erros inatos do metabolismolehtonen/nutricao/12doencas_geneticas... · bloqueia a via que leva da...
TRANSCRIPT

ERROS INATOS DO
METABOLISMO

Archibald Garrod, 1902: Doenças causadas pela
deficiência de uma enzima específica
1) Alcaptonúria: caracterizada pela excreção urinária de
grandes quantidades de ácido homogentísico.
2) Fenilcetonúria (PKU) (Fölling, 1934): primeira
evidência de defeito genético como causa de retardo
mental.
3) Mucopolissacaridoses: grupo de doenças em que há
acúmulo de polissacarídeos nos lisossomos devido à
deficiências enzimáticas. Causa retardo mental,
anormalidades esqueléticas, baixa estatura.

Erros Inatos do Metabolismo
Frequência: cerca de 10% de todas as doenças
genéticas
Cerca de 500 distúrbios: síntese, degradação,
transporte e armazenamento de moléculas
Individualmente raros
1/1.000 recém-nascidos vivos
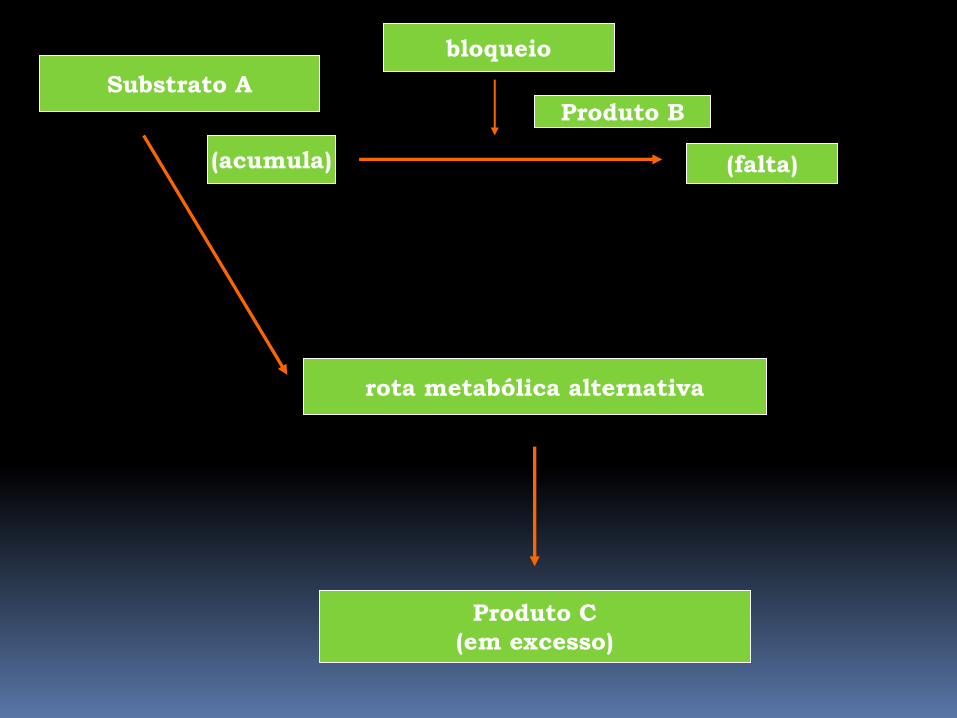
Substrato A
(acumula)
bloqueio
Produto B
(falta)
rota metabólica alternativa
Produto C
(em excesso)

Erros Inatos - Classificação
De acordo com repercussão metabólica do defeito:
1) Distúrbios do transporte: afetam o transporte renal
e/ou intestinal; costumam ser desencadeados pela
dieta; Ex: dissacaridases
2) Distúrbios do armazenamento: depósitos em qt
anormais; terapia por reposição da enzima; Ex:
doenças lisossômicas do depósito
3) Distúrbios da síntese: síntese incompleta ou anormal;
EX: hiperplasia adrenal congênita
4) Distúrbios do metabolismo intermediário:
comprometem a metabolização de pequenas
moléculas; respondem bem à restrição da dieta; Ex:
hiperfenilalaninemias

Erros Inatos -
Consequências Patológicas
1) Ausência do produto final
Ex: albinismo oculocutâneo
Ausência da enzima tirosinase no melanócito que
bloqueia a via que leva da tirosina a melanina
Falta de pigmento na pele, cabelo e íris
Pele branco-leitosa, cabelo amarelado, fotofobia
Suscetibilidade aumentada ao câncer de pele
Cuidados: proteção contra o sol
Heterogeneidade genética - variação clínica

Albinismo oculocutâneo tipo 1
TYROSINASE; TYR
Cytogenetic location: 11q14.3
Tyrosinase (EC 1.14.18.1) catalisa os 2 primeiros passos, e pelo menos um passo subsequente, na conversão de tirosina em melanina
Várias mutações no gene: causam albinismo oculocutâneo tipo 1A e 1B

Erros Inatos- Consequências Patológicas
2) Acúmulo do substrato
a) o próprio substrato acumulado é prejudicial
Ex: galactosemia, intolerância a frutose, alcaptonúria
b) devido ao acúmulo do precursor são utilizadas vias
metabólicas alternativas
superprodução de metabólitos tóxicos
Ex: fenilcetonúria


FENILCETONURIA
Primeira evidência de defeito genético como causa
de retardo mental;
- Causada por mutação na enzima fenilalanina
hidroxilase
- Incidência: 1:10.000
- A fenilalanina se acumula no sangue e é degradada
pela 2ª via toxicidade
- Tratamento: reduzir a ingestão de fenilalanina

FENILCETONURIA
Gene: PHENYLALANINE HYDROXYLASE; PAH
Cytogenetic location: 12q23.2
Fenilalanina hidroxilase catalisa a hidroxilação da fenilalanina em tirosina.

GALACTOSEMIA
Acúmulo de galactose nas células sanguíneas,
fígado, cérebro e rins
- Mutação no gene da enzima galactose-1-fosfato
uridil transferase
- Incidência: 1:10.000 a 1:100.000
- Bebês aparentemente normais - vômito, diarreia,
retardo de crescimento, desnutrição.
- Tratamento: substituição do leite por produtos
sem galactose ou lactose
- Não tratados: retardo mental, catarata, aumento
do fígado.

GALACTOSEMIA
Gene: GALACTOSE-1-PHOSPHATE URIDYLYLTRANSFERASE; GALT
Cytogenetic location: 9p13.3
GALT é a segunda enzima na via metabólica evolutivamente conservada da galactose.
Isso facilita a conversão simultânea de difosfoglucose uridina e galactose-1-fosfato a difosfogalactose uridina e glicose-1-fosfato, respectivamente
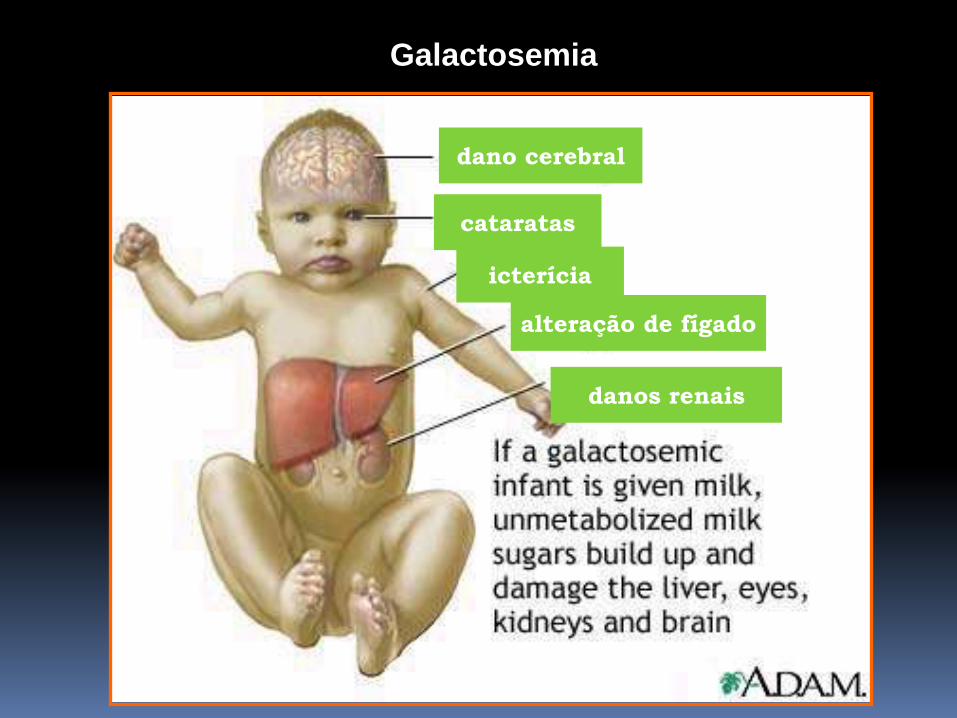
cataratas
dano cerebral
danos renais
alteração de fígado
icterícia
Galactosemia

Intolerância a frutose
- Deficiência da enzima aldolase da frutose-1-
fosfato
- Sintomas leves a graves (retardo, vômitos,
convulsões)
- Tratamento: restrição dietética (mel, frutas e
vegetais)

Intolerância a frutose
Gene ALDOLASE B, FRUCTOSE-BISPHOSPHATE; ALDOB
Cytogenetic location: 9q31.1
A frutose-1,6-bisfosfato-aldolase é uma enzima glicolítica que catalisa a conversão reversível de frutose-1,6-bifosfato em gliceraldeído 3-fosfato e di-hidroxiacetona fosfato.

Mucopolissacaridoses (MPS)
- As manifestações clínicas das MPS afetam
diversos órgãos e são muito variáveis,
existindo formas leves, moderadas e graves.
- Podem afetar o cérebro, olhos, ouvidos,
coração, fígado, ossos e articulações.
- Existem sete tipos de MPS descritas, que são
classificadas de acordo com o tipo
de enzima deficiente na célula.

Mucopolissacaridoses (MPS IV)
Gene: GALACTOSAMINE-6-SULFATE SULFATASE; GALNS
Cytogenetic location: 16q24.3
O gene GALNS codifica a sulfatase N-acetilgalactosamina-sulfato, uma enzima lisossomal envolvida no catabolismo de sulfato de condroitina e de queratano.

Erros Inatos - Suspeita e Investigação
Benefício do diagnóstico precoce: tratamento
adequado (se disponível) e aconselhamento
genético
Início dos sintomas: horas, meses ou anos após
nascimento
Obtenção da história clínica ampla é fundamental
História familiar: consanguinidade dos pais
Sintomas típicos

Erros Inatos- Características sugestivas
RN: coma, convulsões, irritabilidade, vômitos, diarréia
crônica....
Retardo de crescimento
Deficiência mental
Regressão neurológica
Hepato ou esplenomegalia
Deficiência de crescimento
Relato de irmão falecido precocemente
Pais consanguíneos

Erros Inatos - Investigação laboratorial
Estudos moleculares: a partir da década de 90
Aconselhamento genético e diagnóstico pré-natal
Atualmente: identificação de mutações específicas
Útil no prognóstico do paciente
Indicação por suspeita clínica prévia e testes de
triagem
testes que identificam alterações
no gene da enzima responsável

Erros Inatos- Prevenção
1) Triagem neonatal: detecção em fase pré-clínica EX:
fenilcetonúria e hipotireoidismo congênito
2) Diagnóstico pré-natal: para casais de risco como
opção reprodutiva; altamente específico
EX: coleta de vilosidades coriônicas e amniocentese
3) Detecção de portadores: confirmar uma suspeita
diagnóstica, permitindo o Aconselhamento
EX: diagnóstico molecular

Erros Inatos - Tratamento
Para muitos EIM ainda não disponível
Principal estratégia: correção do desbalanço metabólico
- dieta adequada!!!
Controle do acúmulo do substrato
EX: fenilcetonúria e galactosemia
Estratégia: dieta com restrição de fenilalanina ou
galactose previne o RM e o quadro agudo da doença
Terapêutica escolhida de acordo com o diagnóstico
Evitar as consequências fisiopatológicas

Origem das doenças genéticas
• Mitocondriais
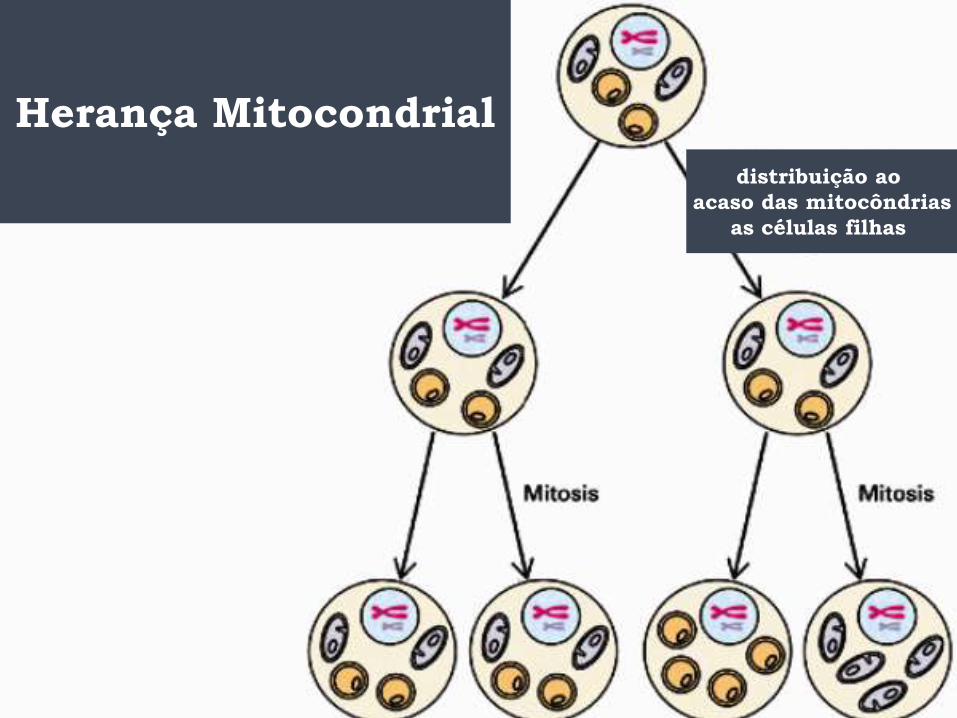
Herança Mitocondrial
distribuição ao
acaso das mitocôndrias
as células filhas

Herança Mitocondrial
- Principais sítios celulares
para a produção do ATP
- Importante função metabólica
- Evoluíram a partir de
bactérias
-Sistema genético secundário
-Contém múltiplas moléculas
de mtDNA

Herança Mitocondrial -
genoma
O genoma mitocondrial é constituído por
uma pequena molécula de DNA circular de cadeia dupla
Contém 37 genes que codificam RNAs transportadores,
ribossômicos e mensageiros
É traduzido em 13 polipeptídeos que compõem a cadeia
respiratória mitocondrial
Genoma: compacto e sem íntrons
Proteínas codificadas: são essenciais no transporte de elétrons
pela membrana intramitocondrial
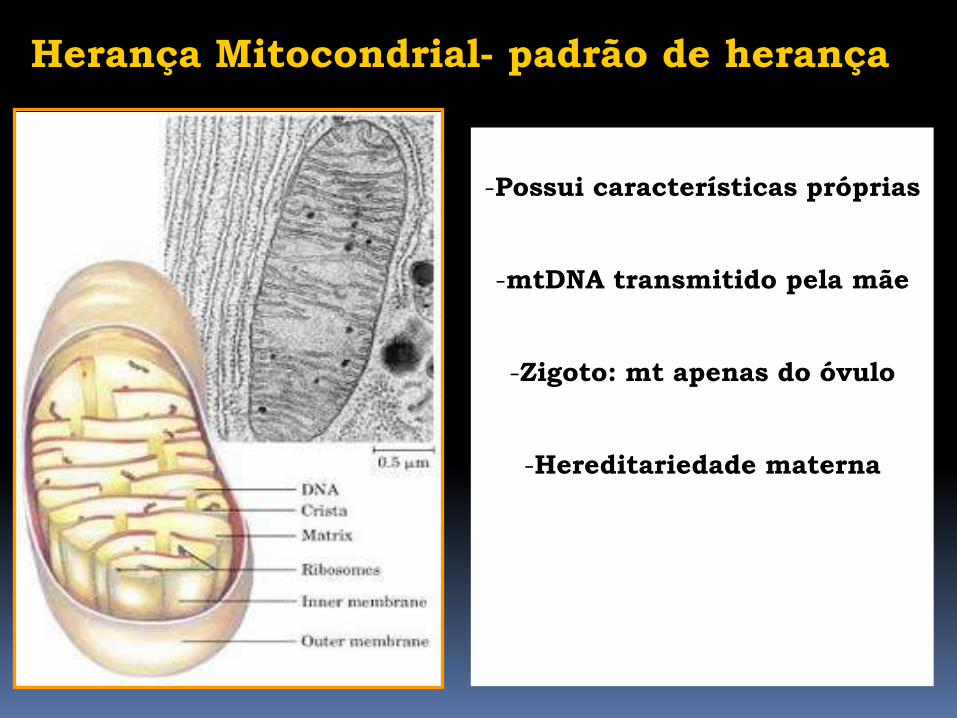
Herança Mitocondrial- padrão de herança
-Possui características próprias
-mtDNA transmitido pela mãe
-Zigoto: mt apenas do óvulo
-Hereditariedade materna

Herança Mitocondrial- homoplasmia
Durante a divisão celular
as mitocôndrias são
distribuídas randomicamente
para as células-filhas
homoplasmia
Somente DNA mutado ou
somente
DNA normal
dentro da mitocôndria
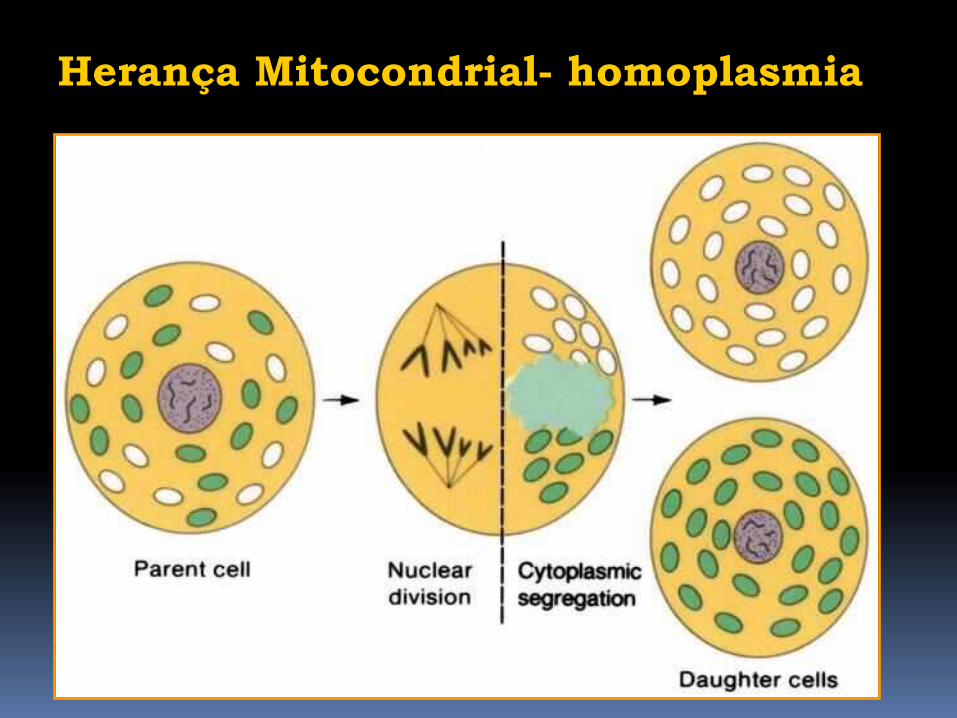
Herança Mitocondrial- homoplasmia

Mitocôndrias- Padrão de herança
Mães: transmitem a deficiência para toda a
descendência
Somente as filhas transmitirão a doença
Alta taxa de divisões e mutações: podem coexistir
dentro da mesma célula- moléculas normais e
mutadas
heteroplasmia
importante na determinação
de doenças

Mitocôndrias- Padrão de herança
Poliploidia mitocondrial durante a mitose-
distribuição aleatória do mtDNA mutado
Fenômeno chamado de segregação mitótica!
Expressão fenotípica: depende da natureza da
mutação, da distribuição tecidual e da necessidade
energética do órgão
Limiar de expressão: proporção de mtDNA normal e
mutado dentro da célula
mtDNA mutado gravidade de manifestações

Pai afetado
Prole afetada
Mãe afetada
Prole não afetada
Padrão de herança: materno

Padrão MATERNO de herança

Primeiro relato de distúrbios: década de 60
Década de 90: avanço na identificação,
classificação e caracterização
Métodos de diagnóstico mais eficientes
Frequência estimada: 1:10.000 nascidos vivos
Geradas por ausência de energia em órgãos como:
Herança Mitocondrial- doenças
Cérebro, músculo, coração, fígado e rins

Grande variabilidade de apresentação clínica
Requer protocolo diferenciado no diagnóstico
(histopatológicos, bioquímicos e moleculares)
Obtenção de material por biópsia muscular
Acometem principalmente cérebro e músculos
Decorrentes de alterações no metabolismo
energético celular
Herança Mitocondrial- doenças

Alguns sintomas: perda visual, características
neurodegenerativas, acidentes vasculares, fraqueza
muscular, regressão psicomotora, demência.....
Tratamento: correção do funcionamento anormal da cadeia
respiratória
Objetivo: aumentar a produção de ATP e o transporte de
elétrons
Substâncias: coenzima Q, vitaminas (C, K3, K1)
Diagnóstico aconselhamento genético: prevenção de
novos casos
Herança Mitocondrial- doenças

Neuropatia óptica de Leber
Perda de visão
18 mutações em 9 genes que codificam proteínas mitocondriais causam o fenótipo
ND1, ND2, CO1, ATP6, CO3, ND4, ND5, ND6, CYTB