consequences of failing to comply with cgmps – an fda perspective steven lynn, ms, cmq/oe director...
TRANSCRIPT

Consequences of Failing to Comply with CGMPs –
An FDA Perspective
Steven Lynn, MS, CMQ/OESteven Lynn, MS, CMQ/OEDirector Director
Office of Manufacturing and Product Quality Office of Manufacturing and Product Quality Office of ComplianceOffice of Compliance
CDER/FDACDER/FDA
FDLI FDLI July 2013July 2013

TOPICS
• Our Common Goal: To avoid unnecessary risks to the Patient – achieved via a Strong, Robust Quality Systemachieved via a Strong, Robust Quality System
• What Can Happen? • Indicators • Inspectional Trends• Go on an Inspection with Me
2

21st Century: Industry Evolution
• An industrial paradigm shift is in progress:– Scientific risk management throughout
lifecycle, with better process understanding and continual improvement
– Quality Systems approach, e.g.• Senior management responsibility• Robust quality monitoring programs
– Proactive quality culture detects problems promptly
3

FDA and Industry Have a Common Interest in…
• Assuring consistently safe and effective drugs are available to the public
• Building quality in throughout the lifecycle and supply chain to prevent risks
• Finding science-based solutions and using contemporary technologies/approaches to address problems
• Adapting to globalization4

The Patient is the Customer • Quality should be patient-focused
– Emphasis on minimizing consumer risk to assure patients receive safe and effective medicines (see ICH Q9)
• Quality is achieved through a robust Quality System– Starts with senior management Commitment to Quality, and is
underpinned by Quality Risk Management (QRM) & Knowledge Management (KM)
– Quality is better assured when management recognizes and leads with the understanding that upstream control and prevention make good business sense
– Sustainable compliance is readily realized when a Quality Assurance (proactive) culture replaces the antiquated Quality Control (reactive) paradigm
– QRM and KM are used to identify potential failure modes, and address variability in facilities, processes, and materials throughout the lifecycle
5

6
What Can Happen?The consequences of
non-compliance

7
Why Follow CGMPS? – It’s the law – Food Drug and Cosmetic Act (FD&C):
• 501(a)(2)(b): requires conformity w/ CGMP– “A drug... shall be deemed to be adulterated if... the methods used in, or the facilities
or controls used for, its manufacture, processing, packing, or holding do not conform to or are not operated or administered in conformity with current good manufacturing practice...”
– “to assure that such drug meets the requirements of this Act as to safety and has the identity and strength, and meets the quality and purity characteristics, which it purports or is represented to possess.”
• Codified in 21 CFR 210 & 211– Not following CGMP regulations constitutes adulteration under the Act
– Scope of CGMPs• Ingredients• Finished dosage forms
– OTC, Rx products– Biologics, veterinary drugs– Drugs undergoing study (IND, e.g.)
• Manufacturers, test laboratories, packagers (including pharmacies)

8
• FDA’s Compliance Policy Guides (CPG)• Explains FDA policy on regulatory issues related to the FDA laws
or regulations. • http://www.fda.gov/ICECI/ComplianceManuals/
CompliancePolicyGuidanceManual/ucm116271.htm
• FDA’s Compliance Program Guidance Manual (CPGM)• Provide instructions to FDA personnel for conducting activities to
evaluate industry compliance with the Federal Food, Drug, and Cosmetic Act and other laws administered by FDA.
• http://www.fda.gov/ICECI/ComplianceManuals/ComplianceProgramManual/default.htm
Note: Neither the CPGs or CPGMs create or confer any rights for or on any person and does not operate to bind FDA or the public.
• An alternative approach may be used as long as the approach satisfies the requirements of the applicable statutes and regulations.
Consequences of Non-Compliance – con’t

9
• FDA’s Regulatory Procedures Manual (RPM)• http://www.fda.gov/ICECI/ComplianceManuals/
RegulatoryProceduresManual/ucm176446.htm
• Outline for all of FDA’s Actions– A reference manual for FDA personnel. – Provides FDA personnel with information on internal
procedures to be used in processing domestic and import regulatory and enforcement matters.
Note: It does not create or confer any rights for or on any person and does not operate to bind FDA or the public.
Consequences of Non-Compliance – con’t

10
Consequences of Non-Compliance – con’t– Who is Responsible for Adulterated Products?
• Everybody in a firm and across the ENTIRE SUPPLY CHAIN• Top executives of the company, operations managers, and
quality managers are examples of individuals who have fundamental roles in preventing these violations
– Commitment to Quality• Cannot settle on meeting perceived “regulator’s minimum
standard”• Must meet YOUR standards to reliably produce high quality
products• What Dr. Woodcock said….
– proactively identify & promptly correct issues– design/qualify robust operations– maintain equipment and facilities– Implement robust quality systems

11
Consequences of Non-Compliance– Form 483 Citations– Regulatory Meetings– Untitled Letters– Warning Letters– Withholds for Pending Applications– Recalls– Market Withdrawals– Consent Decrees– Seizures– Site Shutdowns due to inability to consistently produce
a quality product(s)– Firm shuts itself down – no longer in business– Criminal Investigations

12
Indicators:Field Alert Reports (FARS)

13
Field Alert Reports (FARs) = Quality Defects
21 CFR and FD&C Act basis for requirement– 21 CFR 314.81 Other Postmarketing Reports – 21 CFR 314.98 (c) Postmarketing reports– FD&C Act, Sec. 505(k)
• NDA and ANDA holders are responsible for filing FARs. • Foreign application holders are required to have a US agent registered
in the US per 21 CFR 314.50(a)(5). The US agent will report FARs.

14
What is Reported?• Application holders are required to report to the FDA any incident that
causes the distributed drug product or its labeling to be mistaken for, or applied to, another article – in other words, instances of adulteration or misbranding.
• In addition, they must also report any:• Bacteriological contamination• Significant chemical, physical or other change• Product deterioration• Out-of-specification result
• If firm cannot invalidate problem within 3 days, Field Alert must be reported
• Quality defects serve as an early signal of problems with product quality

15
The number of FARs received from a company/sponsor is usually is a good indicator of general CGMP compliance Quality defects can take many forms (products with missing labels,
sub-potent products, etc.)
However, just the number of quality defects by itself may be misleading large firms with a larger portfolio of drugs, as opposed to smaller
firms with smaller portfolios complexity of the dosage forms, for example:
transdermals vs. capsules sterile dosage forms vs. non-sterile dosage forms
FARs continued….

16
Indicators:Recall Trends

Recall Classifications: OTC and Rx
Total OTC Products Recalled by Year and Classification
15 24 52 25
141
26 61 36 27 22 5 955
213
322
1431
129
262
787
1471
100
409
23 5777 7121
10757
270
97154
3886
15 31
0
200
400
600
800
1000
1200
1400
1600
OTC Rx OTC Rx OTC Rx OTC Rx OTC Rx OTC Rx
2008 2009 2010 2011 2012 2013
Class I
Class II
Class III
17
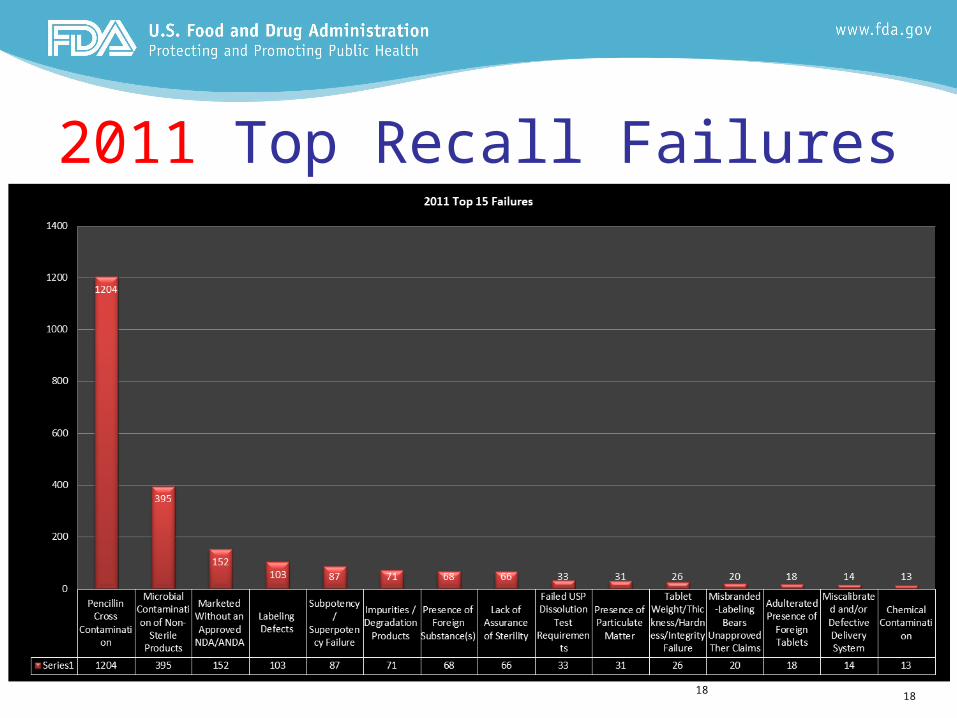
2011 Top Recall Failures
1818
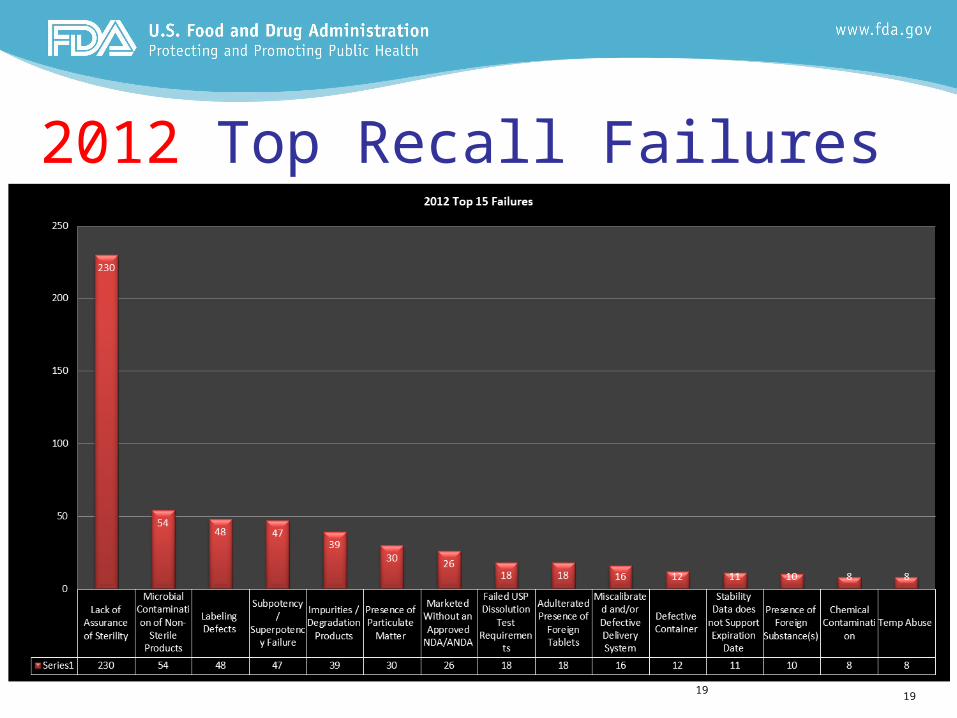
2012 Top Recall Failures
1919

20
Indicators– Inspection Trends
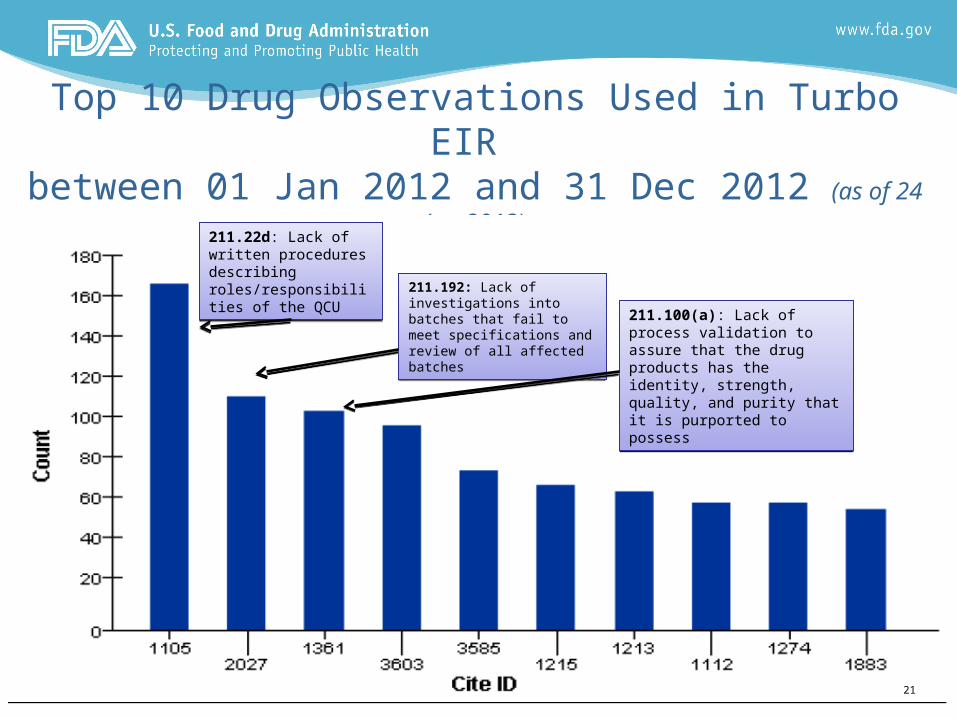
Top 10 Drug Observations Used in Turbo EIR between 01 Jan 2012 and 31 Dec 2012 (as of 24 Jan 2013)
211.22d: Lack of written procedures describing roles/responsibilities of the QCU
211.22d: Lack of written procedures describing roles/responsibilities of the QCU 211.192: Lack of investigations
into batches that fail to meet specifications and review of all affected batches
211.192: Lack of investigations into batches that fail to meet specifications and review of all affected batches
211.100(a): Lack of process validation to assure that the drug products has the identity, strength, quality, and purity that it is purported to possess
211.100(a): Lack of process validation to assure that the drug products has the identity, strength, quality, and purity that it is purported to possess
21
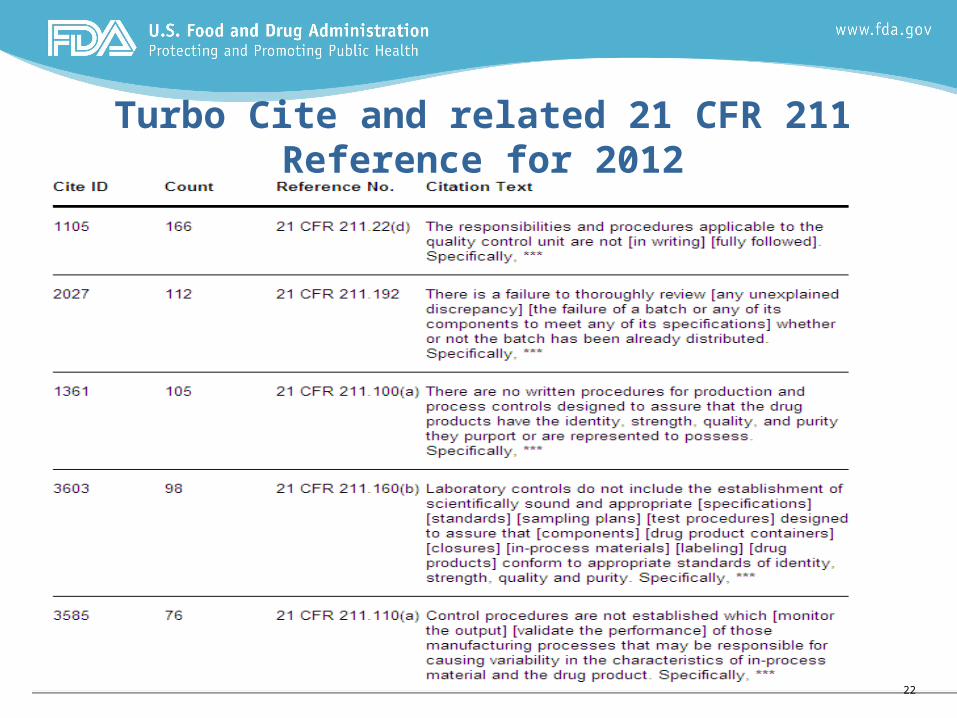
Turbo Cite and related 21 CFR 211 Reference for 2012
22

Turbo Cite and related 21 CFR 211 Reference
23

Summary of Top 10 Cites – 01 Jan to 31 Dec 2012
1. 211.22d: Lack of written procedures describing roles/responsibilities of the QCU
2. 211.192: Lack of investigations into batches that fail to meet specifications and review of all affected batches
3. 211.100(a): Lack of process validation to assure that the drug products has the identity, strength, quality, and purity that it is purported to possess (e.g., the production process is not validated for it’s intended use throughout the life cycle of the product)
4. 211.160(b): Lack of scientifically sound laboratory controls, such as specifications, standards, test methods, sampling plans, to assure that components, containers/closures, in-process materials, labeling, and drug products conform to standards of identity, purity, quality and strength
24

25
Summary of Top 10 Cites – 01 Jan to 31 Dec 2012 (cont’d.)
5. 211.110(a): Lack of in-process monitoring of CPPs that validate the performance of manufacturing processes that cause variability of the in-process materials or drug product
6. 211.67(b): Lack of written procedures for cleaning and maintenance equipment used in the manufacturing, process, packing or holding of a drug product
7. 211.67(a): Equipment and utensils not cleaned, maintained, or sanitized at appropriate intervals to prevent contamination or malfunctions, which would alter the safety, identity, quality, purity and strength of a drug product
8. 211.25(a): Lack of CGMP training and written procedures
9. 211.68(a): Routine calibration of equipment not performed according to a written procedure designed to assure proper performance
10. 211.165(a): Lack of testing drug products to assure products meet final specs and identity/strength of active ingredients prior to release
25
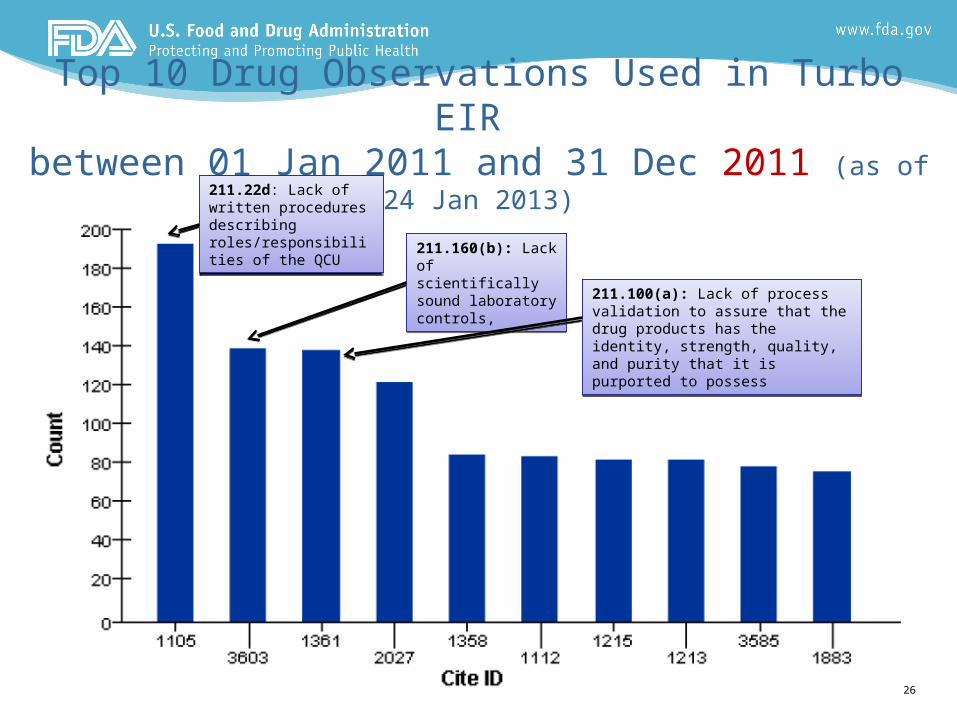
26
Top 10 Drug Observations Used in Turbo EIR between 01 Jan 2011 and 31 Dec 2011 (as of 24 Jan 2013)
211.22d: Lack of written procedures describing roles/responsibilities of the QCU
211.22d: Lack of written procedures describing roles/responsibilities of the QCU 211.160(b): Lack of
scientifically sound laboratory controls,
211.160(b): Lack of scientifically sound laboratory controls,
211.100(a): Lack of process validation to assure that the drug products has the identity, strength, quality, and purity that it is purported to possess
211.100(a): Lack of process validation to assure that the drug products has the identity, strength, quality, and purity that it is purported to possess
26
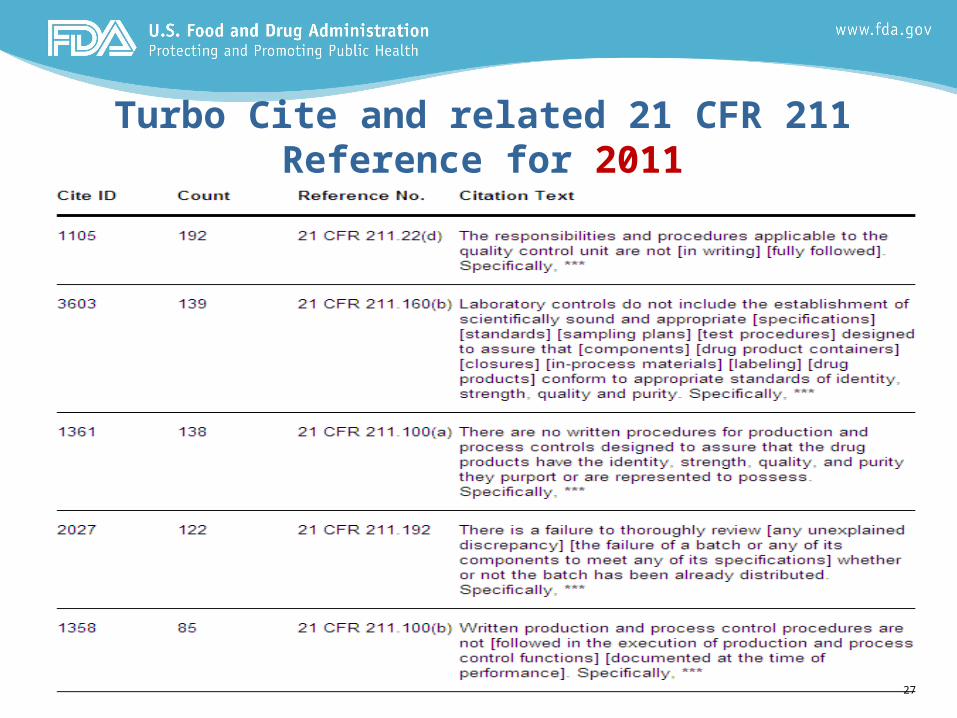
Turbo Cite and related 21 CFR 211 Reference for 2011
27
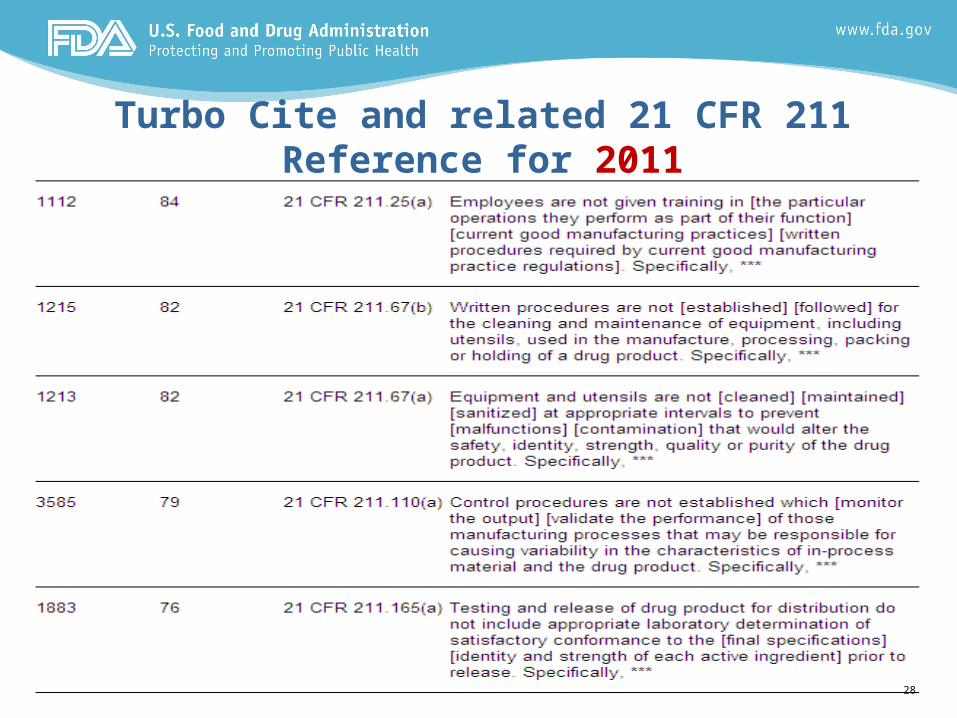
Turbo Cite and related 21 CFR 211 Reference for 2011
28

Summary of Top 10 Cites – 01 Jan to 31 Dec 20111. 211.22d: Lack of written procedures describing roles/responsibilities
of the QCU
2. 211.160(b): Lack of scientifically sound laboratory controls, such as specifications, standards, test methods, sampling plans, to assure that components, containers/closures, in-process materials, labeling, and drug products conform to standards of identity, purity, quality and strength
3. 211.100(a): Lack of process validation to assure that the drug products has the identity, strength, quality, and purity that it is purported to possess (e.g., the production process is not validated for it’s intended use throughout the life cycle of the product)
4. 211.192: Lack of investigations into batches that fail to meet specifications and review of all affected batches
5. 211.100(b): Written production procedures not followed and/or documented at the time of performance
29

Summary of Top 10 Cites – 01 Jan to 31 Dec 2011 (cont’d.)
6. 211.25(a): Lack of CGMP training and written procedures
7. 211.67(b): Lack of written procedures for cleaning and maintenance equipment used in the manufacturing, process, packing or holding of a drug product
8. 211.67(a): Equipment and utensils not cleaned, maintained, or sanitized at appropriate intervals to prevent contamination or malfunctions, which would alter the safety, identity, quality, purity and strength of a drug product
9. 211.110(a): Lack of in-process monitoring of CPPs that validate the performance of manufacturing processes that cause variability of the in-process materials or drug product
10. 211.165(a): Lack of testing drug products to assure products meet final specs and identity/strength of active ingredients prior to release
30

Trends for Top 10 Turbo Citations2011 – 2012 2008 – 2010 2004 - 2007
211.22(d) 211.192 211.160(b) 211.100(b) 211.110(a) 211.100(a) 211.67(a) 211.67(b) 211.25(a) 211.188
211.22(d) 211.110(a) 211.100(b) 211.160(b) 211.192 211.100(a) 211.165(a) 211.25(a) 211.188 211.67(b)
211.22(d) 211.100(a) 211.160(b) 211.192 211.110(a) 211.67(b) 211.67(a) 211.25(a) 211.100(b) 211.165(a)
There are MULTIPLE Repeat Citations!!!31

GMP Issues Found on International Inspections International Inspections FY 2012
CONTAMINATION1%
EQUIPMENT CLEANING/MAINTENANCE, CLEANING
VALIDATION9%
ORGANIZATION/PERSONNAL1%
PACKAGING/LABELING1%
QA SYSTEMS9%
REPROCESSING/REWORKS0%
SYSTEM QUALIFICATION (IQ/OQ)2%
UNVALIDATED LAB TEST METHODS2%
STABILITY PROGRAM4%
PRODUCTION/PROCESS CONTROLS5%
NONE (NO FD-483 ISSUED) 11%
NO PROCESS VALIDATION PROTOCOL1% LACK OF/INADEQUATE SOPS
6%INADEQUATE VALIDATION PROTOCOL
1% INADEQUATE PROCESS VALIDATION3%
INADEQUATE LAB CONTROLS12%
INADEQUATE ENVIRONMENTAL CONTROLS
4%
INADEQUATE COMPLAINT INVESTIGATIONS
4%
HOLDING/DISTRIBUTION CONTROLS0%
EQUIPMENT DESIGN, SIZE, LOCATION1%
DEVIATION FROM MONOGRAPH0%
DEVIATION FROM DMF/NDA/ANDA0%DEVIATION FROM ANTIBIOTIC
REQUIREMENTS0%
DEFICIENCIES IN MEDIA FILL1%
CONTROL OF WATER SYSTEMS2%
CONTROL COMPONENTS, INTERMEDIATES, RAW MATERIALS
6%COMPUTER VALIDATION1%
BUILDINGS/FACILITIES2%
DEFICIENCIES IN RECORDS/REPORTS8%
*Data derived from DIDQ compliance officer reviews of inspection information, for inspections occurring in FY2010-2012. Data pulled Febuary 19th, 2013 from CMS. FY 2012 may not be complete.
32

GMP Issues Found on International Inspections International Inspections FY 2011
0%0%CONTROL OF WATER SYSTEMS
2%
DEFICIENCIES IN RECORDS/REPORTS
9%ORGANIZATION/PERSONNAL
1%
PACKAGING/LABELING2%
PRODUCTION/PROCESS CONTROLS
5%
QA SYSTEMS10%
REPROCESSING/REWORKS1%STABILITY PROGRAM
3%
BUILDINGS/FACILITIES3%
COMPUTER VAL1%
CONTAMINATION1%
CONTROL COMPONENTS, INTERMEDIATES, RAW MATERIALS
5%DEFICIENCIES IN MEDIA FILLS
1% DEVIATION FR ANTIBIOTIC REQ 0%DEVIATION FROM DMF/NDA/ANDA
1%DEVIATION FROM MONOGRAPH0%
EQ DESIGN-SIZE-LOC.1%
HOLDING/DIST.CONTROLS1%
INADEQUATE COMP INV.3%
INADEQUATE ENV. CONT 4%
INADEQUATE LAB CONTROLS13%
INADEQUATE PROCESS VALIDATION
4%
INADEQUATE VALIDATION PROTOCO
1%
LACK OF/INADEQUATE SOPS7%
NO PROCESS VAL PROTOCOL1%
NONE (NO FD-483 ISSUED)10%
EQ.CLEAN/MAINT-CLEAN VAL9%
*Data derived from DIDQ compliance officer reviews of inspection information, for inspections occurring in FY2010-2012. Data pulled Febuary 19th, 2013 from CMS. FY 2012 may not be complete
33

34
Inspections

35
The Quality System: Foundation for Assuring an Ongoing State of Control
FDA Inspection Program Includes:
• Materials System• Equipment & Facilities• Production• Laboratory• Packaging & Labeling• Quality System
Investigations Operations Manual (IOM): http://www.fda.gov/ICECI/Inspections/IOM/default.htm

36
• Evaluate the firm’s Quality Control Unit (QCU)
• Evaluate production and control systems• Evaluate avenues of potential adulteration• Evaluate any potential misbranding• Evaluate SOPs and if firm follows SOPs
What Investigators dodo
36

What Investigators look forlook for?• Marketed products that do not meet USP specifications• Products or processes not validated• Changes made outside validation processes/parameters • Process validated throughout the life cycle of the product?• Product list – high risk products, products manufactured
infrequently• How a firm handles OOSs• How firm conducts investigations and follow-up. Did firm
determine the “root cause”. Are other affected lots eliminated and/or not evaluated as part of the investigation?
• Complaints - only looking at each batch or looking at batch to batch variations/trends?
• Recalls• Overall - Systems which allow inferior product to the
market!37

38
• Validation protocols and reports• Product Development Reports& Summary• Annual Product Reports• Annual Product Reviews• Field Alert Reports (FARs)• Change Controls• Deviations, non-conformances, incidents• CAPAs
What do Investigators look at/reviewlook at/review?
38

39
• Storage/manufacturing practices that may result in potential contamination
• Poor employee practices, esp. in aseptic areas• Rejected/reworked/reprocessed batches• Equipment in disrepair or improperly designed• Material and equipment identification system• Potential products for product/label mix-ups• Utilities with inappropriate plumbing connections
What do Investigators look atlook at?
39

40
• Maintenance records• Equipment usage logs• Cleaning status records• Observed practices vs. validated practices• Personnel observed vs. expected behavior (may
interview employees, but not interfere with their duties)
• Batch records in-process
What do Investigators look atlook at?
40

41
• SOPs present in areas• Calibration stickers on gauges and devices• Proper identification of materials in laboratories• Inappropriate or non-validated analytical methods• Multiple sets of data
What do Investigators look forlook for?
41

42
Systems Inspection Evaluation
• The basic principle of QA:- a drug should be produced so that it is fit for “its intended use”.
• A firm is considered out of control if any one system is out of control.
• A system is out of control if the quality, identity, strength and purity of the products resulting from that system(s) cannot be assured adequately.
• Effective process validation contributes significantly to assuring drug product quality.
42

Benefits of an EffectiveEffective Quality System
• Support and Ownership of Quality Goes Beyond the Quality/Compliance Units
• Scientific Risk Management Throughout the process Lifecycle Yields Many Benefits:– Enhanced Process Stability Drives Productivity and
Performance.– Prevention Reduces Compliance Risks and Costs.– Fewer Significant Complaints and Investigations and
Therefore More Efficient QA Release of Batches. – Protection of Your Client’s Brand– Etc., Etc., Etc.
43

Potential Risks from a Product (or Ingredient) Problem
• Corporate/business Risk
– Poor Quality– Interruption in
manufacturing or supply.– Disqualification of
supplier and need to qualify new supplier.
– Customer complaints and returned goods.
– Loss of reputation, profits, market share – hurt the brand.
– Decline in stock value.
• Patient/consumer Risk– Lessened, excessive or
inconsistent therapeutic effect.
– Side effects and transient adverse events.
– Permanent adverse effects.– Loss of confidence in safety
and quality of drug(s).– Needed drug is unavailable.
44
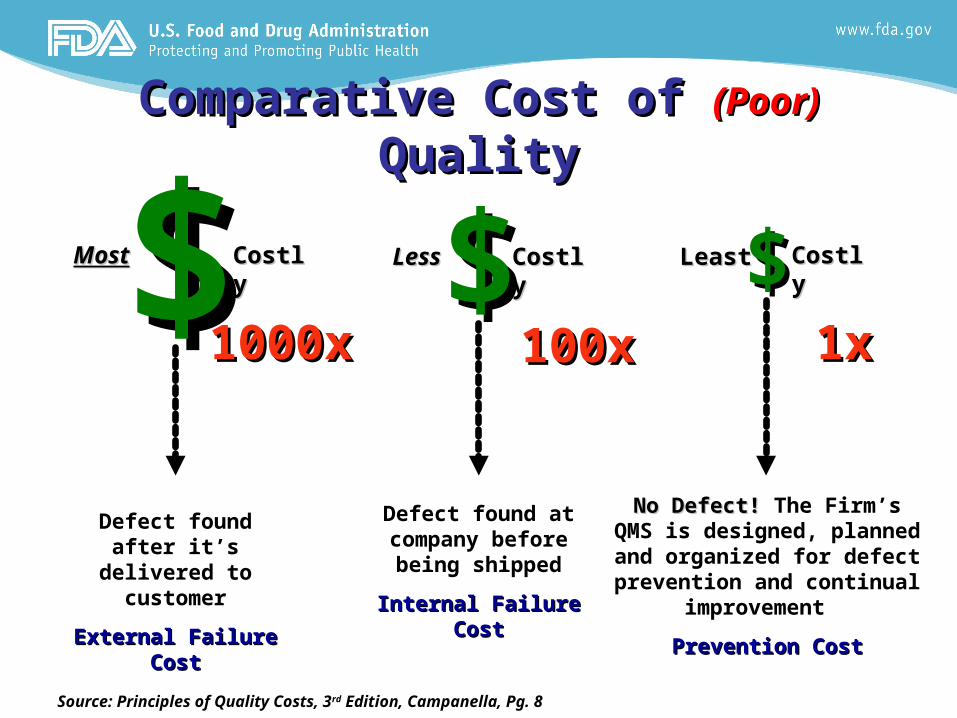
Comparative Cost of Comparative Cost of (Poor) (Poor) QualityQuality
$$ $$ $$MostMost CostlyCostly LessLess CostlyCostly LeastLeast CostlyCostly
Defect found after it’s delivered to
customer
External Failure External Failure CostCost
Defect found at company before being shipped
Internal Failure Internal Failure CostCost
No Defect! No Defect! The Firm’s QMS is designed, planned and
organized for defect prevention and continual
improvement
Prevention CostPrevention Cost
Source: Principles of Quality Costs, 3rd Edition, Campanella, Pg. 8
1x1x100x100x1000x1000x

A Quality System Creates REAL Fixes• A robust quality system is:
– Science and risk-based– At the core of Good Manufacturing Practice – Vigilant and Proactive – Culture-focused – Able to identify issues while they are still small– Responsible for assuring any contracted site is qualified to do the
function, and performed it satisfactorily– Supportive of business needs because it creates dependability and
sustainability, as well as efficiency and effectiveness!
• It should not be:– Reactive or defensive (issues should be surfaced)– Solely a procedural approach (only “plan-do”)
46

Conclusion• A strong and vigilant QS is the offense to counteract a firm
from traveling down the cascading trail of enforcement.
• Maintaining a strong and vigilant quality system throughout the product lifecycle will enable an ongoing state of control.
• An effective quality system allows distributors and manufacturers to assure that consistently high quality drugs are provided to consumers (quality throughout the supply chain)
• The intensity of FDA oversight is related to a firm’s ability to manage risk(s) associated with product quality.
47

Acknowledgments• Rick Friedman,
FDA/CDER/OC
• Arun Shakya, FDA/CDER/OC
• Monica Caphart, FDA/ORA/OO/OMPTO
• Sharon Thoma, FDA/ORA/OO/OMPTO
• Kevin Starry, FDA/ORA/OO/OEIO
48
