a microfluidic platform based on robust gas and liquid
TRANSCRIPT

ANALYTICAL SCIENCES OCTOBER 2019, VOL. 35 1141
Introduction
Microfluidic devices are important platforms to culture biological tissues for observation of tissue functions. Compared with conventional setups, such as culturing dishes, microfluidic devices have advantages for enhanced deliveries of nutrients and gases to tissues, quick exchanges of culturing media, programable culturing operations, and stable observations under a microscope.1–4 Thus, microfluidic devices have been incorporated in studies on biological tissues.
The suprachiasmatic nucleus (SCN) is a small brain region, which functions as the central clock system for generating daily biological rhythms in mammals. Neuronal cells constituting the SCN form a robust circadian clock by exchanging and synchronizing phase information using peptides and small molecules moving between each cell. Neurons producing vasoactive intestinal peptide (VIP), arginine vasopressin (AVP), and/or gastrin releasing peptide (GRP) are localized at different sites in the SCN, and it has been suggested that these molecules are responsible for intercellular interactions.5 For example, Harmar et al.6 showed that synchronization between the SCN constituting cells disappeared in mice lacking VPAC2, a VIP receptor. Additionally, Yamaguchi et al.7 reported that intercellular synchronization in SCN is attenuated in mice deficient of AVP receptors, V1a and V1b, and they suggested that neurotransmission by AVP neurons has a key role in
synchronizing the SCN cells. However, there are still many unclear points in the molecular mechanisms that show how SCN stably keeps rhythms of approximately a 24-h period, even when there is no external input.
In order to elucidate the principle of the SCN in which multicellular cells are synchronized, it is necessary to observe the response of the SCN under various perturbations. Tissue explant culturing is an essential technique to preserve the complex cell–cell interactions in the SCN. Hence, this technique is more suitable to observe the nature of the SCN as a biological functional unit, compared with a dispersed cell culture, which is difficult to reproduce the same degree of cell–cell interactions. On the other hand, explanted tissues present a challenge for long-term observations of more than a few days because no appropriate culturing procedure has been established for explanted tissues to keep them alive and functional.8,9 SCN and other explanted tissues die in a dry condition while perfusion in which tissue explants are immersed in a culture medium can severely damage these tissues by an insufficient exchange of gases. To avoid these conditions, it is desirable to circulate the culture medium so as to cover tissues lightly for the exchange of nutrients and gases. However, conventional culturing dishes contain a bulk culture medium which is difficult to control the amount of precisely, or in a programmed manner. This difficulty causes a deviation in the surface level of the culture medium, affecting tissue viability and microscopic focus for clear observations. To overcome this challenge, microfluidic devices are suitable to control the amount of circulating culture medium precisely.1
However, culturing in reported microfluidic devices10–12 is inferior to conventional tissue culturing in terms of tissue
2019 © The Japan Society for Analytical Chemistry
N. O., G. N. K., and H. M. contributed equally to this work.† To whom correspondence should be addressed.E-mail: [email protected]
A Microfluidic Platform Based on Robust Gas and Liquid Exchange for Long-term Culturing of Explanted Tissues
Nobutoshi OTA,* Genki N. KANDA,* Hiroyuki MORIGUCHI,* Yusufu AISHAN,* Yigang SHEN,* Rikuhiro G. YAMADA,* Hiroki R. UEDA,*,** and Yo TANAKA*†
* Center for Biosystems Dynamics Research, RIKEN, 1-3 Yamadaoka, Suita, Osaka 565–0871, Japan ** Department of Systems Pharmacology, Graduate School of Medicine, The University of Tokyo, 7-3-1 Hongo,
Bunkyo, Tokyo 113–0033, Japan
Microfluidic devices are important platforms to culture and observe biological tissues. Compared with conventional setups, microfluidic devices have advantages in perfusion, including an enhanced delivery of nutrients and gases to tissues. However, explanted tissues can maintain their functions for only hours to days in microfluidic devices, although their observations are desired for weeks. The suprachiasmatic nucleus (SCN) is a brain region composed of heterogeneous cells to control the biological clock system through synchronizing individual cells in this region. The synchronized and complicated cell–cell interactions of SCN cells are difficult to reproduce from seeded cells. Thus, the viability of explanted SCN contributes to the study of SCN functions. In this paper, we propose a new perfusion platform combining a PDMS microfluidic device with a porous membrane to culture an explanted SCN for 25 days. We expect that this platform will provide a universal interface for microfluidic manipulation of tissue explants.
Keywords Organotypic culture, porous membrane, brain slice, suprachiasmatic nucleus, circadian rhythm
(Received March 22, 2019; Accepted June 25, 2019; Published October 10, 2019)

1142 ANALYTICAL SCIENCES OCTOBER 2019, VOL. 35
viability, a sustainable culturing period, and ease of tissue handling and observation. Some studies have described perfusion on microfluidic devices that include microchannel, micropattern or supportive microstructures.3,4,13 Microfluidic devices with a simple microchannel structure could precisely control the flow of the culture medium, although the sustainable culture period was limited to a few days.14 Supportive microstructures, for example, micropillars for holding an explanted tissue at the surface level of the culture medium15 and microneedles for modifying flow of culture medium11,16,17 were also reported. These structures enhance the exchange of gases and nutrients although the explanted tissues on the microstructures were functional for periods of a few hours to a few days. Microfluidic devices are also applicable to microtissue culturing, and the microtissue cultures are sustainable for a longer period than that of explanted tissues due to the larger area-to-volume ratio, by holding them in microwells for days.2 However, observation of complex biological functions in a microtissue is limited because the microtissue contains a smaller number of cells (103 cells) than does in a monolayer tissue (104 – 106 cells).18
Three-dimensional microfluidic devices have also been investigated so as to improve the culturing conditions. For an explanted tissue confined in a microfabricated container, it is difficult to perform tissue introduction and culturing; thus, special skills and a setup such as a system for circulation of gas-mixed culture medium are required.11 Organ-on-chips are also alternative culturing platforms in which models of biological organs are constructed. These devices have advantages in the patterning of cells to investigate the complex nature of target organs such as vascular models in healthy4 and diseased19 states, kidney,20 and gastrointestinal tract.21 Although an appropriate perfusion of culture medium elongates the cell viability in these devices, there are technical challenges concerning in device fabrications and operations, such as cell seeding in microfluidic channels.22 Moreover, organoids in microfluidic devices are delicate things to form into a desired tissue structure and pattern from seeded cells.23
In this study, we demonstrated culturing tissue for 25 days by using a simple microfluidic device with a semi-permeable channel. This long culturing period is due to the use of minimally wet SCN slices for robust exchanges of gases and nutrients, while other devices immersed tissues in the medium that may severely damage them due to an insufficient exchange of gases.9–11,16 Our device was composed of commercially available materials: polydimethylsiloxane (PDMS) and a culture insert of porous hydrophilic polytetrafluoroethylene (PTFE) membrane. After applying PDMS to part of the culture insert, we formed an impermeable channel using the PDMS microstructure and the PDMS-applied part of the PTFE membrane to prevent flooding of the culture medium into unintended areas. Meanwhile, a semi-permeable channel was formed by the PDMS microstructure and the PDMS-free part of the PTFE membrane to deliver the culture medium for an explanted tissue. Porous membranes are widely found in organs-on-chips for culturing a single layer or a few layers of cells, corresponding to a few to tens of μm thickness, as the base to grow cells immersed in culture medium via perfusion.3,4,20,23 On the other hand, porous membranes are also employed for conventional culturing to keep tissue slices with hundreds of μm thickness on the surface of the culture medium.1,11 Similar to conventional culture setups, our setup uses a porous membrane to keep tissue slices on the surface of culture medium. However, unlike conventional culture setups, ours continuously perfuses tissue slices via the microfluidic
system. In addition, out device only requires a simple operation of continuous infusion and withdrawal of the culture medium at a constant flow rate to keep the explanted tissues viable.
Experimental
Reagents and chemicalsDeionized water was generated with the TW-300RU pure
water system (Nomura Micro Science, Kanagawa, Japan). The following items were purchased from Sigma Aldrich (St. Louis, MO): NaHCO3, HEPES, apo-transferrin, putrescine, progesterone and sodium selenite. Kanamycin and DMEM/F12 (basal culture medium) were purchased from Life Technologies (Carlsbad, CA). Insulin and luciferin were purchased from Promega (Madison, WI). Glass slides (76 × 56 × 1 mm) were purchased from Matsunami Glass (Osaka, Japan). PDMS (Silpot 184) was purchased from Dow Corning Toray (Tokyo, Japan).
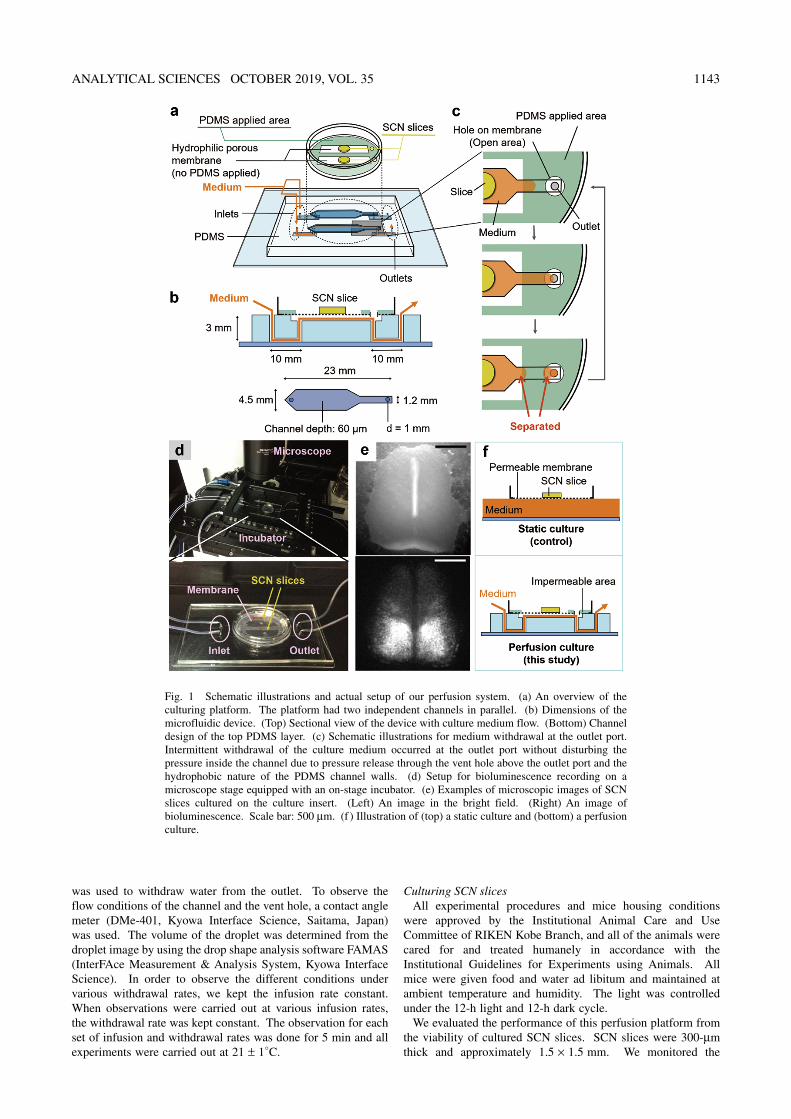
Device fabricationTo construct our culturing system (Fig. 1a), we fabricated a
microfluidic device made of PDMS and a culture insert of hydrophilic PTFE membrane (Millicell, Millipore, Burlington, MA) that were both commercially available. The PTFE membrane was hydrophilic and had 0.4-μm pores. PDMS channels were cast with a polyimide-tape mold of 60-μm thickness. The tape was placed on a glass slide, and was cut into patterns of the channels using a fine scalpel. For the top PDMS layer, the mold was shaped into two nonagonal patterns described in Fig. 1b. For the bottom PDMS layer, the mold was shaped into four rectangular patterns of 10 × 2 mm. PDMS was prepared by mixing elastomer and a curing agent at a 10:1 ratio. Then, PDMS was poured on the glass slide with the polyimide mold to cure through 3-h baking at 70°C. The cured PDMS was peeled off from the glass slide to obtain the cast pattern. The patterned PDMS was bonded with another glass slide by an O2-plasma treatment. A biopsy trepan punch (Kai Industries, Gifu, Japan) was used to inlet and outlet holes on the bonded PDMS; two holes worked as intel and outlet ports, and two holes connected the top and bottom channels (Fig. 1b).
The culture insert membrane was partially covered with PDMS, and this covered area is denoted as the “PDMS applied area” in Fig. 1a. To apply PDMS on the desired part of the membrane, the membrane was covered by tape (Magic Tape, 3M, St. Paul, MN), which was cut with the desired pattern. Then, the PDMS mixture was applied to the uncovered area of the membrane using a spatula, and was left overnight to dry at 21 ± 1°C. The PDMS-applied area of the PTFE membrane could be attached directly to the PDMS portion of the microfluidic platform by natural sealing between the two PDMS layers. A vent hole of 2-mm diameter was made on the PDMS-applied membrane area just above the outlet port on the PDMS channel (Fig. 1c). This hole played an important role for keeping the channel under the membrane open, or the channel could be closed by natural sealing of the PDMS layers when a vacuum pressure was applied from the outlet. The whole setup of the culturing and observation system is shown in Fig. 1d.
Investigation of the constructed perfusion systemWe investigated the constructed perfusion system to find
appropriate rates of infusion and withdrawal. For controlled infusion and withdrawal, two syringe pumps (Fusion 400, Chemyx, Stafford, TX) were used. One pump was applied to inject deionized water into the perfusion system, and the other
ANALYTICAL SCIENCES OCTOBER 2019, VOL. 35 1143
was used to withdraw water from the outlet. To observe the flow conditions of the channel and the vent hole, a contact angle meter (DMe-401, Kyowa Interface Science, Saitama, Japan) was used. The volume of the droplet was determined from the droplet image by using the drop shape analysis software FAMAS (InterFAce Measurement & Analysis System, Kyowa Interface Science). In order to observe the different conditions under various withdrawal rates, we kept the infusion rate constant. When observations were carried out at various infusion rates, the withdrawal rate was kept constant. The observation for each set of infusion and withdrawal rates was done for 5 min and all experiments were carried out at 21 ± 1°C.
Culturing SCN slicesAll experimental procedures and mice housing conditions
were approved by the Institutional Animal Care and Use Committee of RIKEN Kobe Branch, and all of the animals were cared for and treated humanely in accordance with the Institutional Guidelines for Experiments using Animals. All mice were given food and water ad libitum and maintained at ambient temperature and humidity. The light was controlled under the 12-h light and 12-h dark cycle.
We evaluated the performance of this perfusion platform from the viability of cultured SCN slices. SCN slices were 300-μm thick and approximately 1.5 × 1.5 mm. We monitored the
Fig. 1 Schematic illustrations and actual setup of our perfusion system. (a) An overview of the culturing platform. The platform had two independent channels in parallel. (b) Dimensions of the microfluidic device. (Top) Sectional view of the device with culture medium flow. (Bottom) Channel design of the top PDMS layer. (c) Schematic illustrations for medium withdrawal at the outlet port. Intermittent withdrawal of the culture medium occurred at the outlet port without disturbing the pressure inside the channel due to pressure release through the vent hole above the outlet port and the hydrophobic nature of the PDMS channel walls. (d) Setup for bioluminescence recording on a microscope stage equipped with an on-stage incubator. (e) Examples of microscopic images of SCN slices cultured on the culture insert. (Left) An image in the bright field. (Right) An image of bioluminescence. Scale bar: 500 μm. (f ) Illustration of (top) a static culture and (bottom) a perfusion culture.

1144 ANALYTICAL SCIENCES OCTOBER 2019, VOL. 35
activity of SCN slices derived from newborn (postnatal day 7) PER2::LUC knock-in mice,8 in which a clock gene (Period2) was fused with a luciferase gene. The expression level of Period2, representing the clock activity, could be monitored by bioluminescence under the supplementation of luciferin in the culture medium (Fig. 1e). For static cultures, SCN slices were cultured on culture inserts in sealed 35-mm dishes at 37°C. In each dish, a 1.2-mL culture medium was supplied for each slice (Fig. 1f ). The culture medium was DMEM/F12 supplemented with 300 mg/L NaHCO3, 20 mg/L kanamycin, 100 mg/L apo-transferrin, 100 μM putrescine, 20 nM progesterone, 30 nM sodium selenite, 5 μg/L insulin, and 0.1 mM luciferin. The medium was kept unchanged for static cultures during bioluminescence measurements because the abrupt change induced by medium exchange in a large volume works as an undesired stimulation on the SCN slice.24 Bioluminescence was measured with photomultiplier tube detector assemblies (LM2400R, Hamamatsu Photonics, Shizuoka, Japan). For perfusion cultures, SCN slices were cultured on the perfusion platform placed in an on-stage incubator (Tokai Hit, Shizuoka, Japan) that was saturated with water vapor, supplemented with 5% CO2 and kept at 37°C. The culture medium (DMEM/F12 supplemented with 1.2 g/L NaHCO3, 3.6 g/L HEPES, 20 mg/L kanamycin, 100 mg/L apo-transferrin, 100 μM putrescine, 20 nM progesterone, 30 nM sodium selenite, 5 μg/L insulin, and 0.1 mM luciferin) was infused at 2 μL/min and withdrawn at 2.5 μL/min. Bioluminescence was measured with a microscope (MVX10, Olympus, Tokyo, Japan) and an EM-CCD camera (ImagEM,
Hamamatsu Photonics) operated by Metamorph software (Molecular Devices, San Jose, CA). Because not all slices were identical, background signals of each sample were excluded by limiting the luminescence integration to a region of interest.
Results and Discussion
Validation of the constructed perfusion systemWe investigated liquid flow on the constructed perfusion
platform to establish robust culturing of SCN slices. SCN slices must be wet for proper nutritional supply, while the slices cannot survive for a long period if the culture medium completely covers them because their respiration is interfered with. The PDMS-applied membrane area of the culture insert enabled these culturing conditions. The PDMS-applied area worked as the top layer of the PDMS channel. Meanwhile, the untreated area was wet to deliver the medium to the slice, but the medium did not flood and cover the slice unless an excessive medium flow rate was applied. Thus, appropriate rates of infusion and withdrawal were important to ensure that SCN slices were constantly in contact with the culture medium, but not to submerge them in the medium. To balance the rates of infusion and withdrawal easily, a vent hole of 2-mm diameter for releasing pressure was placed just above the outlet (Figs. 1b and 1c). This hole vented excess pressure accumulated in the channel and prevented the overflow of culture medium onto the membrane. Thus, this setup allowed a range of infusion and
Fig. 2 Droplet formation on the membrane of a culture insert. (a) Illustration of droplet formation. (b) Experimental setup and observed droplet formation. (i) Droplet formation when the medium withdrawal rate was smaller than the infusion rate. The droplet volume increased, and eventually the droplet collapsed to submerge the cultured tissue. (ii) No droplet formation occurred when the withdrawal rate was equal to, or slightly greater, than the infusion rate. (iii) Formation of a small droplet when the withdrawal rate was greater than the infusion rate. A small volume of liquid was accumulated and was repeatedly withdrawn into the outlet channel.

ANALYTICAL SCIENCES OCTOBER 2019, VOL. 35 1145
withdrawal rates to be obtained to provide continuous medium flow. This enabled robust conditioning of the tissue on the membrane that was maintained with appropriate wetting.
The present system exchanges nutrients for the tissue by circulating the culture medium while gas exchanges of the tissue occur spontaneously unless the tissue is submerged in excess medium. In order to find an appropriate balance between the infusion and withdrawal rates, we observed the formation of a water droplet at the outlet port on the membrane of the culture insert (Fig. 2). First, we fixed the infusion rate at 2 μL/min and increased withdrawal rate from 0 to 10 μL/min with 0.5 μL/min increments. At the withdrawal rate boundary greater or less than 2 μL/min, different droplet formation behaviors were observed (Fig. 3). When the withdrawal rate was set in the range of 0 to 1.5 μL/min, the droplet was formed on the outlet port and its volume gradually increased as time passed. This growing droplet could eventually flow over the tissue on the membrane, and that would destroy the entire culture by submerging the explanted tissue. When the withdrawal rate was 2.0 μL/min or larger, the formed droplet could be regularly withdrawn into the outlet channel to prevent flooding of culture medium over the explanted tissue. For withdrawal rates of between 2.5 to 3 μL/min, no droplet was formed, and the whole microfluidic system established an equilibrium state of proper medium supply for long-term tissue culturing.
Then, in the same way, we fixed the withdrawal rate at 2.5 μL/min and set the infusion rate at from 0 to 2.5 μL/min with 0.5 μL/min increments. For infusion rates between 0 to 2 μL/min, no droplet formed at the outlet port. But when the infusion rate was equal to 2.5 μL/min, a droplet was formed and its volume constantly increased. From these experiments, we concluded that the withdrawal rate should be slightly greater than the infusion rate in order to avoid droplet formation at the outlet port. For further experiments, we employed an infusion rate of 2.0 μL/min and a withdrawal rate of 2.5 μL/min to culture explanted tissues on our perfusion platform.
Long-term culturing and observation of explanted SCN slicesWe compared the decrease in intensity of bioluminescence
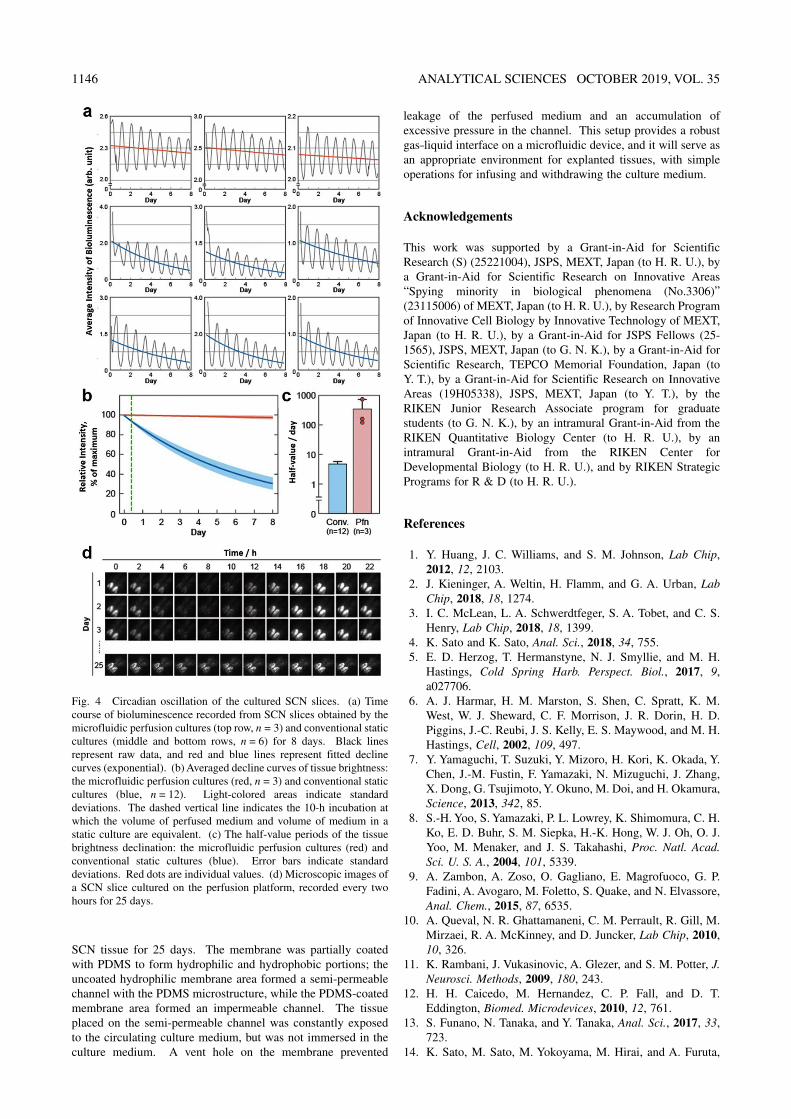
generated in the SCN slices cultured by our microfluidic perfusion cultures and conventional static cultures, assuming that the minimal decline of the bioluminescent intensity should represent a high viability of the cultured tissues. Whereas slices in both cultures showed clear circadian oscillations in bioluminescent intensity (Fig. 4a), the half-value time of the fitted decline curve (Fig. 4b) showed a dramatic improvement in the microfluidic perfusion cultures relative to the static cultures (>100 days in the microfluidic perfusion cultures, compared to 4.69 ± 0.84 days in static cultures, Fig. 4c). Comparing the bioluminescence of SCN slices for the 10-h incubation, where the volume of the perfused medium and the volume of the medium in a static culture were equivalent, the average relative intensity of the slices in the static culture decreased by 0.15 ± 0.10% (n = 3) from 0 h incubation, while that in the static culture decreased by 6.13 ± 1.00% (n = 12) (Fig. 4b). We also performed long-term culturing to confirm improved tissue viability on the perfusion platform, and could record clear oscillations in the bioluminescence for 25 days (Fig. 4d). Although bioluminescence measurement was halted after 25 days of SCN culturing for a demonstration, it was expected that the SCN culturing period could be longer than 25 days, possibly 100 days or more, based on the calculated half-values of the SCN bioluminescence in our perfusion culture (Fig. 4c). These results suggested that microfluidic perfusion can provide an improved platform for explanted tissues not only in the aspects of chemical consumption or device integration, but also in terms of tissue viability and the sustainable culture period.
Conclusions
In this study, we fabricated a PDMS device incorporated with a porous hydrophilic PTFE membrane to culture an explanted
Fig. 3 Performance of our perfusion system evaluated through droplet formation during 5 min at a constant infusion rate of 2.0 μL/min. (a) Low withdrawal rate. The volume of the droplet continuously increased at the outlet port. At withdrawal rates of 0 and 0.5 μL/min, the volume of the droplets was recorded before 5 min had passed; otherwise, the droplets had collapsed. (b) Withdrawal rate close to infusion rate. A small droplet or no droplet was formed. (c) High withdrawal rate. Formation of a small droplet and its withdrawal into the outlet port occurred repeatedly. (b) and (c) were suitable for long-term tissue culturing on the perfusion system with no flooding occurrence.
1146 ANALYTICAL SCIENCES OCTOBER 2019, VOL. 35
SCN tissue for 25 days. The membrane was partially coated with PDMS to form hydrophilic and hydrophobic portions; the uncoated hydrophilic membrane area formed a semi-permeable channel with the PDMS microstructure, while the PDMS-coated membrane area formed an impermeable channel. The tissue placed on the semi-permeable channel was constantly exposed to the circulating culture medium, but was not immersed in the culture medium. A vent hole on the membrane prevented
leakage of the perfused medium and an accumulation of excessive pressure in the channel. This setup provides a robust gas–liquid interface on a microfluidic device, and it will serve as an appropriate environment for explanted tissues, with simple operations for infusing and withdrawing the culture medium.
Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research (S) (25221004), JSPS, MEXT, Japan (to H. R. U.), by a Grant-in-Aid for Scientific Research on Innovative Areas “Spying minority in biological phenomena (No.3306)” (23115006) of MEXT, Japan (to H. R. U.), by Research Program of Innovative Cell Biology by Innovative Technology of MEXT, Japan (to H. R. U.), by a Grant-in-Aid for JSPS Fellows (25-1565), JSPS, MEXT, Japan (to G. N. K.), by a Grant-in-Aid for Scientific Research, TEPCO Memorial Foundation, Japan (to Y. T.), by a Grant-in-Aid for Scientific Research on Innovative Areas (19H05338), JSPS, MEXT, Japan (to Y. T.), by the RIKEN Junior Research Associate program for graduate students (to G. N. K.), by an intramural Grant-in-Aid from the RIKEN Quantitative Biology Center (to H. R. U.), by an intramural Grant-in-Aid from the RIKEN Center for Developmental Biology (to H. R. U.), and by RIKEN Strategic Programs for R & D (to H. R. U.).
References
1. Y. Huang, J. C. Williams, and S. M. Johnson, Lab Chip, 2012, 12, 2103.
2. J. Kieninger, A. Weltin, H. Flamm, and G. A. Urban, Lab Chip, 2018, 18, 1274.
3. I. C. McLean, L. A. Schwerdtfeger, S. A. Tobet, and C. S. Henry, Lab Chip, 2018, 18, 1399.
4. K. Sato and K. Sato, Anal. Sci., 2018, 34, 755. 5. E. D. Herzog, T. Hermanstyne, N. J. Smyllie, and M. H.
Hastings, Cold Spring Harb. Perspect. Biol., 2017, 9, a027706.
6. A. J. Harmar, H. M. Marston, S. Shen, C. Spratt, K. M. West, W. J. Sheward, C. F. Morrison, J. R. Dorin, H. D. Piggins, J.-C. Reubi, J. S. Kelly, E. S. Maywood, and M. H. Hastings, Cell, 2002, 109, 497.
7. Y. Yamaguchi, T. Suzuki, Y. Mizoro, H. Kori, K. Okada, Y. Chen, J.-M. Fustin, F. Yamazaki, N. Mizuguchi, J. Zhang, X. Dong, G. Tsujimoto, Y. Okuno, M. Doi, and H. Okamura, Science, 2013, 342, 85.
8. S.-H. Yoo, S. Yamazaki, P. L. Lowrey, K. Shimomura, C. H. Ko, E. D. Buhr, S. M. Siepka, H.-K. Hong, W. J. Oh, O. J. Yoo, M. Menaker, and J. S. Takahashi, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5339.
9. A. Zambon, A. Zoso, O. Gagliano, E. Magrofuoco, G. P. Fadini, A. Avogaro, M. Foletto, S. Quake, and N. Elvassore, Anal. Chem., 2015, 87, 6535.
10. A. Queval, N. R. Ghattamaneni, C. M. Perrault, R. Gill, M. Mirzaei, R. A. McKinney, and D. Juncker, Lab Chip, 2010, 10, 326.
11. K. Rambani, J. Vukasinovic, A. Glezer, and S. M. Potter, J. Neurosci. Methods, 2009, 180, 243.
12. H. H. Caicedo, M. Hernandez, C. P. Fall, and D. T. Eddington, Biomed. Microdevices, 2010, 12, 761.
13. S. Funano, N. Tanaka, and Y. Tanaka, Anal. Sci., 2017, 33, 723.
14. K. Sato, M. Sato, M. Yokoyama, M. Hirai, and A. Furuta,
Fig. 4 Circadian oscillation of the cultured SCN slices. (a) Time course of bioluminescence recorded from SCN slices obtained by the microfluidic perfusion cultures (top row, n = 3) and conventional static cultures (middle and bottom rows, n = 6) for 8 days. Black lines represent raw data, and red and blue lines represent fitted decline curves (exponential). (b) Averaged decline curves of tissue brightness: the microfluidic perfusion cultures (red, n = 3) and conventional static cultures (blue, n = 12). Light-colored areas indicate standard deviations. The dashed vertical line indicates the 10-h incubation at which the volume of perfused medium and volume of medium in a static culture are equivalent. (c) The half-value periods of the tissue brightness declination: the microfluidic perfusion cultures (red) and conventional static cultures (blue). Error bars indicate standard deviations. Red dots are individual values. (d) Microscopic images of a SCN slice cultured on the perfusion platform, recorded every two hours for 25 days.

ANALYTICAL SCIENCES OCTOBER 2019, VOL. 35 1147
Anal. Sci., 2019, 35, 49. 15. P. A. Passeraub, A. C. Almeida, and N. V. Thakor, Biomed.
Microdevices, 2003, 5, 147. 16. Y. Choi, M. A. McClain, M. C. LaPlaca, A. B. Frazier, and
M. G. Allen, Biomed. Microdevices, 2007, 9, 7. 17. D. K. Cullen, J. Vukasinovic, A. Glezer, and M. C. LaPlaca,
J. Neural Eng., 2007, 4, 159. 18. M. Simian and M. J. Bissell, J. Cell Biol., 2017, 216, 31. 19. K. Sato, M. Nakajima, S. Tokuda, and A. Ogawa, Anal.
Sci., 2016, 32, 1217. 20. Y. Sakuta, I. Takehara, K. Tsunoda, and K. Sato, Anal. Sci.,
2018, 34, 1073. 21. L. A. Schwerdtfeger, E. P. Ryan, and S. A. Tobet, Am. J.
Physiol.:Gastrointest. Liver Physiol., 2016, 310, G240.
22. S. Xiao, J. R. Coppeta, H. B. Rogers, B. C. Isenberg, J. Zhu, S. A. Olalekan, K. E. McKinnon, D. Dokic, A. S. Rashedi, D. J. Haisenleder, S. S. Malpani, C. A. Arnold-Murray, K. Chen, M. Jiang, L. Bai, C. T. Nguyen, J. Zhang, M. M. Laronda, T. J. Hope, K. P. Maniar, M. E. Pavone, M. J. Avram, E. C. Sefton, S. Getsios, J. E. Burdette, J. J. Kim, J. T. Borenstein, and T. K. Woodruff, Nat. Commun., 2017, 8, 14584.
23. S. N. Bhatia and D. E. Ingber, Nat. Biotechnol., 2014, 32, 760.
24. A. C. Liu, D. K. Welsh, C. H. Ko, H. G. Tran, E. E. Zhang, A. A. Priest, E. D. Buhr, O. Singer, K. Meeker, I. M. Verma, F. J. Doyle, J. S. Takahashi, and S. A. Kay, Cell, 2007, 129, 605.