alois fürstner group meeting o'malley 2/29/2005 · pdf filegroup meeting...
TRANSCRIPT
Group Meeting2/29/2005O'Malley Alois Fürstner
Prof. Alois Fürstner was born in 1962 in Bruck an der Mur, Austria. He received his Ph.D. in 1987 from the Technical University of Graz under Prof. Weidmann. In 1990-1991, he was engaged in postdoctoral studies with Prof. Oppolzer at theUniversity of Geneva. He completed his habilitation in 1992 at the Technical University of Graz and obtained a position as a professor at the Max-Planck-Institut für Kohlenforschung. In 1998, he was promoted to director. He is also affiliated with theUniversity of Dortmund. His contributions to chemistry have been recognized with numerous awards, including the Dozentenstipendium of the Fonds der Chemischen Industrie (1994), the Arthur C. Cope Scholar Award (2002), and the Otto Bayer Prize (2006).
Research in the Fürstner group focuses on organometallic chemistry and its application to the synthesis of complex natural products. Specific areas of focus include alkene and alkyne metathesis, development of new metal catalyzed and mediated reactions, and the preparation and use of active metals.
Alkene MetathesisThe Fürstner group has made impressive contributions in the field of ring-closing alkene metathesis, particularly the areas of: (i) extension to the synthesis of medium and macrocyclic rings (ii) control of product stereochemistry (iii) development of new catalyst systems (iv) application to the total synthesisof complex natural products.
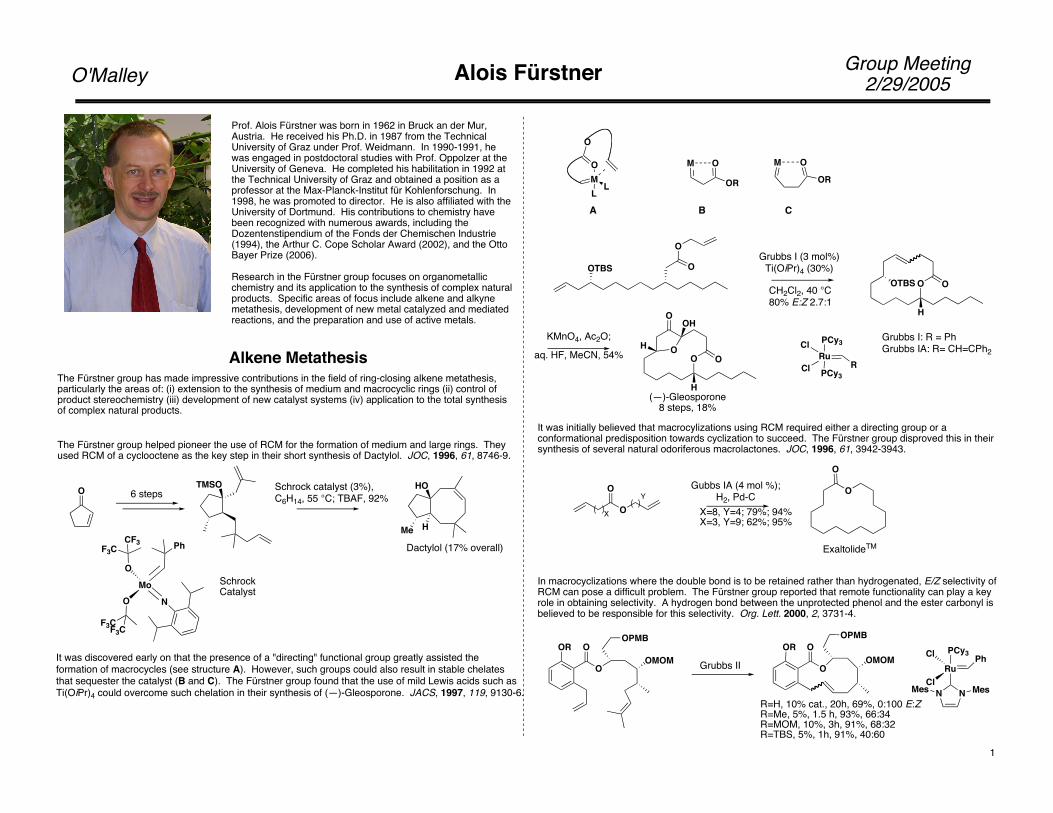
The Fürstner group helped pioneer the use of RCM for the formation of medium and large rings. Theyused RCM of a cyclooctene as the key step in their short synthesis of Dactylol. JOC, 1996, 61, 8746-9.
O TMSO HO
Me H
6 steps Schrock catalyst (3%), C6H14, 55 °C; TBAF, 92%
Dactylol (17% overall)
MoO
O
N
PhF3CCF3
F3CF3C
Schrock Catalyst
It was discovered early on that the presence of a "directing" functional group greatly assisted the formation of macrocycles (see structure A). However, such groups could also result in stable chelatesthat sequester the catalyst (B and C). The Fürstner group found that the use of mild Lewis acids such asTi(OiPr)4 could overcome such chelation in their synthesis of (—)-Gleosporone. JACS, 1997, 119, 9130-6.
ML
O
L
O
M O M O
OR OR
A B C
OTBS O
O
OOTBS O
H
O O
H
OH
OHO
Grubbs I (3 mol%)Ti(OiPr)4 (30%)
CH2Cl2, 40 °C80% E:Z 2.7:1
KMnO4, Ac2O;
aq. HF, MeCN, 54%
(—)-Gleosporone8 steps, 18%
It was initially believed that macrocylizations using RCM required either a directing group or a conformational predisposition towards cyclization to succeed. The Fürstner group disproved this in their synthesis of several natural odoriferous macrolactones. JOC, 1996, 61, 3942-3943.
O
OX
Y O
O
RuPCy3
PCy3
Cl
Cl R
Grubbs I: R = PhGrubbs IA: R= CH=CPh2
Gubbs IA (4 mol %);H2, Pd-C
X=8, Y=4; 79%; 94%X=3, Y=9; 62%; 95%
ExaltolideTM
In macrocyclizations where the double bond is to be retained rather than hydrogenated, E/Z selectivity of RCM can pose a difficult problem. The Fürstner group reported that remote functionality can play a key role in obtaining selectivity. A hydrogen bond between the unprotected phenol and the ester carbonyl is believed to be responsible for this selectivity. Org. Lett. 2000, 2, 3731-4.
OR O
OOMOM
OPMB
OOMOM
OPMBOOR
Grubbs II Ru
PCy3Cl
ClN N MesMes
Ph
R=H, 10% cat., 20h, 69%, 0:100 E:ZR=Me, 5%, 1.5 h, 93%, 66:34R=MOM, 10%, 3h, 91%, 68:32R=TBS, 5%, 1h, 91%, 40:60
1
Group Meeting2/29/2005O'Malley Alois Fürstner
2
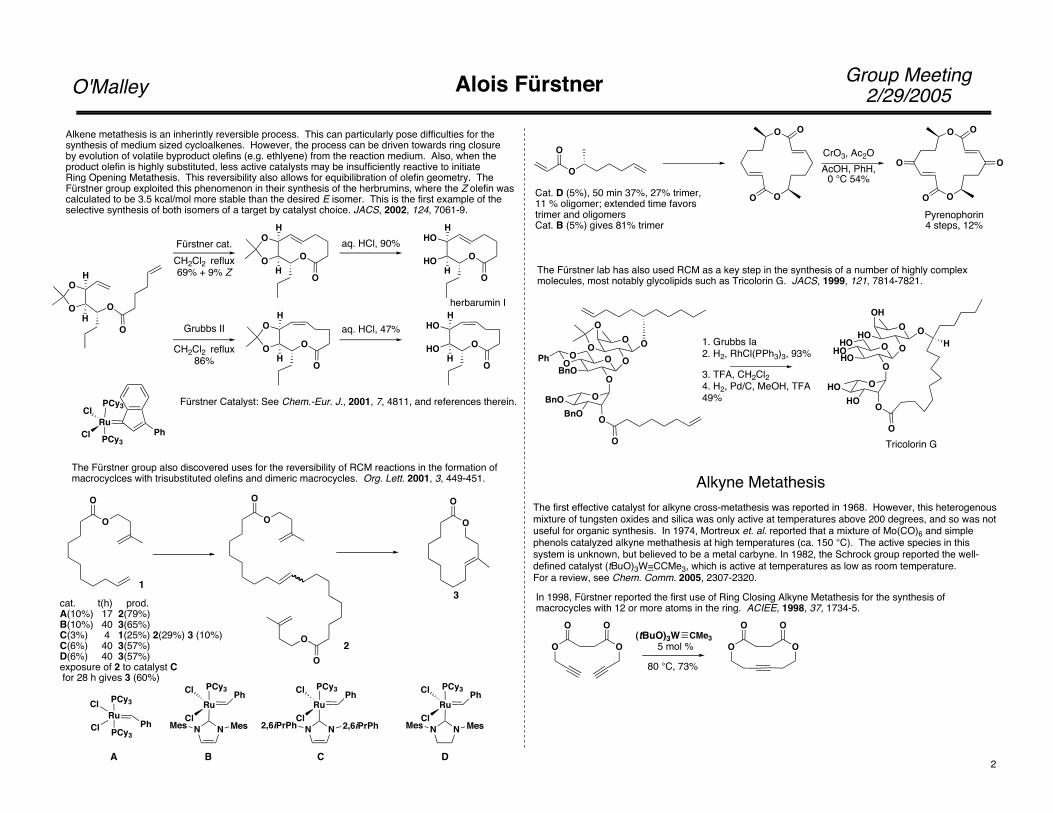
Alkene metathesis is an inherintly reversible process. This can particularly pose difficulties for thesynthesis of medium sized cycloalkenes. However, the process can be driven towards ring closureby evolution of volatile byproduct olefins (e.g. ethlyene) from the reaction medium. Also, when the product olefin is highly substituted, less active catalysts may be insufficiently reactive to initiate Ring Opening Metathesis. This reversibility also allows for equibilibration of olefin geometry. The Fürstner group exploited this phenomenon in their synthesis of the herbrumins, where the Z olefin wascalculated to be 3.5 kcal/mol more stable than the desired E isomer. This is the first example of theselective synthesis of both isomers of a target by catalyst choice. JACS, 2002, 124, 7061-9.
O
O
O
OH
HO
O
O
O
H
H
O
O
O
O
H
H
HO
HO
O
O
H
H
HO
HO
O
O
H
H
Grubbs II
CH2Cl2 reflux86%
Fürstner cat.CH2Cl2 reflux69% + 9% Z
aq. HCl, 90%
aq. HCl, 47%
herbarumin I
Ru
PCy3
PCy3
Cl
Cl Ph
Fürstner Catalyst: See Chem.-Eur. J., 2001, 7, 4811, and references therein.
The Fürstner group also discovered uses for the reversibility of RCM reactions in the formation of macrocyclces with trisubstituted olefins and dimeric macrocycles. Org. Lett. 2001, 3, 449-451.
O
O
O
O
O
O
O
O
Ru
PCy3Cl
ClN N MesMes
PhRu
PCy3Cl
ClN N MesMes
PhRu
PCy3Cl
ClN N 2,6iPrPh2,6iPrPh
Ph
A B C
cat. t(h) prod.A(10%) 17 2(79%)B(10%) 40 3(65%)C(3%) 4 1(25%) 2(29%) 3 (10%)C(6%) 40 3(57%)D(6%) 40 3(57%)exposure of 2 to catalyst C for 28 h gives 3 (60%)
RuPCy3
PCy3
Cl
Cl Ph
D
1
2
3
O
O
O
O
O
O
O
O
O
O
O OCrO3, Ac2OAcOH, PhH,
0 °C 54%Cat. D (5%), 50 min 37%, 27% trimer, 11 % oligomer; extended time favorstrimer and oligomersCat. B (5%) gives 81% trimer
Pyrenophorin4 steps, 12%
The Fürstner lab has also used RCM as a key step in the synthesis of a number of highly complex molecules, most notably glycolipids such as Tricolorin G. JACS, 1999, 121, 7814-7821.
O
OO
O
O
OO
O
OOPh
BnO
BnOBnO O
O
O
OO
O
O
OHO
OH
HOHO
HO
HOHO O
O
H1. Grubbs Ia2. H2, RhCl(PPh3)3, 93%
3. TFA, CH2Cl24. H2, Pd/C, MeOH, TFA49%
Tricolorin G
Alkyne MetathesisThe first effective catalyst for alkyne cross-metathesis was reported in 1968. However, this heterogenous mixture of tungsten oxides and silica was only active at temperatures above 200 degrees, and so was not useful for organic synthesis. In 1974, Mortreux et. al. reported that a mixture of Mo(CO)6 and simple phenols catalyzed alkyne methathesis at high temperatures (ca. 150 °C). The active species in this system is unknown, but believed to be a metal carbyne. In 1982, the Schrock group reported the well-defined catalyst (tBuO)3W=CCMe3, which is active at temperatures as low as room temperature.For a review, see Chem. Comm. 2005, 2307-2320.
In 1998, Fürstner reported the first use of Ring Closing Alkyne Metathesis for the synthesis of macrocycles with 12 or more atoms in the ring. ACIEE, 1998, 37, 1734-5.
OO
O O
OO
O O(tBuO)3W CMe3
80 °C, 73%
5 mol %
Group Meeting2/29/2005O'Malley Alois Fürstner
3
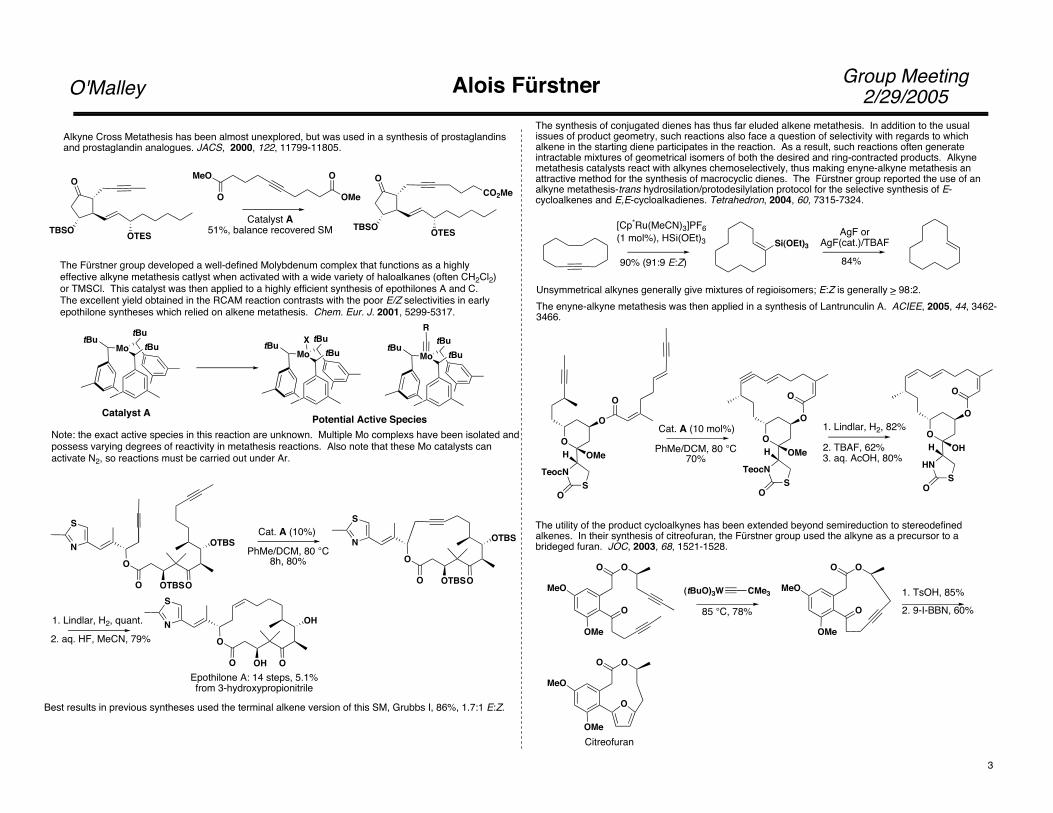
The Fürstner group developed a well-defined Molybdenum complex that functions as a highly effective alkyne metathesis catlyst when activated with a wide variety of haloalkanes (often CH2Cl2)or TMSCl. This catalyst was then applied to a highly efficient synthesis of epothilones A and C.The excellent yield obtained in the RCAM reaction contrasts with the poor E/Z selectivities in earlyepothilone syntheses which relied on alkene metathesis. Chem. Eur. J. 2001, 5299-5317.
Alkyne Cross Metathesis has been almost unexplored, but was used in a synthesis of prostaglandinsand prostaglandin analogues. JACS, 2000, 122, 11799-11805.
O
TBSO OTES
OMe
MeO
O
O
Catalyst A51%, balance recovered SM
O
TBSO OTES
CO2Me
O
S
N
OTBSO
OTBS
O
O
S
N
OTBSO
OTBS
O
MotBu
tButBu
MotBu
tButBu Mo
tButBu
tBuX
R
Catalyst A Potential Active SpeciesNote: the exact active species in this reaction are unknown. Multiple Mo complexs have been isolated andpossess varying degrees of reactivity in metathesis reactions. Also note that these Mo catalysts can activate N2, so reactions must be carried out under Ar.
Cat. A (10%)
PhMe/DCM, 80 °C8h, 80%
Best results in previous syntheses used the terminal alkene version of this SM, Grubbs I, 86%, 1.7:1 E:Z.
O
S
N
OHO
OH
OEpothilone A: 14 steps, 5.1% from 3-hydroxypropionitrile
1. Lindlar, H2, quant.
2. aq. HF, MeCN, 79%
The synthesis of conjugated dienes has thus far eluded alkene metathesis. In addition to the usual issues of product geometry, such reactions also face a question of selectivity with regards to which alkene in the starting diene participates in the reaction. As a result, such reactions often generate intractable mixtures of geometrical isomers of both the desired and ring-contracted products. Alkyne metathesis catalysts react with alkynes chemoselectively, thus making enyne-alkyne metathesis anattractive method for the synthesis of macrocyclic dienes. The Fürstner group reported the use of an alkyne metathesis-trans hydrosilation/protodesilylation protocol for the selective synthesis of E-cycloalkenes and E,E-cycloalkadienes. Tetrahedron, 2004, 60, 7315-7324.
Si(OEt)3
[Cp*Ru(MeCN)3]PF6(1 mol%), HSi(OEt)3
90% (91:9 E:Z)
AgF or AgF(cat.)/TBAF
84%
Unsymmetrical alkynes generally give mixtures of regioisomers; E:Z is generally > 98:2.The enyne-alkyne metathesis was then applied in a synthesis of Lantrunculin A. ACIEE, 2005, 44, 3462-3466.
O
TeocNS
O
H OMe
O
O
O
TeocNS
O
H OMe
O
O
O
HNS
O
H OH
O
O
Cat. A (10 mol%)
PhMe/DCM, 80 °C70%
1. Lindlar, H2, 82%
2. TBAF, 62%3. aq. AcOH, 80%
The utility of the product cycloalkynes has been extended beyond semireduction to stereodefined alkenes. In their synthesis of citreofuran, the Fürstner group used the alkyne as a precursor to a brideged furan. JOC, 2003, 68, 1521-1528.
O
OO
MeO
OMe
O
OO
MeO
OMe
OO
MeO
OMe
O
(tBuO)3W CMe3
85 °C, 78%
1. TsOH, 85%2. 9-I-BBN, 60%
Citreofuran

Group Meeting2/29/2005O'Malley Alois Fürstner
4
Development of Novel Metal Catalyzed and Mediated ReactionsThe Fürstner group has developed procedures for the Ti-mediated synthesis of a number of heterocycles.Tetrahedron 1992, 48, 5991-6010. As background, it is important to note that, although the "McMurry"reagents formed from a variety of Ti precursors and reducing agents are often thought of simply as "Ti(0)",there are important differences in the composition and oxidation potentials of these reagents. SeeFürstner,A. "The McMurry Reaction and Related Transformations" in Transition Metals for Organic Synthesis, 2nd ed. Wiley, 2004, 449-468, for a detailed discussion.
K + 8C(graphite) 150 °C, 10 min KC8 (potassium-graphite laminate)
TiCl3 + 3KC8THF reflux Titanium on graphite
O
OO
R
R
R
R
OOR Ph
R R
Rand
Ti-Graphite
NH
OO
R
R
HN
R
RTi-Graphite
"Instant" low-valent Ti conditions involving the reduction of complexed Ti salts with Zn in situ were also found to give excellent selectivity in this transformation. JOC, 1994, 5215-5229.
NH
NO
CF3
OPh
O
NH
NO
CF3
PhTiCl3, Zn, THF
reflux, 90%
complete retention of chirality
Depending on the nature of the substrate and transformation, low-valent Ti mediated reactions often require several equivalents of Ti salts, this can be inconvenient particularly on large scale or when highlyreactive reducing agents e.g. K or LiAlH4 are used. A procedure for the use of catalytic Ti is highly desirable, but the strength of Ti-O bonds generally procludes the reduction of the titanium oxides formedin this reaction. Using chlorosilanes and stoichiometric Zn, the Fürstner group has developed a procedurefor McMurray reactions catalytic in TiCl3. JACS, 1995, 117, 4468-4475.
NH
OPh
O
O
OEt NH
Ph
O
OEt
TiCl3 (10 mol%)Zn (5 eq.), TMSCl (5 eq.)
MeCN reflux, 30 min73%
Cl Cl
Ideally, commercially avaiable titanium dust would be used as the source of Ti for these reactions. Unfortunately, the passivating layer of titanium oxides which coat titanium metal has defied all attempts at activation. However, the successful development of the catalytic protocol led to the discovery that TMSCl could be used as an activating agent.
NH
OO
Ph
Ph
HN
Ph
PhTi dust (3 eq),TMSCl (3 eq.)
DME reflux, 92%
The use of commercial Ti powder was found to be particularly advantageous for macrocyclizations; a 36 membered ring could be formed in excellent yield without the normal precautions (extremely high dilution, slow addition of substrate).
(CH2)26
O
Ph
O
Ph
(CH2)26
Ti (65 eq.)TMSCl (65 eq.)preactivate 68 h
DME (0.02 M) reflux 6h, 90%
The coupling of an amide and a ketone to form a pyrrole was used as the centerpiece of an expedient synthesis of Lukianol A. JOC, 1995, 60, 6637-6641.
ONH
Ar Ar
OOOMe
NH
Ar Ar
O
OMe
Ar = p-OMeC6H4
N
Ar Ar
O
OMe
O
Ar
NO
O
OH
HO OH
Ti-graphiteDME reflux, 52%
p-MeO-C6H4OCH2Br
K2CO3, 91%
1. tBuOK; Ac2O, NaOAc, reflux 59%2. BBr3, -78 0 °C
Lukianol A8 steps, 12%
Ph
Ph

Group Meeting2/29/2005O'Malley Alois Fürstner
5
O NO2
OH
O NO2OiPr
MeO
N
O NH2OiPr
MeO
R
R=CH2CH2C6H4OMe
Ti mediated indole synthesis was the key step in the recently reported synthesis of Dictyodendrin B.JACS, 2005, 127, 11620-11621.
N
O HNOiPr
MeO
R
O
MeO
OMe
N
NH
OMe
OMe
MeO
R
N
NH
OMe
OMe
MeO
OMe
N
NH
OMe
OMe
MeO
R
N
NH
OMe
OMe
MeO
R
HO
MeO
O
MeO
N
NH
OH
OH
HO
OH
O
HO
OSO3NaOiPr
OiPr OiPr
OiPr
1. iPrBr, K2CO3, 99%2. pMBCHO, NaOMe, 70 °C 74%
1.TosMIC, NaH-30 °C;
pMBCH2Br, 83%2. Fe, HCl, 96%
OMeCl
O
OMe
DMAP, NEt3
89%
hu, Pd/C, PhNO2 81% 1. NBS, 0 °C, 69%
2. MeLi; BuLi; pMBCHO-78 °C to rt, 97%
TiCl3/2 KC8
DME/py reflux71-93%
TPAP (10%), NMO
66%
1. BCl3, TBAI, -20 °C, 85%
3. BCl3, TBAI, 0 °C to rt;Zn,HCO2NH4,58%
2. Cl3CCH2OSO2Cl,DABCO, 92%
Dictyodendrin B13 steps, 8%
The Nozaki-Hiyama-Kishi coupling is the addition of organochromium(III) reagents to electrophiles, usually aldehydes. The organochroumium reagents are generally derived from the Ni or Pd catalyzed reduction of active (allyl, alkenyl, aryl, etc.) halides with Cr(II). The excellent selectivity and functional group tolerance of this reaction has caused it to become the coupling method of choice for the synthesis of many complex natural products, most notably Kishi's synthesis of Palytoxin. However, there are a number drawbacks. The Cr(II) salts, which must often be used in significant excess are highly toxic. Moreover, the requirement to use excess chromium is prohibitive to efforts to find an enantioselective version of this reaction. While the strength of the Cr-O bond formed in the product gives this reaction a powerful thermodynamic driving force, it also complicates the task of liberating the Cr to react further.Drawing on their experience with developing catalytic Ti carbonyl couplings, the Fürstner group developed the first Nozaki-Hiyama-Kishi coupling reaction using catalytic Cr(II). Zn proved to be unsuitable due to the sensitivity of many aldehydes to the Lewis acidic ZnX2 generated by the reduction of Cr(III). Mn proved to be a suitable substitute, and the Cr(II)/Mn/TMSCl conditions have provided the basis for several subsequent asymmetric variants of this reaction. JACS,1996, 118, 2533-2534, 12349-12357.
I
H
O+
OH
400 mol %CrCl2, 20 °C, DMF, 78%15 mol% CrCl2,1.7 eq Mn, 2.4 eq. TMSCl 50 °C, DMF/DME, 67%15 mol% CrCl2,1.7 eq Mn, 2.4 eq. ClMe2Si(CH2)3CN 50 °C, DMF/DME, 72%CrCl2 is doped with NiCl2 (ca. 15%)
Group Meeting2/29/2005O'Malley Alois Fürstner
6
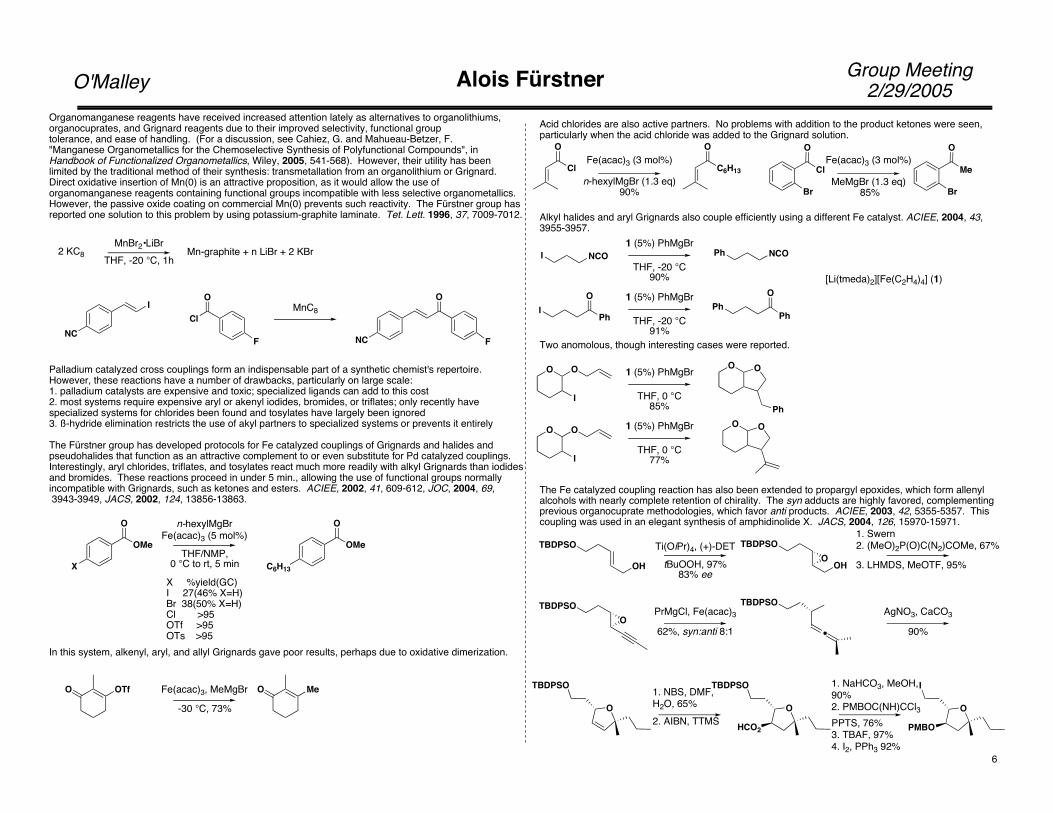
Organomanganese reagents have received increased attention lately as alternatives to organolithiums, organocuprates, and Grignard reagents due to their improved selectivity, functional grouptolerance, and ease of handling. (For a discussion, see Cahiez, G. and Mahueau-Betzer, F. "Manganese Organometallics for the Chemoselective Synthesis of Polyfunctional Compounds", in Handbook of Functionalized Organometallics, Wiley, 2005, 541-568). However, their utility has been limited by the traditional method of their synthesis: transmetallation from an organolithium or Grignard.Direct oxidative insertion of Mn(0) is an attractive proposition, as it would allow the use of organomanganese reagents containing functional groups incompatible with less selective organometallics.However, the passive oxide coating on commercial Mn(0) prevents such reactivity. The Fürstner group hasreported one solution to this problem by using potassium-graphite laminate. Tet. Lett. 1996, 37, 7009-7012.
2 KC8MnBr2•LiBr
THF, -20 °C, 1hMn-graphite + n LiBr + 2 KBr
I
NC
O
Cl
F NC
O
F
MnC8
Palladium catalyzed cross couplings form an indispensable part of a synthetic chemist's repertoire. However, these reactions have a number of drawbacks, particularly on large scale:1. palladium catalysts are expensive and toxic; specialized ligands can add to this cost2. most systems require expensive aryl or akenyl iodides, bromides, or triflates; only recently have specialized systems for chlorides been found and tosylates have largely been ignored3. ß-hydride elimination restricts the use of akyl partners to specialized systems or prevents it entirely
The Fürstner group has developed protocols for Fe catalyzed couplings of Grignards and halides and pseudohalides that function as an attractive complement to or even substitute for Pd catalyzed couplings.Interestingly, aryl chlorides, triflates, and tosylates react much more readily with alkyl Grignards than iodides and bromides. These reactions proceed in under 5 min., allowing the use of functional groups normally incompatible with Grignards, such as ketones and esters. ACIEE, 2002, 41, 609-612, JOC, 2004, 69, 3943-3949, JACS, 2002, 124, 13856-13863.
X
O
OMe
C6H13
O
OMe
n-hexylMgBrFe(acac)3 (5 mol%)
THF/NMP, 0 °C to rt, 5 min
X %yield(GC)I 27(46% X=H)Br 38(50% X=H)Cl >95OTf >95OTs >95
In this system, alkenyl, aryl, and allyl Grignards gave poor results, perhaps due to oxidative dimerization.
Acid chlorides are also active partners. No problems with addition to the product ketones were seen, particularly when the acid chloride was added to the Grignard solution.
O
ClFe(acac)3 (3 mol%)
n-hexylMgBr (1.3 eq)90%
O
C6H13
O
Cl
Br
O
Me
Br
Fe(acac)3 (3 mol%)
MeMgBr (1.3 eq) 85%
Alkyl halides and aryl Grignards also couple efficiently using a different Fe catalyst. ACIEE, 2004, 43, 3955-3957.
O OTf O MeFe(acac)3, MeMgBr
-30 °C, 73%
I NCO Ph NCO
[Li(tmeda)2][Fe(C2H4)4] (1)
1 (5%) PhMgBr
THF, -20 °C90%
I Ph1 (5%) PhMgBr
THF, -20 °C91%
O
Ph
O
Ph
Two anomolous, though interesting cases were reported.
O
I
O
O
I
O
1 (5%) PhMgBr
THF, 0 °C85%
1 (5%) PhMgBr
THF, 0 °C77%
O O
PhO O
The Fe catalyzed coupling reaction has also been extended to propargyl epoxides, which form allenyl alcohols with nearly complete retention of chirality. The syn adducts are highly favored, complementing previous organocuprate methodologies, which favor anti products. ACIEE, 2003, 42, 5355-5357. This coupling was used in an elegant synthesis of amphidinolide X. JACS, 2004, 126, 15970-15971.
TBDPSO
OH
TBDPSO
OHO
TBDPSOO
•
TBDPSO
O
TBDPSO
O
TBDPSO
HCO2
O
I
PMBO
Ti(OiPr)4, (+)-DETtBuOOH, 97%
83% ee
1. Swern2. (MeO)2P(O)C(N2)COMe, 67%
3. LHMDS, MeOTF, 95%
PrMgCl, Fe(acac)362%, syn:anti 8:1
AgNO3, CaCO3
90%
1. NBS, DMF,H2O, 65%2. AIBN, TTMS
1. NaHCO3, MeOH, 90%2. PMBOC(NH)CCl3PPTS, 76%3. TBAF, 97%4. I2, PPh3 92%
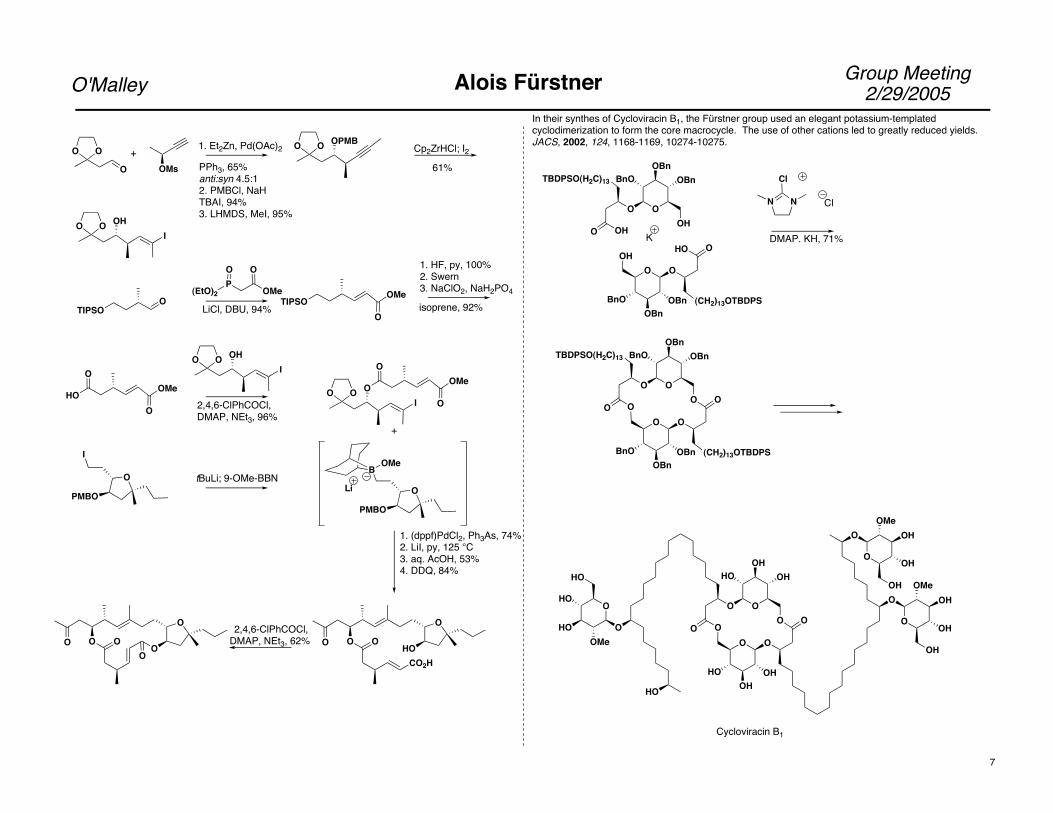
Group Meeting2/29/2005O'Malley Alois Fürstner
7
OO
O OMs+
OO OPMB
OO OHI
TIPSOO TIPSO
O
OMe
HOO
OMeO
OOI
OO
OMeO
O
I
PMBO O
PMBO
BOMe
Li
+
O OHO
CO2H
O
OO OO
O
OO
OO OHI
1. Et2Zn, Pd(OAc)2
PPh3, 65%anti:syn 4.5:12. PMBCl, NaHTBAI, 94%3. LHMDS, MeI, 95%
Cp2ZrHCl; I261%
(EtO)2P
OMe
O O
LiCl, DBU, 94%
1. HF, py, 100%2. Swern3. NaClO2, NaH2PO4
isoprene, 92%
2,4,6-ClPhCOCl,DMAP, NEt3, 96%
tBuLi; 9-OMe-BBN
1. (dppf)PdCl2, Ph3As, 74%2. LiI, py, 125 °C3. aq. AcOH, 53%4. DDQ, 84%
2,4,6-ClPhCOCl,DMAP, NEt3, 62%
O O
OHOH
OBnTBDPSO(H2C)13 OBnBnO
O
OO
HOOH
OBn(CH2)13OTBDPSBnO OBn
OK
In their synthes of Cycloviracin B1, the Fürstner group used an elegant potassium-templated cyclodimerization to form the core macrocycle. The use of other cations led to greatly reduced yields. JACS, 2002, 124, 1168-1169, 10274-10275.
O O
OO
OBnTBDPSO(H2C)13 OBnBnO
OOO
OBn(CH2)13OTBDPSBnO OBn
O
NN
Cl
Cl
DMAP. KH, 71%
O O
OO
OHOHHO
OOO
OHHO OH
OO
O
O
O
OOH
O
HO
OH
HO
HO
OH
OH
HO
OMe
OMe
OH
OMe
OH
Cycloviracin B1
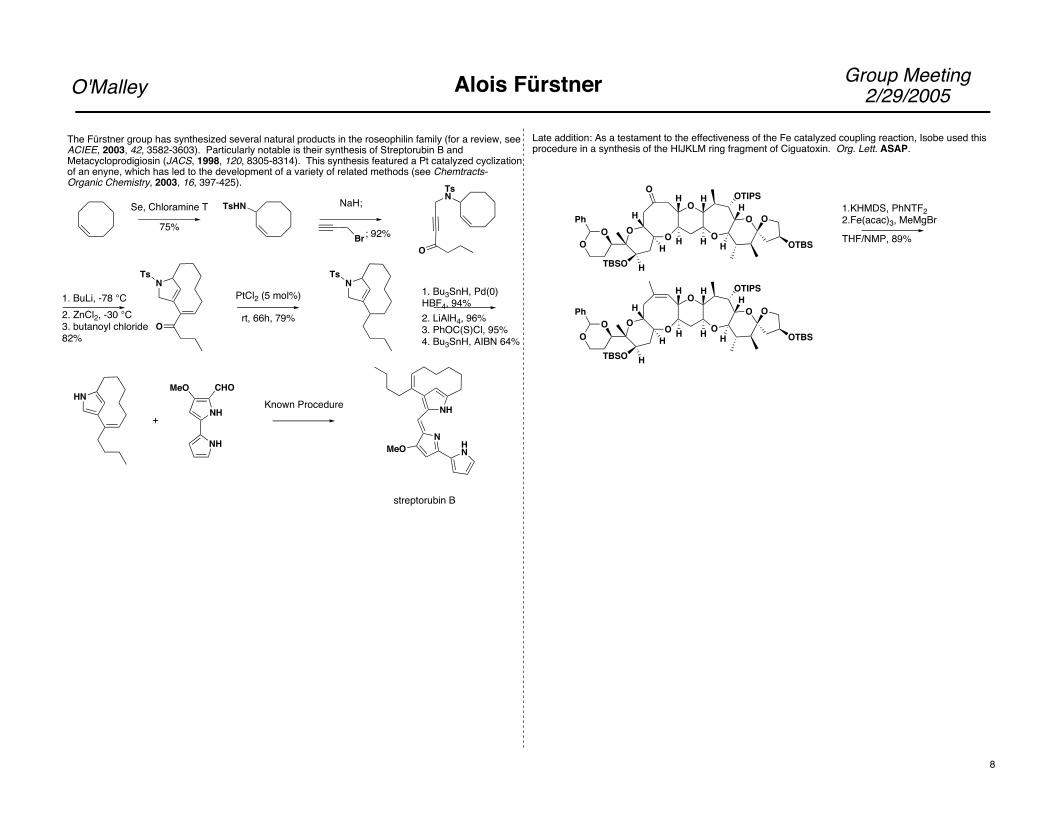
Group Meeting2/29/2005O'Malley Alois Fürstner
8
The Fürstner group has synthesized several natural products in the roseophilin family (for a review, seeACIEE, 2003, 42, 3582-3603). Particularly notable is their synthesis of Streptorubin B and Metacycloprodigiosin (JACS, 1998, 120, 8305-8314). This synthesis featured a Pt catalyzed cyclizationof an enyne, which has led to the development of a variety of related methods (see Chemtracts-Organic Chemistry, 2003, 16, 397-425).
TsHN
TsN
O
N
O
TsN
Ts
HN
NH
NH
MeO CHO
NH
HN
NMeO
Se, Chloramine T
75%
NaH;
Br ; 92%
1. BuLi, -78 °C2. ZnCl2, -30 °C3. butanoyl chloride82%
PtCl2 (5 mol%)
rt, 66h, 79%
1. Bu3SnH, Pd(0)HBF4, 94%2. LiAlH4, 96%3. PhOC(S)Cl, 95%4. Bu3SnH, AIBN 64%
+Known Procedure
streptorubin B
Late addition: As a testament to the effectiveness of the Fe catalyzed coupling reaction, Isobe used this procedure in a synthesis of the HIJKLM ring fragment of Ciguatoxin. Org. Lett. ASAP.
O
O
O O
O OO
O
H
H HH
OTBS
TBSO H
HH H H
OTIPSO
Ph
O
O
O O
O OO
O
H
H HH
OTBS
TBSO H
HH H H
OTIPS
Ph
1.KHMDS, PhNTF22.Fe(acac)3, MeMgBr
THF/NMP, 89%