development and characterization of lipid nanoparticles ... · transporte de fármacos, absorção...
TRANSCRIPT

December 2014
Development and Characterization of Lipid
Nanoparticles prepared by Miniemulsion Technique
Clara Patrícia Andrade Lopes
Thesis to obtain the Master of Science Degree in
Biotechnology
Supervisor: Professor Luís Joaquim Pina da Fonseca
Examination Committee
Chairperson: Professor Duarte Miguel de França Teixeira dos Prazeres
Supervisor: Professor Luís Joaquim Pina da Fonseca
Member of the committee: Doctor Dragana Popovic Correia de Barros

i
Acknowledgments
I would like to start by thanking my supervisor, Professor Luis Fonseca who accepted me
and provides me all the means to developed this research thesis, sharing his knowledge with
me and guiding me through this project with optimism and patience (even through the rough
times and the dangerously close deadlines) and for always being present when needed.
I would like to express my sincere gratitude to all BEBL members, professors and
colleagues, for all opinions, questions, answers and suggestions to improve my project. To all
my lab partners, for making me feel so welcome, giving me all the scientific and moral support
and especially for contributing with the good environment which in my opinion is truly important
because provides a greater motivation and commitment of the people.
I would like to acknowledge all my friends (leirienses, escuteiros and fculianos) that in all
my life, in one way or another, have helped and support me with their truly friendship.
Finally, a deep acknowledgment goes to my family, my mother and sisters, who never
stopped to support me through all my academic years, always encouraging my success and
putting up with my bad mood and bad retorts on the endless hours of work. And last, but
certainly not the least, to my father who taught me to work hard, ever smile and never give up!

ii
Abstract
Lipid nanoparticles are a promising alternative to traditional colloidal drug carriers. The
main reason for this is related with the well-known conception that lipids promote oral drug
absorption, because they undergo the same physiological mechanisms of food lipid digestion.
Lipid nanoparticles also offer unique properties such as small size, large surface area,
increased drug loading and stability (especially for lipophilic drugs), possibility of controlled drug
release, no toxicity and high bioavailability.
In the present work a medium chain fatty acid (MCFA) SLNs and NLCs based on MCFA
and a natural oil were successfully prepared by sonication technique and employing a nonionic
surfactant. The two anti-TB drugs, rifampicin and pyrazinamide, are entrapped into obtained
lipid nanoparticles. The developed particles were characterized in terms of particle size,
polydispersity index, zeta potential, morphology and encapsulation efficiency. Mean particle size
of all formulations ranged between 69 ± 5 nm to 601 ± 100 nm, showing a suitable size for oral
administration. The zeta potential obtained was negatively enough to ensure a good physical
stability of the particles. Morphological studies by TEM showed spherical to oval SLNs and RIF-
NLCs with well-defined periphery. The rifampicin encapsulation efficiency range between 67 ±
7% and 85 ± 5%, while pyrazinamide encapsulation efficiency range between 14 ± 8% to
29±15%. In conclusion, despite being essential several further studies to ensure the efficacy of
obtained lipid nanoparticles, the results from the present work pose a strong argument for lipid
nanoparticles as a promising strategy for the oral delivery of anti-TB drugs.
Keywords: Oral Administration; Solid Lipid Nanoparticles; Medium Chain Fatty Acid; Nonionic
Surfactant; Tuberculosis

iii
Resumo
As nanopartículas lipídicas são uma promissora alternativa aos sistemas coloidais de
transporte de fármacos, absorção oral de fármacos, dado que sofrem os mesmos mecanismos
fisiológicos da digestão dos lípidos provenientes dos alimentos. As nanopartículas lipídicas
também oferecem propriedades únicas como tamanho reduzido, grande área de superfície,
aumento da capacidade de carga, estabilidade e libertação controlada do agente activo dentro
da partícula (especialmente para fármacos lipofílicos), aumento da biodisponibilidade e
reduzida toxicidade.
No presente trabalho, nanopartículas lipídicas sólidas (SLN) de um ácido gordo de
cadeia média (MCFA) e vectores lipídicos nanoestruturados (NLC) baseados na combinação
desse MCFA e óleo natural foram preparados com sucesso através da técnica de sonicação e
recorrendo a um tensioactivo não iónico. Dois fármacos anti-TB, a rifampicina e a pirazinamida,
foram internalizados dentro das partículas lipídicas. Estas foram caracterizadas em termos de
dimensão, índice de polidispersão, potencial zeta, morfologia e eficiência de encapsulação. A
dimensão média de todas as formulações variou entre 69 ± 5 nm e 601 ± 100 nm, exibindo
dimensões apropriadas para administração oral. O potencial zeta obtido foi suficientemente
negativo para garantir uma boa estabilidade física das partículas. Os estudos de morfologia
feitos através de Microscopia Electrónica de Transmissão (TEM) mostram que tanto as SLNs
como as RIF-NLC apresentam uma forma esférica ou oval e uma periferia bem definida. A
eficiência de encapsulação da rifampicina variou entre 67 ± 7% e 85 ± 5%, enquanto a da
pirazinamida variou entre 14 ± 8% e 29±15%. Concluindo, apesar de ser necessário realizar
outros estudos, os resultados apresentados neste trabalho permitem prever que as
nanopartículas lipídicas obtidas são uma alternativa promissora para o transporte de fármacos
anti-TB por via oral.
Palavras-chave: Administração Oral; Nanopartículas Lipídicas Sólidas; Ácidos Gordos de
Cadeia Média; Tensioactivo Não-Iónico, Tuberculose

iv
Table of Contents
Acknowledgments .......................................................................................................................... i
Abstract .......................................................................................................................................... ii
Resumo ......................................................................................................................................... iii
Table of Contents .......................................................................................................................... iv
List of Figures ................................................................................................................................ vi
List of Tables ................................................................................................................................ vii
List of Abbreviations .................................................................................................................... viii
Motivation and Aims of the Thesis ................................................................................................ ix
1. Introduction ............................................................................................................................ 1
1.1. Lipid Nanoparticles ............................................................................................................ 2
1.1.1. Lipid Nanoparticles as Oral Drug Delivery System ....................................................... 5
1.1.1.1. Toxicological Concerns ........................................................................................... 11
1.1.2. Preparation Techniques for Lipid Nanoparticles ......................................................... 13
1.1.2.1. High Pressure Homogenization Technique ............................................................. 13
1.1.2.1.1. Hot Homogenization Technique .......................................................................... 14
1.1.2.1.2. Cold Homogenization Technique ........................................................................ 14
1.1.2.2. Microemulsion Technique ....................................................................................... 15
1.1.2.3. Ultrasonication Technique ....................................................................................... 15
1.1.2.4. Solvent Emulsification Evaporation Technique ....................................................... 16
1.1.2.5. Double Emulsion Technique ................................................................................... 16
1.1.2.6. Solvent Emulsification Diffusion Technique ............................................................ 17
1.1.2.7. Solvent Injection Technique .................................................................................... 17
1.1.2.8. Phase Inversion Temperature Technique ............................................................... 18
1.1.2.9. Microchannel/Microfluidic Technique ...................................................................... 18
1.1.3. Characterization of Lipid Nanoparticles....................................................................... 19
1.1.3.1. Measurement of Particle Size ................................................................................. 19
1.1.3.1.1. Dynamic Light Scattering..................................................................................... 19
1.1.3.2. Measurement of Zeta Potential ............................................................................... 20
1.1.3.3. Encapsulation Efficiency.......................................................................................... 22
1.1.3.4. Morphology .............................................................................................................. 22
1.1.3.4.1. Transmission Electron Microscopy ...................................................................... 23
1.1.3.5. Lipid Crystallinity ...................................................................................................... 23
1.2. Tuberculosis – Review .................................................................................................... 24
2. Materials and Methods ........................................................................................................ 28
2.1. Materials .......................................................................................................................... 28
2.1.1. Lipids ........................................................................................................................... 28

v
2.1.2. Surfactant .................................................................................................................... 28
2.1.2.1. Hexadecane ............................................................................................................ 29
2.1.3. Bioactive Compounds ................................................................................................. 29
2.1.3.1. Rifampicin ................................................................................................................ 29
2.1.3.2. Pyrazinamide ........................................................................................................... 30
2.1.3.3. β-carotene ............................................................................................................... 30
2.1.4. Water ........................................................................................................................... 31
2.2. Methods ........................................................................................................................... 31
2.2.1. Preparation of Lipid Nanoparticles .............................................................................. 31
2.2.1.1. Lyophilisation of Lipid Nanoparticles ....................................................................... 33
2.2.2. Characterization of Lipid Nanoparticles....................................................................... 33
2.2.2.1. Particle Size ............................................................................................................. 33
2.2.2.2. Zeta Potential .......................................................................................................... 33
2.2.2.3. Particle Morphology ................................................................................................. 34
2.2.2.4. Encapsulation Efficiency.......................................................................................... 34
3. Results and Discussion ....................................................................................................... 37
3.1. Study of Fabrication Parameters ..................................................................................... 37
3.1.1. Empty SLN .................................................................................................................. 37
3.1.1.1. Influence of Temperature ........................................................................................ 41
3.1.1.2. Influence of Lipid Content ........................................................................................ 41
3.1.1.3. Influence of Sonication ............................................................................................ 42
3.1.1.4. Lyophilisation ........................................................................................................... 43
3.1.1.5. Long-Term Stability ................................................................................................. 44
3.1.2. Empty NLC .................................................................................................................. 46
3.2. Bioactive Compounds Loaded SLNs and NLCs ............................................................. 47
3.2.1. β-carotene ................................................................................................................... 48
3.2.2. Rifampicin .................................................................................................................... 49
3.2.3. Pyrazinamide ............................................................................................................... 50
4. Conclusions and Further Works .......................................................................................... 52
References .................................................................................................................................. 54
vi
List of Figures
Figure 1 – Schematic structure of solid lipid nanoparticle (a) and nanostructured lipid carrier (b).
....................................................................................................................................................... 2
Figure 2 - Models of drug incorporation in Solid Lipid Nanoparticles. From [32]. ......................... 3
Figure 3 - Models of drug incorporation in Nanostructured Lipid Carriers. From [50]. .................. 4
Figure 4 - Various mechanisms of enhancement of drug bioavailability in the presence of lipids.
....................................................................................................................................................... 7
Figure 5 - Schematic diagram of lipid nanoparticles formation in the microchannel system. From
[113] ............................................................................................................................................. 18
Figure 6 -Correlation differences between small and large particles. From [126]. ..................... 20
Figure 7 - Zeta-potential of a nanoparticle in solution. From [126]. ............................................ 21
Figure 8 – Schematic structure of M. tuberculosis cell wall and action site of first-line anti-TB
drugs. Intercalation of hydrophilic arabinogalactan and hydrophobic mycolate containing layers
creates an extremely impermeable envelope for antibiotic penetration. Small molecules and
nutrients are transported through porin channels that are deposited through these layers.
Adapted from [140]. ..................................................................................................................... 26
Figure 9- Structural formula of hexadecane ................................................................................ 29
Figure 10- Structural formula of rifampicin .................................................................................. 29
Figure 11 - Structural formula of pyrazinamide ........................................................................... 30
Figure 12 – Structural formula of β-carotene .............................................................................. 30
Figure 13 – Lipid nanoparticles production. Left: Lipid phase and aqueous phase in the warm
water bath under same conditions (temperature and agitation). Top Right: Lipid phase (on the
right) and aqueous phase (on the left) after weight. Middle Right: Addition of hot aqueous phase
in melt lipid phase. Bottom Right: Sonication of hot pre-emulsion. ............................................. 32
Figure 14 – Pyrazinamide standard solutions used in constructing of the calibration curve, after
reacting with alkaline sodium nitroprusside. Pyrazinamide concentration increases from left to
right. ............................................................................................................................................. 35
Figure 15 - Rifampicin calibration curve ...................................................................................... 35
Figure 16 - β-carotene calibration curve ..................................................................................... 36
Figure 17 - Pyrazinamide calibration curve ................................................................................. 36
Figure 18- Z-ave and PDI of empty SLNs with different content of MCFA (SLN_1 represents the
lowest lipid phase content and SLN_6 represents the highest lipid phase content), ranging from
a lowest temperature (a) to highest temperature (d). .................................................................. 39
Figure 19 - Zeta Potential of empty SLNs with different content of MCFA (SLN_1 represents the
lowest lipid phase content and SLN_6 represents the highest lipid phase content), ranging from
a lowest temperature (a) to highest temperature (d). .................................................................. 40

vii
Figure 20 - Macroscopic aspect of empty SLNs: (left) - non-sonicated sample; (right) –
sonicated sample ........................................................................................................................ 42
Figure 21 – Powder resulting of SLN_1b_II lyophilisation .......................................................... 43
Figure 22 – Particle Size Distribution by Intensity of SLN_1b_II before and after lyophilisation. 44
Figure 23 - Particle Size Distribution by Intensity of SLN_4b_I after one day and 7 months after
production. ................................................................................................................................... 45
Figure 24 – TEM image of SLN_4b_I after 7 months. ................................................................ 46
Figure 25 – Differences in macroscopic appearance of empty and loaded NLCs immediately
before and after sonication. ......................................................................................................... 47
Figure 26- TEM image of β-carotene loaded NLC. ..................................................................... 48
Figure 27 - TEM image of rifampicin loaded NLC. ...................................................................... 50
Figure 28 - TEM image of pyrazinamide loaded NLC ................................................................. 51
List of Tables
Table 1 – Examples of different Drug-Loaded Lipid Nanoparticles and their potential benefits for
application in oral administration route. ......................................................................................... 8
Table 2 - Dosage forms of TB nanomedicine. From [149]. ......................................................... 27
Table 3 - Composition % (w/w) of bioactive compounds loaded to lipid nanoparticles. ............. 31
Table 4 - Physicochemical characteristics of empty SLN at different conditions (mean ± SD, n =
6). ................................................................................................................................................ 38
Table 5 – Physicochemical characteristics of the SLN_1b_II. Values are mean ± SD, n = 3 ..... 44
Table 6 - Physicochemical characteristics of the SLN_4b_I. Values are mean ± SD, n = 3. ..... 45
Table 7 – Physicochemical characteristics of the empty NLC. Values are mean ± SD, n = 3. ... 46
Table 8 - Physicochemical characteristics of the β-carotene lipid nanoparticles. Values are
mean ± SD, n = 3. ....................................................................................................................... 48
Table 9 - Physicochemical characteristics of the rifampicin lipid nanoparticles. Values are mean
± SD, n = 3. ................................................................................................................................. 49
Table 10 - Physicochemical characteristics of the pyrazinamide lipid nanoparticles. Values are
mean ± SD, n = 3. ....................................................................................................................... 51

viii
List of Abbreviations
ATM Atomic Force Microscopy RES Reticulo-Endothelial System
CMC Critical Micellar Concentration RIF Rifampicin
CNS Central Nervous System SD Standard Deviation
DLS Dynamic Light Scattering SEM Scanning Electron Microscopy
DSC Differential Scanning Calorimetry SLN Solid Lipid Nanoparticle
EE Encapsulation Efficiency STM Scanning Tunnelling Microscopy
ETB Ethambutol TB Tuberculosis
FDA Food and Drug Administration TEM Transmission Electron Microscopy
FFEM Freeze Fracture Electron Microscopy UV Ultraviolet
GI Gastrointestinal Vis Visible
GRAS Generally Recognized as Safe w/o Water-in Oil
HHT Hot Homogenization Technique w/o/w Water-in Oil-in Water
HLB Hydrophilic-Lipophilic Balance w/w Weight/Weight
HPH High Pressure Homogenization WHO World Health Organization
INH Isoniazid XDR Extremely Drug Resistant
KatG Catalase Peroxidase Z-Ave Z-Average
LC Loading Capacity ZP Zeta Potential
MCFA Medium Chain Fatty Acid
MDR Multi-Drug Resistant
MTT (4,5-imethylthiazol-2-yl)2,5-dyphenyl-
tetrazolium bromide
NLC Nanostructured Lipid Carrier
NP Nanoparticle
o/w Oil-in Water
OSPS Optical Single Particle Sizing
PCS Photon Correlation Spectroscopy
PDI Polydispersity Index
PIT Phase Inversion Temperature
PLG Poly(lactide-co-glycolide)
PYZ Pyrazinamide
R2 Determination Coefficient

ix
Motivation and Aims of the Thesis
Tuberculosis (TB) remains one of major health problems in the world. In 2012, there were
8.7 million new cases of TB globally, with 1.4 million deaths in the same year. Treatment of TB
is typically a 6-month regimen comprising an initial intensive phase of rifampicin (RIF), isoniazid
(INH), pyrazinamide (PYZ), and ethambutol (ETB) daily for 2-months, followed by a 4-month
continuation phase of RIF and INH given 3 times a week. The complexity of the treatment and
patient non-compliance result in an incomplete or inadequate treatment and subsequently in the
emergence of multi-drug-resistance. The epidemic of drug-resistant tuberculosis has spread
quickly in some areas due to the convergence of resistant strains of M. tuberculosis in high-risk
patients (e.g., those with human immunodeficiency virus/acquired immunodeficiency syndrome)
and high-risk environments (e.g., hospitals and prisons). Moreover, the administration of anti-
TB drugs for a long period of time can lead to the incidence of unpleasant side effects, which
decreases patient adherence and, therefore, increases costs of therapy. To overcome these
drawbacks, new oral anti-TB formulations are required.
Apart from some particular situations, the oral route is the first choice for drug
administration. This preference is related with its easy access and non-invasive nature, which
improves patient compliance and, therefore, facilitates treatments. However, the poor water
solubility of several drug molecules and/or the risk of degradation throughout the
gastrointestinal tract, turn into impossible their oral administration. In this perspective, efforts
have been done in order to improve the oral bioavailability of poorly water-soluble drugs, by
means of developing new colloidal delivery carriers. Among these systems are lipid
nanoparticles, which have been showing promising results. The main reason for this is related
with the well-known conception that lipids promote oral drug absorption, because they undergo
the same physiological mechanisms of food lipid digestion. Lipid nanoparticles also offer unique
properties such as small size, large surface area, increased drug loading and stability
(especially for lipophilic drugs), possibility of controlled drug release, no toxicity and high
bioavailability.
Therefore, the first objective of the present work was to perform the production,
characterization and study of a medium chain fatty acid (MCFA) SLNs and MCFA/natural oil
NCLs prepared by sonication and employing a nonionic surfactant.
The second objective of the project was to study the obtained SLNs and NCLs as an
alternative system to improve the oral delivery of two anti-TB drugs, rifampicin and
pyrazinamide. Furthermore, β-carotene was also used as lipophilic model. Its delivery has a
great potential for food and cosmetic industry.

1
1. Introduction
Nanotechnology continues to play a growing and tremendous interest, both on academic
and industrial aspects. Such interest relies on the fact that it is now possible to manipulate
nanometer-length atoms and molecules in order to create, according to a bottom-up technology,
larger structures with outstanding properties. The conceptual underpinnings of
nanotechnologies were first laid out in 1959 by the physicist Richard Feynman in his lecture,
“There's plenty of room at the bottom” [1]. Feynman explored the possibility of manipulating
material at the scale of individual atoms and molecules, imagining the whole of the
Encyclopedia Britannica written on the head of a pin and foreseeing the increasing ability to
examine and control matter at the nanoscale. However, the term 'nanotechnology' was not
created until 1974, when Professor Norio Taniguchi of Tokyo Science University used it to refer
to the ability to engineer materials precisely at the nanometer level [2].
In the last decade, drug delivery research is clearly moving from the micro to the
nanometer scale. In simple terms, a drug delivery system is defined as a formulation or a device
that enables the introduction of a therapeutic substance in the body and improves its efficacy
and safety by controlling the rate, time, and place of release of this substance in the body. The
process of drug delivery includes the administration of the therapeutic product, the release of
the active ingredients by the product, and the subsequent transport of the active ingredients
across the biological membranes to the site of action [3].
The new technologies employed in drug discovery lead to find many new powerful
substances. However, the development of new drugs alone is not sufficient to ensure progress
in drug therapy. Poor water solubility and insufficient bioavailability of the new drug molecules
are main and common problems [4]. Therefore, there is an increasing need to develop a drug
carrier system that overcomes these drawbacks. This carrier system should have no toxicity
(acute and chronic), have a sufficient drug loading capacity and the possibility of drug targeting
and controlled release characteristics. It should also provide chemical and physical stability for
the incorporated drug. The feasibility of production scaling up with reasonable overall costs
should also be required [5,6].
Colloidal drug delivery systems, particularly those in the nanosize range, have been
increasingly investigated in the last years because they can fulfil the requirements mentioned
above[7–9]. Size reduction is one of the methods to increase the solubility and hence the
bioavailability of poorly-water soluble actives. Examples of such colloidal carriers are liposomes
[10], nanoemulsions [11], micelles [12–15], polymeric nanoparticles[16–18], lipid nanoparticles
[19,20], dendrimers [21–24] and drug nanocrystals [25–27]. Corresponding to the broad
diversity of colloidal carriers, there are many possible administration routes: dermal [28–31], oral
[32–36], parenteral [37,38], ocular [39], pulmonary [40–43] and intravenous [34,44]. According
to the aim of this thesis, the work will only focus along of the text on lipid nanoparticles. The
interested reader can find in the previous references high-quality reviews regarding the other
carriers.

2
1.1. Lipid Nanoparticles
Lipid nanoparticles were invented in the beginning of the nineties by two different
researchers and co-workers, regarding their different production methods: the R.H. Müller and
J.S. Lucks from Germany [45], and M.R. Gasco from Italy [46]. Nowadays, despite the number
of academic groups interested in the study of these systems have been increased, R.H. Müller
and co-workers are still the leaders of this research area [20].
In general, there are two types of lipid nanoparticles with a solid matrix, the solid lipid
nanoparticles (SLNs) and the nanostructured lipid carriers (NLCs), which differ in their inner lipid
structure (Figure 1).
Figure 1 – Schematic structure of solid lipid nanoparticle (a) and nanostructured lipid carrier (b).
SLNs consist of a solid lipid matrix that is solid at both room and body temperatures and
that are prepared in a similar manner to an oil-in-water (o/w) emulsion, except that the oil phase
of the emulsion is replaced by a solid lipid or a blend of solid lipids at room temperature [10].
Consequently, SLNs may be composed of a lipid or a mixture of lipids dispersed in an aqueous
phase at high temperature and if necessary stabilized with a surfactant or combination of
surfactants and then solidify at room temperature [10]. Particle size of SLNs range between 50-
1000 nm [10].
The incorporation of drugs in SLNs can be described by three different models: the
homogenous matrix model (in which drug is either molecularly dispersed or present as
amorphous clusters in the lipid matrix); the drug-enriched shell model (outer lipid shell
containing drug with lipid core) and the drug-enriched core model (drug core surrounded by lipid
layer or reservoir type system) [10,32]. The three different models are depicted in Figure 2.

3
Figure 2 - Models of drug incorporation in Solid Lipid Nanoparticles. From [32].
The morphological differences between those models depend on the properties of the
drug, on the matrix composition (in terms of lipids and surfactants), as well as on the method
selected for their production. In many cases, a mixture of the three types of systems is obtained
and, depending on the partition coefficient of the drug between the lipid and the aqueous
phases, the drug molecules can also be adsorbed onto the surface of the systems being
therefore not physicochemically protected by the matrix. The homogenous matrix model is
assumed for entrapped drugs that can show prolonged release from SLNs. The drug-enriched
shell model is obtained when phase separation occurs during the cooling process from the
liquid oil droplet to the formation of SLNs. The lipid precipitates first, forming an almost drug-free
lipid core. At the same time, the concentration of drug in the remaining liquid lipid increases
continuously. Finally, the compound-enriched shell crystallizes. The drug-enriched core model
is formed when the opposite mechanism as described for the former model occurs. In this case,
the drug precipitates first and the lipid shell formed around this core will have distinctly less
drug. This leads to a membrane-controlled release governed by Fick’s law of diffusion. This
model is formed when the drug concentration is close to its saturation solubility in the melted
lipid [10,32].
SLNs combine the advantages of early controlled drug delivery systems while minimise
their shortcomings. In general, a solid core offers many advantages in comparison to a liquid
core. Emulsions and liposomes usually show lack of protection of encapsulated drugs and drug
release as a burst (emulsions) or noncontrolled (from liposomes). SLNs allow for a controlled
release effect, whilst protect the drug against degradations, have good long term stability and
higher drug loading capacity. Polymeric nanoparticles also possess a solid matrix identical to
SLNs. However, SLNs can be manufactured using physiologically acceptable and
biodegradable lipid materials that have a GRAS (Generally Recognized as Safe) status and
require the use of lower amounts of organic solvents during production, which decreases the
toxicity [28]. Finally, and by contrast with liposomes and polymeric nanoparticles, SLNs have
low cost excipients and production techniques and are easily transferred to large scale.
Nonetheless, certain drawbacks have been associated with the use of SLNs such as:
SLNs dispersions contain high amount of water, drug loading capacity of SLNs are limited due

4
to crystalline structure of solid lipid, expulsion of encapsulated drug may take place during
storage due to formation of a perfect crystalline lattice especially when SLNs are prepared from
high purified lipid, drug release profile may change with storage time, polymorphic transitions
are possible, particle growth is also possible during the storage, and gelation of the dispersion
may take place during storage [47].
SLNs prepared from one highly purified lipid can crystallize in a perfect crystalline lattice
that allows very small space for the incorporation of drugs (Figure 1a). Lipids crystallize in high
energetic lipid modifications, α and β′, immediately after preparation of SLN. However, the lipid
molecules undergo a time-dependent restructuring process leading to formation of the low-
energetic modifications, βi and β, during storage. Formation of this perfect lipid crystalline
structure leads to expulsion of drug. Therefore, despite SLNs being interesting delivery
systems, relatively low drug loading capacity and potential expulsion of the drug during storage
led scientists to think about new strategies.
Consequently, a second generation of lipid nanocarriers, referred to as nanostructured
lipid carriers (NLCs) were developed at the turn of the 21st century [48]. NLCs matrices consist
of a less ordered lipid matrix with imperfections due to the mixtures of solid and liquid lipids
(Figure 1b) [38].
There are three types of NLC: the imperfect type, the multiple type, and the amorphous
type (Figure 3). The imperfect type is achieved by mixing solid lipids with small amounts of
liquid lipids. If higher amounts of oil are mixed with the solid lipid, a different type of
nanostructure is present. Here, the solubility of the oil molecules in the solid lipid is exceeded;
this leads to phase separation and the formation of oily nanocompartments within the solid lipid
matrix [48,49]. Many drugs show a higher solubility in oils than in solid lipids so that they can be
dissolved in the oil and still be protected from degradation by the surrounding solid lipids. This
type of NLC is called the multiple type, and can be regarded as an analogue to w/o/w emulsions
since it is an oil-in-solid lipid-in-water dispersion. Since drug expulsion is caused by continuing
crystallization or transformation of the solid lipid, this can be minimized by the formation of a
third type, the amorphous type. Here, the particles are solid but crystallization upon cooling is
avoided by mixing special lipids (e.g., hydroxyoctacosanylhydroxy-stearate and
isopropylmyristate) [48,49].
Figure 3 - Models of drug incorporation in Nanostructured Lipid Carriers. From [50].

5
In fact, the different NLC lead to the possibility of incorporating a high amount of drug
inside these systems, and moreover, control of drug release can still be observed because NLC
matrix is still solid. As a result, NLCs have several advantages such as: NLCs dispersions with
higher solid content can be produced, drug loading capacity is better than SLNs, drug release
profile can be easily modulated, drug leakage during storage is lower than SLNs, and
production of final dosage forms (e.g., tablets, capsules) is possible.
1.1.1. Lipid Nanoparticles as Oral Drug Delivery System
Oral route is the most preferred route for drug administration due to greater convenience,
less pain, high patient compliance, reduced risk of cross-infection, and needle stick injuries.
Major portion of the drug delivery market is occupied by oral drug delivery systems. However,
oral drug delivery is continuously looking for new strategies due to factors such as low drug
solubility, poor gastrointestinal (GI) absorption, rapid metabolism, high fluctuation in the drug
plasma level, and variability due to food effects. In addition, the acidic environment and the
presence of several enzymes in distinct parts of the GI tract increase the risk of occurrence drug
degradation [51]. These factors may cause disappointing in vivo results leading to failure of the
conventional delivery systems [52].
Intake of a high-fat meal leads to prolongation of GI tract residence time, stimulation of
biliary and pancreatic secretions, stimulation of lymphatic transport, enhancement of intestinal
wall permeability, reduction of metabolism and efflux activity, and alteration in mesenteric and
liver blood flow, which significantly contribute to improve oral bioavailability of drugs [53,54].
Therefore, lipid-based delivery systems may reduce the inherent limitations of slow and
incomplete dissolution of poorly soluble drugs and facilitate the formation of solubilised phase
from which absorption may occur [55,56].
A normal healthy adult GI tract is able to daily hydrolyse about 100-140 g of dietary lipids
(mainly in the form of triglycerides). Despite the exact body mechanisms for lipid processing
remains unclear, the procedure can be divided in digestive, absorption and systemic blood
uptake phases. Lipids digestion generally begins in the stomach where triglycerides are
hydrolysed to diglycerides and fatty acids by the acid-stable lipases such as lingual lipase and
gastric lipase. Acid lipases have a greater affinity for medium chain triglycerides when
compared with long chain triglycerides and do not hydrolyse phospholipids or cholesterol esters.
Acid lipases are also inhibited by long chain fatty acid digestion products, (which are mostly
protonated at gastric pH), and therefore digestion via acid lipases accounts for only
approximately 10 to 30% of the overall hydrolysis of ingested triglycerides in food. Afterwards,
gastric contents reach the duodenum, the first section of small intestine, where the presence of
lipids stimulates both productions of lipase/co-lipase enzymes by pancreas and bile salts
(phospholipids and cholesterol) by the gall bladder. Bile salts adhere to the surface of emulsion
droplets promoting the lipase/co-lipase action and originating free fatty acids and colloidal

6
species like micelles, mixed micelles, vesicles. Micelles are composed by surfactant molecules
which self-assemble in aqueous solution above a determinate concentration, the so-called
critical micellar concentration (CMC). Mixed micelles are similar to micelles but are composed of
various surfactants molecules. Vesicles are formed by the self-assembling of insoluble
phospholipids. Absorption occurs mostly in the small intestine where occurs the passage of
substances directly to systemic circulation, or firstly to lymphatic circulation and subsequently to
blood [57,58].
Concerning the typical lipid nanoparticles triglyceride-based composition, is expected that
after oral administration, they undergo similar mechanisms of food-ingested lipids. The size of
nanoparticle is an important factor for uptake into the epithelial of GI. Intestinal cells cannot
absorb nanoparticles larger than 400 nm [59]. Furthermore, lipid nanoparticles have adhesive
properties, which permit their adherence to the enterocytes (intestinal epithelial cells) surface.
Therefore, the drug release from the nanoparticles is immediately followed by direct absorption
within the enterocytes. In parallel, the presence of lipid nanoparticles in the duodenum promotes
both lipase/co- lipase activities and bile salts secretion. The former hydrolyse the triglycerides in
monoglycerides and fatty acids forming micelles, which undertake (i.e. re-solubilise) the drug
meanwhile it is released during the degradation of the nanoparticles. Additionally, the bile salts
interact with micelles and form mixed micelles. Subsequently, drug is absorbed together with
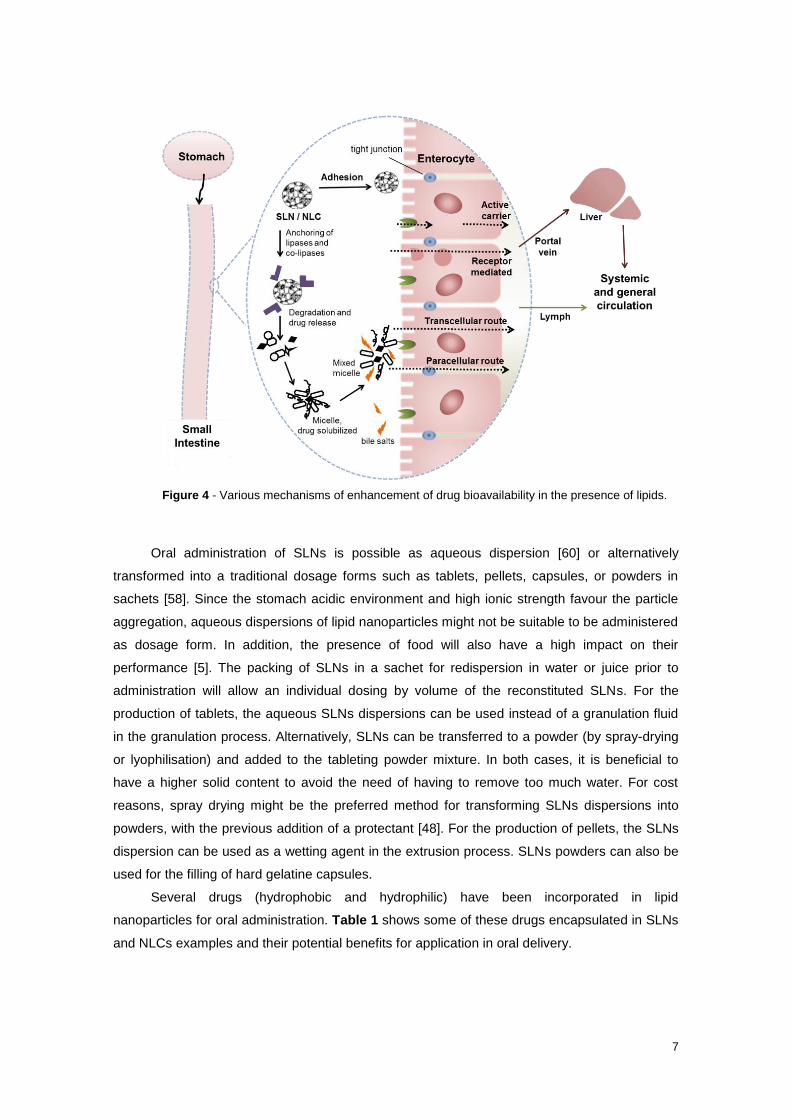
these colloidal species, by one or more of the following transport mechanisms (Figure 4):
transcellular, which is elected by small lipophilic molecules and involve the passive passage
throughout the intestinal epithelial cells; paracellular implies the passive passage through the
aqueous pores existing between enterocytes (tight junctions) and, therefore, is the selected
path by hydrophilic drug molecules; active carrier mediated require the association of drug
molecules with a specific transporter or carrier, which undertake the passage through
enterocytes; receptor mediated comprise cell internalization of drug molecules by means of
processes like endocytosis, phagocytosis and pinocytosis.
After crossing intestinal epithelia, the most part of drug molecules pass directly to the
hepatic portal vein, which carries them to the liver and afterwards to the systemic circulation.
This process constitutes a problem when drugs undertake first-pass metabolism, since they can
be inactivated before reach their local of action. Nonetheless, highly lipophilic drugs (log P > 5)
tend to entry mostly by lymphatic circulation, when comparing to portal circulation, which avoids
the risk of occurring liver first-pass metabolism. Hence, lipid can augment lymphatic uptake of
several drugs, especially lipophilic drugs or large molecular weight macromolecules.
Furthermore, lymphatic capillaries are significantly more permeable to nanoparticles than the
blood capillaries.
However, lymphatic absorption depends on the length of the fatty acid chains. Small- and
medium-chain lipids across the enterocytes by diffusion and enter directly to systemic
circulation, while long-chain lipids pass first to endoplasmic reticulum where they are re-
esterified into triglycerides and associated with lipoproteins. Following they pass to lymphatic
circulation and finally enter into systemic circulation.
7
Figure 4 - Various mechanisms of enhancement of drug bioavailability in the presence of lipids.
Oral administration of SLNs is possible as aqueous dispersion [60] or alternatively
transformed into a traditional dosage forms such as tablets, pellets, capsules, or powders in
sachets [58]. Since the stomach acidic environment and high ionic strength favour the particle
aggregation, aqueous dispersions of lipid nanoparticles might not be suitable to be administered
as dosage form. In addition, the presence of food will also have a high impact on their
performance [5]. The packing of SLNs in a sachet for redispersion in water or juice prior to
administration will allow an individual dosing by volume of the reconstituted SLNs. For the
production of tablets, the aqueous SLNs dispersions can be used instead of a granulation fluid
in the granulation process. Alternatively, SLNs can be transferred to a powder (by spray-drying
or lyophilisation) and added to the tableting powder mixture. In both cases, it is beneficial to
have a higher solid content to avoid the need of having to remove too much water. For cost
reasons, spray drying might be the preferred method for transforming SLNs dispersions into
powders, with the previous addition of a protectant [48]. For the production of pellets, the SLNs
dispersion can be used as a wetting agent in the extrusion process. SLNs powders can also be
used for the filling of hard gelatine capsules.
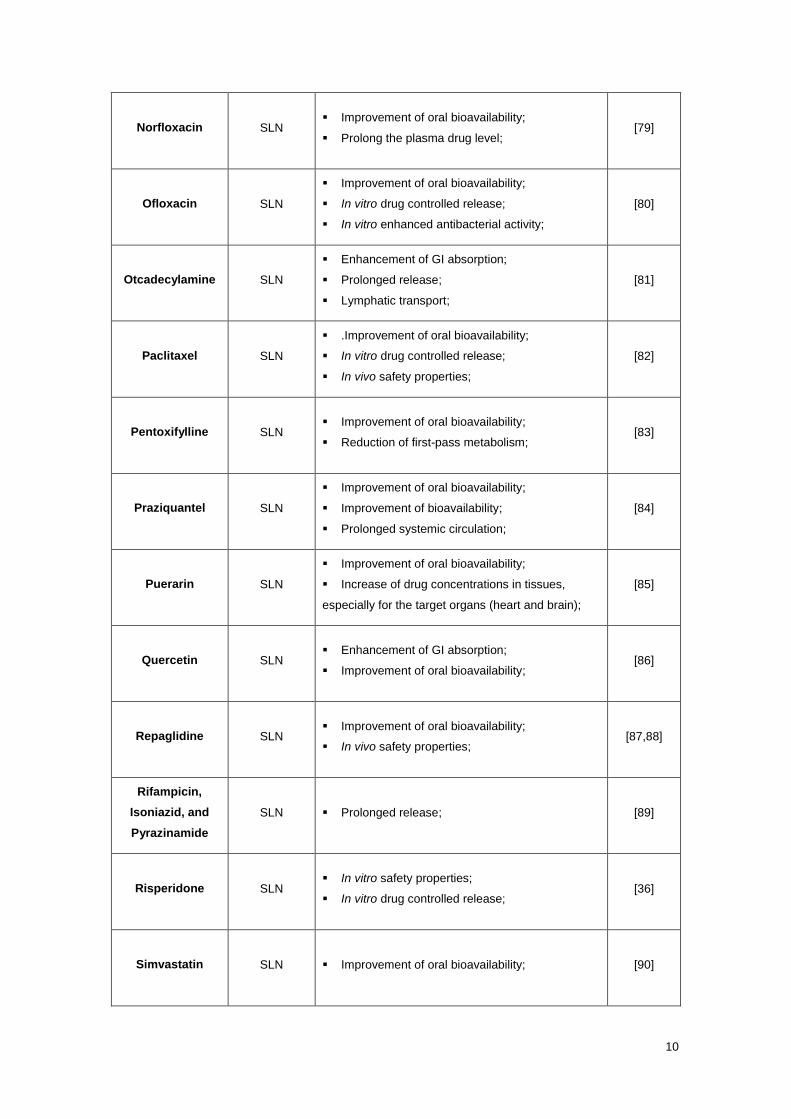
Several drugs (hydrophobic and hydrophilic) have been incorporated in lipid
nanoparticles for oral administration. Table 1 shows some of these drugs encapsulated in SLNs
and NLCs examples and their potential benefits for application in oral delivery.
8
Table 1 – Examples of different Drug-Loaded Lipid Nanoparticles and their potential benefits for
application in oral administration route.
Drug Lipid
Nanoparticle Relevant effects Reference
Amphotericin B SLN
Improvement of oral bioavailability;
Drug protection;
In vivo drug controlled release;
[61]
Apomorphine SLN
Improvement of oral bioavailability;
Successful targeted drug to the local of
therapeutic action (brain striatum);
[62]
Camptothecin SLN Body distribution;
Sustained release and tissue targeting; [63]
Candesartan
cilexetil SLN Improvement of oral bioavailability; [64]
Carvedilol SLN
Enhancement of oral bioavailability;
Lymphatic uptake;
Bypass hepatic first-pass metabolism;
[65]
Clozapine SLN Improvement of oral bioavailability;
Tissue distribution; [34]
Cryptotanshinone SLN
Enhancement of GI absorption;
Improvement of oral bioavailability;
Change in metabolism;
[66]
Curcumin SLN Improvement of oral bioavailability
Prolonged release; [67]
Cyclosporine A SLN Low variation in bioavailability; [68]
Digoxin SLN Enhancement of GI absorption
Improvement of oral bioavailability; [69]

9
Edelfosine SLN
High accumulation of drug in the local of
therapeutic action (brain);
In vitro antiproliferative effects on glioma cells;
In vivo reduction of tumor growth;
[70]
Etoposide NLC
Improvement of oral bioavailability;
In vitro antiproliferative effects on lung
carcinoma cells;
[71]
Fenofibrate SLN Improvement of oral bioavailability [72]
Insulin Lectin-
modified SLN
Protection of insulin degradation by enzyme;
Improvement of oral bioavailability;
Enhancement of GI absorption;
[33]
Insulin SLN Enhancement of GI absorption;
Importance of delivery site; [73]
Lopinavir SLN
Improvement of oral bioavailability;
Effective target of the drug to the local of
therapeutic action (CNS);
Prolong the blood circulation time of the drug;
[74]
Lovastatin NLC
Enhancement of encapsulation efficiency;
Improvement of oral bioavailability;
Improvement of stability in GI environment;
[75]
Melatonin SLN Sustained delivery [76]
Methotrexate SLN
Improvement of oral bioavailability;
Lymphatic transport;
Enhancement of GI absorption;
[23]
N3-O-toluyl-
fluorouracil Cationic SLN
Enhancement of GI absorption;
Improvement of oral bioavailability; [77]
N3-O-toluyl-
fluorouracil Anionic SLN Improvement of intestinal transport; [60]
Nitrendipine SLN Improvement of oral bioavailability;
Reduction of first-pass metabolism; [78]
10
Norfloxacin SLN Improvement of oral bioavailability;
Prolong the plasma drug level; [79]
Ofloxacin SLN
Improvement of oral bioavailability;
In vitro drug controlled release;
In vitro enhanced antibacterial activity;
[80]
Otcadecylamine SLN
Enhancement of GI absorption;
Prolonged release;
Lymphatic transport;
[81]
Paclitaxel SLN
.Improvement of oral bioavailability;
In vitro drug controlled release;
In vivo safety properties;
[82]
Pentoxifylline SLN Improvement of oral bioavailability;
Reduction of first-pass metabolism; [83]
Praziquantel SLN
Improvement of oral bioavailability;
Improvement of bioavailability;
Prolonged systemic circulation;
[84]
Puerarin SLN
Improvement of oral bioavailability;
Increase of drug concentrations in tissues,
especially for the target organs (heart and brain);
[85]
Quercetin SLN Enhancement of GI absorption;
Improvement of oral bioavailability; [86]
Repaglidine SLN Improvement of oral bioavailability;
In vivo safety properties; [87,88]
Rifampicin,
Isoniazid, and
Pyrazinamide
SLN Prolonged release; [89]
Risperidone SLN In vitro safety properties;
In vitro drug controlled release; [36]
Simvastatin SLN Improvement of oral bioavailability; [90]

11
Spironolactone SLN Improvement of oral bioavailability; [91]
Tretinoin SLN In vitro drug controlled release; [92]
Vinpocetine NLC Improvement of oral bioavailability; [93]
Vinpocetine SLN Enhancement of GI absorption;
Improvement of oral bioavailability; [94]
α-Asarone SLN
Enhancement of GI absorption;
Improvement of oral bioavailability;
Improvement of tissue uptake and distribution;
[95]
γ -Tocotrienol SLN Improvement of oral bioavailability. [96]
1.1.1.1. Toxicological Concerns
Lipid nanoparticles are generally made of physiological and GRAS excipients and
therefore, metabolic pathways exist decreasing the risk of acute and chronic toxicity. Their
degradation is relatively fast in non-toxic compounds that are easily eliminated through
physiologic metabolic pathways. The lipid matrix degradation occurs mostly by lipases whereas
only a minor part is degraded by non-enzymatic hydrolytic processes [97]. Nonetheless, even
for biodegradable nanoparticles, the use of high concentrations of the carriers can lead to
toxicological concerns. Therefore, the fate of the carriers in the body should be clarified.
The human immune system, by means of macrophages, recognize all external
nanoparticles as hostile matter and quickly phagocyte and clearance them from the body.
Nonetheless, these immunological specialized cells are present in limited areas of the body
(e.g. lungs), which decrease the risks of interference with oral administered systems.
Furthermore, the nanoparticles with sizes below to 100 nm can be internalized by all body cells
[20,98]. Regarding the typical lipid nanoparticles sizes of 100-300 nm (size above 100 nm),
which means that their cellular uptake is not expected and, therefore, no significant toxicological
concerns exist [25].

12
Toxicological studies should be first performed in vitro, using cell models that mimic the
body conditions, in order to minimize the number of animal studies and to have an idea of the
cytotoxicity of the systems in an early stage. Nonetheless, in vivo studies must always be
performed before pass to human clinical trials, since in vitro studies sometimes use short time
periods and small concentration ranges, which not allow for realistic conclusions about the
cytotoxicity of the nanocarrier systems [99].
Silva et al. [36] studied the toxicity of SLN and risperidone loaded SLNs with Caco-2 cells
by (4,5-dimthylthiazol-2-yl)2,5-diphenyl-tetrazolium bromide (MTT) assay. The Caco-2 cell line
is a human colon epithelial cancer cell line often used as a model to mimic the GI tract
conditions for toxicological and pharmacological studies [100]. MTT assay evaluates the
mitochondrial function as a measurement of cell viability. Therefore, only live cells could react
with the MTT reagent and the cell viability is detected in a very early stage. The results suggest
that all formulations evaluated are biocompatible with Caco-2 cells and well tolerated by the GI
tract. Similar results have been reported elsewhere [101,102]. The amount of viable cells after
SLNs exposure was performed by the MTT assay with Caco-2 cell models. Other authors have
also reported that SLN show biocompatibility, which increase their attractiveness for drug-
delivery applications [101].
Several studies have reported that lipid nanoparticles systems are well tolerated and
demonstrate low cytotoxicity, compared with other conventional nanocarriers [101,103]. Lipid
carriers prepared with several lipids and emulsifying agents did not exhibit substantial cytotoxic
effects in vitro. SLN prepared up to concentrations of 2.5% lipid do not exhibit any cytotoxic
effects in vitro [104]. In fact, it has been shown that even concentrations higher than 10% of lipid
led to a viability of 80% in cultures of human granulocytes [105]. In contrast, some polymeric
nanoparticles showed complete cell death at concentrations of 0.5%. It can be assumed that the
cytotoxicity of the SLNs can be mainly attributed to emulsifiers and preservative compounds
which are used in the production of these systems. Additionally, depending on the route of
administration, the toxicity requirements of the nanoparticles systems can change, being less
for dermal, moderate for oral and limited for intravenous administration [20].
In conclusion, from the data obtained until now, the lipid nanoparticles formulations
appear to fulfil the essential prerequisite to the clinical use of an oral colloidal carrier that means
low cytotoxicity.

13
1.1.2. Preparation Techniques for Lipid Nanoparticles
Since lipid nanoparticles dispersions at high temperatures are miniemulsions,
miniemulsions preparation techniques can be applied or modified for lipid nanoparticles
production. A miniemulsion, also referred as nanoemulsion or ultrafine emulsions, compose a
particular class of emulsions consisting of colloidal dispersions with a droplet size between 20-
500 nm. The physical appearance of nanoemulsion is transparent or bluish for the smallest
droplet sizes between 20–100 nm, or milky for sizes up to 500 nm [106]. Nanoemulsions are
non-equilibrium systems and cannot be formed spontaneously. Nanoemulsion can be produced
through two ways. There is either high-energy emulsification method or low-energy
emulsification method. High-energy emulsification method includes high-pressure
homogenization, sonication or high-shear mixing, whereas condensation method such as phase
inversion temperature (PIT) is an example of low-energy emulsification method [107].
As several techniques have been developed for the production of lipid nanoparticles,
resulting in different particles sizes and shapes, some parameter must be considered in order to
choose the suitable preparation method. The size can influence the pharmacological properties
of the particles, but it is not the unique parameter considered to compare the various
techniques. Toxicological issues are also very important. The materials used must be
biocompatible and biodegradable, while the use of solvents can be a relevant drawback, since
they can remain in traces in the final product. From a technological point of view, the possibility
to scale up the process is very important, but also the feasibility of the method is relevant. In
fact, the use of expensive and complex machine can hamper the production on lab scale.
Finally, the drug entrapment is very important. Nowadays more and more complex molecules
are entrapped within lipid nanoparticles. These molecules have different physical and chemical
properties (solubility, hydrophobicity, etc.) and stability issues (temperature, pH, etc.). The
chosen preparation technique should be the most appropriate to enhance drug loading and
encapsulation efficiency within the nanoparticles, without hampering the chemical stability of the
molecule itself.
The following sections describe different existing approaches for SLNs and NLCs
formulations. However, in some instances combination of different methods has been utilized to
prepare the lipid nanoparticles.
1.1.2.1. High Pressure Homogenization Technique
Müller and Lucks were the first to prepare SLNs by applying high pressure
homogenization (HPH) technique [108]. It makes use of high pressure homogenizer which is
accessible from several manufacturers. In this technique, melted lipid solution is forced at a high
pressure (100-2000 bar) through a narrow gap of few microns. The resulting high shear stress
and cavitation forces decrease the particle size. A homogeneous dispersion with narrow size

14
distribution is desirable to increase the physical stability of the aqueous dispersion. If the
particles localized at different positions in the dispersion volume, experience different forces
then the degree of particle disruption will vary. There are two basic production methods to HPH:
the hot and the cold homogenization techniques. In both the techniques, the lipid contents are in
the range of 5-10%, but even higher concentration of lipid (40%) can be homogenized to
nanodispersion [109].
1.1.2.1.1. Hot Homogenization Technique
In this method, the drug is first dissolved or solubilized in the lipid being melted at 5-10ºC
above its melting point. The drug-containing melt is dispersed under stirring in a hot aqueous
surfactant solution of identical temperature [110]. The pre-emulsion is then passed through a
high pressure homogenizer (e.g. Micron LAB40) for 3 to 5 cycles and applying a pressure of
about 500-1500 bar [111]. The obtained nanoemulsion is cooled down to room temperature or
lower. The lipid recrystallizes and leads to SLNs. Homogenization pressure and the number of
cycles should not be higher than that required to achieve the desired effects because this
increases the cost of production and the chances of metal contamination as well as in some
cases it would result in increase in particle size due to aggregation as a result of the high
surface free energy of the particles [112]. This technique is performed at a high temperature
and thus cannot be used for temperature sensitive drugs. So, to avoid heat-accelerated drug
degradation, the length of time for drug exposure should be shortened [10]. This method is also
not suitable for hydrophilic drugs because they would partition between the melted lipid and
aqueous phase during hot homogenization [10]. Either medium scale or large scale production
is possible for SLNs by HHT and it is the most extensively technique for the preparation of
SLNs.
1.1.2.1.2. Cold Homogenization Technique
In this process, the drug is also dissolved or solubilized in lipid being melted at 5-10ºC
above its melting point. The mixture is rapidly cooled down with help of liquid nitrogen or dry ice.
Rapid cooling procedure helps in the homogeneous distribution of the active ingredient. This
solidified mixture is milled to about 50-100 μm particles using a ball or mortar mill and followed
by dispersion of the lipid microparticles in a cold surfactant aqueous solution to obtain a
suspension. This suspension is then passed through a high pressure homogenization at or
below room temperature. Here, the cavitation forces are strong enough to break the lipid
microparticles to SLNs. The cold homogenization technique reduces the chances of drug
degradation induced by temperature and thus thermo label drugs can be used. Since the solid
lipid is milled the complexity arising due to lipid modification can be avoided [112]. Chances of

15
drug distribution into the aqueous phase are limited and hence this method can be used for
hydrophilic drugs as well as lipophilic drugs. Lipid nanoparticles prepared via this technique
possess a slightly larger particle size and polydispersity when compared to the ones obtained
by hot homogenization technique, using the same lipid at similar homogenization parameters
(pressure, temperature and the number of cycles) but a higher number of homogenization
cycles can be applied to reduce the particle size [10].
1.1.2.2. Microemulsion Technique
Gasco and co-workers were the first to develop SLNs based on the dilution of
microemulsions [46]. Microemulsions are thermodynamically stable, clear and isotropic mixtures
usually composed of an oil or lipid, emulsifier and/or co-emulsifier and water. In this process,
the solid lipids are melted above their melting point. Separately, a mixture of emulsifier, co-
emulsifier and water is heated at same temperature. Both the lipid and the aqueous phase
containing the emulsifier are mixed in appropriate ratios and stirred in order to produce a
microemulsion. The hot microemulsion is then diluted with cold water (2-8°C) while stirring. The
ratio of the hot microemulsion to cold water is usually in the range of 1:10 to 1:50 [113]. It has
been noted that a droplet structure is already present in the microemulsion and therefore, no
external energy is required to achieve the small particle size. When the microemulsion is diluted
by cold water, the lipid droplets solidify as the temperature decreases. The temperature gradient
and pH value determine the quality of the particles in addition to the composition of the
microemulsion. Subsequently, the produced SLNs are washed three times with distilled water
and followed by membrane filtration in order to remove any unwanted bigger lipid particle. Due
to the dilution of microemulsion the concentrations of particle content are below 1% and,
therefore, large amount of water has to be removed either by ultrafiltration or by lyophilisation in
order to increase the particle concentration. The major limitation of this technique is its
sensitivity to minor changes in composition or thermodynamic variables, which can lead to
phase transitions. Lack of robustness of the microemulsion technique can lead to high
production costs. Moreover, lipids solidification shifts the system to a thermodynamically
unstable state. The high concentration of surfactants used may produce toxicity.
Ultracentrifugation, ultrafiltration or dialysis can be applied to remove excess surfactants [113].
1.1.2.3. Ultrasonication Technique
In this technique, solid lipids are melted at 5–10°C above their melting points and drug is
dissolved or dispersed in melted lipids. Then a hot aqueous surfactant solution (preheated at
the same temperature) is added to the drug-lipid melt and homogeneously dispersed by a high
shear mixing device. The coarse hot o/w emulsion obtained is ultrasonicated using a probe

16
sonicator till the desired sized nanoemulsion is formed. Finally, lipid nanoparticles are obtained
by allowing hot nanoemulsion to cool down to room temperature.
Two mechanisms are proposed for ultrasonic emulsification. First, the application of an
acoustic field produces interfacial waves resulting in the dispersion of the oil phase in the
continuous phase in the form of droplets [114]. Secondly, the application of ultrasound induces
acoustic cavitation causing the formation and subsequent collapse of microbubbles by the
pressure fluctuations of a simple sound wave, which creates extreme levels of highly localized
turbulence. Therefore, the turbulent micro-implosions break up primary droplets into sub-micron
size [115].
This technique is a simple and reproducible way to prepared nanoparticles without the
need of organic solvents or any sophisticated instruments and has the potential to easily scale
up for large scale production. However, metallic contamination of the product may occur during
sonication by probe sonicator [116].
1.1.2.4. Solvent Emulsification Evaporation Technique
Sjöström and Bergenståhl were the first to describe the production of SLNs by solvent
emulsification-evaporation technique [117]. The solid lipid is dissolved in a water immiscible
organic solvent (e.g. cyclohexane, dichloromethane, toluene, chloroform, etc.) and the drug is
dissolved or dispersed in the solution [118]. This organic phase containing the drug is emulsified
in an aqueous surfactant solution by mechanical stirring. The organic solvent is then removed
from the emulsion under mechanical stirring or reduced pressure (40-60 mbar) (e.g. rotary
evaporator) [118]. Lipid nanoparticle dispersion is formed by the precipitation of the lipid phase
in the aqueous surfactant medium. The mean particle size depends on the concentration of lipid
in organic phase. Very small particle size can be obtained with low lipid load (5%) related to
organic solvent. Aggregation of the particles can be avoided by removing the solvent at a faster
rate [10]. Thermo labile drugs can be incorporated via this technique as it avoids thermal stress.
Trace amounts of organic solvent remaining in the final product can potentially create toxicity
problems. A large quantity of water has also to be removed during the final step of the
formulation by means of ultrafiltration or evaporation [119,120].
1.1.2.5. Double Emulsion Technique
The double emulsion (w/o/w) method is based on solvent emulsification–evaporation
method [113]. This method is mainly for the production of lipid nanoparticles loaded with
hydrophilic drugs. In this case, drug is firstly dissolved in aqueous solvent (inner aqueous
phase) and then is dispersed in lipid containing emulsifier/stabilizer to produce primary emulsion
(w/o). A double emulsion (w/o/w) is formed after addition of an aqueous solution with a

17
hydrophilic emulsifier followed by stirring. Each emulsification step results in a highly
polydisperse droplet distribution, exacerbating the polydispersity of the final double emulsion.
Thus, lipid nanoparticles formed from double emulsions technique are, by nature, poorly
controlled in both size and structure.
1.1.2.6. Solvent Emulsification Diffusion Technique
Quintanar-Guerrero et al. were the first to describe this technique for the preparation of
polymeric nanoparticles [121]. Recently this technique has been modified by various research
groups for the preparation of lipid nanoparticles [122,123]. In solvent emulsification diffusion
technique, the lipid is dissolved in a partially water miscible solvent (e.g. benzyl alcohol,
isobutyric acid, or tetrahydrofuran) which is previously saturated with water at room temperature
or at a controlled temperature in order to ensure the initial thermodynamic equilibrium [113]. The
mixture is then emulsified in an aqueous solution of a surfactant by mechanical stirring at the
temperature used to dissolve the lipid producing an o/w emulsion. This o/w emulsion is then
diluted with excess water, in typical ratio ranges from 1:5 to 1:10 [113], at a controlled
temperature which causes the diffusion of the solvent into the external phase and subsequent
precipitation of the lipid nanoparticles. The diffused solvent can be removed either by vacuum
distillation or ultrafiltration. The concentration and the nature of the lipid and surfactant, stirring
rate and the processing temperature are critical variables in this technique. This technique
avoids the necessity to melt the lipid which is very useful for preparation of protein and peptide-
loaded lipid nanoparticles.
1.1.2.7. Solvent Injection Technique
The basic principle of the solvent injection method is similar to the solvent emulsification
diffusion technique. In case of solvent injection method, lipids are dissolved in a water-miscible
solvent (e.g., acetone, isopropanol, and methanol) or water-miscible solvent mixture and quickly
injected into an aqueous solution of surfactants through an injection needle [113]. The
advantages of this method are the easy handling and fast production process without
technically sophisticated equipment (e.g., high-pressure homogenizer). However, the main
disadvantage is the use of organic solvents. The resulted dispersion is filtered through a filter
paper in order to remove any excess lipid [113]. The presence of emulsifier within the aqueous
phase helps to produce lipid droplets at the site of injection and stabilize lipid nanoparticles until
solvent diffusion was complete by reducing the surface tension between water and solvent.

18
1.1.2.8. Phase Inversion Temperature Technique
The phase inversion temperature (PIT) method is commonly used for the preparation of
nanoemulsions. The PIT concept uses the specific ability of some polyethoxylated surfactants to
modify their affinities for water and oil as a function of the temperature. In the PIT nanoemulsion
preparation method, the use of such surfactant type leads to an emulsion inversion from o/w
macroemulsion to a w/o emulsion when temperature is increased above the PIT, and to the
formation of an o/w nanoemulsion when the temperature is next lowered below the PIT [124].
Recently it has been adapted for the preparation of SLN. In this case two main components are
used: an oil phase, constituted by solid lipids and non-ionic surfactants and an aqueous phase
containing NaCl. The aqueous phase and the oil phase are separately heated above the PIT;
then the aqueous phase is added dropwise, at constant temperature and under agitation, to the
oil phase, in order to obtain a w/o emulsion. The mixture is then cooled to room temperature
under slow and continuous stirring. At the PIT, the turbid mixture becomes clear, then below the
PIT an o/w nanoemulsion is formed, which turns in lipid nanoparticles below the lipid melting
point [125].
1.1.2.9. Microchannel/Microfluidic Technique
A novel microchannel system with a cross-shaped junction was developed by a main
microchannel and two branches as shown in the Figure 5. The lipid dissolved in a water-
miscible organic solvent is passed through the main channel, while simultaneously an aqueous
surfactant solution is introduced into the branches. These two liquids met together at the cross-
shaped junction and passed along the main channel. The solvent diffused from the lipid solution
stream into the aqueous phase, which resulted in the local supersaturation of lipid and thus led
to the formation of lipid nanoparticles. The size of lipid nanoparticles prepared in the
microchannel system was influenced either by velocities of the lipid and aqueous phases and by
lipid and surfactant concentrations [113]. This technique allows precise control of the outer and
inner drop sizes as well as the number of droplets encapsulated in each larger drop [113].
However, in a continuous production process, lipid nanoparticles contained in the liquid may
block or deposit inside the microchannel, which could result in the break down and failure of
particle preparation [113].
Figure 5 - Schematic diagram of lipid nanoparticles formation in the microchannel system. From [113]

19
1.1.3. Characterization of Lipid Nanoparticles
Characterization of lipid nanoparticles is a challenge due to the small size of these
colloidal carriers and complexity of the system. Numerous parameters need to be considered,
such as mean particle size, zeta potential, drug association efficiency, degree of lipid
crystallinity and lipid modification. For each parameter can be applied several techniques.
However, only methodologies selected in this work will be further described.
1.1.3.1. Measurement of Particle Size
Particle size might be determined by dynamic light scattering (DLS), optical single particle
sizing (OSPS), laser diffraction (LD), transmission electron microscopy (TEM), scanning
electron microscopy (SEM), atomic force microscopy (AFM), scanning tunnelling microscopy
(STM) and freeze fracture electron microscopy (FFEM) [112,113].
1.1.3.1.1. Dynamic Light Scattering
Dynamic Light Scattering (DLS) also referred as Photo Correlation Spectroscopy (PCS) is
a technique used to determine the mean particle size and the width of the particle size
distribution expressed as polydispersity index (PDI). The particle size is the diameter of the
sphere that diffuses at the same speed as the particle being measured.
The measurement using DLS is based on the light scattering phenomena in which the
statistical intensity fluctuations of the scattered light from the particles in the measuring cell are
measured. These fluctuations are due to the random movement of the particles in the
dispersion medium. This movement is called Brownian motion.
Usually a DLS device consists of a laser light which illuminates a small volume of the
sample composed by a dilute suspension of particles. The light scattered from these particles is
collected by a lens and its intensity is measured by a photomultiplier at a certain angle (90º or
173º). The diffusion rate of the particles depends on their size (at a known fluid viscosity and
temperature). Hence, the size of these particles can be calculated from the rate of fluctuation of
the scattered light intensity. When the suspended particles are small, they diffuse relatively fast,
and the fluctuations in the scattered light are rapid. On the other hand, if the particles are large,
they move slowly, and the fluctuations in the scattered light are slow. The detected intensity
signals are used by a correlator to calculate the auto-correlation function G(τ). A correlator is
basically a signal comparator designed to measure the degree of similarity between two signals,
or one signal with itself at varying time intervals. From the decay of correlation function, the
diffusion coefficient (D) of the particles is obtained. Once the diffusion coefficient is known, the

20
hydrodynamic diameter of a spherical particle can be calculated applying the Stokes-Einstein
equation (Eq.1),
d(H) = 𝐾𝑇
3πηD Eq.1
where, d(H) is the hydrodynamic diameter, D is translational diffusion coefficient which
measures the velocity of the Brownian motion, k is the Boltzmann’s constant, T is the absolute
temperature and η is the viscosity of the solution.
As it was previously mentioned, small particles diffuse faster than large ones, causing a
stronger fluctuation in the scattering signal and a more rapid decaying G(τ) (Figure 6). For a
monodisperse particle population, G(τ) is a single exponential, but if more than one size of
particles is present the function is polyexponential. Deviation from a single exponential is used
to calculate the PDI, which is a measure of the width of the size distribution. The PDI value is
zero when a monodisperse particles population is measured. PDI values of around 0.10-0.30
indicate a relatively narrow distribution, values of 0.5 and higher are obtained in case of very
broad distributions [126].
Figure 6 -Correlation differences between small and large particles. From [126].
1.1.3.2. Measurement of Zeta Potential
Almost all particles in contact with a liquid acquire an electric charge on their surface. The
development of a nett charge at the particle surface affects the distribution of ions in the
surrounding interfacial region, resulting in an increased concentration of counter ions close to
the surface. Thus an electrical double layer exists around each particle (Figure 7). The liquid

21
layer surrounding the particle has an inner region called the Stern layer, where the ions are
strongly bound and an outer and diffuse region, where the ions are less firmly attached. Within
the diffuse layer there is an imaginary boundary inside which the ions and particles form a
stable entity. When a particle moves, ions within the boundary move with it, but any ions beyond
the boundary do not move with the particle. This boundary is called the surface of hydrodynamic
shear or slipping plane. Zeta potential (ζ) is the potential that exists at this boundary and it is a
parameter which is very useful for the assessment of the physical stability of colloidal
dispersions [126].
Figure 7 - Zeta-potential of a nanoparticle in solution. From [126].
Charged particles suspended in the electrolyte are attracted towards the electrode of
opposite charge, when an electric field is applied. Viscous forces acting on the particles tend to
oppose this movement. When equilibrium between these two opposing forces is achieved, the
particles move with a constant velocity. This velocity of the particle per unit electric field strength
(E) is called the electrophoretic mobility (μ), which is expressed in micrometers per second per
volt per centimeter (μm/s)/(V/cm). Zeta Potential can be measured by electrophoretic mobility
through Henry’s equation (Eq. 2),
μ = 2εζf(ka)
3η Eq. 2
where η is the viscosity of the dispersion medium, ε the permittivity of the environment (the
dielectric constant) and f(ka) is the Henry’s function. Electrophoretic determinations of zeta

22
potential are most commonly made in aqueous media and moderate electrolyte concentration.
In this case, f(Ka) is 1.5 and is referred to as the Smoluchowski approximation. Non-aqueous
meassurements generally use the Huckel approximation and f(Ka) is 1 [126].
The storage stability of a colloid system can be predicted by the measurement of the zeta
potential. It is currently accepted that absolute zeta potentials values higher than 30 mV are
required for full electrostatic stabilization; potentials between 5 and 15 mV are in the region of
limited flocculation; and lower than 3 mV of maximum flocculation. As a result, particle
aggregation is less expected to occur for charged particles (high zeta potential) due to electric
repulsion [113].
Nanoparticles with a zeta potential between -10 and +10 mV are considered
approximately neutral, while nanoparticles with zeta potentials of greater than +30 mV or less
than -30 mV are considered strongly cationic and strongly anionic, respectively. Since most
cellular membranes are negatively charged, zeta potential can affect a nanoparticle's tendency
to permeate membranes, with cationic particles generally displaying more toxicity associated
with cell wall disruption. Positive charge possesses a membrane destabilizing and
concomitantly destructive effect resulting from an interaction of positive charge and negative
charge of membrane. Thus, positively charged lipids are not approved by Food and Drug
Administration (FDA) for clinical use [127].
1.1.3.3. Encapsulation Efficiency
According to literature, the association efficiency of a drug in SLNs is most frequently
assessed indirectly, meaning that the aqueous phase is separated from the lipid particles and
subsequently drug content of the aqueous phase is determined [113]. The separation of the
SLNs from the aqueous phase can be done by e.g. ultrafiltration, ultracentrifugation and gel
separation. The amount of drug measured in the aqueous phase is considered the amount of
drug not incorporated. The following equation (Eq. 3) is used to calculate the association
efficiency:
EE% = 𝑇𝐷−𝐹𝐷
TD × 100 Eq.3
where TD is total drug weighted at the beginning and FD is the free drug dissolved in aqueous
medium.
1.1.3.4. Morphology
Images of lipid nanoparticles could be achieved through several microscopic techniques,
such as the scanning electron microscopy (SEM), transmission electron microscopy (TEM),

23
atomic force microscopy (AFM), CryoSEM and CryoTEM [112,113]. These techniques are very
useful since allow the visualization and measurement of particles much smaller than 1 μm.
1.1.3.4.1. Transmission Electron Microscopy
TEM utilizes energetic electrons to provide morphologic, compositional and
crystallographic information of samples. At a maximum potential magnification of 1 nm, TEM is
the most powerful microscope, producing high-resolution and two-dimensional images, allowing
the investigation of micro- and nanostructures. Going through the several parts of the TEM
system, first an electron gun generates a high energy electron beam by accelerating electrons
emitted from a cathode. This acceleration voltage, usually greater than 100 kV, determines the
microscope’s resolution. After the accelerator, there are condenser lenses to control beam
diameter and convergence angles of the incident beam on a specimen. The TEM has three
lenses to ensure good magnification capability, and the intermediate lens is used to switch the
TEM between an image mode and a diffraction mode. TEM specimens must be thin foils (about
100 nm) because they should be able to transmit electrons, and this thin specimen is mounted
in a 3 mm diameter grid. The specimen preparation depends on the type of sample.
1.1.3.5. Lipid Crystallinity
Lipid crystallinity and polymorphic changes during storage are strongly related with drug
incorporation and release rates [112,113]. Due to the small size of the SLN and the presence of
surfactants, lipid crystallization and modification changes can be highly retarded. Consequently,
special attention should be paid on the characterisation of such parameters. Lipid crystallinity
and polymorphic forms are mainly investigated by differential scanning calorimetry (DSC) and
X-ray scattering techniques [112,113].
DSC is a thermal analytical technique commonly used to characterize the physical state,
the degree of crystallinity, polymorphism, crystal ordering, eutectic mixtures or glass transition
processes in SLNs dispersions [112,113]. Additionally, DSC experiments are useful to
understand drug and lipid interactions within SLNs.

24
1.2. Tuberculosis – Review
Tuberculosis (TB) is an infectious disease caused by bacilli belonging to the
Mycobacterium tuberculosis complex [128] and this complex consists of seven species
including Mycobacterium tuberculosis, Mycobacterium canetti, Mycobacterium africanum,
Mycobacterium pinnipedii, Mycobacterium microti, Mycobacterium caprae and Mycobacterium
bovis [129].
TB is predominantly a disease of the lung, with pulmonary TB accounting for about 70%
of the cases. Extra-pulmonary disease sites include lymph nodes, bone, and meninges [130] .
The bacilli remain viable in the air for a long time and, eventually, they are inhaled during
breathing, leading the bacteria directly to the lung where they are phagocyted by alveolar
macrophages (white blood cells). Inside the macrophages, the bacilli reside in a membrane-
bound vacuole, and for this reason some are able to avoid fusion with lysosomes and posterior
digestion [131], ending up co-existing with the macrophages [132]. They multiply and eventually
escape the lung through the bloodstream and lymphatic system, spreading to other organs of
the body, resulting in the extra-pulmonary TB [133]. Moreover, M. tuberculosis may exist within
a granulomas consisting of macrophages and giant cells, T cells, B cells, and fibroblasts, and
these granulomas can prevail not only in the lung, but in other organs as well. In latent
infections, the state of the bacteria within the granuloma is unknown.
Despite 100 years of research, infection with M. tuberculosis is still a major health
problem worldwide and one of the main causes of human death by a particular infectious agent.
Inappropriate monotherapy and intermittent treatment are the main causes of drug resistance,
and facilitate the selection and transmission of resistant strains within communities. In 2012,
there were 8.7 million new cases of TB globally, with 1.4 million deaths in the same year, which
confirms TB as a serious global health issue [128]. Of the 8.7 million incident TB cases in 2012,
1.1 million (13%) were among people living with HIV [128].
Currently available chemotherapy includes first-line drugs such as isoniazid (INH),
rifampicin (RIF), ethambutol (EMB) and pyrazinamide (PYZ) [134]. These drugs are
administered orally and are shown to have excellent potency against M. tuberculosis. Isoniazid,
a prodrug activated by the catalase peroxidase (KatG) enzyme, is the most well-known and
used drug to treat TB [135]. Activated isoniazid inhibits NADH-dependent enoyl-ACP reductase,
which plays a role in the synthesis and elongation of mycolic acids on the cell wall [135].
Rifampicin binds to the β-subunit of the RNA polymerase, preventing the formation of the
messenger RNA chain [136]. Pyrazinamide is a prodrug converted into pyrazinoic acid by the
pyrazinamidase enzyme [135]. The protonated pyrazinoic acid accumulates in the cell and
causes cytoplasmic acidification and reduces cell membrane energy, disrupting the proton
motive force and affecting membrane transport [136]. Ethambutol is a bacteriostatic agent that
inhibits the polymerization of arabinogalactan, consequently preventing its biogenesis formation
on the cell wall [137].

25
The second-line drugs used for the treatment of TB are aminoglycosides (e.g. amikacin,
kanamycin), polypeptides (e.g. capreomycin, viomycin, enviomycin), fluroquinolones (e.g.
ciprofloxacin, levofloxacin, moxifloxacin), thioamides (e.g. ethionamide, prothioamide,
cycloserine) [134]. They are used as alternative agents to treat TB when the bacilli are resistant
to first-line drugs and they are less effective, more toxic, and unavailable in many countries due
to high costs [138].
The effect of anti-TB drugs resistance has been around since the first anti-TB agent,
streptomycin, appeared on the market as a monotherapy. Multi-drug resistant (MDR) TB is
defined by resistance to, at least, rifampicin and isoniazid, whereas extensively drug-resistant
(XDR) TB is defined as MDR-TB with additional resistance to any fluoroquinolone and to, at
least, one of the injectable drugs (capreomycin, kanamycin and amikacin). Drug resistance in
TB is the result of spontaneous mutation in combination with selection by poor programmatic
and individual care performance [139]. Globally, WHO estimates that 3.7% of new tuberculosis
cases and 20% of re-treatment cases have MDR-TB [128]. By the end of 2012, 84 countries
had reported at least one case of extensively drug resistant strains (XDR-TB) [128].
M. tuberculosis is intrinsically resistant to numerous antibiotics, and only a few drugs are
effective for treatment [140]. This intrinsic resistance is partially due to the thick, lipid-rich cell
wall, which is an important characteristic of mycobacteria, limiting the penetration of antibiotics.
The cell wall consists of a layer of peptidoglycan surrounding the cell’s basic lipid bilayer. A
second layer of arabinogalactin runs parallel to the peptidoglycan layer, surrounding it in an
intricate, sugary shell. A third layer extends perpendicular to the arabinogalactin shell and
consists of a complex network of mycolic acids. These long and “sticky” mycolic acids are tightly
bound in a final exterior layer, rendering the bacilli virtually impermeable and almost entirely
waterproof. This complex armor allows M. tuberculosis to be resistant to many antibiotics, avoid
death by acidic and alkaline compounds, and prevent cellular phagolysosomal fusion, allowing it
to successfully evade lysis. Figure 8 depicts a simple visual representation of M. tuberculosis
cell wall. Moreover, mycobacteria possess various defence systems against the activity of
antibiotics, including potent β-lactamases and other drug-neutralizing enzymes [140]. Unlike
many other bacteria, M. tuberculosis harbors no resistance plasmids, and drug resistance
arises through the acquisition of specific chromosomal mutations [141].
The current short-course treatment guideline aims for a complete elimination of active
and dormant bacilli involves two phases. During the initial phase four drugs (usually isoniazid,
rifampicin, pyrazinamide and ethambutol) are given daily for two months. The continuation
phase in which fewer drugs (usually isoniazid and rifampicin) are administered for an additional
4 months, either daily or 3 times per week, targets the killing of any remaining or dormant bacilli
in order to prevent relapse [128].

26
Figure 8 – Schematic structure of M. tuberculosis cell wall and action site of first-line anti-TB drugs.
Intercalation of hydrophilic arabinogalactan and hydrophobic mycolate containing layers creates an
extremely impermeable envelope for antibiotic penetration. Small molecules and nutrients are transported
through porin channels that are deposited through these layers. Adapted from [140].
The search for new anti-TB drugs is an important key in this fight, but searching for new
drug delivery strategies may also play an important role. Alternative delivery systems, such as
nanocarriers for anti-TB drugs, may reduce administration frequency and shorten periods of
treatment, hence improving patient compliance and efficacy of treatment, and reduce drug
related toxicity. For example, isoniazid, rifampicin and pyrazinamide are limited by
hepatotoxicity [142], a side effect due to the action of the drug on hepatocytes rather than
macrophages, the primary host cells that harbor M. tuberculosis. Thus, a delivery mechanism
that introduces these antibiotics selectively into macrophages would greatly increase their
therapeutic efficacy by achieving higher concentrations of the antibiotics in site of infection
without exposing the patient to high systemic concentrations that cause toxicities. Because M.
tuberculosis resides and multiplies within host mononuclear phagocytes and because
mononuclear phagocytes internalize particles more efficiently than other host cells,
encapsulation of anti-TB drugs in nanoparticles offers a mechanism for specific targeting of M.
tuberculosis-infected cells. Indeed, because nanoparticles have been shown to be taken up by
macrophages of the reticuloendothelial system and to accumulate in the liver, spleen and lung
[143–145], they are ideally suited to treat M. tuberculosis, which infects macrophages in these
organs. As macrophages exhibit a number of receptors, it is also possible to modify the surface
of these nanocarriers to achieve active targeting of macrophages. Sugars, such as mannose

27
[146] and lactose [147], are among the most commonly used for this purpose, since these
receptors are highly expressed in macrophages. Particle size is also an important characteristic
in passive targeting of macrophages, since they affect the success of internalization within
these cells. In this regard, particles with diameters of about 500 nm have been reported as ideal
to undergo phagocytosis by alveolar macrophages [148].
A variety of different nanocarriers have been produced and tested in vitro and in vivo as
delivery platforms for anti-TB drugs. Table 2 summarizes major data on anti-TB nanoparticulate
formulations.
Table 2 - Dosage forms of TB nanomedicine. From [149].
Mode of delivery and
dosage forms Drug evaluated Animal model
Sustained release
(days)
Plasma Organs
Oral
PLG NPs
Lectin-PLG NPs
Alginate NPs
SLNs
Inhalable
PLG NPs, nebulized
PLG NPs, insufflated
Lectin-PLG NPs
Alginate NPs
SLNs
Injectable
PLG NPs
RIF, INH, PZA
EMB
Streptomycin
Ethionamide
Econazole
Moxifloxacin
RIF, INH, PZA
RIF, INH, PZA, EMB
Econazole
RIF, INH, PZA
RIF, INH, PZA
RIF
RIF, INH, PZA
RIF, INH, PZA, EMB
RIF, INH, PZA
RIF, INH, PZA
Mice, guinea pigs
Mice
Mice
Mice
Mice
Mice
Guinea pigs
Mice, guinea pigs
Mice
Mice
Guinea pigs
Guinea pigs
Guinea pigs
Guinea pigs
Guinea pigs
Mice
6−8
3
4
6
5
4
6−14
7−11
6
8
6−8
0.25
6−14
7−11
5
32
9−11
7
7
5−7
6
6
15
15
8
10
9−11
0.3
15
15
7
36

28
2. Materials and Methods
2.1. Materials
2.1.1. Lipids
All lipids used in this work are well tolerated for human use and recognized as safe
(GRAS status) by FDA.
2.1.2. Surfactant
In this work was used a nonionic surfactant. Surfactants (also known as surface-active
agents or emulsifiers) are essential to stabilize lipid nanoparticles dispersions and prevent
particle agglomeration. Surfactants are amphipathic molecules that possess a hydrophilic
moiety (polar) and a lipophilic moiety (non-polar), which together form the typical head and the
tail of surfactants. At low concentrations, surfactants adsorb onto the surface of a system or
interface. They reduce the surface or interfacial free energy and consequently reduce the
surface or interfacial tension between the two phases [153]. The relative and effective
proportions of these two moieties are reflected in Hydrophilic-Lipophilic Balance, or HLB value.
The HLB scale, defined by Griffin, ranges in value from 1 to 20. Relative hydrophilicity increases
with increasing HLB value. A value of 1 represents a strongly hydrophobic surfactant.
Surfactants with HLB values in the range of 8–18 are suitable for the preparation of lipid
nanoparticles through oil-in-water dispersion medium [154].
The surfactants used include ionic, nonionic and amphoteric surfactants. Ionic surfactants
are traditionally thought to infer electrostatic stability, whilst nonionic surfactants are traditionally
thought to infer steric repulsion stability [153]. Nonionic surfactants are preferred for oral and
parenteral preparations over those with ionic properties because of their lower toxicity and
irritancy. In general, the toxicity of surfactants decreases in the order cationic > anionic >
nonionic > amphoteric [154].

29
2.1.2.1. Hexadecane
Hexadecane 99% was purchased from Sigma-Aldrich, and it was used as hydrophobic
agent for osmotic stabilization of the lipid nanoparticles. Hexadecane, also called cetane, is a
straight chain, saturated alkane hydrocarbon with molecular formula C16H34 and molecular
weight 226.4 g/mol. Its melting point is 18ºC, thus is a liquid at room temperature with a density
of 0.773 g/ml. Its research applications include the preparation of emulsions in water [157]. Its
structure is shown in Figure 9.
Figure 9- Structural formula of hexadecane
2.1.3. Bioactive Compounds
2.1.3.1. Rifampicin
Rifampicin is a semisynthetic antibiotic derivative of rifamycin B and it was purchased
from Sigma-Aldrich. Rifampicin is an orange-brown to red-brown crystalline powder very slightly
soluble in water at neutral pH (2.5 mg/ml, 25ºC) [158]. It is soluble in dimethylsulfoxide
(~100mg/mL), dimethylformamide, methanol (16 mg/ml, 25ºC), chloroform (349 mg/ml, 25°C),
ethyl acetate (108 mg/ml, 25°C), and acetone (14 mg/ml, 25°C) [158]. Its molecular weight is
822.95 g/mol and its chemical formula is C43H58N4O12 [158]. Its structure is shown in Figure 10.
Figure 10- Structural formula of rifampicin

30
2.1.3.2. Pyrazinamide
Pyrazinamide, the pyrazine analogue of nicotinamide, is a white, crystalline powder,
stable at room temperature and it was purchased from Sigma-Aldrich. Pyrazinamide is also
known as pyrazinecarboxamide and its molecular weight is 123.11 g/mol [159]. Its chemical
formula is C5H5N3O and its structural formula is shown in Figure 11 [159]. It is soluble in water
(15 mg/mL) and chloroform, slightly soluble in ethanol (95%) and very slightly soluble in polar
organic solvents [159]. Pyrazinamide is used to form polymeric copper complexes, create
pyrazine carboxamide scaffolds useful as a component of mycobacteria identification kits.
Figure 11 - Structural formula of pyrazinamide
2.1.3.3. β-carotene
For this work, β-carotene was also selected as model lipophile. β-carotene delivery is of
interest in the food industry, the nutraceutical industry, the skin care industry, and marketers of
antioxidant medications. β-carotene possesses a molecular weight of 536.87 g/mol and a
molecular formula of C40H56. It is a strongly-coloured red-orange pigment which belongs to a
group of plant compounds called carotenoids. Carotenoids consist of an isoprene polymer
backbone. β-carotene is composed of two retinyl groups, and is broken down in the mucosa of
the small intestine to retinal, which is a form of vitamin A. β-carotene can be stored in the liver
and converted in two molecules of vitamin A, being a provitamin. β-carotene is insoluble in
water, making it an excellent model small molecule lipophile drug. Figure 12 shows its
molecular structure.
Figure 12 – Structural formula of β-carotene
31
2.1.4. Water
The water used in all experiments was purified water by reverse osmosis and was
obtained from a Milli-Q Plus, Millipore system. The water had an electrical conductivity of 0.5-
1.5 μS/cm.
2.2. Methods
2.2.1. Preparation of Lipid Nanoparticles
Lipid nanoparticles were prepared by ultrasonication and simple magnetic stirring
methods. Both aqueous and lipid phases are separately prepared before mixing. The aqueous
phase is composed of water (≥ 80.0% w/w), non-ionic surfactant (≤8.0% w/w) and hexadecane
(≤3.5% w/w). Six formulations of empty SLNs was prepared ranging lipid content: SLN_1
represents the formulation with the lowest lipid phase content and SLN_6 represents the
formulation with the highest lipid phase content. For NLC formulations was used a combination
of natural oil with the MCFA. The content of bioactive compounds incorporated in lipid
nanoparticles are shown at Table 3.
Table 3 - Composition % (w/w) of bioactive compounds loaded to lipid nanoparticles.
In detail, the lipids are melted in a warm water bath above their melting points. Different
temperatures conditions up than 50ºC were tested. When the MCFA or the MCFA/natural oil
combination was fully melt, a hot aqueous solution with nonionic surfactant and hexadecane
Formulation β RIF PYZ
2RIF_SLN - 0.06 -
2RIF_NLC - 0.06 -
4RIF_SLN - 0.11 -
4RIF_NLC - 0.11 -
7RIF_SLN - 0.20 -
7RIF_NLC - 0.20 -
4PYZ_SLN - - 0.11
4PYZ_NLC - - 0.11
β10-SLN 0.3 - -
β10-NLC 0.3 - -

32
preheated at the same conditions was added. A hot coarse o/w was obtained under magnetic
stirring at 750 rpm for 1min and was subjected to sonication using a probe (MS72) sonicator
(Bandelin, Germany) during 5 minutes. Hot o/w emulsion was allowed to cool to room
temperature under magnetic stirring at 750 rpm to obtain the lipid nanoparticles. After 2h, the
samples of each formulation were stored both in a refrigerator at 4ºC and at room temperature.
For active agents-loaded lipid nanoparticles, the active agent was added to the lipid phase, 15
minutes earlier the addition of aqueous solution. Figure 13 shows the procedures.
Figure 13 – Lipid nanoparticles production. Left: Lipid phase and aqueous phase in the warm water bath
under same conditions (temperature and agitation). Top Right: Lipid phase (on the right) and aqueous
phase (on the left) after weight. Middle Right: Addition of hot aqueous phase in melt lipid phase. Bottom
Right: Sonication of hot pre-emulsion.

33
2.2.1.1. Lyophilisation of Lipid Nanoparticles
Lyophilisation (or freeze-drying) was the final step of the production process. This
process promotes water removal from a frozen sample, using sublimation and desorption under
vacuum. Many pharmaceutical products, mainly heat sensitive compounds, are dried using this
technique, since it improves the long-term physic-chemical stability and prevents degradation
reactions such as hydrolysis. Lyophilisation is also used with nanoparticles to prevent particle
aggregation.
The aqueous samples of lipid nanoparticles were fast frozen under −80 °C in a deep
freeze for 5h in ultra-low refrigerator. Then, the samples were moved to the freeze-drier (Christ
Alpha 1-2 LD) during 48h in order to get powder of lipid nanoparticles.
2.2.2. Characterization of Lipid Nanoparticles
2.2.2.1. Particle Size
In this work, a Zetasizer Nano ZS, Malvern Instruments (Malvern, UK) was used to
measure the size of lipid nanoparticles produced. Particle size measurements were made in
disposable cells at 25°C, by non-invasive back scatter, with dynamic light scattering detected at
an angle of 173°. One ml of samples were used and 1:10 dilutions were performed (900μL
ultrapure Milli-Q water + 100μL sample), to avoid multiple scattering phenomenon. The effective
hydrodynamic diameter was calculated from the diffusion coefficient by the Stokes-Einstein
equation (Eq.1) using the method of cumulants as an alternative to distribution analysis. In
summary, the cumulants analysis gives a good description of the size that is comparable with
other methods of analysis for spherical, reasonably narrow monomodal samples, with PDI value
below 0.1. For samples with a slightly increased width, the Z-average size and PDI will give
values that can be used for comparative purposes. All measurements were performed in
triplicates with 20 runs by measurement.
2.2.2.2. Zeta Potential
Zeta potential measurements were also performed using a Malvern Zetasizer Nano ZS,
Malvern Instruments (Malvern, UK) at 25ºC. The samples were diluted 1:10 in ultrapure Milli-Q
water and placed at a folded capillary cell (DTS1060) where an alternating voltage of ±150mV
was applied. The measured electrophoretic mobilities were converted into zeta potential values
using the Smoluchowski approximation. All measurements are performed in triplicate with 20
runs per measurement.

34
2.2.2.3. Particle Morphology
Morphological observations of the SLNs and NLCs were performed using a transmission
electron microscope (TEM). In this work, 20 µL of samples of with lipid nanoparticles previous
analysed by DLS was deposited over a carbon covered TEM copper grid and then dried at room
temperature. TEM images were collected using a microscope H-8100 Hitachi, operated with
200 kV of acceleration voltage incorporated with a CCD MegaView II bottom-mounted camera.
2.2.2.4. Encapsulation Efficiency
An indirect method was used to determine the encapsulation efficiency of lipid
nanoparticles using Eq.3 as explained in chapter 1.
For all three active compounds loaded within lipid nanoparticles the same following process
was used. After production, lipid nanoparticles were diluted and washed in milli-Q water. The
dispersion was vortexed and then centrifuged at 12000 rpm for 30 min at 4ºC using centrifuge
(Centrifuge 5810 R, Eppendorf). The supernatant was collected and the pellet was resuspended
in fresh milli-Q water. This procedure was repeated three times. After washes, the amount of
active agents in the supernatant was determined from its absorption using a double beam UV–
VIS spectrophotometer (Model U-2000, Hitachi) and 1cm quartz cells.
The amount of drug dissolved in supernatant can be quantified by spectrophotometer,
since absorbance of a solution at a specific wavelength is proportional to the solute's
concentration, as is expressed by Beer-Lambert's law:
A= εlC Eq.4
where ε is the molar absorptivity (a constant that indicates how well the solute absorbs light of a
particular wavelength, in units of M-1cm-1), l is the solution’s length the beam has to cross (1.0
cm for the quartz cell), and C is the solute’s concentration (mol/L).
In this work, the measures of absorbance were done at 333 nm for rifampicin and at 548
nm for β-carotene. Pyrazinamide was analysed based on the formation of a coloured complex
between the drug and sodium nitroprusside at alkaline pH with a mixing ratios of 4:1, which
could be measured at 495nm (Figure 14). The solution of sodium nitroprusside at alkaline pH
was prepared mixing a solution of 2% sodium nitroprusside in milli-Q water with a 2N sodium
hydroxide solution in a ratio of 1:1 [160].
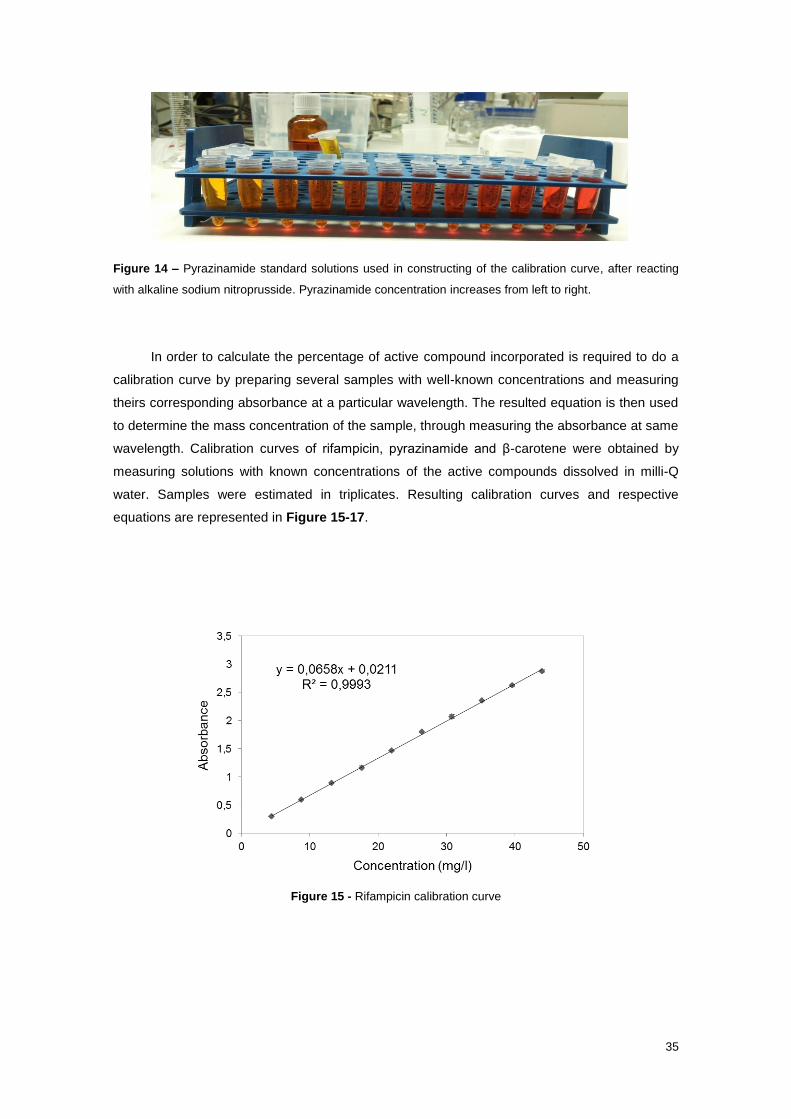
35
Figure 14 – Pyrazinamide standard solutions used in constructing of the calibration curve, after reacting
with alkaline sodium nitroprusside. Pyrazinamide concentration increases from left to right.
In order to calculate the percentage of active compound incorporated is required to do a
calibration curve by preparing several samples with well-known concentrations and measuring
theirs corresponding absorbance at a particular wavelength. The resulted equation is then used
to determine the mass concentration of the sample, through measuring the absorbance at same
wavelength. Calibration curves of rifampicin, pyrazinamide and β-carotene were obtained by
measuring solutions with known concentrations of the active compounds dissolved in milli-Q
water. Samples were estimated in triplicates. Resulting calibration curves and respective
equations are represented in Figure 15-17.
Figure 15 - Rifampicin calibration curve

36
Figure 16 - β-carotene calibration curve
Figure 17 - Pyrazinamide calibration curve

37
3. Results and Discussion
3.1. Study of Fabrication Parameters
3.1.1. Empty SLN
The first objective of this work was to study SLNs at room temperature and their
production conditions by miniemulsion using ultrasonication, based on a MCFA as solid lipid.
The greatest advantage of this procedure is the easily production of SLNs without using organic
solvents. In this work, a nonionic surfactant was employed, providing a steric hindrance,
avoiding emulsion droplets from coming close to each other and thus preventing flocculation
and coalescence. In this process, the MCFA is heated above its melting point. Aqueous solution
of surfactant is mixed with lipid melt at the same temperature. Shear force from sonication will
facilitate the formation of miniemulsion. The droplet size is governed by energy dissipation,
temperature, surfactant and lipid concentration and other factors. The hot emulsion is then
cooled down to form solid particles.
Since the purpose of SLNs produced are for oral administration, the particles must be an
average size less than 400 nm to easily cross intestinal cells [59]. Furthermore, the fabrication
of particles comprises 100-200 nm it would be better, since it was reported particles smaller
than 200 nm usually remain invisible to the reticulo-endothelial system (RES) and keep on
circulation system over a prolonged period of time [35]. For this purpose, it was tested the
influence of different parameters, such as applied lipid amount, temperature and sonication step
on mean particle size expressed as Z-average mean size (Z-ave), polydispersity index (PDI)
and zeta potential (ZP) (Table 4, Figure 18 and Figure 19) .
Mean particle size of all formulations ranged from 69 ± 5 nm to 601 ± 100 nm and PDI
from 0.87 ± 0.09 to 0.11 ± 0.02 (Table 4 and Figure 18). Since all the formulations shows a
PDI higher than 0.1, this indicates that none are monodisperse. Finally, the ZP range from -35.8
± 0.8 mV to -22.7 ± 0.7 mV (Table 4 and Figure 19), indicating good physical stability since
particles aggregation was not likely to occur due to the electrostatic repulsion between the
particles. For long-term stability a balance between electrostatic repulsion and steric effect of
surfactant must be obtained.
.

38
Table 4 - Physicochemical characteristics of empty SLN at different conditions (mean ± SD, n = 6).
Non-sonicated Sonicated
T (ºC)
Formulation Z-Ave (d.nm)
PdI ZP (mV) Z-Ave (d.nm)
PdI ZP (mV)
a
SLN_1 601 ± 100 0.70 ± 0.08 -29.6 ± 0.6 146 ± 2 0.18 ± 0.05 -25.8 ± 0.7
SLN_2 250 ± 6 0.56 ± 0.03 -27.5 ± 0.2 130 ± 9 0.18 ± 0.02 -27.8 ± 1.5
SLN_3 224 ± 28 0.46 ± 0.11 -28.8 ± 0.1 105 ± 1 0.18 ± 0.02 -27.8 ± 0.8
SLN_4 284 ± 19 0.87 ± 0.09 -30.4 ± 0.8 115 ± 1 0.18 ± 0.04 -29.4 ± 0.7
SLN_5 249 ± 16 0.70 ± 0.07 -29.3 ± 0.3 121 ± 3 0.15 ± 0.02 -28.6 ± 0.6
SLN_6 326 ± 6 0.85 ± 0.09 -32.4 ± 0.3 121 ± 1 0.18 ± 0.05 -28.1 ± 1.2
b
SLN_1 299 ± 8 0.55 ± 0.06 -31.0 ± 0.4 139 ± 2 0.17 ± 0.03 -27.2 ± 2.0
SLN_2 262 ± 13 0.44 ± 0.07 -29.9 ± 0.5 134 ± 23 0.18 ± 0.02 -25.1 ± 1.0
SLN_3 231 ± 25 0.54 ± 0.16 -28.3 ± 0.6 121 ± 25 0.20 ± 0.04 -30.3 ± 0.7
SLN_4 255 ± 8 0.47 ± 0.09 -30.0 ± 0.8 122 ± 10 0.17 ± 0.04 -28.6 ± 0.6
SLN_5 411 ± 22 0.51 ± 0.15 -26.5 ± 0.2 122 ± 11 0.19 ± 0.02 -27.2 ± 0.9
SLN_6 330 ± 55 0.45 ± 0.35 -35.8 ± 0.8 109 ± 19 0.18 ± 0.04 -31.7 ± 0.9
c
SLN_1 515 ± 140 0.52 ± 0.13 -30.6 ± 0.6 141 ± 1 0.14± 0.03 -28.4 ± 0.8
SLN_2 155 ± 4 0.26 ± 0.01 -28.3 ± 0.7 126 ± 7 0.15± 0.02 -26.8 ± 0.9
SLN_3 140 ± 1 0.20 ± 0.01 -29.3 ± 0.6 97 ± 30 0.23 ± 0.09 -28.9 ± 1.0
SLN_4 196 ± 2 0.30 ± 0.03 -31.5 ± 0.2 119 ± 1 0.13 ± 0.02 -29.2 ± 0.6
SLN_5 149 ± 1 0.29 ± 0.02 -29.9 ± 0.5 120 ± 1 0.17 ± 0.05 -28.5 ± 3.9
SLN_6 179 ± 1 0.25 ± 0.01 -25.5 ± 0.7 122 ± 2 0.18 ± 0.01 -28.2 ± 0.7
d
SLN_1 333 ± 50 0.41 ± 0.03 -30.0 ± 0.5 139 ± 1 0.11 ± 0.02 -26.6 ± 0.6
SLN_2 93 ± 7 0.30 ± 0.08 -29.1 ± 0.6 87 ± 12 0.21 ± 0.02 -22.7 ± 0.7
SLN_3 144 ± 44 0.87 ± 0.05 -12.3 ± 0.6 105 ± 1 0.23 ± 0.03 -28.4 ± 0.6
SLN_4 90 ± 3 0.46 ± 0.03 -29.3 ± 0.3 69 ± 5 0.19 ± 0.03 -29.4 ± 0.7
SLN_5 99 ± 2 0.49 ± 0.05 -34.7± 1.5 97 ± 30 0.23 ± 0.09 -28.9 ± 1.0
SLN_6 121 ± 2 0.40 ± 0.02 -30.6 ± 0.3 126 ± 7 0.19 ± 0.03 -26.0 ± 0.2

39
Figure 18- Z-ave and PDI of empty SLNs with different content of MCFA (SLN_1 represents the lowest
lipid phase content and SLN_6 represents the highest lipid phase content), ranging from a lowest
temperature (a) to highest temperature (d).

40
Figure 19 - Zeta Potential of empty SLNs with different content of MCFA (SLN_1 represents the lowest
lipid phase content and SLN_6 represents the highest lipid phase content), ranging from a lowest
temperature (a) to highest temperature (d).

41
3.1.1.1. Influence of Temperature
The effect of temperature on pre-emulsion was investigated. Four different temperatures
up than 50ºC were applied. The main influence of temperature was observed on non-sonicated
samples. Table 4 shows a notorious size reduction of non-sonicated particles with the increase
of temperature, while PDI and ZP values remain with non-significant variation. These results are
expected since in general, the increase of temperature usually decreases the viscosity of the
lipid and aqueous phases and hence the efficiency of the agitation and mixture is increased. In
other words, increasing the fusion temperature of the lipids results in smaller droplet/particle
sizes [113].
3.1.1.2. Influence of Lipid Content
The influence of MCFA content on SLNs properties were also investigated (Table 4 and
Figure 18-19). From analysis of non-sonicated samples in a, c and d of Figure 18, it seems
evident that particle size of SLNs prepared with the lowest concentration of MCFA are larger
when compared with other SLNs. In addition, PDI of these samples are also high. However, ZP
appears not be affected as well as all features considered of lipid particles obtained by
sonication.
As PDI of non-sonicated SLNs produced with lowest lipid content are also very high, the
Ostwald ripening effects could be an explanation for their larger sizes. Ostwald ripening is a
thermodynamically driven process, in which smaller particles dissolve and redeposit onto the
surface of larger particles. This process occurs because smaller particles have larger surface
area and higher surface energy and hence higher Gibbs free energy than the larger particles.
All systems tend to attain lowest Gibbs free energy. In other words, larger particles are more
energetically stable and thus, preferred over smaller particles. Ostwald ripening can be reduced
by minimizing PDI in the particle size but it cannot be prevented.
In conclusion, although in general low lipid content decreases the aggregation
phenomenon, it may also increase the Ostwald ripening effects and causes the migration of
surfactant molecules from the particle interface to aqueous medium decreasing the electrostatic
repulsive force and leading particle aggregation.

42
3.1.1.3. Influence of Sonication
The influence of sonication on SLNs features was represented in Table 4 and Figures
18-19.
Figure 20 - Macroscopic aspect of empty SLNs: (left) - non-sonicated sample; (right) –
sonicated sample
Comparing the macroscopic aspect between non-sonicated and sonicated SLNs
suspension (Figure 20), it is possible to see that non-sonicated SLNs dispersion has a milky
aspect, while sonicated SLNs dispersion are more clear and bluish. From literature, usually
colloidal dispersions with droplet sizes between 20–100 nm are transparent or bluish, while
colloidal dispersions with larger droplets sizes up to 500 nm has a milky aspect [107]. Thus,
through macroscopic analysis, it can be expected a decreased on mean SLNs particle size of
sonicated samples.
The particle size reduction is confirmed by DLS results. At Figure 18, it is clear the effect
of sonication in both Z-ave and PDI of MCFA SLNs. All sonicated SLNs show a mean Z-ave
lower than 150 nm. This is due to shear stress, which is resultant from the application of
ultrasound energy that promotes a shear on MCFA molecules, impairing aggregation, thus,
avoiding the formation of larger particles. Despite of this significant effect on particle size,
ultrasonication does not appear to play an essential role in physical stability of SLNs, since ZP
is very similar between non-sonicated and sonicated SLNs, although there is an insignificant
decrease in absolute ZP value (Figure 19).
In conclusion, the application of ultrasound energy seems to decrease the mean SLNs
particle size and also homogenize the SLNs in suspension, since PDI values decrease when
sonication is applied.

43
3.1.1.4. Lyophilisation
Figure 21 – Powder resulting of SLN_1b_II lyophilisation
Lyophilisation was applied to some SLNs samples in order to remove the excess water
and also to enhance stability of powder SLNs, since Ostwald ripening can be avoided and
particles aggregation decreased. The obtained powder of SLN_1b_II is shown at Figure 21.
Table 5 presents the Z-ave, PDI and ZP of the SLN_1b_II before and after lyophilisation.
Z-ave of lyophilised SLNs is slightly larger when compared to liquid dispersion, increasing from
153 ± 31 nm to 191 ± 4 nm. However, PDI of lyophilised SLNs increased from 0.13 ± 0.04 to
0.49 ± 0.10, resulting on a sample far polydisperse. Examining particle size distribution of
SLN_1b_II before and after lyophilisation (Figure 22), it is possible to realize the appearance of
a second particles population larger than 1 µm in reconstituted lyophilised sample. This larger
population could be result of aggregation phenomenon, indicating physical instability of SLNs
and explaining the increase of PDI. However, contrary as expected, absolute ZP value
increased from 30 ± 2 mV to 35 ± 2 mV, meaning an increase on particles stability. An
explanation for these results could be an insufficient stirring force/time applied on resuspension
of lyophilised SLNs in milli-Q water. If the attractive forces holding the aggregate together are
very large compared with the typical forces used in stirring, mixing or ultrasonic probes, the
system is regarded as permanently aggregated. Thus, the larger population of SLNs on
lyophilised SLN_1b_II could be a result of flocculation. To explore this hypothesis, it could be
done a comparative study between different forces used to resuspension of lyophilised SLNs.
In addition, it could be investigated the combination of a cryoprotector previously to
lyophilisation since it was found that cryoprotective agents preserve the physicochemical
properties of SLNs and decrease SLNs aggregation. Typical cryoprotectors are sorbitol,
mannose, trehalose, glucose, and polyvinylpyrrolidone. The best results occurred when
cryoprotector is in concentrations of 10-15% [162].

44
Table 5 – Physicochemical characteristics of the SLN_1b_II. Values are mean ± SD, n = 3
Formulation Z-Average
(nm) PDI
ZP
(mV)
SLN_1b_II 153 ± 32 0.13 ± 0.04 -30 ± 2
Lyophilised SLN_1b_II 191 ± 4 0.49 ± 0.10 -35 ± 2
Figure 22 – Particle Size Distribution by Intensity of SLN_1b_II before and after lyophilisation.
3.1.1.5. Long-Term Stability
In this work, SLNs formulations were stored at room temperature (25ºC) and in the
refrigerator (4ºC), during seven months. The long-term stability of both formulations was
assessed by means of particle size (Z-ave and PDI) and ZP measurements. The stability was
also monitored by macroscopic observations.
The samples stored at room temperature remained with a milky-like appearance,
although some of samples show particle sedimentation. However, during storage for more than
one week some unpredictable gelation occurred in SLNs containing samples stored at 4ºC.
Gels are more structured than liquids. SLNs are able to organize themselves in superstructures.
The change in morphology of lipid nanoparticles from spheres to platelets is responsible for the
gelation of SLN dispersions [163]. Depending on the composition, especially of the emulsifier(s)
and the amount of lipid matrix, a gelation of the normally liquid dispersions can be observed on
storage [163].
Table 6 shows the obtained Z-ave, PDI and ZP results for SLN_4b_I, an example of a
macroscopic milky-like colloidal SLNs dispersion stored at room temperature. From results
analysis, it can notice a decrease in the absolute ZP value from 39.1 ± 0.9 mV to 28.3 ± 0.6 mV,
indicating changes in the particles structure. This can be confirmed by the DLS measurements
where particle size growth was observed (Table 6 and Figure 23). The mean particle size

45
remained lower than 400 nm, although there was an increasing on the size from 154 ± 25 nm to
223 ± 54 nm. Since PDI and particle size remain relatively low, homogeneously sized SLNs
displayed a satisfactory long-term stability.
Table 6 - Physicochemical characteristics of the SLN_4b_I. Values are mean ± SD, n = 3.
Formulation Z-Average
(nm) PDI
ZP
(mV)
SLN_4b_I 154 ± 25 0.07 ± 0.03 -39.1 ± 0.9
SLN_4b_I after 7months 223 ± 54 0.05 ± 0.02 -28.3 ± 0.6
Figure 23 - Particle Size Distribution by Intensity of SLN_4b_I after one day and 7 months after
production.
The shape and surface morphology of empty SLNs after seven months were also
analysed by TEM and the resulting images are represented in Figure 24. In the TEM study, the
size of the SLNs was found to be in agreement with the DLS. All the particles were found to be
smoothly spherical or oval in shape with a well-defined periphery. No obvious aggregation of the
SLNs was observed. It should be noted, however, that the particles align in three dimensions,
whereas the micrographs only allow particle presentation in two dimensions so that the given
particle dimensions are only estimates due to the fact that a particle observation exactly top-on
and edge-on, respectively, is rare and cannot be discriminated from slight particle twists, in
addition.

46
Figure 24 – TEM image of SLN_4b_I after 7 months.
3.1.2. Empty NLC
For NLC production, the fabrication procedure was similar to the SLNs fabrication and
was described in chapter 2.
Table 7 presents the Z-ave, PDI and ZP of the NLC. The Z-ave of NLC is larger when
compared to SLN_4b. Since both lipid nanoparticles have an equal percentage of lipid content
and were produced at same conditions, the increase of the size from 119 ± 0 nm to 282 ± 56 nm
are related with the use of natural oil. The combination of different chain fatty acid triglycerides
promotes changes in structure of lipid nanoparticles. These changes on particles surface are
also supported by the slightly reduction of absolute ZP value from -29.2 ± 0.6 mV to -26.4 ± 0.7
mV. In conclusion, despite the growth of particle size, NLC size remains lower than 400 nm, as
required to easily cross intestinal cells.
Table 7 – Physicochemical characteristics of the empty NLC. Values are mean ± SD, n = 3.
Formulation Z-Average
(nm) PDI
ZP
(mV)
NLC 282 ± 56 0.43 ± 0.09 -26.4 ± 0.7

47
3.2. Bioactive Compounds Loaded SLNs and NLCs
The second main goal of this project was to investigate the entrapment efficiency of some
bioactive compounds in MCFA based lipid nanoparticles. The anti-TB drugs rifampicin and
pyrazinamide were chosen to encapsulate within lipid nanoparticles and β-carotene was also
used as lipophilic compound model.
From the first part of this study, it had to be considered a compromise between the
adequate size, a low PDI and also a good stability of the samples for the final choice of the
parameters to be used in loaded lipid nanoparticles production. As mentioned above, the
desired formulations have to be lower than 400 nm. Furthermore, the fabrication of particles
comprises 100-200 nm it would be better, since it was reported particles smaller than 200 nm
usually remain invisible to the reticulo-endothelial system (RES) and keep on circulation system
over a prolonged period of time [35]. Taking all this considerations into account, the final
parameters chosen to lipid nanoparticles production were those that correspond to the SLN_4
formulation and the temperature of lipid melting must be up 60ºC, followed by 5 minutes of
sonication. Optimal temperature also depends of drug characteristics.
Figure 25 shows the difference in macroscopic appearance of empty and loaded NLCs
before and after sonication. After sonication, all the dispersions are significantly more
transparent.
Figure 25 – Differences in macroscopic appearance of empty and loaded NLCs immediately before and
after sonication.

48
3.2.1. β-carotene
β-carotene was incorporated in SLNs and NLCs. The Z-ave, PDI and ZP of loaded
particles were measured one day after production and the results are represented in Table 8.
The %EE of the prepared lipid nanoparticles were determined indirectly, by calculating the
amount of free β-carotene (non-encapsulated) present in the aqueous phase of dispersions, as
explained in chapter 2 and was also represented in Table 8. As expected, SLNs and NLCs
loaded with β-carotene are larger when compared with empty SLNs (Table 4) and empty NLCs
(Table 7). Particle sizes remain lower than 400 nm, as required. No important changes seem to
occur on stability of both loaded SLNs and NLCs, since ZP values remain similar from empty
SLNs and NLCs. In addition, comparing with rifampicin (Table 9) and pyrazinamide (Table 10)
loaded-particles, β-carotene shows the highest percentage of encapsulation efficiency. This
result was predictable, since β-carotene is the most lipophilic of these three active compounds.
Also, since β-carotene is less soluble in oil then in fat, the %EE of β10-SLN is higher than β10-
NLC.
Table 8 - Physicochemical characteristics of the β-carotene lipid nanoparticles. Values are mean ±
SD, n = 3.
Formulation Z-Average
(nm) PDI
ZP
(mV)
EE
(%)
β10-SLN 281 ± 19 0.34 ± 0.03 -29 ± 5 95 ± 5
β10-NLC 328 ± 65 0.28 ± 0.06 -25 ± 3 83 ± 6
Morphological analysis of β-NLC was performed by TEM and the resulting images are
represented in Figure 26. The size of the particles was in agreement with DLS results. TEM
revealed nanoparticles with almost spherical shapes and a roughness surface. However, it was
also reported the presence of some aggregates and some particles with an irregular shape.
Figure 26- TEM image of β-carotene loaded NLC.

49
3.2.2. Rifampicin
The Z-ave, PDI, ZP and %EE of RIF loaded lipid nanoparticles are presented in Table 9.
As expected RIF-NLCs samples are larger than RIF-SLNs. In addition, RIF loaded particles
show a reduction in the electrical charge at the surface to mean values below -20 mV. These
results could be taken as an indication that RIF is entrapped in the lipid matrix of lipid
nanoparticles. Due to the high solubility of RIF in the lipid core, the encapsulation efficiency was
good enough for all the formulations, ranging between 67 ± 7% and 85 ± 5%. In literature, it was
found that NLCs can encapsulate slightly more amount of drug than SLNs. However, none
significant differences in %EE it seems to exist between RIF-SLNs and RIF-NLCs. Muller et al.
also reported that the presence of liquid lipid in the solid matrix avoids the drug expulsion during
storage that can occur when the lipid matrix undergoes polymorphic transformations from
unstable to more stable configurations [28]. In this work, no long-term stability study was done
to verify this event. And, since, it is obvious that polymorphic crystal transformation during
storage can affect the encapsulation efficiency and the drug release profile of lipid
nanoparticles, additional thermal and crystallization studies must be done in order to a better
control of encapsulation efficiency and the drug release.
Table 9 - Physicochemical characteristics of the rifampicin lipid nanoparticles. Values are mean ±
SD, n = 3.
Formulation Z-Average
(nm) PDI
ZP
(mV)
EE
(%)
2RIF_SLN 341 ± 78 0.45 ± 0.12 -20.3 ± 2.2 74 ± 10
2RIF_NLC 391 ± 210 0.62 ± 0.10 -18.5 ± 1.3 67 ± 7
4RIF_SLN 84 ± 1 0.23 ± 0.01 -17.8 ± 0.7 69 ± 1
4RIF_NLC 180 ± 90 0.36 ± 0.08 -19.5 ± 0.9 69 ± 1
7RIF_SLN 181 ± 31 0.34 ± 0.03 -19.2 ± 1.3 79 ± 7
7RIF_NLC 215 ± 5 0.28 ± 0.03 -18.8 ± 0.6 85 ± 5
From TEM analysis represented at Figure 27, it can be observed that RIF-NLCs also
exhibit spherical shapes, which indicates drug loading is not lead to morphological changes.
Again, the particle size was in concordance with particle size analysis data obtained from
dynamic light scattering

50
Figure 27 - TEM image of rifampicin loaded NLC.
In literature, it was found two studies involving the encapsulation of RIF in lipid
nanoparticles. In first one, RIF was loaded into Compritol ATO 888 SLNs fabricated by a
modified microemulsion technique. The particles sizes obtained were 141 ± 13nm, ZP value
was −3.5 ± 0.8mV and %EE was 65 ± 3% [164]. In second one, RIF was encapsulated in group
with pyrazinamide and isoniazid in a stearic acid SLNs produced by emulsion solvent diffusion
with a %EE of 51 ± 5% [89]. Perhaps due to RIF was been encapsulated combined with other
anti-TB drugs the %EE was significant lower than results described in this work. In general,
results obtained in this project seem to be very good, by comparison with reported results by
literature. For one hand, it was obtained higher encapsulation efficiency and for another hand,
fabrication method applied is free-use of organic solvent, meaning less toxicity of the obtained
particles.
3.2.3. Pyrazinamide
The results of Z-ave, PDI, ZP and %EE of pyrazinamide loaded lipid nanoparticles are
represented in Table 10. The %EE value obtained range from 14 ± 8% for PYZ-SLN to 29±15%
for PYZ-NLC. These low results are expected since pyrazinamide is the most hydrophilic
compound in that study, being soluble in water (15 mg/ml). Thus, pyrazinamide partitioned
between the melted lipid and aqueous phase.
However, in literature pyrazinamide was encapsulated in a stearic acid SLNs combined
with isoniazid and rifampicin and %EE reported was 41 ± 7% [89]. Since pyrazinamide is one of

51
anti-TB drugs administered in current chemotherapy, in further studies could be investigated the
combine incorporation of pyrazinamide with other anti-TB drugs such as rifampicin, isoniazid or
ethambutol. Furthermore, the process of cooling from high to room temperature could be
optimized in order to control crystallization processes and consequently the encapsulation
efficiency.
Table 10 - Physicochemical characteristics of the pyrazinamide lipid nanoparticles. Values are
mean ± SD, n = 3.
Formulation Z-Average
(nm) PDI
ZP
(mV)
EE
(%)
4PYZ-SLN 217 ± 87 0.31 ± 0.08 -21.0 ± 0.7 14 ± 8
4PYZ-NLC 313 ± 214 0.34 ± 0.17 -22.7 ± 2.4 29 ± 15
The resulting images of TEM analysis of PYZ-NLC are represented in Figure 28. The
size of the particles was in agreement with DLS results. However, TEM revealed nanoparticles
with poorly spherical and irregular shapes. It was also reported the presence of a lot of
aggregates, indicating important alterations on PYZ loaded particles structure.
Figure 28 - TEM image of pyrazinamide loaded NLC

52
4. Conclusions and Further Works
In the present work MCFA SLNs and NCLs with a combination of MCFA and natural oil
as lipid phase were successfully prepared by sonication technique. This technique was simple,
reproducible, prepared nanoparticles without the need of organic solvents or any sophisticated
instruments and has the potential to easily scale up for large scale production.
Furthermore, the lipid nanoparticles obtained in the present work are suitable carrier
systems for the incorporation of different bioactive compounds intended for oral administration,
since mean particle size of all formulations ranged from 69 ± 5 nm to 601 ± 100 nm and the
great part of the formulations revealed a particle size less than 400 nm which easily cross
intestinal cells. The presence of a small quantity of bigger particles was not seen as critical,
because the preparations were intended for oral use and not for intravenous administration.
Also, it was obtained a lot of formulations with a particle size less than 200 nm, which is an even
better result since they will remain invisible to the reticulo-endothelial system (RES) and keep
on circulation system over a prolonged period of time. The Zeta Potential obtained was
negatively enough to ensure a good physical stability of the particles, indicating that the
nonionic surfactant used is a good surfactant for lipid nanoparticles produced with this particular
formulation. In general, the formulations show a PDI higher than 0.1, indicating that none are
monodisperse. To obtain formulations with lower PDI values, such that they could be
considered monodisperse, filtration could be done with porous of low diameters, such as 450
nm or even 200 nm. In industry, other methods to produce lipid nanoparticles could be used,
and some of them, like high pressure homogenization, will naturally produce monodisperse
populations of particles.
Morphological studies using TEM images showed spherical to oval SLNs and RIF-NLCs
with well-defined periphery. The particle size was in concordance with particle size analysis
data obtained from dynamic light scattering. However, the β-NLCs and PYZ-NLCs show an
irregular shape and some tendency to aggregate.
Lyophilisation of lipid nanoparticles seems to be a good alternative to remove excess of
water and to enhance stability of the particles during the storage. Also, the obtained powders
can be used to produce classic solid dosage forms, can be redispersed in water or juice prior to
administration, and can also be used for the filling of hard gelatine capsules. However, further
studies must be done to prevent aggregation of lyophilised particles. It could be explored the
use of an additional cryoprotector previously lyophilisation. Furthermore, other alternatives to
lyophilisation could be investigated such spray drying. This method has been used scarcely for
SLNs formulation, although it is cheaper than lyophilisation
Rifampicin, Pyrazinamide and β-carotene were successful loaded at lipid nanoparticles.
Rifampicin encapsulation efficiencies obtained in this work were higher than the values reported
in literature for encapsulation of rifampicin in lipid nanoparticles. Pyrazinamide shows low
encapsulation efficiencies due to its hydrophilicity leading to partition between lipid phase and

53
aqueous phase. However, pyrazinamide could be encapsulated combined with other anti-TB or
could be done a stearic modification in order to improve its entrapment into solid matrix. In
further studies could be explored the encapsulation of isoniazid and ethambutol in produced
lipid nanoparticles. Nevertheless, further analysis such as DSC must be performed to
characterize the state and the degree of crystallinity of lipid dispersions provide guidance to
better control polymorphic transition of lipid nanoparticles, which have profound impact on
increasing loading capacity, encapsulation efficiency, manipulating release pattern and even
improving long term stability of lipid nanoparticles.
Furthermore, the next step on the characterization of produced lipid nanoparticles must
be in vitro releases and antibacterial activity studies. In addition, further studies related with
cytotoxicity and uptake of lipid nanoparticles could be done using Caco-2 cell line since it is
often used as a model to mimic the GI tract conditions.
In conclusion, despite being essential several further studies to ensure the efficacy of
obtained lipid nanoparticles as drug delivery system for oral route, these initial results are very
promising, and so they should encourage us to go farther.

54
References
[1] Feynman RP (1960) Engineering and Science. 22–36.
[2] Taniguchi N (1974) On the Basic Concept of “Nano-Technology.” Bull. Japan Soc.
Precis. Eng. 18–23.
[3] Attama AA, Momoh MA, Builders PF (2012) Lipid Nanoparticulate Drug Delivery
Systems : A Revolution in Dosage Form Design and Development. In Recent Advances
in Novel Drug Carrier Systems.
[4] Fahr A, Liu X (2007) Drug delivery strategies for poorly water-soluble drugs. Expert
Opin. Drug Deliv. 4, 403–16.
[5] Mehnert W, Mäder K (2001) Solid lipid nanoparticles: production, characterization and
applications. Adv. Drug Deliv. Rev. 47, 165–96.
[6] Barratt GM (2000) Therapeutic applications of colloidal drug carriers. Pharm Sci
Technolo Today 3, 163–71.
[7] Van de Waterbeemd H (1998) The fundamental variables of the biopharmaceutics
classification system (BCS): a commentary. Eur J Pharm Sci 7, 1–3.
[8] Yadav N, Khatak S, Vir U, Sara S (2013) Solid Lipid Nanoparticles - A review. Int. J.
Appl. Pharm. 5,.
[9] Wilczewska AZ, Niemirowicz K, Markiewicz KH, Car H (2012) Nanoparticles as drug
delivery systems. Pharmacol. Reports 64, 1020–1037.
[10] Müller RH, Mäder K, Gohla S (2000) Solid lipid nanoparticles (SLN) for controlled drug
delivery - a review of the state of the art. Eur. J. Pharm. Biopharm. 50, 161–77.
[11] Ahlin P, Kristl J, Šentjurc M, Štrancar J, Pečar S (2000) Influence of spin probe structure
on its distribution in SLN dispersions. Int. J. Pharm. 196, 241–244.
[12] Gaucher G, Satturwar P, Jones M-C, Furtos A, Leroux J-C (2010) Polymeric micelles for
oral drug delivery. Eur. J. Pharm. Biopharm. 76, 147–158.
[13] Gaucher G, Marchessault RH, Leroux J-C (2010) Polyester-based micelles and
nanoparticles for the parenteral delivery of taxanes. J. Control. Release 143, 2–12.
[14] Kamimura M, Nagasaki Y (2014) Chapter 14 - PEGylated polymer micelles for
anticancer drug delivery carrier. In, Makino HOBT-C and IS in PR and D, ed. Elsevier,
Amsterdam, pp. 285–298.
[15] Taha EI, Badran MM, El-Anazi MH, Bayomi MA, El-Bagory IM (2014) Role of Pluronic
F127 micelles in enhancing ocular delivery of ciprofloxacin. J. Mol. Liq. 199, 251–256.
[16] He C, Yin L, Tang C, Yin C (2013) Multifunctional polymeric nanoparticles for oral
delivery of TNF-α siRNA to macrophages. Biomaterials 34, 2843–2854.
[17] Menon JU, Ravikumar P, Pise A, Gyawali D, Hsia CCW, Nguyen KT (2014) Polymeric
nanoparticles for pulmonary protein and DNA delivery. Acta Biomater. 10, 2643–2652.
[18] Patel T, Zhou J, Piepmeier JM, Saltzman WM (2012) Polymeric nanoparticles for drug
delivery to the central nervous system. Adv. Drug Deliv. Rev. 64, 701–705.

55
[19] Kovacevic a, Savic S, Vuleta G, Müller RH, Keck CM (2011) Polyhydroxy surfactants for
the formulation of lipid nanoparticles (SLN and NLC): effects on size, physical stability
and particle matrix structure. Int. J. Pharm. 406, 163–72.
[20] Müller RH, Shegokar R, Keck CM (2011) 20 years of lipid nanoparticles (SLN and NLC):
present state of development and industrial applications. In Current Drug Discovery
Technologies, pp. 207–227.
[21] Sadekar S, Ghandehari H (2012) Transepithelial transport and toxicity of PAMAM
dendrimers: Implications for oral drug delivery. Adv. Drug Deliv. Rev. 64, 571–588.
[22] Nasr M, Najlah M, D’Emanuele A, Elhissi A (2014) PAMAM dendrimers as aerosol drug
nanocarriers for pulmonary delivery via nebulization. Int. J. Pharm. 461, 242–250.
[23] Patri AK, Kukowska-Latallo JF, Baker Jr. JR (2005) Targeted drug delivery with
dendrimers: Comparison of the release kinetics of covalently conjugated drug and non-
covalent drug inclusion complex. Adv. Drug Deliv. Rev. 57, 2203–2214.
[24] Spataro G, Malecaze F, Turrin C-O, Soler V, Duhayon C, Elena P-P, Majoral J-P,
Caminade A-M (2010) Designing dendrimers for ocular drug delivery. Eur. J. Med.
Chem. 45, 326–334.
[25] Müller RH, Gohla S, Keck CM (2011) State of the art of nanocrystals – Special features,
production, nanotoxicology aspects and intracellular delivery. Eur. J. Pharm. Biopharm.
78, 1–9.
[26] Junyaprasert VB, Morakul B (2014) Nanocrystals for enhancement of oral bioavailability
of poorly water-soluble drugs. Asian J. Pharm. Sci. (in press).
[27] Müller RH, Runge S, Ravelli V, Mehnert W, Thünemann a F, Souto EB (2006) Oral
bioavailability of cyclosporine: solid lipid nanoparticles (SLN) versus drug nanocrystals.
Int. J. Pharm. 317, 82–9.
[28] Müller RH, Radtke M, Wissing SA (2002) Solid lipid nanoparticles (SLN) and
nanostructured lipid carriers (NLC) in cosmetic and dermatological preparations. Adv.
Drug Deliv. Rev. 54, Supple, S131–S155.
[29] Schubert R (1991) Liposomes in topical application and their mode of action in the skin.
Arch. Pharm 324, 627–633.
[30] Küchler S, Herrmann W, Panek-Minkin G, Blaschke T, Zoschke C, Kramer KD, Bittl R,
Schäfer-Korting M (2010) SLN for topical application in skin diseases--characterization of
drug-carrier and carrier-target interactions. Int. J. Pharm. 390, 225–33.
[31] Chen Y-C, Liu D-Z, Liu J-J, Chang T-W, Ho H-O, Sheu M-T (2012) Development of
terbinafine solid lipid nanoparticles as a topical delivery system. Int. J. Nanomedicine 7,
4409–18.
[32] Muchow M, Maincent P, Muller RH (2008) Lipid nanoparticles with a solid matrix (SLN,
NLC, LDC) for oral drug delivery. Drug Dev. Ind. Pharm. 34, 1394–405.
[33] Zhang N, Ping Q, Huang G, Xu W, Cheng Y, Han X (2006) Lectin-modified solid lipid
nanoparticles as carriers for oral administration of insulin. Int. J. Pharm. 327, 153–9.

56
[34] Venishetty VK, Chede R, Komuravelli R, Adepu L, Sistla R, Diwan P V (2012) Design
and evaluation of polymer coated carvedilol loaded solid lipid nanoparticles to improve
the oral bioavailability: a novel strategy to avoid intraduodenal administration. Colloids
Surf. B. Biointerfaces 95, 1–9.
[35] Bhandari R, Kaur IP (2013) Pharmacokinetics, tissue distribution and relative
bioavailability of isoniazid-solid lipid nanoparticles. Int. J. Pharm. 441, 202–12.
[36] Silva C, González-Mira E, García ML, Egea M, Fonseca J, Silva R, Santos D, Souto EB,
Ferreira D (2011) Preparation, characterization and biocompatibility studies on
risperidone-loaded solid lipid nanoparticles (SLN): high pressure homogenization versus
ultrasound. Colloids Surf. B. Biointerfaces 86, 158–65.
[37] Reitmeier H, Herrmann J, Gopferich A (2001) Lipid microparticles as a parenteral
controlled release device for peptides. J. Control. Release 73, 339–350.
[38] Wissing S a, Kayser O, Müller RH (2004) Solid lipid nanoparticles for parenteral drug
delivery. Adv. Drug Deliv. Rev. 56, 1257–72.
[39] Merodio M, Irache JM, Valamanesh F, Mirshahi M (2002) Ocular disposition and
tolerance of ganciclovir- loaded albumin nanoparticles after intravitreal injection in rats.
Biomaterials 23, 1587–1594.
[40] Paranjpe M, Müller-Goymann CC (2014) Nanoparticle-mediated pulmonary drug
delivery: a review. Int. J. Mol. Sci. 15, 5852–73.
[41] Liu J, Gong T, Fu H (2008) Solid lipid nanoparticles for pulmonary delivery of insulin. Int
J Pharm 356, 333–44.
[42] Muttil P, Wang C, Hickey AJ (2009) Inhaled drug delivery for tuberculosis therapy.
Pharm. Res. 26, 2401–16.
[43] Pandey R, Khuller GK (2005) Solid lipid particle-based inhalable sustained drug delivery
system against experimental tuberculosis. Tuberculosis (Edinb). 85, 227–34.
[44] Fang Y-P, Wu P-C, Huang Y-B, Tzeng C-C, Chen Y-L, Hung Y-H, Tsai M-J, Tsai Y-H
(2012) Modification of polyethylene glycol onto solid lipid nanoparticles encapsulating a
novel chemotherapeutic agent (PK-L4) to enhance solubility for injection delivery. Int. J.
Nanomedicine 7, 4995–5005.
[45] Lucks JS, Müller RH (1993) Medication vehicles made of solid lipid particles (Solid Lipid
Nanospheres - SLN).
[46] Gasco M (1993) Method for producing solid lipid microspheres having a narrow size
distribution. US Pat. 5,250,236.
[47] Westesen K, Bunjes H, Koch MHJ (1997) Physicochemical characterization of lipid
nanoparticles and evaluation of their drug loading capacity and sustained release
potential. J. Control. Release 48, 223–236.
[48] Jenning V, Thünemann AF, Gohla SH (2000) Characterisation of a novel solid lipid
nanoparticle carrier system based on binary mixtures of liquid and solid lipids. Int. J.
Pharm. 199, 167–177.

57
[49] Müller RH, Radtke M, Wissing SA (2002) Solid lipid nanoparticles (SLN) and
nanostructured lipid carriers (NLC) in cosmetic and dermatological preparations. Adv.
Drug Deliv. Rev. 54, 131–155.
[50] Tamjidi F, Shahedi M, Varshosaz J, Nasirpour A (2013) Nanostructured lipid carriers
(NLC): A potential delivery system for bioactive food molecules. Innov. Food Sci. Emerg.
Technol. 19, 29–43.
[51] Mudie DM, Amidon GL, Amidon GE (2012) Physiological Parameters for Oral Delivery
and in Vitro Testing. Mol. Pharm. 7, 1388–1405.
[52] Das S, Chaudhury A (2011) Recent advances in lipid nanoparticle formulations with solid
matrix for oral drug delivery. AAPS PharmSciTech 12, 62–76.
[53] Wagner D, Spahn-Langguth H, Hanafy A, Koggel A, Langguth P (2001) Intestinal drug
efflux: formulation and food effects. Adv Drug Deliv Rev 50, S13–31.
[54] Touitou E, Barry B, eds. (2006) Enhancement in drug delivery, CRC Press, Florida.
[55] Crounse R (1961) Human pharmacology of griseofulvin: the effect of fat intake on
gastrointestinal absorption. J Invest Dermatol 37, 529–33.
[56] Horter D, Dressman J (2001) Influence of physicochemical properties on dissolution of
drugs in the gastrointestinal tract. Adv Drug Deliv Rev 46, 75–87.
[57] Trevaskis NL, Charman WN, Porter CJH (2008) Lipid-based delivery systems and
intestinal lymphatic drug transport: a mechanistic update. Adv. Drug Deliv. Rev. 60, 702–
16.
[58] Chakraborty S, Shukla D, Mishra B, Singh S (2009) Lipid--an emerging platform for oral
delivery of drugs with poor bioavailability. Eur. J. Pharm. Biopharm. 73, 1–15.
[59] Hyuk Suh W, Suslick S, Stucky D, Suh Y-H (2009) Nanotechnology, nanotoxicology, and
neuroscience. Pro Neurobiol 87, 133–170.
[60] Liu C, Liu D, Bai F, Zhang J, Zhang N (2010) In vitro and in vivo studies of lipid-based
nanocarriers for oral N3-o-toluyl-fluorouracil delivery. Drug Deliv. 17, 352–363.
[61] Patel PA, Patravale VB (2011) AmbiOnp: Solid Lipid Nanoparticles of Amphotericin B for
Oral Administration. J. Biomed. Nanotechnol. 7, 632–639.
[62] Tsai M-J, Huang Y-B, Wu P-C, Fu Y-S, Kao Y-R, Fang J-Y, Tsai Y-H (2011) Oral
apomorphine delivery from solid lipid nanoparticleswith different monostearate
emulsifiers: Pharmacokinetic and behavioral evaluations. J. Pharm. Sci. 547–557.
[63] Yang S, Zhu J, Lu Y, Liang B, Yang C (1999) Body distribution of camptothecin solid
lipid nanoparticles after oral administration. Pharm Res 16, 751–7.
[64] Zhang Z, Gao F, Bu H, Xiao J, Li Y (2012) Solid lipid nanoparticles loading candesartan
cilexetil enhance oral bioavailability: in vitro characteristics and absorption mechanism in
rats. Nanomedicine 8, 740–7.
[65] Sanjula B, Shah F, Javed A, Alka A (2009) Effect of poloxamer 188 on lymphatic uptake
of carvedilol-loaded solid lipid nanoparticles for bioavailability enhancement. J Drug
Target 17., 249–56.

58
[66] Hu L, Xing Q, Meng J, Shang C (2010) Preparation and enhanced oral bioavailability of
cryptotanshinone-loaded solid lipid nanoparticles. AAPS PharmSciTech 11, 582–7.
[67] Tiyaboonchai W, Tungpradit W, Plianbangchang P (2007) Formulation and
characterization of curcuminoids loaded solid lipid nanoparticles. Int. J. Pharm. 337,
299–306.
[68] Zhang Q, Yie G, Li Y, Yang Q, Nagai T (2000) Studies on the cyclosporine A loaded
stearic acid nanoparticles. Int J Pharm 200, 153–9.
[69] Hu L, Jia H, Luo Z, Liu C, Xing Q (2010) Improvement of digoxin oral absorption in
rabbits by incorporation into solid lipid nanoparticles. Pharmazie 65, 110–3.
[70] Estella-Hermoso de Mendoza A, Préat V, Mollinedo F, Blanco-Prieto MJ (2011) In vitro
and in vivo efficacy of edelfosine-loaded lipid nanoparticles against glioma. J. Control.
Release 156, 421–6.
[71] Zhang T, Chen J, Zhang Y, Shen Q, Pan W (2011) Characterization and evaluation of
nanostructured lipid carrier as a vehicle for oral delivery of etoposide. Eur. J. Pharm. Sci.
43, 174–179.
[72] Hanafy A, Spahn-Langguth H, Vergnault G, Grenier P, Tubic Grozdanis M, Lenhardt T,
Langguth P (2007) Pharmacokinetic evaluation of oral fenofibrate nanosuspensions and
SLN in comparison to conventional suspensions of micronized drug. Adv. Drug Deliv.
Rev. 59, 419–26.
[73] Sarmento B, Martins S, Ferreira D, Souto E (2007) Oral insulin delivery by means of
solid lipid nanoparticles. Int J Nanomedicine 2, 743–9.
[74] Alex A, Paul W, Chacko AJ, Sharma CP (2012) Enhanced delivery of lopinavir to the
CNS using Compritol®-based solid lipid nanoparticles. Ther. Deliv. 2, 25–35.
[75] Chen C-C, Tsai T-H, Huang Z-R, Fang J-Y (2010) Effects of lipophilic emulsifiers on the
oral administration of lovastatin from nanostructured lipid carriers: physicochemical
characterization and pharmacokinetics. Eur. J. Pharm. Biopharm. 74, 474–82.
[76] Priano L, Esposti D, Esposti R, Castagna G, De Medici C, Fraschini F, Gasco MR,
Mauro A (2007) Solid lipid nanoparticles incorporating melatonin as new model for
sustained oral and transdermal delivery systems. J. Nanosci. Nanotechnol. 7, 3596–601.
[77] Liu D, Liu C, Zou W, Zhang N (2009) Enhanced gastrointestinal absorption of N3-O-
toluyl-fluorouracil by cationic solid lipid nanoparticles. J. Nanoparticle Res. 12, 975–984.
[78] Kumar VV, Chandrasekar D, Ramakrishna S, Kishan V, Rao YM, Diwan PV (2007)
Development and evaluation of nitrendipine loaded solid lipid nanoparticles: influence of
wax and glyceride lipids on plasma pharmacokinetics. Int. J. Pharm. 335, 167–75.
[79] Dong Z, Xie S, Zhu L, Wang Y, Wang X, Zhou W (2011) Preparation and in vitro, in vivo
evaluations of norfloxacin-loaded solid lipid nanopartices for oral delivery. Drug Deliv. 18,
441–450.
[80] Xie S, Zhu L, Dong Z, Wang Y, Wang X, Zhou W (2011) Preparation and evaluation of
ofloxacin-loaded palmitic acid solid lipid nanoparticles. Int J Nanomedicine 6, 547–555.

59
[81] Yuan H, Chen J, Du Y-Z, Hu F-Q, Zeng S, Zhao H-L (2007) Studies on oral absorption of
stearic acid SLN by a novel fluorometric method. Colloids Surf. B. Biointerfaces 58, 157–
64.
[82] Lee M-K, Lim S-J, Kim C-K (2007) Preparation, characterization and in vitro cytotoxicity
of paclitaxel-loaded sterically stabilized solid lipid nanoparticles. Biomaterials 28, 2137–
46.
[83] Varshosaz J, Minayian M, Moazen E (2010) Enhancement of oral bioavailability of
pentoxifylline by solid lipid nanoparticles. J. Liposome Res. 20, 115–23.
[84] Xie S, Pan B, Wang M, Zhu L, Wang F, Dong Z, Wang X, Zhou W (2010) Formulation,
characterization and pharmacokinetics of praziquantel-loaded hydrogenated castor oil
solid lipid nanoparticles. Nanomedicine (Lond). 5, 693–701.
[85] Luo C-F, Yuan M, Chen M-S, Liu S-M, Zhu L, Huang B-Y, Liu X-W, Xiong W (2011)
Pharmacokinetics, tissue distribution and relative bioavailability of puerarin solid lipid
nanoparticles following oral administration. Int. J. Pharm. 410, 138–44.
[86] Li H, Zhao X, Ma Y, Zhai G, Li L, Lou H (2009) Enhancement of gastrointestinal
absorption of quercetin by solid lipid nanoparticles. J. Control. Release 133, 238–44.
[87] Rawat MK, Jain A, Singh S (2011) In vivo and cytotoxicity evaluation of repaglinide-
loaded binary solid lipid nanoparticles after oral administration to rats. J. Pharm. Sci.
100, 2406–17.
[88] Rawat MK, Jain A, Singh S (2011) Studies on binary lipid matrix based solid lipid
nanoparticles of repaglinide: in vitro and in vivo evaluation. J. Pharm. Sci. 100, 2366–78.
[89] Pandey R, Sharma S, Khuller GK (2005) Oral solid lipid nanoparticle-based
antitubercular chemotherapy. Tuberculosis (Edinb). 85, 415–20.
[90] Shah M, Chuttani K, Mishra AK, Pathak K (2011) Oral solid compritol 888 ATO
nanosuspension of simvastatin: optimization and biodistribution studies. Drug Dev. Ind.
Pharm. 37, 526–537.
[91] Langguth P, Hanafy A, Frenzel D, Grenier P, Nhamias A, Ohlig T, Vergnault G, Spahn-
Langguth H (2005) Nanosuspension formulations for low-soluble drugs: pharmacokinetic
evaluation using spironolactone as model compound. Drug Dev. Ind. Pharm. 31, 319–
29.
[92] Das S, Ng WK, Kanaujia P, Kim S, Tan RBH (2011) Formulation design, preparation and
physicochemical characterizations of solid lipid nanoparticles containing a hydrophobic
drug: Effects of process variables. Colloids Surfaces B Biointerfaces 88, 483–489.
[93] Zhuang C-Y, Li N, Wang M, Zhang X-N, Pan W-S, Peng J-J, Pan Y-S, Tang X (2010)
Preparation and characterization of vinpocetine loaded nanostructured lipid carriers
(NLC) for improved oral bioavailability. Int. J. Pharm. 394, 179–85.
[94] Luo Y, Chen D, Ren L, Zhao X, Qin J (2006) Solid lipid nanoparticles for enhancing
vinpocetine’s oral bioavailability. J. Control. Release 114, 53–9.
[95] Wang D, Wang X, Li X, Ye L Preparation and characterization of solid lipid nanoparticles
loaded with alpha-Asarone. PDA J. Pharm. Sci. Technol. 62, 56–65.

60
[96] Abuasal B, Lucas C, Peyton B, Alayoubi A, Nazzal S, Sylvester P, Kaddoumi A (2012)
Enhancement of Intestinal Permeability Utilizing Solid Lipid Nanoparticles Increases γ-
Tocotrienol Oral Bioavailability. Lipids 47, 461–469.
[97] Olbrich C, Mehnert W, Müller R (1998) In vitro degradation properties of Solid Lipid
Nanoparticles SLNTM
. In 2nd World Meeting APGI/APV , Paris.
[98] Vega-Villa KR, Takemoto JK, Yáñez J a, Remsberg CM, Forrest ML, Davies NM (2008)
Clinical toxicities of nanocarrier systems. Adv. Drug Deliv. Rev. 60, 929–38.
[99] Souto EB, Martins-Lopes P, Lopes CM, Gaivao I, Silva AM, Guedes-Pinto H (2009) A
note on regulatory concerns and toxicity assessment in lipid-based delivery systems
(LDS). J. Biomed. Nanotechnol. 5, 317–322.
[100] Ferruzza S, Rossi C, Scarino ML, Sambuy Y (2012) A protocol for differentiation of
human intestinal Caco-2 cells in asymmetric serum-containing medium. Toxicol. In Vitro
26, 1252–5.
[101] Müller RH, Rühl D, Runge S, Schulze-Forster K, Mehnert W (1997) Cytotoxicity of Solid
Lipid Nanoparticles as a Function of the Lipid Matrix and the Surfactant. Pharm. Res. 14,
458–462.
[102] Nassimi M, Schleh C, Lauenstein HD (2010) A toxicological evaluation of inhaled solid
lipid nanoparticles used as a potential drug delivery system for the lung. Eur. J. Pharm.
Biopharm. 75, 107–116.
[103] Serpe L, Catalano MG, Cavalli R, Ugazio E, Bosco O, Canaparo R, Muntoni E, Frairia R,
Gasco MR, Eandi M, Zara GP (2004) Cytotoxicity of anticancer drugs incorporated in
solid lipid nanoparticles on HT-29 colorectal cancer cell line. Eur. J. Pharm. Biopharm.
58, 673–680.
[104] Schubert M a, Müller-Goymann CC (2005) Characterisation of surface-modified solid
lipid nanoparticles (SLN): influence of lecithin and nonionic emulsifier. Eur. J. Pharm.
Biopharm. 61, 77–86.
[105] Muller RH, Maaßen S, Weyhers H, Specht F, Lucks JS (1996) Cytotoxicity of magnetite-
loaded polylactide, polylactide/ glycolide particles and solid lipid nanoparticles. Int. J.
Pharm. 138, 85–94.
[106] Solans C, Esquena J, Forgiarini AM, Uson N, Morales D, Izquierdo P (2002)
Nanoemulsions: Formation and Properties. In Surfactants in Solution: Fundamentals and
Applications, Mittal KL, Shah DO, eds. , New York, p. 525.
[107] Sadtler V, Galindo-Alvarez J, Marie -Bégué E (2012) Low Energy Emulsification
Methods for Nanoparticles Synthesis. In The Delivery of Nanoparticles, Hashim AA, ed.
InTech.
[108] Lucks S, Müller R (1999) Medication vehicles made of solid lipid particles (solid lipid
nanospheres-SLN). EP Patent 0,605,497.
[109] Lippacher A, Müller R, Mader K (2000) Investigation on the viscoelastic properties of
lipid based colloidal drug carriers. Int J Pharm 196, 227–30.

61
[110] Ahlin P, Kristl J, Smid-Korbar J (1998) Optimization of procedure parameters and
physical stability of solid lipid nanoparticles in dispersions. Acta Pharm. 40, 259–267.
[111] Schwarz C, Mehnert W, Müller R (1994) Influence of production parameters of solid lipid
nanoparticles (SLN) on the suitability for intravenous injection. Eur. J. Pharm. Biopharm
40, 24S.
[112] Mehnert W, Mäder K (2001) Solid lipid nanoparticles: Production, characterization and
applications. Adv. Drug Deliv. Rev. 47, 165–196.
[113] Parhi R, Suresh P (2012) Preparation and characterization of solid lipid nanoparticles-a
review. Curr. Drug Discov. Technol. 9, 2–16.
[114] Li M, Fogler H (1978) Acoustic emulsification. Part 1. The instability of the oil –water
interface to form the initial droplets. J. Fluid Mech 88, 499–511.
[115] Li M, Fogler H (1978) Acoustic emulsification. Part 2. Break-up of the large primary oil
droplets in a water medium. J. Fluid Mech 88, 513–528.
[116] Kim B-D, Na K, Choi H-K (2005) Preparation and characterization of solid lipid
nanoparticles (SLN) made of cacao butter and curdlan. Eur. J. Pharm. Sci. 24, 199–205.
[117] Sjöström B, Bergenståhl B (1992) Preparation of submicron drug particles in lecithin-
stabilized o/w emulsions I. Model studies of the precipitation of cholesteryl acetate. Int. J.
Pharm. 88, 53–62.
[118] Siekmann B, Westesen K (1996) Investigations on solid lipid nanoparticles prepared by
precipitation in o/w emulsions. Eur. J. Pharm. Biopharm. 42, 104–109.
[119] Sjöström B, Bergenståhl B, Kronberg B (1993) A method for the preparation of
submicron particles of sparingly water‐soluble drugs by precipitation in oil in water
emulsions. II: Influence of the emulsifier, the solvent, and the drug substance. J. Pharm.
Sci. 82, 584–589.
[120] Sjöstrom B, Kronberg B, Carlfors J (1993) A method for the preparation of submicron
particles of sparingly water‐soluble drugs by precipitation in oil‐in‐water emulsions. I:
Influence of emulsification and surfactant concentration. J. Pharm. Sci. 82, 579–583.
[121] Quintanar-Guerrero D (1999) Pseudolatex preparation using a novel emulsion-diffusion
process involving direct displacement of partially water-miscible solvents by distillation.
Int. J. Pharm. 188, 155–164.
[122] Trotta M, Debernardi F, Caputo O (2003) Preparation of solid lipid nanoparticles by a
solvent emulsification–diffusion technique. Int JPharm 257, 153–60.
[123] Shahgaldian P (2003) A study of the freeze-drying conditions of calixarene based solid
lipid nanoparticles. Eur. J. Pharm. Biopharm. 55, 181–184.
[124] K. S, H. S (1969) The stability of O/W type emulsions as a function of temperature and
the HLB of emulsifiers: the emulsification by PIT-method. J Colloid Interface Sci 30, 258–
263.
[125] Montenegro L, Campisi A, Sarpietro MG, Carbone C, Acquaviva R, Raciti G, Puglisi G
(2011) In vitro evaluation of idebenone-loaded solid lipid nanoparticles for drug delivery
to the brain. Drug Dev. Ind. Pharm. 37, 737–46.

62
[126] Zetasizer Nano Series User Manual. MAN 0317 Issue 1.1.
[127] Honary S, Zahir F (2013) Effect of Zeta Potential on the Properties of Nano-Drug
Delivery Systems - A Review (Part 2). Trop. J. Pharm. Res. 12, 265–273.
[128] WHO (2013) Global Tuberculosis Report.
[129] Dye C, CJ W, DM B, Hosseini S, MC R (2005) EVolution of tuberculosis control and
prospects for reducing tuberculosis incidence, prevalence, and deaths globally. JAMA
293, 2767–2775.
[130] O’Garra A, Redford PS, McNab FW, Bloom CI, Wilkinson RJ, Berry MPR (2013) The
Immune Response in Tuberculosis. Annu. Rev. Immunol. 31, 475–527.
[131] Aderem A, Underhill DM (1999) Mechanisms of phagocytosis in macrophages. Annu.
Rev. Immunol 17, 593–623.
[132] Armstrong JA, Hart PD (1971) Response of Cultured Macrophages to Mycobacterium
Tuberculosis, with Observations on Fusion of Lysosomes with Phagosomes. J. Exp. Med
134, 713–740.
[133] Krishnan N, Robertson BD, Thwaites G (2010) The mechanisms and consequences of
the extra-pulmonary dissemination of Mycobacterium tuberculosis. Tuberculosis 90,
361–366.
[134] Grange JM, Zumla A (2002) The global emergency of tuberculosis: what is the cause? J.
R. Soc. Promot. Heal. 122 , 78–81.
[135] Almeida Da Silva PEA, Palomino JC (2011) Molecular basis and mechanisms of drug
resistance in Mycobacterium tuberculosis: classical and new drugs. J. Antimicrob.
Chemother. 66, 1417–30.
[136] Zhang Y, Yew WW Mechanisms of drug resistance in <i
xmlns=“http://pub2web.metastore.ingenta.com/ns/”>Mycobacterium tuberculosis</i>
[State of the art series. Drug-resistant tuberculosis. Edited by C-Y. Chiang. Number 1 in
the series]. Int. J. Tuberc. Lung Dis. 13, 1320–1330.
[137] Mikusová K, Slayden R a, Besra GS, Brennan PJ (1995) Biogenesis of the
mycobacterial cell wall and the site of action of ethambutol. Antimicrob. Agents
Chemother. 39, 2484–9.
[138] Espinal MA, Kim SJ, Suarez PG, Kam KM, Khomenko AG, Migliori GB, Baez J, Kochi A,
Dye C, Raviglione MC (2000) Standard short-course chemotherapy for drug-resistant
tuberculosis - Treatment outcomes in 6 countries. JAMA-J. Am. Med. Assoc. 283, 2537–
2545.
[139] Müller B, Borrell S, Rose G, Gagneux S (2013) The heterogeneous evolution of
multidrug-resistant Mycobacterium tuberculosis. Trends Genet. 29, 160–9.
[140] Nguyen L, Pieters J (2009) Mycobacterial subversion of chemotherapeutic reagents and
host defense tactics: challenges in tuberculosis drug development. Annu. Rev.
Pharmacol. Toxicol. 49, 427–53.
[141] Ramaswamy S, Musser JM (1998) Molecular genetic basis of antimicrobial agent
resistance in Mycobacterium tuberculosis: 1998 update. Tuber. Lung Dis. 79, 3–29.

63
[142] Van Hest R, Baars H, Kik S, van Gerven P, Trompenaars M-C, Kalisvaart N, Keizer S,
Borgdorff M, Mensen M, Cobelens F (2004) Hepatotoxicity of Rifampin-Pyrazinamide
and Isoniazid Preventive Therapy and Tuberculosis Treatment. Clin. Infect. Dis. 39 ,
488–496.
[143] He Q, Zhang Z, Gao F, Li Y, Shi J (2011) In vivo Biodistribution and Urinary Excretion of
Mesoporous Silica Nanoparticles: Effects of Particle Size and PEGylation. Small 7, 271–
280.
[144] Lee JE, Lee N, Kim H, Kim J, Choi SH, Kim JH, Kim T, Song IC, Park SP, Moon WK,
Hyeon T (2009) Uniform Mesoporous Dye-Doped Silica Nanoparticles Decorated with
Multiple Magnetite Nanocrystals for Simultaneous Enhanced Magnetic Resonance
Imaging, Fluorescence Imaging, and Drug Delivery. J. Am. Chem. Soc. 132, 552–557.
[145] Chen H, Wang L, Yeh J, Wu X, Cao Z, Wang YA, Zhang M, Yang L, Mao H (2010)
Reducing non-specific binding and uptake of nanoparticles and improving cell targeting
with an antifouling PEO-b-PγMPS copolymer coating. Biomaterials 31, 5397–5407.
[146] Nimje N, Agarwal A, Saraogi GK, Lariya N, Rai G, Agrawal H, Agrawal GP (2009)
Mannosylated nanoparticulate carriers of rifabutin for alveolar targeting. J. Drug Target
17, 777–787.
[147] Jain SK, Gupta Y, Ramalingam L, Jain A, Khare P, Bhargava D (2010) Lactose-
Conjugated PLGA Nanoparticles for Enhanced Delivery of Rifampicin to the Lung for
Effective Treatment of Pulmonary Tuberculosis. PDA J. Pharm. Sci. Technol. 64, 278–
287.
[148] Muttil P, Wang C, Hickey A (2009) Inhaled Drug Delivery for Tuberculosis Therapy.
Pharm. Res. 26, 2401–2416.
[149] Pandey R, Ahmad Z (2011) Nanomedicine and experimental tuberculosis: facts, flaws,
and future. Nanomedicine 7, 259–72.
[150] Sigma’s Product Information.
[151] Seidell A, Linke WF (1952) Solubilities of Inorganic and Organic Compounds, , New
York.
[152] Rahman HIHA (2000) The Chemistry of Natural oil. Sci. Bruneiana 9–15.
[153] Shah R, Eldridge D, Palombo E, Harding I (2015) Composition and Structure. In Lipid
Nanoparticles: Production, Characterization and Stability Springer International
Publishing, pp. 11–22.
[154] Souto EB (2011) Lipid Nanocarriers in Cancer Diagnosis and Therapy, i Smithers -
Creative Publishing Solutions, London.
[155] BASF (2008) Nonionic surfactant AT types - technical information.
[156] Lorenz MR (2006) Uptake of functionalized, fluorescent-labeled polymeric particles in
different cell lines and stem cells. Biomaterials 27, 2820–2828.
[157] Sigma Hexadecane - Product Information Sheet.
[158] Sigma’s Product Information Sheet.
[159] Sigma Pyrazinecarboxamide - product information sheet.

64
[160] Gurumurthy P, Nair NG, Sarma GR (1980) Methods for the estimation of pyrazinamide
and pyrazinoic acid in body fluids. Indian J. Med. Res. 71, 129–34.
[161] Siekmann B, Westesen K (1994) Melt-homogenized solid lipid nanoparticles stabilized
by the nonionic surfactant tyloxapol. I. Preparation and particle size determination.
Pharm. Pharmacol. 3, 194–197.
[162] Schwarz C, Mehnert W (1997) Freeze-drying of drug-free and drug-loaded solid lipid
nanoparticles. Int. J. Pharm. 157, 171–179.
[163] Westesen K, Siekmann B (1997) Investigation of the gel formation of phospholipid
stabilized solid lipid nanoparticles. Int J Pharm 151, 35–45.
[164] Singh H, Bhandari R, Kaur IP (2013) Encapsulation of Rifampicin in a solid lipid
nanoparticulate system to limit its degradation and interaction with Isoniazid at acidic pH.
Int. J. Pharm. 446, 106–11.