a comprehensive network and pathway analysis of human...
TRANSCRIPT

A Comprehensive Network and Pathway Analysisof Human Deafness Genes
*Georgios A. Stamatiou and †Konstantina M. Stankovic
*Department of Otolaryngology, Hippokration General Hospital, University of Athens, Athens, Greece; andÞDepartment of Otology and Laryngology, Harvard Medical School and Department of Otolaryngology,
Massachusetts Eye and Ear Infirmary, Boston, Massachusetts, U.S.A.
Objective: To perform comprehensive network and pathwayanalyses of the genes known to cause genetic hearing loss.Study Design: In silico analysis of deafness genes using inge-nuity pathway analysis (IPA).Methods: Genes relevant for hearing and deafness were iden-tified through PubMed literature searches and the HereditaryHearing Loss Homepage. The genes were assembled into 3 groups:63 genes that cause nonsyndromic deafness, 107 genes that causenonsyndromic or syndromic sensorineural deafness, and 112 genesassociated with otic capsule development and malformations. Eachgroup of genes was analyzed using IPA to discover the mostinterconnected, that is, ‘‘nodal’’ molecules, within the most statis-tically significant networks (p G 10j45).Results: The number of networks that met our criterion for sig-nificance was 1 for Group 1 and 2 for Groups 2 and 3. Nodalmolecules of these networks were as follows: transforming growth
factor beta1 (TGFB1) for Group 1, MAPK3/MAPK1 MAP kinase(ERK 1/2) and the G protein coupled receptors (GPCR) forGroup 2, and TGFB1 and hepatocyte nuclear factor 4 alpha (HNF4A)for Group 3. The nodal molecules included not only those knownto be associated with deafness (GPCR), or with predisposition tootosclerosis (TGFB1), but also novel genes that have not beendescribed in the cochlea (HNF4A) and signaling kinases (ERK 1/2).Conclusion: A number of molecules that are likely to be keymediators of genetic hearing loss were identified through threedifferent network and pathway analyses. The molecules includednew candidate genes for deafness. Therapies targeting thesemoleculesmay be useful to treat deafness.KeyWords: Deafness genesVERK1/2VGPCRVHNF4AVMAPK1VMolecular pathways analysisVTGFB1.
Otol Neurotol 34:961Y970, 2013.
Hearing loss is the most common sensory deficit in theworld, affecting almost 600 million people (1), and themost common congenital anomaly, affecting 2 to 6 per1,000 newborns (1,2). At least two-thirds of cases ofprelingual deafness in the developed countries are dueto genetic factors; the remaining one-third is attributedto environmental and unidentified genetic factors (3).
In general, genetic hearing loss can be classified assyndromic or nonsyndromic, depending on whether otherdistinguishing physical features are present or absent,respectively. Nonsyndromic hearing loss is typically
sensorineural and believed to account for 70% of cases(4). The remaining 30% of hearing loss is syndromicand can be sensorineural, conductive, or mixed. Morethan 400 syndromes include hearing loss as a part oftheir phenotypic signature (3). Typically, genetic hear-ing loss is monogenic. Based on the mode of inheri-tance, monogenic hearing loss is classified as autosomalrecessive, accounting for 80% of cases; autosomal domi-nant, accounting for almost 20% of cases; X-linked; andmitochondrial; the latter two account for less than 1% ofcases (5).
Hearing loss is one of the most genetically hetero-geneous disorders, with more than 100 mapped loci andmore than 60 causally implicated genes within these loci(6). Additional complexity exists because mutations in thesame gene may cause syndromic or nonsyndromic hearingloss, and hearing loss may be oligogenic (7). Given theintricacy of normal hearing, which requires interaction ofmany diverse molecules, it is estimated that approximately1% of human genes play a role in hearing (8).
Although the shear number of genes involved inhearing and deafness may seem daunting, this complexity
Address correspondence and reprint requests to Konstantina Stankovic,M.D., Ph.D., Massachusetts Eye and Ear Infirmary, 243 Charles St,Boston, MA 02114; E-mail: [email protected] conducted at: Department of Otolaryngology, Massachusetts
Eye and Ear Infirmary, Harvard Medical School, Boston, MA, USA.Source of Funding: NIDCD K08 DC010419 (K. M. S.) and the
Bertarelli Foundation (K. M. S.).The study was presented at the AOS Spring Meeting, April 21Y22,
2012, San Diego, CA.The authors disclose no conflicts of interest.Supplemental digital content is available in the text.
Otology & Neurotology34:961Y970 � 2013, Otology & Neurotology, Inc.
961
Copyright © 2013 Otology & Neurotology, Inc. Unauthorized reproduction of this article is prohibited.

may be simplified by looking at well-characterized cellsignaling and metabolic pathways through which thesegenes are known to interact in various tissues. A commonassumption of pathway analysis is that genes whose dys-function contributes to a disease phenotype tend to befunctionally related (9,10). Unraveling these pathways isessential to understanding biological mechanisms, diseasestates, and the function of drugs that affect them. Wetherefore undertook the first comprehensive pathway andnetwork analysis of all known genes implicated in hu-man deafness. We used tools of bioinformatics to analyzeinteractions across multiple biological dimensions includ-ing molecular interactions, cellular processes, and diseaseprocesses. We focused on 3 groups of genes: those causingnonsyndromic hearing loss, those causing nonsyndromicor syndromic sensorineural hearing loss, and those asso-ciated with malformation of the otic capsule. The thirdgroup of genes was augmented with genes implicated inotic capsule development based on animal studies inmammals. Our analyses suggest new candidate genes fordeafness within the known deafness loci and highlightseveral genes as potential novel targets for diagnosis andtreatment of genetic hearing loss.
MATERIALS AND METHODS
A PubMed English search was performed to identify genesand molecules associated with human hearing loss and withmammalian development and malformation of the otic capsule.All genes listed on the Hereditary Hearing Loss Homepage en-tered our analyses. The pertinent genes were identified throughlinkage analysis, immunohistochemistry, quantitative reversetranscription-polymerase chain reaction, or microarray ana-lyses. Only studies with relevant controls or appropriate sta-tistical analyses were included; studies without control groupsor with unspecified statistical significance were excluded.Genes that met our criteria were classified based on their estab-lished role in nonsyndromic or syndromic sensorineural hear-ing loss, or association with otic capsule development ormalformation. The number of genes that entered our anal-yses was 63 genes that cause nonsyndromic deafness, 107 genesthat cause nonsyndromic or syndromic sensorineural deaf-ness, and 112 genes associated with otic capsule developmentand malformations (Table S1, Supplemental Digital Content 1,http://links.lww.com/MAO/A149, alphabetically lists genes usedin the analyses, along with their associated clinical pheno-types). The second analysis was done to enrich for genes asso-ciated with sensorineural hearing loss, whereas the third analysiswas done to enrich for genes that contribute to mixed conductiveand sensorineural hearing loss.For each of the 3 groups of genes, a different analysis was
performed using ingenuity pathway analysis software (IPA;Ingenuity Systems, Redwood City, CA, USA), which is basedon the world’s largest curated and highest quality knowledgebase of biological networks created from millions of individu-ally modeled relationships among proteins, genes, complexes,cells, tissues, drugs, and diseases to understand how candidategenes may work together as molecular modules that impactcellular processes and disease phenotypes. Within IPA, pathwayrefers to well-characterized cell signaling and metabolic path-ways based on molecular and biochemical studies, whereasnetwork refers to regulatory relationships that exist among user-
specified molecules. We refer to the most interconnected mol-ecule (or class of molecules) in a network as key or central node.Networks of up to 140 molecules were studied to allow for thepossibility that all genes within a group belonged to the samenetwork. Statistical analysis of networks and pathways wasperformed as part of the overall IPA using the right-tailedFisher’s exact test (11). The p value reflects the likelihood thatthe association between the input genes and a given pathway ornetwork is due to chance. Only networks with p G 10j45 wereconsidered significant. Less stringent criterion of p G 0.05 wasused for pathway significance to facilitate discovery ofpotentially novel signaling pathways.Expression of select genes was validated by summarizing
their reported expression in the mouse cochlear hair cells basedon RNASeq (12) or mouse spiral ganglion neurons based onGeneChips (13). Both databases catalog changes in gene expres-sion during embryonic and early postnatal development.
RESULTS
Network analysis of the nonsyndromic deafnessgenes revealed 6 networks. The top network was veryhighly significant (p = 10j105) and included nearlyall (50 of 63) genes currently implicated in nonsyndro-mic HL, suggesting close coupling of deafness geneswithin the cacophony of human interactome. This networkis shown in Figure S1, Supplemental Digital Content 2,http://links.lww.com/MAO/A150, and all molecules ofthe network are listed in Table S2, Supplemental DigitalContent 7, http://links.lww.com/MAO/A155. The remain-ing 5 networks did not meet our criterion for significance(p = 10j2). The central node of the top pathway wastransforming growth factor beta1 (TGFB1), having 35 con-nections with other genes in the network (Fig. 1). Thesecond and third central nodes were beta-estradiol andfilamentous actin (F-Actin), with 29 and 24 gene inter-connections, respectively (Table 1). Pathway analysisrevealed 5 statistically significant pathways (Table 2),highlighting the importance of actin cytoskeleton sig-naling, which is known to be critical for stereociliar andhair cell function. The second ranked pathway was celljunction signaling (Table 2), consistent with the obser-vations that about 50% of cases of nonsyndromic HL aredue to mutations in GJB2 encoding a gap junction protein(14). The other top pathways point to previously under-studied pathways in the context of deafness, that is,hepatic fibrosis, and signaling via sildenafil (Viagra) orintegrin-linked kinase (Table 2).
Network analysis of genes causing both nonsyndromicand syndromic sensorineural deafness generated 10 net-works, two of which met our criterion for statistical sig-nificance (with p = 10j103 and p = 10j46); the othernetworks had p e 10j2. The central node of the topnetwork was MAPK3/MAPK1 MAP kinase (ERK 1/2)with 33 connections, closely followed by collagens with32 connections, and hepatocyte growth factor (HGF)with 30 connections (Table 1, Fig. 2, Figure S2, Supple-mental Digital Content 3, http://links.lww.com/MAO/A151,showing the top network of the second pathway anal-ysis and Table S3, Supplemental Digital Content 8,
962 G. A. STAMATIOU AND K. M. STANKOVIC
Otology & Neurotology, Vol. 34, No. 5, 2013
Copyright © 2013 Otology & Neurotology, Inc. Unauthorized reproduction of this article is prohibited.

http://links.lww.com/MAO/A156, listing all moleculesof the network). The central node of the second mostsignificant network was G Protein Coupled Receptors(GPCR) with 74 interconnections (Figure S3, Supple-mental Digital Content 4, http://links.lww.com/MAO/A152,showing network 2 of the second pathway anal-ysis, and Table S4, Supplemental Digital Content 9,http://links.lww.com/MAO/A157, listing all moleculesof this network). This node was more than 3 times moreinterconnected than the second and third key nodesVbetaestradiol with 24 connections and TGFB1 with 21 con-nections. Pathway analysis pointed to the potential rele-vance of signaling pathways in melanocytes, melanoma,and renal cell carcinoma for sensorineural deafness (Table 2),in addition to the pathways already highlighted in thefirst analysis.
Network analysis of genes involved in otic capsule devel-opment and malformation syndromes generated 4 net-works, 2 of which met our criterion for statistical significance(with p = 10j105 and p = 10j91); the other 2 had p = 10j2.The central node of the top ranked network was TGFB1with 70 connections, followed by ERK 1/2 and bonemorphogenetic protein 4 (BMP4) with 54 and 44 connec-tions, respectively (Table 1, Fig. 3, Figure S4, Supple-mental Digital Content 5, http://links.lww.com/MAO/A153,showing the top network of the third pathway anal-ysis, and Table S5, Supplemental Digital Content 10,
http://links.lww.com/MAO/A158, listing all molecules ofthis network). The top 3 nodes of the second most sig-nificant network were hepatocyte nuclear factor 4 alpha(HNF4A) with 51 connections, TGFB1 with 48 connec-tions, and tumor necrosis factor (TNF) with 31 connec-tions (Table 1, Figure S5, Supplemental Digital Content 6,http://links.lww.com/MAO/A154, showing the second net-work of the third pathway analysis, and Table S6, Supple-mental Digital Content 11, http://links.lww.com/MAO/A159,listing all molecules of this network). Pathway analysispointed tomultiple signaling pathways that may be relevant
FIG. 1. A part of the most significant network generated by IPA network analysis of genes implicated in nonsyndromic hearing loss(Analysis 1). The full network is shown in Supplemental Figure 1. Molecules with yellow frames are the top nodes of the network. The inputgenes are highlighted with blue frames. Molecules within white symbols were provided by the ingenuity knowledge base. Solid and dashedlines between molecules represent direct and indirect interactions, respectively.
TABLE 1. The top 3 nodes of the statistically significantnetworks identified through 3 different types of analyses
Analysis
Network 1 Network 2
Molecule(no. connections)
Molecule(no. connections)
1: Nonsyndromic HL TGFB1 (35) VEstradiol (29) VF-actin (24) V
2: Syndromic andnonsyndromic HL
ERK 1/2 (33) GPCR (74)Collagen (32) Estradiol (24)
HGF (30) TGFB1 (21)3: Otic capsule development
and malformationTGFB1 (70) HFN4a (51)
ERK 1/2 (54) TGFB1 (48)BMP4 (44) TNF (31)
HL indicates hearing loss.
963IPA OF HUMAN DEAFNESS GENES
Otology & Neurotology, Vol. 34, No. 5, 2013
Copyright © 2013 Otology & Neurotology, Inc. Unauthorized reproduction of this article is prohibited.

for deafness and cochlear malformation, including sig-naling pathways in rheumatoid arthritis, cardiogenesis,stem cells pluripotency, chromosomal replication, andBMP signaling (Table 2).
Chromosomal locations of the top nodes listed inTable 1 are provided in Table 3, along with the corre-sponding human and mouse deafness loci, and thereported expression in the mouse cochlea. Similar infor-mation is provided for all genes in Supplemental TablesS2YS6. Of the 10 nodal molecules in Table 3, six repre-sent novel candidate genes for human deafness because oftheir location within human deafness loci with unknowncausative genes (MAPK1, HNF4A, BMP4, TNF) or withinhuman deafness loci where another gene has been identi-
fied as causative (TGFB1, CYP19A1), and their docu-mented expression in the mouse cochlea. When analyzingall genes comprehensively across the studied networks(Supplemental Tables S2-S6), many represent additionalcandidate genes for human deafness because of their chro-mosomal location within deafness loci with unknowncausative genes or within deafness loci where knowncausative genes are different than the candidate genes.Focusing on the former, we have selected 25 geneswhose expression has been unambiguously documentedin murine cochlear hair cells (12) or cochlear neurons (13)(Table 4). In addition, the genes that are known to causesyndromic hearing loss in humans are attractive candi-dates for human nonsyndromic hearing loss when they
TABLE 2. The top canonical pathways discovered by IPA for each of the 3 analyses
Analysis Top canonical pathways p
1: Nonsyndromic HL Actin cytoskeleton signaling 1.22E-04Tight junction signaling 2.34E-04
Hepatic fibrosis/hepatic stellate cell activation 1.37E-02Cellular effects of sildenafil (Viagra) 1.47E-02
ILK signaling 2.92E-022: Nonsyndromic and syndromic sensorineural HL Melanocyte development and pigmentation signaling 1.19E-05
Actin cytoskeleton signaling 2.86E-04Melanoma signaling 2.16E-03
Tight junction signaling 2.23E-03Renal cell carcinoma signaling 8.66E-03
3: Otic capsule development and malformation Role of osteoblasts, osteoclasts and chondrocytes in rheumatoid arthritis 1.4E-15Factors promoting cardiogenesis in vertebrates 1.24E-10
Human embryonic stem cell pluripotency 6.27E-10BMP signaling pathway 2.76E-07
Cell cycle control of chromosomal replication 5.22E-07
FIG. 2. The central part of the most significant network generated by IPA network analysis of genes implicated in nonsyndromic orsyndromic sensorineural hearing loss (Analysis 2).
964 G. A. STAMATIOU AND K. M. STANKOVIC
Otology & Neurotology, Vol. 34, No. 5, 2013
Copyright © 2013 Otology & Neurotology, Inc. Unauthorized reproduction of this article is prohibited.

fall within the loci for nonsyndromic deafness (Supple-mental Tables S1YS6) because several well-studiedgenes, such asMYO7A (15,16) and SLC26A4 (17,18), areknown to cause syndromic or nonsyndromic hearing loss,depending on the location and severity of a mutation.
DISCUSSION
A comprehensive network and pathway analysis ofthe human deafness genes has revealed that these genesform a small and well-interconnected neighborhood inthe cacophony of the human interactome, as reflected inthe very highly significant top networks for each of the 3analyses that we performed. Using musical analogy, anode is a note or a chord, a pathway is an instrumentaltune linking interconnected notes and chords, and a net-work is a symphony involving interconnected tunes frommany different instruments. In this harmony of sound,TGFB1 stands out as the only note of central significancein all 3 analyses (Table 1). Although mutations in thisgene are known to cause Camurati-Engelmann disease(19) and a coding polymorphism in TGFB1 is associatedwith otosclerosis (20), TGFB1 has not been causally
linked to nonsyndromic sensorineural hearing loss; ourresults strongly support future exploration of this poten-tial causal link. TGFB1 is a multifunctional peptide thatcontrols proliferation, differentiation, extracellular matrixproduction, adhesion, migration, and other functions inmany cell types (21). In the inner ear, TGFB1 stimulatesotic capsule chondrogenesis during early development (22)and controls remodeling of the developing otic capsule byinhibiting osteoclast activation and stimulating osteoblastdifferentiation (23). Our results suggest that TGFB1 mayplay additional, as of yet unrecognized, roles in the phy-siology of the cochlear sensorineural structures, in additionto its established role in otic capsule development and re-modeling. Interestingly, BMP4, which we have identifiedas another candidate gene for human deafness (Table 1),is a member of the TGFB superfamily of signalingmolecules (24).
Beta-estradiol is another prominent node in the analy-ses of nonsyndromic and syndromic sensorineural hearingloss (Table 1). This molecule is a sex hormone derivedfrom cholesterol, present not only in women but also inmen because it is derived from testosterone by the action ofaromatase. In addition to its critical role in reproduction
FIG. 3. An area of the most significant network generated by IPA network analysis of genes implicated in otic capsule development andmalformation (Analysis 3).
965IPA OF HUMAN DEAFNESS GENES
Otology & Neurotology, Vol. 34, No. 5, 2013
Copyright © 2013 Otology & Neurotology, Inc. Unauthorized reproduction of this article is prohibited.
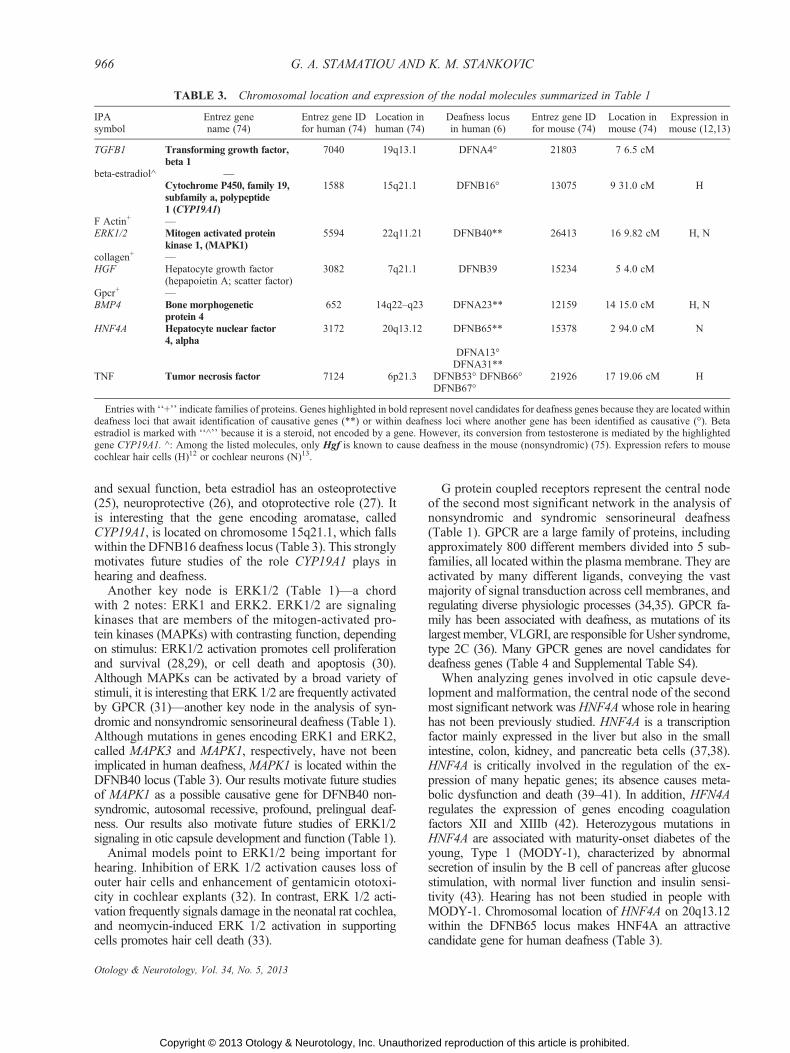
and sexual function, beta estradiol has an osteoprotective(25), neuroprotective (26), and otoprotective role (27). Itis interesting that the gene encoding aromatase, calledCYP19A1, is located on chromosome 15q21.1, which fallswithin the DFNB16 deafness locus (Table 3). This stronglymotivates future studies of the role CYP19A1 plays inhearing and deafness.
Another key node is ERK1/2 (Table 1)Va chordwith 2 notes: ERK1 and ERK2. ERK1/2 are signalingkinases that are members of the mitogen-activated pro-tein kinases (MAPKs) with contrasting function, dependingon stimulus: ERK1/2 activation promotes cell proliferationand survival (28,29), or cell death and apoptosis (30).Although MAPKs can be activated by a broad variety ofstimuli, it is interesting that ERK 1/2 are frequently activatedby GPCR (31)Vanother key node in the analysis of syn-dromic and nonsyndromic sensorineural deafness (Table 1).Although mutations in genes encoding ERK1 and ERK2,called MAPK3 and MAPK1, respectively, have not beenimplicated in human deafness, MAPK1 is located within theDFNB40 locus (Table 3). Our results motivate future studiesof MAPK1 as a possible causative gene for DFNB40 non-syndromic, autosomal recessive, profound, prelingual deaf-ness. Our results also motivate future studies of ERK1/2signaling in otic capsule development and function (Table 1).
Animal models point to ERK1/2 being important forhearing. Inhibition of ERK 1/2 activation causes loss ofouter hair cells and enhancement of gentamicin ototoxi-city in cochlear explants (32). In contrast, ERK 1/2 acti-vation frequently signals damage in the neonatal rat cochlea,and neomycin-induced ERK 1/2 activation in supportingcells promotes hair cell death (33).
G protein coupled receptors represent the central nodeof the second most significant network in the analysis ofnonsyndromic and syndromic sensorineural deafness(Table 1). GPCR are a large family of proteins, includingapproximately 800 different members divided into 5 sub-families, all located within the plasma membrane. They areactivated by many different ligands, conveying the vastmajority of signal transduction across cell membranes, andregulating diverse physiologic processes (34,35). GPCR fa-mily has been associated with deafness, as mutations of itslargest member, VLGRI, are responsible for Usher syndrome,type 2C (36). Many GPCR genes are novel candidates fordeafness genes (Table 4 and Supplemental Table S4).
When analyzing genes involved in otic capsule deve-lopment and malformation, the central node of the secondmost significant network wasHNF4Awhose role in hearinghas not been previously studied. HNF4A is a transcriptionfactor mainly expressed in the liver but also in the smallintestine, colon, kidney, and pancreatic beta cells (37,38).HNF4A is critically involved in the regulation of the ex-pression of many hepatic genes; its absence causes meta-bolic dysfunction and death (39Y41). In addition, HFN4Aregulates the expression of genes encoding coagulationfactors XII and XIIIb (42). Heterozygous mutations inHNF4A are associated with maturity-onset diabetes of theyoung, Type 1 (MODY-1), characterized by abnormalsecretion of insulin by the B cell of pancreas after glucosestimulation, with normal liver function and insulin sensi-tivity (43). Hearing has not been studied in people withMODY-1. Chromosomal location of HNF4A on 20q13.12within the DFNB65 locus makes HNF4A an attractivecandidate gene for human deafness (Table 3).
TABLE 3. Chromosomal location and expression of the nodal molecules summarized in Table 1
IPAsymbol
Entrez genename (74)
Entrez gene IDfor human (74)
Location inhuman (74)
Deafness locusin human (6)
Entrez gene IDfor mouse (74)
Location inmouse (74)
Expression inmouse (12,13)
TGFB1 Transforming growth factor,beta 1
7040 19q13.1 DFNA4- 21803 7 6.5 cM
beta-estradiol^ VCytochrome P450, family 19,subfamily a, polypeptide1 (CYP19A1)
1588 15q21.1 DFNB16- 13075 9 31.0 cM H
F Actin+ VERK1/2 Mitogen activated protein
kinase 1, (MAPK1)5594 22q11.21 DFNB40** 26413 16 9.82 cM H, N
collagen+ VHGF Hepatocyte growth factor
(hepapoietin A; scatter factor)3082 7q21.1 DFNB39 15234 5 4.0 cM
Gpcr+ VBMP4 Bone morphogenetic
protein 4652 14q22Yq23 DFNA23** 12159 14 15.0 cM H, N
HNF4A Hepatocyte nuclear factor4, alpha
3172 20q13.12 DFNB65** 15378 2 94.0 cM N
DFNA13-DFNA31**
TNF Tumor necrosis factor 7124 6p21.3 DFNB53- DFNB66-DFNB67-
21926 17 19.06 cM H
Entries with ‘‘+’’ indicate families of proteins. Genes highlighted in bold represent novel candidates for deafness genes because they are located withindeafness loci that await identification of causative genes (**) or within deafness loci where another gene has been identified as causative (-). Betaestradiol is marked with ‘‘^’’ because it is a steroid, not encoded by a gene. However, its conversion from testosterone is mediated by the highlightedgene CYP19A1. ^: Among the listed molecules, only Hgf is known to cause deafness in the mouse (nonsyndromic) (75). Expression refers to mousecochlear hair cells (H)12 or cochlear neurons (N)13.
966 G. A. STAMATIOU AND K. M. STANKOVIC
Otology & Neurotology, Vol. 34, No. 5, 2013
Copyright © 2013 Otology & Neurotology, Inc. Unauthorized reproduction of this article is prohibited.

TABLE 4. Additional candidate genes for human deafness based on their presence in the networks studied(Table 1, and Supplemental Tables S2Y26), unreported association with human deafness (Supplemental Table S1),
and chromosomal location within deafness loci whose causative genes remain unidentified (**)
IPA symbolEntrez genename (74)
Entrez gene ID forhuman (74)
Location inhuman (74)
Deafness locusin human (6)
Entrez gene IDfor mouse (74)
Expression inmouse (12,13)
ATP6V1B1 ATPase, H+ transporting,lysosomal 56/58kDa,
V1 subunit B1
525 2p13.1 DFNA58** 110935 H
BMP6 Bone morphogeneticprotein 6
654 6p24-p23 DFNA21** 12161 H
CA14 Carbonic anhydrase XIV 23632 1q21 DFNA7** DFNA49** 23831 H, NCALML4 Calmodulin-like 4 91860 15q23 DFNB48** 75600 H, NCALCA Calcitonin-related
polypeptide alpha796 11p15.2 DFNA32** H, N
CLDN10 Claudin 10 9071 13q31-q34 DFNA33** 58187 H, NCNST Consortin, connexin
sorting protein163882 1q44 DFNB45** 226744 H, N
ELOVL2 ELOVL fattyacid elongase 2
54898 6p24.2 DFNA21** 54326 H, N
EPS8L2 EPS8-like 2 64787 11p15.5 DFNA32** 98845 HGPR33 G protein-coupled
receptor 33(gene/pseudogene)
2856 14q12 DFNA9- DFNA53**, DFNB5** H, N
GPR85 G protein-coupledreceptor 85
54329 7q31 DFNB4- 64450 H, N
DFNB14**DFNB17**GPR141 G protein-coupled
receptor 141353345 7p14.1 DFNB44** 353346 H
GPR155 G protein-coupledreceptor 155
151556 2q31.1 DFNB27**DFNB59- 68526 H, N
GJA1 Gap junction protein,alpha 1, 43kDa
2697 6q21-q23.2 DFNB38** 14609 H, N
ITGA10 Integrin, alpha 10 8515 1q21 DFNA7** 213119 H, NDFNA49**
LDHA Lactate dehydrogenase A 3939 11p15.4 DFNA32** 16828 H, NLGR4 Leucine-rich repeat
containing Gprotein-coupled
receptor 4
55366 11p14-p13 DFNB18- 107515 H, N
DFNB51**MFHAS1 Malignant fibrous
histiocytomaamplified sequence 1
9258 8p23.1 DFNM2** 52065 H, N
MYCN v-myc myelocytomatosisviral related oncogene,
neuroblastomaderived (avian)
4613 2p24.3 DFNB47**DFNB83** 18109 H, N
PLIN2 Perilipin 2 123 9p22.1 DFNA47** 11520 H, NPTPN11 Protein tyrosine
phosphatase,non-receptor type 11
5781 12q24 DFNA41**DFNA64- 19247 H
PRDX2 Peroxiredoxin 2 7001 19p13.2 DFNA57** DFNB15- 21672 H, NDFNB68**DFNB72-DFNB81**
SLC39A1 Solute carrier family 39(zinc transporter),
member 1
27173 1q21 DFNA7** 30791 H,N
DFNA49**SMARCA4 SWI/SNF-related,
matrix-associated,actin-dependent regulator
of chromatin,subfamily a, member 4
6597 19p13.2 DFNA57**DFNB15- 20586 H,N
DFNB68**DFNB72-DFNB81**
TMEM17 Transmembraneprotein 17
200728 2p15 DFNA58** 103765 H
Some of these genes also fall within deafness loci where other causative genes have been identified (-). Mouse orthologs of these genes have beenreported in cochlear hair cells (H)12 or cochlear neurons (N)13.
967IPA OF HUMAN DEAFNESS GENES
Otology & Neurotology, Vol. 34, No. 5, 2013
Copyright © 2013 Otology & Neurotology, Inc. Unauthorized reproduction of this article is prohibited.

Emergence of the proinflammatory cytokine, TNF, asa candidate gene for deafness is intriguing because di-verse inflammatory conditions, such as otitis media andautoimmune disease, can cause hearing loss (44), whereasanti-inflammatory medications, such as steroids (45,46)and TNF blockers (44), can ameliorate hearing loss. How-ever, chronic use of some nonsteroidal anti-inflammatorymedications has been associated with hearing loss, in a sex-dependent manner (47,48), highlighting the importanceof understanding genetic underpinning of cochlear responseto inflammation.
By exploring topologic characteristics of deafness genesin the human interactome, we have also discovered bio-logic processes that provide a global prospective on deaf-ness (Table 2). Some of these processes provide surprisingnew insight into mechanisms that may be shared betweenthe process of hearing and diverse processes such asmelanoma signaling, cardiogenesis, and hepatic fibrosis.Our finding that the canonical pathway for cellular effectsof sildenafil (Viagra) is relevant for sensorineural hearingloss is consistent with the reported sudden hearing lossfrom phosphodiesterase-5 inhibitors used to treat erectiledysfunction (49).
In addition to identifying molecules and pathways ofkey relevance for hearing and hearing loss, our analyseshave predicted drug targets to treat or prevent hearingloss. Although emerging therapies for deafness are likelyto benefit from rapid developments of pharmacother-apies in seemingly unrelated fields, such as cancer biologyor sexual dysfunction, we need to be cognizant of poten-tially harmful effects of these therapies on hearing. Manydifferent drugs are known to target the top nodal mole-cules of the 5 networks that we analyzed (Table 1).
Majority of the existing drugs that target TGFB1 focuson enhancing TGFB1 activation to promote chon-drogenesis or new bone formation. However, severalstudies propose inhibition of gene function using agentssuch as neutralizing antibodies, soluble receptors, recep-tor kinase antagonists, antisense reagents, and specificdrugs (50). The commonly used drugs that are capableof blocking TGFB1, among their other functions, includeantagonists of angiotensin II, Type 1 receptors (e.g.,losartan), angiotensin-converting enzyme (ACE) inhibi-tors (51Y56), statins (especially simvastatin) (57Y60),the oral antimicrobial pirfenidone (61,62) (InterMuneInc., CA, USA), and the antifibrotic drug tranilast (63)(Kissei Pharma, Japan). Furthermore, a number of spe-cific TGFB1 inhibitors are being investigated, includingthe pan-TGF-b monoclonal antibody (64), the solubleTGF-ARII receptor, antisense oligonucleotides, and inhi-bitors of ALK5 (50). Interestingly, long-term treatmentwith the anti-TGF-b mAb was not found to result in sig-nificant pathologic changes in major organs and tissues inmice (64), although other studies have raised concernsregarding the potential acceleration of tumor progressionafter TGF-b inhibition (65).
Regarding GPCR, which are involved in nearly allhuman physiologic processes, about half of the cur-rently available drugs on the market nonspecifically
target members of this superfamily (34). Therefore,specific targeting of GPCR most relevant for hearingmay be challenging.
Blockage of the ERK1/2 pathway has been of greatinterest for cancer biology because of the pathway’sknown role in cancer aggressiveness. Although power-ful inhibitors of ERK 1/2 pathway have been describedwith promising results (66,67), the production of specificdrugs targeting these signaling kinases remains challen-ging. Given the reported dual role of ERK1/2 in cochlearphysiology and pathophysiology, any therapeutic manip-ulation of the ERK1/2 pathway will have to be leveragedagainst the risk of worsening or accelerating hearing losswith such manipulation.
Interestingly, HNF4A expression is inhibited by cyclos-porine (68), a commonly used drug whose chronic use isassociated with hearing loss (69). This further highlightsa likely important role of HNF4A in hearing. Other sub-stances reported to inhibit HNF4A gene expression andfunction include the tumor suppressor P53 (70), cytokines,and the peroxisome-proliferatorYactivated receptor-F coac-tivator 1> (71). Curiously, fasting upregulates and feedingdownregulates HNF4A expression (72), suggesting futuredietary measures to modulate the levels and activity ofHNF4A (73).
CONCLUSION
This study is the first systematic network and pathwayanalysis of human deafness genes. By studying geneticchords, tunes, and symphonies we have identified novelcandidate genes for deafness and highlighted biologicprocesses that may provide better understanding of hear-ing and hearing loss. Our analyses have implications fordesign of new drugs and therapies to prevent or treatdeafness and for strategies to benefit from parallel devel-opments of pharmacotherapies in other fields of medicine.A caveat of the study is that it is based on the knowledgedatabases that are always evolving because of new dis-coveries that are integrated into the databases. Therefore,our analyses inevitably represent a snapshot in time.
Acknowledgments: The authors thank the National Instituteon Deafness and Other Communication Disorders (NIHYNIDCDK08 DC010419 to K. M. S.), and the Bertarelli Foundation(K. M. S.) for the support. The authors also thank Dr. Joe Adamsfor helpful comments on earlier versions of the article.
REFERENCES
1. Traynor B, ‘‘The incidence of hearing loss around the world.’’Hearing International. Available at: http://hearinghealthmatters.org/hearinginternational/2011/incidence-of-hearing-loss-around-the-world/ (2012). Accessed March 1, 2012.
2. Morton NE. Genetic epidemiology of hearing impairment. Ann NY Acad Sci 1991;630:16Y31.
3. Hilgert N, Smith RJ, Van Camp G. Forty-six genes causing non-syndromic hearing impairment: which ones should be analyzed inDNA diagnostics? Mutat Res 2009;681:189Y96.
4. Bitner-Glindzics M. Hereditary deafness and phenotyping in humans.Br Med Bull 2002;63:73Y94.
968 G. A. STAMATIOU AND K. M. STANKOVIC
Otology & Neurotology, Vol. 34, No. 5, 2013
Copyright © 2013 Otology & Neurotology, Inc. Unauthorized reproduction of this article is prohibited.

5. Cryns K, Van Camp G. Deafness genes and their diagnostic appli-cations. Audiol Neurootol 2004;9:2Y22.
6. Hereditary Hearing Loss Homepage. Available at: http://hereditaryhearingloss.org/. Accessed March 29, 2012.
7. Schrijver I. Hereditary non-syndromic sensorineural hearing loss:transforming silence to sound. J Mol Diagn 2004;6:275Y84.
8. Friedman TB, Griffith AJ. Human nonsyndromic sensorineural deaf-ness. Annu Rev Genomics Hum Genet 2003;4:341Y402.
9. Linghu B, Snitkin ES, Hu Z, Xia Y, DeLisi C. Genome-wideprioritization of disease genes and identification of disease-diseaseassociations from an integrated human functional linkage network.Genome Biol 2009;10:R91.
10. Accetturo M, Creanza TM, Santoro C, et al. Finding new genes fornon-syndromic hearing loss through an in silico prioritization study.PLoS One 2010;5:pii e 12742.
11. Fisher RA. On the interpretation of x2 from contingency tables, andthe calculation of P. J R Stat Soc 1922;85:81Y94.
12. Scheffer D, Shen J, Mingqian Huang, Corey D, Chen ZY. Cell-specificgene expression in the inner ear with FACS and RNA-Seq (in pre-paration). Available at: http://shield.hms.harvard.edu/gene_search.html.Accessed March 29, 2012.
13. Lu CC, Appler JM, Houseman EA, Goodrich LV. Developmentalprofiling of spiral ganglion neurons reveals insights into auditorycircuit assembly. J Neurosci 2011;31:10903Y18.
14. Kenneson A, Van Naarden Braun K, Boyle C. GJB2 (connexin 26)variants and nonsyndromic sensorineural hearing loss: a HuGEreview. Genet Med 2002;4:258Y74.
15. Weil D, Blanchard S, Kaplan J, et al. Defective myosin VIIA generesponsible for Usher syndrome type 1B. Nature 1995;374:60Y1.
16. Liu XZ, Walsh J, Mburu P, et al. Mutations in the myosin VIIAgene cause non-syndromic recessive deafness. Nat Genet 1997;16:188Y90.
17. Everett LA, Glaser B, Beck JC, et al. Pendred syndrome is causedby mutations in a putative sulphate transporter gene (PDS). NatGenet 1997;17:411Y22.
18. Li XC, Everett LA, Lalwani AK, et al. A mutation in PDS causesnon-syndromic recessive deafness. Nat Genet 1998;18:215Y7.
19. Campos-Xavier AB, Saraiva JM, Savarirayan R, et al. Phenotypicvariability at the TGF-beta-1 locus in Camurati-Engelmann dis-ease. Hum Genet 2001;109:653Y8.
20. Thys M, Schrauwen I, Vanderstraeten K, et al. The coding poly-morphism T263I in TGF-b1 is associated with otosclerosis in twoindependent populations. Hum Mol Genet 2007;16:2021Y30.
21. Janssens K, ten Dijke P, Janssens S, Van Hul W. Transforminggrowth factor-beta1 to the bone. Endocr Rev 2005;26:743Y74.
22. Frenz DA, Galinovic-Schwartz V, Liu W, Flanders KC, Van deWater TR. Transforming growth factor beta 1 is an epithelial-derived signal peptide that influences otic capsule formation. DevBiol 1992;153:324Y36.
23. Oreffo ROC, Mundy GR, Seyedin SM, Bonewald LF. Activation ofthe bone-derived latent Tgf beta-complex by isolated osteoclasts.Biochem Biophys Res Commun 1989;158:817Y23.
24. Bakrania P, Efthimiou M, Klein JC, et al. Mutations in BMP4 causeeye, brain, and digit developmental anomalies: overlap between theBMP4 and hedgehog signaling pathways. Am J Hum Genet 2008;82:304Y19.
25. Gennari L, Merlotti D, Nuti R. Aromatase activity and bone loss.Adv Clin Chem 2011;54:129Y64.
26. Behl C, Widman M, Trapp T, Holsboer F. 17-beta estradiol protectsneurons from oxidative stress-induced cell death in vitro. BiochemBiophys Res Commun 1995;216:473Y82.
27. Hultcrantz M, Simonoska R, Stenberg AE. Estrogen and hearing: asummary of recent investigations. Acta Otolaryngol 2006;126:10Y4.
28. Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposingeffects of ERK and JNK-p38MAPkinases on apoptosis. Science1995;270:1326Y31.
29. Arthur DB, Georgi S, Akassoglou K, Insel PA. Inhibition of apoptosisby P2Y2 receptor activation: novel pathways for neuronal survival. JNeurosci 2006;26:3798Y804.
30. Pearson G, Robinson F, Beers Gibson T, et al. Mitogen activatedprotein (MAP) kinase pathways: regulation and physiologicalfunctions. Endocr Rev 2001;22:153Y83.
31. Wood K, Sarnecki C, Roberts TM, Blenis J. c-RAS mediates nervegrowth factor receptor modulation of three signal-transducing pro-tein kinases: MAP kinase, Raf-1 and RSK. Cell 1992;68:1041Y50.
32. Battaglia A, Pak K, Brors D, Bodmer D, Frangos JA, Ryan AF.Involvement of ras activation in toxic hair cell damage of themammalian cochlea. Neuroscience 2003;122:1025Y35.
33. Lahne M, Gale JE. Damaged-induced activation of ERK 1/2 incochlear supporting cells is a hair cell death-promoting signal thatdepends on extracellular ATP and calcium. J Neurosci 2008;28:4918Y28.
34. Rozenfeld R, Devi LA. Exploring a role for heteromerization inGPCR signalling specificity. Biochem J 2010;433:11Y8.
35. Millar RP, Newton CL. The year in G protein-coupled receptorresearch. Mol Endocrinol 2010;24:261Y74.
36. McMillan DR, White PC. Studies on the very large G protein-coupled receptor: from initial discovery to determining its role insensorineural deafness in higher animals. Adv Exp Med Biol 2010;706:76Y86.
37. Drewes T, Senkel S, Holewa B, Ryffel GU. Human hepatocytenuclear factor 4 isoforms are encoded by distinct and differentiallyexpressed genes. Mol Cell Biol 1996;16:925Y31.
38. Jiang S, Tanaka T, Iwanari H, et al. Expression and localization ofP1 promoter-driven hepatocyte nuclear factor-4alpha (HNF4alpha)isoforms in humans and rats. Nucl Recept 2003;1:5.
39. Hayhurst GP, Lee YH, Lambert G, Ward JM, Gonzalez FJ. Hepa-tocyte nuclear factor 4alpha (nuclear receptor 2A1) is essential formaintenance of hepatic gene expression and lipid homeostasis. MolCell Biol 2001;21:1393Y403.
40. Inoue Y, Hayhurst GP, Inoue J, Mori M, Gonzalez FJ. Defectiveureagenesis in mice carrying a liver-specific disruption of hepatocytenuclear factor 4alpha (HNF4alpha). HNF4alpha regulates ornithinetranscarbamylase in vivo. J Biol Chem 2002;277:25257Y65.
41. Inoue Y, Yu AM, Inoue J, Gonzalez FJ. Hepatocyte nuclear factor4alpha is a central regulator of bile acid conjugation. J Biol Chem2004;279:2480Y9.
42. Inoue Y, Peters LL, Yim SH, Inoue J, Gonzalez FJ. Role of hepa-tocyte nuclear factor 4alpha in control of blood coagulation factorgene expression. J Mol Med 2006;84:334Y44.
43. Gupta RK, Vatamaniuk MZ, Lee CS, et al. The MODY1 gene HNF-4alpha regulates selected genes involved in insulin secretion. J ClinInvest 2005;115:1006Y15.
44. Satoh H, Firestein GS, Billings PB, Harris JP, Keithley EM. Tumornecrosis factor-alpha, an initiator, and etanercept, an inhibitor ofcochlear inflammation. Laryngoscope 2002;112:1627Y34.
45. McCabe BF. Autoimmune sensorineural hearing loss. Ann OtolRhinol Laryngol 1979;88:585Y9
46. Parnes LS, Sun AH, Freeman DJ. Corticosteroid pharmacokineticsin the inner ear fluids: an animal study followed by clinical appli-cation. Laryngoscope 1999;109:1Y17.
47. Curhan SG, Eavey R, Shargorodsky J, Curhan GC. Analgesic useand the risk of hearing loss in men. Am J Med 2010;123:231Y7.
48. Curhan SG, Shargorodsky J, Eavey R, Curhan GC. Analgesic use andthe risk of hearing loss in women. Am J Epidemiol 2012;176:544Y54.
49. Maddox PT, Saunders J, Chandrasekhar SS. Sudden hearing lossfrom PDE-5 inhibitors: a possible cellular stress etiology. Lar-yngoscope 2009;119:1586Y9.
50. Prud’homme GJ. Pathobiology of transforming growth factor betain cancer, fibrosis and immunologic disease, and therapeutic con-siderations. Lab Invest 2007;87:1077Y91.
51. Wolf G. Renal injury due to rennin-angiotensin-aldosterone sysyemactivation of the transforming growth factor-beta pathway. KidneyInt 2006;70:1914Y9.
52. Tox U, Steffen HM. Impact of inhibitors of the renin-angiotensin-aldosterone system on liver fibrosis and portal hypertension. CurrMed Chem 2006;13:3649Y61.
53. Yao HW, Zhu JP, Zhao MH, Lu Y. Losartan attenuates bleomycininduced pulmonary fibrosis in rats. Respiration 2006;73:236Y42.
54. Bujak M, Frangogiannis NG. The role of TGF-beta signaling inmyocardial infarction and cardiac remodeling. Cardiovasc Res2007;74:184Y95.
55. Cohn RD, Van Erp C, Habashi JP, et al. Angiotensin II type 1receptor blockade attenuates TGF-beta-induced failure of muscle
969IPA OF HUMAN DEAFNESS GENES
Otology & Neurotology, Vol. 34, No. 5, 2013
Copyright © 2013 Otology & Neurotology, Inc. Unauthorized reproduction of this article is prohibited.

regeneration in multiple myopathic states. Nat Med 2007;13:204Y10.
56. Dussaule JC, Chatziantoniou C. Reversal of renal disease: is itenough to inhibit the action of angiotensin II? Cell Death Differ2007;14:1343Y9.
57. Kim SI, Kim HJ, Han DC, Lee HB. Effect of lovastatin on smallGTP binding proteins an on TGF-beta1 and fibronectin expression.Kidney Int Suppl 2000;77:S88YS92.
58. Watts KL, Spiteri MA. Connective tissue growth factor expressionand induction by transforming growth factor-beta is abrogated bysimvastatin via a Rho signaling mechanism. Am J Physiol Lung CellMol Physiol 2004;287:L1323Y32.
59. Vieira JM Jr, Mantovani E, Rodrigues LT, et al. Simvastatinattenuates renal inflammation, tubular transdifferentiation andinterstitial fibrosis in rats with unilateral uteral obstruction. NephrolDial Transplant 2005;20:1582Y91.
60. Redondo S, Santos-Gallego CG, Tejerina T. TGF-beta1: a noveltarget for cardiovascular pharmacology. Cytokine Growth FactorRev 2007;18:279Y86.
61. Lasky J. Pirfenidone. IDrugs 2004;7:166Y72.62. Antoniu SA. Pirfenidone for the treatment of idiopathic pulmonary
fibrosis. Expert Opin Investig Drugs 2006;15:823Y8.63. Wojtowicz-Praga S. Reversal of tumor-induced immunosuppres-
sion by TGF-beta inhibitors. Invest New Drugs 2003;2:21Y32.64. Ruzek MC, Hawes M, Pratt B, et al. Minimal effects on immune
parameters following chronic anti-TGF-beta monoclonal antibodyadministration to normal mice. Immunopharmacol Immunotoxicol2003;25:235Y57.
65. Forrester E, Chytil A, Bierie B, et al. Effect of conditional knockoutof the type II TGF-beta receptor gene in mammary epithelia onmammary gland development and polyomavirus middle T antigen
induced tumor formation and metastasis. Cancer Res 2005;65:2296Y302.
66. Tanimura S, Asato K, Fujishiro SH, Kohno M. Specific blockade ofthe ERK pathway inhibits the invasiveness of tumor cells: down-regulation of matrix metalloproteinase -3/-9/-14 and CD44. Bio-chem Biophys Res Commun 2003;304:801Y6.
67. Kohno M, Poyssegur J. Pharmacological inhibitors of the ERKsignaling pathway: applications as anticancer drugs. Prog CellCycle Res 2003;5:219Y24.
68. Borlak J, Niehof M. HNF4alpha and HNF1alpha dysfunction as amolecular rational for cyclosporine induced posttransplantationdiabetes mellitus. PLoS ONE 2009;4:e4662.
69. Marioni G, Perin N, Tregnaghi A, Bellemo B, Staffieri D, deFilippis C. Progressive sensorineural hearing loss probably inducedby chronic cyclosporin A treatment after renal transplantation forfocal glomerulosclerosis. Acta Otolaryngol 2004;124:603Y7.
70. Maeda Y, Hwang-Verslues WW, Wei G, et al. Tumor suppressorp53 down-regulates the expression of the human hepatocyte nuclearfactor 4> (HNF4>) gene. Biochem J 2008;415:289Y96.
71. Wang Z, Burke PA. Modulation of hepatocyte nuclear factor-4afunction by the peroxisome-proliferator-activated receptor-F co-activator-1> in the acute phase response. Biochem J 2008;415:289Y96.
72. Xie X, Liao H, Dang H, et al. Down-regulation of hepaticHNF4alpha gene expression during hyperinsulinemia via SREBPs.Mol Endocrinol 2009;23:434Y43.
73. Hwang-Veslues W, Sladek FM. HFN4aVrole in drug metabolismand potential drug target? Curr Opin Pharmacol 2010;10:698Y705.
74. Online Mendelian Inheritance in Man Database Homepage (OMIM).Available at: http://omim.org/. Accessed September 19, 2012.
75. The Jackson Laboratory Homepage. Available at: http://hearingimpairment.jax.org/. Accessed September 19, 2012.
Erratum
Long-Term Benefit and Sound Localization in Patients With Single-Sided DeafnessRehabilitated With an Osseointegrated Bone-Conduction Device: Erratum
In the article that appeared on page 111 of the January 2013 (Volume 34, Issue 1) issue of Otology &Neurotology, errors have been noted in the author names. They should have appeared as:
Nicolas Saroul, Mohamed Akkari, Yoann Pavier, Laurent Gilain, and Thierry Mom.
REFERENCE
Nicolas S, Mohamed A, Yoann P, Laurent G, Thierry M. Long-term benefit and sound localization in patients with single-sided deafnessrehabilitated with an osseointegrated bone-conduction device. Otol Neurotol 2013;34:111Y4.
970 G. A. STAMATIOU AND K. M. STANKOVIC
Otology & Neurotology, Vol. 34, No. 5, 2013
Copyright © 2013 Otology & Neurotology, Inc. Unauthorized reproduction of this article is prohibited.