clinical study protocol - university of oxford · (dtu), uk, professor da yi hu from peking...
TRANSCRIPT

Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 1 of 45
CLINICAL STUDY PROTOCOL
Title: The Acarbose Cardiovascular Evaluation (ACE) Trial.
A Long-term, Multicentre, Double-blind, Randomised
Parallel-group Trial to Determine Whether Reducing
Post-prandial Glycaemia can Reduce Cardiovascular-
related Morbidity and Mortality in Patients with
Established Coronary Heart Disease or Acute Coronary
Syndrome who have Impaired Glucose Tolerance.
Study Number/Version/Date: 11232 / Version 10 / Amendment 7 / July 28th
2016
Development Phase: Phase IV
Test Drug: Acarbose
Sponsoring Institution: The University of Oxford
The University Offices
Wellington Square
Oxford OX1 2JD
UK
Chief Investigator: Professor Rury Holman
Diabetes Trials Unit
Oxford Centre for Diabetes, Endocrinology and
Metabolism (OCDEM)
Churchill Hospital
Old Road
Headington
Oxford OX3 7LJ
Tel: +44 (0) 1865 857240
Confidentiality
statement:
The following confidential information is the property of the ACE Trial Steering Committee. As long as
the information contained in this protocol has not been published, it may only be used when permission
has been obtained from the ACE Trial Steering Committee. It is not possible to make reproductions of all
or sections of this protocol. Commercial use of the information is only possible with the permission of the
proprietor and is subject to a license fee.


Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 3 of 45
Acceptance of Protocol Version 10 / Amendment 7
Principal Investigator:
I have read this protocol and agree to abide by all provisions set forth therein.
I agree to comply with the International Conference on Harmonisation Tripartite Guideline on Good
Clinical Practice.
______________________________________
Print Name of Principal Investigator
______________________________________ __________________
Signature of Principal Investigator Date

Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 4 of 45
Table of Contents
1. INTRODUCTION ................................................................................................. 8
2. STUDY OBJECTIVES ........................................................................................ 9
3. INVESTIGATORS AND OTHER STUDY PARTICIPANTS ................................ 9
3.1 Honoured Advisor ....................................................................................................................................... 9
3.2 Chief Investigator ...................................................................................................................................... 10
3.3 Co-Chairs ................................................................................................................................................... 10
3.4 Steering Committee ................................................................................................................................... 10
3.5 Data Safety Monitoring Board (DSMB) .................................................................................................. 11
3.6 Endpoint Adjudication Committee .......................................................................................................... 11
3.7 Publications ............................................................................................................................................... 12
4. INVESTIGATIONAL PLAN ............................................................................... 12
4.1 Study Design and Plan .............................................................................................................................. 12
4.2 Selection of Study Population................................................................................................................... 13 4.2.1 Inclusion Criteria ............................................................................................................................... 13 4.2.2 Exclusion Criteria .............................................................................................................................. 14 4.2.3 Diabetes Diagnosed on Screening ..................................................................................................... 14
4.3 Removal of Patients from Study .............................................................................................................. 15
4.4 Premature Closure of Centre ................................................................................................................... 15
4.5 Premature Termination of Study ............................................................................................................. 15
4.6 Study Medication ...................................................................................................................................... 16 4.6.1 Study Medication to be Administered ............................................................................................... 16 4.6.2 Identity of Investigational Product(s) ................................................................................................ 16 4.6.3 Method of Assigning Patients to Treatment Groups .......................................................................... 17 4.6.4 Selection of Doses in the Study ......................................................................................................... 17 4.6.5 Timing and Titration of Dose for Each Patient .................................................................................. 17 4.6.6 Blinding ............................................................................................................................................. 18 4.6.7 Concomitant Therapy ........................................................................................................................ 18 4.6.8 Treatment Adherence ........................................................................................................................ 18 4.6.9 Assessment Periods ........................................................................................................................... 19 4.6.10 New Onset Diabetes During Study ............................................................................................... 20
4.7 Observations and Measurements: Assessment for Treatment Effects ................................................. 21 Note: patients who experience any of the non-fatal endpoints musts remain on study medication and continue
the study. Any subsequent endpoint should also be reported........................................................................... 21

Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 5 of 45
4.7.1 Screening Visit (Visit 1, -5 weeks) .................................................................................................... 21 4.7.2 Run-in Visit (Visit 2, -4 weeks) ......................................................................................................... 22 4.7.3 Randomisation Visit (Visit 3, week 0)............................................................................................... 23 4.7.4 Follow up Visits ................................................................................................................................ 24 4.7.5 Annual Visits ..................................................................................................................................... 24 4.7.6 Closeout Visit .................................................................................................................................... 25 4.7.7 Final Visit/Telephone Contact ........................................................................................................... 26
4.8 Data Quality .............................................................................................................................................. 26
4.9 Documentation .......................................................................................................................................... 27
5. ETHICAL AND LEGAL ASPECTS ................................................................... 28
5.1 Ethics Committee or Institutional Review Board................................................................................... 28
5.2 Ethical Conduct of the Study ................................................................................................................... 28
5.3 Regulatory Authority Approvals/Authorizations ................................................................................... 28
5.4 Informed Consent ..................................................................................................................................... 28
5.5 Insurance ................................................................................................................................................... 29
5.6 Confidentiality ........................................................................................................................................... 29
6. STATISTICAL METHODS AND DETERMINATION OF SAMPLE SIZE .......... 29
6.1 Description of Patient Groups for Analysis ............................................................................................ 29
6.2 Study Objectives ........................................................................................................................................ 29 6.2.1 Primary Endpoint ............................................................................................................................... 29 6.2.2 Secondary Endpoints ......................................................................................................................... 30
6.3 Statistical and Analytical Plans ................................................................................................................ 30
6.4 Interim Analysis/Analyses ........................................................................................................................ 31
6.5 Sample Size Estimation............................................................................................................................. 31 6.5.1 Event Rate ......................................................................................................................................... 31 6.5.2 Number of Patients ............................................................................................................................ 31
6.6 Health Economic Evaluation .................................................................................................................... 31 6.6.1 Resource use and costs ...................................................................................................................... 32 6.6.2 Economic outcomes ........................................................................................................................... 32 6.6.3 Cost-effectiveness .............................................................................................................................. 32 6.6.4 Analysis and presentation of economic data ...................................................................................... 33
7. ADVERSE EVENTS ......................................................................................... 33
7.1 Adverse Event Monitoring ....................................................................................................................... 33
7.2 Adverse Event Definitions ........................................................................................................................ 34 7.2.1 Adverse Event (AE)........................................................................................................................... 34

Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 6 of 45
7.2.2 Serious Adverse Event (SAE) ........................................................................................................... 34 7.2.3 Suspected Serious Adverse Reaction (SSAR) ................................................................................... 34 7.2.4 Suspected Unexpected Serious Adverse Reaction (SUSAR) ............................................................ 34 7.2.5 Relationship to Study Medication ..................................................................................................... 35 7.2.6 Severity of the Adverse Event ........................................................................................................... 35
7.3 Collection and Reporting of Adverse Events .......................................................................................... 35
7.4 Reporting of Pregnancy ............................................................................................................................ 36
7.5 Warnings/Precautions for Acarbose ........................................................................................................ 36
7.6 Overdosage ................................................................................................................................................ 37
8. USE OF DATA AND PUBLICATION ................................................................ 37
9. APPENDIX 1 ..................................................................................................... 38
9.1 Study Flow Chart and/or Schedule Procedure ....................................................................................... 38
10. APPENDIX 2: NYHA GRADING CRITERIA FOR HEART FAILURE .......... 40
11. APPENDIX 3: OGTT PROCEDURE ............................................................. 41
12. REFERENCES .............................................................................................. 43

Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 7 of 45
Glossary and Abbreviations
2HPG
ACS
AE
ALT
CABG
CHD
CVD
2-hour post-challenge plasma glucose
Acute Coronary Syndrome
Adverse Event
Alanine Transaminase
Coronary Artery Bypass Graft
Coronary Heart Disease
Cardiovascular Disease
CRF
DSMB
DTU
Case Report Form (either paper or electronic)
Data Safety Monitoring Board
Diabetes Trials Unit
EC
ECG
eCRF
FPG
Ethics Committee
Electrocardiogram
Electronic Case Report Form
Fasting Plasma Glucose
GCP Good Clinical Practice
ICH
IFG
IGT
International Conference on Harmonization
Impaired Fasting Glucose
Impaired Glucose Tolerance
IRB
MDRD
MI
Institutional Review Board
Modification of Diet in Renal Disease
Myocardial Infarction
MRR
NAFLD
NYHA
OGTT
PCI
Medical Research Report
Non Alcoholic Fatty Liver Disease
New York Heart Association classification
Oral Glucose Tolerance Test
Percutaneous Coronary Intervention
SAE
T2DM
TIA
TMS
ULN
Serious Adverse Event
Type 2 Diabetes Mellitus
Transient Ischaemic Attack
Trial Management System
Upper Limit of Normal

Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 8 of 45
1. INTRODUCTION
The number of people worldwide with diabetes is predicted to exceed 300 million by
the year 2025, more than all of the people living currently in North America [1]. Over 90% of
those with diabetes have type 2 diabetes (T2DM), which confers a two to four times greater
risk of heart disease and stroke than in the general population and a concomitant reduction in
life expectancy of five to ten years [2]. Given that around 80% of all diabetes-related deaths
are associated with cardiovascular disease (CVD), it is of paramount importance that
everything possible is done to reduce coronary heart disease (CHD) risk in these people.
Equally, previously undiagnosed diabetes and impaired glucose tolerance are common in
patients with an acute myocardial infarction [3] and the Euro Heart Survey showed that
normal glucose regulation was less common than abnormal glucose regulation in patients
with coronary artery disease [4]. A meta analysis [2] of people with impaired glucose
tolerance (IGT) has shown that this prediabetic state is associated also with a substantially
increased CVD risk with some evidence that postprandial hyperglycaemia may have the
greatest deleterious effect [5]. China has one of the highest rates of new-onset T2DM in the
world today [6] and the China Heart Survey has shown that the prevalence of IGT is
significantly higher in Chinese patients with CHD, compared with those without CHD [7].
The rate of development of new-onset diabetes has been shown in patients with IGT
to be reduced by 25% with acarbose in the international STOP NIDDM trial [8], 88% in a
Chinese multicentre trial [9] and to be reduced non-significantly in a smaller Chinese study
[10]. Reductions of 31% with metformin were seen in the Diabetes Prevention Program [11]
and 77% in a Chinese multicentre trial [9]. Improved lifestyle achieved a 58% reduction in
both the Diabetes Prevention Program [11] and the Finnish Diabetes Prevention Study [12].
In a mixed population of IGT and/or IFG patients the rate of development of new-onset
diabetes was reduced by 60% with rosiglitazone therapy [13]. Epidemiological analyses of
UK Prospective Diabetes Study (UKPDS) data for patients with newly diagnosed T2DM
have confirmed that, in addition to age and gender, the potentially modifiable risk factors for
CHD include the “deadly quintet” of hyperglycaemia, a raised LDL cholesterol, a low HDL
cholesterol, hypertension and smoking [14]. Whilst the excess CHD risk seen in those with
diabetes is not explained fully by these conventional risk factors, there is increasing evidence
of the benefits that can be obtained from a number of different cardiovascular interventions.
The UKPDS showed that intensive control of glycaemia with sulphonylurea or insulin, and
with metformin in overweight patients, can reduce substantially the risk of diabetic
complications [15], although the true extent to which reducing glycaemia can impact on CVD
is still to be established. The UKPDS showed that intensive control of blood pressure with an
ACE inhibitor or a beta-blocker can also reduce substantially the risk of diabetic
complications [16]. The HPS (Heart Protection Study) [17] and the CARDS (Collaborative
Atorvastatin Diabetes Study) [18] trials have shown definitively that lowering LDL
cholesterol levels can reduce cardiovascular risk to a major extent. However, despite
confirmation that lowering glycaemic, blood pressure and LDL cholesterol levels improves
clinical outcomes substantially, the residual risk of further cardiovascular events or premature
death in people with T2DM remains unacceptably elevated.
It is clear that additional approaches to cardiovascular risk reduction are required, as
well as strategies to reduce the incidence of new-onset T2DM. The STOP NIDDM study

Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 9 of 45
demonstrated not only that acarbose can reduce the risk of T2DM in patients with IGT but
suggested also that acarbose administration to IGT patients may reduce cardiovascular
morbidity and mortality [19]. This latter suggestion is supported by a study by Hanefeld and
colleagues, which showed acarbose, can reduce the rate of intima-media thickness (IMT)
progression [20].
Despite these findings, there remains a genuine uncertainty as to whether acarbose
can reduce CVD morbidity and mortality in IGT patients with CHD. Accordingly, it is
proposed to evaluate whether these potential benefits of acarbose can be confirmed in this
population by undertaking a multicentre clinical outcome study in Chinese patients with
established CHD or an acute coronary syndrome (ACS) who also have IGT.
2. STUDY OBJECTIVES
The primary objective of the Acarbose Cardiovascular Evaluation (ACE) Trial is to
determine whether acarbose therapy can reduce cardiovascular-related morbidity and
mortality in patients with IGT who have established CHD or ACS (secondary cardiovascular
prevention).
A secondary objective of the ACE Trial is to determine if acarbose therapy can prevent or
delay transition to T2DM in this patient population (primary diabetes prevention).
3. INVESTIGATORS AND OTHER STUDY PARTICIPANTS
The ACE Trial is a multicentre trial, which will be conducted, in around 150 cardiovascular
units in Mainland China and Hong Kong. ACE is an investigator-initiated, academic study
designed by Professor Rury Holman from the University of Oxford Diabetes Trials Unit
(DTU), UK, Professor Da Yi Hu from Peking University and Professor Chang Yu Pan from
the Chinese PLA General Hospital, Beijing, China.
The trial will be sponsored, run and analysed by the DTU. The ACE Coordinating Centre,
located in the DTU, will set up and oversee a regional Project Office located in Beijing that
will handle the day-to-day operations of the trial.
Cardiovascular endpoint adjudication was performed by the University of Glasgow until July
2014. Thereafter, the ACE Steering Committee mandated that this process would be
transferred to a Chinese Cardiovascular Endpoint Adjudication Committee, led by senior
cardiologists from Peking University People’s Hospital, Beijing, and Shanghai Tenth
People's Hospital, Tongji University, Shanghai.
Diabetes endpoint adjudication will be performed by the University of Glasgow.
Health Economic Evaluation will be undertaken by the University of Oxford Health
Economic Research Centre (HERC), led by Professor Alastair Gray.
3.1 Honoured Advisor
Professor Jialun Chen, Shanghai Institute of Endocrine and Metabolic Diseases.

Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 10 of 45
3.2 Chief Investigator
The ACE Trial Chief Investigator and Chair of the study will be Professor Rury Holman.
3.3 Co-Chairs
The ACE Trial Co-Chairs will be Professor Da Yi Hu and Professor Chang Yu Pan.
3.4 Steering Committee
The ACE Steering Committee will have overall responsibility for finalising the protocol and
supervising the conduct of the study. It will meet six-monthly, or more often if necessary,
with at least one face-to-face meeting each year to consider issues raised during the progress
of the study, to review relevant information from other sources e.g. related studies, and to
consider recommendations from the Data Safety Monitoring Board (DSMB). The Steering
Committee will comprise twelve independent academics, the DTU Head of Clinical Research
and two representatives from Bayer.
The Steering Committee will oversee the operation of the study in accordance with current
Good Clinical Practice (GCP) guidelines, including production of the electronic CRFs
(eCRFs), site monitoring, data management, statistical analyses and final submission of
publications.
Steering Committee members, in alphabetical order, will be:
Juliana Chan Hong Kong Endocrinologist
Jean Louis Chiasson Canada Endocrinologist
Junbo Ge China Cardiologist
Hertzel Gerstein Canada Endocrinologist
Rury Holman UK Endocrinologist (Chair)
Da Yi Hu China Cardiologist (Co-Chair)
John McMurray UK Cardiologist
Chang Yu Pan China Endocrinologist (Co-Chair)
Lars Rydén Sweden Cardiologist
Michal Tendera Poland Cardiologist
Jaakko Tuomilehto Finland Epidemiologist
Wenying Yang China Endocrinologist
Yong Huo China Cardiologist

Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 11 of 45
UK DTU Head of Clinical Research
China/Germany Bayer Representative
China/Germany Bayer Representative
3.5 Data Safety Monitoring Board (DSMB)
The DSMB will report to the Steering Committee. It will consist of seven members with
proven DSMB experience to include a Chairman, two cardiologists, two diabetologists, a
hepatologist and a statistician, at least two of which will be practicing currently in China.
ACE will commission a statistical group independent of Bayer and the DTU to provide
confidential interim analyses for the DSMB, under the direction of the DSMB statistician.
DSMB members will remain independent of the study staff, the Steering Committee and of
Bayer. At DSMB meetings, senior ACE Coordinating staff may present the updated status of
the study to the committee but will not remain in attendance whilst the DSMB statistician
presents unmasked study data, or for any subsequent discussions.
Anybody associated with the study may write to the DSMB chairman drawing attention to
any worries they may have e.g. about the possibility of particular side-effects, or about
particular categories of patient requiring special consideration, or about any other matters that
may be relevant.
The remit of the DSMB is to:
Consider unmasked interim data and relevant information from other sources
including endpoints and serious adverse events.
Determine how frequently interim safety analyses of study data should be undertaken.
These analyses will be itemised in a statistical analysis plan (SAP), agreed in advance
with the DSMB and modified by them as appropriate during the trial.
Report to the Steering Committee following each DSMB meeting recommending
whether the trial should continue in accordance with the protocol, the protocol should
be modified or the study should be stopped.
To highlight any areas of concern with respect to the trial.
To feedback any suggestions they might have for improving the conduct of the trial.
To consider any requests for unmasking and release of interim study data and to
recommend the importance of this to the Steering Committee. The Steering
Committee will then decide whether to modify the trial (or to seek extra data). Unless
this happens or the trial is stopped, the Steering Committee, study investigators, study
staff and Bayer will remain ignorant of interim trial results.
3.6 Endpoint Adjudication Committee
The Endpoint Adjudication Committee will be responsible for reviewing and coding all
major clinical outcomes pertinent to the study, identified from adverse events, biochemistry
or electrocardiogram (ECG) results. They will also be required to adjudicate any instances

Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 12 of 45
where diabetes has been diagnosed other than by the protocol defined process. All potential
primary and secondary endpoints will be reviewed and adjudicated by two members,
independently, according to pre-determined criteria (see the Endpoint Committee Charter). If
both members agree, the event will be considered adjudicated and coded accordingly. In the
case of disagreement, the potential endpoint will be reviewed by the full endpoint committee
and adjudicated either by agreement of all committee members (usually) or, if consensus is
not reached (rarely), a majority vote.
3.7 Publications
The Steering Committee will recommend a writing group for each manuscript with a lead
author and approve the production of all ACE Trial manuscripts. Writing groups will
comprise typically of three to seven individuals identified usually from trial members. The
lead author will be responsible for ensuring that the paper is written in a timely manner,
liaising with the trial statistician as necessary, and keeping the ACE Trial Manager informed
of progress. For papers involving analysis of data generated by the ACE Trial, at least one
member of the writing group will be from the DTU’s Statistics and Modelling Group.
Major publications will be authored “on behalf of the ACE Trial group” and produced in
accordance with CONSORT guidelines [21]. The first substantive ACE publication will list
all trial staff and Committee members. Individual authorship will be governed by the
international standards for publication of academic research [22]. The Steering Committee
will review and comment on drafts of papers to ensure accurate, uniform, timely and high
quality reporting of the ACE Trial and will be responsible for the final approval and
submission of all ACE publications.
4. INVESTIGATIONAL PLAN
4.1 Study Design and Plan
ACE is a double blind, randomised, multi-centre, prospective, cardiovascular intervention
trial planned to start in 2008. Six thousand five hundred patients will be randomised in
around 150 Chinese clinical centres. Recruitment will be competitive, with no cap on the
number of patients that any one site can randomise, and will close once 6500 patients in total
have been randomised.
Patients will be allocated randomly 1:1 to double blind therapy with acarbose (50 mg) or
matching placebo three times daily with meals.
Patients will be followed up until there have been 728 adjudicated primary composite
endpoints, unless the trial should be terminated earlier. All patients that cease study
medication will be followed up, if at all possible, for the full study period. All patients that
withdraw from the trial will have their vital status ascertained, if at all possible, at the end of
the trial.
Potential participants will be preselected by clinical centre investigators by reviewing
cardiology outpatient medical records and inviting any patients thought suitable to attend a
screening visit. Those who fulfill all of the inclusion criteria and violate none of the exclusion
criteria, and who have provided written informed consent, will undergo an oral glucose

Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 13 of 45
tolerance test (OGTT). Only those patients found to have impaired glucose tolerance (IGT)
will be entered into the Trial.
Enrolled patients will undergo a four-week single-blind placebo run-in period during which
time their existing therapy for coronary heart disease (CHD) will be optimised if it does not
conform already to internationally accepted guidelines for the treatment of patients with
established CHD i.e. antiplatelet therapy (unless contraindicated or not tolerated), a statin
(unless contraindicated or not tolerated) and, if considered indicated by the investigator, an
ACE inhibitor, a beta-blocker and/or antihypertensive therapy.
ACE follow up visits will be performed at one month, at two months, at four months post-
randomisation, and then every four months.
4.2 Selection of Study Population
Approximately 6,500 patients found to have IGT and with established coronary heart disease
(CHD), including those with a recent acute coronary syndrome (ACS).
4.2.1 Inclusion Criteria
1. Male or female, aged 50 years or more.
2. Definite CHD, defined as a, b or c below:
a) Previous myocardial infarction (MI) or Acute Coronary Syndrome (ACS), but not
within the last 3 months, with any two of the following:
i) Typical clinical presentation
ii) Confirmatory ECG changes
iii) Appropriate elevation of cardiac enzymes/biomarkers
If original reports are unavailable then alternative documentation e.g. discharge
summary or a clinical note from the study Investigator describing the evidence for a
previous MI will be accepted.
Note: Patients with stents are eligible.
b) Previous unstable angina (UA) or Acute Coronary Syndrome (ACS), but not within
the last 3 months, with any two of the following:
i) Typical clinical presentation
ii) Confirmatory ECG changes
iii) Either elevation of a cardiac biomarker or a >50% stenosis in ≥1 major epicardial
coronary artery shown on coronary angiography or CT angiography. Where
stenosis is reported in a qualitative manner, the categories “moderate” and
“severe” will be taken as equating to >50% stenosis.
c) Current stable angina defined as:
i) Typical clinical history with symptoms occurring within the last month, and
ii) A >50% stenosis in ≥1 major epicardial coronary artery shown on coronary
angiography or CT angiography. Where stenosis is reported in a qualitative

Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 14 of 45
manner, the categories “moderate” and “severe” will be taken as equating to
>50% stenosis.
3. Impaired glucose tolerance diagnosed on a single standard OGTT, defined as a 2-hour
plasma glucose (2HPG) value ≥7.8 but <11.1 mmol/l and a fasting plasma glucose
(FPG) <7.0 mmol/l within six months prior to enrollment.
4. Optimised cardiovascular drug therapy.
5. At least 80% adherent to single blind placebo Study Medication during the run-in period.
6. Provision of written informed consent.
4.2.2 Exclusion Criteria
1. Previous history of diabetes, other than gestational diabetes.
2. MI, unstable angina, stroke or a transient ischaemic attack (TIA) within the previous
three months.
3. Planned or anticipated coronary, cerebrovascular or peripheral arterial revascularisation
or other major surgical intervention, at the time of randomisation.
4. NYHA class III or IV heart failure. NYHA classification criteria are listed in Section 10,
Appendix 2.
5. Evidence of severe hepatic disease.
6. Evidence of severe renal impairment or an eGFR <30 ml/min/1.73m2
(derived using the
MDRD Chinese equation [23])
7. Any other condition likely to reduce adherence to the protocol e.g. alcoholism, major
active psychiatric disorder, cognitive impairment or a condition likely to markedly limit
life expectancy e.g. malignancy.
8. Pregnancy (or planned pregnancy within the next five years).
9. Concurrent participation in any other clinical interventional trial. Note: Patients who
were treated previously with an alphaglucosidase inhibitor must have at least a three-
month washout period before being randomised into the ACE trial.
10. Known intolerance to alpha glucosidase inhibitors or gastrointestinal problems.
11. Thought by the investigator for any reason to be unsuitable for participation in this
clinical study.
4.2.3 Diabetes Diagnosed on Screening
Patients diagnosed with diabetes during screening or run-in are not eligible to join the trial
and will be referred for medical care in line with local practice. Note: for the purposes of this
study any diagnosis prior to randomisation only requires a single test result in the diabetic
range and does not need to be confirmed.

Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 15 of 45
4.3 Removal of Patients from Study
Patients will be excluded from entry to the Study if they take less than 80% of their single
blind placebo run-in medication.
Once randomised, all patients should be followed wherever possible until the end of the trial
even if no longer taking study medication.
In accordance with the World Medical Association Declaration of Helsinki (1996 version), as
amended from time to time, all patients have the right to withdraw from the study at any time
and for any reason, without prejudice to his or her future medical care by the physician or
institution, and is not obliged to give his or her reasons for doing so. Without compromising
the right of patients to withdraw from the trial, every effort will be made to understand and
accommodate their concerns and avoid withdrawal wherever possible. This will include, for
instance, down-titrating Study Medication if side effects are a concern, agreeing to a Study
Medication interruption if circumstances dictate, flexibility with regard to follow up intervals
and continued automatic follow-up of patients to end of trial even if they are not taking Study
Medication.
In the event that a patient is unwilling to attend follow up visits, as outlined in 4.6.8, they
will be asked if they will permit the investigator to contact them, while the study is ongoing,
to collect as much data as possible and/or try to establish their vital status at the end of the
trial either by direct contact with them or the people that they have listed as contacts (in
person, by telephone or by letter) or through records (hospital records, public records etc.).
4.4 Premature Closure of Centre
An individual clinical centre may be closed under the following circumstances:
Failure to recruit
Non-compliance with the protocol
Non-compliance with Good Clinical Practice
Unethical practices
Should a clinical centre be closed prematurely, all study materials (except documentation that
has to remain stored at site) must be returned to the sponsor. The investigator will retain all
other documents until notification given by the sponsor for destruction.
4.5 Premature Termination of Study
ACE is an event driven trial and will continue until a total of 728 adjudicated primary events
have been accumulated. The study may be terminated at any time by the Steering Committee
if, in the opinion of the DSMB continuation of the study represents a serious medical risk to
the patients.
During the period of the study, interim analyses of primary endpoints, mortality and of any
other information that is available on major events along with any other analyses that the
Steering Committee may request, will be supplied in strict confidence, to the chairman of the
independent DSMB. In the light of these analyses, and the results of any other relevant

Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 16 of 45
studies, the DSMB will advise the Steering Committee if, in their view, the randomised
comparisons in the study have provided “proof beyond reasonable doubt” that for all, or for
some specific types, of patient, really prolonged use of either treatment is clearly indicated or
clearly contra-indicated in terms of a net difference in all-cause mortality.
The DSMB may recommend premature termination of the study if recruitment is so low that
it becomes unrealistic to consider completion of the protocol in an acceptable period of
time.
4.6 Study Medication
4.6.1 Study Medication to be Administered
At week -4, all patients who complete all screening procedures and who meet the eligibility
criteria to enter the study will be commenced on single blind once-daily placebo tablets.
At Week 0, all patients who complete all screening procedures, who meet the eligibility
criteria to enter the study and who have taken at least 80% of their single-blind placebo run-
in study medication are to be randomized into one of the following two double-blind
treatment groups:
Acarbose tablets (50 mg)
Matching placebo tablets
For selection and timing of the dose please refer to respective section of the study protocol
(4.6.5.).
Investigators will dispense Study Medication only to patients included in this trial following
the procedures set out in the study protocol.
4.6.2 Identity of Investigational Product(s)
Bayer will be responsible for the packaging of the study drug and placebo according to
applicable regulatory requirements. All drug supplies must be stored in accordance with the
manufacturer’s instructions. Label text will be approved according to agreed Bayer
procedures, and a copy of the labels will be made available to study sites upon request.
For the study Acarbose tablets (Bayer ID number: BAY g 5421; trade name: Glucobay)
manufactured by Bayer HealthCare Company Ltd, China, will be used. Matching placebo
tablets will be manufactured and provided by Bayer HealthCare AG, Germany.
Run-in tablets will be packaged in aluminium-aluminium strips. Within study Acarbose and
placebo will be packaged in PVC/PVDC foil.
All Study Medication is to be stored in a securely locked, limited access area, protected from
light and at temperatures below 25˚C, until dispensed to the patient. After the drug is

Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 17 of 45
dispensed it is desirable, but not mandatory, to store at temperatures below 25oC. (Note:
Patients should be made aware that some discolouration of the tablets might occur).
4.6.3 Method of Assigning Patients to Treatment Groups
Each clinical centre will be assigned a unique three-digit centre code when they are enrolled.
Participants considered for entry to the Study at their Screening Visit (Visit 1) will be
identified by a unique within-centre serially assigned participant number, by their initials and
their date of birth. Participant numbers will be assigned automatically by the TMS and no
attempt should be made to allocate numbers outside the system.
At their Randomisation Visit (Visit 3), participants who fulfil all of the inclusion criteria,
violate none of the exclusion criteria and have demonstrated satisfactory compliance during
the run-in period, will be assigned a unique Randomisation number that will allow
subsequent identification of their randomised treatment group allocation. These
randomisation numbers will be generated by Bayer using a computerised procedure. In order
to maintain balance between treatment groups within each Clinical Centre, the randomisation
sequence will allocate patients 1:1 to acarbose or to matching placebo in permuted blocks of
an appropriate size on a centre-by-centre basis.
Any errors made concerning randomisation, such as omitting a number or dispensing
incorrect medication should be documented by the investigator in the study file and on the
eCRF.
4.6.4 Selection of Doses in the Study
The two study treatment regimens are as follows:
Treatment group Tablet content
Acarbose
Acarbose 50 mg
One tablet three times a day
No treatment
Placebo to match acarbose 50 mg tablet
One tablet three times a day
4.6.5 Timing and Titration of Dose for Each Patient
A “Start low, Go slow” dose titration scheme will be used. Patients will be instructed to
commence Study Medication by taking one tablet a day with their smallest meal during the
single-blind placebo run-in period and then for one week following allocation to randomised
Study Medication. During the second week post-randomisation they will be instructed to
increase their Study Medication by taking a second tablet with their next smallest meal.
During the third week they will be instructed to add a third tablet with their third meal and
continue with three tablets thereafter. Study medication must only be taken with meals; if
patients habitually take two meals per day then only two tablets of Study Medication per day
should be taken.
Reduction of Study Medication doses will be allowed in the event of side effects. Patients
may discontinue Study Medication temporarily e.g. for intercurrent illness or hospital

Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 18 of 45
admission. Patients who temporarily discontinue drug or reduce their dose to one tablet will
be required to repeat the “Start low, Go slow” dose titration scheme when it is restarted or up
titrated. In the event of Study Medication over-dosage, please refer to the guidelines in
Section 7.6. Note: Patients who discontinue study drug should be asked at every visit to
consider restarting it. In the case where a patient is unable to attend a clinic visit, the
drug may be mailed to the patient; contact ACPO for details.
4.6.6 Blinding
Copies of the code break will be held by the Bayer Drug Surveillance Group in China. They
will maintain a 24-hour telephone help line (010-5979 9788) should emergency unmasking of
a participant’s Study Medication become necessary. If unmasking is necessary because of a
serious, unexpected and related adverse event or if a medical emergency occurs which
requires knowledge of the exact therapy, the investigator may contact the Bayer help line.
The ACE Trial Manager must be notified of the code break as soon as possible. The date and
reason of the code break must be documented and signed and this information sent to the Co-
ordinating Centre. The patient should be withdrawn from Study Medication but be followed
up according to the protocol.
4.6.7 Concomitant Therapy
A list of concomitant medications relevant to the Study Medication (i.e. cardiac/control of
glucose) will be itemised on an eCRF. The Investigator should record if a patient on entry
and/or given is taking any of these medications in addition to Study Medication during the
course of the study.
4.6.8 Treatment Adherence
Treatment Cessation
After the Randomisation (Visit 3) patients may cease taking Study Medication for any of the
following reasons:
Voluntarily
Safety reasons
Patients diagnosed with diabetes requiring open-label acarbose therapy
Patients whose creatinine clearance falls below 25mls/min/1.73m2 (derived using the
MDRD Chinese equation [23])
Adherence will be assessed by a Study Medication tablet count at relevant visits. Patients
who discontinue Study Medication should be encouraged at every visit, wherever
appropriate, to restart it, using the “Start low, Go slow” dose titration scheme. Patients may
take a reduced dose of Study Medication if limited by side effects. For patients who become
unwilling to take any Study Medication, the reason for discontinuation must be clearly stated.
Patients who withdraw permanently from double blind Study Medication should continue to
be followed as normal and undergo all protocol determined assessments until study closeout,
provided that they have not withdrawn consent. In the event that a patient is unwilling or

Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 19 of 45
unable to attend clinic visits but has not withdrawn consent, arrangements must be made to
obtain annual and study closeout updates on the patient’s vital status and evidence of putative
study endpoints.
4.6.9 Assessment Periods
Patients with established CHD or hospitalized for ACS will be asked to consider joining the
study. They will be provided with written information about the Study and the procedures
involved. They will be given the opportunity to ask questions and given ample time to
consider participation.
Patients will be invited to a Screening Visit (Visit 1) as soon as the treating physician
considers their condition to have stabilized and asked to provide written informed consent;
those who fulfill all of the inclusion criteria and violate none of the exclusion criteria, will
have an OGTT performed. Patients found to have IGT and who continue to be eligible will
attend a Run-in Visit (Visit 2) approximately one week later and, if there are no
contraindications, be entered into the trial. (Note: Visits 1 and 2 may be conducted at the
same time if the patient has a valid OGTT result that establishes eligibility at Visit 1)
Demographic characteristics of patients screened but not able to join the trial will be recorded.
Patients with a recent MI, Unstable Angina, Stoke or TIA, may be screened once stable and
enter run-in if eligible but must not be randomised until 3 months after the
MI/UA/Stroke/TIA.
Following enrolment, patients will have a four-week single-blind placebo run-in during
which time their physician will be asked to optimise their CHD therapy if it does not conform
to international guidelines for treating patients with established CHD i.e. antiplatelet therapy
and a statin unless contraindicated or not tolerated and, if considered indicated by the
investigator, an ACE inhibitor, a beta-blocker and/or antihypertensive therapy. Visits will be
performed one, two and four months post-randomisation and subsequently every four
months. A final visit/telephone contact will be performed approximately one month after the
patient completes the study.
Patients who have not returned to site for >120 days during the pre-randomisation period (i.e.
screening and run-in) should be discontinued in the rTMS.
All routine blood samples will be analysed by the Clinical Centre’s local laboratory. The
biomarker and DNA samples will be stored in a central laboratory in China. Any analyses of
these samples will be performed in China. They can only be undertaken at the request of the
Steering Committee and with any approvals required by the Chinese Authorities.
It should be stated in the patient’s medical notes that the patient is participating in the ACE
Trial.
If a patient misses a visit they should be contacted as soon as possible to re-schedule the visit.
If a patient has missed visits, and returns to the clinic, data should be entered in the most
recent due visit. Consider carrying out any tests or procedures that were missing in the
interim. If you are unsure how to record missed data contact ACPO.

Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 20 of 45
4.6.10 New Onset Diabetes During Study
For diabetes to be diagnosed during the Study (i.e. at a study site), abnormal plasma glucose
values (a FPG ≥7.0 mmol/l and/or a 2HPG ≥11.1 mmol/l) need to be recorded on two
successive occasions.
These tests may be a FPG alone or an OGTT but must be carried out on different days.
Patients found to have an FPG value ≥7.0 mmol/l, or a two-hour plasma glucose (2HPG)
≥11.1 mmol/l after a 75g oral glucose challenge, should have an OGTT performed at their
next scheduled clinic visit to confirm possible diagnosis of diabetes.
A diagnosis of diabetes will be confirmed if there is a second abnormal plasma glucose value
(FPG ≥7.0 mmol/l and/or 2HPG ≥11.1 mmol/l). The date of diagnosis of diabetes will be
taken as the date of the first abnormal glucose value. If the OGTT does not confirm a
diagnosis of diabetes the patient will continue to be followed as normal and the process is
restarted.
A diabetes diagnosis adjudication process will be put in place to review patients who are
diagnosed as having diabetes or have been commenced on antidiabetic therapy outside the
trial.
Patients diagnosed with diabetes will remain in the Study and continue on double blind Study
Medication. They will not require an OGTT at subsequent visits but centres will continue to
record FPG as documented in section 9.1 of this protocol. Their diabetes will be treated by
adding open label metformin therapy, commencing with 250 mg daily and titrating as
necessary to 1000 mg twice daily, aiming to maintain fasting plasma glucose levels <7.0
mmol/l. In the event that FPG values are >7.0 mmol/l despite maximally tolerated metformin
therapy, additional anti-diabetic therapy should be added as deemed appropriate by the
clinician but avoiding the use of alpha-glucosidase inhibitors or prandial glucose regulators.
Double-blind acarbose Study Medication is continued even after diabetes is diagnosed in
order to continue to evaluate the potential impact of acarbose on the primary cardiovascular
endpoint. If a patient is commenced on an alpha-glucosidase inhibitor by a non study
physician the possibility of replacing it with another antidiabetic agent. e.g. metformin should
be explored. In the event that the patient continues to take an open-label alpha-glucosidase
inhibitor, Study Medication should be discontinued.

Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 21 of 45
4.7 Observations and Measurements: Assessment for Treatment Effects
Patients should attend all visits, except the run-in visit, fasting (apart from water) from 22:00
the previous evening.
Note: patients who experience any of the non-fatal endpoints musts remain on study
medication and continue the study. Any subsequent endpoint should also be
reported.
4.7.1 Screening Visit (Visit 1, -5 weeks)
Patients who fulfil all of the inclusion and violate none of the exclusion criteria, and who
have provided written informed consent, will be asked to attend fasting for a Screening Visit
(Visit 1). Patients with a recent MI, UA, stroke or TIA may be screened once the patient is
considered stable and enter run-in, but must not be randomized until 3 months after the MI,
UA, stroke or TIA. The following actions will be completed:
Obtain written informed consent
Record eligibility criteria status
Record relevant concomitant medication
Record age, gender, alcohol consumption, smoking status and self-reported ethnic group
Record past medical history
Note: If a patient has had any tests that are required to establish eligibility as part
of standard care within the last 6 months, then the results may be used as part of
the eligibility check.
Note: Documentation of previous cardiac disease to establish eligibility should include a
thorough clinical history detailing the patients signs and symptoms, 12 lead ECGs,
cardiac markers such as CK-MB and/or troponins and details of any treatments or
procedures relevant to their disease.
Record menopausal status for women

Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 22 of 45
Undertake physical examination (full external examination with emphasis on CVD-
related issues e.g. evidence of heart failure or previous stroke)
Record height
Record waist/hip circumference
Record body weight
Record blood pressure (right arm in sitting position after five minutes rest)
Perform 12 lead ECG (read locally)
Perform 75g OGTT with 0 and 2 hour plasma samples for assay of glucose
Note: The OGTT may be omitted if the patient has had a standard oral glucose tolerance
test, demonstrating IGT within the last 6 months.
Note: For patients that have had a recent acute event, defined as AMI, Unstable Angina
or Stroke, an OGTT may be carried out at any time, at the physician’s discretion, but the
patient cannot be randomized until at least 3 months after the event occurred.
Take fasting serum sample for assay of creatinine
Take fasting serum sample for assay of lipid profile
Take EDTA whole blood sample for full blood count
Provide appropriate lifestyle advice with respect to diet, exercise and smoking
Agree date and time of next study follow up appointment
Patients may be rescreened on one occasion only, but not within one week of the first screen.
The Registration eCRF provides an option for entering the additional data for patients who
are screened a second time. If the patient has a MI/UA/Stroke/TIA after screening but before
randomization they remain eligible to join the study, but cannot be randomized until three
months after the event and undergoing a second run-in period if the first was not completed
satisfactorily. Patients who have experienced any other acute cardiac event may be screened
at any time, once the treating physician considers their condition is stable. Patients who are
screened a second time do not need to re-sign an informed consent form.
4.7.2 Run-in Visit (Visit 2, -4 weeks)
Patients who continue to be eligible will attend a Run-in Visit (Visit 2) one week later (±7
days), at which time the following assessments will be completed:
Recheck patient eligibility
Dispense single-blind run-in placebo Study Medication
Agree date and time of next study follow up appointment
All patients will commence a four-week single-blind placebo run-in and will be instructed to
take this Study Medication once a day with their smallest meal of the day. Patients requiring
CHD therapy optimisation will be prescribed additional open-label medication, titrated as
necessary during this time period. Optimised CHD therapy should include: Antiplatelet

Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 23 of 45
therapy, a statin, unless contraindicated or not tolerated, an ACE inhibitor, and a beta-
blocker and/or antihypertensive therapy, if considered indicated by the investigator.
4.7.3 Randomisation Visit (Visit 3, week 0)
Patients who continue to be eligible will be asked to attend fasting for a Randomisation Visit
(Visit 3) four weeks later, at which time the following actions will be completed:
Recheck patient eligibility
Check patient has taken at least 80% of single-blind placebo run-in study medication
Record relevant concomitant medication
Record any relevant adverse events
Record body weight
Record blood pressure (right arm in sitting position after five minutes rest)
Take fasting plasma sample for assay of glucose. Patients found to have an FPG value
≥7.0 mmol/l should have an OGTT performed at their next clinic visit to confirm possible
diagnosis of diabetes. If there is clinical concern, an OGTT should be performed within
one month. If diabetes is confirmed treat, as outlined in section 4.6.10
Take fasting serum sample for assay of alanine transaminase (ALT)
Record Hepatitis B status, if available
Take fasting serum sample for assay of lipid profile
Take fasting whole blood sample for assay of HbA1c
Take fasting serum and plasma samples for later biochemical tests (sent to central
laboratory). The maximum volume of these samples will not exceed 14ml. If these
samples are not taken, or found to be unsatisfactory, further samples cannot be taken once
patients are taking study medication.
Take whole blood sample for later molecular analyses (sent to central laboratory). The
maximum volume of these samples will not exceed 9ml. If these samples are not taken, or
found to be unsatisfactory, a further sample can be taken (but only while recruitment for
the study is ongoing).
Randomise patient and dispense four months’ double-blind Study Medication
Provide “Start low, Go Slow” instructions
Administer Quality of Life (QoL) questionnaire
Agree date and time of next study follow up appointment
If a follow up visit is delayed or missed, subsequent follow-up visits will still take place as
scheduled originally from the date of randomisation, to ensure timely completion of the
study.

Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 24 of 45
4.7.4 Follow up Visits
Follow up telephone visits 4 and 5 will be scheduled for one and two months post-
randomisation respectively. All telephone contacts must be recorded in the patient’s notes.
The following items will be addressed:
Record any relevant adverse events
Record any Study Endpoints
(Note, perform 12 lead ECG (read locally) if it is suspected that a CV event occurred
since the last visit)
Review Study Medication and assess need for Study Medication to be titrated
Agree date and time of next appointment
Visit 6 will be scheduled four months post-randomisation with participants asked to attend
fasting. The following actions will be performed:
Check Study Medication adherence
Record any relevant adverse events
Record any Study Endpoints
Record blood pressure (right arm in sitting position after five minutes rest)
Take fasting plasma sample for assay of glucose. Patients found to have an FPG value
≥7.0 mmol/l should have an OGTT performed at their next clinic visit to confirm possible
diagnosis of diabetes. If there is clinical concern, an OGTT should be performed within
one month. If diabetes is confirmed treat as outlined in section 4.6.10
Assess need for Study Medication to be titrated
Dispense four months’ Study Medication
Agree date and time of next study follow up appointment
Visit 6 and all subsequent scheduled study visits will be at four-monthly intervals with
participants asked to attend fasting. The assessments performed at these four-monthly visits
will be the same as at Visit 6.
4.7.5 Annual Visits
At annual visits (Visit 8, 11, 14, 17 & 20 etc. and annually thereafter) participants will be
asked to attend fasting. The following actions will be performed:
Record relevant concomitant medication
Check Study Medication adherence
Record any relevant adverse events
Record any Study Endpoints

Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 25 of 45
(Note, perform 12 lead ECG (read locally) if it is suspected that a CV event occurred
since the last visit)
Record smoking status
Record menopausal status for women
Record alcohol consumption
Record body weight
Record waist/hip circumference
Record blood pressure (right arm in sitting position after five minutes rest)
Perform ECG (read locally)
Undertake physical examination (full external examination with emphasis on CVD-
related issues e.g. evidence of heart failure or previous stroke)
Perform 75g OGTT with 0 and 2 hour plasma samples for assay of glucose.
Note: OGTT must be performed using the glucose sachets provided for the study.
Patients found to have an FPG value ≥7.0 mmol/l or a 2HPG ≥11.1 mmol/l should have
an OGTT performed at their next clinic visit to confirm possible diagnosis of diabetes. If
there is clinical concern, an OGTT should be performed within one month. If diabetes is
confirmed treat at outlined in section 4.6.10
Take fasting serum sample for assay of ALT
Take fasting serum sample for assay of creatinine
Take fasting whole blood sample for assay of HbA1c
Take fasting serum sample for assay of lipid profile
Assess need for Study Medication to be titrated
Dispense four months’ Study Medication
Administer Quality of Life (QoL) questionnaire
Agree date and time of next study follow up appointment
4.7.6 Closeout Visit
Participants will be asked to attend the closeout visit in a fasting state and the following
actions will be performed:
Record relevant concomitant medication
Check Study Medication adherence
Collect any remaining Study Medication
Record any relevant adverse events
Record any Study Endpoints
Record smoking status

Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 26 of 45
Record menopausal status for women
Record alcohol consumption
Record body weight
Record waist/hip circumference
Record blood pressure (right arm in sitting position after five minutes rest)
Perform ECG (read locally)
Undertake physical examination (full external examination with emphasis on CVD-
related issues e.g. evidence of heart failure or previous stroke)
Take fasting plasma sample for assay of fasting plasma glucose (FPG). If the FPG is ≥7.0
mmol/l arrange appropriate follow-up outside of the study to confirm whether the patient
has developed diabetes and, if so, receives appropriate therapy.
Take fasting serum sample for assay of ALT
Take fasting serum sample for assay of creatinine
Take fasting serum sample for assay of lipid profile
Take fasting whole blood sample for assay of HbA1c
Administer Quality of Life (QoL) questionnaire
Agree date and time of final visit/telephone contact (if applicable)
Thank the patient for their participation in the study and inform them that they will be
notified of the main results after they are published.
4.7.7 Final Visit/Telephone Contact
A final visit/telephone contact will be conducted only for patients who:
a) Stop taking study drug at their close-out visit
OR
b) Have stopped taking study drug within 28 days prior to their close-out visit.
This final visit/telephone contact, which should take place a minimum of 28 days after drug
discontinuation, is intended to identify any AEs, SAEs, hospitalisations or study endpoints
that have occurred in this period.
4.8 Data Quality
The ACE Co-ordinating Centre will be based in the Diabetes Trials Unit at the University of
Oxford (DTU). Co-ordinating Centre staff will set up, supervise and track the progress of the
study and establish a Chinese Project Office in Beijing to monitor and provide local support
for the Clinical Centres.

Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 27 of 45
The Chinese Project Office will assess all Clinical Centres prior to their initiation to verify
the qualifications of each investigator, check the site facilities and inform the investigators of
their responsibilities and the procedures for ensuring adequate and correct documentation.
Investigator training meetings will be held to introduce investigators and their personnel to
the study protocol, eCRF and procedures. Centre initiation visits will also be performed.
During the course of the Study, top level monitoring will be performed centrally using the
DTU’s electronic Trial Management System (TMS). A Study Monitor will conduct a site
initiation visit and conduct the first monitoring visit(s) shortly after the first patient is
screened/randomised. Monitoring visits will be conducted periodically throughout the study,
culminating in a study closeout visit. Direct access to original source data will be required to
check the accuracy of eCRF completion and for inspections/audits. The TMS will be used to
perform data capture, data validation, data and process audit, monitor protocol adherence and
facilitate statistical analyses.
Data will be entered directly onto electronic Case Report Forms (eCRFs) wherever feasible to
avoid unnecessary transcription. Mandarin or English can be used for free text entries. The
eCRFs will be completed using a laptop supplied solely for use in the ACE Study. All data
will be entered online by an ADSL or telephone dial-up Internet connection.
Data added to the TMS will be checked for internal consistency (within range and known
categories), external consistency (data is consistent with other data items known for that
patient) and trend consistency (data values have not changed by more than allowable amounts
e.g. excessive weight gain or loss which might indicate digit transposition). Study data audits
will be performed weekly. These will include checks of the number of data items obtained
from each centre, the number found to have anomalies and the number of data items overdue
to permit progress chasing.
A rule-based version of the study protocol, encapsulated within the TMS, will allow
automated checking of each patient's progress through the study. A system log will be
maintained which identifies every database edit made, the date it was made and the operator
who made the change. Data backups will be made daily with rotating on and off site storage.
4.9 Documentation
Entries made in the eCRF must be either verifiable against source documents, or have been
directly entered into the eCRF, in which case the entry in the eCRF will be considered as the
source data for verification purposes. All data necessary for the continuity of patient care
should also be maintained as normal in patients’ notes. Site staff should clearly document in a
patient’s notes that the patient is in a clinical study, and they should provide contact
information with a request that any other medical staff who might treat the patient should
contact the trial site staff. In cases where the patient retains ownership of their notes, copies
of all trial related data should be retained at site. Unless data are entered directly on to an
eCRF, they must be written in the patient’s notes (or a study file) and transferred to the
relevant eCRF at the earliest opportunity. The study file and all source data should be
retained until notification is given by the sponsor for their destruction.
If a patient has been seen by another department or institution, every effort should be made to
obtain copies of discharge summaries, cardiac biomarkers and ECGs to enable adjudication

Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 28 of 45
of events/safety data. For laboratory parameters quantitative information should be sourced,
wherever possible, rather than qualitative information.
In the event of temporary lack or delayed access to the rTMS, the use of paper CRFs is
allowed on a temporary basis. These should be sourced from ACPO.
5. ETHICAL AND LEGAL ASPECTS
5.1 Ethics Committee or Institutional Review Board
Documented approval from appropriate Ethics Committee(s) or Institutional Review Board(s)
must be obtained for all participating centres prior to study start, according to Good Clinical
Practice (GCP), local laws, regulations and organizations. When necessary, an extension,
amendment or renewal of the Ethics Committee approval must also be obtained. Ethics
Committees must supply upon request, a list of the Ethics Committee members involved in
the vote and a statement to confirm that the Ethics Committee is organized and operates
according to GCP and applicable laws and regulations.
Modifications to the study protocol will not be implemented without the agreement of the
Steering Committee and appropriate ethical approval.
5.2 Ethical Conduct of the Study
The procedures set out in this protocol, pertaining to the conduct, evaluation, and
documentation of this study, are designed to ensure that the sponsor and investigator abide by
GCP Guidelines. The study will also be carried out in keeping with applicable local law(s)
and regulation(s). This may include an inspection by the sponsor representatives and/or
Regulatory Authority representatives at any time. The investigator must agree to the
inspection of study-related records by the Regulatory Authority/sponsor representatives, and
must allow direct access to source documents to the Regulatory Authority/sponsor
representatives.
5.3 Regulatory Authority Approvals/Authorizations
Regulatory Authority approvals/authorizations/notifications, where required, must be in place
and fully documented prior to study start.
5.4 Informed Consent
Written and verbal versions of Informed Consent will be presented to the patient, detailing no
less than: the research objective, the methodology and duration, the anticipated benefits and
the limits and risks associated. It will be clearly stated that the patient is free to withdraw
from the study at any time for any reason without prejudice to future care, and with no
obligation to give the reason for withdrawal. Written Informed Consent must be obtained
before any study specific procedure takes place. If a patient is illiterate, he/she can nominate
someone to sign on their behalf and the patient can make a mark beside the signature, such a
process should be clearly documented. A copy of the signed Informed Consent will be given
to the patient. The original signed form will be retained at the study site.

Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 29 of 45
5.5 Insurance
All patients participating in the study will have insurance coverage arranged by the sponsor,
which is in line with applicable laws and/or regulations.
5.6 Confidentiality
All records identifying the patient will be kept confidential and, to the extent permitted by the
applicable laws and/or regulations, will not be made publicly available. The patient will be
identified only by their date of birth, initials and a unique patient ID number on the eCRF.
All documents will be stored securely and kept in strict confidence in compliance with the
Data Protection Act.
Study findings stored on a computer will be stored in accordance with local data protection
laws. The patients will be informed in writing that representatives of the sponsor, Ethical
Committees, Institutional Review Boards or Regulatory Authorities may inspect their
medical records to verify the information collected, and that all personal information made
available for inspection will be handled in strictest confidence and in accordance with local
data protection laws.
When the results of the study are published, the patient’s identity will remain confidential.
The investigator will maintain a list to enable patients’ records to be identified.
6. STATISTICAL METHODS AND DETERMINATION OF SAMPLE
SIZE
6.1 Description of Patient Groups for Analysis
All patients will be analysed according to the randomised treatment that they first received
i.e. the primary population for analysis is intention-to-treat (ITT), irrespective of whether
they took randomised Study Medication. Patients lost to follow-up will be censored at their
last contact point. Patients who do not have an event in the given study period will be
censored at their last follow-up visit. Further analyses to be undertaken will include Valid
Case (Per Protocol) and Safety Analyses.
6.2 Study Objectives
6.2.1 Primary Endpoint
This will be a composite cardiovascular outcome defined as the time after randomisation to
the first occurrence of any one of the following:
Cardiovascular death
Non-fatal MI
Non-fatal stroke

Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 30 of 45
Hospitalisation for unstable angina
Hospitalisation for heart failure
6.2.2 Secondary Endpoints
Transition to type 2 diabetes confirmed by two successive diagnostic plasma glucose
values (FPG ≥7.0 mmol/l and/or 2HPG ≥11.1 mmol/l), with no intervening non-
diagnostic values.
All cause mortality.
Each of the components of the primary composite cardiovascular outcome will also be
analysed individually, both as first and as total events.
MACE composite cardiovascular outcome, defined as the time after randomisation to the
first occurrence of any one of the following:
Cardiovascular death
Non-fatal MI
Non-fatal stroke
Proportion of patients with an impaired renal function as evidenced by:
A reduced eGFR (<30 ml/minute/ 1.73 m2) estimated using the Chinese
MDRD formula [23]
A doubling of the baseline plasma creatinine level
A halving of the baseline eGFR
Resource use, costs and cost effectiveness.
6.3 Statistical and Analytical Plans
The primary outcome will be evaluated by time-to-event analysis methods using a log-rank
test. It will be performed using an intent-to-treat approach, and will be supplemented by per-
protocol and on-treatment analyses. Given that secondary outcome, the progression to
diabetes, can be established in all patients only at the 4-month visits (that is, the endpoint is
interval-censored), the log-rank test for this endpoint will be based on a discrete time
proportional odds model [Cox et al 1989]. Evaluations of baseline characteristics, safety
assessments, and additional measurements during the trial (including treatment
interruption/discontinuation, lifestyle evaluation, and use of concomitant medications) will be
based on appropriate summary statistics. The effects of demographic and other baseline
characteristics on the primary outcomes will be explored using appropriate multivariate
methods. Full details of analyses to be performed will be itemised in a separate Statistical
Analysis Plan (SAP).

Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 31 of 45
6.4 Interim Analysis/Analyses
No interim analyses are planned, other than those commissioned by the Data Safety and
Monitoring Board.
6.5 Sample Size Estimation
6.5.1 Event Rate
The event rate for the original primary cardiovascular (MACE) outcome was estimated to be
3.5% per year for the population to be enrolled.
6.5.2 Number of Patients
Sample size calculations assumed a relative risk reduction of 20% in the primary composite
endpoint event rate with acarbose, an accrual period of 18 months, a minimum follow up of 4
years and an alpha of 5%. A total of 7,268 patients (3634 per group) were be required to
obtain 90% power with a total of 904 participants having an adjudicated primary event. To
allow for a possible 3% loss-to-follow up, a total of 7500 patients were planned be
randomised. No allowance was been made in this calculation for discontinuation of Study
Medication.
The sample size was reviewed by the ACE Steering Committee when they met face-to-face
on 15 February 2014. At that time the Committee noted that the substantially extended
recruitment period meant that sufficient patient years of follow up to achieve a total of 904
participants with an adjudicated primary event could be accumulated with fewer patients
randomised and revised the target down from 7,500 to 6,500. As the trial remained event
driven with the 904 primary event target unchanged, no adjustment was required for the
power calculations.
The sample size was reviewed again by the ACE Steering Committee when they met by
teleconference on 19 May 2016. As event rates for the primary composite cardiovascular
outcome remained much lower than originally anticipated, they agreed to reduce the power of
the study from 90% to 85%. This decision reflected the increasing participant and site
fatigue, and financial constraints. Revised power calculations, using the formula published by
Stuart Pocock et al. [24], showed that 728 participants with a confirmed primary composite
cardiovascular outcome would be required for 85% power.
6.6 Health Economic Evaluation
An economic evaluation will be performed, to assess the cost-effectiveness of acarbose
compared with placebo. The evaluation will consist of three main elements:
collection of resource use information and unit costs;
collection of outcome data appropriate to economic evaluation; and

Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 32 of 45
calculation of within-trial and extrapolated cost-effectiveness.
6.6.1 Resource use and costs
Resource use will be recorded on eCRFs, consisting mainly of number, type and dose of
medications including trial therapy; number of hospital clinic visits; and in-patient
hospitalisations associated with complications/adverse events. The duration of
hospitalisations will be recorded on the eCRF, and whether the patient underwent
angiography, angioplasty, coronary artery bypass grafting or other major procedure.
Unit costs will be then attached to these resource items, using published national average
costs, tariff averages for procedures, and drug costs to the Chinese Health Service prevailing
at the end of the study, to derive a cost per patient for each year or part of year in the study.
6.6.2 Economic outcomes
For the purposes of the economic evaluation, outcomes will be defined and measured in
terms of:
number of primary endpoints
number of fatal primary endpoints
quality adjusted life years
To assess quality of life, the EQ-5D in official Chinese language version will be administered
at randomisation, at annual visits and at closeout. Descriptive information from the EQ-5D
questionnaire will be reported and analysed in the form of distributions across response levels
on each question at each survey point, and published valuation tariffs will be used to attach
quality of life valuation to each reported health state and calculate quality adjusted survival.
6.6.3 Cost-effectiveness
Cost-effectiveness will be estimated as the net costs of therapy with acarbose compared with
placebo expressed as a ratio of the net effectiveness of acarbose compared with placebo.
Effectiveness will be measured in terms of life-years gained and quality adjusted life years
(QALYS) gained, first within trial, and secondly over lifetime using a parametric
extrapolation model that will estimate time to events, primarily from trial data.
Within trial cost-effectiveness will be reported as the incremental cost per:
Primary endpoint averted
Fatal primary endpoint averted
Quality adjusted life year gained
Lifetime cost-effectiveness will be in terms of:
Life years gained

Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 33 of 45
Quality adjusted life years gained.
6.6.4 Analysis and presentation of economic data
The primary analysis of all economic data will be on an intention-to-treat basis. Mean values
and 95% confidence intervals will be reported for each component of resource use and cost
and for total costs and effectiveness. Uncertainty around the cost-effectiveness ratios reported
will be estimated using the non-parametric bootstrap, and reported using the cost-
effectiveness plane and the cost-effectiveness acceptability curve.
All costs and outcomes will be discounted to present values at recommended annual rates in
the baseline analyses, with a range of discount rates used in the sensitivity analyses.
Sensitivity analyses will be performed on all aspects of the economic evaluation, which are
subject to uncertainty. Missing data will be handled by multiple imputations.
When the EQ-5D is administered, a small number of additional questions will be asked about
household type, household income and expenditure, health insurance status, and expenditure
on hospital visits and admissions. These data will be reported descriptively, and used as
control variables in multivariate analyses of costs, outcomes and cost-effectiveness.
7. ADVERSE EVENTS
7.1 Adverse Event Monitoring
As acarbose is already licensed in China for the treatment of IGT, ACE is a Phase IV trial. As
such, there is no requirement to collect adverse events routinely. However, an adverse event
(AE) will be recorded where study medication is reduced or stopped as a result of the adverse
event if the adverse event is thought to be related to study medication administration.
Serious Adverse Events (SAEs), including SSARs and SUSARs, that are thought to be
possible Study Endpoints (SEs) will not be reported as SAEs during the trial.
SAEs, SSARS and SUSARS that are not thought to be possible Study Endpoints will be
reported by the Investigator to the Sponsor, CFDA and EC according to the relevant
regulations.
Possible Study Endpoints (including those that are SAEs SSARs or SUSARs) will be
reported to the Diabetes Endpoint Adjudication Committee or the Cardiovascular Endpoint
Adjudication Committee as rapidly as possible.
The DSMB will review all Study Endpoints (unadjudicated and adjudicated) and all reported
SAEs by allocated therapy at least twice yearly.

Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 34 of 45
7.2 Adverse Event Definitions
7.2.1 Adverse Event (AE)
An adverse event (AE) is any undesirable medical event occurring to a patient in a clinical
trial, whether or not related to the trial product(s). An AE will be recorded where study
medication is reduced or stopped as a result of the adverse event and where the adverse event
is thought to be related to study medication administration.
7.2.2 Serious Adverse Event (SAE)
A serious adverse event or serious adverse drug reaction is any untoward medical occurrence
that results in:
Death.
A life threatening* experience.
Hospitalisation: any adverse event leading to hospitalisation or prolongation of
hospitalisation will be considered as Serious, UNLESS the admission results in a hospital
stay of less than 12 hours or the admission is pre-planned i.e. elective or scheduled
surgery arranged prior to the start of the study.
A persistent or significant disability/incapacity (disability means a substantial disruption
of a person’s ability to conduct normal life’s functions).
A congenital anomaly/birth defect.
Important medical events that may not result in death, be life threatening*, or require
hospitalisation may be considered a serious adverse event when, based upon appropriate
medical judgment, they may jeopardise the patient and may require medical or surgical
intervention to prevent one of the outcomes listed in this definition.
* The term life threatening in the definition of serious adverse event refers to an event in
which the patient was at risk of death at the time of the event. It does not refer to an event
that hypothetically might have caused death if it was more severe.
7.2.3 Suspected Serious Adverse Reaction (SSAR)
This is defined as a serious adverse reaction, the nature or severity of which is consistent with
the known study treatment information (e.g. Summary of Medicinal Product Characteristics
(SmPC), Investigator Brochure (IB) or Investigator Medicinal Product Dossier (IMPD)).
7.2.4 Suspected Unexpected Serious Adverse Reaction (SUSAR)
This is defined as a serious adverse reaction, the nature or severity of which is not consistent
with the known study treatment information. A serious event or reaction is not defined as a
SUSAR when:
‘it is serious but expected’ or it does not fit the definition of an SAE, whether expected or not.

Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 35 of 45
7.2.5 Relationship to Study Medication
Probable: Good reasons and sufficient documentation to assume a causal
relationship.
Possible: A causal relationship is conceivable and cannot be dismissed.
Unlikely: The event is most likely related to aetiology other than the trial
product.
7.2.6 Severity of the Adverse Event
The severity of adverse events should be graded as follows:
Mild: usually transient in nature and generally not interfering with normal
activities.
Moderate: sufficiently discomforting to interfere with normal activities
Severe: prevents normal activities.
7.3 Collection and Reporting of Adverse Events
Adverse event data will be collected from the first trial-related activity after the patient
provides informed consent up until 28 days after the last administration of study medication.
AEs that meet the definition listed in Section 7.2.1 of this protocol (and where study
medication is reduced or stopped as a result of the adverse event and where the adverse event
is thought to be related to study medication administration), SAEs, SSARs, SUSARs and
events thought to be Study Endpoints will be captured on Adverse Event/SAE or
Cardiovascular Event eCRFs that collect detailed data on the event including an assessment
of causality and standard safety data, and will be reported according to the timelines detailed
below.
All SAEs that are also thought to be Study Endpoints will be reported to the Cardiovascular
Endpoint Adjudication Committee as rapidly as possible.
All SAEs that are considered to be related to study drug (SSARs and SUSARS), including
Study Endpoints, will be reported via the standard paper forms to the Safety Officer within
24hrs of the site staff being aware of the event and will be forwarded to the Beijing ADR
centre by the Safety Officer, via the electronic system in the required timeline. The Endpoint
eCRF will collect detailed data on the event and also includes an assessment of causality, to
enable Study Endpoints that are considered to be Serious Adverse Drug Reactions to be
reported.
New-onset diabetes during the study will not be considered an adverse event but will be
reviewed by the Diabetes Endpoint Adjudication Committee as rapidly as possible.

Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 36 of 45
The DSMB will review all safety data, including Study Endpoints (unadjudicated and
adjudicated) and all reported SAEs by allocated therapy at least twice yearly.
SAEs, SSARs and SUSARs that are not thought to be possible Study Endpoints will be
reported by the Investigator to the Sponsor, CFDA and EC according to the relevant
regulations.
The sponsor will report all SSARs and SUSARs to:
1) Beijing ADR immediately, ideally within 3 days in the event of death; within 15 days
for all other events.
2) Bayer Global Pharmacovigilance within 24 hrs in the event of death or a life-
threatening event, within 2 working days for all others.
3) An Annual Safety Report (ASR) will be submitted to Beijing ADR and to the Oxford
Tropical Research Ethics Committee (OXTREC).
4) Local occurring SSARs and will be reported to the HK regulatory authorities as
follows;
Fatal or life-threatening unexpected Adverse Drug Reactions (ADRs) should be
reported as soon as possible but no later than 7 calendar days after first knowledge by
the sponsor that a case qualifies, followed by as complete a report as possible within 8
additional calendar days. Other serious, unexpected ADRs that are not fatal or life
threatening should be reported as soon as possible but no later than 15 calendar days
after first knowledge by the sponsor that the case meets the minimum criteria for
expedited reporting.
In addition, at the end of the trial a brief summary of local non-serious ADRs and
serious ADRs that are expected will also be submitted.
5) The Sponsor will provide the ASR, which includes all Serious Adverse Drug
Reactions, to the sites. Sites will submit the ASR to their Ethics Committee, if
required.
7.4 Reporting of Pregnancy
Pregnancy occurring during a clinical investigation, although not considered a serious
adverse event, must be reported within the same timelines as a SAE on a Pregnancy
Monitoring Form. The patient should stop the study medication but continue to attend
follow–up visits until the end of the trial. The outcome of a pregnancy should be followed up
carefully and any abnormal outcome of the mother or the child should be reported as an SAE
if it meets the SAE definition in 7.2.2.
7.5 Warnings/Precautions for Acarbose
In this trial, annual ALT monitoring will be performed. These ALT levels will be reviewed
by the DSMB as asymptomatic elevation of liver enzymes, sometimes even to a clinically

Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 37 of 45
relevant degree (up to three times the upper limit of normal), has been observed very rarely
but was generally reversible on ceasing treatment
Other precautions
Regular treatment with acarbose should not be interrupted without the doctor’s
knowledge, as this may lead to a rise in blood glucose.
Acarbose does not cause hypoglycaemia in patients treated with diet alone. Should
signs of hypoglycaemia occur during acarbose therapy as a result of the lower insulin
requirement in patients being treated with insulin, sulfonylureas or metformin, glucose
(not table sugar [cane sugar]) should be taken.
Possible treatment with acarbose should be included in the patient’s records as well as
their inclusion in the ACE Trial.
7.6 Overdosage
If acarbose is taken with drinks and/or meals containing carbohydrates (polysaccharides,
oligosaccharides, disaccharides), overdose may lead to meteorism, flatulence and diarrhoea.
If an overdose of acarbose is taken in the absence of food, excessive intestinal symptoms are
not to be expected.
In the event of overdose, no drinks or meals containing carbohydrates (polysaccharides,
oligosaccharides, disaccharides) should be consumed for the next 4 - 6 hours.
8. USE OF DATA AND PUBLICATION
During the Trial all data derived from the Trial will be held by the Diabetes Trials Unit
(DTU) but with access for Bayer to any data required for safety and regulatory purposes.
At the time database lock occurs, Bayer will provide the DTU with an electronic file
containing the full randomisation codes for upload to the TMS. The DTU will undertake the
planned analyses and prepare and submit manuscripts for publication and presentations to
academic meetings, as agreed by the Steering Committee. Bayer will have the right to
comment on these but the final editorial control remains with the Steering Committee.
Following publication of the main trial results, a copy of the database will be transferred to
Bayer.
Study data will be archived in the DTU in the data warehouse and each site will receive a CD
with their site’s data in PDF or html format.
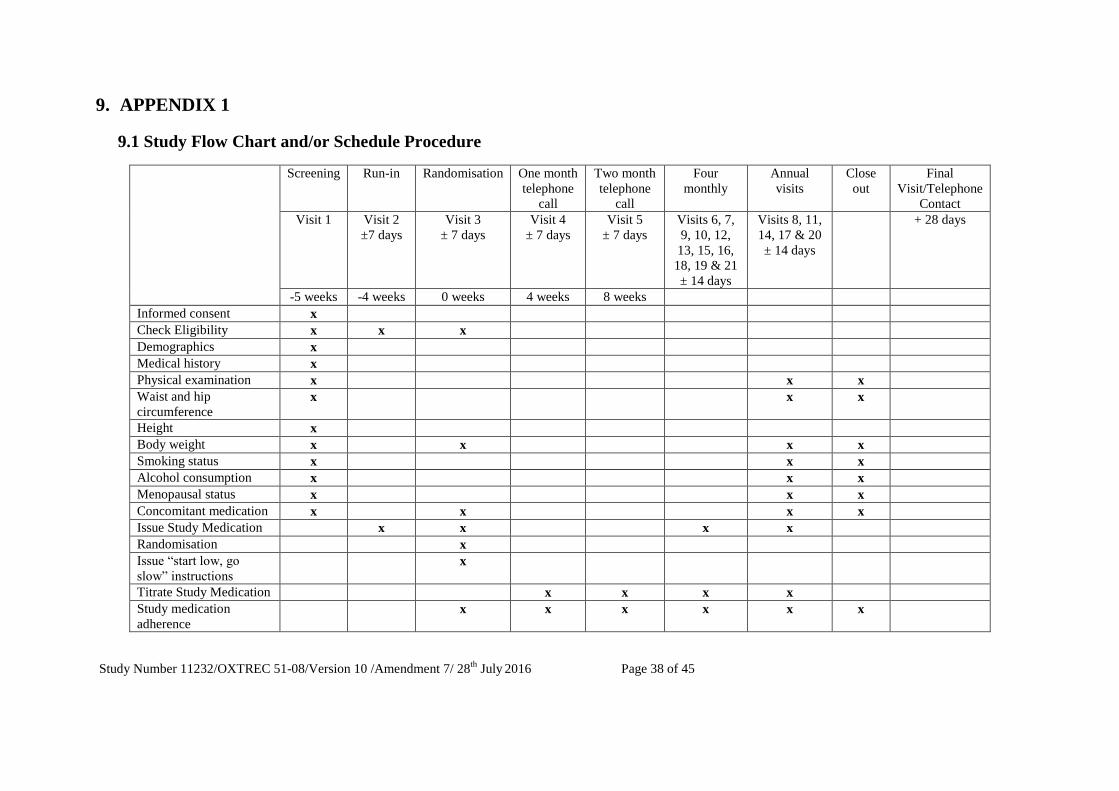
Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 38 of 45
9. APPENDIX 1
9.1 Study Flow Chart and/or Schedule Procedure
Screening Run-in Randomisation
One month
telephone
call
Two month
telephone
call
Four
monthly
Annual
visits
Close
out
Final
Visit/Telephone
Contact
Visit 1 Visit 2
±7 days
Visit 3
± 7 days
Visit 4
± 7 days
Visit 5
± 7 days
Visits 6, 7,
9, 10, 12,
13, 15, 16,
18, 19 & 21
± 14 days
Visits 8, 11,
14, 17 & 20
± 14 days
+ 28 days
-5 weeks -4 weeks 0 weeks 4 weeks 8 weeks
Informed consent x
Check Eligibility x x x
Demographics x
Medical history x
Physical examination x x x
Waist and hip
circumference x x x
Height x
Body weight x x x x
Smoking status x x x
Alcohol consumption x x x
Menopausal status x x x
Concomitant medication x x x x
Issue Study Medication x x x x
Randomisation x
Issue “start low, go
slow” instructions x
Titrate Study Medication x x x x
Study medication
adherence x x x x x x

Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 39 of 45
Lifestyle advice x
AE/SAE enquiry x x x x x x x
Endpoint enquiry x x x x x x
Blood pressure x x x x x
ECG x x x
Fasting plasma glucose* x x x
OGTT* x x
Serum ALT* x x x
Serum creatinine* x x x
Hepatitis B status
(HBsAg) ***
x
x
Lipid profile* x x x x
Full blood count* x
HbA1c x x x
Blood volume per visit
(blood samples for local
analysis) (ml)
8.5 8.5 3.5 8.5 8.5
Serum and plasma
samples for later
biochemical tests**
x
Volume of whole blood
taken for later
biochemical tests (ml)
14
Whole blood sample for
later molecular analyses §
x
Volume of whole blood
for later molecular
analyses (ml)
9
Completion of QoL
questionnaires x x x
* local assay; ** store centrally; *** If available § If samples for molecular analysis are not taken, or found to be unsatisfactory, a further sample can be taken (but only whilst recruitment for the study is ongoing).

Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 40 of 45
10. APPENDIX 2: NYHA GRADING CRITERIA FOR HEART FAILURE
Class Patient Symptoms
Class I (Mild) No limitation of physical activity. Ordinary physical activity does
not cause undue fatigue, palpitation, or dyspnea (shortness of
breath).
Class II (Mild) Slight limitation of physical activity. Comfortable at rest, but
ordinary physical activity results in fatigue, palpitation, or
dyspnea.
Class III
(Moderate)
Marked limitation of physical activity. Comfortable at rest, but less
than ordinary activity causes fatigue, palpitation, or dyspnea.
Class IV (Severe) Unable to carry out any physical activity without discomfort.
Symptoms of cardiac insufficiency at rest. If any physical activity
is undertaken, discomfort is increased.

Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 41 of 45
11. APPENDIX 3: OGTT PROCEDURE
For ALL OGTT tests performed within the study, the trial-specific sachets provided by trial
must be used. Commercially produced sachets containing 82.5g of dextrose monohydrate
powder are used for the OGTTs. The sachets are distributed to sites by the ACPO.
Note: the OGTT to establish eligibility, may be omitted if the patient has had a standard oral
glucose tolerance test within the last 6 months that demonstrated IGT.
This test should not be carried out on patients who are known to be diabetic.
The day before the OGTT, the participant must:
1 Eat a normal diet on the day prior to the test and refrain from drinking alcohol, in
the evening they should refrain from doing any strenuous exercise.
2 Fast from 22.00 hours on the evening prior to the test. The participant may only
drink water after 22.00 hours of the evening prior to the test..
3 Refrain from smoking the morning of the test.
The day of the test:
1 As the patient is fasting, the test should be scheduled for early in the morning.
2 The participant must be seated or lying down for the whole period of the test.
3 Prepare the OGTT drink by dissolving the contents of the trial-specific sachet in
300ml warm water (20-30oC is preferred). Note: a range of 250ml – 300ml is
permissible; < 250ml or > 300ml will affect the result.
4 Collect a fasting sample (basal) using the local procedure for collecting venous
blood samples. This sample will be time zero. Then, ask the patient to drink the
glucose drink, the drink should be consumed at a steady rate over 4-5 minutes.
5 After 120 minutes collect another venous blood sample according to local
procedure. This will be time 120min.
6 A finger stick blood glucose measurement is recommended for the safety of the
patients at the termination of OGTT.
a. If glucose < 3.1mmol/L or there are symptoms of hypoglycaemia;
administer a light carbohydrate snack (~15g of carbohydrate)
Re-test after 30 mins
Repeat if necessary until glucose > 4.0mmol/L; then discharge from clinic
b. If glucose ≥ 3.1mmol/L and there are no signs of hypoglycaemia;
discharge from clinic
7 A light meal or snack is also recommended before the patients leave the clinic.
8 If the participant vomits while drinking the glucose drink, the test should be
stopped. A finger stick blood glucose measurement should be obtained and the
investigator should follow the procedure outlined in 6. The investigator should
then reschedule an OGTT as soon as possible (on a different day).

Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 42 of 45
Processing samples for the OGTT
To ensure an accurate result, it is critical that the OGTT blood samples are processed as soon
as possible after sampling. Ideally, the OGTT samples should be separated immediately after
each patient visit due to the time-critical biochemical reaction. If this is not possible the
samples should be stored on ice prior to processing.
For the plasma glucose test, there are three options for the storage of OGTT blood samples
prior to analysis depending on the local laboratory procedures.
(A) The use of fluoride containing tubes (the most accurate and preferred option).
The samples should be spun and sent to the local laboratory for analysis within one hour.
For sites where processing times exceed one hour, the Investigator should store the whole
blood samples immediately after collection on crushed ice or on a 4oC ‘cold tray’ until
analysis. The whole process must be completed as soon as possible with an absolute time
limit of 4 hours.
(B) The use of tubes with an anticoagulant other than fluoride, for example heparin.
These samples must be spun and sent to the local laboratory for analysis within one hour.
(C) The use of tubes without anticoagulant.
This last option should only be used if options A and B above are not possible. The blood
is collected in tubes without anticoagulant and must be sent to the local laboratory for
analysis within one hour. The local laboratory should use appropriate
adjustment for the difference between plasma and whole blood.
Send processed labeled samples to the local hospital laboratory for testing.
As soon as the participant’s test results are available, enter the data onto the ‘Oral Glucose
Tolerance Test’ eCRF and add a printed copy of the results to the participant’s study file for
future monitoring purposes.

Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 43 of 45
12. REFERENCES
1. The burden of mortality attributable to diabetes: realistic estimates for the year 2000.
Roglic G, Unwin N, Bennett PH, Mathers C, Tuomilehto J, Nag S, Connolly V, King H.
Diabetes Care. 2005; 28(9): 2130-5
2. Glucose tolerance and cardiovascular mortality: comparison of fasting and 2-hour
diagnostic criteria.
DECODE Study Group, the European Diabetes Epidemiology Group.
Arch Intern Med 2001; 161: 397-405
3. Glucose metabolism in patients with acute myocardial infarction and no previous
diagnosis of diabetes mellitus: a prospective study.
Norhammar A, Tenerz A, Nilsson G, Hamsten A, Efendic S, Ryden L, Malmberg K.
Lancet 2002; 359: 2140-4.
4. The prevalence of abnormal glucose regulation in patients with coronary artery disease
across Europe.
Bartnik M, Ryden L, Ferrari R, Malmberg K, Pyorala K, Simoons M, Standl E, Soler-
Soler J, Ohrvik J.
The Euro Heart Survey on diabetes and the heart. Eur Heart J. 2004; 25(21): 1880-90.
5. Postprandial hyperglycaemia: noxious effects on the vessel wall.
Hanefeld M.
Int J Clin Pract Suppl. 2002; (129): 45-50
6. International Diabetes Federation e-Atlas
7. The relationship between coronary artery disease and abnormal glucose regulation in
China: the China Heart Survey.
Hu DY, Pan CY, Yu JM.
Eur Heart J. 2006; 27(21): 2573-9.
8. Acarbose for prevention of type 2 diabetes mellitus: the STOP-NIDDM randomised
trial.
Chiasson JL, Josse RG, Gomis R, Hanefeld M, Karasik A, Laakso M.
Lancet. 2002; 359(9323): 2072-7.
9. The preventive effect of acarbose and metformin on the progression to diabetes mellitus
in the IGT population: a 3-year multicentre prospective study.
Yang W, Lin L, Qi J, Yu Z, Pei H, He G, Yang Z, Wan F, Li G, Pan X.
Chinese Journal of Endocrinology and Metabolism 2001; 17: 131-6
10. Efficacy of acarbose in Chinese subjects with impaired glucose tolerance.
Pan CY, Gao Y, Chen JW, Luo BY, Fu ZZ, Lu JM, Guo XH, Cheng H.
Diabetes Research and Clinical Practice 2003; 61: 183-90
11. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin.
Knowler WC, Barrett-Connor E, Fowler SE, Hamman RF, Lachin JM, Walker EA,
Nathan DM.
N Engl J Med. 2002; 346(6): 393-403
12. Prevention of type 2 diabetes mellitus by changes in lifestyle among subjects with
impaired glucose tolerance.

Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 44 of 45
Tuomilehto J, Lindstrom J, Eriksson JG, Valle TT, Hamalainen H, Ilanne-Parikka P,
Keinanen-Kiukaanniemi S,Laakso M, Louheranta A, Rastas M, Salminen V, Uusitupa
M.
N Engl J Med. 2001; 344(18): 1343-50.
13. Effect of rosiglitazone on the frequency of diabetes in patients with impaired glucose
tolerance or impaired fasting glucose: a randomised controlled trial.
H C Gerstein, S Yusuf, J Bosch, J Pogue, P Sheridan, N Dinccag, M Hanefeld, B
Hoogwerf, M Laakso, V Mohan, J Shaw, B Zinman, R R Holman.
Lancet 2006; 368: 1096-105
14. Risk factors for coronary artery disease in non-insulin dependent diabetes
R C Turner, H Millns, H A W Neil, I M Stratton, S E Manley, D R Matthews, R R
Holman.
BMJ 1998; 316: 823-828
15. Intensive blood glucose control with sulphonylureas or insulin compared with
conventional treatment and risk of complications in patients with type 2 diabetes.
Robert C Turner, Rury R Holman, Carole A Cull, Irene M Stratton, David R Matthews,
Valeria Frighi, Susan E Manley, Andrew Neil, Heather McElroy, David Wright, Eva
Kohner, Charles Fox, and David Hadden.
Lancet 1998; 352: 837-853
16. Tight blood pressure control and risk of macrovascular and microvascular
complications in type 2 diabetes.
Robert Turner, Rury Holman, Irene Stratton, Carole Cull, Valeria Frighi, Susan Manley,
David Matthews, Andrew Neil, Heather McElroy, Eva Kohner, Charles Fox, David
Hadden, and David Wright.
BMJ 1998; 317: 703-713
17. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536
high-risk individuals: a randomised placebo-controlled trial.
Rory Collins, Jane Armitage, Sarah Parish, Peter Sleight, Richard Peto.
Lancet. 2002; 360(9326): 7-22
18. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the
Collaborative Atorvastatin Diabetes Study (CARDS): multicentre randomised placebo-
controlled trial.
Colhoun HM, Betteridge DJ, Durrington PN, Hitman GA, Neil HA, Livingstone SJ,
Thomason MJ, Mackness MI, Charlton-Menys V, Fuller JH.
Lancet. 2004; 364(9435): 685-96
19. Acarbose treatment and the risk of cardiovascular disease and hypertension in patients
with impaired glucose tolerance: the STOP-NIDDM trial.
Chiasson JL, Josse RG, Gomis R, Hanefeld M, Karasik A, Laakso M.
JAMA. 2003; 290(4): 486-94
20. Acarbose slows progression of intima-media thickness of the carotid arteries in subjects
with impaired glucose tolerance.
Hanefeld M, Chiasson JL, Koehler C, Henkel E, Schaper F, Temelkova-Kurktschiev T.
Stroke. 2004; 35(5): 1073-8

Study Number 11232/OXTREC 51-08/Version 10 /Amendment 7/ 28th
July 2016 Page 45 of 45
21. The CONSORT statement: revised recommendations for improving the quality of
reports of parallel-group randomised trials.
Moher D, Schulz KF, Altman DG.
Lancet. 2001; 357(9263): 1191-4
22. Uniform Requirements for Manuscripts Submitted to Biomedical Journals. 2006
ICMJE Guidelines.
23. Modified glomerular filtration rate estimating equation for Chinese patients with
chronic kidney disease.
Ma YC, Zuo L, Chen JH.
J Am Soc Nephrol 2006;17:2937-44.
24. Design of Major Randomized Trials. Part 3 of a 4-Part Series on Statistics for Clinical
Trials.
Pocock SJ, Clayton TC, Stone GW.
J Am Coll Cardiol. 2015;66(24):2757-2766.