeta1-Mediated SMAD3 Enhances PD-1 Expression on Antigen-Specific T Cells in Cancer. Cancer Discov 2016;6(12):1366-81 doi 10.1158/2159-8290.CD-15-1347.
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

34
43. Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity 2009;30(6):899-911 doi 10.1016/j.immuni.2009.03.019.
44. Pan F, Yu H, Dang EV, Barbi J, Pan X, Grosso JF, et al. Eos mediates Foxp3-dependent gene silencing in CD4+ regulatory T cells. Science 2009;325(5944):1142-6 doi 10.1126/science.1176077.
45. Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 2012;7(3):562-78 doi 10.1038/nprot.2012.016.
46. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102(43):15545-50 doi 10.1073/pnas.0506580102.
47. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell 2011;146(5):772-84 doi 10.1016/j.cell.2011.07.033.
FIGURE LEGENDS
Figure 1. Expression of YAP mRNA and protein by Thelper subsets. Naïve CD4+ T cells
(CD4+ CD25- CD62L+) were isolated from the spleen and lymph nodes of wild-type (WT)
C57BL/6 mice and activated under polarizing conditions to generate the indicated Thelper
subset. The cells were harvested for different time points and mRNA or protein levels of YAP
were assessed by (A) qRT-PCR and (B) western blot. (C) Human Tregs (CD3+/CD4+/CD8–
/CD25HIGH/CD127low/CD39+) and non-Treg CD4+ T cells were obtained from the peripheral
blood of healthy donors by FACS after Ficoll-Paque PLUS gradient centrifugation and magnetic
bead enrichment of CD4+ T cells. YAP mRNA was measured by qRT-PCR. For A and B, shown
are representative findings from at least 3 independent experiments (mean +/-SEM of triplicates
for A). For C, mean expression of YAP mRNA is shown for 10 healthy human donor samples.
The data were analyzed using the Student t test and considered significant if *, p<0.05;**,
p<0.01; ****, p<0.001.
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

35
Figure 2. The effects of YAP-deficiency on CD4+ T cell subsets. (A-C) Naïve CD4+ T cells
(CD4+ CD25- CD62L+) were isolated from wild-type (WT) YAPfl/fl CD4Cre- or YAP fl/fl
CD4Cre+ (YAP cKO) mice (n= 5/group/experiment) and were activated under the indicated
polarizing conditions for 4 days. The cells were harvested and signature cytokines and
transcription factors for each Th subset were assessed by flow cytometry and qRT-PCR. Shown
in A and B are representative flow cytometry results and the mean percentage of cells +/- SEM
from at least 3 independent experiments. C depicts the relative expression level of IL-17A
transcript during the early stages of Th17 cell differentiation for WT and YAP cKO derive cells
+/-SEM. (D) iTreg differentiation of WT (YAPfl/fl Foxp3Cre-), and Foxp3Cre-driven YAP
knockout mice (YAPfl/fl Foxp3Cre+). Naïve CD4+ T cells were isolated from the indicated mice
as above before activation in the presence of IL-2 and varying concentrations of TGFβ. Treg
differentiation was assessed by intracellular staining for Foxp3 and flow cytometry analysis.
Shown are representative histograms (left panels) and the mean fluorescence intensity of Foxp3
staining (MFI) was found in at least 3 independent experiments average MFI +/-SEM are
shown. (E) The suppressive function of WT or YAP cKO derived Tregs (CD4+ CD25HIGH T cells
FACS isolated from lymph node and spleen cell suspensions) was determined using an in vitro
suppression assay. Naïve CD4+ T cells (responders) and Tregs were isolated from the
indicated mice (n= 5/group/experiment). WT responders were pre-stained with CFSE and co-
cultured with WT and YAP cKO derived Tregs at the indicated ratios. The cultures were
activated with anti-CD3/anti-CD28-conjugated beads at a cell to bead ratio of 1:1. The
percentage of proliferating (CFSELOW) responder cells in each culture was determined by flow
cytometry. Shown are representative histograms (upper panel) and the mean percentages of
proliferating cells +/-SEM over at least 3 independent experiments (lower panel). For A-E,
significant differences were determined by the Student t test (*p<0.05; **p<0.02; ***p<0.002; ns:
not significant).
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

36
Figure 3. The impact of T cell- and Treg- restricted YAP-deficiency and YAP inhibition on
the anti-tumor response. Wild-type (WT; n= 5) or YAP fl/fl CD4Cre+ (YAP cKO; n= 5) mice
were challenged with 5x105 B16-melanoma cells (s.c.) and the tumor dimensions were
measured every 2 days and tumor volume was calculated (A, B). On Day 21, the mice were
euthanized and tumor-infiltrating lymphocytes (TILs) were isolated from the excised tumors. (C)
TILs were gated on CD4+ and CD8+ T cells and effector cytokines IFN-γ and TNF-α levels were
measured by flow cytometry. (D) Tumor challenge of YAPfl/fl Foxp3Cre+ mice (n= 4) and WT
controls (n= 6) was carried out as above, and the frequencies of IFNγ- and IL-17-producing
leukocytes within the B16 TILs of these mice were determined by flow cytometry (E, left). Mean
frequencies of IFNγ+ TILs as well as the number of IFNγ+/CD4+ and IFNγ+/CD8+ per gram of
tumor tissue (+/- SEM) are also shown (E, middle and right panels, respectively) from at least 3
independent experiments. (F) Proportions of Foxp3+ Tregs within the TILs of WT and YAPfl/fl
Foxp3Cre+ mice (left) were also found by intracellular staining followed by flow cytometry
analysis. Average Treg frequencies amongst CD4+ TILs were found as where and the ratio of
tumor CD8+ T cell-to-Foxp3+ Treg numbers are also shown (center and right, respectively). (G)
Targeting YAP improves the anti-tumor effects of immunotherapies. C57BL/6 mice were
challenged with B16-melanoma cells and tumor progression was monitored as above
mentioned. Cohorts of mice were treated with i.p. injected VP, GM-Vac, anti-PD1 antibody, VP
and anti-PD-1, or VP and GM-Vac beginning day 7 post-tumor injection. Control mice were left
untreated (n=5/group). Shown are the mean tumor volumes for the groups +/- SEM. (H)
Characterization of TILs from treated and control mice were also determined by flow cytometry.
The frequencies of IFNγ-producing CD4+ and CD8+ T cells as well as the ratio of tumor CD4+
and CD8+ T cell numbers to Foxp3+ Treg numbers were also. For A, D, and G, the mean tumor
volumes for the groups are shown over time +/- SEM. Bar graphs in C, E, F, and H depict the
mean frequency (%), ratio or absolute number/gram tumor of the indicated immune cell subset
+/- SEM in 3 independent experiments. All other findings are representative of at least two
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

37
independent experiments. Statistically significant differences were determined by t test for all
panels except for G where a two-way ANOVA was used.
Figure 4. RNASeq analysis of wild type and YAP-deficient Treg transcriptomes. Genes
that had their expression level significantly changed by YAP knockout in naive CD4+ T cells,
unstimulated Tregs, or stimulated Tregs were determined by RNASeq. Results are presented as
a heat map. Genes are arranged based on the Fold Change in expression between WT and
YAP-deficient Tregs (Genes with a Fold Change of more than 3 are shown). The color
representation from green to red denotes log2-transformed FPKM from -2 to 2.
Figure 5. YAP drives ACVR1C transcription through molecular cooperation with TEAD.
(A) Shown is a portion (1.2kb) of the murine AcVR1c promoter with TEAD binding sites
determined by transcription factor prediction software. (B) Design schema of luciferase-based
AcVR1c reporter constructs (C) AcVR1c Reporter Assays. The mAcVR1c-wt reporter construct
was co-transfected into Jurkat T cells along with the indicated YAP and TEAD expression
constructs or an Empty Vector control. Cells were cultured with or without PMA/ionomycin
activation for 8 hrs prior to harvest and cell lysis. Luciferase activity was determined as
previously described (47). (D) As in C, YAP and TEAD expression were delivered to Jurkat T
cells, except cells received variants of the mAcVR1c reporter possessing one, both, or none of
the identified TEAD binding sites. Reporter activity was determined as in C. For both C and D,
the mean relative luciferase values +/-SEM are shown for the results of 3 independent
experiments. (E) A chromatin immunoprecipitation (ChIP) assay was carried out in iTregs
generated from WT- and YAP cKO-derived naïve CD4+ T cells. The ability of antibodies against
TEAD1 and YAP to pull down the indicated factors along with the AcVR1c promoter region was
calculated based upon qPCR relative to a control immunoglobulin G (IgG). The relative
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

38
enrichment for each factor over 3 experiments are shown (+/-SEM). Significant differences for
all experiments shown were determined by the Student t test.
Figure 6. The YAP/Activin/ACVR1C pathway enhances SMAD activation, Treg generation
and function, and tumor progression. (A) Freshly isolated CD4+CD25- T cells were isolated
from the lymphoid tissues of WT mice (n= 6/experiment), cultured with plate bound anti-CD3
(2µg/ml) and soluble anti-CD28 (2µg/ml) for 24 hrs, followed by treatment with different
concentrations of Activin A and TGFβ as indicated for an additional 12 hours. Cells were
harvested and subjected to SDS-PAGE and Western blot with the indicated antibodies (left
panel). Band densities indicating protein amount were quantified by using Image J software,
normalized to β-actin loading controls, and the mean density +/-SEM across 3 independent
experiments were found (right panel). (B) Naïve CD4+ T cells from YAPfl/fl, Foxp3Cre+ and
YAPwt/wt, Foxp3Cre+ (WT) mice (n= 6/group/experiment) were stimulated with andti-
CD3/CD28 antibodies (1 and 4 µg/ml, respectively) for 3 days in the presence of IL-2 (100U/ml)
and the indicated doses of TGF and exogenous Activin A. Activin was dosed at 50ng/ml on
day 0 and day 2. Treg induction was assessed by flow cytometric detection of intracellular
Foxp3. Shown are representative Foxp3 stainings (left panel) and the mean results of 3
independent experiments +/- SEM (right) (C) Effect of ectopic AcVR1c expression on YAP-
deficient Tregs. As before, WT responder T cells and Tregs were isolated from the indicated
mice (n= 6/group/experiment). Following lentiviral delivery of an ACVR1C over-expression
construct or an empty vector control construct to YAPfl/fl Foxp3Cre+ Tregs (activated ex vivo
overnight with anti-CD3/CD28 antibodies and IL-2), the functional capacity of these cells were
assessed in vitro. The transduced Tregs were co-cultured with CFSE-stained CD45.1+ naïve
CD4+ T cells (responders) at the indicated ratio and antigen presenting cells (T cell-depleted
splenocytes). After 5 days of activation, responder cell proliferation was assessed by flow
cytometry. Shown at left are representative plots of responder cell gated (CD45.1+/CD4+)
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

39
events from one of two independent experiments with like results. The immunoblot at right
confirms expression levels of AcVR1c in transduced Tregs, and the bar graph (lower right
panel) depicts the mean fraction of proliferating responder cells over all experiments +/-SEM.
Where indicated by asterisks, significant differences were found by the Student t test.
Figure 7. Activin blockade and AcVR1c-deficiency slows B16 tumor growth and
enhances the anti-tumor immune response. (A) B16 melanoma cells were injected into
individual female C57BL/6 mice (8-12 weeks of age). Tumor-bearing mice were randomly
assigned into treatment groups once tumors were palpable ~7 days post-injection. Anti-activin A
antibodies (R&D Systems) were administered (100 µg/mouse/injection) intraperitoneally twice a
week once to one group. Another group received like does of control IgG1. Other cohorts of
tumor-bearing mice received GM-vaccine (100 µl of 1x106 lethally irradiated (150Gy) B16 GM-
vaccine cells or combined anti-Activin/GM-vaccine treatment (n=10 mice per group). (B) Treg
frequencies among the TILs of treated mice. Intracellular staining of Foxp3 in CD4+ TILs from
the indicated treatment groups were determined by flow cytometry. (C) IFNγ-producing CD4+
and CD8+ T cells recovered from tumor cell suspensions were similarly assessed. (D) The right
flank of 8-week WT and AcVR1cKO female mice (C57BL/6 background; n=8/group) were
injected with 4x105 B16 cells in 100 μl PBS. (E) The proportions of IFNγ- and TNFα-expressing
T cells (CD3+) with the TILs of these mice were determined by flow cytometry (F) as where the
frequencies of Foxp3 and IL-17+ CD4+ T cells. For (A) and (D) Tumor development and
changes in tumor volume were recorded for all groups and the mean volume +/-SEM for each
are displayed. For B, C, E and F, representative flow plots from a single mice from each group
are depicted (left panels) alongside the mean cell frequencies across 3 independent
experiments (right panels). All experiments were repeated at least three times. Significant
differences were determined by a Student t test for all panels, except A were a two-way ANOVA
was used.
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

A B
Figure 1
Th1 Th2 Treg Th17
Anti-YAP
anti-actin
C
YAP
mR
NA
exp
ress
ion
****
N a iv e T h 0 T h 1 T h 2 T h 1 7 T r e g
0
2 0
4 0
6 0
8 0
1 0 0
**
CD4+Non Treg Treg
YAP
mR
NA
exp
ress
ion
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

A B
C
D
E
Figure 2
WT
YAP cKO
CFSE
8:1 4:1 2:1 1:1 Unstimulated 1:0 Teff:Treg
1 :1 2 :1 4 :1 8 :1
0
2 0
4 0
6 0
8 0
1 0 0
% P
rolif
erat
ion
Teff:Treg
*
****
****
ns WT YAP cKO
Foxp3
Co
un
t
0 0.05 0.1 0.5 5 TGF-β (ng/ml)
WT
YAP fl/fl Foxp3Cre+
Isotype control
0 0 .0 5 0 .1 0 .5 5
0
1 0 0
2 0 0
3 0 0
4 0 0
Foxp
3 M
FI
TGF-β (ng/ml)
* *
* ns ns
WT YAP fl/fl Foxp3Cre+
IL -2 IF N -
0
2 0
4 0
6 0
8 0
1 0 0
ns
*
% c
ells
WT YAP cKO
WT YAP cKO
IL-2
IFN
-
CD4+ WT YAP cKO
Foxp3
IL-1
7A
CD4+
IL -1 7 A F o x p 3
0
1 0
2 0
3 0
4 0
5 0WT YAP cKO
*
***
% c
ells
W T N a iv e 6 h r s 1 2 h r s
0
1 0 0
2 0 0
3 0 0
4 0 0
5 0 0
IL-1
7A
Rel
ativ
e Ex
pre
ssio
n
**
*** WT YAP cKO
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

D E
F G
H
IL-17
IFNγ
WT
YAP fl/fl Foxp3Cre+
CD4+ TILs
WT
YAP fl/fl Foxp3Cre+
CD4
Foxp
3
0
500
1000
1500
2000
2500
3000
4 7 10 13 16 17 19 21
YapFl/Fl Foxp3Cre-
YapFl/Fl Foxp3Cre+
Tum
or
Vo
lum
e (m
m^3
)
Days post-injection
*
*
A B C
Figure 3
0
2
4
6
8
CD
4+
Fo
xp
3-/
CD
4+
Fo
xp
3+
* *
* *
CD
4+F
oxp
3-/
CD
4+Fo
xp3+
0
2
4
6
8
1 0
1 2
CD
8+
/C
D4
+F
ox
p3
+
* *
* *
CD
8+/
CD
4+F
oxp
3+
0
1 0
2 0
3 0
4 0
%IF
N-
+C
D4
+
* *
* *
%C
D4
+IFN
γ
0
1 0
2 0
3 0
4 0
5 0
6 0
7 0
%IF
N+
CD
8+
*
* *
%C
D8
+IFN
γ
0
2
4
6
8
10
12
14
16
18
% T
ILs IFNγ+
** **
**
WT YAP fl/fl Foxp3Cre+
****
0
5
10
15
20
25
30
35*
% F
oxp
3+
of
CD
4+
TILs
0
1
2
3
4
5
6
7
8
CD
8+/
Treg
Rat
io
*
WT YAP fl/fl Foxp3cre+
C D 8+
IF N +
T N F+
C D 4+ I
F N +
T N F+
0
1 0
2 0
3 0
4 0
% c
ells
WT YAP cKO
* *
CD8+IFN-γ+
TNF-α+ CD4+IFN-γ+
TNF-α+
WT
YAP cKO
1 1 1 3 1 5 1 7 1 9 2 1
0
4 0 0
8 0 0
1 2 0 0
Tum
or
Vo
lum
e (m
m3)
*
****
ns ns ns
WT YAP cKO
Days
0
1
2
3
4
5
6
Nu
mb
er o
f IF
NΥ+
CD
4+
ce
lls/
g
of
tum
or
(x1
0^4
)
0
5
10
15
20
25
Nu
mb
er o
f IF
NΥ+
CD
8+
cel
ls/
g o
f tu
mo
r (x
10
^4)
Tum
or
Vo
lum
e (m
m3)
Days post-injection
*
*
*
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

B
WT YAP cKO WT WT YAP cKO YAP cKO
Naïve T cells Unstim nTreg Stim nTreg
log2(fold change) KO Treg/WT Treg
Figure 4
A
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124
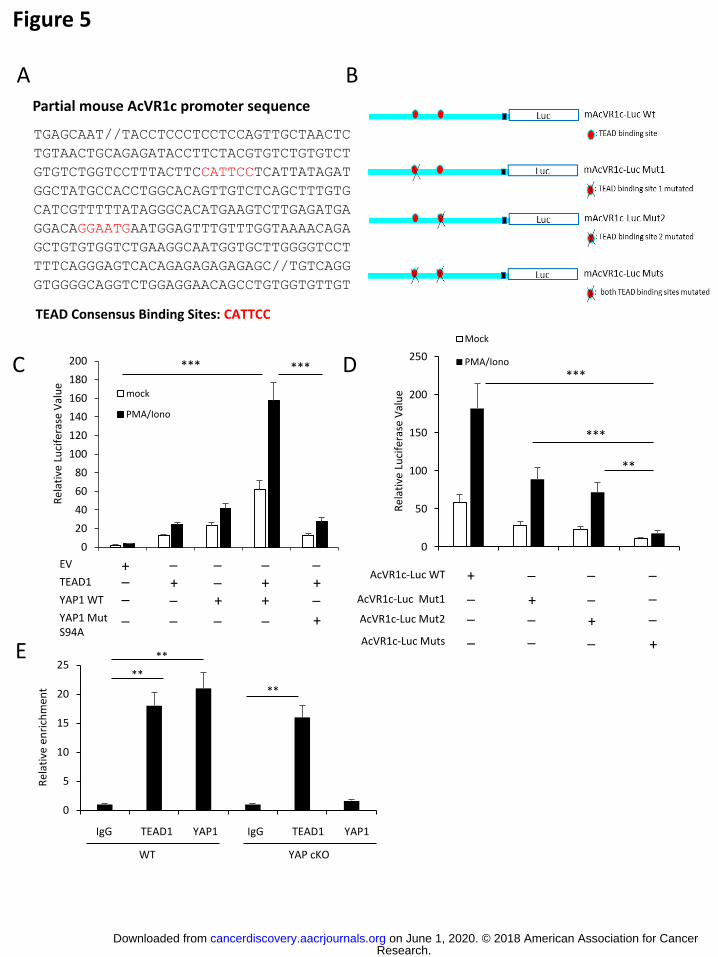
TEAD Consensus Binding Sites: CATTCC
Partial mouse AcVR1c promoter sequence
TGAGCAAT//TACCTCCCTCCTCCAGTTGCTAACTC
TGTAACTGCAGAGATACCTTCTACGTGTCTGTGTCT
GTGTCTGGTCCTTTACTTCCATTCCTCATTATAGAT
GGCTATGCCACCTGGCACAGTTGTCTCAGCTTTGTG
CATCGTTTTTATAGGGCACATGAAGTCTTGAGATGA
GGACAGGAATGAATGGAGTTTGTTTGGTAAAACAGA
GCTGTGTGGTCTGAAGGCAATGGTGCTTGGGGTCCT
TTTCAGGGAGTCACAGAGAGAGAGAGC//TGTCAGG
GTGGGGCAGGTCTGGAGGAACAGCCTGTGGTGTTGT
A B
Figure 5
C
E
D
0
20
40
60
80
100
120
140
160
180
200
mock
PMA/Iono
Rel
ativ
e Lu
cife
rase
Val
ue
EV
TEAD1
YAP1 WT
YAP1 Mut S94A
+ +
+ + + +
+
_ _ _ _
_ _ _ _
_ _ _ _
_
*** ***
0
50
100
150
200
250
Mock
PMA/Iono
Rel
ativ
e Lu
cife
rase
Val
ue
AcVR1c-Luc WT
AcVR1c-Luc Mut1
AcVR1c-Luc Mut2
AcVR1c-Luc Muts
+
+
+
+
_ _ _
_ _ _
_
_ _ _
_ _
**
***
***
0
5
10
15
20
25
Rel
ativ
e en
rich
me
nt
IgG TEAD1 YAP1 IgG TEAD1 YAP1
WT YAP cKO
**
**
**
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124
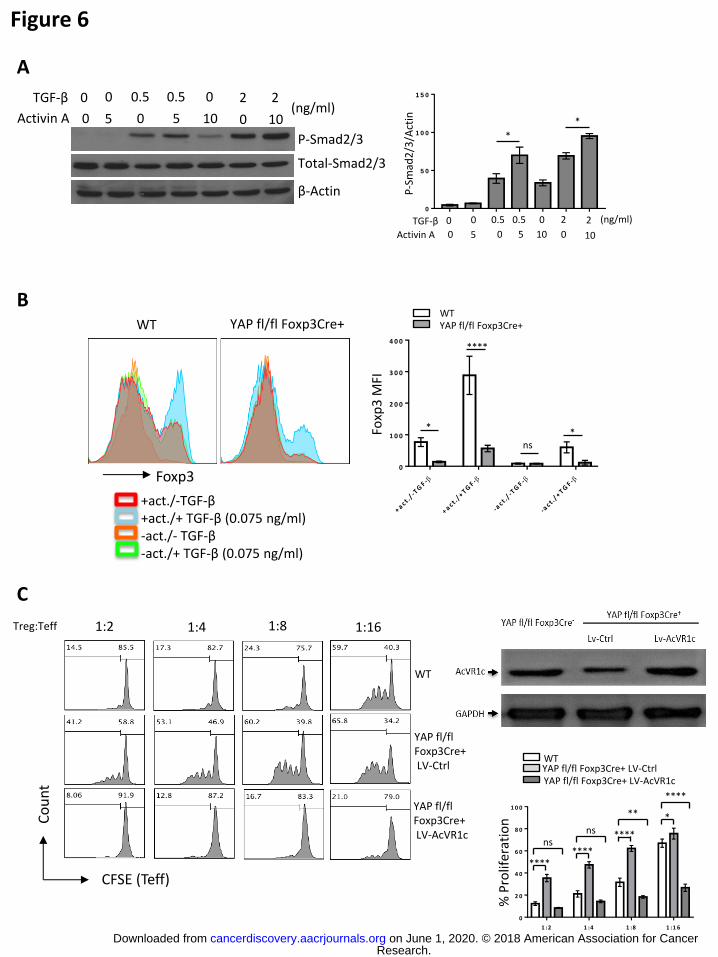
A
C
B
Figure 6 C
ou
nt
CFSE (Teff)
YAP fl/fl Foxp3Cre+ LV-Ctrl
YAP fl/fl Foxp3Cre+ LV-AcVR1c
WT
1:2 1:4 1:8 1:16 Treg:Teff
Foxp3
WT YAP fl/fl Foxp3Cre+
+act./-TGF-β +act./+ TGF-β (0.075 ng/ml) -act./- TGF-β -act./+ TGF-β (0.075 ng/ml)
+ a c t ./ -T
GF -
+ a c t ./ + T G
F -
-ac t .
/ -TG
F -
-ac t .
/ + T GF -
0
1 0 0
2 0 0
3 0 0
4 0 0
Foxp
3 M
FI
WT YAP fl/fl Foxp3Cre+
****
*
ns *
WT YAP fl/fl Foxp3Cre+ LV-Ctrl
1 :2 1 :4 1 :8 1 :1 6
0
2 0
4 0
6 0
8 0
1 0 0
YAP fl/fl Foxp3Cre+ LV-AcVR1c
% P
rolif
erat
ion
****
ns ****
ns ****
** *
****
Activin A
P-Smad2/3
Total-Smad2/3
β-Actin
TGF-β
0
0 0
5
0.5
0
0.5
5
0
10
2
0
2
10 (ng/ml)
Activin A
TGF-β
0
0 0
5
0.5
0
0.5
5
0
10
2
0
2
10
(ng/ml) 0
5 0
1 0 0
1 5 0
*
*
P-S
mad
2/3
/Act
in
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

A
C
B
D
Figure 7
E
Days
Tu
mo
r V
olu
me
(mm
3)
0
100
200
300
400
500
600
7 9 11 13 15 17 19 21 23 25
IgG1
anti-activin A
anti-activin A+GM-Vac
GM-Vac
* *
*
*
CD3+ TIL
lgG
1
a nt i
-ac t i
v inA
GM
-Va c
a nt i
-ac t i
v inA
+ GM
-Va c
0
2 0
4 0
6 0
8 0*
ns
**
ns
ns
*
% F
oxp
3+
of
CD
4+
lgG1 anti-activinA GM-Vac anti-activinA+GM-Vac
C D 3+
C D 8+
C D 3+
C D 4+
0
2 0
4 0
6 0
% IF
N-γ
*
*
ns
ns ns ns
** **
**** **
ns *
F
IFN
-γ
CD4
IFN
-γ
CD8
lgG1 anti-activin A GM-Vac anti-activin A+GM-Vac
CD3+CD4+ TIL
Foxp
3
CD4
Foxp
3
CD4
lgG1 anti-activin A
GM-Vac anti-activin A+GM-Vac
% IF
N-γ
+
WT AcVR1c KO
*
*
C D 3+
C D 4+
C D 3+
C D 8+
0
2 0
4 0
6 0
8 0
CD3+CD4+ CD3+CD8+
CD3+ TIL
IFN
-γ
TNF-α
WT AcVR1c KO
*
% F
oxp
3+
of
CD
4+
W t K o
0
1 0
2 0
3 0
4 0
5 0
WT AcVR1c KO
Foxp
3
IL-17
WT AcVR1c KO CD4+
1 3 1 5 1 7 1 9 2 1 2 3
0
5 0 0
1 0 0 0
1 5 0 0
Tum
or
Vo
lum
e (m
m3)
*
**
ns ns
ns
WT
AcVR1c KO
Days
*
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124

Published OnlineFirst June 15, 2018.Cancer Discov Xuhao Ni, Jinhui Tao, Joseph Barbi, et al. immunityYAP is essential for Treg mediated suppression of anti-tumor
Updated version
10.1158/2159-8290.CD-17-1124doi:
Access the most recent version of this article at:
Material
Supplementary
http://cancerdiscovery.aacrjournals.org/content/suppl/2018/06/15/2159-8290.CD-17-1124.DC1
Access the most recent supplemental material at:
Manuscript
Authoredited. Author manuscripts have been peer reviewed and accepted for publication but have not yet been
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerdiscovery.aacrjournals.org/content/early/2018/06/15/2159-8290.CD-17-1124To request permission to re-use all or part of this article, use this link
Research. on June 1, 2020. © 2018 American Association for Cancercancerdiscovery.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on June 15, 2018; DOI: 10.1158/2159-8290.CD-17-1124