1 division of oncology drug products presentation nda 21-649 genasense (oblimersen) for metastatic...
TRANSCRIPT

1
Division of Oncology Drug Products
Presentation NDA 21-649
Genasense (Oblimersen) for metastatic melanoma
ODAC May 3, 2004

2
FDA Review Team for Genasense (G3139)
• Project ManagementNicholette Hemingway, MPH
• Medical ReviewRobert Kane, MD
Ann Farrell, MD
• Statistical ReviewPeiling Yang, Ph.D.
Rajeshwari Sridhara, Ph.D.
• PharmacologyLilliam Rosario, Ph.D.
David E. Morse, Ph.D
• Chemistry Haripada Sarker, Ph.D.
Hasmukh Patel, Ph.D.
• Clin. Pharm/BiopharmGene Williams, Ph.D.
Brian Booth, Ph.D.

3
Presentation Outline 1. Requirements for FDA approval
2. ODAC Review of Temozolomide
3. Genasense (Oblimersen) NDA 21-649• Trial Design (GM 301)• Primary Endpoint – Survival • Secondary Endpoints
4. Summary

4
Requirements - New Drug Approval
• FD&C Act 1962 - Substantial evidence of effectiveness required by Congress
– Adequate & well-controlled investigations
– Generally understood to mean evidence from at least 2 adequate and well-controlled studies

5
Requirements - New Drug Approval
• FDAMA - 1997 One trial may suffice with other confirmatory evidence
• Effectiveness Guidance Document - 1998 A single trial should be of excellent design, internally consistent, and demonstrate a compelling result – statistically strong evidence of an important clinical benefit such as survival.

6
New Drug Approval Efficacy Requirement
• Regular approval – clinical benefit or established surrogate
• Accelerated Approval– uses a surrogate endpoint reasonably likely to
predict clinical benefit– confirmation of clinical benefit required

7
Approved drugs - Metastatic Melanoma
• Hydroxyurea (1967) - 10% response rate• Dacarbazine (1975) – DTIC single arm studies
Response Rate (RR) = 23% (6%CR) Survival times range 5 – 9 months
– To date, no evidence for Survival or Progression-free Survival (PFS) benefit for DTIC– RR: 5% - 24% in other studiesNo evidence for survival advantage for any
combination over DTIC alone • Aldesleukin (1998) – IL-2
– RR: 16% (6% CR with duration 2-5 yrs)

8
Selected Non-approved drugs for Metastatic Melanoma
• Interferon– 1997 Interferon alfa-2b approval for adjuvant
therapy of melanoma
• Temozolomide– ODAC 1999

9
Temozolomide (TMZ)
• One main study: open label, 305 patients with metastatic melanoma randomized to
DTIC IV each 3 weeks versus TMZ p.o. each 4 weeks
• Primary Endpoint – Survival (superiority) from median 6 mo-DTIC to 9 mo-TMZ
• Secondary endpoints: PFS and RR
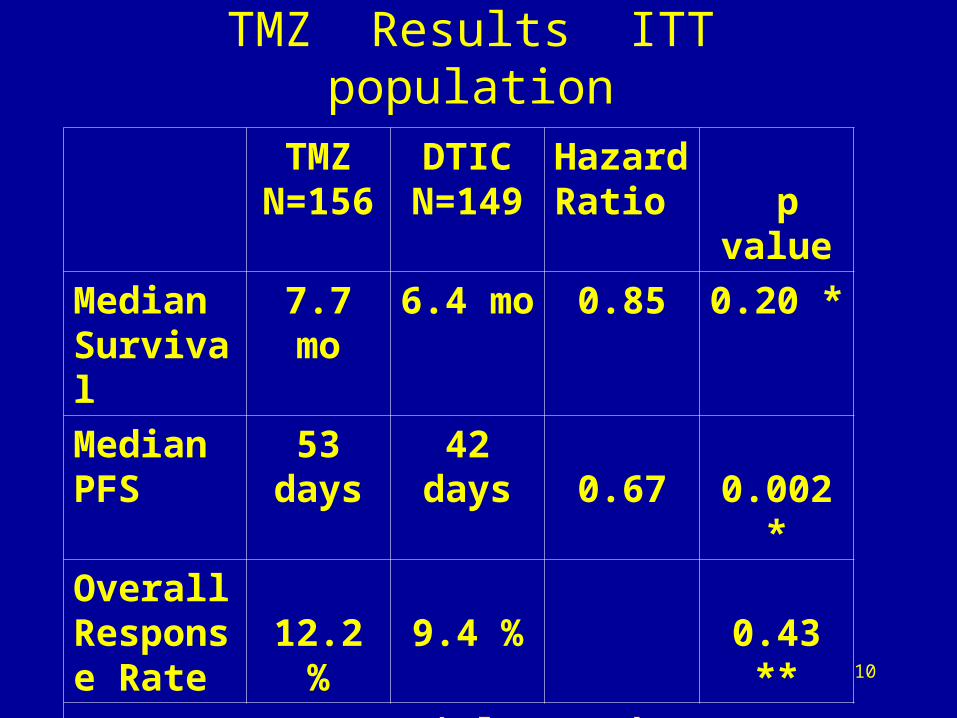
10
TMZ Results ITT population
TMZN=156
DTICN=149
Hazard Ratio
p value
Median Survival
7.7 mo 6.4 mo 0.85 0.20 *
Median PFS
53days
42days 0.67 0.002 *
Overall Response Rate
12.2 % 9.4 % 0.43 **
* log rank ** Chi square

11
TMZ was not approved
• Failed Primary Endpoint: No Survival benefit
• PFS, a secondary endpoint, small magnitude
• ODAC questioned the PFS difference
• No symptomatic benefit demonstrated
• A post-hoc survival analysis using a 6 month endpoint was not convincing

12
Genasense GM301 TrialRegulatory History
• July 2000 - Phase 3 protocol began
• August 1, 2003 – Data Cutoff Date
• December 8, 2003 - NDA submission

13
Genasense GM301 Trial Design
• Large, multicenter, unblinded study
• Prolonged central venous access required for Genasense (G)
• Protocol specified IRC for responders
• Intellectual, emotional, and physical ability to maintain an ambulatory infusion pump
(for patients assigned to G + DTIC)

14
Genasense GM301 Trial Design
• Primary Endpoint – Survival Improvement Secondary endpoints:
1. Progression-free survival (PFS)
2. Response rate (RR)
3. Duration of response
4. Durable response rate (RR at 6 months)
5. Performance status (PS)
6. Tumor-related symptoms
7. Safety

15
Genasense Trial Design - GM301
• Trial design - to show a 2 month median improvement in survival from 6 months with DTIC alone to 8 months with Genasense plus DTIC; 90% power
• Trial Primary endpoint – survival
• Trial Primary analysis – unadjusted log rank analysis of the intention-to-treat population

16
ITT G + D D Total
Number of Patients Randomized
386 385 771
Less number not treated 15 25 40
Still on study after day 42 (1st assessment)
186(48%)
151(39%)
337 (44%)
Discontinued early -
( < 8 cycles)
326
(84%)
328 (85%)
654 (85%)
Patient Disposition – GM301

17
Genasense study GM301
• Data cutoff date August 1, 2003
• Analysis occurred at 535 deaths (70%)
• Primary Endpoint Analysis:
Using the protocol specified analysis with the ITT population, no survival benefit was demonstrated by adding Genasense to DTIC treatment versus DTIC alone.

18
GM 301: Primary Analysis Survival Time ITT population
Number ( %)
died
Median Survival Time
HR *
Log rank
p
G + DTIC
n = 386
266 (69%)
274 days 9.0 mo 0.89 0.18
DTIC
n = 385
269 (70%)
238 days 7.8 mo
* 95% C.I. (0.75, 1.06)

19
Statistical Review of Statistical Review of Efficacy:Efficacy:
Progression-Free Survival (PFS) Progression-Free Survival (PFS) (Secondary Endpoint)(Secondary Endpoint)

20
Results
• Failed to demonstrate efficacy in the primary endpoint, overall survival– at two-sided alpha level of 0.05
• Strength of efficacy findings in the secondary endpoint, progression-free survival, is uncertain

21
Outline
• Review of Applicant’s Analyses and Results
• Major FDA Concern: Lesion Assessment Times
• Additional FDA Concerns

22
Review of Applicant’s PFS Analyses and Results
• PFS – Time from date of randomization to
date of disease progression/death
• Recorded Date of Disease Progression– The assessment date– Assessment date in each cycle:
• The latest date among different lesion assessments in that cycle

23
Review of Applicant’s PFS Analyses and Results
• Protocol-Specified Analysis: – Logrank: p-value = 0.0003– Median: 74 (G3139 + DTIC) vs. 49 days (DTIC)– Cox Model (supportive): Hazard Ratio = 0.73
• Alternative Approach:– Logrank: p-value = 0.0006– Median: 61 (G3139 + DTIC) vs. 48 days (DTIC)
– Cox model (supportive): Hazard Ratio = 0.75

24
Review of Applicant’s PFS Analyses and Results
• Question:Is this a true finding?

25
Major FDA Concern: Lesion Assessment Times
• Imbalance in Observed Lesion Assessment Times between Treatment Arms

26
Lesion Assessment Times
• Planned Timing for Lesion Assessments
• In Practice: – Not always as planned.– Even when assessed in planned cycles,
there were differences in timing between arms.

27
Visit 1 Visit 2Randomization
= Date of Death or actual tumor progression
Survival Event Date
Visit 1 Visit 2Randomization
PFS Event Date
Survival Analysis
PFS Analysis
Determining Event Dates

28
Summary of Time to First 3 Observed Lesion Assessments
(Actual Trial Data)
AssessmentNumber
Median in days Logrank p-valueG3139 +
DTICDTIC
1 48 43 <0.001
2 94 87 <0.001
3 137 129 <0.001
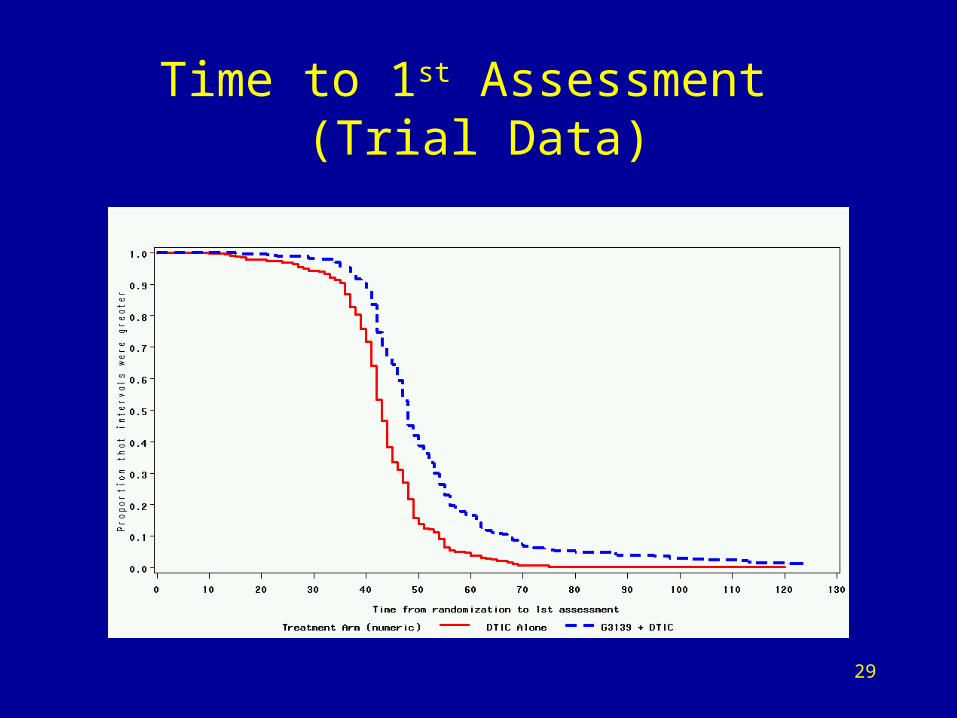
29
Time to 1st Assessment (Trial Data)

30
Time to 2nd Assessment (Trial Data)

31
Time to 3rd Assessment (Trial Data)

32
Lesion Assessment Times
• Impact of Imbalance in Assessment Times on PFS Analysis:
– Bias may be introduced in estimating PFS
– Even a small imbalance may lead to incorrect conclusion

33
Lesion Assessment Times
How Bias May Be Introduced: A Hypothetical Example
Patient 001Control
Patient 002Experiment
al
Actual day of DP Day 35
1st time of assessment
Day 42 Day 48
Recorded day of DP Day 42 Day 48

34
Lesion Assessment Times
Impact of systematic bias: Simulation study
– Distribution identical in both arms (Median PFS = 50 days, 300 subjects in each arm)
– Systematic increase by 2 days in assessment time in one arm
– In 98% of the 5000 simulations p-values were < 0.05 (average p-value = 0.004)

35
Additional FDA Concern
• Missing data was observed – Missing assessments visits– Missing individual lesion
measurements
• In presence of missing data– Bias could be introduced, especially
in an open-label study

36
Summary of PFS Finding
The claimed PFS benefit may not be a true finding because of:
• Difference in assessment intervals may explain observed PFS effect
• Questions regarding reliability of data collected in an open-label study

37
Summary of Statistical Review
• Study failed to achieve the primary objective of the study – No Overall Survival Benefit
• Secondary endpoints – PFS analysis:•Existence of effect ?•Magnitude of effect ?•Multiplicity Issues

38
PFS analysis - continued
• Assessments done only at 6 week intervals, however PFS difference only 2-3 weeks
• PFS difference is highly statistically significant, but may be fully accounted for by asymmetry in timing of assessments between arms
• Magnitude of the effect size is uncertain
• PFS: Real problem: what is the clinical relevance

39
Primary Endpoint – Survival No Advantage for Genasense
• Secondary endpoints:1. Progression-free survival (PFS)
2. Response rate (RR)
3. Response duration
4. Durable response rate
5. Performance status (PS)
6. Tumor-related symptoms
7. Safety

40
G + DTIC
(N=386)
DTIC
(N=385)
difference
P value
Genta / Investigator
45
(11.7%)
26
(6.8%) 4.9%
0.018
Blinded Independent
26
(6.7%)
14 (3.6%) 3.1%
0.056
Comparison of RR data as of original NDA
submission (12/8/03)

41
Response Concordance• 5 CRs identified by Genta/site investigators
– 3 in G + DTIC arm and 2 in DTIC arm
• None were adjudicated as CRs by Independent Review
• 44% of responders (CR or PR) by Genta/site investigators were determined not assessable or unconfirmed by Independent Review.
• 49% full concordance rate for response category between Genta and Independent Review.

42
Genta New Response Data provided April 9, 2004
• New data is being examined
• Problems with data developed outside of the study protocol:– Ascertainment bias between arms can occur when
analysis is not prospectively planned– Subsequent therapies, e.g. surgery, not part of the
protocol treatment may not be applied symmetrically

43
Duration of Response – Genta analysis
Intent-to-Treat Population
G + DTIC DTIC Alone
Number of patients N = 45 N = 26
Mean (SD) Days 175.7 (136.0) 153.5 ( 99.2)
Median Days 126.0 127.5
Range (Days) (41 , 565) (42 , 390)

44
Durable response rateGenta Analysis
• Genta has pre-specified a response of ≥ 6 months as a durable response.• Durable response rate for G + D = 3.4%• Durable response rate for D = 1.3%• Difference - not significant

45
ECOG Performance Status
• There were no differences in performance status observed between study arms
during treatment

46
Tumor-related symptoms
• There were no differences in symptoms observed between study arms
during treatment

47
Category G + DTICN (%)
DTICN (%)
Number of patients receiving any therapy
371 360
At least 1 Grade 3 or 4 adverse event (AE)
249 (67%)
154 (43%)
At least 1 serious adverse event (SAE)
149 (40%)
97 (27%)
At least 1 adverse event and discontinued permanently
69 (19%)
39 (11%)
Adverse Events – Toxicity

48
G + DTIC (N = 371)
n (%)
DTIC (N = 360)
n (%)
Grade 3 - 4 Neutropenia 79 (21%) 45 (12%)
Grade 3 - 4 Thrombocytopenia 58 (16%) 23 (6%)
Adverse Events Hematologic Toxicity

49
Adverse Events ≥ 5%Non-hematologic Toxicity
AE G + DTIC DTICNausea 231 (62%) 169 (47%)
Pyrexia 197 (53%) 63 (18%)
Fatigue 170 (46%) 142 (39%)
Vomiting 139 (38%) 75 (21%)
Infections 123 (33%) 65 (18%)
Anorexia 114 (31%) 56 (16%)
Headache 97 (26%) 47 (13%)

50
AE G + DTIC DTICRigors 76 (21%) 16 (4%)
Pruritus and Rash 57 (15% ) 18 (5%)
Injection site infection 24 (7%) 4 (1%)
Injection-site Reactions 21 (6%) 5 (1%)
Influenza-like Illness 19 (5%) 3 (0.8%)
Upper extremity thrombosis
18 (5%) 3 (0.8%)
*Axillary vein, Subclavian vein, Jugular vein, injection site thrombosis and superior vena caval syndrome
Adverse Events ≥ 5%Non-hematologic Toxicity

51
Genasense Summary
• GM301Trial failed its primary protocol-specified endpoint
• No Survival Benefit demonstrated with the addition of Genasense to DTIC
• The efficacy of the control arm, DTIC alone, is consistent with other studies.

52
Secondary Endpoints - PFS
1. PFS: No precedent for PFS as evidence of
clinical benefit for metastatic melanoma– May not be a true finding– PFS difference may be 13 or 25 days
depending on censoring technique chosen for missing data
– Clinical relevance ?

53
Secondary Endpoints - Response
2. Response rate:– Difference from DTIC alone: 3 - 5%– No CRs Confirmed by IRC– Clinical relevance ?
3. Durable response rate: No sig. difference (3.4% (G+DTIC) vs. 1.3% (DTIC alone))
4. Response duration: 126 days (G+DTIC) versus 127.5 days (DTIC alone)

54
Secondary endpoints- Genasense
5. Performance status:- No benefit observed from Genasense
6. Symptomatic benefit: Not observed
7. Safety: Greater toxicity for the combination of G + DTIC