defective lipid remodeling of gpi anchors in peroxisomal ... · defects affecting peroxisome...
TRANSCRIPT

This article is available online at http://www.jlr.org Journal of Lipid Research Volume 53, 2012 653
Copyright © 2012 by the American Society for Biochemistry and Molecular Biology, Inc.
Supplementary key words glycosylphosphatidylinositol • plasmalo-gens • peroxisomal disorder • rhizomelic chondrodysplasia punctata
The peroxisome, a single membrane-bounded organ-elle, contains over 50 different enzymes catalyzing various metabolic pathways important for human physiology ( 1–3 ). Biosynthesis of plasmalogens and � -oxidation of very long chain fatty acids are among its essential functions. Genetic defects affecting peroxisome biogenesis or one of the enzymes localized in peroxisomes cause peroxisomal dis-orders ( 1, 3–6 ).
Zellweger syndrome (ZS), the most severe peroxisomal disorder, is characterized by very early lethality, neonatal neurodegeneration, mental retardation, seizures, en-larged liver, and characteristic facial features ( 3, 7, 8 ). In ZS, either biogenesis of the organelle membrane or im-port of the major group of enzymes bearing a type 1 per-oxisome targeting signal (PTS1) into the organelle is defective. The former defect is caused by mutation in PEX3 , PEX16 , or PEX19 , which are involved in protein incorporation into the peroxisomal membrane ( 9 ). The latter defect is caused by mutations in other PEX genes, such as PEX1 , - 5 , - 6 , - 12 , and - 26 . Among them, PEX5 is a receptor for PTS1 ( Fig. 1 ).
Abstract Many cell surface proteins in mammalian cells are anchored to the plasma membrane via glycosylphos-phatidylinositol (GPI). The predominant form of mamma-lian GPI contains 1-alkyl-2-acyl phosphatidylinositol (PI), which is generated by lipid remodeling from diacyl PI. The conversion of diacyl PI to 1-alkyl-2-acyl PI occurs in the ER at the third intermediate in the GPI biosynthetic pathway. This lipid remodeling requires the alkyl-phospholipid bio-synthetic pathway in peroxisome. Indeed, cells defective in dihydroxyacetone phosphate acyltransferase (DHAP-AT) or alkyl-DHAP synthase express only the diacyl form of GPI-anchored proteins. A defect in the alkyl-phospholipid bio-synthetic pathway causes a peroxisomal disorder, rhizomelic chondrodysplasia punctata (RCDP), and defective biogene-sis of peroxisomes causes Zellweger syndrome, both of which are lethal genetic diseases with multiple clinical phe-notypes such as psychomotor defects, mental retardation, and skeletal abnormalities. Here, we report that GPI lipid remodeling is defective in cells from patients with Zellweger syndrome having mutations in the peroxisomal biogenesis factors PEX5, PEX16, and PEX19 and in cells from patients with RCDP types 1, 2, and 3 caused by mutations in PEX7, DHAP-AT, and alkyl-DHAP synthase, respectively. Absence of the 1-alkyl-2-acyl form of GPI-anchored proteins might account for some of the complex phenotypes of these two major peroxisomal disorders . —Kanzawa, N., N. Shimozawa, R. J. A. Wanders, K. Ikeda, Y. Murakami, H. R. Waterham, S. Mukai, M. Fujita, Y. Maeda, R. Taguchi, Y. Fujiki, and T. Kinoshita. Defective lipid remodeling of GPI anchors in peroxisomal disorders, Zellweger syndrome, and rhi-zomelic chondrodysplasia punctate. J. Lipid Res . 2012. 53: 653–663.
This work was supported by grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan and by the Osaka University Global COE program.
Manuscript received 1 October 2011 and in revised form 15 January 2012.
Published, JLR Papers in Press, January 17, 2012 DOI 10.1194/jlr.M021204
Defective lipid remodeling of GPI anchors in peroxisomal disorders, Zellweger syndrome, and rhizomelic chondrodysplasia punctata
Noriyuki Kanzawa , * Nobuyuki Shimozawa , † Ronald J. A. Wanders , § Kazutaka Ikeda , ** Yoshiko Murakami , * Hans R. Waterham , § Satoru Mukai , †† Morihisa Fujita , * Yusuke Maeda , * Ryo Taguchi , ** Yukio Fujiki , †† and Taroh Kinoshita 1, * , §§
Department of Immunoregulation,* Research Institute for Microbial Diseases, Osaka University , Suita, Osaka 565-0871, Japan ; Division of Genomics Research, † Life Science Research Center, Gifu University , Gifu, Gifu 501-1193, Japan ; Academic Medical Center, § University of Amsterdam , Amsterdam 1105 AZ, The Netherlands ; Graduate School of Medicine,** University of Tokyo , Bunkyo-ku, Tokyo 113-0033, Japan ; Department of Biology, †† Faculty of Sciences, Kyushu University , Fukuoka, Fukuoka 812-8581, Japan ; WPI Immunology Frontier Research Center, §§ Osaka University , Suita, Osaka 565-0871, Japan
Abbreviations: alkyl-DHAP synthase, alkyl-dihydroxyacetone phos-phate synthase; DHAP-AT, dihydroxyacetone phosphate acyltrans-ferase; GlcN, glucosamine; Man, mannose; GlcNAc, N -acetyl glucosamine; GPI, glycosylphosphatidylinositol; GPI-AP, GPI-anchored protein; GST, glutathione S -transferase; PI, phosphatidylinositol; PTS, peroxi-some targeting signal; RCDP, rhizomelic chondrodysplasia punc-tata; uPAR, urokinase-type plasminogen activator receptor; ZS, Zellweger syndrome.
1 To whom correspondence should be addressed. email: [email protected]
by guest, on June 5, 2018w
ww
.jlr.orgD
ownloaded from

654 Journal of Lipid Research Volume 53, 2012
enzymes (such as alkaline phosphatase and 5 ′ -nucleotidase), and complement regulatory proteins (CD55 and CD59), playing roles in cell-cell and cell-environment interactions ( 13–15 ). GPI, consisting of phosphatidylinositol (PI), glu-cosamine (GlcN), three mannoses (Man), and three etha-nolamine phosphates, is synthesized from PI in the ER, and preassembled GPI is transferred to the protein’s car-boxyl terminus ( 14–16 ). In mammalian cells, the majority of protein-linked GPI contains 1-alkyl-2-acyl PI, whereas most free PIs from which GPIs are generated are in the diacyl form. The fi rst and second intermediates of GPI precursors, N -acetyl glucosaminyl (GlcNAc)-PI and GlcN-PI, are mostly diacyl forms, whereas about 60% of the third intermediate, GlcN-(inositol-acylated)-PI (GlcN-acyl-PI), contain 1-alkyl-2-acyl PI, indicating that the 1-alkyl chain is generated after the third biosynthetic step in the ER ( 17 ). We reported that generation of the 1-alkyl-2-acyl form of GPI is dependent upon the peroxisomal alkyl-phospho-lipid biosynthetic pathway ( 18 ): Two different Chinese hamster ovary (CHO) mutant cell lines, NRel-4 and NZel-1 cells, which were defective in DHAP-AT or alkyl-DHAP synthase, respectively ( 19, 20 ), did not produce a detect-able amount of the 1-alkyl-2-acyl form of GPI ( 18 ). It is conceivable that a putative alkyl-containing lipid donor is generated in the ER from alkyl-DHAP and is used in the remodeling reaction that converts the diacyl form GlcN-acyl-PI to the 1-alkyl-2-acyl form ( Fig. 1 ).
These results suggested that remodeling of the diacyl form of GPI to the 1-alkyl-2-acyl form would not occur in cells from patients with RCDP types 2 and 3. Cells from
In rhizomelic chondrodysplasia punctata (RCDP) type 1, which is characterized by lethality between 2–3 years of age, a typical facial appearance, cataracts, skeletal dyspla-sia, microcephaly, and severe psychomotor defects, biosyn-thesis of alkyl-phospholipids and plasmalogens, and degradation of phytanic acid derived from phytol in chlo-rophyl are defective, whereas peroxisome membrane bio-genesis per se and incorporation of PTS1-bearing proteins are not severely affected. RCDP type 1 is caused by muta-tion in PEX7 that is essential for transferring the minor group of enzymes bearing a PTS2, such as 3-ketoacyl-CoA thiolase of fatty-acid � -oxidation pathway, alkyl-dihy-droxyacetone phosphate synthase (alkyl-DHAP synthase), which is required for synthesis of alkyl-phospholipids, and phytanoyl-CoA 2-hydroxylase, which is required for degradation of phytanic acid ( 3 ). Specifi c defects in DHAP-acyltransferase (DHAP-AT) and alkyl-DHAP synthase (also called alkylglycerone phosphate synthase), the fi rst two enzymes in the alkyl-phospholipid biosynthetic pathway, cause disorders similar to RCDP type 1, termed RCDP type 2 and type 3, respectively ( 10–12 ), indicating that the ma-jor symptoms of RCDP are due to defective biosynthesis of plasmalogens and/or other alkyl-phospholipids, such as platelet activating factor.
More than 150 different cell surface proteins in mam-malian cells are anchored to the plasma membrane via glycosylphosphatidylinositol (GPI). GPI-anchored pro-teins (GPI-APs) include receptors (such as folate receptor and urokinase-type plasminogen activator receptor), ad-hesion molecules (such as neural cell adhesion molecule),
Fig. 1. Peroxisome-dependent lipid remodeling of GPI and biosynthesis of plasmalogens. Plasmalogens are synthesized from DHAP through multiple reaction steps. The fi rst two steps, mediated by DHAP-AT and alkyl-DHAP synthase, occur in the peroxisome; further reactions occur in the ER. Lipid remodeling of GPI, namely conversion of diacyl GPI into 1-alkyl-2-acyl GPI, occurs in the ER, requiring a putative alkyl donor lipid that is derived from 1-alkyl-DHAP. Import of DHAP-AT and alkyl-DHAP synthase into the peroxisome is dependent upon PEX5 and PEX7, respectively. Incorporation of membrane proteins into the peroxisomal membrane is dependent upon PEX3, PEX16, and PEX19. Defi ciencies of PEX3, PEX5, PEX16, or PEX19 cause ZS, whereas those of PEX7, DHAP-AT, and alkyl-DHAP synthase cause RCDP types 1, 2, and 3, respectively.
by guest, on June 5, 2018w
ww
.jlr.orgD
ownloaded from

GPI remodeling in peroxisomal disorders Q1 655
ZPG207/FLAG-ClPEX7, and ZP139 complemented with HA-tagged hamster Pex5L , termed 139/5L-HA, were kind gifts from Drs. Yasuhiro Miyauchi and Kanji Okumoto, Kyushu University, respectively. For mass analysis of GPI, we generated ZPG207 cells stably expressing His-FLAG-GST-FLAG-tagged CD59 (HFGF-CD59). The CHO mutant cells NRel-4 and NZel-1 defective in DHAP-AT and alkyl-DHAP synthase, respectively, are gifts from R. A. Zoeller (Boston University, MA) ( 19, 20 ). pcDNA3.1/Zeo(+)-DHAP-AT plasmid was electroporated into NRel-4 and NZel-1 cells under the conditions of 1000 � F/260V. After 3 days, the cells were stained by mouse IgG1 monoclonal antibody clone 5D6 against hamster uPAR ( 40 ) and phycoerythrin-conjugated goat anti-mouse IgG (BD Biosciences, Franklin Lakes, NJ), and analyzed by FACS Calibur (BD Biosciences). In Western blotting, antibodies for Calnexin and Caveolin-1 (BD Biosciences), � -Tubulin (Sigma-Aldrich, St. Louis, MO), and glyceraldehyde-phosphate dehydrogenase (GAPDH, Life Technologies, Carlsbad, CA) were used.
Mass spectrometric analysis of PI from GPI-AP and HFGF-CD59
We used approximately 1 nmol (50 � g) of HFGF-CD59 for mass analysis of the PI moiety. The PI portion was isolated from affi nity-purifi ed HFGF-CD59 by deamination with sodium nitrite as de-scribed previously ( 18 ). PI derived from HFGF-CD59 was stored at –80°C until MS analysis. In CHO cells, data are cited from refer-ence 18, and the method was described in same reference. For analysis of PI in ZPG207 cells, Nano ESI-MS/MS analysis was per-formed in negative ion mode using a 4000Q TRAP (AB Sciex, Foster City, CA) with a chip-based ionization source, TriVersa NanoMate (Advion BioSystems, Ithaca, NY). The ion spray voltage was set at –1.2 kV, the gas pressure at 0.3 psi, and the fl ow rate at 200 nl/min. The parameter settings were m/z 400–1,200 for scan range, –100 V for declustering potential, –50 to –60 volts for colli-sion energy, and Q1/Q3 “unit” resolution. Samples were dissolved in C/M (1:2) containing 5 mM ammonium formate for injection into the mass spectrometer. The molecular species of PI that were liberated from HFGF-CD59 were directly subjected to fl ow injec-tion and selectively analyzed by precursor ion scanning of the phosphoryl inositol part ( 59 ). The structure of each PI molecular species was confi rmed by MS/MS analysis of the precursor ion.
In vivo labeling of cells with 3 H-mannose and a test for the alkali resistance of GPI
Before labeling, cells (3 × 10 6 in a 60-mm dish) were cultured in medium containing 10 � M BE49385A/YW3548, a PIG-N
patients with RCDP type 1 might also be defective in this remodeling. It is also likely that cells from patients with ZS are defective in generating 1-alkyl-2-acyl GPI. In this study, we investigated these hypotheses using CHO cells defec-tive in PEX7, PEX5, or PEX19 and fi broblasts from pa-tients with RCDP types 1, 2, or 3, or ZS ( 21–23 ). Our results indicate that these cells generate only the diacyl form of GPI.
MATERIALS AND METHODS
Cells and materials Fibroblast cell lines defective in PEX7, termed Gifu R01 and
R03, were derived from two patients with RCDP type 1; those de-fective in PEX5, termed 2-07, and those defective in PEX16, termed D-01, were from patients with ZS; those defective in DHAP-AT, termed NL-#2 and #3, and those defective in alkyl-DHAP synthase, termed NL-#5 and #6, were from patients with RCDP type 2 and patients with RCDP type 3, respectively ( 24, 54 ). This study was approved by internal review committees in the Re-search Institute for Microbial Diseases of Osaka University, Gifu University School of Medicine, and University of Amsterdam. Written informed consent for specifi c research use was not ob-tained because all patients died within a few years after birth. The research use of their fi broblast cells that were taken for diagnos-tic purposes and stored frozen was approved by the internal re-view committees because these are rare lethal diseases for which investigation of the disease mechanism is important for the well-ness of patients with ZS and RCDP and their families. To gener-ate immortalized cell lines, we introduced cDNAs of human telomerase reverse transcriptase and SV40-T into fi broblast cell lines using a retrovirus vector ( 55–58 ). The immortalized cells were cultured in MEM medium with 10% FBS supplemented with 100 � g/ml of G418 and 0.5 � g/ml of puromycin to main-tain plasmids. To complement RCDP fi broblasts, human PEX7 and DHAP-AT cDNAs were transduced using a retrovirus vector with a blasticidin resistance gene, and the cells were maintained in 10 � g/ml of blasticidin (InvivoGen, San Diego, CA). The PEX mutant and complemented CHO cells have been reported previ-ously ( 21, 22, 34 ). ZPG207, ZP139, and ZP119 are defective in PEX7, PEX5, and PEX19, respectively. We used ZPG207 comple-mented with human PEX7 cDNA, termed ZPG207P7 ( 22 ), and ZP119 complemented with human PEX19 , termed 119/P19 ( 34 ). ZPG207 complemented with FLAG-tagged hamster Pex7 , termed
Fig. 2. Transfection of human PEX7 restored the localization of PTS2-bearing protein at the peroxi-some in PEX7-defective CHO cells. PTS2-GFP is dif-fuse in PEX7-defective ZPG207 cells (top panels), whereas a punctate distribution of PTS2-GFP was re-stored by transfection with human PEX7 (bottom pan-els). Cells were observed for GFP (left-hand panels) and stained with anti-catalase antibody (middle pan-els). Merged profi les are shown in the right-hand pan-els. Scale bars, 20 � m.
by guest, on June 5, 2018w
ww
.jlr.orgD
ownloaded from

656 Journal of Lipid Research Volume 53, 2012
Fig. 3. GPI lipid remodeling to generate 1-alkyl-2-acyl GPI is defective in RCDP type1 cells. A: Schematic showing the alkali-sensitive and -resistant characteristics of diacyl and 1-alkyl-2-acyl GPI intermediates, respectively. After KOH treatment, 1-alkyl-2-acyl GPI is converted to 1-alkyl-2-lyso GPI, which is retained in the lipid fraction. B: Alkali-sensitivity of GPI mannolipids derived from PEX7-defective ZPG207 cells.
by guest, on June 5, 2018w
ww
.jlr.orgD
ownloaded from

GPI remodeling in peroxisomal disorders Q1 657
by human PEX7 cDNA was confi rmed by the punctate pat-tern of PTS2-GFP staining that coincided with the staining pattern of catalase, an endogenous peroxisome enzyme ( Fig. 2 ). We then analyzed production of the 1-alkyl-2-acyl form of GPI by testing alkali sensitivity ( Fig. 3A ). Due to the sensitivity of ester linkage to KOH treatment, diacyl form GPI is lost from the organic phase, whereas 1-alkyl-2-lyso GPI generated from 1-alkyl-2-acyl from GPI remained in the organic phase and can be detected by TLC analysis. In ZPG207 cells, all spots of GPI biosynthetic intermedi-ates labeled with mannose disappeared after KOH treat-ment ( Fig. 3B , lane 4), and expression of PEX7 restored the alkali-resistant 1-alkyl-2-lyso forms of GPI intermedi-ates (Fig. 3B, lanes 7 and 8). To determine the structure of the PI moiety of protein-linked GPI, we generated ZPG207 cells stably expressing CD59, a human GPI-anchored pro-tein, bearing tandem His-, FLAG-, glutathione S -transferase (GST)-, and FLAG-tags at the N-terminus (HFGF-CD59). We affi nity-purifi ed HFGF-CD59 with glutathione-Sepharose and released the PI moiety from approximately 1 nmol of protein by treatment with sodium nitrite ( 28, 29 ) and sub-jected it to nano ESI-MS analysis ( Fig. 3C ). All PIs in HF-GF-CD59 from ZPG207 cells were the diacyl form ( Fig. 3C , left bottom). MS/MS analysis of the major species of m/z = 865.80 demonstrated 18:0 fatty acids representing stearic acid, cyclic lyso phosphatidic acid bearing 18:0 chain (18:0 cPA), and inositol-phosphate ( m/z = 241.0), indicating that it is di-stearoyl-PI (right bottom). Similarly, MS/MS analysis of a minor species of m/z = 837.70 demonstrated that it is 1-palmitoyl-2-stearoyl-PI (Fig. 3C, right top). In contrast, wild-type CHO-K1 cells contained mostly 1-alkyl-2-acyl PI ( Fig. 3C , left top) ( 18 ). Therefore, PEX7 is re-quired for the production of the 1-alkyl-2-acyl form of GPI anchors.
We next analyzed fi broblast cell lines previously estab-lished from patients with RCDP type 1. As in CHO cells, signifi cant fractions of GPI intermediates in wild-type fi -broblasts were resistant to alkali, indicating generation of 1-alkyl-2-acyl GPI ( Fig. 3D , lanes 1 and 4). Alkali-resistant GPI was greatly decreased in patients’ cells (Fig. 3D, lanes 5 and 6). After complementation with human PEX7 cDNA, the alkali-resistant GPIs were generated at levels compara-ble to those in wild-type cells (Fig. 3D, lanes 9 and 10), es-tablishing that generation of 1-alkyl-2-acyl GPI anchors is defective in RCDP type 1.
inhibitor (a gift from Banyu Pharmaceutical) for 12–16 h ( 60, 61 ). When PIG-N, which transfers ethanolamine-phosphate side-branch to the fi rst Man, is inhibited, Man-containing GPI inter-mediates lacking the side-branch are accumulated, and effi cient radio-labeling is achieved ( 60 ). Cells were then incubated in 2.5 ml of glucose-free RPMI-1640 medium (Gibco/Invitrogen) con-taining 10 � g/ml tunicamycin (Wako Pure Chemical Industries, Ltd.), 10% dialyzed FBS, 20 mM HEPES, and 100 � g/ml D-glu-cose for 1 h. After tunicamycin treatment, 40 � Ci/ml for CHO cells or 10 � Ci/ml for fi broblasts of D-[2- 3 H(N)]mannose (Amer-ican Radiolabeled Chemicals) was added, and incubation was continued for 1 h. Lipids were extracted from the cell pellet us-ing two 300- � l aliquots of water-saturated butanol (BuOH). The extracted lipids were treated with 500 � l of 0.1 N KOH in metha-nol (MeOH) or MeOH only for 1 h at 37°C. To neutralize the solutions, 50 � l of 1 M acetic acid or water was added, and the samples were evaporated to dryness using a speed-vac. The lipids were extracted using two 300- � l aliquots of water-saturated BuOH and back-washed with 200 � l of BuOH-saturated water. After evaporation, the extracted lipids were used as samples for TLC on activated high-performance TLC silica gel 60 plates (Merck) with a developing solvent system of chloroform/MeOH/H 2 O (10:10:3).
Immunofl uorescence microscopy Cells were washed with PBS, fi xed with PBS containing 4%
paraformaldehyde for 20 min at room temperature, and permea-bilized with PBS containing 1% BSA, 0.1% NaN 3 , and 0.1% Triton X-100. Cells were stained with combinations of rabbit anti-human catalase serum (1:3,000) and Alexa 594-conjugated goat secondary antibodies. For uPAR staining, cells were permeabi-lized with PBS containing 0.1% saponin instead of Triton X-100 and stained with 5 � g/ml anti-uPAR mAb (clone 5D6) and Alexa 488-conjugated goat secondary antibodies (Invitrogen). Images were acquired on a FluoView FV1000 (Olympus).
RESULTS
GPI lipid remodeling to generate 1-alkyl-2-acyl GPI is defective in PEX7 mutant CHO cells and fi broblasts from patients with RCDP type 1
PEX7 is required for transporting proteins bearing a PTS2, such as alkyl-DHAP synthase, into the peroxisome and is a causal gene for RCDP type 1 ( 24–27 ). We tested whether a defect in PEX7 affects the lipid remodeling of GPI. We fi rst tested the PEX7 -mutant CHO cell line ZPG207 and ZPG207 transfected with human PEX7 cDNA ( 22 ). Normalization of the PTS2-bearing protein distribution
Cells were metabolically labeled with D-[2- 3 H] mannose in the presence of BE49385A to accumulate late GPI intermediates. The extracted lipids were treated with 0.1 N KOH in MeOH or with MeOH alone and analyzed by TLC. H3-H7’, mannose-containing GPI intermediates; H3*-H7’*, alkaline-resistant part of H3-H7’. Lanes 1 and 3, wild-type CHO-K1; lanes 2 and 4, ZPG207; lanes 5 and 7, ZPG207 comple-mented with human PEX7; lanes 6 and 8, ZPG207 complemented with hamster Pex7; lanes 1, 2, 5, and 6, MeOH-treated cells; lanes 3, 4, 7, and 8, KOH-treated cells. C: Mass analysis of PI moieties in HFGF-CD59 from the plasma membrane of wild-type CHO-K1 and PEX7-defective ZPG207 cells. In CHO-K1 cells, 1-alkyl-2-acyl species (16:0e-18:0, 18:1e-18:0, and 18:0e-18:0) were dominant (left top, data cited from reference 18 ), whereas only diacyl species were found in ZPG207 cells (left bottom). Fatty chain compositions of PI species are indi-cated as A:B-C:D, where A and C are the number of carbons, B and D are the number of unsaturated bonds, and A:B and C:D are sn1 and sn2 chain, respectively. e indicates 1-alkyl form. Fatty acid chain compositions of the major ( m/z = 865.80) and minor ( m/z = 837.70) species of PI in ZPG207 were determined by MS/MS analysis (right bottom and right top, respectively). FA, fatty acid; cPA, cyclic lyso phosphatidic acid; Ino-P, phosphoryl inositol. The 18:1e-18:0 species seen in CHO-K1 cells might contain a plasmalogen type. The exact structure is yet to be determined. D: Alkali sensitivity of GPI mannolipids derived from RCDP type 1 fi broblasts from two patients. Lanes 1 and 4, healthy controls; lanes 2 and 5, RCDP type1 cell line R01; lanes 3 and 6, another RCDP type1 cell line R03; lanes 7 and 9, R01 complemented with PEX7; lanes 8 and 10, R03 complemented with PEX7.
by guest, on June 5, 2018w
ww
.jlr.orgD
ownloaded from

658 Journal of Lipid Research Volume 53, 2012
Fig. 4. TLC analysis of alkali sensitivity of GPI mannolipids derived from fi broblasts from patients with RCDP types 2 and 3 and their complemented counterparts. Lanes 1 and 6, fi broblasts from a healthy control; lanes 2 and 7, NL-#2 (RCDP type 2); lanes 3 and 8, NL-#3 (RCDP type 2); lanes 4 and 9, NL-#5 (RCDP type 3); lanes 5 and 10, NL-#6 (RCDP type 3); lanes 11 and 13, NL-#2 complemented with DHAP-AT cDNA; lanes 12 and 14, NL-#3 complemented with DHAP-AT cDNA; lanes 1–5, 11, and 12, MeOH-treated cells; lanes 6–10, 13, and 14, KOH-treated cells.
Fibroblasts from patients with RCDP types 2 and 3 are defective in 1-alkyl-2-acyl GPIs
We next assessed generation of alkali-resistant GPI in fi -broblast cell lines from patients with RCDP types 2 and 3. As expected from our previous results with mutant CHO cells defective in DHAP-AT and alkyl-DHAP synthase, all spots of GPI-mannolipids generated in these cells from RCDP type 2 and type 3 patients disappeared after alkali treatment ( Fig. 4 , lanes 7–10). When the responsible DHAP-AT cDNA was transfected into RCDP type 2 fi broblasts, spots of the alkali-resistant lyso-forms appeared (Fig. 4, lanes 13 and 14). It is, therefore, evident that generation of 1-alkyl-2-acyl GPI-APs is defective in RCDP types 2 and 3.
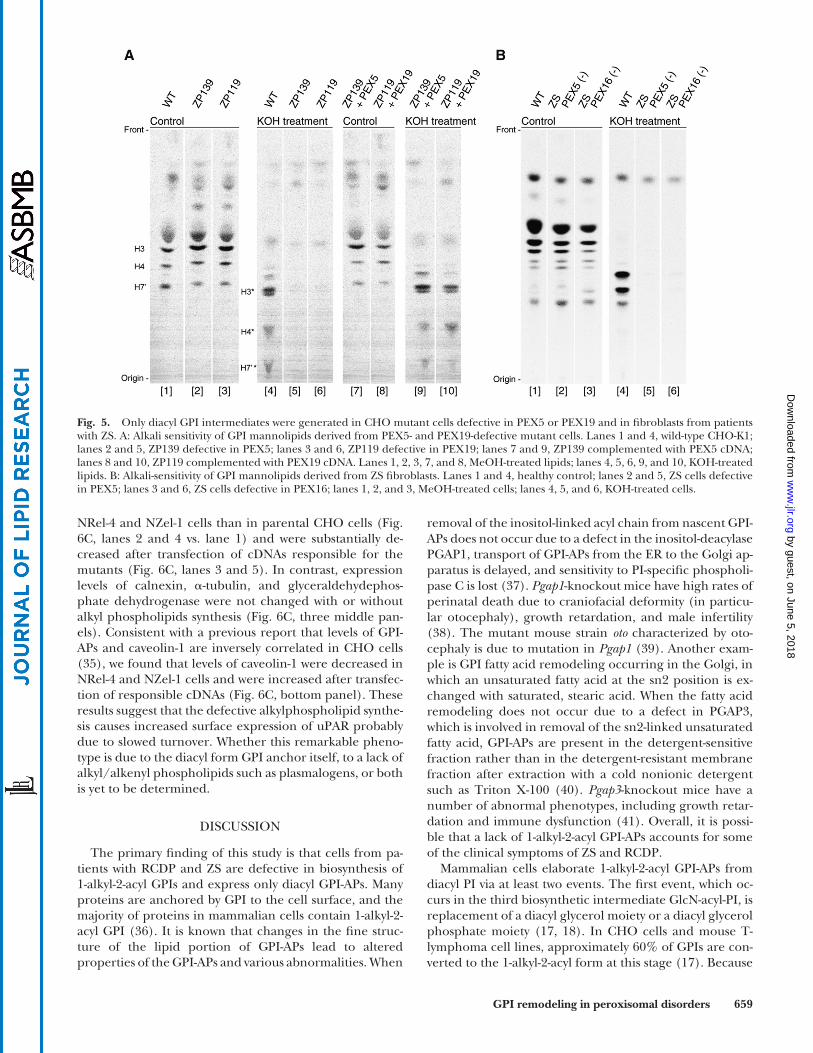
GPI lipid remodeling to generate 1-alkyl-2-acyl GPI is defective in PEX5- and PEX19-mutant CHO cells and fi broblasts from patients with ZS
PEX5 recognizes the PTS1 signal and imports many pro-teins bearing a PTS1 into the peroxisome. A defect in PEX5 causes ZS. PEX16 and PEX19 are required for incorporation of peroxisomal membrane proteins into the membrane ( 3, 30–32 ). Defects in PEX16 and PEX19 also cause ZS ( 3, 33 ). First, we analyzed PEX5-defective CHO cells ( Fig. 5A , lanes 2 and 5) and PEX19-defective CHO cells (Fig. 5A, lanes 3 and 6) ( 21, 34 ). Man-labeled GPIs generated in these mutant CHO cells disappeared after KOH treatment (Fig. 5A, lanes 5 and 6). Expression of PEX5 and PEX19 restored the produc-tion of alkali-resistant GPIs (Fig. 5A, lanes 9 and 10). Next, we tested fi broblast cell lines from patients with ZS ( Fig. 5B ). Cells from PEX5-defective (Fig. 5B, lanes 2 and 5) and PEX16-defective (Fig. 5B, lanes 3 and 6) patients did not produce alkali-resistant GPIs (Fig. 5B, lanes 3 and 6). These results in-dicate that PEX5, PEX16, and PEX19 are required for the
generation of 1-alkyl-2-acyl GPI anchors and that patients with ZS are defective in generation of 1-alkyl-2-acyl GPI-APs.
Surface expression of urokinase-type plasminogen activator receptor, a GPI-AP, signifi cantly increase when alkylphospholipid biosynthesis is defective
We next asked if a defect in peroxisome-dependent alkylphospholipid biosynthesis affects any properties of GPI-APs other than lipid remodeling to 1-alkyl-2-acyl form. We focused on urokinase-type plasminogen activator recep-tor (uPAR), an endogenous GPI-AP of CHO cells. We used NRel-4 and NRel-1 cells defective in DHAP-AT and alkyl-DHAP synthase, respectively ( 19, 20 ). The surface expression of uPAR on NRel-4 cells was 4-times of that on wild-type CHO-K1 cells and returned to 1.5-times of normal level by transfection of DHAP-AT cDNA ( Fig. 6A ). Similarly, uPAR on NZel-1 cells was 2-times of that on CHO-K1 cells and re-turned to 1.4-times of normal level by transfection of alkyl-DHAP synthase cDNA ( Fig. 6A ). To see if the increased surface expression of uPAR was due to an increase in total cellular uPAR level or to a skew in a ratio of surface to in-tracellular uPAR fractions, we used confocal fl uorescence microscopy. The total cellular level of uPAR was higher in NRel-4 and NZel-1 cells than in CHO-K1 cells and was sub-stantially decreased after transfection of DHAP-AT and alkyl-DHAP synthase cDNAs, respectively (Fig. 6B). We as-sessed the amounts of uPAR and some nonGPI-APs, such as calnexin, � -tubulin, glyceraldehydephosphate dehydro-genase, and caveolin-1, with Western blotting before and after PI-PLC treatment ( Fig. 6C ). Most of the uPAR was lost by PI-PLC treatment, indicating that most cellular uPAR was on the cell surface (Fig. 6C, top panel, lanes 1–5 vs. lanes 6–10). Levels of uPAR were clearly higher in
by guest, on June 5, 2018w
ww
.jlr.orgD
ownloaded from
GPI remodeling in peroxisomal disorders Q1 659
Fig. 5. Only diacyl GPI intermediates were generated in CHO mutant cells defective in PEX5 or PEX19 and in fi broblasts from patients with ZS. A: Alkali sensitivity of GPI mannolipids derived from PEX5- and PEX19-defective mutant cells. Lanes 1 and 4, wild-type CHO-K1; lanes 2 and 5, ZP139 defective in PEX5; lanes 3 and 6, ZP119 defective in PEX19; lanes 7 and 9, ZP139 complemented with PEX5 cDNA; lanes 8 and 10, ZP119 complemented with PEX19 cDNA. Lanes 1, 2, 3, 7, and 8, MeOH-treated lipids; lanes 4, 5, 6, 9, and 10, KOH-treated lipids. B: Alkali-sensitivity of GPI mannolipids derived from ZS fi broblasts. Lanes 1 and 4, healthy control; lanes 2 and 5, ZS cells defective in PEX5; lanes 3 and 6, ZS cells defective in PEX16; lanes 1, 2, and 3, MeOH-treated cells; lanes 4, 5, and 6, KOH-treated cells.
removal of the inositol-linked acyl chain from nascent GPI-APs does not occur due to a defect in the inositol-deacylase PGAP1, transport of GPI-APs from the ER to the Golgi ap-paratus is delayed, and sensitivity to PI-specifi c phospholi-pase C is lost ( 37 ). Pgap1 -knockout mice have high rates of perinatal death due to craniofacial deformity (in particu-lar otocephaly), growth retardation, and male infertility ( 38 ). The mutant mouse strain oto characterized by oto-cephaly is due to mutation in Pgap1 ( 39 ). Another exam-ple is GPI fatty acid remodeling occurring in the Golgi, in which an unsaturated fatty acid at the sn2 position is ex-changed with saturated, stearic acid. When the fatty acid remodeling does not occur due to a defect in PGAP3, which is involved in removal of the sn2-linked unsaturated fatty acid, GPI-APs are present in the detergent-sensitive fraction rather than in the detergent-resistant membrane fraction after extraction with a cold nonionic detergent such as Triton X-100 ( 40 ). Pgap3 -knockout mice have a number of abnormal phenotypes, including growth retar-dation and immune dysfunction ( 41 ). Overall, it is possi-ble that a lack of 1-alkyl-2-acyl GPI-APs accounts for some of the clinical symptoms of ZS and RCDP.
Mammalian cells elaborate 1-alkyl-2-acyl GPI-APs from diacyl PI via at least two events. The fi rst event, which oc-curs in the third biosynthetic intermediate GlcN-acyl-PI, is replacement of a diacyl glycerol moiety or a diacyl glycerol phosphate moiety ( 17, 18 ). In CHO cells and mouse T-lymphoma cell lines, approximately 60% of GPIs are con-verted to the 1-alkyl-2-acyl form at this stage ( 17 ). Because
NRel-4 and NZel-1 cells than in parental CHO cells (Fig. 6C, lanes 2 and 4 vs. lane 1) and were substantially de-creased after transfection of cDNAs responsible for the mutants (Fig. 6C, lanes 3 and 5). In contrast, expression levels of calnexin, � -tubulin, and glyceraldehydephos-phate dehydrogenase were not changed with or without alkyl phospholipids synthesis (Fig. 6C, three middle pan-els). Consistent with a previous report that levels of GPI-APs and caveolin-1 are inversely correlated in CHO cells ( 35 ), we found that levels of caveolin-1 were decreased in NRel-4 and NZel-1 cells and were increased after transfec-tion of responsible cDNAs (Fig. 6C, bottom panel). These results suggest that the defective alkylphospholipid synthe-sis causes increased surface expression of uPAR probably due to slowed turnover. Whether this remarkable pheno-type is due to the diacyl form GPI anchor itself, to a lack of alkyl/alkenyl phospholipids such as plasmalogens, or both is yet to be determined.
DISCUSSION
The primary fi nding of this study is that cells from pa-tients with RCDP and ZS are defective in biosynthesis of 1-alkyl-2-acyl GPIs and express only diacyl GPI-APs. Many proteins are anchored by GPI to the cell surface, and the majority of proteins in mammalian cells contain 1-alkyl-2-acyl GPI ( 36 ). It is known that changes in the fi ne struc-ture of the lipid portion of GPI-APs lead to altered properties of the GPI-APs and various abnormalities. When
by guest, on June 5, 2018w
ww
.jlr.orgD
ownloaded from

660 Journal of Lipid Research Volume 53, 2012
Fig. 6. Increased expression of uPAR in CHO cells defective in DHAP-AT or alkyl-DHAP synthase. A: FACS analysis of endogenous GPI-AP, uPAR in CHO mutant cells defective in DHAP-AT (NRel-4 cells, top), or al-kyl-DHAP synthase (NZel-1 cells, bottom) and their complemented counterparts. Mutant cells were trans-fected with DHAP-AT or alkyl-DHAP synthase cDNA stained 72 h later for uPAR and analyzed for FACS. Dark gray areas, CHO-K1 transfected with an empty vector; dotted lines, mutant cells transfected with an empty vector; solid lines, complemented counterparts; light gray areas, isotype control staining of mutant cells. B: Confocal immunofl uorescence microscopic analysis of uPAR in CHO mutant NRel-4 and NZel-1 cells. Cells were fi xed with paraformaldehyde, permeabilized with 0.1% saponin, and stained for uPAR. The uPAR stain-ing in two mutant cells was stronger (left middle and bottom) than in wild-type CHO-K1 cells (right top). The uPAR expression in mutant cells was reduced after transfection with DHAP-AT or alkyl-DHAP synthase cDNA (right middle and bottom). C: Western blotting of uPAR and nonGPI-APs in CHO mutant NRel-4 and NZel-1 cells before and after PI-PLC treatment. Cells (1 × 10 6 ) were harvested with EDTA, incubated with or without 1 U/ml of PI-PLC in 100 � l of Ham F-12 medium for 2 h at 20°C, and subjected to SDS/PAGE and Western blotting under nonreducing conditions for uPAR (top) and Caveolin-1 (bottom) and under reduc-ing conditions for Calnexin, � -Tubulin, and GAPDH (middle).
by guest, on June 5, 2018w
ww
.jlr.orgD
ownloaded from

GPI remodeling in peroxisomal disorders Q1 661
phosphatase, a GPI-AP that is expressed on the membrane of osteoblasts and that is secreted to the blood during bone formation ( 14, 46–50 ), might be responsible for the abnormal bone formation or chondrodysplasia. Delayed cerebellar development is a hallmark in RCDP and ZS pa-tients, and these patients suffer from severe neonatal neu-rodegenerative disorders. The severity is related to the importance of peroxisomes in the maturation of the cen-tral nervous system. Peroxisomes are abundant at the ter-mini of developing neurons and have been implicated in the early determination of neural polarity ( 51, 52 ). Al-though the mechanisms of neuropathology are unknown, ZS leads to loss of essential peroxisomal function and causes problems in cerebellum formation. Neural cell ad-hesion molecule, a GPI-AP, is expressed on the surface of neural cells and contributes to cell polarization ( 53 ). The abnormal diacyl form GPI might be causally related to the neurodegenerative phenotypes. Although the functional importance of the alkyl-acyl form of GPI-APs is yet to be determined, our results suggest the possibility that the ab-sence of alkyl-acyl GPI contributes to some of the symp-toms of RCDP and ZS.
The authors thank Drs. Y. Miyauchi and K. Okumoto for cells; K. Kinoshita, K. Miyanagi, and Y. Onoe for excellent technical assistance; K. Nakamura for help with cell sorting; and Drs. N. Inoue and Y. Morita for helpful discussions.
REFERENCES
1 . Wanders , R. J. , S. Ferdinandusse , P. Brites , and S. Kemp . 2010 . Peroxisomes, lipid metabolism and lipotoxicity. Biochim. Biophys. Acta . 1801 : 272 – 280 .
2 . Wanders , R. J. , and H. R. Waterham . 2006 . Peroxisomal disorders: the single peroxisomal enzyme defi ciencies. Biochim. Biophys. Acta . 1763 : 1707 – 1720 .
3 . Steinberg , S. J. , G. Dodt , G. V. Raymond , N. E. Braverman , A. B. Moser , and H. W. Moser . 2006 . Peroxisome biogenesis disorders. Biochim. Biophys. Acta . 1763 : 1733 – 1748 .
4 . Fujiki , Y. 2000 . Peroxisome biogenesis and peroxisome biogenesis disorders. FEBS Lett. 476 : 42 – 46 .
5 . Fujiki , Y. , K. Okumoto , N. Kinoshita , and K. Ghaedi . 2006 . Lessons from peroxisome-defi cient Chinese hamster ovary (CHO) cell mu-tants. Biochim. Biophys. Acta . 1763 : 1374 – 1381 .
6 . Fujiki , Y. 2011 . Peroxisome biogenesis disorders. In Encyclopedia of Life Sciences. John Wiley and Sons, Hoboken, NJ. 1–9.
7 . Shimozawa , N. , T. Tsukamoto , Y. Suzuki , T. Orii , Y. Shirayoshi , T. Mori , and Y. Fujiki . 1992 . A human gene responsible for Zellweger syndrome that affects peroxisome assembly. Science . 255 : 1132 – 1134 .
8 . Goldfi scher , S. , C. L. Moore , A. B. Johnson , A. J. Spiro , M. P. Valsamis , H. K. Wisniewski , R. H. Ritch , W. T. Norton , I. Rapin , and L. M. Gartner . 1973 . Peroxisomal and mitochondrial defects in the cerebro-hepato-renal syndrome. Science . 182 : 62 – 64 .
9 . Shimozawa , N. 2011 . Molecular and clinical fi ndings and diagnos-tic fl owchart of peroxisomal diseases. Brain Dev. 33 : 770 – 776 .
10 . Wanders , R. J. , H. Schumacher , J. Heikoop , R. B. Schutgens , and J. M. Tager . 1992 . Human dihydroxyacetonephosphate acyltrans-ferase defi ciency: a new peroxisomal disorder. J. Inherit. Metab. Dis. 15 : 389 – 391 .
11 . Wanders , R. J. , C. Dekker , V. A. Hovarth , R. B. Schutgens , J. M. Tager , P. Van Laer , and D. Lecoutere . 1994 . Human alkyldihy-droxyacetonephosphate synthase defi ciency: a new peroxisomal disorder. J. Inherit. Metab. Dis. 17 : 315 – 318 .
12 . Barr , D. G. , J. M. Kirk , M. al Howasi , R. J. Wanders , and R. B. Schutgens . 1993 . Rhizomelic chondrodysplasia punctata with iso-lated DHAP-AT defi ciency. Arch. Dis. Child. 68 : 415 – 417 .
fatty acid composition of the diacyl form of GlcN-acyl-PI was different from those of earlier GPI intermediates and PI, even the diacyl form must be subjected to replacement reaction ( 18 ). Therefore, a putative donor lipid for the remodeling presumably comprises 60% of the 1-alkyl-2-acyl form and 40% of the diacyl form ( 18 ). The exact reac-tion mechanism of this GPI lipid remodeling is yet to be elucidated. The second event occurs during the following biosynthetic step(s), in which enrichment of the 1-alkyl-2-acyl form proceeds further, leading to occupation of 80–90% of cell surface GPI-APs in CHO cells ( 18 ). A pos-sible mechanism might be selection by biosynthetic en-zymes, selective compartmentalization of the 1-alkyl-2-acyl form, or, although less likely, degradation of diacyl GPI intermediates.
To understand the functional signifi cance of 1-alkyl-2-acyl GPI-APs, the enzyme required for the GPI lipid re-modeling needs to be identifi ed and disrupted. If the gene for the putative enzyme that catalyzes diacyl to 1-alkyl-2-acyl exchange in GlcN-acyl-PI is disrupted in mice, such mice would not express 1-alkyl-2-acyl GPI-APs but would have normal plasmalogens; this should allow us to deter-mine the phenotypes that result from defective generation of 1-alkyl-2-acyl GPI-APs. Until this is achieved, informa-tion derived from the cells of patients with RCDP types 2 and 3 and CHO mutant cell lines defective in DHAP-AT and alkyl-DHAP synthase is useful. Cells from patients with RCDP types 2 and 3 are defective only in alkyl-phospho-lipid biosynthesis, resulting in defects in several end-prod-ucts, namely, plasmalogens, platelet activating factor, and 1-alkyl-2-acyl GPI-APs, whereas cells from patients with RCDP type 1 and ZS are also defective in other peroxi-somal pathways. We found that the surface expression of uPAR is 2- to 4-times elevated on mutant cells defective in DHAP-AT or alkyl-DHAP synthase ( Fig. 6 ). It was reported that these mutant CHO cells are ineffi cient in the trans-port of cholesterol for the cell surface or endocytic com-partment to the ER ( 42 ). Fibroblasts from patients with RCDP types 2 and 3 display a number of morphological and cell biological abnormalities, such as the common presence of enlarged and multipolar cells, dilated ER and Golgi cisternae, a reduced number of caveolae associated with a 50–60% reduction in caveolin 1, accumulation of cholesterol in perinuclear structures, an increased level of clathrin, and ineffi cient endocytosis of transferrin ( 43 ). Whether the elevated uPAR expression is related to these cellular abnormalities, including affected cholesterol traf-fi cking, is unclear.
Dhap-at -knockout mice display a variety of phenotypes, including male infertility, defects in eye development, cat-aract, optic nerve hypoplasia, and cerebellum anomalies including impaired Purkinje cell innervation, defective myelination, and disturbances in paranode organization ( 44, 45 ). Patients with RCDP types 2 and 3 have craniofa-cial dysmorphism, cataract, severe psychomotor retarda-tion, as well as rhizomelia and chondrodysplasia punctata ( 3, 10–12 ). It is tempting to speculate that the lack of a particular 1-alkyl-2-acyl GPI-AP is responsible for some of these abnormalities. For example, abnormal alkaline
by guest, on June 5, 2018w
ww
.jlr.orgD
ownloaded from

662 Journal of Lipid Research Volume 53, 2012
32 . Matsuzaki , T. , and Y. Fujiki . 2008 . The peroxisomal membrane pro-tein import receptor Pex3p is directly transported to peroxisomes by a novel Pex19p- and Pex16p-dependent pathway. J. Cell Biol. 183 : 1275 – 1286 .
33 . Ebberink , M. S. , J. Kofster , R. J. Wanders , and H. R. Waterham . 2010 . Spectrum of PEX6 mutations in Zellweger syndrome spec-trum patients. Hum. Mutat. 31 : E1058 – E1070 .
34 . Matsuzono , Y. , N. Kinoshita , S. Tamura , N. Shimozawa , M. Hamasaki , K. Ghaedi , R. J. Wanders , Y. Suzuki , N. Kondo , and Y. Fujiki . 1999 . Human PEX19: cDNA cloning by functional comple-mentation, mutation analysis in a patient with Zellweger syndrome, and potential role in peroxisomal membrane assembly. Proc. Natl. Acad. Sci. USA . 96 : 2116 – 2121 .
35 . Abrami , L. , M. Fivaz , T. Kobayashi , T. Kinoshita , R. G. Parton , and F. G. van der Goot . 2001 . Cross-talk between caveolae and glycosylphosphatidylinositol-rich domains. J. Biol. Chem. 276 : 30729 – 30736 .
36 . Chatterjee , S. , and S. Mayor . 2001 . The GPI-anchor and protein sorting. Cell. Mol. Life Sci. 58 : 1969 – 1987 .
37 . Tanaka , S. , Y. Maeda , Y. Tashima , and T. Kinoshita . 2004 . Inositol deacylation of glycosylphosphatidylinositol-anchored proteins is mediated by mammalian PGAP1 and yeast Bst1p. J. Biol. Chem. 279 : 14256 – 14263 .
38 . Ueda , Y. , R. Yamaguchi , M. Ikawa , M. Okabe , E. Morii , Y. Maeda , and T. Kinoshita . 2007 . PGAP1 knock-out mice show otocephaly and male infertility. J. Biol. Chem. 282 : 30373 – 30380 .
39 . Zoltewicz , J. S. , A. M. Ashique , Y. Choe , G. Lee , S. Taylor , K. Phamluong , M. Solloway , and A. S. Peterson . 2009 . Wnt signaling is regulated by endoplasmic reticulum retention. PLoS ONE . 4 : e6191 .
40 . Maeda , Y. , Y. Tashima , T. Houjou , M. Fujita , T. Yoko-o , Y. Jigami , R. Taguchi , and T. Kinoshita . 2007 . Fatty acid remodeling of GPI-anchored proteins is required for their raft association. Mol. Biol. Cell . 18 : 1497 – 1506 .
41 . Murakami , H. , Y. Wang , H. Hasuwa , Y. Maeda , T. Kinoshita , and Y. Murakami . 2011 . Enhanced response of T lymphocytes from Pgap3 knockout mouse: insight into roles of fatty acid remodeling of GPI anchored proteins. Biochem. Biophys. Res. Commun. 417 : 1235 – 1241 .
42 . Munn , N. J. , E. Arnio , D. Liu , R. A. Zoeller , and L. Liscum . 2003 . Defi ciency in ethanolamine plasmalogen leads to altered choles-terol transport. J. Lipid Res. 44 : 182 – 192 .
43 . Thai , T. P. , C. Rodemer , A. Jauch , A. Hunziker , A. Moser , K. Gorgas , and W. W. Just . 2001 . Impaired membrane traffi c in defective ether lipid biosynthesis. Hum. Mol. Genet. 10 : 127 – 136 .
44 . Rodemer , C. , T. P. Thai , B. Brugger , T. Kaercher , H. Werner , K. A. Nave , F. Wieland , K. Gorgas , and W. W. Just . 2003 . Inactivation of ether lipid biosynthesis causes male infertility, defects in eye devel-opment and optic nerve hypoplasia in mice. Hum. Mol. Genet. 12 : 1881 – 1895 .
45 . Teigler , A. , D. Komljenovic , A. Draguhn , K. Gorgas , and W. W. Just . 2009 . Defects in myelination, paranode organization and Purkinje cell innervation in the ether lipid-defi cient mouse cerebellum. Hum. Mol. Genet. 18 : 1897 – 1908 .
46 . Fedde , K. N. , C. C. Lane , and M. P. Whyte . 1988 . Alkaline phos-phatase is an ectoenzyme that acts on micromolar concentrations of natural substrates at physiologic pH in human osteosarcoma (SAOS-2) cells. Arch. Biochem. Biophys. 264 : 400 – 409 .
47 . Hooper , N. M. 1997 . Glycosyl-phosphatidylinositol anchored mem-brane enzymes. Clin. Chim. Acta . 266 : 3 – 12 .
48 . Bellows , C. G. , J. E. Aubin , and J. N. Heersche . 1991 . Initiation and progression of mineralization of bone nodules formed in vitro: the role of alkaline phosphatase and organic phosphate. Bone Miner. 14 : 27 – 40 .
49 . Barling , P. M. , D. K. Gupta , and C. E. Lim . 1999 . Involvement of phosphodiesterase I in mineralization: histochemical studies using antler from red deer (Cervus elaphus) as a model. Calcif. Tissue Int. 65 : 384 – 389 .
50 . Wennberg , C. , L. Hessle , P. Lundberg , S. Mauro , S. Narisawa , U. H. Lerner , and J. L. Millan . 2000 . Functional characterization of os-teoblasts and osteoclasts from alkaline phosphatase knockout mice. J. Bone Miner. Res. 15 : 1879 – 1888 .
51 . Ishikawa , T. , C. Kawai , M. Sano , and Y. Minatogawa . 2001 . Peroxisomes exist in growth cones and move anterogradely and ret-rogradely in neurites of PC12D cells. Exp. Cell Res. 266 : 260 – 269 .
52 . Bradke , F. , and C. G. Dotti . 1997 . Neuronal polarity: vectorial cyto-plasmic fl ow precedes axon formation. Neuron . 19 : 1175 – 1186 .
53 . Powell , S. K. , B. A. Cunningham , G. M. Edelman , and E. Rodriguez-Boulan . 1991 . Targeting of transmembrane and GPI-anchored
13 . McConville , M. J. , and A. K. Menon . 2000 . Recent developments in the cell biology and biochemistry of glycosylphosphatidylinositol lipids [review]. Mol. Membr. Biol. 17 : 1 – 16 .
14 . Ikezawa , H. 2002 . Glycosylphosphatidylinositol (GPI)-anchored proteins. Biol. Pharm. Bull. 25 : 409 – 417 .
15 . Kinoshita , T. , M. Fujita , and Y. Maeda . 2008 . Biosynthesis, remodel-ling and functions of mammalian GPI-anchored proteins: recent progress. J. Biochem. 144 : 287 – 294 .
16 . Fujita , M. , and T. Kinoshita . 2010 . Structural remodeling of GPI anchors during biosynthesis and after attachment to proteins. FEBS Lett. 584 : 1670 – 1677 .
17 . Houjou , T. , J. Hayakawa , R. Watanabe , Y. Tashima , Y. Maeda , T. Kinoshita , and R. Taguchi . 2007 . Changes in molecular species profi les of glycosylphosphatidylinositol anchor precursors in early stages of biosynthesis. J. Lipid Res. 48 : 1599 – 1606 .
18 . Kanzawa , N. , Y. Maeda , H. Ogiso , Y. Murakami , R. Taguchi , and T. Kinoshita . 2009 . Peroxisome dependency of alkyl-containing GPI-anchor biosynthesis in the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA . 106 : 17711 – 17716 .
19 . Nagan , N. , A. K. Hajra , L. K. Larkins , P. Lazarow , P. E. Purdue , W. B. Rizzo , and R. A. Zoeller . 1998 . Isolation of a Chinese hamster fi -broblast variant defective in dihydroxyacetonephosphate acyltrans-ferase activity and plasmalogen biosynthesis: use of a novel two-step selection protocol. Biochem. J. 332 : 273 – 279 .
20 . Nagan , N. , A. K. Hajra , A. K. Das , H. W. Moser , A. Moser , P. Lazarow , P. E. Purdue , and R. A. Zoeller . 1997 . A fi broblast cell line defective in alkyl-dihydroxyacetone phosphate synthase: a novel defect in plasmalogen biosynthesis. Proc. Natl. Acad. Sci. USA . 94 : 4475 – 4480 .
21 . Otera , H. , K. Okumoto , K. Tateishi , Y. Ikoma , E. Matsuda , M. Nishimura , T. Tsukamoto , T. Osumi , K. Ohashi , O. Higuchi , et al . 1998 . Peroxisome targeting signal type 1 (PTS1) receptor is involved in import of both PTS1 and PTS2: studies with PEX5-defective CHO cell mutants. Mol. Cell. Biol. 18 : 388 – 399 .
22 . Mukai , S. , K. Ghaedi , and Y. Fujiki . 2002 . Intracellular localization, function, and dysfunction of the peroxisome-targeting signal type 2 receptor, Pex7p, in mammalian cells. J. Biol. Chem. 277 : 9548 – 9561 .
23 . Kinoshita , N. , K. Ghaedi , N. Shimozawa , R. J. Wanders , Y. Matsuzono , T. Imanaka , K. Okumoto , Y. Suzuki , N. Kondo , and Y. Fujiki . 1998 . Newly identifi ed Chinese hamster ovary cell mu-tants are defective in biogenesis of peroxisomal membrane vesi-cles (Peroxisomal ghosts), representing a novel complementation group in mammals. J. Biol. Chem. 273 : 24122 – 24130 .
24 . Shimozawa , N. , Y. Suzuki , Z. Zhang , K. Miura , A. Matsumoto , M. Nagaya , S. Castillo-Taucher , and N. Kondo . 1999 . A novel nonsense mutation of the PEX7 gene in a patient with rhizomelic chon-drodysplasia punctata. J. Hum. Genet. 44 : 123 – 125 .
25 . Braverman , N. , G. Steel , C. Obie , A. Moser , H. Moser , S. J. Gould , and D. Valle . 1997 . Human PEX7 encodes the peroxisomal PTS2 receptor and is responsible for rhizomelic chondrodysplasia punc-tata. Nat. Genet. 15 : 369 – 376 .
26 . Motley , A. M. , E. H. Hettema , E. M. Hogenhout , P. Brites , A. L. ten Asbroek , F. A. Wijburg , F. Baas , H. S. Heijmans , H. F. Tabak , R. J. Wanders , et al . 1997 . Rhizomelic chondrodysplasia punctata is a peroxisomal protein targeting disease caused by a non-functional PTS2 receptor. Nat. Genet. 15 : 377 – 380 .
27 . Purdue , P. E. , J. W. Zhang , M. Skoneczny , and P. B. Lazarow . 1997 . Rhizomelic chondrodysplasia punctata is caused by defi ciency of human PEX7, a homologue of the yeast PTS2 receptor. Nat. Genet. 15 : 381 – 384 .
28 . Serrano , A. A. , S. Schenkman , N. Yoshida , A. Mehlert , J. M. Richardson , and M. A. Ferguson . 1995 . The lipid structure of the glycosylphosphatidylinositol-anchored mucin-like sialic acid accep-tors of Trypanosoma cruzi changes during parasite differentiation from epimastigotes to infective metacyclic trypomastigote forms. J. Biol. Chem. 270 : 27244 – 27253 .
29 . Fontaine , T. , T. Magnin , A. Melhert , D. Lamont , J. P. Latge , and M. A. Ferguson . 2003 . Structures of the glycosylphosphatidylinositol membrane anchors from Aspergillus fumigatus membrane pro-teins. Glycobiology . 13 : 169 – 177 .
30 . Titorenko , V. I. , and R. T. Mullen . 2006 . Peroxisome biogenesis: the peroxisomal endomembrane system and the role of the ER. J. Cell Biol. 174 : 11 – 17 .
31 . Fujiki , Y. , Y. Matsuzono , T. Matsuzaki , and M. Fransen . 2006 . Import of peroxisomal membrane proteins: the interplay of Pex3p- and Pex19p-mediated interactions. Biochim. Biophys. Acta . 1763 : 1639 – 1646 .
by guest, on June 5, 2018w
ww
.jlr.orgD
ownloaded from

GPI remodeling in peroxisomal disorders Q1 663
forms of N-CAM to opposite domains of a polarized epithelial cell. Nature . 353 : 76 – 77 .
54 . Honsho , M. , S. Tamura , N. Shimozawa , Y. Suzuki , N. Kondo , and Y. Fujiki . 1998 . Mutation in PEX16 is causal in the peroxisome-de-fi cient Zellweger syndrome of complementation group D. Am. J. Hum. Genet. 63 : 1622 – 1630 .
55 . Blasco , M. A. , H. W. Lee , M. P. Hande , E. Samper , P. M. Lansdorp , R. A. DePinho , and C. W. Greider . 1997 . Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell . 91 : 25 – 34 .
56 . Bodnar , A. G. , M. Ouellette , M. Frolkis , S. E. Holt , C. P. Chiu , G. B. Morin , C. B. Harley , J. W. Shay , S. Lichtsteiner , and W. E. Wright . 1998 . Extension of life-span by introduction of telomerase into nor-mal human cells. Science . 279 : 349 – 352 .
57 . Manfredi , J. J. , and C. Prives . 1994 . The transforming activity of simian virus 40 large tumor antigen. Biochim. Biophys. Acta . 1198 : 65 – 83 .
58 . Counter , C. M. , W. C. Hahn , W. Wei , S. D. Caddle , R. L. Beijersbergen , P. M. Lansdorp , J. M. Sedivy , and R. A. Weinberg . 1998 . Dissociation among in vitro telomerase activity, telomere maintenance, and cellular immortalization. Proc. Natl. Acad. Sci. USA . 95 : 14723 – 14728 .
59 . Taguchi , R. , T. Houjou , H. Nakanishi , T. Yamazaki , M. Ishida , M. Imagawa , and T. Shimizu . 2005 . Focused lipidomics by tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 823 : 26 – 36 .
60 . Hong , Y. , Y. Maeda , R. Watanabe , K. Ohishi , M. Mishkind , H. Riezman , and T. Kinoshita . 1999 . Pig-n, a mammalian homologue of yeast Mcd4p, is involved in transferring phosphoethanolamine to the fi rst mannose of the glycosylphosphatidylinositol. J. Biol. Chem. 274 : 35099 – 35106 .
61 . Sutterlin , C. , A. Horvath , P. Gerold , R. T. Schwarz , Y. Wang , M. Dreyfuss , and H. Riezman . 1997 . Identifi cation of a species-specifi c inhibitor of glycosylphosphatidylinositol synthesis. EMBO J. 16 : 6374 – 6383 .
by guest, on June 5, 2018w
ww
.jlr.orgD
ownloaded from