genetic and pharmacological inhibition of trem-1 limits ... · genetic and pharmacological...
TRANSCRIPT

Listen to this manuscript’s
audio summary by
JACC Editor-in-Chief
Dr. Valentin Fuster.
J O U R N A L O F T H E A M E R I C A N C O L L E G E O F C A R D I O L O G Y V O L . 6 8 , N O . 2 5 , 2 0 1 6
ª 2 0 1 6 B Y T H E A M E R I C A N C O L L E G E O F C A R D I O L O G Y F O U N D A T I O N
P U B L I S H E D B Y E L S E V I E R
I S S N 0 7 3 5 - 1 0 9 7 / $ 3 6 . 0 0
h t t p : / / d x . d o i . o r g / 1 0 . 1 0 1 6 / j . j a c c . 2 0 1 6 . 1 0 . 0 1 5
Genetic and Pharmacological Inhibition ofTREM-1 Limits the Development ofExperimental Atherosclerosis
Jeremie Joffre, MD,a,b Stephane Potteaux, PHD,a,b Lynda Zeboudj, MSC,a,b Xavier Loyer, PHD,a,b Amir Boufenzer, PHD,cLudivine Laurans, MSC,a,b Bruno Esposito, BSC,a,b Marie Vandestienne, MSC,a,b Saskia C.A. de Jager, MD, PHD,d
Carole Hénique, PHD,a,b Ivana Zlatanova, MSC,a,b Soraya Taleb, PHD,a,b Patrick Bruneval, MD, PHD,a,b,e
Alain Tedgui, PHD,a,b Ziad Mallat, MD, PHD,a,b,f Sebastien Gibot, MD, PHD,g Hafid Ait-Oufella, MD, PHDa,b,h
ABSTRACT
Fro
Pa
Ne
Pa
Kin
Hô
Pa
Re
Re
au
Ma
BACKGROUND Innate immune responses activated through myeloid cells contribute to the initiation, progression, and
complications of atherosclerosis in experimental models. However, the critical upstream pathways that link innate
immune activation to foam cell formation are still poorly identified.
OBJECTIVES This study sought to investigate the hypothesis that activation of the triggering receptor expressed on
myeloid cells (TREM-1) plays a determinant role in macrophage atherogenic responses.
METHODS After genetically invalidating Trem-1 in chimeric Ldlr�/� Trem-1�/� mice and double knockout ApoE�/�
Trem-1�/� mice, we pharmacologically inhibited Trem-1 using LR12 peptide.
RESULTS Ldlr�/� mice reconstituted with bone marrow deficient for Trem-1 (Trem-1�/�) showed a strong reduction of
atherosclerotic plaque size in both the aortic sinus and the thoracoabdominal aorta, and were less inflammatory
compared to plaques of Trem-1þ/þ chimeric mice. Genetic invalidation of Trem-1 led to alteration of monocyte
recruitment into atherosclerotic lesions and inhibited toll-like receptor 4 (TLR 4)-initiated proinflammatory macrophage
responses. We identified a critical role for Trem-1 in the upregulation of cluster of differentiation 36 (CD36), thereby
promoting the formation of inflammatory foam cells. Genetic invalidation of Trem-1 in ApoE�/�/Trem-1�/� mice or
pharmacological blockade of Trem-1 in ApoE�/� mice using LR-12 peptide also significantly reduced the development of
atherosclerosis throughout the vascular tree, and lessened plaque inflammation. TREM-1 was expressed in human
atherosclerotic lesions, mainly in lipid-rich areas with significantly higher levels of expression in atheromatous than in
fibrous plaques.
CONCLUSIONS We identified TREM-1 as a major upstream proatherogenic receptor. We propose that TREM-1
activation orchestrates monocyte/macrophage proinflammatory responses and foam cell formation through coordinated
and combined activation of CD36 and TLR4. Blockade of TREM-1 signaling may constitute an attractive novel and double-
hit approach for the treatment of atherosclerosis. (J Am Coll Cardiol 2016;68:2776–93) © 2016 by the American College
of Cardiology Foundation.
m the aINSERM U970, Paris Cardiovascular Research Center, Paris, France; bUniversité Paris Descartes, Sorbonne Paris Cité,
ris, France; cINOTREM SA, Nancy, France; dLaboratory for Experimental Cardiology, University Medical Center, Utrecht, the
therlands; eDepartment of Anatomopathology, Hôpital Européen Georges Pompidou, Assistance Publique-Hopitaux de Paris,
ris, France; fDepartment of Medicine, Division of Cardiovascular Medicine, University of Cambridge, Cambridge, United
gdom; gINSERM Unité mixte de Recherche-S1116, Faculté de Médecine, Université de Lorraine, Medical Intensive Care Unit,
pital Central, Nancy, France; and the hMedical Intensive Care Unit, Hôpital Saint-Antoine, Assistance Publique-Hopitaux de
ris, Université Pierre-et-Marie Curie, Paris, France. This research was supported by Institut National de la Santé et de la
cherche Médicale, Agence Nationale Recherche 2014 Physiopathologie des maladies humaines program, the Fondation pour la
cherche Médicale, the European Research Council, and the British Heart Foundation. Dr. Gibot is co-founder of INOTREM. All
thors have reported that they have no relationships relevant to the contents of this paper to disclose.
nuscript received June 2, 2016; revised manuscript received September 12, 2016, accepted October 4, 2016.

AB BR E V I A T I O N S
AND ACRONYM S
Apo = apolipoprotein
BMDM = bone marrow-derived
cells
CD = cluster of differentiation
IFN = interferon
IL = interleukin
LDL = low-density lipoprotein
LPS = lipopolysaccharide
PBS = phosphate-buffered
saline
SMC = smooth muscle cell
TLR = toll-like receptor
TNF = tumor necrosis factor
TREM = triggering receptors
expressed on myeloid cells
TUNEL = terminal dUTP nick
end labeling
J A C C V O L . 6 8 , N O . 2 5 , 2 0 1 6 Joffre et al.D E C E M B E R 2 7 , 2 0 1 6 : 2 7 7 6 – 9 3 TREM-1 Inhibition Reduces Atherosclerosis
2777
I schemic cardiovascular diseases, largely due toatherosclerosis, are expected to remain themain cause of death globally for the next 15
years, forcing us to revisit their basic mechanismsand consider new strategies for prevention and treat-ment (1–3). There is a large body of human and exper-imental evidence indicating that innate immunecells, particularly monocytes/macrophages, areinvolved in the initiation and progression of athero-sclerosis and its complications, such as plaquerupture and consecutive acute myocardial infarction(AMI) (4,5). Activation of the vascular wall after sub-endothelial retention and modification of apolipopro-tein B (ApoB) containing low-density lipoproteins(LDL) promotes the recruitment of circulating mono-cytes into the intima. Accumulating monocytessignificantly contribute to the pool of macrophagesin lesions (6). Monocytes differentiate into macro-phages and lipid-laden foam cells and promote pla-que development and vulnerability throughcytokine, chemokine, and matrix metalloproteaseproduction and through direct interactions with sur-rounding inflammatory and vascular cells. Macro-phage differentiation, activation, and proliferationare necessary steps for atherosclerosis and are associ-ated with upregulation of pattern recognition recep-tors for innate immunity, including scavengerreceptors, cluster of differentiation 36 (CD36), andtoll-like receptors (TLR) (7–9). Macrophages inter-nalize and are activated by a broad range of moleculesand particles bearing danger-associated molecularpatterns (e.g., oxidized LDL, apoptotic debris) and ul-timately are transformed into proinflammatory foamcells. However, the critical upstream pathways thatlink inflammatory cell activation and foam cell forma-tion and lead to the generation of proinflammatorylipid-laden macrophages are still poorly defined.
SEE PAGE 2794
Direct interactions between scavenger receptorsinvolved in lipid body formation (e.g., CD36), andTLR involved in the induction of inflammatoryresponses (e.g., TLR2, TLR4, and TLR6) have beenidentified (10,11) and proposed to play a role in thisprocess (11). However, the independent in vivoimpact of such pathways on lesion development andthe accumulation of proinflammatory foamy macro-phages remains modest. For example, deletion ofCD36 does not limit lesion development in LDL recep-tor knockout (Ldlr�/�) mice (12) and reduced athero-sclerosis only in the descending aorta of ApoE�/�
mice (13). Moreover, lesion development was notsignificantly altered in Ldlr�/� mice, despite com-bined deficiency of TLR2 and TLR4 in macrophages
(10). Thus, the critical receptor or combina-tion of receptors that play nonredundantroles in orchestrating the development ofproinflammatory foamy macrophages are stillto be defined.
Triggering receptors expressed on myeloidcells (TREM) proteins are a family of cell sur-face receptors, members of the immunoglob-ulin family, discovered 15 years ago byBouchon et al. (14,15). TREM-1 expression isconstitutive on neutrophils and monocytes/macrophages and increases in response tolipopolysaccharide or other microbial prod-ucts (15). Engagement of TREMs, after associ-ation with the adapter protein DAP12, hasbeen shown to stimulate the production ofproinflammatory cytokines and chemokines,such as interleukin (IL)-8 and CCL-2 andCCL-7, as well as rapid neutrophil degranula-tion. Activation of TREM-1 in the presence of
TLR2 or TLR4 ligands amplifies the production ofproinflammatory cytokines (tumor necrosis factor[TNF]-a, IL-1b) while it inhibits the release of anti-inflammatory IL-10 (16). TREM-1 also regulatesmonocyte and neutrophil migration to inflammatorysites (15,17). TREM-1 has been studied mainly duringseptic shock, but it also is critical during asepticinflammation in both acute (pancreatitis) (18) andchronic (rheumatoid arthritis) conditions (19). Arecently designed dodecapeptide named LR12 (LQEE-DAGEYGCM), derived from TREM-1-like transcript 1,strongly inhibits TREM-1 engagement by competingwith its still unknown endogenous ligand. LR12 mod-ulates the innate immune response following bacterialaggression in rodents (20), pigs (21), and primates (22),translating into an attenuation of organ dysfunctionand improved outcome (20,21). We recently reportedthat TREM-1 is involved is post-ischemic myocardialremodeling by orchestrating leukocyte recruitment tothe ischemic heart and subsequent inflammatory re-sponses (23). Additionally, in a multicenter cohort ofpatients with acute coronary syndromes, we showedthat plasma TREM-1 level was an independent pre-dictor of major adverse cardiovascular events (23).Here, we evaluated the effects of TREM-1 in murinemonocytes/macrophages on inflammatory cell acti-vation, trafficking, and foam cell formation to assessTREM-1 as an upstream target in atherosclerosis.
METHODS
HUMAN CAROTID PLAQUES. The processing andexamination of dissected atherosclerotic plaqueshave been previously described (24). All stained

Joffre et al. J A C C V O L . 6 8 , N O . 2 5 , 2 0 1 6
TREM-1 Inhibition Reduces Atherosclerosis D E C E M B E R 2 7 , 2 0 1 6 : 2 7 7 6 – 9 3
2778
samples were examined microscopically and plaquecharacteristics scored semiquantitatively (24). Semi-quantitative analysis of atherosclerotic plaque his-tology is reproducible, both intraobserver andinterobserver (25). Plaque classification was based onthe size of the lipid core and cell population dominance(macrophages or smooth muscle cells [SMC]). Fibrousplaqueswere characterized by a small lipid core (<10%)with a dominance of SMCs, whereas atheromatousplaques had an important lipid core (>40%) dominatedby macrophages. For these human plaques, immuno-staining studies were performed in vascular tissues(normal aorta, atherosclerotic plaques from carotidartery) obtained post-mortem at autopsy. We used aTREM-1 goat polyclonal antibody in formalin-fixedparaffin-embedded tissues after antigen retrieval byheating in citrate buffer and ABC peroxidase techniqueas previously described (26). Polyclonal goat anti-P57antibody was used as a nonrelevant antibody fornegative control procedure. Additionally, we stainedmacrophages (anti-CD68), SMCs (a-actinþ SMCs), CD3þ
T cells, and neutrophils (myeloperoxidase).Total proteins were extracted from fresh human
atherosclerotic carotid plaques after endarterectomy,and TREM-1 protein levels were determined (LuminexxMAP technology, Thermo Fisher Scientific, Inc.,Waltham, Massachusetts). The plaques were obtainedfrom theAthero-Express study, a longitudinal vascularbiobank study that was approved by theMedical EthicsCommittee of the University Medical Center Utrecht,Utrecht, the Netherlands, and for which participantsprovided written informed consent.ANIMALS. Experiments were conducted according tothe guidelines formulated by the European Commu-nity for experimental animal use (L358-86/609EEC)and approved by the Ethical Committee of INSERMand the French Ministry of Agriculture (agreementA75-15-32). Trem-1�/� mice (null for the Trem-1 gene)were generated (GenOway, Lyon, France) (23) andbackcrossed for more than 10 generations into aC57BL/6J background. Ten-week-old male C57BL/6JLdlr�/� mice were subjected to medullar aplasia bylethal total body irradiation (9.5 Gy). The mice wererepopulated with an intravenous injection of bonemarrow cells isolated from femurs and tibias ofsex-matched C57BL/6J Trem-1�/� mice or Trem-1þ/þ
littermates. After 4 weeks of recovery, mice were feda proatherogenic diet containing 15% fat, 1.25%cholesterol, and 0% cholate for 4, 8, or 14 weeks.Eight-week old male ApoE�/� mice were treated dailyby intraperitoneal injection of the Trem-1 inhibitorLR12 (100 mg/day) or the peptide Scramble during4 weeks and were put on either a chow or a high-fatdiet (15% fat, 1.25% cholesterol).
LR12 (LQEEDAGEYGCM) and LR12-scramble (theinactive control peptide) were chemically synthesizedas COOH terminally amidated peptides. The correctpeptides were obtained with >99% yields and werehomogeneous after preparative purification, asconfirmed by mass spectrometry and analyticreversed-phase high-performance liquid chromatog-raphy. These peptides were free of endotoxin. Ani-mals were blindly randomized to receive 5 mg/kg LR12or LR12-scramble peptides intraperitoneally once aday for 28 days. We used the same dose of peptide asin studies of septic shock (21,22) and AMI (23).
EXTENT AND COMPOSITION OF ATHEROSCLEROTIC
LESIONS. Plasma cholesterol was measured using acommercial cholesterol kit. Quantification of lesionsize was performed as described previously (27).Briefly, the basal half of the ventricles and ascendingaorta were perfusion-fixed in situ with 4% para-formaldehyde. Afterward, they were removed,transferred to a phosphate-buffered saline (PBS)-30%sucrose solution, embedded in frozen optimal cuttingtemperature compound and stored at �70� C. Serial10-mm sections of the aortic sinus with valves (80 permouse) were cut on a cryostat as previously described(28). One of every 5 sections was kept for plaque sizequantification after Oil Red O (Sigma-Aldrich, St.Louis, Missouri) staining. Thus, 16 sections, spanningan 800-mm length of the aortic root, were used todetermine mean lesion area for each mouse. Oil RedO-positive lipid contents were quantified by a blindedoperator using HistoLab software (MicrovisionsInstruments, Paris France), which was also used formorphometric studies (29). En face quantification wasused for atherosclerotic plaques along the thoraco-abdominal aorta. The aorta was flushed with PBSthrough the left ventricle and removed from the rootto the iliac bifurcation. Then, the aorta was fixed with10% neutral-buffered formalin. After a thoroughwashing, adventitial tissue was removed, and theaorta opened longitudinally to expose the luminalsurface. Afterward, the aorta was stained with OilRed O for visualizing with the atherosclerotic lesionsquantified, as just noted, by a blinded operator.Collagen was detected using Sirius red stain, andnecrotic core was quantified after Masson’s tri-chrome staining. Macrophage presence was deter-mined using specific antibodies, as previouslydescribed (29). At least 4 sections per mouse wereexamined for each immunostaining, and appropriatenegative controls were used. For immunostaining ofmouse atherosclerotic plaques, we used antibodiesagainst Trem-1 (Bs 4886R), macrophage/monocyteantibody (MOMA)-2 (specifically MAB1852), Ly6G

J A C C V O L . 6 8 , N O . 2 5 , 2 0 1 6 Joffre et al.D E C E M B E R 2 7 , 2 0 1 6 : 2 7 7 6 – 9 3 TREM-1 Inhibition Reduces Atherosclerosis
2779
(1A8), and CD3 (A0452). Terminal dUTP nick end-labeling (TUNEL) staining was performed using his-tochemistry and fluorescent staining. Total proteinswere extracted from human atherosclerotic plaque,and TREM-1 protein level was quantified by Luminex(Thermo Fischer Scientific).
Cells were cultured in RPMI 1640 medium supple-mented with L-alanyl-L-glutamine dipeptide (Gluta-max, Thermo Fisher Scientific), 10% fetal calf serum,0.02 mM b-mercaptoethanol, and antibiotics. Forcytokine measurements, splenocytes were stimulatedwith lipopolysaccharide (LPS) (10 mg/ml) and inter-feron (IFN)-g (100 UI/ml) for 24 or 48 h. IL-10, IL-12,and TNF-a production in the supernatants wasmeasured using specific enzyme-linked immunosor-bent assays.
Primary macrophages were derived from mousebone marrow-derived cells (BMDM). Tibias andfemurs of C57Bl6/J male mice were dissected, andtheir marrow was flushed out. Cells were grown for 7days at 37�C in a solution of RPMI 1640 medium, 20%neonatal calf serum, and 20% macrophage–colony-stimulating factor-rich L929-conditioned medium. Toanalyze oxidized LDL (oxLDL) uptake, BMDMs wereexposed to human oxLDL (25 mg/ml) for 24 and 48 h.Cells were washed, fixed, and stained using Red Oil.Foam cells were quantified blindly on 6 to 8 fields,and the mean was recorded. To analyze macro-phage phenotype, BMDMs were stimulated withLPS (10 mg/ml) and IFN-g (100 UI/ml) for 24 h. IL-10,IL-12, IL-1b, and TNF-a production in the superna-tant was measured using specific enzyme-linkedimmunosorbent assays. To analyze apoptosis sus-ceptibility, macrophages were incubated with TNF-a(10 ng/ml) and cycloheximide (10 mmol/l) for 6 h oretoposide (50 mmol/l) for 12 h, or in a fetal calf serum-free medium. Apoptosis was determined by inde-pendent experiments using Annexin V fluoresceinisothiocyanate apoptosis detection kit with 7-AAD(APC, BD Biosciences, San Jose, California) accord-ing to the manufacturer’s instructions.
Human monocytes were isolated using anti-CD14microbeads from healthy donors. Cells werecultured with macrophage–colony-stimulating factor(50 ng/ml) for 7 days to induce mature macrophages.
Nonclassical monocytes were labeled in vivo byretro-orbital intravenous injection of 1 mm fluorescentmicrosphere diluted to one-quarter in sterile PBS.Chimeric Ldlr�/� mice were euthanized 48 h later,and cell labeling was checked by flow cytometry (30).Beads that reflect monocyte recruitment were quan-tified in 8 aortic sinus sections per mouse. Additionalinformation regarding methods used is provided inthe Online Appendix.
STATISTICAL ANALYSIS. Values are mean � SE ofthe mean. Differences between values were examinedusing the nonparametric Mann-Whitney U test andwere considered significant at a p value of <0.05.
RESULTS
To address the role of myeloid Trem-1 in the devel-opment of atherosclerosis, we performed bonemarrow transplantation experiments using eitherTrem-1þ/þ or Trem-1�/� bone marrow to repopulatelethally irradiated Ldlr�/� mice. In the chimericLdlr�/�/Trem-1þ/þ control group, we confirmed thatTrem-1 was expressed by circulating nonclassicalmonocytes (Figures 1A and 1B) and also within thethoracoabdominal aorta (Figure 1C). Trem-1 expres-sion was almost abolished in circulating monocytesand atherosclerotic aorta of chimeric Ldlr�/�/Trem-1�/� (Figures 1A to 1C). Leukocyte populationswere not different between groups in the spleen,blood, or bone marrow, except for a slight increase ofCd4þ T cells in chimeric Ldlr�/�/Trem-1�/� mice(Online Figure 1). As shown in Figure 1, Trem-1 defi-ciency in the bone marrow was associated with asignificant decrease in lesion development comparedwith controls; 42% decrease after 8 weeks of a fat diet(p < 0.01) (Figures 1D to 1F) and 60% decrease after 14weeks of a fat diet (p < 0.01) (Figures 1G and 1H).Additionally, Trem-1 deletion induced a less inflam-matory plaque phenotype with significant reductionsin both macrophage accumulation (Figures 1I to IK)and necrotic core size (Figures 1L to 1N) but no dif-ferences regarding collagen content (Online Figure 2).We observed no differences in body weight or serumcholesterol levels (Online Figure 2) between the 2groups of mice.
TREM-1 DEFICIENCY EFFECTS. In order to gaininsight into the mechanisms of reduction of macro-phage and necrotic core content in Ldlr�/� micereconstituted with Trem-1�/� bone marrow, we firstevaluated apoptosis. In vitro, apoptosis susceptibilityof Trem-1þ/þ and Trem-1�/� macrophages was com-parable between the 2 genetic backgrounds afterserum deprivation or exposure to etoposide. How-ever, apoptosis was slightly reduced in Trem-1�/�
macrophages after stimulation with TNF-a andcycloheximide stimulation (Online Figure 3). In vivo,we found that Trem-1 deficiency led to a significantreduction in TUNELþ cells in the atherosclerotic pla-ques (Figures 2A to 2C). This observation mightexplain the decrease of necrotic core size but did notreconcile the apparent decrease in macrophages,suggesting that other mechanisms prevail. We thentested the possibility that Trem-1 controls monocyte

FIGURE 1 Hematopoietic Trem-1 Deficiency Reduces Atherosclerosis
Trem-1+/+/LdIr-/-
105
105
104
104
103
103
102
102
0
105
104
103
102
0
Gr-1
Trem-10 1051041031020
Trem-1-/-/LdIr-/-A B0.0075
Trem-1+/+/LdIr-/- Trem-1-/-/LdIr-/-
Trem-1 mRNA
0.0050
0.0025
0.0000
***
C
600
800Plaque size (aortic sinus)
400X1
03 μm
2
200
0Trem-1+/+/LdIr-/- Trem-1-/-/LdIr-/-
**
Trem-1+/+/LdIr-/- Trem-1-/-/LdIr-/-
D E F15
10
0
5
Plaque size (thoracic aortic)
%
Trem-1+/+/LdIr-/- Trem-1-/-/LdIr-/-
**2000
1500
1000
500
0
Plaque size (aortic sinus)
Trem-1+/+/LdIr-/- Trem-1-/-/LdIr-/-
**
X103 μ
m2
G H
300
200
100
0
Macrophage area
Trem-1+/+/LdIr-/- Trem-1-/-/LdIr-/-
X103 μ
m2
*
Trem-1+/+/LdIr-/- Trem-1-/-/LdIr-/-
I J K300
200
100
0
Necrotic core*
Trem-1+/+/LdIr-/- Trem-1-/-/LdIr-/-
X103 μ
m2
Trem-1+/+/LdIr-/- Trem-1-/-/LdIr-/-
L M N
In Ldlr�/� mice, Trem-1 deficiency in myeloid cells reduced development of atherosclerosis and induced a stable plaque phenotype. As seen in flow cytometry in blood leukocytes gated on CD11bþCD115high monocytes,
TREM-1 was expressed by nonclassical Gr1low monocytes in the control (Ldlr�/�/Trem-1þ/þ) group (A) but was absent in Ldlr�/�/Trem-1�/� mice (B). (C) Using quantitative polymerase chain reaction assays of aortas of
chimeric Ldlr�/� mice after 6 weeks of fat diet, Trem-1 mRNA was detected in the control group, but its expression was almost abolished in the Ldlr�/�/Trem-1�/� group. Representative photomicrographs of controls (D)
and Trem-1-deficient mice (E) and quantitative analysis (F) of atherosclerotic lesions of chimeric Ldlr�/� mice after 8 weeks of fat diet are shown. Quantitative analysis of atherosclerotic lesions of chimeric Ldlr�/� mice
after 14 weeks of fat diet in the (G) left aortic sinus and (H) thoracoabdominal aorta are shown. (E) Representative photomicrographs of controls (I) and Trem-1-deficient mice (J) and quantitative analysis (K) of
macrophage accumulation (macrophage plus monocyte antibody [MOMA] staining) are shown in atherosclerotic lesions of chimeric Ldlr�/� mice. Representative photomicrographs of controls (L) and Trem-1-deficient
mice (M) and quantitative analysis (N) of acellular areas (Masson’s trichrome staining) of chimeric Ldlr�/� mice are shown. *p < 0.05; **p < 0.01; ***p < 0.001 Scale bar ¼ 200 mm. CD ¼ cluster of differentiation;
Ldlr�/� ¼ low-density lipoprotein receptor; Trem-1 ¼ triggering receptors expressed on myeloid cells.
Joffreet
al.JACC
VOL.68,NO.25,2016
TREM
-1Inhibition
Reduces
Atherosclerosis
DECEMBER
27,2016
:2776–93
2780

FIGURE 2 Hematopoietic Trem-1 Deficiency Limits Pro-Inflammatory Responses
Trem-1+/+/LdIr-/- Trem-1-/-/LdIr-/-
0.04
0.03
0.02
0.01
0.00
%
LdIr-/-/Trem-1+/+ LdIr-/-/Trem-1-/-
*TUNEL area
A B C
Trem-1+/+/LdIr-/- Trem-1-/-/LdIr-/-
20
15
10
5
0
nb B
eads
/ Se
ctio
n
Trem-1+/+/LdIr-/- Trem-1-/-/LdIr-/-
*Monocyte recruitment
D E F
80
60
40
20
0
**
TNF-a/IL-10 IL-12p70/IL-10
Lps/ifn-γ stimulated splenocytes
G
0.15
0.10
0.05
0.00
***
II-12 mRNA0.05
0.04
0.03
0.02
0.01
0.00
***
IL-1b mRNA0.25
0.20
0.15
0.10
0.05
0.00
***
II-6 mRNATrem-1+/+/LdIr-/- Trem-1-/-/LdIr-/-
0.015
0.010
0.005
0.000
*
II-10 mRNA
H I J K
**
* **
Trem-1 +/+
Trem-1 -/-
Cytokines by macrophagesL
22500
12500
2500
2000
1000
0
pg/m
l
TNF-a IL-12p70 IL-10IL-1b
0.005
0.002
0.004
0.003
0.001
0.000
NS
Arginase 1 mRNA
0.0008
0.0004
0.0006
0.0002
0.0000
NSiNos mRNA
M NTrem-1+/+/LdIr-/- Trem-1-/-/LdIr-/-
TREM-1 deficiency reduced monocyte migration into atherosclerotic lesions and limited macrophage inflammatory response. Representative photomicrographs of controls (A) and Trem-1-deficient mice (B) and
quantitative analysis (C) of TUNELþ apoptotic area (red/brown, arrows) within atherosclerotic lesions of chimeric Ldlr�/� mice after 8 weeks of fat diet. Representative photomicrographs of controls (D) and Trem-1-
deficient mice (E) and quantitative analysis (F) of beads within atherosclerotic lesions of chimeric Ldlr�/� mice after 4 weeks of fat diet. (G) Cytokine production by lipopolysaccharide (LPS)/interferon (IFN)-g-stimulated
splenocytes isolated from chimeric Ldlr�/� mice. Quantification of interleukin (IL) (H) IL-10, (I) IL-12p70, (J) IL-1b, and (K) IL-6 mRNA expression in the spleens of chimeric Ldlr�/� mice. (L) Cytokine production by
LPS/IFNg-stimulated bone marrow-derived macrophages (BMDM) isolated from Ldlr�/� chimeric mice, 6 weeks after transplantation. Quantification of (M) iNOS and (N) Arg1mRNA expression in abdominal aortas from
chimeric Ldlr�/� mice. *p < 0.05; **p < 0.01; ***p < 0.001. Scale bar ¼ 200 mm. Data are median and percentiles (5th to 95th). IFN ¼ interferon; LPS ¼ lipopolysaccharide; TUNEL ¼ terminal dUTP nick end-labeling;
other abbreviations as in Figure 1.
JACC
VOL.68,NO.25,2016
Joffreet
al.DECEMBER
27,2016:2
776–93
TREM
-1Inhibition
Reduces
Atherosclerosis
2781

Joffre et al. J A C C V O L . 6 8 , N O . 2 5 , 2 0 1 6
TREM-1 Inhibition Reduces Atherosclerosis D E C E M B E R 2 7 , 2 0 1 6 : 2 7 7 6 – 9 3
2782
infiltration into the lesions. We quantified monocyterecruitment to atherosclerotic plaque by using avalidated bead-labeling technique (30). After 4 weeksof fat diet, chimeric Ldlr�/� mice were intravenouslyinjected with fluorescent beads and euthanized 48 hlater. We confirmed the reduction of atherosclerosisdevelopment at early stages (data not shown) inLdlr�/�/Trem-1�/� mice and observed a significantreduction of the recruitment of Trem-1�/� bead-positive monocytes within the lesions in comparisonwith Trem-1þ/þ monocytes (Figures 2D to 2F). In par-allel, we evaluated the peritoneal recruitment ofnonclassical monocytes following a septic and anonseptic injury. After intraperitoneal injection ofLPS or thioglycollate, nonclassical monocyte recruit-ment was significantly reduced within the peritonealcavity in Trem-1 deficient animals (Online Figure 4).These findings showed that Trem-1�/� circulatingmonocytes have reduced ability to migrate into in-flammatory sites and colonize plaques, corroboratinga previously identified role of Trem-1 in promotingneutrophil migration during acute inflammation (31).
Because Trem-1 is reported to amplify cytokineproduction, particularly in response to TLR ago-nists (32,33), we next investigated the immunoin-flammatory response in chimeric Ldlr�/� mice. LPSand IFN-g-stimulated splenocytes showed a devia-tion toward an anti-inflammatory profile in Ldlr�/�/Trem-1�/� chimeric mice with a reduction of TNF-a/IL-10 and IL-12p70/IL-10 ratios (Figure 2G). Theexpression of IL-10, IL-12p70, Il1b, and IL-6messenger ribonucleic acids (mRNA) was also signif-icantly reduced in the spleens of Ldlr�/�/Trem-1�/�
mice (Figures 2H to 2K). Furthermore, Trem-1�/�
BMDMs were less prone to polarize toward aproinflammatory phenotype after LPS/IFN-g, as theyproduced less TNF-a, IL-12p70, and IL-1b and moreIL-10 compared to Trem-1þ/þ BMDMs (Figure 2L).However, expressions of Nos2 and Arg1 in theabdominal aorta (as markers of M1 and M2 macro-phages, respectively) were not different betweenchimeric Ldlr�/�/Trem-1�/� and Ldlr�/�/Trem-1þ/þ
mice after 6 weeks of fat diet (Figures 2M and 2N).Finally, we investigated macrophage metabolism,including mitochondrial respiration and glycolysis;those functions were very similar between Trem-1þ/þ
and Trem-1�/� macrophages (Online Figure 5).We speculated that the marked reduction of Oil Red
Oþ lesions in Ldlr�/�/Trem-1�/� mice could be due atleast in part to reduced foam cell formation. To explorethis hypothesis, we performed in vitro experiments toexamine the uptake of oxLDL by BMDMs and theirability to accumulate intracellular lipids. Interestingly,foam cell formation was significantly reduced in
Trem-1�/� cultured macrophages compared to that inTrem-1þ/þmacrophages after 24 and 48 h of incubationwith oxLDL (Figures 3A to 3F). In contrast, phagocytosisof apoptotic cells or zymosan beads was not alteredby Trem-1 deficiency (Online Figure 6), suggesting aselective role in lipid uptake. We found no differencesamong Sr-b1, Abca1, or Abcg1 mRNA expression andonly a slight and delayed reduction of Sr-a mRNAexpression in Trem-1�/� cells (Figures 3G to 3I) (data notshown). However, we observed a marked reduction ofCd36 mRNA expression in Trem-1�/� cultured macro-phages at baseline and after stimulation with oxLDL(Figures 3G to 3I). A reduction of Cd36 protein wasfurther confirmed by flow cytometry (Figures 3J and 3K).WealsoquantifiedCd36proteinexpression in theplaquesof chimeric Ldlr�/� mice and observed a profounddecrease of Cd36 staining in the absence of bonemarrow-derived Trem-1 (Figures 3L to 3N). To begin to address themechanisms of Trem-1-dependent upregulation of Cd36,we investigated signaling pathways downstream ofTrem-1 that may control Cd36 expression. BMDMsco-incubated with oxLDL were treated with differentpharmacological inhibitors, and Cd36 expression wasquantified 16 h later by using flow cytometry. Pharma-cological blockade of Erk1/2 (PD98059 [Sigma-Aldrich]) orNF-kB (PDTC [ammonium pyrrolidinedithiocarbamate,Abcam, Cambridge, Massachusetts]) had no effect onCd36 expression (data not shown). However, incubationwith wortmaninn significantly reduced oxLDL-inducedCd36 expression on Trem-1þ/þ BMDMs down to levelsseen in Trem-1�/� BMDMs (Figure 3O), suggesting a rolefor phosphatidylinositol 3 kinase (PI3K) activation inTREM-1-dependent upregulation of Cd36 followingexposure to oxLDL.GENETIC INVALIDATION OF TREM-1. We nextaddressed the role of TREM-1 in ApoE�/� mice. Wefound an elevated expression of Trem-1 mRNAexpression in the aorta of ApoE�/�mice on fat diet and,to a lesser extent, in ApoE�/� mice on chow dietcompared to that in healthy C57Bl6 aortas, whichshowed no expression of Trem-1 (Online Figure 7A).Trem-1 expression levels within the aorta significantlycorrelated with plaque size (r ¼ 0.71; p ¼ 0.0003) andmacrophage infiltration (r ¼ 0.87; p < 0.0001) in theaortic sinus (Online Figure 7B). Immunostainingconfirmed that TREM-1 was expressed in atheroscle-rotic lesions of ApoE�/� mice and co-localized mainlywith MOMAþ macrophages (Figure 4A). We alsodetected a few TREM-1þ Ly6Gþ neutrophils in athero-sclerotic lesions at that stage of lesion development(Figure 4B). To investigate the role of TREM-1 in thismodel, we generated ApoE�/�/Trem-1�/� mice. Trem-1deficiency in myeloid population was confirmed byflow cytometry (Figures 4C and 4D) and by
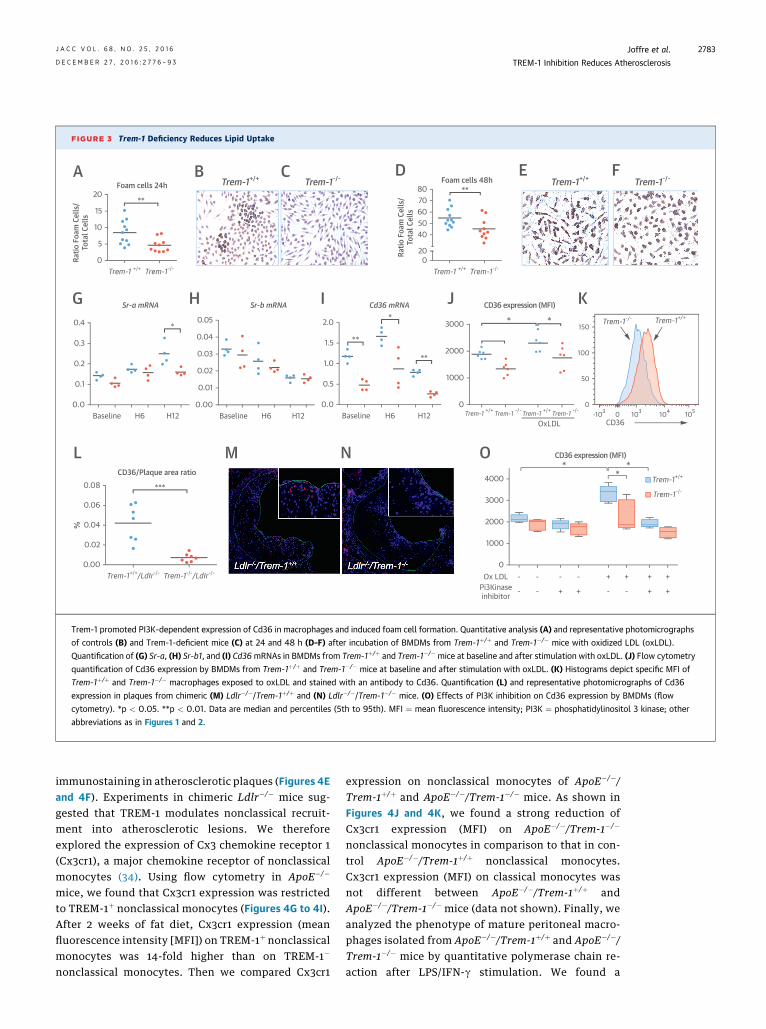
FIGURE 3 Trem-1 Deficiency Reduces Lipid Uptake
Trem-1+/+ Trem-1-/- Trem-1+/+ Trem-1-/-
20
15
10
5
0Ratio
Foa
m C
ells
/To
tal C
ells
Trem-1 +/+ Trem-1-/-
**
Foam cells 24h
0.4
0.3
0.2
0.1
0.0Baseline H6 H12
**
**
**
Sr-a mRNA
0.05
0.04
0.03
0.02
0.01
0.00Baseline H6 H12
Sr-b mRNA
2.0
1.5
1.0
0.5
0.0Baseline H6 H12
Cd36 mRNA
* *3000
2000
1000
0
OxLDL CD36
0
50
-103 103 104 1050
100
150
CD36 expression (MFI)
A B C8070605040
200
Ratio
Foa
m C
ells
/To
tal C
ells
Trem-1 +/+
Trem-1+/+
Trem-1-/-
Trem-1-/-
Trem-1+/+
Trem-1-/-
Trem-1 +/+ Trem-1 -/- Trem-1 +/+ Trem-1 -/-
**Foam cells 48h
D E F
J K
OL M N
G H I
4000
3000
2000
1000
0Ox LDL -
-
-
-
-
+
-
+
+
-
+
-
+
+
+
+Pi3Kinaseinhibitor
* **
CD36 expression (MFI)
0.08
0.06
0.04
0.02
0.00
%
Trem-1+/+/LdIr-/- Trem-1-/-/LdIr-/-
***
CD36/Plaque area ratio
Trem-1 promoted PI3K-dependent expression of Cd36 in macrophages and induced foam cell formation. Quantitative analysis (A) and representative photomicrographs
of controls (B) and Trem-1-deficient mice (C) at 24 and 48 h (D–F) after incubation of BMDMs from Trem-1þ/þ and Trem-1�/� mice with oxidized LDL (oxLDL).
Quantification of (G) Sr-a, (H) Sr-b1, and (I) Cd36mRNAs in BMDMs from Trem-1þ/þ and Trem-1�/� mice at baseline and after stimulation with oxLDL. (J) Flow cytometry
quantification of Cd36 expression by BMDMs from Trem-1þ/þ and Trem-1�/� mice at baseline and after stimulation with oxLDL. (K) Histograms depict specific MFI of
Trem-1þ/þ and Trem-1�/� macrophages exposed to oxLDL and stained with an antibody to Cd36. Quantification (L) and representative photomicrographs of Cd36
expression in plaques from chimeric (M) Ldlr�/�/Trem-1þ/þ and (N) Ldlr�/�/Trem-1�/� mice. (O) Effects of PI3K inhibition on Cd36 expression by BMDMs (flow
cytometry). *p < 0.05. **p < 0.01. Data are median and percentiles (5th to 95th). MFI ¼ mean fluorescence intensity; PI3K ¼ phosphatidylinositol 3 kinase; other
abbreviations as in Figures 1 and 2.
J A C C V O L . 6 8 , N O . 2 5 , 2 0 1 6 Joffre et al.D E C E M B E R 2 7 , 2 0 1 6 : 2 7 7 6 – 9 3 TREM-1 Inhibition Reduces Atherosclerosis
2783
immunostaining in atherosclerotic plaques (Figures 4Eand 4F). Experiments in chimeric Ldlr�/� mice sug-gested that TREM-1 modulates nonclassical recruit-ment into atherosclerotic lesions. We thereforeexplored the expression of Cx3 chemokine receptor 1(Cx3cr1), a major chemokine receptor of nonclassicalmonocytes (34). Using flow cytometry in ApoE�/�
mice, we found that Cx3cr1 expression was restrictedto TREM-1þ nonclassical monocytes (Figures 4G to 4I).After 2 weeks of fat diet, Cx3cr1 expression (meanfluorescence intensity [MFI]) on TREM-1þ nonclassicalmonocytes was 14-fold higher than on TREM-1�
nonclassical monocytes. Then we compared Cx3cr1
expression on nonclassical monocytes of ApoE�/�/Trem-1þ/þ and ApoE�/�/Trem-1�/� mice. As shown inFigures 4J and 4K, we found a strong reduction ofCx3cr1 expression (MFI) on ApoE�/�/Trem-1�/�
nonclassical monocytes in comparison to that in con-trol ApoE�/�/Trem-1þ/þ nonclassical monocytes.Cx3cr1 expression (MFI) on classical monocytes wasnot different between ApoE�/�/Trem-1þ/þ andApoE�/�/Trem-1�/� mice (data not shown). Finally, weanalyzed the phenotype of mature peritoneal macro-phages isolated from ApoE�/�/Trem-1þ/þ and ApoE�/�/Trem-1�/� mice by quantitative polymerase chain re-action after LPS/IFN-g stimulation. We found a
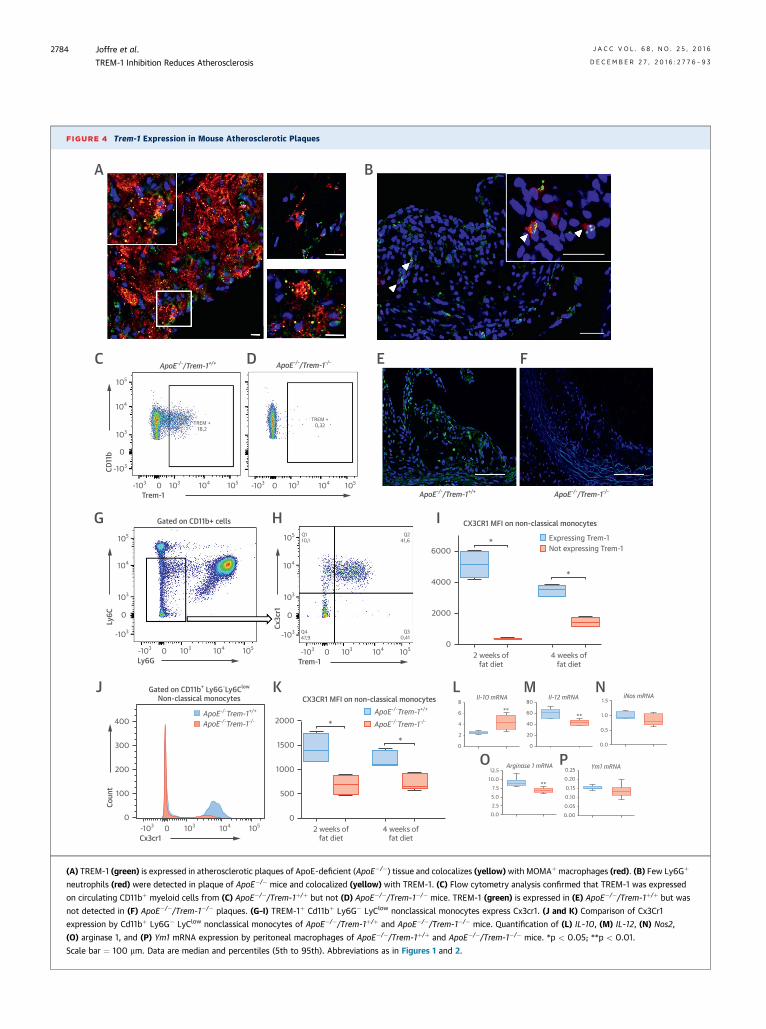
FIGURE 4 Trem-1 Expression in Mouse Atherosclerotic Plaques
A B
ApoE-/-/Trem-1+/+ ApoE-/-/Trem-1-/-
105
104
103
0
-103
-103 0 103 104 105 -103 0 103 104 105
Trem-1
CD11
b
TREM +18,2
TREM +0,32
C D
ApoE-/-/Trem-1+/+ ApoE-/-/Trem-1-/-
E F
6000
4000
2000
02 weeks of
fat diet4 weeks of
fat diet
*
*
Expressing Trem-1Not expressing Trem-1
CX3CR1 MFI on non-classical monocytesGated on CD11b+ cells
105
104
103
0
-103
Ly6C
105
104
103
0
-103
Cx3c
r1
-103 0 103 104 105
Ly6G-103 0 103 104 105
Trem-1
Q110,1
Q241,6
Q447,9
Q30,41
G H I
2000
1000
1500
500
02 weeks of
fat diet4 weeks of
fat diet
*
CX3CR1 MFI on non-classical monocytes
*
ApoE-/-Trem-1+/+
ApoE-/-Trem-1-/-ApoE-/-Trem-1+/+
ApoE-/-Trem-1-/-400
300
200
100
0
Coun
t
-103 0 103 104 105
Cx3cr1
Gated on CD11b+ Ly6G-Ly6Clow
Non-classical monocytesJ K
8
6
4
2
0
**
II-10 mRNA80
60
40
20
0
**
II-12 mRNA 1.5
1.0
0.5
0.0
iNos mRNA
12.5
10.0
7.5
2.5
5.0
0.0
**
Arginase 1 mRNA0.25
0.20
0.15
0.05
0.10
0.00
Ym1 mRNA
L M
O P
N
(A) TREM-1 (green) is expressed in atherosclerotic plaques of ApoE-deficient (ApoE�/�) tissue and colocalizes (yellow) with MOMAþ macrophages (red). (B) Few Ly6Gþ
neutrophils (red) were detected in plaque of ApoE�/� mice and colocalized (yellow) with TREM-1. (C) Flow cytometry analysis confirmed that TREM-1 was expressed
on circulating CD11bþ myeloid cells from (C) ApoE�/�/Trem-1þ/þ but not (D) ApoE�/�/Trem-1�/� mice. TREM-1 (green) is expressed in (E) ApoE�/�/Trem-1þ/þ but was
not detected in (F) ApoE�/�/Trem-1�/� plaques. (G–I) TREM-1þ Cd11bþ Ly6G� LyClow nonclassical monocytes express Cx3cr1. (J and K) Comparison of Cx3Cr1
expression by Cd11bþ Ly6G� LyClow nonclassical monocytes of ApoE�/�/Trem-1þ/þ and ApoE�/�/Trem-1�/� mice. Quantification of (L) IL-10, (M) IL-12, (N) Nos2,
(O) arginase 1, and (P) Ym1 mRNA expression by peritoneal macrophages of ApoE�/�/Trem-1þ/þ and ApoE�/�/Trem-1�/� mice. *p < 0.05; **p < 0.01.
Scale bar ¼ 100 mm. Data are median and percentiles (5th to 95th). Abbreviations as in Figures 1 and 2.
Joffre et al. J A C C V O L . 6 8 , N O . 2 5 , 2 0 1 6
TREM-1 Inhibition Reduces Atherosclerosis D E C E M B E R 2 7 , 2 0 1 6 : 2 7 7 6 – 9 3
2784

J A C C V O L . 6 8 , N O . 2 5 , 2 0 1 6 Joffre et al.D E C E M B E R 2 7 , 2 0 1 6 : 2 7 7 6 – 9 3 TREM-1 Inhibition Reduces Atherosclerosis
2785
significant increase of IL-10 mRNA expression and asignificant decrease of IL-12p70 and Arg1 by ApoE�/�/Trem-1�/� macrophages (Figures 4L to 4P).
There were no significant differences in plasmacholesterol levels between ApoE�/�/Trem-1þ/þ andApoE�/�/Trem-1�/� groups (Figure 5A). At 10 weeks ofage, animals were put on a fat diet for 6 weeks toaccelerate plaque formation. ApoE�/�/Trem-1�/� miceshowed a significant reduction of atheroscleroticlesion formation in the aortic sinus (�41%; p < 0.05)(Figures 5B to 5D) and along the thoracoabdominalaorta (�56%; p ¼ 0.005) (Figures 5E to 5G). Addition-ally, Trem-1 deletion in ApoE�/� background induceda switch toward a less inflammatory plaque pheno-type with significant reductions in macrophageaccumulation (Figures 5H to 5K), necrotic core size(Figures 5L to 5O), and TUNELþ area (only absolutearea) (Figures 5P to 5S). We did not observe any dif-ferences in T-cell accumulation (Online Figure 8A)and plaque collagen content (Online Figure 8B)between the 2 groups of mice.
PHARMACOLOGIC BLOCKADE OF TREM-1. To eval-uate a new therapeutic approach in atherosclerosis,we used a dodecapeptide, LR12, a validated inhibitorof TREM-1 activation (21,23). LR12 reduced oxLDLuptake by BMDMs (Figures 6A to 6C) and down-regulated Cd36 expression (Figure 6D). Both ofthese results were confirmed in human monocyte-derived macrophages (Online Figure 9). UsingApoE�/� peritoneal macrophages, we confirmed thatLR12 treatment reduced Cd36 mRNA (Figure 6E) andinduced a deviation of the immune response towardan anti-inflammatory profile with an increase of IL-10 and a reduction of IL-12p70 and TNF-a produc-tion following LPS/IFN-g stimulation (Figure 6F).When we used human monocytes, LR12 also down-regulated IL-12, TNF-a, and CD36 mRNA expressionlevels (Online Figure 10). Next, we investigated theconsequences of TREM-1 pharmacological inhibitionon monocyte trafficking. ApoE�/� mice fed a fat dietand treated with daily intraperitoneal injections ofScramble or LR12 peptide for 2 weeks were intrave-nously injected with fluorescent beads 48 h beforeeuthanasia. We observed a significant reductionof the recruitment of bead-positive monocyteswithin the lesions of LR12-treated ApoE�/� mice(Figures 6G to 6I).
To evaluate the effects of TREM-1 inhibition onatherosclerosis, we treated ApoE�/� male mice for 4weeks with daily intraperitoneal injections of LR12 ora Scramble control peptide. LR12 treatment had noeffect on cholesterolemia (Online Figures 11A and 11B)but significantly reduced atherosclerosis growth
on a chow diet (56,600 � 22,300 mm2 vs. 98,900 �30,900 mm2; p < 0.05) (Figures 6J to 6L) and on ahigh-fat diet, both in the aortic sinus (103,300 �21,100 mm2 vs. 148,700 � 15,200 mm2; p < 0.05)(Figures 6M to 6O) and the thoracoabdominal aorta (6.0� 3.0% vs. 2.5 � 0.9%; p < 0.05) (Figures 6P to 6R).TREM-1 pharmacological inhibition also reducedmacrophage accumulation (Figures 6S to 6U) andTUNELþ cells/debris (Online Figure 11C) in the lesions,but had no effect on T cell infiltration (OnlineFigure 11D) or collagen content (Online Figure 11E). Toinvestigate the role of neutrophils in the atheropro-tection induced by TREM-1 blockage, neutrophil-depleted ApoE�/� mice were treated with daily intra-peritoneal injections of scramble or LR12 peptide for 4weeks. Administration of anti-Ly6G-depleting anti-body every 3 days led to 70% depletion of circulatingneutrophils. As shown in Online Figure 12, the athero-protection induced by pharmacological inhibition ofTREM-1was preserved in neutrophil-depleted animals.
TREM-1 EXPRESSED IN ATHEROSCLEROTIC HUMAN
LESIONS. To evaluate the clinical relevance of ourresults, we examined TREM-1 expression in humanatherosclerotic plaques from carotid arteries. TREM-1was not detected in normal aorta (Figures 7A and 7B).However, TREM-1 was expressed in fatty streak le-sions (Figures 7C and 7D) and in advanced athero-sclerotic plaques (Figures 7E and 7F) in areassurrounding the necrotic core (Figure 7G). Immuno-histochemistry showed TREM-1 staining localized tothe membrane of giant lipid-laden foam cells(Figure 7H). Fluorescent staining confirmed thatTREM-1 expression was confined mostly to CD68þ
intimal macrophages (Figure 7I) and a few neutrophils(Online Figure 13A) but was not detected in a-actinþ
SMCs (Figure 7J) or CD3þ T cells (Online Figure 13B).We quantified TREM-1 protein levels in human pla-que extracts obtained after endarterectomy (OnlineFigure 14) and found TREM-1 expression was signifi-cantly higher in atheromatous lesions than in fibrouslesions (p ¼ 0.002) (Figure 7K).
DISCUSSION
Using 3 complementary approaches, we found thatTREM-1 deficiency/inhibition significantly reducedatherosclerosis growth in mice and induced a lessinflammatory plaque phenotype characterized byreduced macrophage infiltration and necrotic coresize. It is interesting to note that TREM-1 deletion orblockade was associated with significant and pro-found (up to 60%) reduction of the development ofboth early and advanced atherosclerosis and theprotective effect was observed throughout the aorta

FIGURE 5 Genetic Deletion of Trem-1 on ApoE-/- Background Reduces Atherosclerosis
15
10
5
0ApoE-/-/Trem-1+/+ ApoE-/-/Trem-1-/-
400
200
300
100
0ApoE-/-/Trem-1+/+ ApoE-/-/Trem-1-/-
*Cholesterolemia (g/L) Plaque size (aortic sinus, 103μm2)
ApoE-/-/Trem-1+/+ ApoE-/-/Trem-1-/-
20
25
10
15
5
0ApoE-/-/Trem-1+/+ ApoE-/-/Trem-1-/-
Plaque size (thoracic aorta %)
**
80
100
40
60
20
0
H I J K
L M N O
P Q R S
A
E F
G
BC D
ApoE-/-/Trem-1+/+ ApoE-/-/Trem-1-/-
Macrophage area (103μm2)
** 80
40
60
20
0ApoE-/-/Trem-1+/+ ApoE-/-/Trem-1-/-
Macrophage (% plaque)
**
30
20
10
0ApoE-/-/Trem-1+/+ ApoE-/-/Trem-1-/-
Necrotic core (% plaque)
**80
40
60
20
0ApoE-/-/Trem-1+/+ ApoE-/-/Trem-1-/-
Necrotic core area (103μm2)
**
0.2
0.1
0.0ApoE-/-Trem-1+/+ ApoE-/-Trem-1-/-
TUNEL+ area (% plaque)
ns300
200
100
0ApoE-/-Trem-1+/+ ApoE-/-Trem-1-/-
TUNEL+ area (μm2)*
ApoE-/-/Trem-1+/+ ApoE-/-/Trem-1-/-
ApoE-/-/Trem-1+/+ ApoE-/-/Trem-1-/-
ApoE-/-/Trem-1+/+ ApoE-/-/Trem-1-/-
After 6 weeks of a fat diet, various parameters differed in mouse groups. (A) Plasma cholesterol levels; atherosclerotic lesions in (B to D) the aortic sinus and (E to G)
in the thoracoabdominal aorta. Variations are seen in (H to K) macrophage accumulation, (L to O) acellular area (after Masson’s trichrome), and (P to S) TUNELþ
in ApoE�/�/Trem-1þ/þ and ApoE�/�/Trem-1�/� mice. *p < 0.05; **p < 0.01. Scale bar ¼ 200 mm (C and D) and 100 mm (J, K, N, O, R, S). Abbreviations as in
Figures 1 and 2.
Joffre et al. J A C C V O L . 6 8 , N O . 2 5 , 2 0 1 6
TREM-1 Inhibition Reduces Atherosclerosis D E C E M B E R 2 7 , 2 0 1 6 : 2 7 7 6 – 9 3
2786
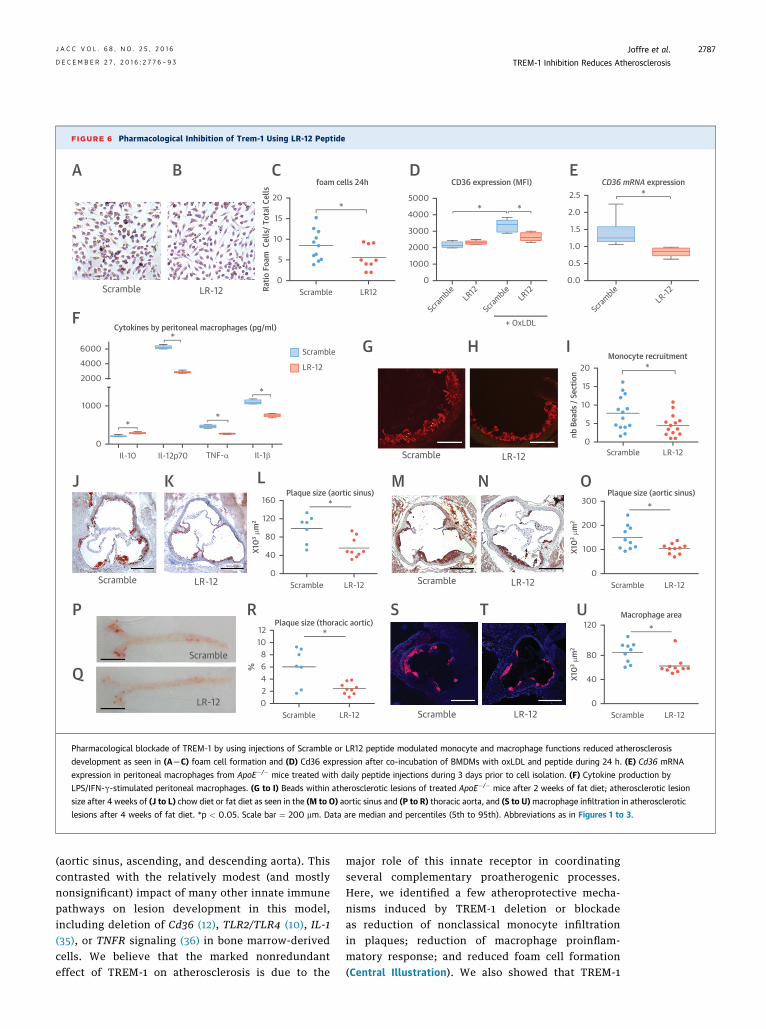
FIGURE 6 Pharmacological Inhibition of Trem-1 Using LR-12 Peptide
20
10
15*
Ratio
Foa
m C
ells
/ Tot
al C
ells
foam cells 24h
5
0Scramble LR12
+ OxLDL
2.5
1.0
2.0
1.5
*CD36 mRNA expression
0.5
0.0
5000
3000
4000 * *
CD36 expression (MFI)
2000
1000
0
Scramble
LR12LR-12
Scramble
Scramble
LR12
20
10
15
*
nb B
eads
/ Se
ctio
n
Monocyte recruitment
5
0ScrambleScramble LR-12LR-12
Scramble LR-12Scramble LR-12
Scramble LR-12
Scramble LR-12
300
100
200
*
X103 μ
m2
Plaque size (aortic sinus)
0Scramble LR-12
160
40
120
80
*
X103 μ
m2
Plaque size (aortic sinus)
0Scramble LR-12
12
2
108
46
*
%
Plaque size (thoracic aortic)
0Scramble LR-12
120
40
80
*
X103 μ
m2
Macrophage area
0Scramble LR-12
6000
2000
4000
Cytokines by peritoneal macrophages (pg/ml)
1000
0Il-10 Il-12p70 TNF-α
LR-12
Scramble
A B C
FG H I
M N OJ K L
P
Q
R S T U
D E
Il-1β
*
*
*
*
Scramble
LR-12
Pharmacological blockade of TREM-1 by using injections of Scramble or LR12 peptide modulated monocyte and macrophage functions reduced atherosclerosis
development as seen in (ALC) foam cell formation and (D) Cd36 expression after co-incubation of BMDMs with oxLDL and peptide during 24 h. (E) Cd36 mRNA
expression in peritoneal macrophages from ApoE�/� mice treated with daily peptide injections during 3 days prior to cell isolation. (F) Cytokine production by
LPS/IFN-g-stimulated peritoneal macrophages. (G to I) Beads within atherosclerotic lesions of treated ApoE�/� mice after 2 weeks of fat diet; atherosclerotic lesion
size after 4 weeks of (J to L) chow diet or fat diet as seen in the (M to O) aortic sinus and (P to R) thoracic aorta, and (S to U)macrophage infiltration in atherosclerotic
lesions after 4 weeks of fat diet. *p < 0.05. Scale bar ¼ 200 mm. Data are median and percentiles (5th to 95th). Abbreviations as in Figures 1 to 3.
J A C C V O L . 6 8 , N O . 2 5 , 2 0 1 6 Joffre et al.D E C E M B E R 2 7 , 2 0 1 6 : 2 7 7 6 – 9 3 TREM-1 Inhibition Reduces Atherosclerosis
2787
(aortic sinus, ascending, and descending aorta). Thiscontrasted with the relatively modest (and mostlynonsignificant) impact of many other innate immunepathways on lesion development in this model,including deletion of Cd36 (12), TLR2/TLR4 (10), IL-1(35), or TNFR signaling (36) in bone marrow-derivedcells. We believe that the marked nonredundanteffect of TREM-1 on atherosclerosis is due to the
major role of this innate receptor in coordinatingseveral complementary proatherogenic processes.Here, we identified a few atheroprotective mecha-nisms induced by TREM-1 deletion or blockadeas reduction of nonclassical monocyte infiltrationin plaques; reduction of macrophage proinflam-matory response; and reduced foam cell formation(Central Illustration). We also showed that TREM-1

FIGURE 7 TREM-1 Expression in Human Atherosclerotic Plaques
4000
3000
2000
1000
0Fibrous Atheromatous
TREM-1 (pg/ml)P=0.002
K
TREM-1 is not expressed in normal aorta (A and B) but is expressed in fatty streak lesions of the aorta (C and D) and in advanced carotid artery
plaques (E and F),mainly around the necrotic core (G). TREM-1 was strongly expressed by cells that engulf lipids and cholesterol crystals and
on the membrane of giant lipid-laden foam cells (H). Fluorescent staining confirmed the fact that TREM-1 was expressed by CD68þ mac-
rophages (I) but not by a-actinþ smooth muscle cells (J). TREM-1 expression was significantly higher in atheromatous lesions than in fibrous
plaques (K). Magnification ¼ �20 (A to F, I, J), �2.5 (G), �40 (H). Lum ¼ lumen; other abbreviations as in Figures 1 and 2.
Joffre et al. J A C C V O L . 6 8 , N O . 2 5 , 2 0 1 6
TREM-1 Inhibition Reduces Atherosclerosis D E C E M B E R 2 7 , 2 0 1 6 : 2 7 7 6 – 9 3
2788
was expressed in advanced human atheroscleroticlesions, mostly close to cholesterol-rich and necrotic-rich areas, and its expression was significantly higherin atheromatous lesions.
TREM-1 is highly conserved during evolution (37)and expressed mainly by bone marrow-derived
leukocytes (15). In our study, TREM-1 was highlyexpressed in circulating neutrophils and Gr1lo but notin Gr1hi monocytes. Deletion of TREM-1 in bonemarrow-derived cells did not significantly alterleukocyte number in blood but limited Gr1lo mono-cyte recruitment into developing atherosclerotic
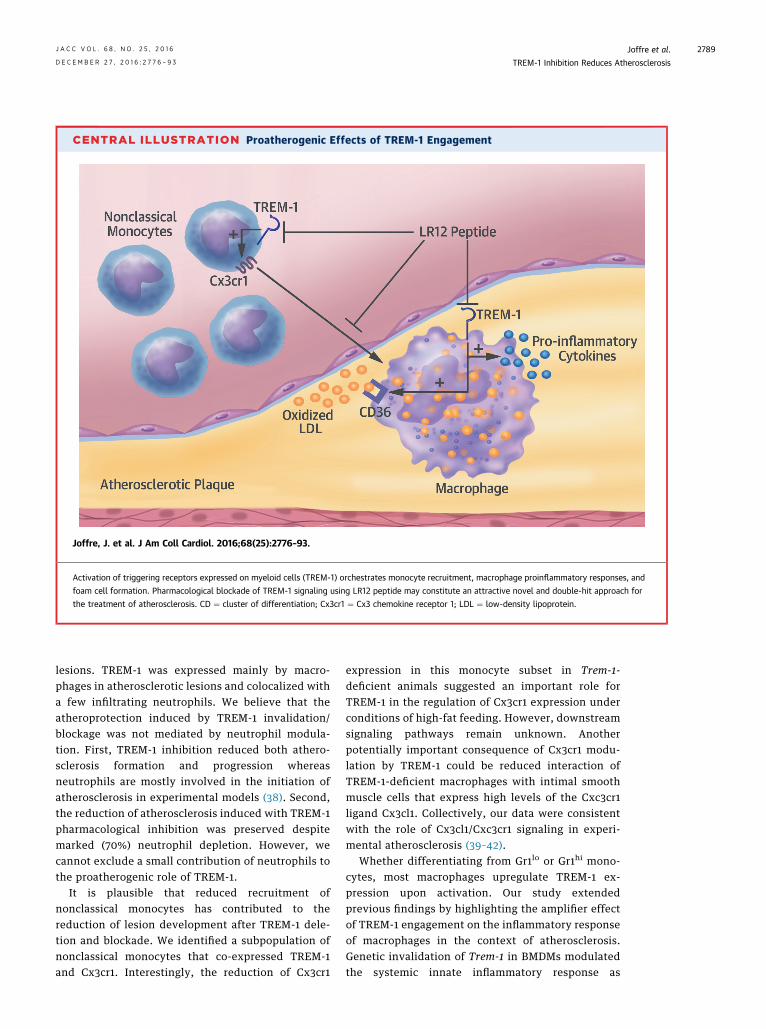
CENTRAL ILLUSTRATION Proatherogenic Effects of TREM-1 Engagement
Joffre, J. et al. J Am Coll Cardiol. 2016;68(25):2776–93.
Activation of triggering receptors expressed on myeloid cells (TREM-1) orchestrates monocyte recruitment, macrophage proinflammatory responses, and
foam cell formation. Pharmacological blockade of TREM-1 signaling using LR12 peptide may constitute an attractive novel and double-hit approach for
the treatment of atherosclerosis. CD ¼ cluster of differentiation; Cx3cr1 ¼ Cx3 chemokine receptor 1; LDL ¼ low-density lipoprotein.
J A C C V O L . 6 8 , N O . 2 5 , 2 0 1 6 Joffre et al.D E C E M B E R 2 7 , 2 0 1 6 : 2 7 7 6 – 9 3 TREM-1 Inhibition Reduces Atherosclerosis
2789
lesions. TREM-1 was expressed mainly by macro-phages in atherosclerotic lesions and colocalized witha few infiltrating neutrophils. We believe that theatheroprotection induced by TREM-1 invalidation/blockage was not mediated by neutrophil modula-tion. First, TREM-1 inhibition reduced both athero-sclerosis formation and progression whereasneutrophils are mostly involved in the initiation ofatherosclerosis in experimental models (38). Second,the reduction of atherosclerosis induced with TREM-1pharmacological inhibition was preserved despitemarked (70%) neutrophil depletion. However, wecannot exclude a small contribution of neutrophils tothe proatherogenic role of TREM-1.
It is plausible that reduced recruitment ofnonclassical monocytes has contributed to thereduction of lesion development after TREM-1 dele-tion and blockade. We identified a subpopulation ofnonclassical monocytes that co-expressed TREM-1and Cx3cr1. Interestingly, the reduction of Cx3cr1
expression in this monocyte subset in Trem-1-deficient animals suggested an important role forTREM-1 in the regulation of Cx3cr1 expression underconditions of high-fat feeding. However, downstreamsignaling pathways remain unknown. Anotherpotentially important consequence of Cx3cr1 modu-lation by TREM-1 could be reduced interaction ofTREM-1-deficient macrophages with intimal smoothmuscle cells that express high levels of the Cxc3cr1ligand Cx3cl1. Collectively, our data were consistentwith the role of Cx3cl1/Cxc3cr1 signaling in experi-mental atherosclerosis (39–42).
Whether differentiating from Gr1lo or Gr1hi mono-cytes, most macrophages upregulate TREM-1 ex-pression upon activation. Our study extendedprevious findings by highlighting the amplifier effectof TREM-1 engagement on the inflammatory responseof macrophages in the context of atherosclerosis.Genetic invalidation of Trem-1 in BMDMs modulatedthe systemic innate inflammatory response as

Joffre et al. J A C C V O L . 6 8 , N O . 2 5 , 2 0 1 6
TREM-1 Inhibition Reduces Atherosclerosis D E C E M B E R 2 7 , 2 0 1 6 : 2 7 7 6 – 9 3
2790
illustrated by the reduction of TNF-a-to-IL-10 and IL-12p70-to-IL-10 ratios in the spleens of Ldlr�/�/Trem-1�/� mice. Trem-1�/� macrophages displayed a moreanti-inflammatory phenotype with less production ofproatherogenic IL-12, IL-1b, and TNF-a (3,8) andincreased production of anti-atherogenic IL-10(43,44). A similar anti-inflammatory phenotype wasobserved after pharmacological inhibition of TREM-1in both human monocytes and mouse macrophages.The regulation of IL-1b and TNF-a production byTREM-1 has previously been reported in vitro usingLPS-stimulated (human and animal) neutrophils,LPS-stimulated monocytes (45), and cultured foamcells (46). Similar regulation of innate immune re-sponses by TREM-1 was described in other asepticcontexts, such as during experimental colitis (47,48)or, more recently by our group, during AMI (23).Furthermore, in a model of ureteral obstruction-induced nephritis, Trem-1 deficiency reduced M1macrophage polarization within the kidney, as illus-trated by a decrease of Nos2 and an increase of Arg1expression (49). We observed no major differencesbetween those 2 markers in the aortas of Ldlr�/�/Trem-1þ/þ and Ldlr�/�/Trem-1�/� mice. The discrep-ancy could be related to the disease setting (differenttypes of sterile inflammation, acute versus chroniccontext) or to differences in local environmentalcues, which are known to affect macrophage pheno-type (50).
Necrotic core size is another major predictor ofplaque vulnerability (51). The formation and pro-gression of the necrotic core is influenced by a varietyof proatherogenic processes, including lipid accu-mulation, inflammatory cell activation, and the ratesof apoptotic cell death and efferocytosis within thelesions. The TREM-1/DAP12 signaling pathwayinvolves extracellular signal-regulated kinase andPI3K/AKT, 2 cascades regulating cell survival (52,53).However, the effect of Trem-1 stimulation on mono-cyte/macrophage survival is not well defined. So far,available studies have reported contradictory resultssuggesting that the role of TREM-1 on cell survivaldepends on the cell type and the experimental con-ditions. For example, Radsak et al. (54) found thatTREM-1 engagement on neutrophils had no effect onapoptosis susceptibility, but promoted apoptosis inthe presence of TLR ligands. In contrast, Cai et al. (55)observed that TREM-1 overexpression on humanmonocytes promoted cell survival in the presence ofstaurosporine. In our study, we used 3 differentapoptotic challenges and observed a slight reductionof apoptosis in Trem-1�/� BMDM only after TNF-a/cycloheximide stimulation. In vivo, we observed areduction of TUNELþ area in atherosclerotic plaques
of Ldlr�/�/Trem-1�/� chimeric mice, ApoE�/�Trem-1�/�
mice, and LR12-treated ApoE�/�mice compared totheir respective controls, consistent with the reducedsusceptibility of Trem-1�/� macrophages to apoptosis.However, the reduction of plaque apoptosis andnecrotic core in the absence of TREM-1 could also besecondary to reduced macrophage accumulation andactivation.
Foam cell formation is a major determinant ofplaque development and progression. Given thedrastic reduction of the area of aortic Oil Red Ostaining in the absence of TREM-1, we investigatedthe role of TREM-1 in macrophage foam cell formationand found a marked reduction of oxLDL uptake andlipid accumulation in Trem-1�/� macrophages. Amongthe major receptors that govern lipid body formationin macrophages (i.e., Sr-a, Sr-b1, Cd36, Abca1, Abcg1),TREM-1 deletion selectively altered the expression ofCd36 both at the gene and cell surface proteinexpression levels and both in vitro and withinatherosclerotic plaque macrophages in vivo. Wefound that signaling through PI3K, which is activateddownstream of TREM-1 (33,56), was required forTREM-1-dependent upregulation of Cd36 in responseto oxLDL, consistent with the role of PI3K activationin the induction of Cd36 (57,58). The profoundreduction of atherosclerosis observed in chimericLdlr�/�/Trem-1�/� mice was independent of thegenetic background (observed under ApoE�/� andLdlr�/� background) and the high-fat diet (observedunder chow-fed ApoE�/� mice and high-fat diet-fedLdlr�/� mice). Thus, it is difficult to attribute thismarked impact on atherosclerosis entirely to thereduction of CD36 expression, given the variableimpact of CD36 on the development of atherosclerosisin mice, which depends on the ApoE/Ldlr backgroundand the presence or absence of cholesterol-enrichedhigh-fat diet (13,59–61). Previous studies haveshown that interactions between Cd36 and severalTLRs were required for optimal induction of sterileinflammation (and macrophage apoptosis) byatherogenic lipids (10,11,62). However, in vivo evi-dence for an additive or synergistic effect of com-bined blockade of Cd36 and TLR pathways is stilllacking. TREM-1 signaling is a major amplifier ofTLR-dependent responses (63), and we demon-strated here that it is also critical for both basal andoxLDL-induced Cd36 expression. Therefore, webelieve that the combined reduction of inflammatorysignaling and Cd36 expression after TREM-1 deletionpromoted a synergistic effect and was ultimatelyresponsible for the marked reduction in inflamma-tory foam cell formation and plaque development.Our data support the combinatorial concept of

PERSPECTIVES
COMPETENCY IN MEDICAL KNOWLEDGE: TREM-1 is an
important factor in the progression of atherosclerosis in both
experimental and clinical models.
TRANSLATIONAL OUTLOOK: Further studies are needed to
assess the efficacy of pharmacological inhibition of TREM-1 to
prevent complications of atherosclerosis.
J A C C V O L . 6 8 , N O . 2 5 , 2 0 1 6 Joffre et al.D E C E M B E R 2 7 , 2 0 1 6 : 2 7 7 6 – 9 3 TREM-1 Inhibition Reduces Atherosclerosis
2791
synergistic Cd36-TLR signaling and show thatblockade of a single cell surface receptor, hereTREM-1, may be equivalent to a double-hit approach,simultaneously influencing 2 major and interdepen-dent proatherogenic processes.
Despite the crucial role of the immune response inatherosclerosis development (1,3), therapeutic stra-tegies targeting innate immune pathways havemostly failed (4,64), although some continue clinicaltesting (65). We believe that a double-hit approachthrough TREM-1 blockade, as is the case with the useof the inhibitory dodecapeptide LR12, may provideextended benefit and will be worthy of clinicaltesting. The relevance of such an approach is furthersupported by our clinical findings of increased levelsof TREM-1 in atheromatous human atheroscleroticlesions and the very significant association betweenhigh circulating levels of soluble TREM-1 and majoradverse cardiovascular events in patients with AMI(23). A first clinical trial to assess the safety, tolera-bility, and pharmacokinetics of TREM-1 blockadeusing LR12 peptide in healthy volunteers is ongoing.We speculate that acute coronary disease patientsrepresent the population of choice that could benefitfrom LR12 treatment because TREM-1 inhibitionwould prevent myeloid cell recruitment to theischemic heart (23) and the atherosclerotic plaques,limiting deleterious post-ischemic cardiac remodel-ing, the acceleration of atherosclerosis after ischemicinjury (66) and the recurrence of atherothromboticevents (67).
STUDY LIMITATIONS. In this study, we found thatTREM-1 inhibition protected against atherosclerosisdevelopment through several anti-inflammatory andanti-atherogenic mechanisms. However, we did notspecifically modulate these pathways (i.e., Cx3cr1 or
Cd36) to decipher their direct roles and relativeimportance in mediating TREM-1 proatherogeniceffects.
CONCLUSIONS
Altogether, our studies identified TREM-1 as a crucialplayer in the development and complications ofatherosclerosis, both in experimental models and inhumans. This was achieved through the unique roleof TREM-1 in connecting and coordinating signalingthrough several pattern recognition receptors (e.g.,CD36 and TLR4), which led to the activation of syn-ergistic proatherogenic pathways, namely innate in-flammatory activation of macrophages and foam cellformation. Targeting TREM-1 may constitute apromising new therapeutic approach to combat car-diovascular diseases.
REPRINT REQUESTS AND CORRESPONDENCE: Dr.Hafid Ait-Oufella, INSERM U970, Paris CardiovascularResearch Center, 56 rue Leblan, Paris, France. E-mail:[email protected].
RE F E RENCE S
1. Libby P, Lichtman AH, Hansson GK. Immuneeffector mechanisms implicated in atherosclerosis:from mice to humans. Immunity 2013;38:1092–104.
2. Hansson GK. Inflammation, atherosclerosis, andcoronary artery disease. N Engl J Med 2005;352:1685–95.
3. Tedgui A, Mallat Z. Cytokines in atherosclerosis:pathogenic and regulatory pathways. Physiol Rev2006;86:515–81.
4. Moore KJ, Tabas I. Macrophages in the path-ogenesis of atherosclerosis. Cell 2011;145:341–55.
5. Weber C, Noels H. Atherosclerosis: currentpathogenesis and therapeutic options. Nat Med2011;17:1410–22.
6. Potteaux S, Ait-Oufella H, Mallat Z. Role ofsplenic monocytes in atherosclerosis. Curr OpinLipidol 2015;26:457–63.
7. Libby P. Inflammation in atherosclerosis. Nature2002;420:868–74.
8. Ait-Oufella H, Taleb S, Mallat Z, Tedgui A.Recent advances on the role of cytokines inatherosclerosis. Arterioscler Thromb Vasc Biol2011;31:969–79.
9. Robbins CS, Hilgendorf I, Weber GF, et al. Localproliferation dominates lesional macrophageaccumulation in atherosclerosis. Nat Med 2013;19:1166–72.
10. Seimon TA, Nadolski MJ, Liao X, et al.Atherogenic lipids and lipoproteins trigger CD36-TLR2-dependent apoptosis in macrophages
undergoing endoplasmic reticulum stress. CellMetab 2010;12:467–82.
11. Stewart CR, Stuart LM, Wilkinson K, et al. CD36ligands promote sterile inflammation through as-sembly of a Toll-like receptor 4 and 6 hetero-dimer. Nat Immunol 2010;11:155–61.
12. Brown PM, Kennedy DJ, Morton RE,Febbraio M. CD36/SR-B2-TLR2 dependent path-ways enhance Porphyromonas gingivalis mediatedatherosclerosis in the Ldlr KO mouse model. PLoSOne 2015;10:e0125126.
13. Moore KJ, Freeman MW. Scavenger re-ceptors in atherosclerosis: beyond lipid uptake.Arterioscler Thromb Vasc Biol 2006;26:1702–11.
14. Bouchon A, Dietrich J, Colonna M. Cuttingedge: inflammatory responses can be triggered by

Joffre et al. J A C C V O L . 6 8 , N O . 2 5 , 2 0 1 6
TREM-1 Inhibition Reduces Atherosclerosis D E C E M B E R 2 7 , 2 0 1 6 : 2 7 7 6 – 9 3
2792
TREM-1, a novel receptor expressed on neutro-phils and monocytes. J Immunol 2000;164:4991–5.
15. Bouchon A, Facchetti F, Weigand MA,Colonna M. TREM-1 amplifies inflammation and isa crucial mediator of septic shock. Nature 2001;410:1103–7.
16. Ford JW, McVicar DW. TREM and TREM-likereceptors in inflammation and disease. Curr OpinImmunol 2009;21:38–46.
17. Klesney-Tait J, Keck K, Li X, et al. Trans-epithelial migration of neutrophils into the lungrequires TREM-1. J Clin Invest 2013;123:138–49.
18. Yasuda T, Takeyama Y, Ueda T, et al. Increasedlevels of soluble triggering receptor expressed onmyeloid cells-1 in patients with acute pancreatitis.Crit Care Med 2008;36:2048–53.
19. Kuai J, Gregory B, Hill A, et al. TREM-1expression is increased in the synovium ofrheumatoid arthritis patients and induces theexpression of pro-inflammatory cytokines. Rheu-matology (Oxford) 2009;48:1352–8.
20. Derive M, Bouazza Y, Sennoun N, et al. SolubleTREM-like transcript-1 regulates leukocyte acti-vation and controls microbial sepsis. J Immunol2012;188:5585–92.
21. Derive M, Boufenzer A, Bouazza Y, et al.Effects of a TREM-like transcript 1-derived peptideduring hypodynamic septic shock in pigs. Shock2013;39:176–82.
22. Derive M, Boufenzer A, Gibot S. Attenuation ofresponses to endotoxin by the triggering receptorexpressed on myeloid cells-1 inhibitor LR12 innonhuman primate. Anesthesiology 2014;120:935–42.
23. Boufenzer A, Lemarie J, Simon T, et al. TREM-1mediates inflammatory injury and cardiac remod-eling following myocardial infarction. Circ Res2015;116:1772–82.
24. Verhoeven BA, Velema E, Schoneveld AH,et al. Athero-express: differential atheroscleroticplaque expression of mRNA and protein in relationto cardiovascular events and patient characteris-tics. Rationale and design. Eur J Epidemiol 2004;19:1127–33.
25. Hellings WE, Pasterkamp G, Vollebregt A,et al. Intraobserver and interobserver variabilityand spatial differences in histologic examination ofcarotid endarterectomy specimens. J Vasc Surg2007;46:1147–54.
26. Bariety J, Mandet C, Hill GS, Bruneval P. Pa-rietal podocytes in normal human glomeruli. J AmSoc Nephrol 2006;17:2770–80.
27. Ait-Oufella H, Herbin O, Bouaziz JD, et al.B cell depletion reduces the development ofatherosclerosis in mice. J Exp Med 2010;207:1579–87.
28. Daugherty A, Whitman SC. Quantification ofatherosclerosis in mice. Methods Mol Biol 2003;209:293–309.
29. Mallat Z, Gojova A, Brun V, et al. Induction of aregulatory T cell type 1 response reduces thedevelopment of atherosclerosis in apolipoproteinE-knockout mice. Circulation 2003;108:1232–7.
30. Potteaux S, Gautier EL, Hutchison SB, et al.Suppressed monocyte recruitment drives macro-phage removal from atherosclerotic plaques ofApoE�/� mice during disease regression. J ClinInvest 2011;121:2025–36.
31. Klesney-Tait J, Keck K, Li X, et al. Trans-epithelial migration of neutrophils into the lungrequires TREM-1. J Clin Invest 2013;123:138–49.
32. Zanzinger K, Schellack C, Nausch N,Cerwenka A. Regulation of triggering receptorexpressed on myeloid cells 1 expression on mouseinflammatory monocytes. Immunology 2009;128:185–95.
33. Dower K, Ellis DK, Saraf K, Jelinsky SA, Lin LL.Innate immune responses to TREM-1 activation:overlap, divergence, and positive and negativecross-talk with bacterial lipopolysaccharide.J Immunol 2008;180:3520–34.
34. Geissmann F, Jung S, Littman DR. Bloodmonocytes consist of two principal subsets withdistinct migratory properties. Immunity 2003;19:71–82.
35. Freigang S, Ampenberger F, Weiss A, et al.Fatty acid-induced mitochondrial uncouplingelicits inflammasome-independent IL-1alpha andsterile vascular inflammation in atherosclerosis.Nat Immunol 2013;14:1045–53.
36. Xanthoulea S, Gijbels MJ, van der Made I, et al.P55 tumour necrosis factor receptor in bonemarrow-derived cells promotes atherosclerosisdevelopment in low-density lipoprotein receptorknock-out mice. Cardiovasc Res 2008;80:309–18.
37. Klesney-Tait J, Turnbull IR, Colonna M. TheTREM receptor family and signal integration. NatImmunol 2006;7:1266–73.
38. Drechsler M, Megens RT, van Zandvoort M,Weber C, Soehnlein O. Hyperlipidemia-triggeredneutrophilia promotes early atherosclerosis. Cir-culation 2010;122:1837–45.
39. Combadiere C, Potteaux S, Gao JL, et al.Decreased atherosclerotic lesion formation inCX3CR1/apolipoprotein E double knockout mice.Circulation 2003;107:1009–16.
40. Lesnik P, Haskell CA, Charo IF. Decreasedatherosclerosis in CX3CR1�/� mice reveals a rolefor fractalkine in atherogenesis. J Clin Invest2003;111:333–40.
41. Combadiere C, Potteaux S, Rodero M, et al.Combined inhibition of CCL2, CX3CR1 and CCR5abrogates Ly6C(hi) and Ly6C(lo) monocytosis andalmost abolishes atherosclerosis in hypercholes-terolemic mice. Circulation 2008;117:1649–57.
42. Cheng C, Tempel D, van Haperen R, et al.Shear stress-induced changes in atheroscleroticplaque composition are modulated by chemokines.J Clin Invest 2007;117:616–26.
43. Mallat Z, Besnard S, Duriez M, et al. Protectiverole of interleukin-10 in atherosclerosis. Circ Res1999;85:e17–24.
44. Potteaux S, Deleuze V, Merval R, et al. In vivoelectrotransfer of interleukin-10 cDNA preventsendothelial upregulation of activated NF-kappaBand adhesion molecules following an atherogenicdiet. Eur Cytokine Netw 2006;17:13–8.
45. Gibot S, Kolopp-Sarda MN, Bene MC, et al.A soluble form of the triggering receptor
expressed on myeloid cells-1 modulates the in-flammatory response in murine sepsis. J Exp Med2004;200:1419–26.
46. Wang YS, Li XJ, Zhao WO. TREM-1 is a positiveregulator of TNF-alpha and IL-8 production inU937 foam cells. Bosn J Basic Med Sci 2012;12:94–101.
47. Schenk M, Bouchon A, Seibold F, Mueller C.TREM-1-expressing intestinal macrophagescrucially amplify chronic inflammation in experi-mental colitis and inflammatory bowel diseases.J Clin Invest 2007;117:3097–106.
48. Weber B, Schuster S, Zysset D, et al. TREM-1deficiency can attenuate disease severity withoutaffecting pathogen clearance. PLoS Pathog 2014;10:e1003900.
49. Lo TH, Tseng KY, Tsao WS, et al. TREM-1regulates macrophage polarization in ureteralobstruction. Kidney Int 2014;86:1174–86.
50. Gosselin D, Link VM, Romanoski CE, et al.Environment drives selection and function of en-hancers controlling tissue-specific macrophageidentities. Cell 2014;159:1327–40.
51. Virmani R, Burke AP, Farb A, Kolodgie FD.Pathology of the vulnerable plaque. J Am CollCardiol 2006;47:C13–8.
52. Colonna M. TREMs in the immune system andbeyond. Nat Rev Immunol 2003;3:445–53.
53. Tessarz AS, Cerwenka A. The TREM-1/DAP12pathway. Immunol Lett 2008;116:111–6.
54. Radsak MP, Salih HR, Rammensee HG,Schild H. Triggering receptor expressed onmyeloid cells-1 in neutrophil inflammatory re-sponses: differential regulation of activation andsurvival. J Immunol 2004;172:4956–63.
55. Cai M, Chen Q, Chen C, et al. Activation oftriggering receptor expressed on myeloid cells-1protects monocyte from apoptosis through regu-lation of myeloid cell leukemia-1. Anesthesiology2013;118:1140–9.
56. Gomez-Pina V, Martinez E, Fernandez-Ruiz I,et al. Role of MMPs in orchestrating inflammatoryresponse in human monocytes via a TREM-1-PI3K-NF-kappaB pathway. J Leukoc Biol 2012;91:933–45.
57. Lin CS, Lin FY, Ho LJ, et al. PKCdelta signallingregulates SR-A and CD36 expression and foam cellformation. Cardiovasc Res 2012;95:346–55.
58. Mwaikambo BR, Yang C, Chemtob S, Hardy P.Hypoxia up-regulates CD36 expression and func-tion via hypoxia-inducible factor-1- and phospha-tidylinositol 3-kinase-dependent mechanisms.J Biol Chem 2009;284:26695–707.
59. Febbraio M, Podrez EA, Smith JD, et al. Tar-geted disruption of the class B scavenger receptorCD36 protects against atherosclerotic lesiondevelopment in mice. J Clin Invest 2000;105:1049–56.
60. Moore KJ, Kunjathoor VV, Koehn SL, et al.Loss of receptor-mediated lipid uptake via scav-enger receptor A or CD36 pathways does notameliorate atherosclerosis in hyperlipidemic mice.J Clin Invest 2005;115:2192–201.
61. Kennedy DJ, Kuchibhotla SD, Guy E, et al.Dietary cholesterol plays a role in CD36-mediated

J A C C V O L . 6 8 , N O . 2 5 , 2 0 1 6 Joffre et al.D E C E M B E R 2 7 , 2 0 1 6 : 2 7 7 6 – 9 3 TREM-1 Inhibition Reduces Atherosclerosis
2793
atherogenesis in LDLR-knockout mice. ArteriosclerThromb Vasc Biol 2009;29:1481–7.
62. Sheedy FJ, Grebe A, Rayner KJ, et al.CD36 coordinates NLRP3 inflammasome acti-vation by facilitating intracellular nucleationof soluble ligands into particulate ligands insterile inflammation. Nat Immunol 2013;14:812–20.
63. Arts RJ, Joosten LA, van der Meer JW,Netea MG. TREM-1: intracellular signalingpathways and interaction with pattern recog-nition receptors. J Leukoc Biol 2013;93:209–15.
64. Weber C, Zernecke A, Libby P. The multifac-eted contributions of leukocyte subsets toatherosclerosis: lessons from mouse models. NatRev Immunol 2008;8:802–15.
65. Ridker PM, Howard CP, Walter V, et al. Effectsof interleukin-1beta inhibition with canakinumabon hemoglobin A1c, lipids, C-reactive protein,interleukin-6, and fibrinogen: a phase IIb ran-domized, placebo-controlled trial. Circulation2012;126:2739–48.
66. Dutta P, Courties G, Wei Y, et al. Myocardialinfarction accelerates atherosclerosis. Nature2012;487:325–9.
67. Keeley EC, Velez CA, O’Neill WW, Safian RD.Long-term clinical outcome and predictors ofmajor adverse cardiac events after percuta-neous interventions on saphenous vein grafts.J Am Coll Cardiol 2001;38:659–65.
KEY WORDS apolipoprotein, foam cells,inflammation, macrophage, toll-like receptor
APPENDIX For an expanded Methods sectionas well as supplemental figures, please see theonline version of this article.