evaluation and automization of phosphopeptide … · characterization of the acidic nutritional...
TRANSCRIPT

Bachelor Thesis Scheikunde
Evaluation and Automization of Phosphopeptide
Enrichment via TiO2 Affinity Chromotography
door
Matthew Delport
28th March 2016
Studentnummer
10452540
Onderzoeksinstituut Verantwoordelijk docent
Van’t Hoff Institute for Molecular Sciences (HIMS) Prof. Garry Corthals
Onderzoeksgroep Begeleider
Analytical Chemistry Dr. Michelle Camenzuli

2
Table of Contents Pupulair Wetenchsappelijke Samenvatting ............................................................................................3
Summary ..................................................................................................................................................4
Introduction .............................................................................................................................................5
The biological importance of phosphorylation ...................................................................................5
A short history of protein phosphorylation .........................................................................................6
Mechanism of Phosphorylation ..........................................................................................................7
Protein Kinases ....................................................................................................................................8
Signal Transduction Cascades .............................................................................................................9
Signal Transduction Cascades .............................................................................................................9
MS of Phosphopeptides .................................................................................................................... 10
Phosphopeptide Enrichment ............................................................................................................ 11
The Aim of This Project ..................................................................................................................... 13
Methodology......................................................................................................................................... 14
Column Packing & Fritting ............................................................................................................... 14
Trypsin Digestion of α-casein From Bovine Milk ............................................................................. 15
Off-line Phosphopeptide Enrichment of Digested α-casein Sample ............................................... 16
Semi On-line Phosphopeptide Enrichment of Digested α-casein Sample ...................................... 19
Semi On-line Phosphopeptide Enrichment of Digested α-casein Sample ...................................... 19
Fully On-line Phosphopeptide Enrichment of Digested α-casein Sample ....................................... 20
Results & Discussion ............................................................................................................................. 21
Offline Phosphopeptide TiO2 Enrichment ......................................................................................... 21
Semi-Online Phosphopeptide TiO2 Enrichment ................................................................................ 26
Online Phosphopeptide TiO2 Enrichment ......................................................................................... 28
Conclusion & Outlook ........................................................................................................................... 29
Acknowledgements .............................................................................................................................. 30
References............................................................................................................................................. 30

3
Populair Wetenschappelijke Samenvatting
Het lichaam gebruikt eiwit fosforylering reacties om bepaalde processen te controleren. Je kunt het
zien als een lichtknopje, wanneer fosforylering plaats vindt word het lichtknopje aangezet en kan er
een bepaald proces plaatsvinden. Dit proces kan ook weer gestopt worden door de reactie terug te
draaien en zo wordt het knopje weer uitgezet. Een voorbeeld van waar fosforylering een grote rol
speelt binnen onze lichaam is bij het ontwikkelen van kanker. Bepaalde eiwitten worden dan
aangezet en vervolgens niet goed uitgezet waardoor een persoon kanker kan krijgen. Fosforylering
speelt niet alleen een rol bij velen andere ziektes, maar ook bij het algemeen functioneren van het
lichaam. Het kunnen inzien van waar en hoe deze reacties plaats vinden binnen het lichaam is dus
bijzonder interessant.
Methodes om eiwit fosforylering te kunnen begrijpen door analyses uit te voeren is over de laatste
decennia sterk ontwikkeld maar alles gebeurd wel nog offline, wat inhoudt dat men de analyses
handmatig uitvoeren in een lab. Deze methodes zijn tijdrovend waardoor er simpelweg niet genoeg
data verkregen kan worden. Ook maken mensen altijd foutjes waardoor deze methodes slecht
reproduceerbaar zijn. Deze redenen zorgen ervoor dat er niet genoeg betrouwbare data verkregen
kan worden om nuttige conclusies te trekken.
Mijn project richt zich op het ontwikkelen en implementeren van een online proces waardoor de
eerdergenoemde uitdagingen verholpen kunnen worden. Een online proces houdt in dat de analyse
d.m.v. geautomatiseerde machines uitgevoerd wordt. Deze machines kunnen dan geprogrammeerd
worden om de hele tijd door analyses uit te voeren waardoor er niet altijd iemand in het lab hoeft te
zijn. Dit maakt het ook mogelijk om veel meer data te verkrijgen in een kortere tijd. Ook zijn
machines vele malen nauwkeuriger dan de meesten van ons, wat resulteert in betrouwbare data wat
door wetenschappers overal ter wereld gereproduceerd kan worden. Dit is een belangrijke stap in
het volledig begrijpen van hoe eiwit fosforylering precies werkt zodat nare ziektes zoals kanker
bestreden kunnen worden.

4
Summary
Cells constantly have to react to signals received by their surroundings, an example being growth-
factor signals which are responsible for cell growth, differentiation and proliferation. The
degradation of the capability to effectively regulate these cellular responses is one of the main
causes of many human diseases such as cancers6. Since the isolation of phosphoserine by Phoebus A.
Levene and Fritz A. Lipmann in 1932, post-translational modifications (PTMs) have been found to
play a major role in the regulation of cellular responses as they strongly influence and control
enzymatic activity, protein conformation, protein-protein interactions, and cellular localization.1,2 In
particular protein phosphorylations have been found to be one of the most important PTMs as it is
believed that there are over 105 phosphorylated sites in the mammalian proteome3. At any given
moment 30-50% of all proteins are phosphorylated4 and the number of genes involved in
phosphorylation processes may be as much 2-3% of the whole eukaryotic genome.5 It is therefore of
great interest to develop and improve methods for the rapid detection of phosphorylation,
phosphorylation site mapping along with protein kinase and phosphatase substrate identification.
This will lead to a better understanding of signalling networks and the proteins involved making it
possible to develop potent and specific pharmacological signalling modulators for therapeutic use.6
In the quest of finding a capable tool for such research mass spectrometry (MS) has proved to be a
powerful option in phosphorylation analysis, however it is still far from being robust due to
challenges in phosphoproteomics such as proteins being present in low-abundance and sub-
stoichiometric phosphorylation.1 Phosphopeptide enrichment prior to MS analysis is therefore of the
essence and an effective method for this is offline titanium dioxide affinity chromatography. This has
led to the question of whether titanium dioxide affinity chromatography can be improved and
implemented into an online analysis method, which is the aim of this project.
This thesis will begin with a complete discussion of protein phosphorylations by explaining their
biological purpose, the chemistry involved and the practical importance of understanding and
mapping phosphorylations. Methods currently used in phosphoproteomics including their strengths
and weaknesses will be described and compared to the results of our in-house developed methods
used in our research. Concluding remarks based on our experimental observations will be presented
along with perspectives for future research.

5
1. Introduction
1.1. The biological importance of phosphorylation
Every form of life is made up of countless gene-encoded proteins which all have their own essential
task in maintaining the organism they are situated in. These proteins are even further diversified via
so called PTMs which enzymatically and covalently modify proteins during or after their biosynthesis,
expanding the number of distinct human protein species by at least one order of magnitude. PTMs
are comprised of many types of reactions such as glycosylation, lipidation, cleavage of peptide bonds
and many more but one of the most important and prevalent PTM is reversible phosphorylation,
catalysed by kinases and phosphatases. Besides covalent modifications, protein functionality is even
further broadened by the formation noncovalent complexes resulting in unique functionality, activity
or cellular localization. Proteins are able to form homo- and heteromeric complexes as well as
complexes with drugs, metabolites or metals. The covalent and noncovalent interactions work side-
by-side as a noncovalent complex’s lifetime, function and localization may all be affected by certain
covalent modifications (Fig. 1)7. This intricate interplay is what makes the proteome extremely
dynamic, in turn providing for the capability of effective sensing and regulation within cells.
Figure 1: Visualization of the expansion of the human proteome by the principles of covalent protein modifications and noncovalent interaction (with proteins, cofactors, metals). The estimated number of human genes (22000 – 25000)
corresponds to a similar basic number of linear protein sequences.7

6
1.2. A short history of protein phosphorylation
The first signs of the proteins containing phosphate were made evident in 1906 with the
characterization of the acidic nutritional proteins, casein and vitellin.8 The following advances took
place nearly 30 years later when phosphoserine was detected in vitellin,9 a protein isolated from egg
yolk. The high levels of phosphorylation within casein grant it calcium binding properties, allowing
phosphate and calcium to be readily available for bone formation. The main component of bone is
calcium hydroxyapatite, a mixture of calcium hydroxide phosphate. By concluding that
phosphoproteins acted as nutritional sources of phosphate and calcium, the first known function of
phosphorylated proteins was discovered.
Following these observations, Carl and Gerty Cori in the late 1940’s, and Earl Sutherland, Edwin Krebs
and Edmond Fischer all contributed to the revolutionary understanding that phosphate attachment
was not only an add-on to nutritional proteins, but that it also controls the function of glycogen
phosphorylase, the key enzyme of energy metabolism. This enzyme cuts α-D-glucose from glycogen,
forming α-D-glucose-1-phosphate after concomitant attachment of inorganic phosphate. The
discovery that glycogen phosphorylase’s activity is regulated by hormones and exists in an active and
inactive form was made by the Coris10. Sutherland set out to study the effects of hormones on
glycogen metabolism and then made the discovery of cAMP being the first ‘second messenger’ in
transmembrane signal transduction. In the same time frame, Krebs and Fischer made the Nobel Prize
winning discovery of reversible phosphorylation being a control mechanism for the activation and
deactivation of glycogen phosphorylase. They observed that without a phosphorylated Ser15,
glycogen phosphorylase was inactive and once phosphorylated, activity was restored.11
The evolution of our understanding of phosphorylations did not stop here, and with the use of cAMP-
dependant protein kinase A as a model enzyme, two breakthroughs were achieved. Firstly, it was the
first kinase to be fully sequenced12 and visualized with a complete 3D structure.13 This also led to the
discovery of tyrosine phosphorylation14 in addition to the previously known serine and threonine
phosphorylation. Certain tyrosine kinases were also found to be viral oncogene products and a class
of receptors with intracellular tyrosine kinase activity was found which paved the road to discovering
a common mechanism for transmembrane signal transduction which will shortly be described. The
biological importance of protein phosphorylation has become extremely evident over the last
century resulting in an ever increasing interest to expand our knowledge in the field and more
revolutionary findings will surely surface.
7
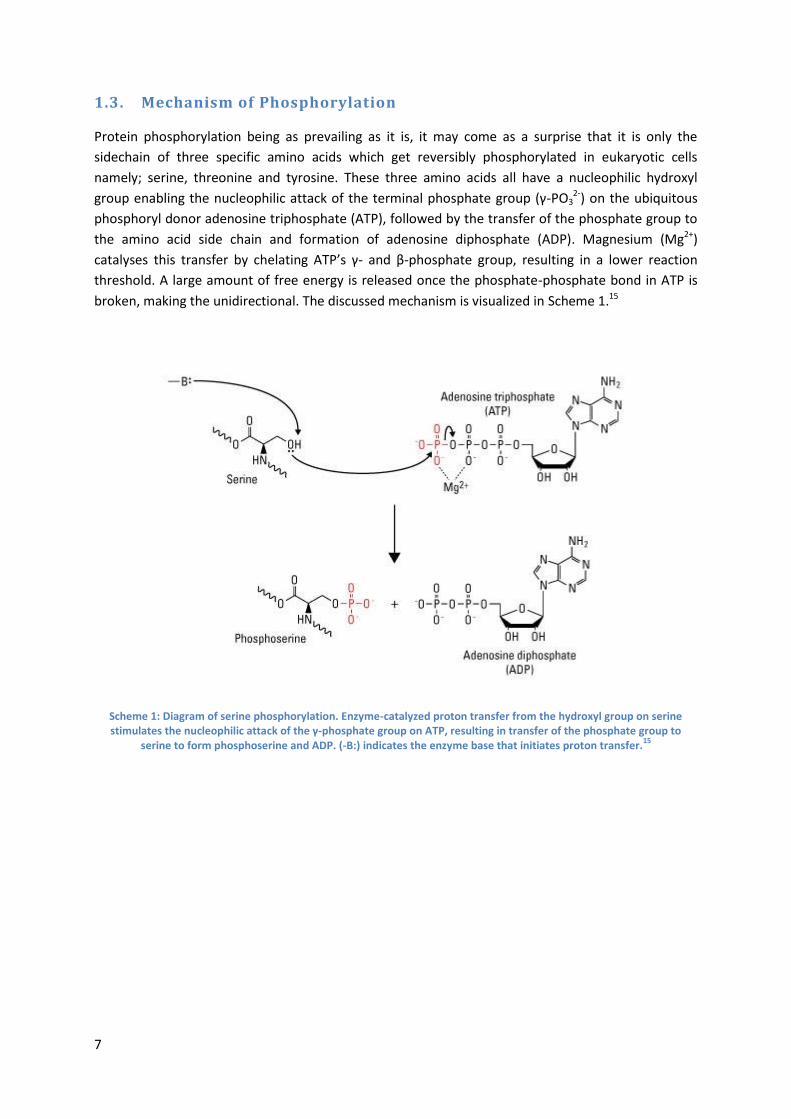
1.3. Mechanism of Phosphorylation
Protein phosphorylation being as prevailing as it is, it may come as a surprise that it is only the
sidechain of three specific amino acids which get reversibly phosphorylated in eukaryotic cells
namely; serine, threonine and tyrosine. These three amino acids all have a nucleophilic hydroxyl
group enabling the nucleophilic attack of the terminal phosphate group (γ-PO32-) on the ubiquitous
phosphoryl donor adenosine triphosphate (ATP), followed by the transfer of the phosphate group to
the amino acid side chain and formation of adenosine diphosphate (ADP). Magnesium (Mg2+)
catalyses this transfer by chelating ATP’s γ- and β-phosphate group, resulting in a lower reaction
threshold. A large amount of free energy is released once the phosphate-phosphate bond in ATP is
broken, making the unidirectional. The discussed mechanism is visualized in Scheme 1.15
Scheme 1: Diagram of serine phosphorylation. Enzyme-catalyzed proton transfer from the hydroxyl group on serine stimulates the nucleophilic attack of the γ-phosphate group on ATP, resulting in transfer of the phosphate group to
serine to form phosphoserine and ADP. (-B:) indicates the enzyme base that initiates proton transfer.15

8
1.4. Protein Kinases
As all reactions in the body are driven by enzymes, so is protein phosphorylation which is catalysed
by kinases. To date there is believed to be more than 500 kinases in the human proteome; this class
of proteins is also referred to as the human kinome16 and substrates are found in various forms such
as carbohydrates, lipids, nucleotides and proteins. With ATP being such an efficient phosphate donor,
nearly all protein kinases use ATP as their co-substrate, with a few exceptions which make use of
guanosine triphosphate. ATP has the ideal structure for the transfer of a α-, β-, γ-phosphate group in
nucleotidyl-, pyrophosphoryl- or phosphoryl transfer, respectively.17 Although kinases vary greatly in
substrate specificity, they do display a highly conserved ATP-binding site.18
The kinome can be divided into subfamilies which display variable catalytic domain specificity,
including tyrosine kinases or serine/threonine kinases. The mammalian kinome is comprised of
roughly 80% serine/threonine kinases, and in line with this statistic, over 90% of the
phosphoproteome consists of pS and pT. This has been backed by studies which have stated the
relative abundance ratio of pS:pT:pY in cells to be 1800:200:1.19 Despite the low abundance of
tyrosine phosphorylation, it is a heavily researched in the biomedical field due to its association to
human diseases by dysregulation of receptor tyrosine kinases. Besides protein kinases showing
specificity towards target amino acids, they also take adjacent consensus sequences in to account.20
These consensus sequences play a role in determining whether a kinase is able to phosphorylate one
or more substrates and also the multiplicity in which a single protein can be phosphorylated.
Kinases themselves also have regulatory subunits that are often controlled by phosphorylation21. The
majority of protein kinases are found to be in their inactive basal state when dephosphorylated and
activated via phosphorylation and the opposite is only true in rare cases. Other kinases may even
require a combination of phosphorylated and dephosphorylated sites in order to be activated, the
proto-oncogene Src serves as an example of this complexly regulated system22.

9
1.5. Signal Transduction Cascades
The key to protein phosphorylations being extremely efficient in signal transduction lies in their
reversibility which makes them capable of swiftly responding to intra- and extracellular stimuli15.
Signal transduction cascades are set off by one or more proteins physically sensing cues through
cleavage, ligand binding or other responses. These proteins then submit a signal to second
messengers and signalling enzymes, which in case of phosphorylation, activate downstream kinases,
resulting in phosphorylation and activation of linked downstream substrates, including additional
kinases. The described process is repeated until the desired physiological response is achieved. Signal
transduction cascades have been found to proceed in two fashions. One being a linear fashion
whereby kinase I activates kinase II which then activates kinase III and so forth. The second form is
the amplification of an initial signal whereby kinase I activates multiple kinases, which in turn also all
activate other kinases (Fig.2)15. The second form of signalling makes it possible for a single molecule,
for example a growth factor, to activate global cellular programs including proliferation.23
Figure 2: Signal transduction cascades amplify the signal output. External and internal stimuli induce a broad range of cellular responses through a series of second messengers and enzymes. Linear signal transduction pathways yield the
sequential activation of a discrete number of downstream effectors, while other stimuli elicit signal cascades that amplify the initial stimulus for large-scale or global cellular responses.
15
An example of how meaningful such cascades are lies in one of the molecular mechanisms of cancer
development in which genomic mutations hijack a signal transduction pathway, resulting in a
dysregulated surplus of cellular proliferation. Recognizing viral oncogene products as tyrosine kinases
or products directly related to the interference of intracellular signal transduction triggered the
widespread acceptance of kinases and phosphatases being involved in cellular growth control. A
recent and rather impressive example is the finding of the chromosomal translocation leading to the
Philadelphia chromosome is the molecular cause of chronic myelogenous leukaemia (CML).24 Fully
understanding these mechanisms and pathways plays an essential role in pharmacology and in order
to do so, effective analysis methods have to be established.

10
1.6. MS of Phosphopeptides
With data claiming that 1/3 of all proteins in a mammalian cell are phosphorylated25, the human
genome contains 500 genome-encoded protein kinases and that phosphorous to sulphur ratios point
towards a high degree of protein phosphorylation26, it is safe to say that phosphorylation appears to
be the most abundant and essential protein modification. This being said, phosphorylations are only
intermittently detected in proteomic analyses not specially adapted for phosphopeptide detection.
Additionally, a method capable of efficiently detecting multiply phosphorylated peptides has yet to
be developed as it is only singly and some doubly phosphorylated peptides which are readily
detected. The inconsistency between what is expected and what is actually detected regarding
protein phosphorylations has led to the general belief that phosphopeptide analysis is an extremely
challenging practice. Researchers interested in the matter have somewhat united and agreed upon
some of the underlying factors that are apparently making this analysis so challenging. Most reports
on this topic tend to attribute undesired results to these factors which are: (i) the widely sub-
stoichiometric phosphorylation of proteins, (ii) the suppression of phosphopeptide ionization in the
presence of an excess of unmodified peptides, (iii) a generally lower ionization efficiency of
phosphopeptides versus non-phosphopeptides (in the generally applied positive ion mode), and (iv)
the application of data-directed MS/MS analysis which leads to a notorious under sampling of
peptides of low abundance. A recent article has focused on sorting these blindly used arguments into
facts and myths27. The article makes the following conclusion regarding each argument. Argument (i)
is likely true as the majority of phosphopeptide signals detected in digests are accompanied by their
nonphosphorylated analogues which are also more abundant. Argument (ii) is likely false or at least
not always true in the strict sense as stated. LC-MS experiments were executed with a complex
protein digest which was also spiked with variable amounts of phosphopeptides. These experiments
were not able to confirm a decrease of the absolute phosphopeptide signals intensities with the
increasing excess of unmodified peptides28. This being said, it is true that in LC-MS, with electrospray
ionization (ESI), the MS detection usually involves relatively simple mixtures which have already
passed LC separation. However one of ESI’s true weaknesses is its limited dynamic range due to all
analytes competing for the limited number of excess charges on the spray droplets. This makes the
conclusion ‘that when in the presence of a large number of analytes the less abundant species will
fail to be ionized’, plausible. However concrete evidence supporting this general hypothesis is yet to
be developed. Argument (iii) is true for many cases with a couple exceptions, although this effect
doesn’t seem to be highly significant. Finally, argument (iv) was deemed true and can certainly result
in reduced detection of phosphopeptides when combined with a lower ionization efficiency and
phosphopeptide abundance.
Factors leading to phosphopeptide analysis being notoriously challenging which were not discussed
in the article mentioned above but however do play an important role are phosphoprotein
degradation during sample work up, for example by phosphatases, resistance of phosphoproteins to
digestion as well as metal-ion mediated adsorption of multiply phosphorylated phosphoproteins or
phosphopeptides on surfaces, to which the sample is exposed in the analytical process7.

11
1.7. Phosphopeptide Enrichment
Due to the challenging nature of phosphopeptide analysis it is of the essence to make use of
phosphopeptide enrichment techniques. Usually using phosphoprotein rich samples followed by
protease-specific digestion and MS-analysis does not suffice and therefore complex samples require
a second enrichment step in order to accurately determine the sites of phosphorylation. There are
commercial kits available on the market making the enrichment process relatively easy, fast, and
reproducible however is has been shown that these various methods differ in specificity of isolation
and in the set of phosphoproteins and phosphopeptides isolated29, indicating that the use of one
single method may not be sufficient for global phosphoproteome analysis. Various enrichment
methods will be discussed below.
1.7.1. Immunoprecipitation
Total proteins are immunoprecipitated via the use of antibodies specific to phosphorylated residues.
Immunoprecipitation of proteins containing phosphotyrosine is used more often than that of
proteins containing phosphoserine or phosphothreonine as the antibodies specific to the former
tend to be much more reliable than those specific to the latter30,31,32,33. Phosphotyrosine (pTyr)
residues have therefore been studied much more intensely over recent years, despite their low
abundancy compared to phosphoserine (pSer) and phosphotheronine (pThr) residues and specific
pTyr binding domains (PTB) have resulted in the determination of global tyrosine phosphorylation
states of the cell34,35. It is not yet fully understood why pSer- and pThr-specific antibodies fail to work
efficiently in phosphoprotein immunoprecipitation, however these antibodies are renowned to be
extremely specific towards certain consensus motifs. In reality, immunoprecipitation of pSer and
pThr phosphoproteins can only be achieved with an expensive mixture of multiple antibodies,
resulting in only a minor amount of research being reported in this area36.
1.7.2. Immobilized Metal-Ion Affinity Chromatography (IMAC)
Originally being introduced for purification of His-tagged proteins37, IMAC has evolved into one of the
most commonly used technique for phosphopeptide enrichment. The reason for this technique
showing improved success rates is due to the fact that it reduces ion suppression effects that would
otherwise occur in untreated complex samples38. Negatively charged phosphate groups on
phosphopeptides bind to the IMAC stationary phase via electrostatic interactions with positively
charged metal ions, which are bound to the column material via linkers such as nitriloacetic acid
(NTA), iminodiacetic acid (IDA), and Tris(carboxymethyl)ethylenediamine (TED). It was initially found
that immobilized metal ions such as Ni2+, Co2+, and Mn2+ preferentially bind to proteins with a high
His density but more relevantly, immobilized metal ions of Fe3+, Ga3+, and Al3+ have higher specificity
for phosphopeptides. Zr4+ has also recently been reported to bind phosphopeptides with a high
specificity39. A major disadvantage in IMAC methods is they tend to non-specifically bind peptides
containing the acidic amino acids glutamine and aspartate and the strong binding of multiply
phosphorylated peptides. The issue of binding acidic amino acids can be countered via esterification
of the carboxylic acids to methyl esters using hydrochloric acid-saturated, dried methanol40, however
reaction conditions must be chosen with extreme care to prevent the occurrence of side reactions

12
which would increase the complexity of the sample. The experimental conditions (for example; pH,
ionic strength, or organic composition of the solvents) of any IMAC procedure have to also be
carefully considered as slight variations are known to have radical effects on the specificity of the
IMAC stationary phase. Despite these drawbacks, IMAC has become increasingly popular, and one of
the reasons for this is the exceptional compatibility with subsequent separation and detection
techniques such as LC-ESI-MS/MS41.
1.7.3. Titanium Dioxide (TiO2)
Pinkse et al. have recently reported the use of titanium dioxide as a promising alternative to IMAC42.
The method is based on the selective interaction of water-soluble phosphates with porous titanium
dioxide microspheres via bidentate binding at the TiO2 surface. A TiO2 precolumn is used to trap
phosphopeptides under acidic conditions, which are then subsequently desorbed under alkaline
conditions. Even though TiO2 based columns still trap nonphosphorylated acidic peptides, it has been
reported to display increased specificity towards the enrichment of phosphopeptides. The
nonspecific binding of acidic peptides has been found to be able to be diminished to a certain extent
when 2,5-dihydroxy-benzoic acid was used during peptide loading43. This novel procedure has proven
to be more effective than IMAC. An in-depth analysis has recently been reported describing the
relationship between the occurrence of some amino acids and the phospho-specific and nonspecific
binding of peptides using TiO2-based enrichment44. Two well characterized peptide mixtures, each
consisting of either 33 or 8 synthetic phosphopeptides or their non-phosphorylated analogue and
varying in charge and hydrophobicity were tested. The acquired data confirmed that TiO2 is indeed
highly selective for phosphopeptide enrichment. It is noteworthy to mention that a drastic decrease
in phosphopeptide recovery was found in samples containing phosphopeptides with multiple basic
amino acids. Once again being able to be coupled to a LC—ESI-MS/MS workflow has been a major
advantage of this technique.
Figure 3: Schematic discription of the interaction of a phosphopeptide with a TiO2 surface. The directly interacting partners are oxygen atoms of the phosphate group and positively polarized titanium (IV) atoms. It is assumed that the two negatively charged oxygen atoms of a phosphate group may interact either with a single Ti(IV) centre or with two
such centres7.

13
1.7.4. Online Phosphopeptide Enrichment
The enrichment of phosphopeptides is often executed manually as automated procedures have not
yet been adequately developed. These manual offline procedures involve the use hand-made
columns by packing the desired solid phase into pipette tips, which are then used to desalt and
enrich the phosphopeptides. Offline procedures are not only highly labour intensive, but also result
extremely poor reproducibility due to factors such as loading speed which cannot be precisely
controlled45. It is therefore of great interest to establish efficient online enrichment methods if we
are to move forward in the field of phosphoproteomics. Online methods have many advantages
which make it an attractive route to follow. Analytical instrumentation can carry out procedures with
high precision factors such as loading speed and sample handling being easily controlled which would
greatly improve reproducibility. The possibility of automation also serves as a solution for the labour
insensitivity of offline methods as unattended operation and ultimately high throughput analysis is
made possible. In the journey to fully understanding protein phosphorylations, it is essential to
improve the factors mentioned and this can be achieved by creating online enrichment methods.
1.8. The Aim of This Project
The main goal of this project was to establish a fully online method for enrichment of
phosphopeptides using a TiO2 pre-column. Recent attempts have been made however these
procedures were not fully online as they involved offline desalting and sub-fractionation of the
sample prior to the online phosphopeptide enrichment45. On the road to achieving the goal, offline
and semi-online methods will also be executed and analysed in order to predict conditions which will
be successful in online enrichment.

14
2. Methodology
2.1. Column Packing & Fritting
2.1.1. Chemicals, Materials and Apparatus Used
The following chemicals, material and apparatus were used for column packing
Table 1: Chemicals, materials and apparatus used for column fritting and packing
Chemical, material, apparatus Manufacturer
Milli-Q water, 18 MΩcm Merck Millipore TiO2 particles, 5 μm particle diameter Zirchrom Ethanol absolute for analysis Emsure Fused silica cappilaries Polymicro Technologies Kasil 1 potassium silicate NEXT Advance Frit Kit Formamide 25% NEXT Advance Frit Kit GF/C glass microfiber filters, 25 mm diameter Whatman In-house built pressure bomb
2.2.1. General Procedure
Fused silica capillaries were cut with a diamond cutter and inspected under a microscope to ensure
that the ends were smooth, which were otherwise smoothen with a ceramic file. A mixture of 25 %
formamide (100 μL) and milli-q water (75 μL) was made and vortexed. Kasil 1 (100 μL) was then
added to the mixture and vortexed once more. One end of the cut capillaries was fritted by gently
poking holes in the GF/C filters after wetting the penetration area with the prepared kasil /
formamide solution (2 μL). This process was repeated 5 times per frit. Finally the capillaries were left
to bake overnight at 85 °C with the frit-end down.
Once fritted, column packing could be executed. A slurry of TiO2 (4 mg) and EtOH AAS (200 μL) was
made and vortexed. The slurry was then placed into the pressure bomb while being magnetically
stirred. Columns were cleaned by wetting an analytical tissue with EtOH AAS and gently wiping the
column before being placed into the pressure bomb with the frit-end up. The TiO2 particles were
packed under a pressure of 1000 psi. Once the desired bed length had been reached the bomb was
closed and the column was swiftly removed. The stationary phase bed was consolidated to form a
stable bed formation by passing 100% Milli-Q water through the column at a flow rate of 1 uL/min.

15
2.2. Trypsin Digestion of α-casein From Bovine Milk
All percentages used in the methodology are specified as volume/volume (v/v).
2.2.1. Chemicals used
The following chemicals were used for the digestion of alpha-casein.
Table 2: Chemicals used for Trypsin Digestion of α-casein From Bovine Milk
Chemical Purity Manufacturer
Milli-Q water, 18 MΩcm Merck Millipore Ammonium bicarbonate (NH4HCO3) ≥ 99.5 % Fluka DL-Dithiolthreitol (DTT) ≥ 99.0 % Sigma Iodoacetamide (IAA) ≥ 99 % Sigma α-casein from bovine milk ≥ 70 % Sigma Urea Sigma Trypsin BRP European Pharmacopoeia reference Standard
2.2.2. Solutions Prepared
For the digestion of α -casein, the following solutions were prepared:
2,5 M ammonium bicarbonate was prepared by dissolving x g in 20 μL milli-q water
6 M urea solution was prepared by dissolving urea (3.9990 g) in NH4HCO3 (2.5 M, 2.5 mL)
Reducing reagent was prepared by dissolving DTT (59.8 mg) in NH4HCO3 (2.5 M, 1.0 mL)
Alkylating reagent was prepared by dissolving IAA (72.0 mg) in NH4HCO3 (2.5 M, 1.0 mL)
Trypsin solution was prepared by dissolving trypsin (3.4 mg) in NH4HCO3 (2.5 M, 3.4 mL)
2.2.3. General Procedure
α-casein was reconstructed in urea solution (100 μL) in a 2 mL Eppendorf tube. Reducing reagent (5
μL) was added and the mixture was left to reduce for 1 hour at 37 °C. Once reduced, alkylating
reagent (20 μL) was added and left to react for 1 hour in the dark (wrapped in aluminium foil). After 1
hour, reducing reagent (20 μL) was added to consume leftover alkylating reagent present. NH4HCO3
(2.5 M, 900 μL) was then added to dilute the urea and finally trypsin (1:30 trypsin to protein ratio by
weight) was added and left to digest over night at 37 °C. The sample was stored at -20 °C
See Error! Reference source not found. for amounts of α-casein and trypsin solution used.
Table 3: Variable amounts of chemicals used
Date Sample Name
29-04-2016 20160429MDα1
29-04-2016 20160429MDα2
α-casein from bovine milk 3.6 mg 2.7 mg Trypsin solution 120 μL 90 μL

16
2.3. Off-line Phosphopeptide Enrichment of Digested α-casein Sample
2.3.1. Chemicals, materials and apparatus used
The following chemicals, materials and apparatus were used for the desalting and phosphopeptide
enrichment by TiO2 affinity chromatography (Table 4):
Table 4: Chemicals, materials and apparatus used for Off-line Phosphopeptide Enrichment of Digested α-casein Sample
Furthermore, in-house made GF/C and C18 spin columns were made via the following procedures:
The GF/C spin column was made by cutting 3 small discs (approximate radius of 0.5 mm) from a GF/C
sic with the end of a glass pipette. The cut discs were placed inside a 200 μL pipette tip to serve as a
plug. A small hole was then pricked into the lid of a 2 mL Eppendorf tube and the pipette tip was
placed into this hole so that it was firmly held above the 1 mL mark.
The C18 spin column was made by cutting 1 small disc (approximate radius of 0.5 mm) from a C18
disc with the end of a glass pipette. The cut disc was placed inside a 200 μL pipette tip to serve as a
plug. A small hole was then pricked into the lid of a 2 mL Eppendorf tube and the pipette tip was
placed into this hole so that it was firmly held above the 1 mL mark.

17
2.3.2. General Procedure
2.3.2.1. Desalting prior to enrichment:
A previously digested sample 20160429MDα1 thawed at 4 °C and then handled at room
temperature. An empore C18 cartridge was placed in to a 2 mL Eppendorf tube after cutting a small
hole in the tubes lid. The cartridge was then wet with ACN (1 mL) via centrifugation (2000 rpm, 60 s)
and then placed in a new 2 mL eppendorf tube. The digest sample was then loaded (2000 rpm, 60 s)
and the flow-through was recovered and loaded once more. The cartridge was then washed 3 times
with 0.1% TFA (1 mL, 2000 rpm, 60 s) and eluted with 6% TFA / 80% ACN (1 mL, 2000 rpm, 60 s). The
desalted digest was stored at 4 °C for an hour before proceeding to the enrichment.
2.3.2.2 Phosphopeptide enrichment via TiO2 affinity chromatography:
The centrifugation times noted were in insufficient in certain cases. In the case of incomplete flow-
through, centrifuge until complete.
TiO2 particles (5 μm, 11.3 mg) were added to milli-Q water (226 μL) to form slurry. The slurry was
loaded onto the in-house made GF/C spin column via centrifugation (4000 rpm, 120 s). The column
was then equilibrated with 6% TFA / 80% ACN (200 μL, 4000 rpm, 120 s) after which the desalted
sample was loaded in steps of 200 μL (4000 rpm, 180 s). The column was then washed twice with 6%
TFA / 80% ACN (200 μL, 4000 rpm, 180 s) flowed by a 2 washes with 0.1% TFA (200 μL, 4000 rpm, 180
s). After washing, 5 % NH4OH was added to the column and the phosphopeptides were manually
eluted with a plastic syringe (PTFE tape was wrapped around the syringe tip to ensure a tight
connection with the pipette tip.) into a new Eppendorf tube. The large amount of back-pressure
resulted in a lot of man-force to be applied. After elution 10% FA (400 μL) was added to acidify the
sample. The following desalting should be immediately performed to avoid methionine oxidation.
Centrifugation times should be lengthened if incomplete flow-through is observed.
2.3.2.3. Desalting after phosphopeptide enrichment:
A C18 spin column was wet with ACN (50 μL, 4000 rpm) and equilibrated with 0.1% TFA (50 μL, 4000
rpm). The enriched phosphopeptide sample was then loaded onto the column in 200 μL (4000 rpm).
The column was then washed 3 times with 0.1% TFA (50 μL, 4000 rpm). Finally the phosphopeptides
were manually eluted with a syringe into a new tube. As explained in the enrichment step, tape was
used for a secure fit and a large amount of man-power was needed. The eluate was then evaporated
to dryness in a GeneVac DNA concentrator for 30 minutes and then stored at -20 °C. The sample was
named; 20160502 MD2.
Note that centrifugation was executed until complete flow-through was observed which usually took
approximately 10 minutes.
18
2.3.3. LC-MS Method for Phosphopeptide Analysis
LC-MS analysis was performed on an inline setup of a Eksigent nanoLC quarto serie coupled to a
TripleTOF 5600 + (Sciex, Framingham MA, USA).
2.3.3.1. Instrumental conditions
After calibration of the LC-MS system, the C18 trap column (NanoLC, ChromXP trap, C18 3 μm fully
porous particles, 350 μm i.d. x 0.5 mm) was attached to the trapping/injection valve. After purging
the system for 20 cycles in the loading buffer (0.1% TFA 3% acetonitrile, 97% LC-MS grade water v/v)
the trap was equilibrated at 1 μL/min for 45 min. The analytical column was in-house packed with
Magic AQ (New Objective, Woburn MA, USA) C18 stationary phase (5 μm fully porous particles, 100
angstrom pore size). The C18 column was equilibrated at 300 nL/min for 1 hr after purging the A and
B reservoirs for 20 cycles in 0,1% formic acid in LC-MS grade water for A and 0,1% formic acid in LC-
MS grade ACN for B. After equilibration the sample (10 μL) was loaded onto the trap via the LC-MS
autosampler. After washing with loading buffer for 10 min, the sample was eluted off the trap
column by the system automatically switching the valve so that the trap column was inline with the
analytical column, this allowed the gradient mobile phase flow through the trap column, eluting the
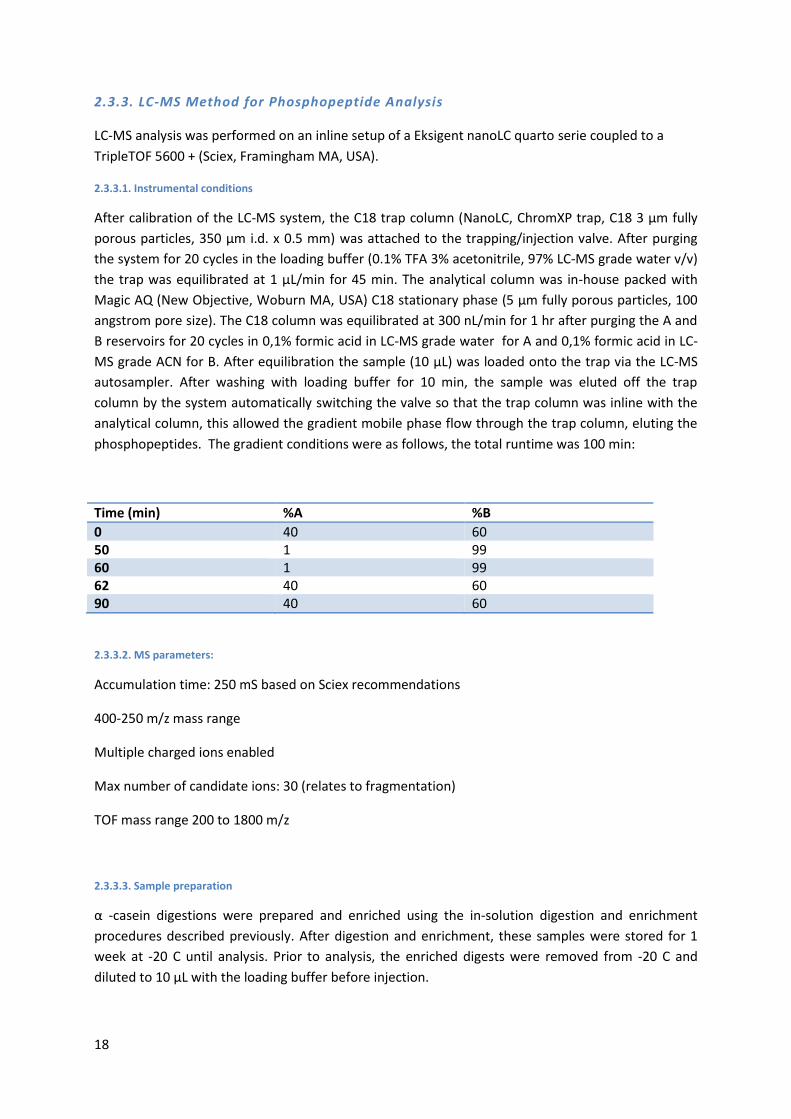
phosphopeptides. The gradient conditions were as follows, the total runtime was 100 min:
Time (min) %A %B
0 40 60 50 1 99 60 1 99 62 40 60 90 40 60
2.3.3.2. MS parameters:
Accumulation time: 250 mS based on Sciex recommendations
400-250 m/z mass range
Multiple charged ions enabled
Max number of candidate ions: 30 (relates to fragmentation)
TOF mass range 200 to 1800 m/z
2.3.3.3. Sample preparation
α -casein digestions were prepared and enriched using the in-solution digestion and enrichment
procedures described previously. After digestion and enrichment, these samples were stored for 1
week at -20 C until analysis. Prior to analysis, the enriched digests were removed from -20 C and
diluted to 10 μL with the loading buffer before injection.

19
2.4. Semi On-line Phosphopeptide Enrichment of Digested α-casein
Sample
2.4.1. Chemicals, Materials and Equipment Used
The same chemicals, materials and apparatus were used as in section 2.2.1., with the addition of the
following apparatus (Table 5: Additional appartus used for the semi-online phosphopeptide
enrichment):
Table 5: Additional appartus used for the semi-online phosphopeptide enrichment
Apparatus
Acquity UPLC Binary solvent manager, M-class, Waters Acquity UPLC μSample manager – FL, Waters Acquity UPLC TUV detector, Waters
2.4.2. General Procedure
The desalting procedures performed on sample 20160429MDα2 were the same as those in section
2.3.2.
2.4.2.1. Online enrichment:
A home packed TiO2 column (75 μm ID, 300 Å pore size) was attached to the M-Class UPLC. 0.1% TFA
was placed in the A1 solvent position and 6% TFA / 80% ACN in the B1 position. The system was
purged and then slowly ramped up to a flow-rate of 0.500 μL/min under a flow of 100% solvent B1.
The pressure started stabilizing around 1000 psi. The column was left to equilibrate for 45 minutes.
The loading method was made and applied which comprised of 40 injections of 5 μL. The column was
then washed with 100% of solvent B1 for 30 minutes followed by a 45 minute wash with 100% of
solvent A1. During this wash, the 6% TFA / 80% ACN in the B1 position was switched with 5% NH4OH.
After washing, the elution took place with 100% of the new B1 solvent for 1 hour. The eluent was
collected in a 2 mL Eppendorf tube. Finally, 70 μL was added to the enriched phosphopeptide sample
and subsequent desalting was executed.
2.4.3. LC-MS Method for Phosphopeptide Analysis
The LC-MS analysis of the sample acquired in the semi on-line phosphopeptide enrichment
procedure was analysed in the same manner as described in section 2.3.3.

20
2.5. Fully On-line Phosphopeptide Enrichment of Digested α-casein
Sample
2.5.1. Instrumental Conditions and MS Parameters
The instrumental conditions and MS parameters of the fully on-line phosphopeptide enrichment
were the same as those described in section 2.3.3. with the following exceptions;
An in-house packed trap column (20 μm TiO2 particles, 300 Å, 200 μm ID) was attached to the
trapping/injection valve instead of the C18 trap column. 5% ammonium hydroxide in LC-MS grade
water was attached to line A and 45% water, 50 % acetonitrile for line B.
2.5.1. Sample Preparation
Separate digestions of yeast and alpha-casein were prepared using the in-solution digestion
procedure described previously. After digestion, these digests were stored for 1 week at -20 C until
analysis. Prior to analysis, the digests were removed from -20 C and injected without dilution to be in
agreement with the offline enrichment procedure where the digest was enriched without dilution.

21
3. Results & discussion
3.1. Offline Phosphopeptide TiO2 Enrichment
3.1.1. Condition Performance
A common drawback of TiO2 enrichment techniques is nonspecific isolation of acidic peptides along
with the desired phosphopeptides. Current techniques devoted to countering this challenge exist,
such as O-methyl esterification of carboxyl-groups40, however result in incomplete yields and
unwanted side reactions such as deamidation and subsequent methylation of asparagine and
glutamine residues43. Another method is the addition of dihydroxybenzoic acid (DHB) during
phosphopeptide binding which leads to a decrease in the amount of non-phosphorylated peptides
being detected43. The addition of DHB may cause problems with LC-MS systems and contaminate the
MS’ ion source making it incompatible with online methods46,47. Due to the drawbacks mentioned
above, these techniques were not explored. Rather, TFA was used as mobile phase during
phosphopeptide binding and washing and NH4OH was used to elute the phosphopeptides. 0.1% TFA
is known to have pH of 1.8 – 2.0 resulting in acidic residues remaining in their neutral state during
phosphopeptide loading minimizing the amount of acidic residues binding to the TiO2 stationary
phase opposed to when acetic acid (pH 2.7-2.9) is used. As the pH of phosphoric acid decreases from
1.8 to 1.1 when methylated48, a similar decrease is expected when bound to a peptide which would
mean that under the acidic conditions provided by a 0.1% TFA mobile phase, the phosphate groups
would be negatively charged and readily bind to the TiO2 stationary phase. Recently presented work
stated that elution with ammonium bicarbonate (pH 9) was not sufficient to elute all
phosphopeptides and that a pH of 10.5 is optimal43. This is why the stronger base ammonium
hydroxide was used as to elute the phosphopeptides.
3.1.2. Peptides Detected
The conditions used resulted in the detection of 537 peptides of which 354 were phosphorylated.
273 of these peptides belonged to α-casein S1 (86.5 % sequence coverage) with 117
phosphopeptides. Mainly single phosphopeptides were isolated with a few doubles and multiples
(Fig. 4). 168 peptides belonged to the α-casein S2 protein (71.17 % sequence coverage), of which 42
were phosphorylated.

22
Figure 4: Number of singly, doubly and multiply phosphopeptides isolated belonging to α-casein S1 The programme searched against the Uniprot database (not species specific). False discovery rate: 1%. Total number of distinct peptides
detected was 273, total number of distinct phosphopeptides detected was 117.
As α-casein is a well-studied and understood protein, it may be used as a benchmark to compare our
method to other studies. According to the literature49 nine phosphorylation sites exist on α-casein S1
and the method used detected five of these sites. This result confirms that the method used is
capable of effectively determining phosphosites. However, α-casein S2 contains ten phosphosites, of
which only two were detected50. This could simply mean that the protein sample predominantly
contains α-casein S1.
The phosphosites detected can be can be validated by manually analysing the obtained spectra for
each peptide. The mass spectra contain b- and y-ion peaks which correlate to cleavages resulting in
the charge being retained by either the amino- or carboxy-terminus respectively. By validating that
cleavage has taken place and a b- or y-ion peak is present after and before a suggested
phosphorylated site, respectively, one can confirm whether that specific phosphorylation is indeed
present. In the case of a confirmation, the preceding ion will have the mass of the corresponding
peptide fragment excluding a phosphate group and the mass of the ion after the phosphosites will
correlate to the mass of the corresponding peptide fragment including a phosphate group51. When
dealing with suggested pThr and pTyr sites, one must take into account that these are much less
common than pSer sites and therefore confirmation of such sites must be done so with caution,
especially when the suggested phosphosite is neighbouring a Ser. The method used to confirm
phosphosites is discussed below using pSer130 as an example:
The mass spectrum (Figure 5: see next page) indicating phosphorylation at Ser 130 corresponds to
the following peptide fragment:
PNS[Pho]AEER
The obtained y6-ion with a mass of 785.2825 corresponds to the peptide fragment NSAEER with
water and a phosphorylation. The y4-ion with a mass of 504.2413 corresponds to the peptide
fragment AEER with water. This data indicates that Ser130 is indeed phosphorylated. This conclusion
can be further validated by also analysing the b2- and b5-ion which also support the existence of
pSer130
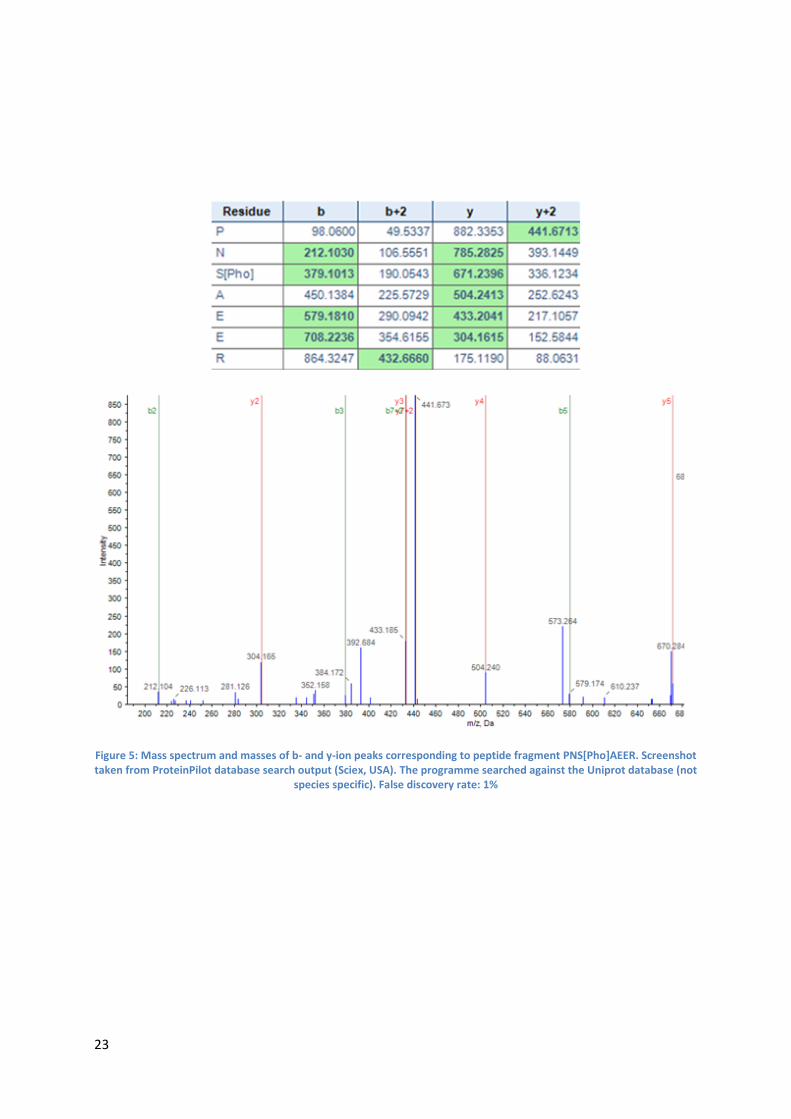
23
Figure 5: Mass spectrum and masses of b- and y-ion peaks corresponding to peptide fragment PNS[Pho]AEER. Screenshot taken from ProteinPilot database search output (Sciex, USA). The programme searched against the Uniprot database (not
species specific). False discovery rate: 1%

24
3.1.3. Novel Phosphosites on α-casein S1
Of the 117 α-casein S1 phosphopeptides detected, according to ProteinPilot, 10 were distinct and 6
of these have not yet been reported in literature (Table 6). Since the phosphorylation of α-casein S1
has been fully characterised, it is expected that the newly observed phosphorylation sites seen in this
study are most likely due to the high concentrations we used, which may not reflect realistic
conditions. In any case, it is prudent to examine the phosphorylation sites reported by ProteinPilot.
The validity of the reported phosphorylation sites were examined by manually interpreting the mass
spectra as shown in the previous section. The results are shown below in Table 1.
Table 6: Summary novel distinct phosphopeptides detected belonging to α-casein S1 and whether they can be confirmed or not.
Position Notes
Thr64 Possible but cannot be confirmed
Ser103 Possible but cannot be confirmed
Ser137 Possible but cannot be confirmed
Ser193 Possible but cannot be confirmed
Ser195 Data suggests existence
Ser203 Possible but cannot be confirmed
25
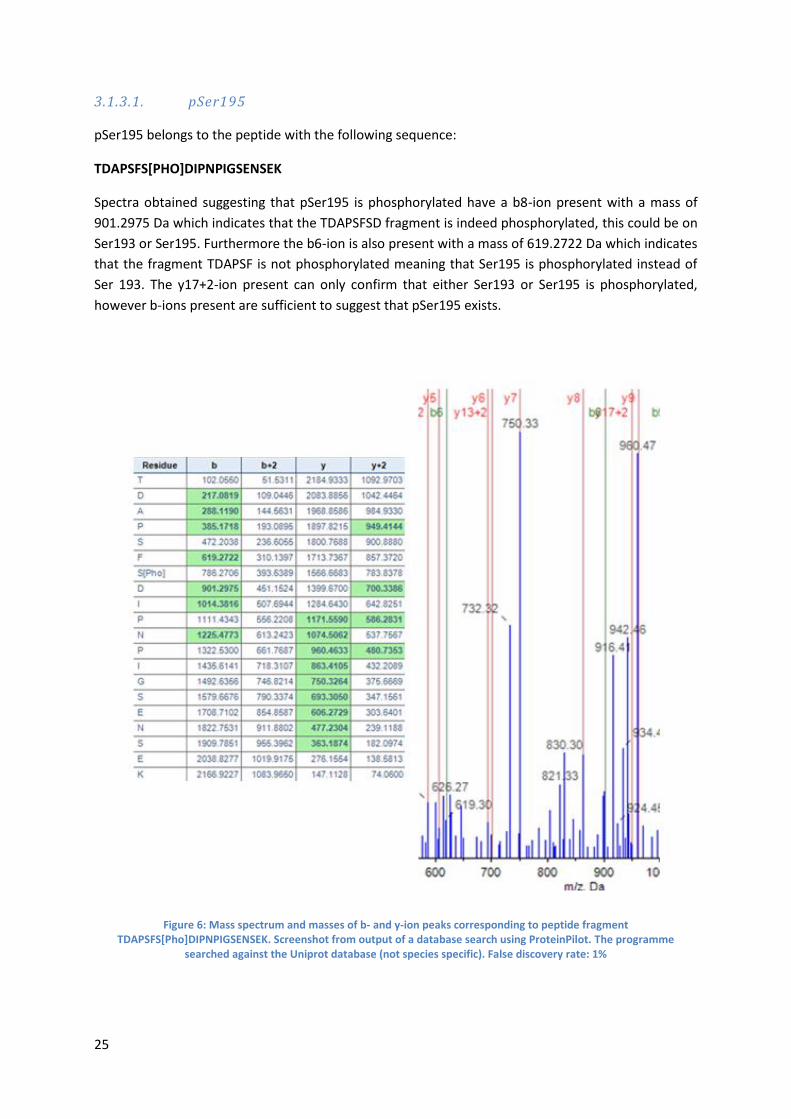
3.1.3.1. pSer195
pSer195 belongs to the peptide with the following sequence:
TDAPSFS[PHO]DIPNPIGSENSEK
Spectra obtained suggesting that pSer195 is phosphorylated have a b8-ion present with a mass of
901.2975 Da which indicates that the TDAPSFSD fragment is indeed phosphorylated, this could be on
Ser193 or Ser195. Furthermore the b6-ion is also present with a mass of 619.2722 Da which indicates
that the fragment TDAPSF is not phosphorylated meaning that Ser195 is phosphorylated instead of
Ser 193. The y17+2-ion present can only confirm that either Ser193 or Ser195 is phosphorylated,
however b-ions present are sufficient to suggest that pSer195 exists.
Figure 6: Mass spectrum and masses of b- and y-ion peaks corresponding to peptide fragment TDAPSFS[Pho]DIPNPIGSENSEK. Screenshot from output of a database search using ProteinPilot. The programme
searched against the Uniprot database (not species specific). False discovery rate: 1%

26
3.1.4. Novel Phosphosites on α-casein S2
Of the 42 phosphorylated peptides belonging to the α-casein S2 protein, 7 were distinct and 5 were
novel phosphorylation not reported in earlier literature (Table 7). These phosphorylations can be
verified in the same manner utilized in section 3.1.2. However all spectra obtained for these novel
phosphosites had insufficient data present to make any confirmations.
Table 7: Summary novel phosphopeptides detected belonging to α-casein S1 and whether they can be confirmed or not.
Position Notes
Thr137 Possible but cannot be confirmed
Ser144 Possible but cannot be confirmed
Thr145 Possible but cannot be confirmed
Ser146 Possible but cannot be confirmed
Thr159 Possible but cannot be confirmed
3.1.5. Conclusion: Offline TiO2 Enrichment
The results acquired under the chosen conditions seemed promising to apply to an online method. A
large amount of peptides were detected, of which 66 % were phosphorylated showing that this
technique is effective in trapping phosphopeptides. The method also proved effective by determining
the new phosphorylation site pSer195 and many other possible novel phosphorylations were found
but lacked sufficient data for confirmation. These results were effective therefore we decided to
choose these conditions as a starting point to carry out the semi-online phosphopeptide TiO2
enrichment.
3.2. Semi-Online Phosphopeptide TiO2 Enrichment
3.2.1. Detected Peptides
After following the semi-online phosphopeptide enrichment procedure 148 distinct phosphopeptides
were detected of which 22 were phosphorylated. 83 of these distinct peptides belonged to α-casein
S1 (80.84 % sequence coverage) and 31 to α-casein S2 (49.55 % sequence coverage). Of these
peptides, 12 belonging to α-casein S1 were phosphorylated and 7 belonging to α-casein S2 were
phosphorylated. From the known phosphosites in α-casein S1 and α-casein S2, only 3 sites were
detected for α-casein S1 and 2 were detected for α-casein S2. The decrease in peptides detected is
related to two factors; the amount of α-casein digested and the amount of sample loaded during
enrichment. In both cases these amounts were lower during the semi-online enrichment. 2.7 mg of
α-casein was digested in the semi-online method opposed to 3.6 mg for the off-line method and
0.200 ml of the desalted sample was used to load the TiO2 column online opposed to 1 ml during the
off-line method. This choice was made due to the injection volume limits of the instrument (20 uL)

27
and instrument flow rate limitations: the maximum flow rate was 2.0 uL/min which would require
200 mins of elution time. Considering the lower amounts used it is no surprise that the number of
detected proteins are lower, if 1 ml was to be loaded and if the number of peptides detected
correlate linearly to sample volume, we could expect that 5 x 148 = 740 peptides would be detected.
It is remarkable that only 15 % of the detected peptides were phosphorylated. This decrease may be
due to the use of an in-house packed column and further column optimization would have to be
executed to improve the phosphopeptide selectivity. The online method has still proven to
specifically isolate singly phosphorylated peptides (Figure 7: Number of singly and doubly
phosphopeptides isolated belonging to α-casein S1).
Figure 7: Number of singly and doubly phosphopeptides isolated belonging to α-casein S1. ProteinPilot searched against the Uniprot database (not species specific). False discovery rate: 1%. Total number of distinct peptides detected was 83,
total number of distinct phosphorylated peptides detected was 12.
3.2.2. Novel Phosphosites on α-casein S2
The semi-online method detected far fewer novel phosphosites compared to that of the offline
method. This could mean that is less efficient but as many of the novel phosphosites in the off-line
method could not be confirmed and are likely false identifications, it can be argued that the semi-
online method is more efficient by displaying less false identification.
One novel phosphosite was however suggested by ProteinPilot; α-casein S2 at Thr159. The
corresponding mass spectra did not display enough data to make a confirmation and pThr159 was
regarded as a false identification.
3.2.3. Conclusion: Semi-online TiO2 Enrichment
Although the phosphopeptide specificity of the semi-online method was not as highly observed for
the off-line method, it still proved selective for phosphopeptide enrichment. Possible reasons lie in
the decreased mass loading capabilities due to instrument restrictions. Less mass was loaded onto
the column since a single injection could only load 20 μL of sample and took approximately 3 minutes
for one injection cycle. The low flow rate also resulted in lengthy analysis times and reducing these
times can lead to incomplete washing of non-phosphorylated peptides and elution of
phosphopeptides. Increasing the washing time could also improve the final phosphopeptide
11
1
# of single
# of double

28
specificity by eliminating more of the non-phosphorylated peptides from the column prior to elution.
However, this may also increase dilution which would decrease sensitivity.
The results under the chosen conditions showed effectiveness to a certain degree and were therefor
also used for the fully on-line method. However, we packed a shorter column and used larger TiO2
particles for increased porosity, both facilitating the use of higher flow rates by reducing the system
backpressure.
3.3. Online Phosphopeptide TiO2 Enrichment
The attempt at on-line failed and no data was able to be obtained. The main cause for this was the
high pH of the two mobile phases which were both approximately 12. This proved to be far too basic
as it was observed that the silica tubing started dissolving. The pH range compatible with a typical LC-
MS system is known to be 2-8 however there are systems which are compatible with a broader range
of pH values. These systems make use of stainless steel tubing which unfortunately would not be
compatible with phosphopeptide enrichment processes as metals are known to have interactions
with phosphopeptides. Additional challenges in online phosphopeptide were already mentions in
3.2.3., such as low injection volumes resulting in low mass loading and low flow rates resulting in
lengthy analysis times or insufficient washing and elution when analysis times are lowered. To
determine whether it was possible to lower the pH to more LC-MS compatible values while
preserving selectivity for phosphopeptide enrichment, a series of offline phosphopeptide
enrichments were carried out at different pH values. Unfortunately, the MS sensitivity was low to
produce any meaningful conclusions.

29
3. Conclusion & Outlook
The results obtained from the offline and semi-online methods clearly show that TiO2 is effective at
enriching phosphopeptides. The offline method did however have better results than the semi-online
method which can be due to numerous factors. These factors include lower mass loading and lower
flow rates. This can be countered by increasing analysis times however one of the reasons for seeking
an online method is to decrease analysis time. The in-house packed TiO2 column could also undergo
optimization. During the semi-online method the column used had an inner diameter of 75 μm and
the particles used had a diameter of 5 μm. By increasing the particle diameter the porosity of the bed
would increase resulting in less backpressure which would facilitate higher flow rates.
Although the in-house packed columns proved promising, it was not able to implement them in a
fully online method. The main reason for failure was the extremely high pH caused by the
ammonium hydroxide mobile phase. As a high pH is needed to specifically elute the phosphopeptides
this is a worrying factor however the pH used was around 12 which led to the exploration of
phosphopeptide elution using different pH values ranging from 9.6 to 11.4. Unfortunately no results
were obtained when analysed via MS due to complications and this is still an area worth addressing
in the future.

30
Acknowledgements
I would like to thank Prof. Garry Corthals for welcoming me into his research group and making it
possible for me to work on the project. I am also very grateful for Dr. Michelle Camenzuli being such
a great day-to-day mentor and supervisor which supported me whenever needed. Furthermore I
would like to thank the whole Corthals group and it was a pleasure working with each and every one
of them. Finally I would like to thank Dr. Leo de Koning for his time and effort in being my second
assessor.
References
1 A. Paradela, J.P. Albar, Advances in the analysis of protein phosphorylation, Journal of proteome research 7 (2008) 1809. 2 J.V. Olsen, B. Blagoev, F. Gnad, B. Macek, C. Kumar, P. Mortensen, M. Mann, Global, In Vivo, and Site- Specific Phosphorylation Dynamics in Signaling Networks, Cell 127 (2006) 635-648. 3 http://www.phosphosite.org 4 D.E. Kalume, H. Molina, A. Pandey, Tackling the phosphoproteome: tools and strategies, Curr. Opin. Chem. Biol. 7 (2003) 64-69. 5 M.J. Hubbard, P. Cohen, On target with a new mechanism for the regulation of protein phosphorylation, Trends Biochem. Sci. 18 (1993) 172-177. 6 T. Hunter, Signaling— 2000 and Beyond, Cell 100 (2000) 113-127. 7 W.D. Lehmann, Protein phosphorylation analysis by electrospray mass spectrometry a guide to concepts and practice / / Wolf D. Lehmann. Cambridge : : Royal Society of Chemistry, Cambridge,
(2010). 8 P.A. Levene, C.L. Alsberg, The cleavage products of vitellin, J. Biol. Chem. 2 (1906) 127–133. 9 F.A. Lipmann, P.A. Levene, Serinephosphoric acid obtained on hydrolysis of vitellinic acid, J. Biol. Chem. 98 (1932) 109–114. 10 C. F. Cori, G. T. Cori, Polysaccharide phosphorylase, In Nobel lectures, physiology or medicine
1942-1962, Amsterdam: Elsevier, (1947) 186-206. 11 E. H. Fischer, D. J. Graces, E. R. Crittender, E. G. Krebs, Structure of the site phosphorylated in the phosphorylase b to a reaction, J. Biol. Chem. 234 (1959) 1698–1704. 12 S. Shoji, D. C. Parmelee, R. D. Wade, S. Kumar, L. H. Ericsson, K. A. Walsh, H. Neurath, G. L. Long, J. G. DeMaille, E. H. Fischer, K. Titani, Complete amino acid sequence of the catalytic subunit of bovine cardiac muscle cyclic AMP-dependent protein kinase, Proc. Natl. Acad. Sci. USA 78 (1981)
848–851. 13 D. R. Knighton, J. Zheng, L. F. T. Eyck, V. A. Ashford, N. H. Xuong, S. S. Taylor, J. M. Sowadski, Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase, Science 253 (1991) 407–414. 14 T. Hunter, B. M. Sefton, Transforming gene product of Rous sarcoma virus phosphorylates tyrosine, Proc. Natl. Acad. Sci. USA 77 (1980) 1311–1315. 15 https://www.thermofisher.com/nl/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/phosphorylation.html# 16 G. Manning et al., The protein kinase complement of the human genome, Science 298 (2002) 1912-34. 17 C. Walsh, Enzymatic reaction mechanisms, San Francisco: W. H. Freeman xv (1979) 978. 18 C. Walsh, Posttranslational modification of proteins: Expanding nature's inventory, Englewood, Colo.: Roberts and Co. Publishers xxi (2006) 490. 19 M. Mann et al., Analysis of protein phosphorylation using mass spectrometry: Deciphering the
phosphoproteome, Trends Biotechnol. 20 (2002) 261-8. 20 T. Pawson, P. Nash, Assembly of cell regulatory systems through protein interaction domains, Science 300 (2003) 445-52 21 L. N. Johnson, R. J. Lewis Structural basis for control by phosphorylation, Chem Rev. 101 (2001) 2209-4

31
22 M. B. Yaffe, Phosphotyrosine-binding domains in signal transduction. Nat. Rev. Mol. Cell Biol. 3
(2002) 177-86. 23 G. L. Johnson, R. Lapadat, Mitogen-activated protein kinase pathways mediated by ERK, JNK,
and p38 protein kinases, Science 298 (2002) 1911-2 24 P. C. Nowell, J. Clin, Discovery of the Philadelphia chromosome: a personal perspective, Invest. 117 (2007) 2033–2035. 25 P. Cohen, The origins of protein phosphorylation, Nat. Cell Biol. 4 (2002) E127–E130. 26 R. Krϋger, D. Kϋbler, R. Pallisse´ , A. Burkovski, W. D. Lehmann, Protein and proteome
phosphorylation stoichiometry analysis by element mass spectrometry, Anal. Chem. 78 (2006) 1987–1994. 27
H. Steen, J.A. Jebanathirajah, J. Rush, N. Morrice, M.W. Kirschner, Phosphorylation analysis by
mass spectrometry: myths, facts, and the consequences for qualitative and quantitative measurements, Molecular & cellular proteomics : MCP 5 (2006) 172. 28
H. Steen, J. A. Jebanathirajah, M. Springer,M.W. Kirschner, Stable isotope-free relative and
absolute quantitation of protein phosphorylation stoichiometry by MS, Proc. Natl. Acad. Sci. USA 102 (2005) 3948–3953. 29
B. Bodenmiller, N.M. Lukas, M. Mueller, B. Domon, R. Aebersold, Reproducible isolation of
distinct, overlapping segments of the phosphoproteome, Nature Methods 4 (2007) 231. 30
A. Pandey, M.M. Fernandez, H. Steen, B. Blagoev, M.M. Nielsen, S. Roche, M. Mann, H.F. Lodish,
Identification of a novel immunoreceptor tyrosine- based activation motif- containing molecule, STAM2, by mass spectrometry and its involvement in growth factor and cytokine receptor signaling pathways, The Journal of biological chemistry 275 (2000) 38633. 31
A. Pandey, A.V. Podtelejnikov, B. Blagoev, X.R. Bustelo, M. Mann, H.F. Lodish, Analysis of
receptor signaling pathways by mass spectrometry: Identification of Vav- 2 as a substrate of the epidermal and platelet- derived growth factor receptors, Proceedings of the National Academy of Sciences of the United States 97 (2000) 179. 32
H. Steen, B. Kuster, M. Fernandez, A. Pandey, M. Mann, Tyrosine phosphorylation mapping of the
epidermal growth factor receptor signaling pathway, The Journal of biological chemistry 277 (2002) 1031. 33
Y.G. Yeung, Y. Wang, D.B. Einstein, P.S. Lee, E.R. Stanley, Colony- stimulating factor- 1
stimulates the formation of multimeric cytosolic complexes of signaling proteins and cytoskeletal components in macrophages, The Journal of biological chemistry 273 (1998) 17128. 34
G. Caratù, D. Allegra, M. Bimonte, G.G. Schiattarella, C. D'Ambrosio, A. Scaloni, M. Napolitano,
T. Russo, N. Zambrano, Identification of the ligands of protein interaction domains through a functional approach, Molecular & cellular proteomics : MCP 6 (2007) 333. 35
K. Machida, C.M. Thompson, K. Dierck, K. Jablonowski, S. Kärkkäinen, B. Liu, H. Zhang, P.D.
Nash, D.K. Newman, P. Nollau, T. Pawson, G.H. Renkema, K. Saksela, M.R. Schiller, D. Shin, B.J. Mayer, High- Throughput Phosphotyrosine Profiling Using SH2 Domains, Mol. Cell 26 (2007) 899-915. 36
M. Grønborg, T.Z. Kristiansen, A. Stensballe, J.S. Andersen, O. Ohara, M. Mann, O.N. Jensen, A.
Pandey, A mass spectrometry-based proteomic approach for identification of serine/threonine-phosphorylated proteins by enrichment with phospho- specific antibodies: identification of a novel
protein, Frigg, as a protein kinase A substrate, Molecular & cellular proteomics : MCP 1 (2002) 517. 37
J. Porath, J. Carlsson, I. Olsson, G. Belfrage, Metal chelate affinity chromatography, a new
approach to protein fractionation, Nature 258 (1975) 598. 38
G.L. Corthals, R. Aebersold, D.R. Goodlett, Identification of Phosphorylation Sites Using
Microimmobilized Metal Affinity Chromatography, Meth. Enzymol. 405 (2005) 66-81. 39
S. Feng, M. Ye, H. Zhou, X. Jiang, X. Jiang, H. Zou, B. Gong, Immobilized zirconium ion affinity
chromatography for specific enrichment of phosphopeptides in phosphoproteome analysis, Molecular & cellular proteomics : MCP 6 (2007) 1656. 40
S.B. Ficarro, M.L. Mccleland, P. Todd Stukenberg, D.J. Burke, M.M. Ross, J. Shabanowitz, D.F.
Hunt, F.M. White, Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae, Nat. Biotechnol. 20 (2002) 301. 41
K. Moser, F.M. White, Phosphoproteomic analysis of rat liver by high capacity IMAC and LC- MS/
MS, Journal of proteome research 5 (2006) 98. 42
M.W.H. Pinkse, P.M. Uitto, M.J. Hilhorst, B. Ooms, A.J.R. Heck, Selective isolation at the
femtomole level of phosphopeptides from proteolytic digests using 2D- NanoLC- ESI- MS/ MS and titanium oxide precolumns, Anal. Chem. 76 (2004) 3935.

32
43
M.R. Larsen, T.E. Thingholm, O.N. Jensen, P. Roepstorff, T. Jørgensen J.D., Highly selective
enrichment of phosphorylated peptides from peptide mixtures using titanium dioxide microcolumns, Molecular & cellular proteomics : MCP 4 (2005) 873. 44
C. Klemm, S. Otto, C. Wolf, R.F. Haseloff, M. Beyermann, E. Krause, Evaluation of the titanium
dioxide approach for MS analysis of phosphopeptides, Journal of Mass Spectrometry 41 (2006) 1623-1632. 45 M.W.H. Pinkse, S. Mohammed, J.W. Gouw, B. van Breukelen, H.R. Vos, A.J.R. Heck, Highly
robust, automated, and sensitive online TiO2-based phosphoproteomics applied to study endogenous phosphorylation in Drosophila melanogaster, Journal of proteome research 7 (2008) 687. 46 T. E. Thingholm, T. J. D. Jorgensen, O. N. Jensen, M. R. Larsen, Highly selective enrichment of phosphorylated peptides using titanium dioxide, Nat. Protocols, 1, 4 (2006) 1929–1935. 47 N. Sugiyama, T. Masuda, K. Shinoda, A. Nakamura, M. Tomita, Y. Ishihama, Phosphopeptide
enrichment by aliphatic hydroxyl acid-modified metal oxide chromatography for NanoLC-MS/MS in proteomics applications, Mol. Cell. Proteomics, 6 (2007) 1103–1109. 48 A. Saha, N. Saha, L. N. Ji, J. Zhao, F. Gregan, S. A. A. Sajadi, B. Song, and H. Sigel, Stability of
metal ion complexes formed with methyl phosphate and hydrogen phosphate, J. Biol. Inorg. Chem.
1 (1996) 231–238 49
http://www.uniprot.org/uniprot/P02662 50
http://www.uniprot.org/uniprot/P02662 51
I.A. Papayannopoulos, ChemInform Abstract: The Interpretation of Collision‐ Induced Dissociation
Tandem Mass Spectra of Peptides, ChemInform 27 (1996) no-no.