chapter 11 reaction of alcohols pdc or pcc h ox h c c ohbiewerm/2325-summary.pdf · conversion of...
TRANSCRIPT

Chapter 11 Reaction of Alcohols
Oxidation of alcohols
Alcohols can be oxidized to carbon-oxygen double bonds (carbonyl compounds)
Alcohols are at the same oxidation level as alkenes
Therefore alkenes can be converted to alcohols with acidic water
H3O+H2C
CH2 H3C OH H3C
O
HOX
H3C
O
OHOX
Primary alcohols will be converted to aldehydes under basic oxidation conditions
Primary alcohols will be converted to carboxylic acids under nonbasic oxidation conditions
PDC or PCC
CrO3, H2SO4

Alcohols as Nucleophiles
To make alcohols more nucleophilic, need to abstract the acidic hydrogen (remember pKa’s!)
With this method, can make nucleophilic oxygen that can react through any SN2 type reaction already studied
Neutral alcohols can react as nucleophiles (often observe this with carboxylic acid derivatives or inorganic analogs)
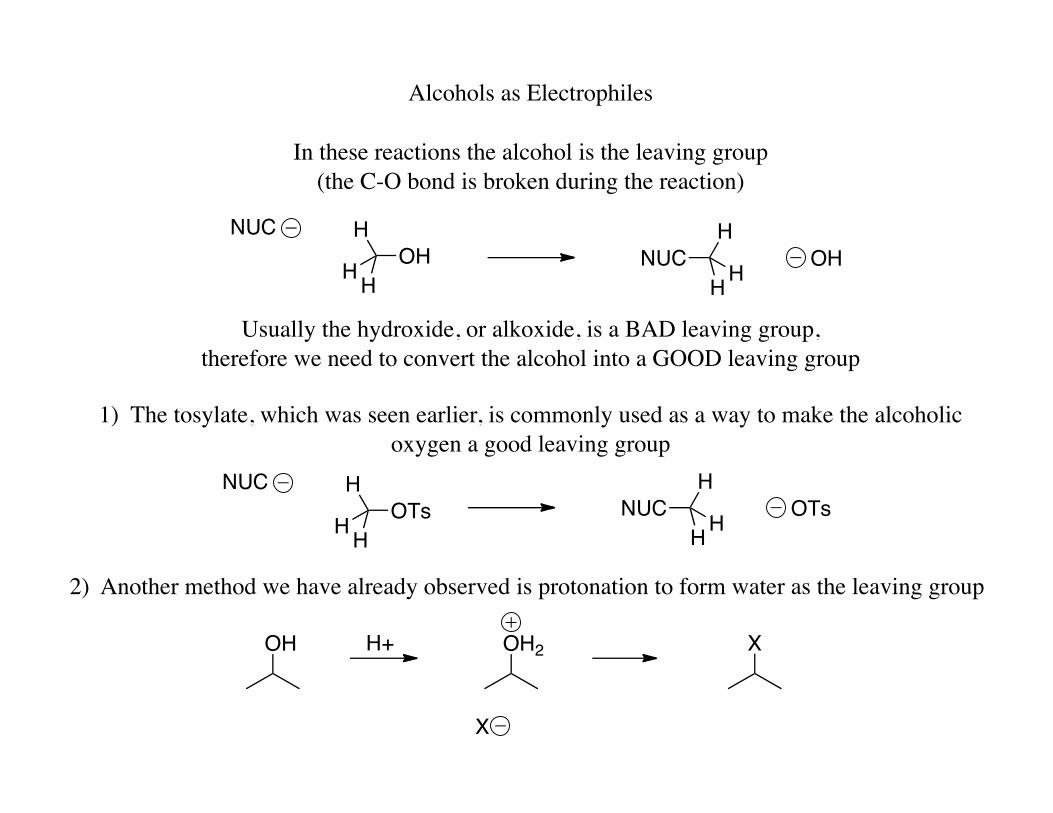
Alcohols as Electrophiles
OHH
HH
NUC
In these reactions the alcohol is the leaving group (the C-O bond is broken during the reaction)
NUCH
HHOH
Usually the hydroxide, or alkoxide, is a BAD leaving group, therefore we need to convert the alcohol into a GOOD leaving group
1) The tosylate, which was seen earlier, is commonly used as a way to make the alcoholic oxygen a good leaving group
OTsH
HH
NUCNUC
H
HHOTs
2) Another method we have already observed is protonation to form water as the leaving group
OH OH2 XH+
X

Conversion of Alcohols into Halides
The methods to convert alcohols into electrophiles are used to transform alcohols into alkyl halides
Can occur with either SN1 or SN2 conditions depending upon alcohol used
Stereochemistry, and possibility of rearrangements, depends on mechanism for each reaction
Type of alcohol chloride bromide iodide primary
secondary tertiary
SOCl2
SOCl2
HCl HBr HI
PBr3
PBr3
P/I2
P/I2
Best reagents for interconversions:

Chapter 12 Infrared Spectroscopy
IR is used for Functional Group Identification
Position of peaks can be used to differentiate all possible carbonyl compounds in addition to distinguishing common functional groups including nitriles, alkynes and aromatic rings
Factors to be considered for peaks: Position of Absorbance
(related to energy needed for absorbance)
C=C double bond CC triple bond
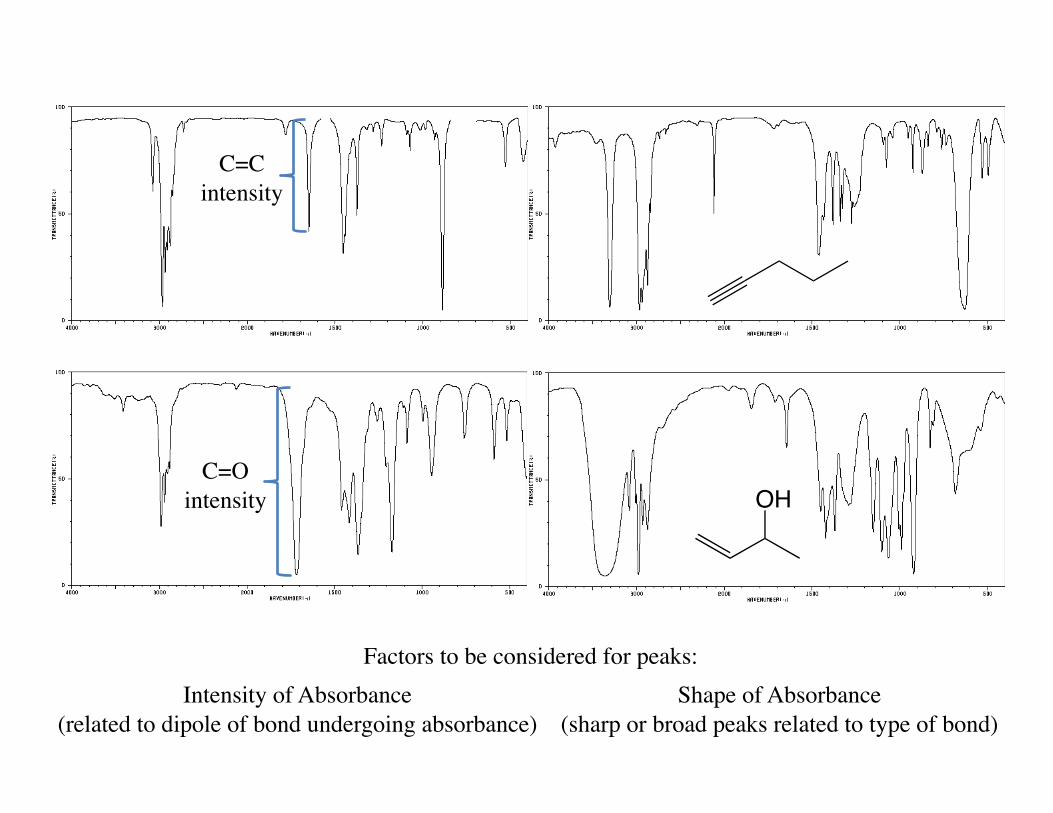
Factors to be considered for peaks: Intensity of Absorbance
(related to dipole of bond undergoing absorbance)
C=C intensity
C=O intensity
O
Shape of Absorbance (sharp or broad peaks related to type of bond)
OH

Chapter 12 Mass Spectrometry
In addition to determining the parent molecular weight for a sample, a MS can also be used to characterize what atoms are present and to differentiate isomers
Isotopic Patterns for natural abundance:
Fragmentation patterns can also be different for isomers

Predicted Mass Spectrometry Differences
Isotope Differences Used to Distinguish Halogen Substituents
I
m/z = 204no m+2 for iodinem+1 = 6.4% of m
Br
m/z = 156m+2 ~ equal to m
Cl
m/z = 112m+2 ~ 1/3 of m
F
m/z = 96no m+2 for fluorine

Fragmentation Pattern to Determine Structure OH e-beam OH
m/z 102OH OH
OH
87 45
CH3 OH!-cleavage
57
or or
OH
84
CH3
69
loss of water Remember:
Only charged forms are observed in a MS

Chapter 13 NMR Spectroscopy
Any nucleus with either an odd atomic number or odd mass has a “nuclear spin”
A charge species that is spinning creates a current loop, which in turn creates magnetic field lines
In solution, however, there are many hydrogens present and the spinning direction is random
In presence of large external magnetic field the spin directions are quantized

Shielding
Need to remember the structure of a compound (consider only an isolated C-H bond)
To reach the nucleus the magnetic field must past through the electron cloud surrounding the nucleus
The electrons surrounding the nucleus are charged species that can rotate in the presence of the external magnetic field
What this means is that the external magnetic field (B0) is effectively reduced by the time it reaches the nucleus (B0 minus the field of the electron cloud)
C HB0
Bnet = B0 - Belectron

In addition to the electron density surrounding the hydrogen causing shielding, each hydrogen acts like its own magnetic field
If one magnet is close to another, it will feel the effect of that magnetic field
This occurs in a 1H NMR a hydrogen will feel the effect of neighboring hydrogens
In the field of one additional hydrogen therefore we would observe two signals
N+1 rule: can predict the splitting that will be observed by counting the number of hydrogens on adjacent carbons (N) and adding one
Splitting
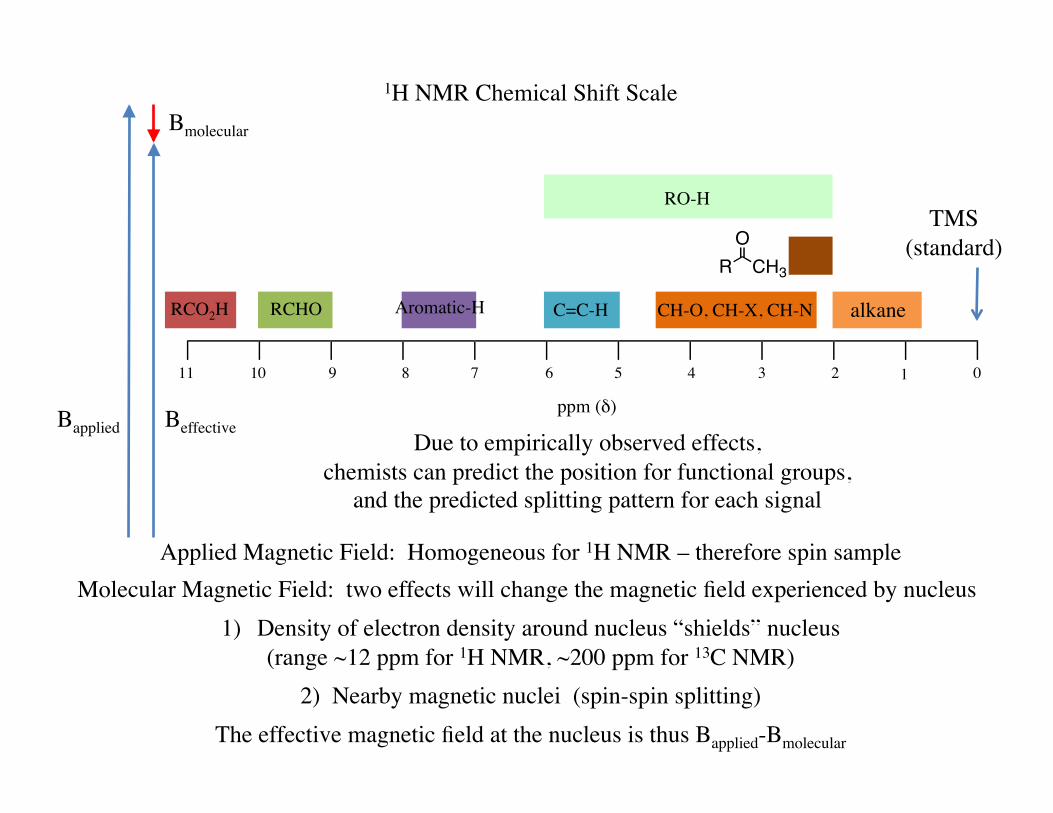
1H NMR Chemical Shift Scale
Bapplied
Bmolecular
Beffective
Applied Magnetic Field: Homogeneous for 1H NMR – therefore spin sample Molecular Magnetic Field: two effects will change the magnetic field experienced by nucleus
1) Density of electron density around nucleus “shields” nucleus (range ~12 ppm for 1H NMR, ~200 ppm for 13C NMR)
2) Nearby magnetic nuclei (spin-spin splitting) The effective magnetic field at the nucleus is thus Bapplied-Bmolecular
Due to empirically observed effects, chemists can predict the position for functional groups,
and the predicted splitting pattern for each signal
01234567891011
ppm (!)
TMS (standard)
alkane CH-O, CH-X, CH-N C=C-H Aromatic-H RCHO RCO2H
RO-H
ROCH3

13C NMR Spectroscopy
Because a 13C atom has an odd mass, it also has inherent magnetic field lines and will display NMR spectroscopy in the presence of a large external magnetic field
The downfield shift is dependent upon the shielding caused by the electrons around the carbon, due to the extra electron density around carbon compared to hydrogen there is a
greater amount of shielding in a 13C NMR
Instead of ~11 ppm range for 1H NMR, 13C NMR is typically in a ~200 ppm range
Due to the low probability of having two 13C isotopes adjacent, there is no splitting observed from the possible spin states of adjacent carbons
Typically observe a spin decoupled 13C NMR spectrum which has only singlets for each carbon
Observe all carbons though, do not need to have a hydrogen attached as with a 1H NMR

020406080100120140160180200220
ppm (!)
01234567891011
ppm (!)
Amount of Downfield Shift is Comparable
1H NMR
13C NMR
alkanes
alkanes
alkyl halide
C X
ether
alkene
aromatic
aldehyde
carboxylicacid
C-O
alkene
aromatic
carbonyl carbons
alkyne
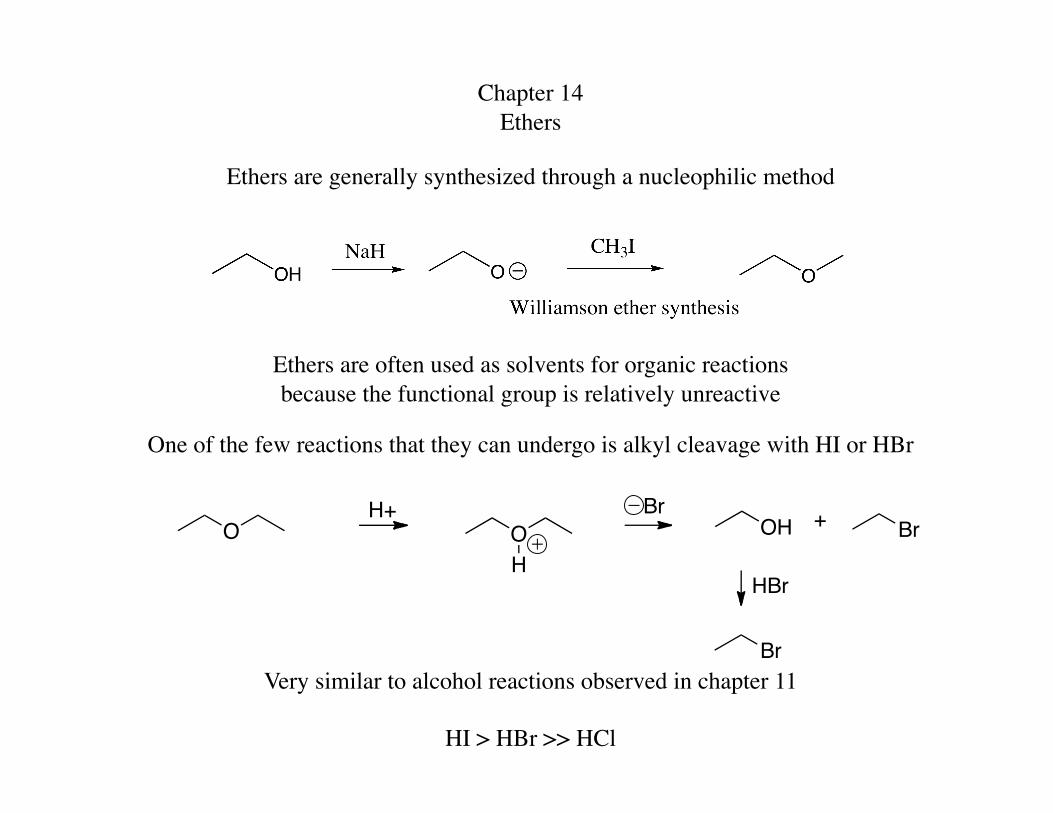
Chapter 14 Ethers
Ethers are generally synthesized through a nucleophilic method
Ethers are often used as solvents for organic reactions because the functional group is relatively unreactive
One of the few reactions that they can undergo is alkyl cleavage with HI or HBr
Very similar to alcohol reactions observed in chapter 11
HI > HBr >> HCl
OH+
OH
BrOH Br+
HBr
Br

Epoxides
One type of ethers that are reactive is a cyclic ether in a 3-membered ring called epoxides
Epoxides can be synthesized in one of two ways:
1) Reacting alkenes with a peracid
2) Reacting halohydrins with a weak base

Regiochemistry in Reaction of Epoxides
The base catalyzed opening of epoxides goes through a common SN2 mechanism, therefore the nucleophile attacks the least hindered carbon of the epoxide
OCH3MgBr
O
O H+ OH
In the acid catalyzed opening of epoxides, the reaction first protonates the oxygen This protonated oxygen can equilibrate to an open form that places more
partial positive charge on more substituted carbon, therefore the more substituted carbon is the preferred reaction site for the nucleophile
CH3OHHO OCH3

Chapter 15 Conjugated Systems
Conjugated systems occur anytime there are p orbitals on adjacent atoms in conjugation
Whenever there are p orbitals in conjugations, molecular orbitals result by the mixing of orbitals
The difference in energy is due to the number of nodes (different phases overlap), more nodes means higher in energy and electrons are filled in lowest energy orbitals first
=
4 p orbitals = 4 MOs
E

Addition to Conjugated Dienes
As we have already seen, alkenes can react with electrophiles to create a carbocation
With conjugated dienes this reaction will create an allylic carbocation
The nucleophile can then react with either resonance form in the second step

Kinetic versus Thermodynamic Control
What forms faster (kinetic product) and what is more stable (thermodynamic product) need not be the same
H+
H2O
Consider the addition to conjugated dienes
Generate allylic cation in first step Allylic cation can have water react at two sites
Reaction at 2˚ cation site has a more stable transition state
!+
!+H2O
H2O
!+
!+
Thus the kinetic product has water reacting at 2˚ site
OH
OH
Reaction a 1˚ site, though, generates more stable product
(more substituted double bond) The thermodynamic product has
water reacting at 1˚ site
E
Reaction at 2˚ site Reaction at 1˚ site
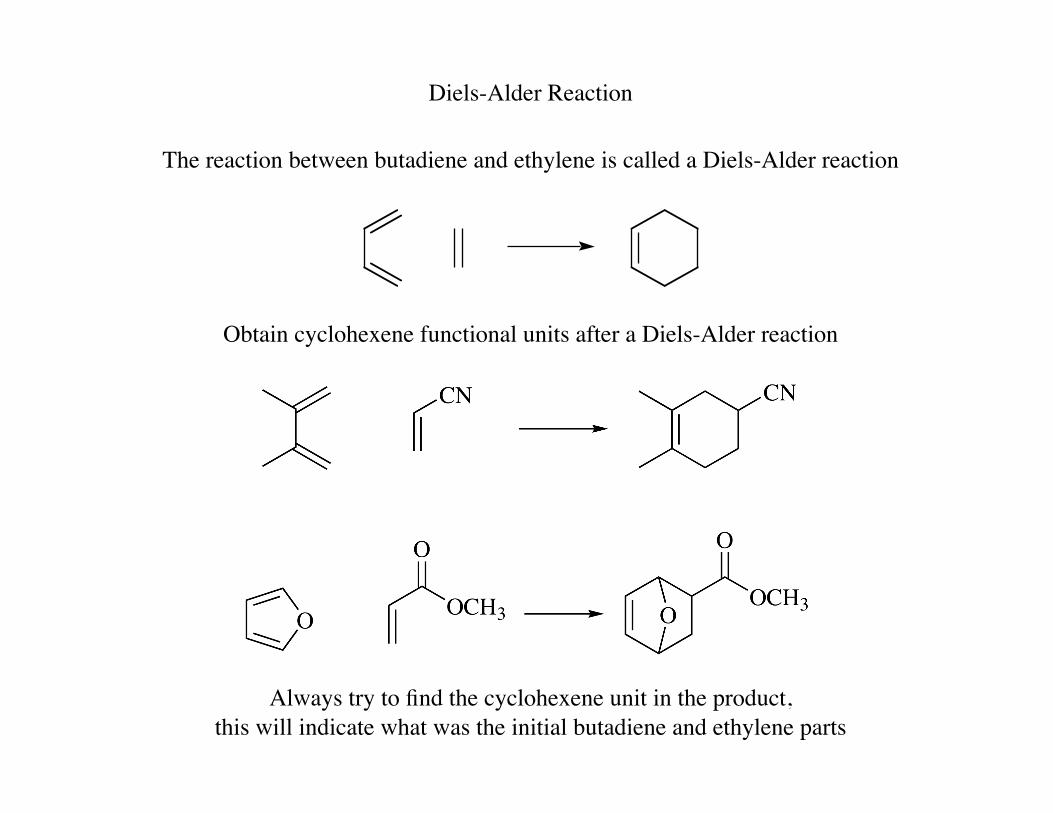
Diels-Alder Reaction
The reaction between butadiene and ethylene is called a Diels-Alder reaction
Obtain cyclohexene functional units after a Diels-Alder reaction
Always try to find the cyclohexene unit in the product, this will indicate what was the initial butadiene and ethylene parts
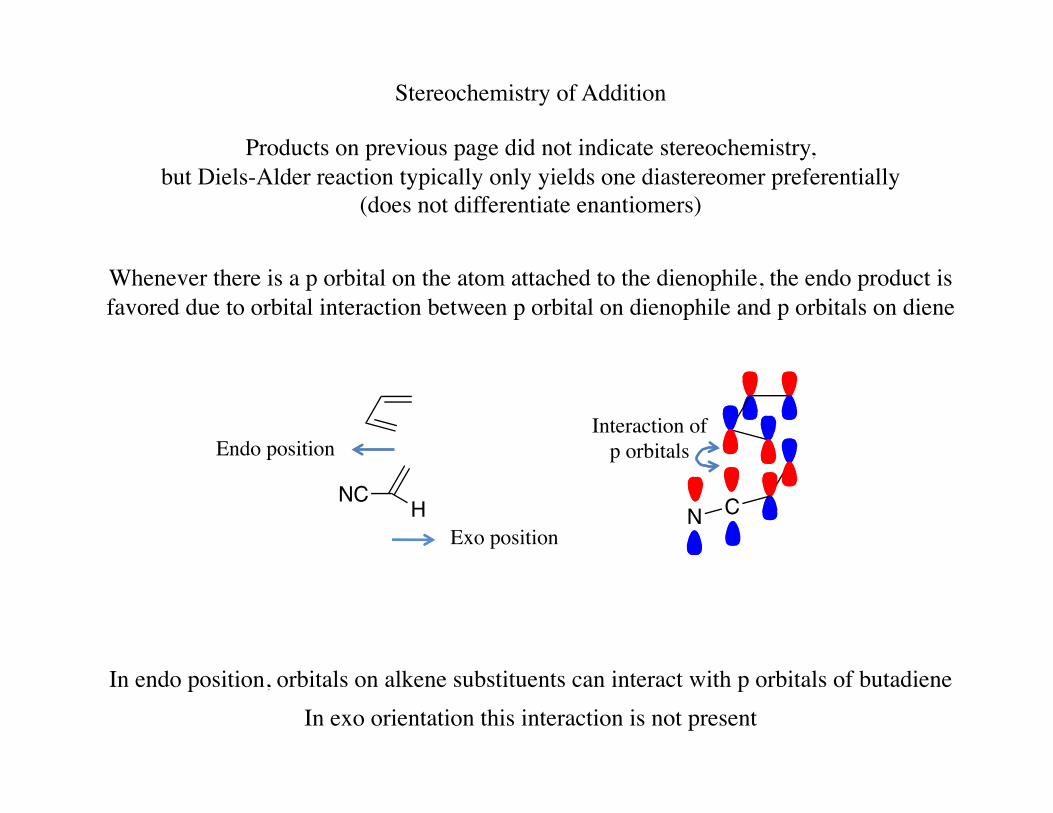
Stereochemistry of Addition
Products on previous page did not indicate stereochemistry, but Diels-Alder reaction typically only yields one diastereomer preferentially
(does not differentiate enantiomers)
Whenever there is a p orbital on the atom attached to the dienophile, the endo product is favored due to orbital interaction between p orbital on dienophile and p orbitals on diene
NC H
Endo position
Exo position CN
Interaction of p orbitals
In exo orientation this interaction is not present In endo position, orbitals on alkene substituents can interact with p orbitals of butadiene

Regiochemistry of Unsymmetrically Substituted Diels-Alder Products
When a monosubstituted butadiene and a monosubstituted alkene react, different regioproducts can be obtained
Can predict favored product by understanding location of charge in molecules
OCH3 OCH3 OCH3
Consider resonance forms Negative charge is located on C2 and C4
OCH3NO2 !
OCH3NO2
OCH3
NO2
or
123
4
NO
O
Consider resonance forms
NO
O
Positive charge located on C2
12
The negative charge will react preferentially with the positive charge to obtain one regioproduct

Using this analysis we can predict regioproducts
A Diels-Alder reaction can therefore control both regio- and stereochemistry

Ultraviolet-Visible (UV-Vis) Spectroscopy
Instead of causing molecular vibrations, UV-VIS light causes electronic excitations
An electron is excited from the HOMO to the LUMO
E h!
Ethylene HOMO
Ethylene LUMO
If the correct amount of energy is applied (i.e. the correct wavelength of light), the excitation of one electron from the HOMO to the LUMO will occur
As the amount of conjugation increases, the energy gap between the HOMO and LUMO decreases
With a lower energy gap, the λmax shifts to a longer wavelength of light to cause excitations
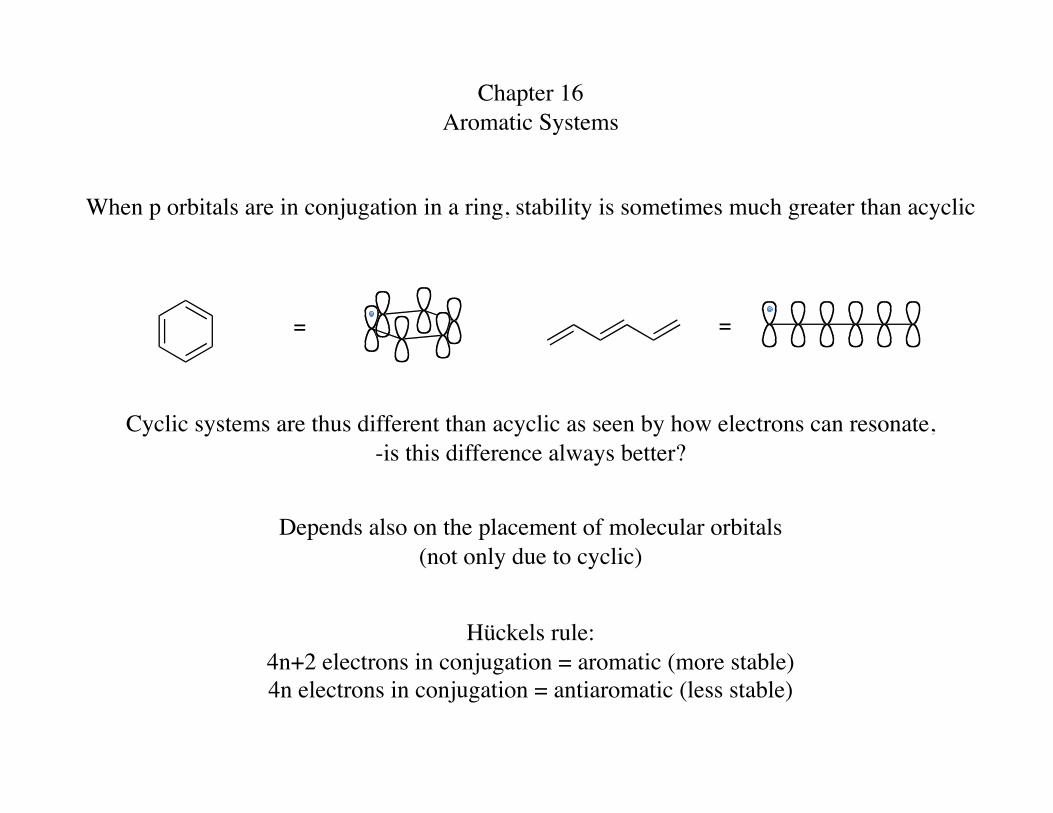
Chapter 16 Aromatic Systems
When p orbitals are in conjugation in a ring, stability is sometimes much greater than acyclic
= =
Cyclic systems are thus different than acyclic as seen by how electrons can resonate, -is this difference always better?
Depends also on the placement of molecular orbitals (not only due to cyclic)
Hückels rule: 4n+2 electrons in conjugation = aromatic (more stable) 4n electrons in conjugation = antiaromatic (less stable)

Aromatic Ions
If p orbitals are in full conjugation in a cyclic system and the number of electrons in conjugation is equal to 4n+2, the compound is more stable regardless of whether the
compound is neutral, negatively charged or positively charged
=After rehybridization, system has 6 electrons in conjugation in a ring, therefore aromatic
pKa is ~16
Cyclopentadiene anion is very stable
Cyclopentadiene cation is very unstable (will not form)
With all cyclic ions, see if p orbitals are conjugated in a ring and then count the number of electrons in conjugation, if 4n+2 then stable, if 4n then unstable

Chapter 17 Aromatic Reactions
Electrophilic Aromatic Substitution
Aromatic compounds react through a unique substitution type reaction
Initially an electrophile reacts with the aromatic compound to generate an arenium ion (also called sigma complex)
The arenium ion has lost aromatic stabilization (one of the carbons of the ring no longer has a conjugated p orbital)
In a second step, the arenium ion loses a proton to regenerate the aromatic stabilization The product is thus a substitution
(the electrophile has substituted for a hydrogen) and is called an Electrophilic Aromatic Substitution

A Variety of Electrophiles Can Be Used
The key is generating an electrophilic reagent that can react with the aromatic ring
Br Br FeBr3!+ !-
Br
CH3
Cl2AlCl3
ortho/para director
CH3
Cl
NO2
HNO3
H2SO4
meta director
NO2O2N
CH3
O
ClAlCl3
OElectrophile:
Br2FeBr3
!+ !-Cl Cl AlCl3 NO2
O

Reactions on Aromatic Substituents
A variety of reactions can be performed on the aromatic substituents
NO2 NH2Sn, Fe, or ZnHCl
deactivating activating
AlCl3
O
Cl
OCl
AlCl3
Friedel-Crafts acylation
Friedel-Crafts alkylation
NH2NH2KOH
Wolf-Kishner
ZnHCl
Clemmensen
CO2H
KMnO4KOH
Br
NBSh!
Carbon side chains:

Nucleophilic Aromatic Substitution
Mechanism
Cl
O2N
NO2
NaCN
NO2
O2N
ClCN
NO2
O2N
ClCN
NO2
O2N
ClCN
The anion is stabilized by electron withdrawing groups ortho/para to leaving group
To regain aromatic stabilization, the chloride leaves to give the substituted product
NO2
O2N
ClCN
NO2
O2N
CN
1) Must have EWG’s ortho/para to leaving group -the more EWG’s present the faster the reaction rate (intermediate is stabilized)
2) The leaving group ability does not parallel SN2 reactions -follows electronegativity trend (F > Cl > Br > I)
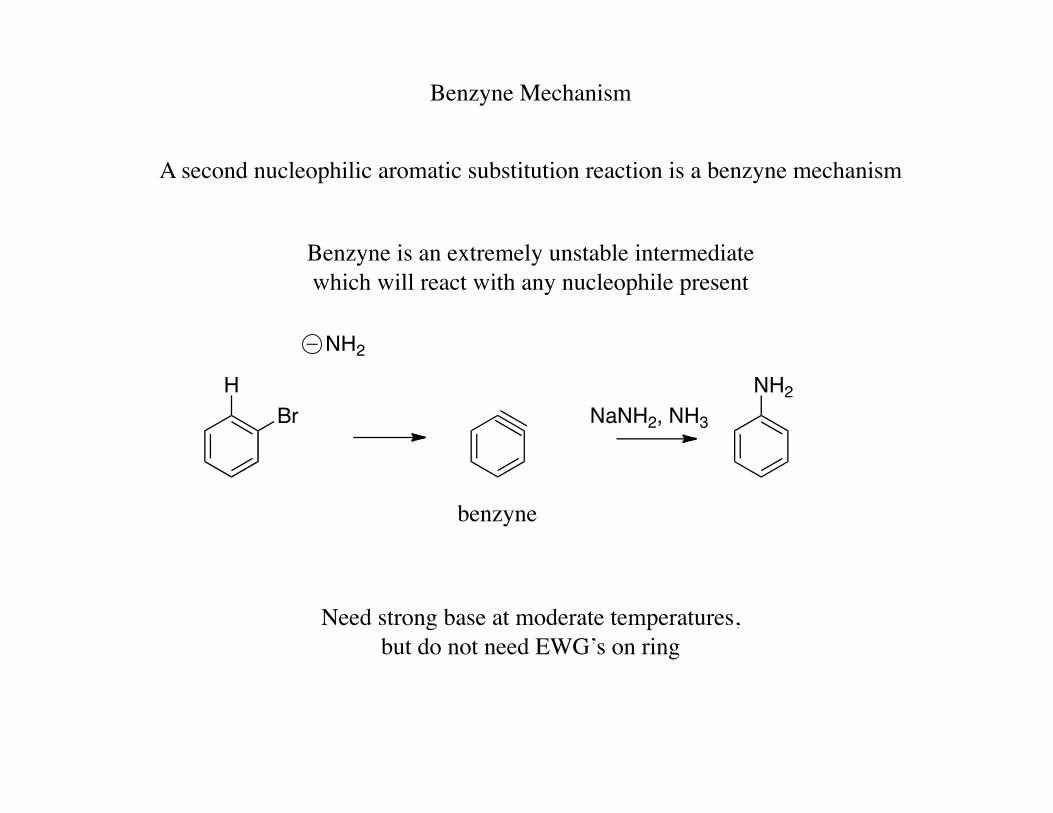
Benzyne Mechanism
A second nucleophilic aromatic substitution reaction is a benzyne mechanism
Benzyne is an extremely unstable intermediate which will react with any nucleophile present
HBr
NH2NH2
NaNH2, NH3
benzyne
Need strong base at moderate temperatures, but do not need EWG’s on ring

Chapter 18 Ketones and Aldehydes
New routes to synthesize ketones and aldehydes
From carboxylic acids:
O
OH
RLi(2 equiv.)
O
R
SOCl2 O
Cl
O
HLiAlH(OtBu)3
R2CuLi
O
RFrom nitriles:
C N1) RMgBr
2) H+, H2O
O
R1) DIBAL
2) H+, H2O
O
H
O
OR
O
H
1) DIBAL
2) H+, H2O
From dithianes:
S S S S
H
BuLiS S
R H
RXH+, HgCl2
H2O
R
O
H

Reactions of Ketones and Aldehydes
R
O
R
NUC O NUCR
O
R
H
R R
O HNUC
R R
HO NUC
Base mechanism Acid mechanism
Types of NUC : RMgBr LAH ylide Types of neutral NUC: cyanide H2O ROH RNH2
Reactivity
As electrophilicity of carbonyl carbon increases, the reactivity increases
R
O
R R
O
H H
O
H Cl3C
O
H< < <

Wittig Reaction
The carbanion of the ylide is nucleophilic and will react with the carbonyl
H3C
O
CH3(Ph)3P
(Ph)3P O
CH3CH3H3C
betaine
oxyphosphetane
The betaine structure will form 4-membered ring between phosporous and oxygen
(Ph)3P O
CH3CH3H3C
(Ph)3P O
H3CCH3
CH3
The oxyphosphetane will collapse to form a second phosphorous-oxygen bond
(Ph)3P O
H3CCH3
CH3 H3C CH3
CH3(Ph)3P O
Overall an alkene is formed from the initial carbonyl compound

Acetals
Acetals are related to hydrates, Instead of geminal dialcohols have geminal ethers
This process is once again an equilibrium process
Aldehydes (which are more reactive than ketones) typically favor acetals
H3C
O
CH3 H+ H3C CH3
ORROROH
When both alcohols to form an acetal are intramolecular (on same molecule) then a cyclic acetal is formed
H3C
O
H H+ H3C H
HO OH OO
Cyclic acetals and ketals are often used because they have a higher equilibrium for the acetal form

Baeyer-Villiger
Allows conversion of ketone to ester
R
O
RRCO3H
R
O
O R
Mechanism of oxygen insertion?
R
O
R H O O
O
R R
O
RO O
O
R
H
R
HO
O
RO
O
RR
O
O R O
O
R
H
Mechanism is not an insertion, but rather a reaction at carbonyl followed by a migration
Weak oxygen-oxygen single bond

Chapter 19 Amines
The basicity, and hence pKb, is determined by the stability of the amine after protonation
The lone pair of electrons on nitrogen can act as an acceptor (hence Brønsted-Lowry base)
R NH
HH2O
KbR N H
H HOH
H3CN
H3CH3C
N
H3C C N
As the percent s character increases for the orbital holding the lone pair of electrons, the electrons are held more closely to the positively charged nucleus
sp3 hybridization
sp2 hybridization
sp hybridization
pKb = 4.26
pKb = 8.75
pKb = 24
Other effects, such as whether lone pair is in resonance or involved in aromatic system, also will affect the pKb for the amine

Synthesis of Amines
Nucleophilic:
NH2CH3I
NH
NCH3I
problem is overalkylation
NH
O
O
1) KOH2) CH3I NH2NH2N
O
O
CH3 H3C NH2
Gabriel allows only 1 addition
Reduction: N OH
O
NHN3
CN
LAH NH2
LAH
LAH
LAH
NH
NH2
NH2
oxime
amide
azide
nitrile

Elimination of Amines Hoffman Elimination
With quaternary ammonium salts, E2 reaction occurs to eliminate trimethylamine
Cope Elimination
With N-oxide, syn elimination occurs to eliminate dimethylhydroxy amine
anti-elimination
syn-elimination
F H(CH3)3N
FH(H3C)2N OH
NFH
N OFH
Both eliminations favor the less substituted alkene to be formed

Arenediazonium Salts
Arenediazonium salts can be generated from aniline derivatives
NH2 NaNO2
HCl
N N
The diazonium salt can then be converted into a number of different functional groups
NN
OH
H2SO4
F
HBF4
I
KI
CN
CuCN
H
H3PO2
Unique phenol derivatives
Unique F substitution
Easier I substitution
Versatile CN (Sandmeyer)
Reduce to H

Chapters 20&21 Carboxylic Acids & Derivatives
Reactions of Acyl Compounds
There is a commonality amongst carbonyl reactions, they have a nucleophile react at the carbonyl carbon
O
X
NUC
O
NUCX
O
NUCX
Generate a tetrahedral intermediate that can expel a leaving group
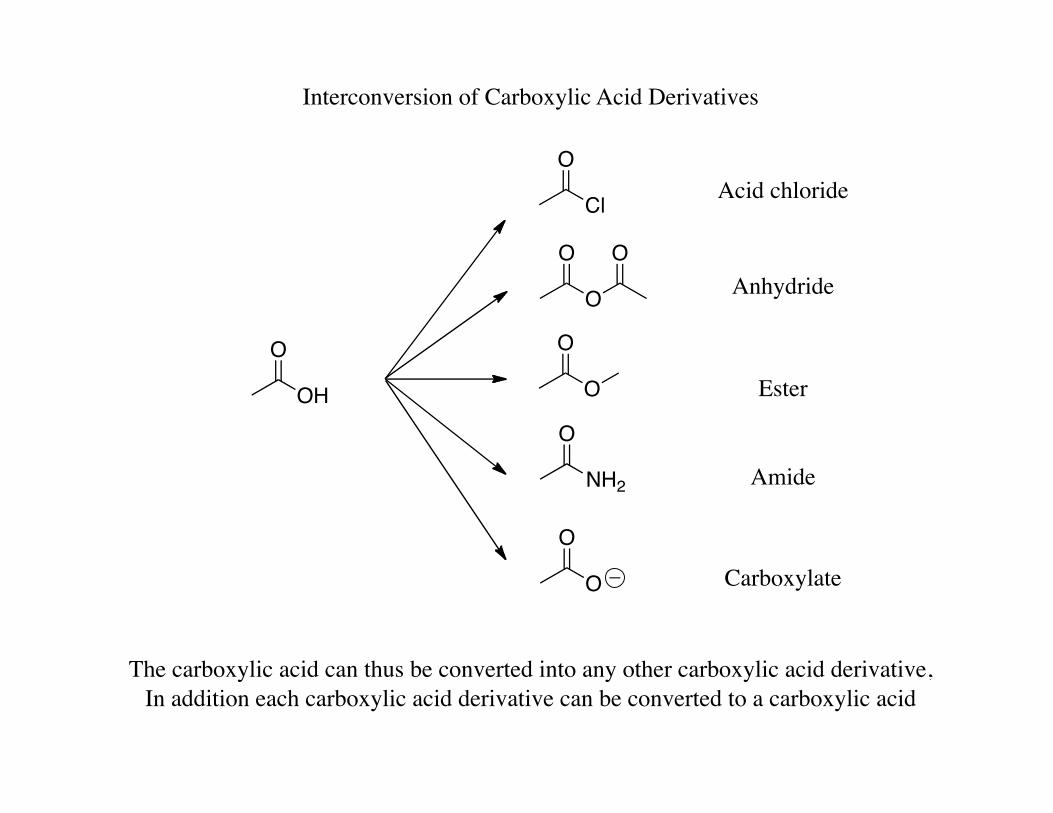
Interconversion of Carboxylic Acid Derivatives
O
OH
O
Cl
The carboxylic acid can thus be converted into any other carboxylic acid derivative, In addition each carboxylic acid derivative can be converted to a carboxylic acid
O
O
O
O
O
O
NH2
O
O
Acid chloride
Anhydride
Ester
Amide
Carboxylate

Reactivity of Carboxylic Acid Derivatives
All carboxylic acid derivatives can also be converted back to the carboxylic acid (by either acidic or basic hydrolysis) or the derivatives can be directly
interconverted to a less reactive form
O
NH2
O
Cl
O
O
O
O
O
O
O
reactivity
Acid chloride
Anhydride
Ester
Amide
Carboxylate
But cannot interconvert a less reactive acyl derivative into a more reactive

Predicting Reactivity Patterns for Carboxylic Acid Derivatives
All of the carboxylic acid derivatives can react with a nucleophile to generate the same carbonyl product – the difference is the leaving group
O
ClNUC O
NUC Cl
O
O
O O
ONUC O
NUC
O
O ONUC O
NUC
O
NH NH
NUC O
NUC
pKa of conjugate for leaving group
-7
~4-5
16
35
The stability of the leaving group affects the reactivity pattern for the acid derivatives

Chapter 22 Reactions at α-Carbon
A type of reaction with carbonyl compounds is an α-substitution (an electrophile adds to the α carbon of a carbonyl)
O O
E
E+
In the preceding chapters, we primarily studied nucleophiles reacting at the electrophilic carbonyl carbon
O NUC OH
NUC

Reactions of Enols
The enol form can react with electrophiles
A common reaction is halogenation
Under basic conditions it is hard to stop at one addition due to hydrogen abstraction of product is more favored than starting material
O OHNaOH Br Br OH
Br
O OHNaOH Br2 O
Br Br Br Br

Enolates
Enolates are similar to enols but they are far more nucleophilic
In order to generate an enolate, need a base to abstract an α-hydrogen
LDA will quantitatively form enolate
H3C
O
CH3 H3C
O
CH2LDA
Using LDA the enolate will be formed quantitatively, with weaker bases will only form the enolate in a small fraction

Enolate as a Nucleophile
Have already observed many reactions with a negatively charged nucleophile (most SN2 reactions)
An enolate is simply another type of nucleophile, it can react in similar manner as other nucleophiles
H3C
O
CH2E+
H3C
O
CH2E
One common reaction is to alkylate the enolate
H3C
O
CH2 H3C
O
CH2CH3
CH3Br
This reaction will place an alkyl substituent at the α-position of a carbonyl Any electrophile that will react in a SN2 reaction can be used

Thermodynamic vs. Kinetic Control
With unsymmetrical ketones, different enolates can be generated
The enolate can be preferentially generated at either site depending upon conditions
O LDAO
H H
O
Kinetic enolate easier hydrogen to abstract
O
Thermodynamic enolate more stable double bond
Lower temperature favors kinetic product
Higher temperatures (in this case usually room temperature and above) favors thermodynamic product

Aldol Condensation
Instead of reacting the enolate with an alkyl halide, we can also react the enolate with a carbonyl compound
The carbonyl can react as an electrophile
OO
O OH
Upon work-up obtain a β-hydroxy ketone
O OH -H2O O
The β-hydroxy ketone that is formed can also lose water to form an α,β-unsaturated ketone
Greater the conjugation, the easier for loss of water

Claisen Condensation
There are many “Name” reactions that are modifications of the aldol condensation, A Claisen condensation is an aldol where one carbonyl compound is an ester
By using an ester, the chemistry is changed due to the presence of a leaving group
OCH3ONaO O
O
Can run reaction with both carbonyls present with weak base due to
differences in pKa (ketones ~20, esters ~24)
O
OO O
O
O O
With ester leaving group, obtain diketone product

Dieckmann
A Dieckmann condensation is an intramolecular Claisen condensation
CH3OO
O O
O O
O O
O
OO
OOCH3
O
O
OConvenient method to
form 5- or 6-membered rings

Michael Addition
If we add stabilized enolates to α,β-unsaturated system, the reaction can occur with 1,4-addition
O1
23
4
ONUC
O
NUC
Michael product (1,4 addition)
Whether a reaction occurs with 1,2- or 1,4-addition, selectively often depends on the stability of the nucleophilic anion. A more stable anion occurs with 1,4 selectivity while a less stable
anion occurs with 1,2 selectivity.
O O O NCH3MgBr (CH3)2CuLi
1,2 1,2 1,4 1,4 1,4
Enolate anions prefer 1,2 but β-diketone or enamines favor 1,4
Grignard reagents prefer 1,2 but Gilman reagents prefer 1,4